

Summary
Spleen dendritic cells (DC) are critical for initiation of adaptive immune responses against blood borne invaders. Key to DC function is their positioning at sites of pathogen entry, and their abilities to selectively capture foreign antigens and promptly engage T cells. Focusing on conventional DC-2 (cDC2), we discuss the contribution of chemoattractant receptors (EBI2 or GPR183, S1PR1, and CCR7) and integrins to cDC2 positioning and function. We give particular attention to a newly identified role in cDC2 for adhesion G-protein coupled receptor E5 (Adgre5 or CD97) and its ligand CD55, detailing how this mechanosensing system contributes to splenic cDC2 positioning and homeostasis. Additional roles of CD97 in the immune system are reviewed. The ability of cDC2 to be activated by circulating missing self-CD47 cells and to integrate multiple red blood cell (RBC)-derived inputs is discussed. Finally, we describe the process of activated cDC2 migration to engage and prime helper T cells. Throughout the review, we consider the insights into cDC function in the spleen that have emerged from imaging studies.
Keywords: Dendritic cells, Cell surface molecules, chemokines, integrins, cell trafficking, sleen
Introduction
The spleen and its DC subsets
The spleen is the largest secondary lymphoid tissue, and it acts as a filter of the blood, surveying for blood-borne pathogens (1, 2). It also has a role in clearing defective red blood cells (RBCs). It is organized into lymphocyte-rich white pulp cords, and surrounding RBC- and macrophage-rich red pulp. The white pulp cords have a T cell-rich core and surrounding B cell follicles, and in rodents they are separated from the red pulp by the marginal zone (MZ), an area densely populated by specialized macrophages and MZ B cells. The spleen is unusual in having an open blood circulation, where terminal arterioles release blood into the MZ, or directly into the red pulp (3, 4). Blood then flows from these regions into red pulp sinuses to return to circulation (1). White pulp cords have a region where the T cells connect to the red pulp, and this is known as the MZ bridging channel (BC). Building from earlier spleen slice studies (5), recent intravital microscopy showed that after their release in the red pulp T cells undergo directional migration on perivascular tracks through the BC to the T zone (6).
Conventional dendritic cells (cDC) are stationed in most tissues of the body where they continually sample their microenvironment for antigens (7–9). The spleen contains two main classes of cDC, cDC1 and cDC2, that have propensities to support CD8 and CD4 T cell responses, respectively (7–9). By promoting CD4 T cell activation and T follicular helper (Tfh) cell generation, cDC2 are important for B cell responses. At homeostasis, cDC2 in the spleen are most enriched in the BC (7, 10) (Fig. 1). Some cDC2 are also present within the MZ, red pulp and T zone. Conventional DC1 are found in the T zone, MZ and red pulp (2).
Figure 1. Section of mouse spleen showing DCIR2 distribution and labeling of blood exposed cells.
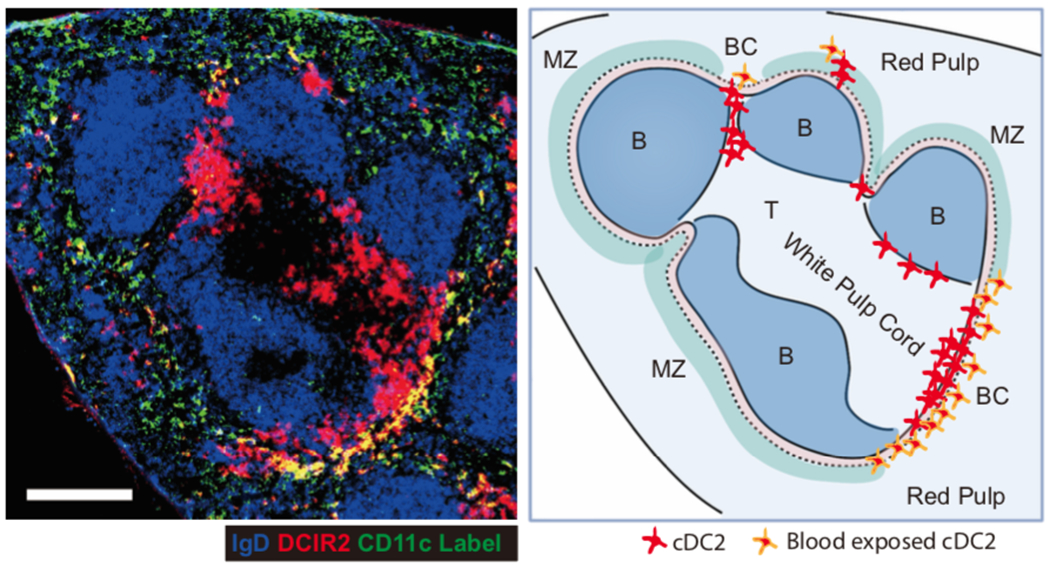
Left panel: Spleen section from a mouse that had been i.v. injected with anti-CD11c-PE (green) 3 minutes before organ isolation, stained to detect IgD (blue) and DCIR2 (red). Right panel: Sketch of the main white pulp cord in the spleen image, with principal zones labeled, distribution of cDC2 shown and cDC2 that are blood exposed highlighted.
The cDC2s that are positioned in the outer regions of the BC are rapidly labeled by intravenously injected antibodies and are readily exposed to circulating cells, including RBCs, and to blood-borne antigens (10–15) (Fig. 1). Following exposure to stimuli that provoke DC activation such as TLR ligands or particulate antigens, cDC rapidly undergo an activation program that includes upregulation of CCR7, MHCII, and CD86. In the spleen, upregulation of CCR7 promotes cDC2 movement into the T zone where they can initiate T cell responses (7–9).
Several surface markers are used to identify splenic cDC2. SIRPα (CD172α) is broadly expressed by cDC2 in the spleen and in other tissues (7–9). In accord with their original designation as myeloid DC, the majority of cDC2 are CD11b+. Heterogeneity in the compartment emerges when the CD4 marker is used, with about a third of splenic cDC2 lacking CD4. The significance of splenic cDC2 heterogeneity is still being determined, as detailed further below. The lectin DCIR2 (Clec4A4) is enriched on the majority of splenic cDC2 (16, 17). Because few other cell types in the spleen express DCIR2 it enables assessment of cDC2 distribution in sections without a requirement for multicolor DC staining. The precise function of DCIR2 is unclear, though cDC2 themselves display oligosaccharides that can bind its lectin domain (18). DCIR2 harbors an immunoreceptor tyrosine-based inhibitory motif (ITIM) in its cytoplasmic domain and DCIR2 knockout (KO) DC show deregulated cytokine responses to TLR ligands and augmented T cell responses to antigen (18). Artificially targeting antigen to DCIR2 using an antibody-antigen conjugate leads to efficient antigen acquisition by B cells and strong extrafollicular T-dependent B cell responses (19).
Flt3l, Lymphotoxin, Notch, and cDC2 heterogeneity
Precursors of splenic cDC travel via the blood from the bone marrow (BM) as pre-DC1 and pre-DC2 and undergo maturation in the spleen (7–9). Transcription factors established to have roles in cDC2 development include IRF4, ZEB2 and KLF4 while cDC1 depend on ID2, BATF3, NFIL3 and IRF8 (7, 8, 20). The cytokine Flt3l is required for the normal development and homeostasis of cDC (9). Sources of Flt3l supporting splenic cDC include both hematopoietic cells, in particular T cells, and non-hematopoietic cells (21, 22) (Fig. 2). The broad expression of Flt3l suggests that access to this cytokine may not be limited to a specific niche, though the differential functions of membrane versus secreted Flt3l are incompletely understood (23). PI3K signaling downstream of Flt3, the Flt3l receptor, is important for maintenance of splenic cDC2 numbers (24). The cDC compartment is a major consumer of peripheral Flt3l and when cDC are depleted, Flt3l availability increases leading to compensatory increases in spleen cDC proliferation (25, 26).
Figure 2. Expression of Flt3l, Dll1, and oxysterol synthesizing enzymes in spleen stromal cells.
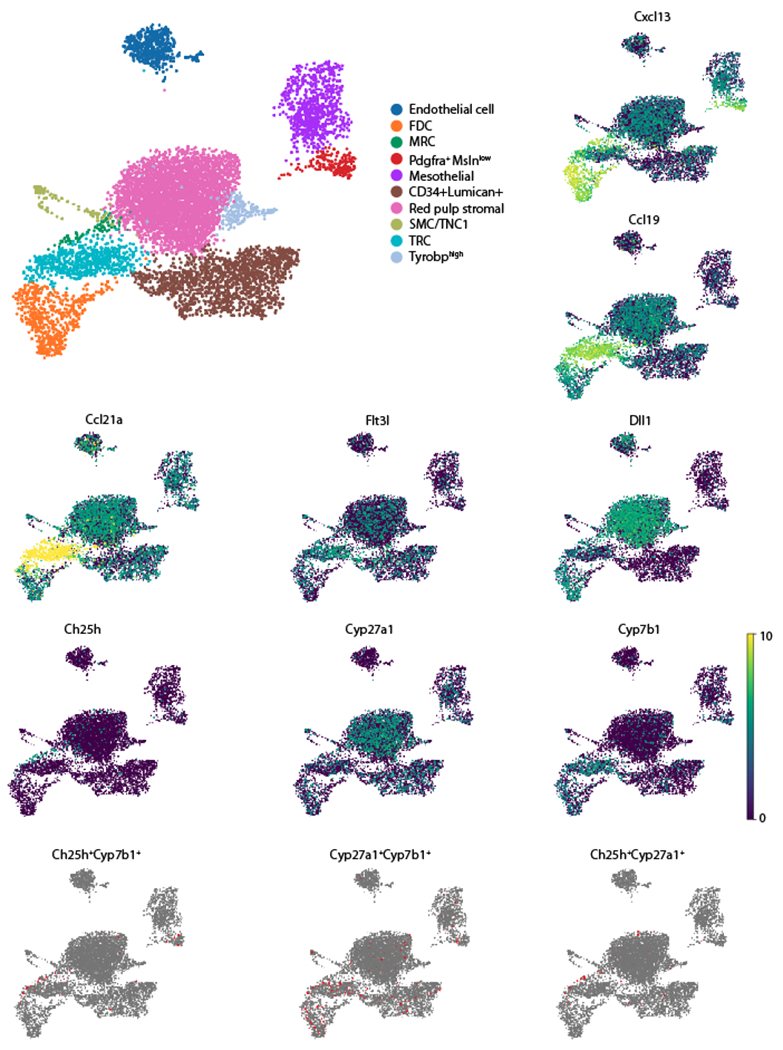
Analysis of mouse splenic fibroblastic stromal cell scRNA-seq data from (38). Umap plots generated using Scanpy (50). Upper left plot shows cluster assignments according to the marker genes expressed by each cluster. The remaining Umap plots show the expression of the indicated individual genes (expression level shown as log normalized count) or co-expression of the two indicated genes. Assignment of co-expression required that each cell has at least 1 UMI count of each gene. FDC, follicular dendritic cells; MRC, marginal reticular cells (likely includes marginal sinus lining cells); SMC, smooth muscle cells (or TNC1, triple negative cells-1 (38)); Mesothelial cells are from the spleen capsule and CD34+Lumican+ cells are thought to be predominantly subcapsular fibroblasts but this cluster may also include perivascular reticular cells (PRC) (38); remaining clusters are labeled by marker genes.
Splenic cDC homeostasis depends on LTα1β2 engagement of DC LTβR and triggering of NFκB activation. The required sources of LTα1β2 include B cells and ILC3 (27, 28). B cells are abundant in the spleen and cDC2 in BC and MZ are situated adjacent to B cells (Fig. 1). ILC3 are rare but imaging showed that splenic ILC3 often colocalize with cDC2 (28). The role of LTα1β2 appears to be multifaceted, helping establish a DC supportive niche and providing a cDC intrinsic signal that is most critical for the cDC2 compartment. LTα1β2 is also important for cDC homeostasis in lymph nodes (LN) and gut (27, 29). RelB deficiency leads to a strong reduction in splenic cDC2 and this NFκB family member is likely to be functioning at least in part downstream of LTβR, though RelB deficiency has a more selective effect on spleen cDC2 than LTβR deficiency (30, 31). A reporter study revealed constitutive RelB activity in splenic cDC2 (32). The key LTβR and RelB regulated genes that are needed for cDC homeostasis remain to be elucidated.
Notch2 is intrinsically required for cDC2 maturation into a state distinguished by expression of the surface marker Esam (33–35). These cells strongly express a range of Notch target genes including Dtx1. The key required Notch ligand, DLL1, is present on stromal cells in both white pulp and red pulp (36, 37). The exact location of the required DLL1+ stromal cells is not defined but thought to be in the marginal sinus and B cell areas, though expression by CCL19+ cells in BC is not excluded (37). Indeed, spleen stromal cell scRNA-seq data (38) indicates DLL1 is expressed by T zone reticular cells (TRC) as well as by marginal reticular cells (MRC), follicular dendritic cells (FDC), red pulp stromal cells and endothelial cells (Fig. 2). Notch activation depends on metalloproteases and ADAM10 is critical for processing Notch while also playing additional still to be defined roles in cDC2 (35, 39, 40). One study suggested this may include processing of Flt3l (39) but this was not supported by another study (35). The kinase TAOK3 is required for Notch2 function in cDC2, possibly acting to augment ADAM10 expression or activity (40).
Recent single cell genomic analysis has more clearly resolved splenic cDC2 into two subsets: cDC2A are marked by Tbet and enriched for Esam and Dtx1, while cDC2B have measurable RORγt and are marked by Clec12A and Mgl2 (CD301b) (41). Differences in TLR, chemokine and cytokine transcript levels between the subsets were also observed. Mgl2 staining of spleen sections showed Mgl2− cDC2A are localized mostly in BC while Mgl2+ cDC2B are more enriched in the red pulp (42). Further analysis is needed to definitively establish if cDC2A and cDC2B have distinct tropism within the spleen, although the suggestion that these states arise due to environmental signals (41), perhaps including spatially restricted DLL1 signals, would be consistent with occupancy of different niches. In accord with cDC2B being Notch independent, in the absence of ADAM10 there was an accumulation of Clec12A+ cDC2B (35). Functionally, cDC2A and cDC2B promote similar extents of CD4 T cell proliferation in vitro, but cDC2A had a reduced ability to polarize T cells toward an IFNγ and IL17A effector state (41). Instead, their higher expression of amphiregulin (Areg) and MMP9 (a potential TGFβ activator) was taken to suggest a role in tissue repair. Discerning how these differing properties influence the types of immune responses in the spleen, for example the ability to support Tfh cell generation and T-dependent antibody responses, requires tools to ablate or modulate the function of one or other cDC2 subset.
Positioning the sentinels
EBI2 and its oxysterol ligands position cDC2 in bridging channels
Enrichment of CD11c+ DC in MZ BC was perhaps first observed by Steinman and coworkers (43). They referred to these DC-rich structures as ‘doors’ since they (rightly) inferred T cells pass through them en route to the T zone (or periarteriolar lymphoid sheath). They also noted a lack of the cDC1 marker DEC205 in this area. Positioning of cDC2 in BC is dependent on the chemoattractant receptor EBI2 (GPR183) (10, 44). This Gi-coupled receptor is highly expressed on cDC2 and present in lower amounts on cDC1, and splenic cDC2 show a vigorous migratory response to EBI2 ligands in vitro (10, 44). When mice lack EBI2 or when it is deleted in CD11c+ cells, there is a significant reduction in splenic cDC2, and they are almost absent from BC. DC from mice lacking ADAM10 have reduced EBI2 function in vitro, an unexplained defect that may contribute to their cDC2 deficiency (35). EBI2 responds to the oxysterol ligand 7α,25-dihydroxycholesterol (7α,25-HC) with nanomolar sensitivity. 7α,25-HC is synthesized from cholesterol by the action of two enzymes, Ch25h and Cyp7b1 (45). Both enzymes are needed for splenic cDC2 homeostasis and positioning in BC (10). A second oxysterol, 7α,27-HC, also contributes to splenic cDC2 positioning and homeostasis (12, 46). EBI2 supports splenic cDC2 homeostasis at least in part by promoting their interaction with cells expressing LTα1β2 (10, 27, 47) though additional pathways are likely involved as EBI2 deficiency and LTβR deficiency do not cause fully overlapping changes in cDC2 gene expression (unpubl. obs.).
By in situ hybridization, Ch25h is expressed by marginal-sinus lining cells as well as by cells in BC and at the follicle-T zone interface (12). Cyp27a1 is expressed more broadly by cells in the T zone but with evidence of concentrated expression in the BC. Cyb7b1 is expressed by a scattering of cells in the white pulp, including cells associated with BC. BM chimera experiments showed that each of these enzymes were required in radiation resistant cells to support cDC2 homeostasis (12). The nature of the BC stromal cells that express Ch25h, Cyp27a1 or Cyp7b1 are not yet well understood. Analysis of spleen stromal cell scRNAseq data (38) confirmed strong expression of Ch25h by a cluster with features of MRC, while Cyp27a1 was broadly expressed and Cyp7b1 showed stronger expression in the TRC and FDC clusters (Fig. 2). Cells co-expressing Ch25h and Cyp7b1, perhaps the most potent EBI2 ligand producers, were enriched in the MRC cluster (Fig. 2). Cyp27a1 Cyp7b1 co-expressing cells were present in this cluster but also in the FDC and TRC clusters (Fig. 2). We suggest that BC stromal cells are encompassed within one or more of these clusters and include enzyme co-expressing cells.
So far, the contribution of 7α,27-HC to cDC2 positioning is the only well-defined in vivo example of this oxysterol acting as a ligand for EBI2. It remains of interest to understand whether 7α,25-HC and 7α,27-HC operate in a fully overlapping (additive) manner, or whether they act in adjacent regions and thus in a complimentary manner. Moreover, it remains to be determined whether EBI2 ligand concentrations are higher in BC than in adjacent zones, or whether it is a composite of signals via EBI2 and other receptors that enable cDC2 to navigate to and reside within BC. Matrix-assisted laser desorption ionization (MALDI)-mass spectrometry imaging approaches are improving in sensitivity and have recently been used to determine metabolite distribution in tissues (48, 49). Further advances in such technology will be needed to define the in situ distribution of oxysterols.
S1P’s indirect influences on splenic DC distribution
The actions of S1P on spleen cDC have been difficult to discern. Gene expression data indicates minimal expression of S1PR1 by spleen cDC1 or cDC2 (Immgen.org and (13)). Treatment with the S1PR1 modulating drug FTY720 causes displacement of cDC, predominantly cDC2, from BC into the MZ (51). However, in accord with the minimal S1PR1 transcripts in spleen cDC2, this action is indirect and appears to be a consequence of displacement of MZ B cells from the MZ (13). FTY720 treatment and genetic studies established S1PR1 is required intrinsically in B cells for positioning in the MZ. When S1PR1 function is lost, MZ B cells become lodged within the follicle in a CXCR5 dependent manner (52, 53). Real-time spleen imaging revealed that MZ B cells continually shuttle between MZ and follicle (14). This shuttling occurs because the blood bathed MZ contains high amounts of S1P, and S1PR1 undergoes rapid ligand-induced desensitization in this zone; after moving into the follicle due to CXCR5 function, the B cells become shielded from S1P and can recover S1PR1 expression and return to the MZ (54). In experiments where MZ B cells were lacking from the MZ due to B cell intrinsic S1PR1 deficiency or other perturbations, there was a redistribution of cDC2 into the MZ (13). In contrast, S1PR1 deficiency in CD11cCre-expressing cells did not alter cDC2 distribution in the spleen (13). Further studies are needed to determine how MZ occupancy by B cells restrains cDC2 access to this compartment. Cannabinoid receptor-2 (CNR2) contributes to MZ B cell retention in the MZ (55) but this receptor was not required for cDC2 access to this zone (13). The high expression of Ch25h by MRC suggested EBI2 may be involved in cDC2 accessing the MZ but it also was not required (13).
The mouse MZ contains a dense compartment of MZ macrophages (MZM) many of which are SIGN-R1+ and CD169+ (1). The relationship between MZM and splenic cDC is complex. One study found that ablation of macrophages from the MZ using either clodronate liposomes or diphtheria toxin (DT) treatment of CD169-DTR mice did not alter splenic cDC2 distribution (13). However, in another study involving clodronate liposome-mediated depletion of MZM, cDC2 occupied the MZ (56). The basis for this discrepancy is unclear. Since the studies were performed in different institutions, the microbiome of the mice was most likely different, and this might have been associated with altered circulating microbiome-derived metabolites that could influence properties of MZ-associated cells.
In neonatal mice there are fewer MZ B cells and cDC2 preferentially occupy the MZ (13). This ‘niche opportunism’ might allow compensation for the lack of MZ B cells during responses to systemic pathogens, or it might have tolerogenic or immunoregulatory roles. Indeed, while displacement of cDC2 into the MZ of adult mice was correlated with augmented early CD4 T cell responses in one study (13), it was correlated with a defective ability to generate a GC-supporting Tfh cell response in another study (56). In adult mice, the role of cDC in the MZ maybe better defined for cDC1. MZ cDC1 functionally interact with CD169hi marginal metallophillic macrophages (MMM) during bacterial infection (57–59) and with NK cells during viral infection (60). CD169hi MMM also facilitate cDC1 capture of circulating apoptotic cells in a process that contributes to maintenance of self-tolerance (61, 62)
In a remarkable recent finding, during influenza infection a subset of cDC emigrated from lung mediastinal LN and trafficked to the spleen to prime splenic CD8 T cell responses (63). These cells may correspond to a Ccr7hi migratory cDC subset detected in the spleen by scRNA-seq analysis (41). FTY720 treatment strongly blocked the migratory cDC trafficking from mediastinal LN to spleen. Migratory cDC in the mediastinal LN of influenza infected mice expressed elevated S1pr1 mRNA compared to nonmigratory resident cDC making it likely that S1PR1 is acting in a cDC intrinsic manner to promote LN egress, though experiments with conditional S1PR1 KO mice will be needed for this conclusion to be definitive. DC trafficking via efferent lymph to reach blood circulation is rare and it might be speculated that this occurs in the case of the lung because of the limited number of LN draining the lung despite it being a frequent site of pathogen encounter. Including the spleen as a priming site for lung antigens achieves a marked increase in the repertoire of recirculating T and B cells available for recruitment into the response.
The spleen may also be a site of cDC homing via blood from the heart. In mouse cardiac allograft studies, heart-derived cDCs are observed within the spleen where they can contribute to the allogeneic response (64, 65). Whether heart cDC that have captured foreign antigens travel to the spleen to engage with T cells in the non-transplant setting needs investigation. In contrast to the mouse where circulating cDC frequencies are very low, in humans they are abundant (9). It seems possible that some of these cells are derived from heart or lung and are en route to the spleen.
Integrins and DC retention
Using intravascular antibody labeling approaches, approximately 40% of splenic cDC2 and 20% of cDC1 are labeled and thus inferred to be exposed to blood flow (10, 11, 13) (Fig. 1). MZ B cells show an even higher proportion of cells that are blood exposed (50-60%). Integrin-mediated adhesion is crucial for MZ B cells to be retained against the shear forces of blood flow in the MZ compartment (14). The MZ contains stromal cells that express ICAM1 and VCAM1 and both ligands contribute to MZ B cell retention (66). LTα1β2 is required for expression of ICAM1 and VCAM1, providing an explanation for the strong LTα1β2 dependence of the MZ compartment (66). Splenic cDC2 retention in blood exposed regions similarly depends on Itgb1 and Itgb2 and blocking antibody data indicate the key integrins are α4β1 and αLβ2. Talin is a cytoskeletal adaptor downstream of many integrins and splenic cDC depend on Talin for positioning in blood exposed regions (13). This contrasts with the lack of a critical requirement for Talin in LN cDC (67). Loss of integrin β1 or β2 or deficiency in Talin are all associated with increased frequencies of cDC2 in the blood, indicating that integrin-mediated adhesion is necessary to retain the cells in the spleen against the shear forces exerted by blood flow (13).
Following activation by signals from, for example, chemokine receptors, some ligand-bound integrins can reach a more stable open configuration during exposure to shear forces (68). Imaging of MZ B cells in a flow chamber system where the cells are exposed to shear forces revealed different roles for α4β1 and αLβ2 (69). Cells adhering via αLβ2-ICAM1 migrated against the direction of shear flow whereas cells adhering via α4β1-VCAM1 tended to move in the direction of flow. In vivo antibody blocking of αLβ2 showed increased movement of MZ B cells towards the red pulp and beyond the normal confines of the MZ, consistent with this integrin promoting migration against blood flow in vivo (69). It will be of interest to determine if cDC2 distribution in flow-exposed regions of the BC as well as the MZ employ these two differentially acting integrins to fine tune cell localization. Real time imaging of CD11c+ cDC in spleen slices indicated that some of the cells in the BC area are motile (57), but determining how cDC behavior is influenced by shear stress will require intravital imaging.
CD97 mechanosensing allows cDC2 to detect blood flow
Adhesion GPCR (aGPCR) are a 33-member family of GPCR that have unique features compared to the much larger rhodopsin family GPCR (70–72). They are typified by a long N-terminal extracellular domain that can contain various motifs and that divides them into 9 subfamilies. This domain is connected to the GPCR domain by a GPCR-autoproteolysis-inducing (GAIN) domain. The GAIN domain facilitates autocatalytic cleavage of the N-terminal fragment (NTF) from the GPCR domain at the GPCR proteolysis site (GPS) within the GAIN domain, and the NTF and GPCR domains remain non-covalently bound as a heterodimer (Fig. 3). Adhesion GPCR have a sequence immediately following the GPS (and before the first transmembrane domain) called the Stachel (German for ‘stinger’) peptide that is thought to function as a tethered ligand (71–73). For several aGPCR, activation is believed to occur due to force being exerted on the NTF that leads to its removal, exposing the Stachel sequence for binding to the GPCR domain (71–75).
Figure 3: CD97 pathway restrains cDC2 motility and promotes retention in the spleen.
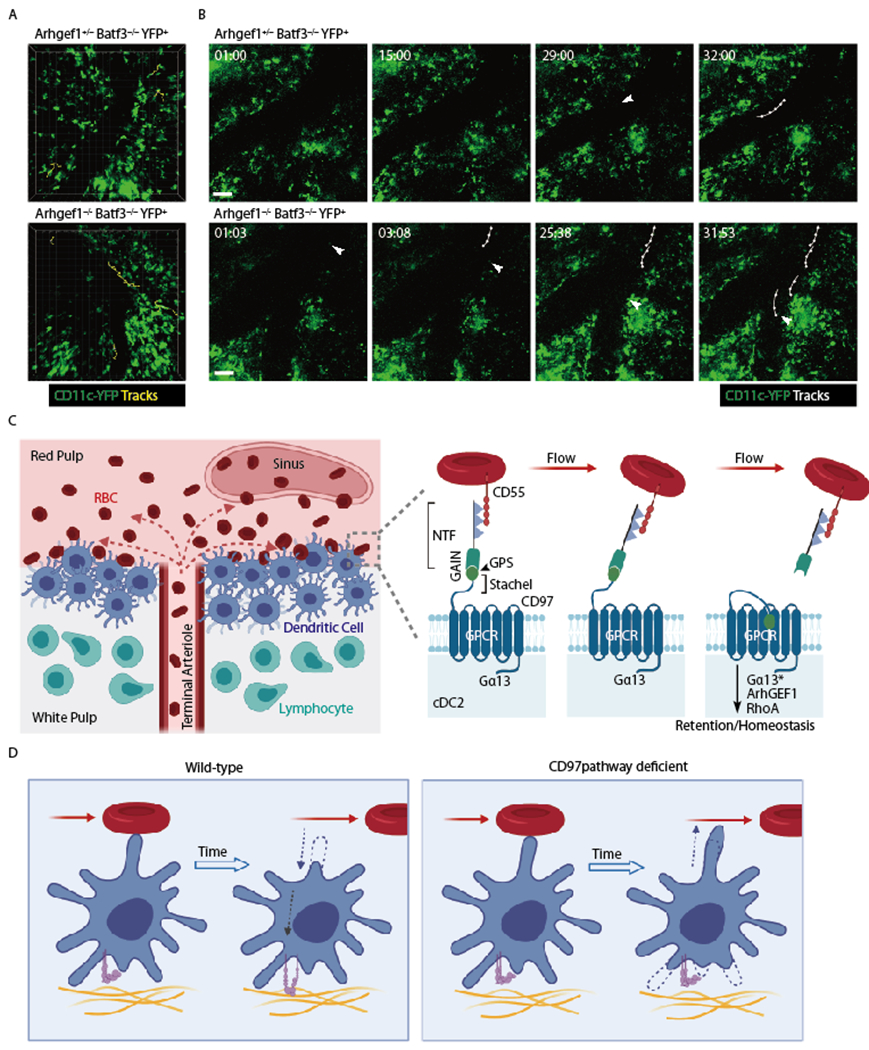
(A, B) Images from intravital two-photon microscopy of CD11c-YFP+ control (Arhgef1+/−) or Arhgef1−/− cDC in the spleen of mice that lack cDC1 (Batf3−/−). The view in A shows tracks of all YFP+ cells (cDC2) in the imaging volume that moved more than 30μm during the ~30min imaging period. The time series in B show YFP+ cells (cDC2) that enter a large sinus (arrowhead), with each such cell then being tracked over time (min:sec). Imaging was performed as in (17). (C) Left diagram shows cDC2 enriched in a bridging channel (BC) at the interface of the white pulp and red pulp, with cells facing the red pulp being exposed to RBC that are released from an open-ended terminal arteriole. Right panel shows a model of CD97 activation by engagement with CD55 on an RBC (not to scale) that is in fluid flow. The pulling force exerted by CD55 on the CD97 NTF causes its extraction and exposure of the putative tethered ligand domain (stachel sequence) and activation of the GPCR leading to Rho activation and cDC2 retention within the spleen. NTF, N-terminal fragment; Gα13*, activated form of Gα13 that has separated from Gβγ and engaged ArhGEF1. (D) Model of WT versus CD97 pathway deficient cDC2 behavior following encounter with an RBC in flow. Left: Contact with RBC triggers cDC2 to retract a membrane extension and increase integrin mediated adhesion to its tissue microenvironment. Right: In the absence of the CD55-CD97 signal, the cDC2 is shown extending into a region of blood flow.
Adhesion GPCR family member E5 (Adgre5), more commonly known as CD97, is abundantly expressed by many immune cell types including mouse and human cDC (76–79). The NTF of CD97 is alternatively spliced and in mouse includes three or four EGF domains (EGF1-2-3-4 or EGF1-2-4) or four EGF domains and a spacer sequence (EGF1-2-x-3-4) while the three human splice variants encode three to five EGF domains (EGF1-2-5, EGF1-2-3-5 and EGF1-2-3-4-5) (80)[ref]. Early studies found that CD97 transfected cells rosetted RBCs and an antibody screen showed that this occurred through RBC CD55 binding CD97 (81). Mapping revealed that CD55 binding to CD97 requires EGF domains 1 and 2. Human CD97 also binds chondroitin sulfate (CS), Thy1 and integrins α5β1 and αvβ3 (70). CS binds the 4th EGF domain of human CD97; the CS binding domain is not conserved in mouse CD97 (82). Thy1 is thought to bind the stalk region between the EGF and GAIN domains, though this has only been shown in one study for human CD97 (83). The integrin binding site is lacking in mouse CD97 (84).
Several studies have provided evidence that CD97 may function as a mechanosensitive receptor. CD55-deficient mice have elevated CD97 surface levels on lymphocytes, monocytes, and neutrophils in blood, spleen and BM (85). Moreover, when lymphocytes from these mice were transferred to WT mice, there was a rapid loss of surface CD97 that was thought to reflect mechanically induced NTF shedding upon contact with CD55+ cells under conditions of shear stress (85). This effect could be mimicked in vitro by incubating CD97+ B cells with CD55+ RBCs and subjecting the cells to vibrational shear stress (85). In accord with CD55-inducing CD97 shedding, soluble CD97 NTF is detectable in circulation (85). In vitro experiments in HEK293T or Cos-7 cell lines provided evidence that expression of CD97 truncated at the GPS to remove the NTF but have an intact Stachel sequence led to constitutive signaling via Rho (86, 87). This and other work provided evidence that CD97 could signal via Gα12/13 (these Gα proteins are highly related) (88). Despite these striking observations, it had been unclear whether CD55 engagement of CD97 triggers functionally important signals in vivo. Exposure of cells expressing WT CD97 to vibrational shear stress in the absence of ligand did not activate the Rho-inducible serum response element (SRE) reporter (87).
Our recent finding that CD97 is required for splenic cDC2 homeostasis (17) emerged from experiments asking whether there were signals that countered chemoattractive receptors to achieve more precise control of cDC distribution. Given the key role of the Gα13 containing heterotrimeric G proteins and the downstream RhoA-activating effector ArhGEF1 in promoting B cell confinement to specific compartments (89), we asked if this pathway had a role in cDC. Mice lacking ArhGEF1 or Gα13 in CD11c+ cells had a selective deficiency in splenic cDC2 but not cDC1 or in cDC at other sites. In accord with a confinement defect, ArhGEF1 and Gα13 deficient cDC2 were reduced in splenic BC and increased in circulation. Intravascular labeling showed that the cDC2 remaining in the spleen were less blood exposed, consistent with loss of the most blood exposed cells into circulation (and their rapid clearance, likely by cell death or phagocytosis). A CRISPR-based knockdown screen of 20 candidate GPCR (selected based on their expression in cDC2 and lack of well-defined function) revealed that knockdown of CD97 caused a reduction in cDC2. When this knockdown was done in cells that were ArhGEF1 deficient, there was no further cDC2 reduction, indicating that these molecules are in the same pathway. Analysis of CD97 KO mice showed a similar cDC2 deficiency to ArhGEF1 and Gα13 conditional KO (cKO) mice. Moreover, RNAseq analysis of sorted cDC2 showed strong similarity in their gene expression profiles. All three mutants also had elevated F-actin content and reduced expression of F4/80 (Adgre1) on their surface. Deficiency in CD55, the best defined and only well conserved CD97 ligand, phenocopied CD97 deficiency in its impact on the splenic cDC2 compartment (17). By contrast, treatment with a Thy1 depleting antibody did not alter cDC2 homeostasis (unpubl. obs.).
CD55 as complement regulator and CD97 ligand
CD55 is the decay accelerating factor (DAF) of complement. First purified from RBCs, it is a glycosylphosphatidylinositol (GPI) anchored protein with four extracellular short consensus repeat (SCR) domains that is widely expressed by hematopoietic and non-hematopoietic cells (90). Notably, CD55 is minimally expressed by cDC1 or cDC2 (17). CD55 protects from complement by accelerating the degradation of both the classical (C4b2b) and alternative (C3bBb) pathway C3 convertases, enzymes involved in complement activation (90). Deficiency in CD55 is a cause of the human condition complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (CHAPLE) (91). RBC CD55 also serves as a receptor for Plasmodium falciparum malaria parasites (90). Paroxysmal nocturnal hemoglobinuria (PNH) occurs due to acquired mutations in the phosphatidylinositol glycan A (PIGA) gene that is needed for generating GPI anchors. This causes a deficiency in both CD55 and CD59 (an inhibitor of the membrane attack complex of the complement system) causing RBCs to become hypersusceptible to complement mediated lysis (90).
Recent structural work has shown that the first 3 SCRs of human CD55 bind to EGF1-2-5 of CD97 via extensive contacts with EGF1 and 2 and a small area of contact with EGF5 (92). These observations help explain why the EGF1-2-5 isoform of human CD97 binds most strongly to CD55 than the EGF1-2-3-5 and EGF1-2-3-4-5 isoforms. In accord with a large interaction surface, CD97 binds to CD55 with a Kd of 3.2μM and with relatively slow on and off rates. The large interface and antiparallel binding mode are suggested to establish a geometry that resists force applied to the protein interface, consistent with an ability of CD55 to extract the CD97 NTF (92).
Several lines of evidence support the conclusion that CD55 on RBC activates CD97 signaling in cDC2 by physical extraction of the NTF. First, transfers of WT RBC into CD55 KO mice restored the cDC2 compartment in matter of days. Cell transfer and genetic experiments showed that CD55 on lymphocytes and platelets was not required for cDC2 homeostasis. Second, CD97 surface levels were elevated on cDC2 (as well as B cells) from CD55 KO mice. When splenocytes were transferred intravenously into mice that then had their inferior vena cava (IVC) tied off, transferred cells in the IVC that lacked blood flow maintained higher CD97 surface levels than cDC2 in regions with flow (17). Third, while transduction with WT CD97 could rescue cDC2 numbers in CD97 KO BM chimeras, a form of CD97 mutated in the GPS motif such that it could not undergo autoproteolytic cleavage (87, 93) was unable to rescue cDC2 numbers despite being expressed equivalently to WT (17). Similar mutations in the GPS of other aGPCR disrupt autoproteolysis and receptor function (94, 95). Fourth, incubation of cDC2 with WT but not CD55 KO RBC under conditions of shear stress led to a reduction in the CD97 NTF on the cell surface without affecting the amount of the CD97 GPCR domain (17).
While the in vivo data indicate that CD97 function in cDC2 depends on Gα13 and ArhGEF1, it has not yet been shown that RhoA is activated in cDC2 following CD55-mediated CD97 NTF extraction. However, in vitro data have shown that an NTF-deleted form of CD97 can activate RhoA in 293T cells (87) and ArhGEF1 is well defined as a GEF only for RhoA (96, 97). Consistent with CD97 acting via RhoA, when RhoA was deleted from CD11c+ cells in vivo there was a reduction in cDC (26). The DC phenotype was more severe than that caused by CD97-deficiency, likely reflecting a requirement for RhoA downstream of additional surface molecules (26). Whether the Stachel peptide in CD97 acts as a tethered ligand in cDC2 needs investigation. Again, in vitro data in a cell line system are consistent with this possibility as mutation of the Stachel sequence in a deltaNTF CD97 construct caused a loss of signaling (87). However, in our in vivo retroviral transduction studies, this mutant form of CD97 failed to restore WT levels of surface expression and thus the data were not interpretable (unpubl. obs.).
Influence of CD97 on splenic cDC2 migration dynamics
Using 2-photon microscopy to image CD11c-YFP+ cells in Batf3−/− mice that lack cDC1 (58), CD11c+ cells (presumed cDC2) could be observed in the red pulp and in regions that were likely proximal to the BC or MZ. Similar to an earlier imaging study (98), DC in these regions showed little motility though occasional cells were highly motile. The fraction of motile cells was increased in the absence of CD97 (17). More striking was the increased frequency of cDC2 detected entering large vessels and migrating or flowing within them (17) (Fig. 3). The spleen imaging data are consistent with CD97 being required to prevent cDC2 loss into blood flow. The model we favor is that when cDC2 extend membrane processes into regions with increased flow, engagement of CD97 by CD55 on passing RBC leads to mechanical removal of the NTF and activation of CD97 and Gα13 signaling and the associated activation of RhoA then causes a membrane retraction event (99–101). The role of RhoA activating receptors (including GPCR, Plexins and Eph receptors) in triggering dendrite retraction is particularly well studied in the nervous system (102–106). Higher resolution in vivo imaging studies are needed to further test the model and observe membrane retraction by WT but not CD97 KO cDC2 following encounter with passing RBC. The imaging also needs improved landmarks to enable better definition of the types of cDC2 under analysis. It will also be valuable to observe cDC2 migration with respect to the perivascular tracts that T cells use to travel from sites of release in the red pulp into the T zone (6).
Beyond promoting membrane retraction, RhoA signaling may contribute to integrin activation. While many studies have shown RhoA activation downstream of integrins, some studies also indicate that RhoA activation can promote integrin activation and adhesion through inside-out signaling (107, 108). Given the integrin dependence of cDC2 retention in the spleen (13), CD97 might act at least in part by promoting integrin- and Talin-mediated adhesion. However, mice lacking Talin in DC did not have all the phenotypic changes detected in CD97-deficient cDC2 (unpubl. obs.) indicating that CD97 cannot only function as an integrin activator.
Pertinent to the possible action of CD97 upstream of integrins are findings for ADGRG1 (GPR56). Several studies reported ADGRG1 can engage Gα12/13 to activate Rho (109, 110) and recent work showed that ADGRG1 in platelets binds to collagen and when this occurs under shear stress, signaling via Gα13 and Rho causes platelet shape change and integrin activation, events important for hemostasis and platelet plug formation (108).
CD97 functions downstream of Irf4
Irf4 is an important transcription factor for the normal accumulation and function of cDC2 (8, 11, 111, 112). Splenic cDC2 in mice lacking IRF4 selectively in CD11c+ cells share many phenotypes with CD97-pathway deficient cDC2, including reduced frequency, reduced blood exposure, increased representation in blood, increased F-actin and reduced F4/80 expression (17). Irf4 binds to the Adgre5 promoter in ChIP-seq analysis (113). Gene expression data showed that genes altered in expression in CD97-deficient cDC2s were enriched in Irf4-regulated genes (17). Moreover, rescue of CD97 expression in Irf4 cKO mice by retroviral gene transduction was able to partially restore cDC2 frequencies (17). Therefore, we propose that CD97 is an essential part of the Irf4-driven gene expression program in splenic cDC2. However, Irf4 must play additional roles in cDC2 since Irf4-deficiency leads to reductions in LN DC whereas CD97 pathway deficiency does not.
Another remaining question is why the action of the CD97 pathway is selective to cDC2 given that some cDC1 also position in the splenic MZ and red pulp and cDC1 also highly express CD97 (17). While the answer to this question is not clear, studies in mixed BM chimeras showed a role for ArhGEF1 but not Gα13 or CD97 in cDC1 homeostasis under competitive conditions (17). ArhGEF1 is a large multi-subunit protein, and it may activate RhoA (or other pathways) in cDC1 in response to distinct inputs. The RhoA dependence of cDC1 would be consistent with such an action (26).
Possible CD97 and CD55 actions at the T cell synapse
Beyond a role in promoting cDC2 retention in blood-exposed regions of the spleen, CD97 and CD55 have been implicated through in vitro studies in T cell – antigen presenting cell interaction. A recent screen in human T cells identified several GPCR, including CD97, that when knocked down led to reduced CD4 T cell activation in response to DC presenting a superantigen (114). This finding is in accord with an earlier study showing that blocking the CD55–CD97 interaction between monocytes and T cell clones reduced the T cell response (115). In a proteomic screen, a Salmonella effector proteolytically reduced CD97 and MHCII in a DC cell line and KO of CD97 reduced the ability of the DC line to stimulate TCR transgenic T cells (116). This study also showed CD97 accumulation at the immune synapse. However, the impact of selective CD97 deficiency on T cell stimulation in vivo was not examined. The degradative targeting of CD97 by Salmonella, an enteric pathogen suggests that CD97 has immune promoting functions in intestinal sites. In the other direction, several studies have shown an ability of antibodies to CD55 to augment human T cell activation and proliferation in vitro, a property they have in common with antibodies against other GPI-anchored T cell surface molecules (117, 118). In some studies, CD97-Fc also had activity. Whether this was a consequence of the plate bound protein improving the extent of T cell engagement by plate bound anti-CD3 or whether it reflected a unique CD55-mediated signal needs more study. Contrary to these findings, in vivo work showed that CD55 deficiency enhanced T cell responses to immunization as determined by in vitro restimulation, and exacerbated experimental autoimmune encephalomyelitis (EAE), effects that were due to deregulated complement activity as they were reversed by C3-deficiency (119).
Taken together, several in vitro studies suggest CD97 may have an intrinsic role in T cells in augmenting their response to antigen presenting cells. Whether CD97 NTF extraction is needed for this function has not been examined. The lack of CD55 on cDC1 and cDC2 (17) suggests that other CD97 ligands would need to be involved during T cell-DC interactions. In the case of human T cells this might be integrins given the presence of an integrin binding site in hCD97 (70). In contrast to DCs, B cells highly express CD55 and CS (17, 82). It will be of interest to compare the impact of CD97 deficiency on the ability of B cells versus DCs to engage with and activate T cells. The role of CD55 signaling intrinsically in T cells is less clear given the contradictory in vitro and in vivo findings and the lack of a CD55 intracellular domain.
It might be asked whether splenic cDC2 function is compromised in CD55-deficient humans with CHAPLE syndrome or PNH. The most striking defects in these patients can be attributed to complement hyperactivity and this conclusion is supported by the ability to overcome the most severe symptoms by treating the patients with the monoclonal antibody Eculizumab to inhibit terminal complement component C5 (90). The complex disease condition that these patients experience makes it difficult to determine whether they also suffer immune defects that arise from reduced splenic cDC2 function. Given the very different organization of the human versus mouse spleen (2), determining the role of the CD55-CD97 pathway in promoting retention of cDC2 in blood-exposed regions of the human spleen will require innovative approaches. Also complicating the mouse-human comparison is that humans but not mice have an aGPCR (ADGRE2 or EMR2) that is very closely related to CD97 though notably, it does not bind CD55 (82, 88, 92).
Further CD97 functions in the immune system and other tissues
A phenotype that has been identified in two lines of CD97-deficient mice is mild granulocytosis (76, 77). Whether this reflects an intrinsic role of CD97 in granulocytes, and by what mechanism CD97 is maintaining normal homeostasis of granulocytes has not been determined. The increased frequency of circulating granulocytes may be associated with improved bacterial defenses (76, 120) although this has not been seen in all studies (77). One study reported that CD55 KO mice had a similar increase in circulating granulocytes and a ligand-receptor interaction was suggested though the cell type that needed to express CD55 was not defined (120). Treatment with antibodies to CD97 reduced granulocyte trafficking to the inflamed peritoneum (77) but this may have been due to an indirect effect of the antibody as a BM chimera study showed that intrinsic CD97-deficiency did not alter granulocyte homing to the peritoneum (76).
CD97 is frequently upregulated in acute myeloid leukemia (AML) cells and high expression correlates with poor prognosis (121). CD97 knockdown human cells and CD97-deficient mouse leukemic cells displayed reduced growth and increased apoptosis and CD97 was suggested to act by inhibiting myeloid cell differentiation, an activity that might contribute to the mild granulocytosis in CD97-deficient mice (121). It will be of interest to understand whether CD97 functions in the BM microenvironment as a sensor of cell exposure to areas with fluid flow.
In the K/BxN serum transfer model of arthritis, disease severity was reduced in CD55 KO and CD97 KO mice (122). Since CD55 is abundant on synoviocytes it was suggested that an interaction between synoviocytes and CD97+ myeloid cells was contributing to disease (122). It might be speculated that shear stresses associated with synovial fluid movement contribute to CD55-CD97 signaling in this microenvironment.
Studies in cell lines have suggested that CD97 signaling can be more complex than selective coupling to Gα13 with evidence that the receptor may in some cases heterodimerize with lysophosphatidic acid (LPA) receptors, although signaling continued to involve Rho activation (86, 123). In one case, LPA from platelets was suggested to induce tumor cell invasiveness via CD97-LPAR signaling (124). CD97 in a colorectal cell line associated via a C-terminal PDZ motif with the cytoskeletal protein DLG1. Mechanical stress or fluid shear stress led to CD97 serine phosphorylation within the PDZ motif, and this prevented DLG1 association. The CD97 ligand requirement for these effects was not determined. Uncoupling of DLG1 from CD97 was suggested to contribute to cell dissociation from other cells and to tumor cell invasion (87). In HT1080 fibrosarcoma cells CD97 expression augmented survival in serum starvation conditions (125) and upregulated N-cadherin causing cell-cell aggregation (126). In these cells, CD97 inhibited cell migration by suppressing membrane metalloprotease activity (93). The ligand requirement for these effects was again unclear. Taken together, these studies suggest that the signaling outputs of CD97 may be context and cell type dependent.
Antigen sensing and DC activation
Spleen cDC are highly responsive to a range of pathogen associated molecular patterns (PAMPs) such as LPS and nucleic acids and to inflammatory cytokines. These stimuli cause potent activation of cDC2 and cDC1 and they have been well discussed in other reviews (9, 20). Here we focus on one pathway of antigen-sensing used uniquely by SIRPα+ cDC2.
DC surveillance for self CD47
The CD47-SIRPα system has been extensively studied for its role as a ‘don’t eat me’ signal that protects living CD47+ cells from engulfment by SIRPα+ macrophages (127, 128). CD47 is widely expressed by hematopoietic cells, including RBC. CD47 binding to SIRPα can result in Src-family kinase (SFK) mediated phosphorylation of tyrosines in the SIRPα ITIMs, leading to recruitment of the protein tyrosine phosphatases (PTPase) SHP1 and SHP2 and antagonism of engulfment. Although CD47 is also the integrin associated protein (IAP), its function in engaging SIRPα does not require its association with integrins as exemplified by the anti-engulfment activity of CD47 on RBC, cells that lack integrins (127). The first of SIRPα’s three extracellular Ig domains is highly polymorphic and often binds CD47 in a species-specific manner (129). The presence of CD47-related molecules in some viral pathogens, such as in Myxoma virus where it can reduce macrophage activation and viral clearance, is thought to have been a driver of the polymorphism (129).
SIRPα is abundantly expressed on cDC2 and minimally expressed on cDC1 (9, 20). During efforts to understand what makes sheep (and other xenogeneic) RBCs potent immunogens in mice we noted that they caused rapid activation of splenic cDC2 (15). After excluding several other mechanisms, we tested the possibility that CD47 engagement of SIRPα on cDC2 was needed to restrain cDC2 activation. Sheep CD47 was unable to engage mouse SIRPα. Removal of CD47 from syngeneic mouse RBC was sufficient to make them potent in vivo activators of cDC2. CD47-deficient lymphocytes could also provoke cDC2 activation. The activation process involved Talin, integrins and SFK (Hck, Fgr and Lyn) and led to cDC2 upregulation of CCR7 and costimulatory molecules and repositioning into the outer T zone (15). Notably, the ‘signature’ integrin of DC, CD11c-containing integrin αXβ2 (also known as CD11c/CD18 or CR4), had a non-redundant role in cDC2 uptake of missing self-CD47 cells (130). CD11b/CD18 (αMβ2, Mac1 or CR3) also contributed to the process. In vitro studies of human blood-derived DC had earlier revealed an ability of SIRPα engagement by CD47-Fc or RBC to diminish DC activation (131, 132). These findings suggest that some human cDC are also likely to respond to missing self-CD47 cells in vivo.
Using a reconstituted membrane system, the Vale group showed that SIRPα was excluded from the contact site between the phagocyte and an IgG coated particle (phagocytic synapse) unless the particle also displayed CD47 (133). When SIRPα was in the phagocytic synapse it was phosphorylated by SFK and could recruit SHP1 and/or SHP2 and antagonize integrin-activating inside-out signals from Fcγ receptors. Chemical reactivation of integrins could bypass CD47-mediated engulfment inhibition (133). Although SIRPα is quite small, having only 3 extracellular Ig domains, the molecule is heavily glycosylated, and its biophysical properties may be sufficient to favor its exclusion from a cell-cell interface unless CD47 is available to cause its accumulation (133).
A key mechanistic question in cDC2 RBC recognition that remains unanswered is what molecular interactions mediate the positive contacts between cDC2 and RBC (Fig. 4). It seems possible that the CD55-CD97 interaction may contribute and this needs to be tested although the lack of cross species conservation of CD55-CD97 binding makes this less likely (92). In addition to the requirements for forming synaptic contact, it remains unclear what target molecules are recognized by CD11c- and CD11b-containing integrins during the activation and uptake process. Several surface molecules expressed by RBCs including ICAM4, have been ruled out as playing a nonredundant role (130). It is notable that these closely related integrins share a diversity of ligands (including iC3b, ICAM1, fibronectin, and fibrinogen) and they can also bind charged polymers, with CD11c favoring negatively charged ones, thus possibly allowing promiscuous interaction with negatively charged cell membranes or glycocalyx (134–136).
Figure 4. Model for missing self-CD47 sensing by splenic cDC2.
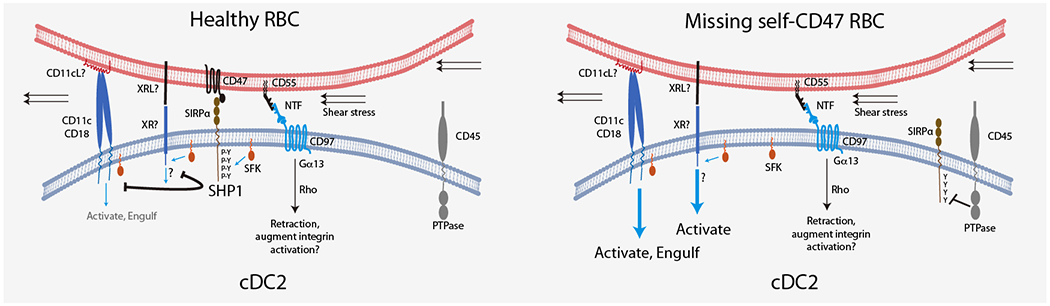
Encounter between cDC2 and healthy RBC leads to close membrane juxtaposition, CD47 binding to SIRPα and exclusion of the bulky CD45 transmembrane PTPase from the interface. SFK-mediated phosphorylation of SIRPα ITIMs allows retention of the SH2-domain containing SHP1 PTPase at the interface and antagonism of SFK-mediated signaling via other receptors, possibly including CD11c/CD18 and an unknown receptor (XR). The existence of XR is postulated because the integrin may be too bulky to locate near SIRPα (and away from CD45) at the cell-cell contact site. The ligands for CD11c/CD18 (CD11cL) and XR (XRL) are unclear, but shear stress may contribute to activation of the CD11c/CD18 integrin. Contemporaneously with CD47 engagement of SIRPα we suggest that CD55 engages CD97 leading to extraction of the CD97 NTF and activation of signaling via Rho to promote cDC2 membrane retraction. This signal may also contribute to inside-out priming of CD11c/CD18 function. In the case of cells with defective or missing self-CD47, SIRPα is not recruited to the interface and remains unphosphorylated due to CD45 PTPase activity. The lack of SHP1 activity at the interface may allow SFK to support signaling via CD11c/CD18 and XR to trigger cDC2 activation and RBC engulfment.
A notable feature of cDC2 missing self-CD47 recognition is that the introduction of CD47 KO RBC into blood circulation such that they constitute less than 1% of total RBC is sufficient to cause swift activation of the majority of splenic cDC2 (15). This indicates continual sensing of passing RBC via CD47. White blood cells are most likely similarly sensed but since RBC in blood outnumber white blood cells by 1000 to 1, the most frequent encounters of blood exposed splenic cDC2 are with RBC. We speculate that the cell-cell interactions taking place under shear stress contributes to achieving full CD11c- and CD11b-integrin activation (137).
Following activation, cDC2 turnover rapidly and continual cDC2 activation may explain the cDC2 deficiency in SIRPα and CD47 deficient mice (15, 138–140). However, the contributions of SIRPα and CD47 to maintenance of cDC2 homeostasis appear to be multifaceted. Mice lacking SIRPα or CD47 also have reduced cDC2 in LN and defects in migration of skin and lung cDC to draining LN (138–140). One study found that both hematopoietic and non-hematopoietic CD47 was needed for SIRPα-dependent maintenance of cDC2 (140). As well as restraining cDC2 activation by engaging SIRPα, CD47 actions intrinsically in the DC as an integrin associated protein may be important for cDC2 migration and function.
Recent analysis of SIRPα and CD47-deficient mice showed a reduced splenic T zone with reduced Pdpn+ fibroblastic reticular cells (FRC) and of CCL19, CCL21 and IL7, and culture of SIRPα+ DC with FRC could prolong FRC survival (141). DC expression of TNF was SIRPα dependent and TNF was involved in maintaining FRC. Interestingly, CD47 KO DC had similar defects to SIRPα KO DC suggesting that CD47 and SIRPα need to engage each other in the same cells or between cDC. The cDC2 CD47-SIRPα was needed for maintenance of FRC in a manner that was not strictly related to cDC2 number since IRF4 deficiency did not affect splenic FRC (141). This suggests that only a fraction of the cDC2 compartment is needed for FRC maintenance. It will be important to understand if this property of cDC2 contributes to development or maintenance of FRC at sites of ectopic lymphoid tissue formation, such as in tumors, especially since there are many efforts underway to antagonize CD47-SIRPα interaction as a cancer immunotherapy (128).
Sensing of RNA via RIG-I-like receptor (RLR)-mitochondrial anti-viral signaling adaptor (MAVS) contributes to cDC2 activation by xenogeneic RBC (142). For such cytosolic nucleic acid sensing to occur it is likely that there first needs to be a missing self-CD47 signal to allow uptake of the RBC. A recent study has suggested that RBC bind circulating bacterial and mitochondrial DNA via TLR9 (143). The magnitude and trajectory of DC activation following RBC uptake may well be influenced by additional properties of RBC beyond their extent of SIRPα engagement. The conditions under which cells lose CD47 function to an extent that leads to physiological activation of cDC2 remains to be established. Malaria infection of RBC can alter CD47 mobility on the cell surface and may be one such condition (144, 145). While loss of MHC class I is a major mechanism of self non-self discrimination for most cell types in the body, RBC lack (or have very low) MHC class I expression and this may give added importance to the CD47-SIRPα system for discrimination of altered self-RBC.
In addition to SIRPα it is notable that cDC2 express other ITIM containing receptors including DCIR2 (18), PirB (LILRB1) (128) and Siglec-G (Immgen.org). LILRB1 binds MHC class I and restrains macrophage phagocytic activity (128). Siglec-G binds the sialoprotein CD24 and has a role in restraining B cell responses against self (146). How these systems contribute to self non-self discrimination or healthy versus malignant-self discrimination by cDC2 needs further investigation. For example, increased sialylation commonly occurs in tumor cells and can engage ITIM-containing Siglecs to repress myeloid cell responses (147).
Migrating to the T zone to sound the alarm
CCR7, EBI2 and T zone positioning
The chemokine receptor CCR7 is rapidly upregulated on activated cDC. Deficiency in CCR7 prevents the movement of activated cDC2 and cDC1 into the splenic T zone and compromises the induction of T cell responses (11, 112). CCR7 has two ligands, CCL21 and CCL19. Mice that lack both chemokines suffer a general defect in T zone organization including a marked reduction in cDC in this region (148). The key ligand appears to be CCL21 since mice lacking CCL19 have intact cDC distribution in the spleen (unpubl. obs.). In vitro evidence has shown that CCL21 associates with reticular fibers and promotes DC haptotaxis while CCL19 diffuses and promotes chemotaxis (149). Whether some features of DC movement in the spleen are CCL19-mediated needs further investigation. In vitro data have indicated an ability of CCL19 but not CCL21 to promote dendrite extensions in splenic DC (150). It will be of interest to determine whether dendrite length or density in vivo is influenced by CCL19. CCL21 can be cleaved such that the highly basic N-terminus is removed, allowing the C-terminal chemokine domain to become more diffusive (149). How important this cleavage event is to DC movements in the spleen is not known. CCR7 in skin cDC depends on polysialylation with the sugars mediating release of CCL21 from autoinhibition by its C-terminal domain (151). Polysialylation was not required for cDC migration within the LN, suggesting that inside lymphoid tissues CCL21 autoinhibition is relieved by another mechanism, perhaps by binding to stromal cell proteoglycans.
Activated cDC2 typically show a strong bias in distribution for the follicle-T zone interface in contrast to the centralized T zone distribution of activated cDC1 (10, 11). Activated cDC2 increase their expression of EBI2 in parallel with induction of CCR7. Based on the enriched expression of Ch25h by stromal cells at the follicle-T zone interface (as well as in BC and MRC) it is inferred that EBI2 ligand abundance is higher in this region than in the central T zone and activated cDC2 are distributed into the region with the strongest combinatorial CCR7 and EBI2 signaling (10, 12). The enzyme that degrades 7α,25-HC and 7α,27-HC, Hsd3b7, is expressed by white pulp stromal cells but is also highly expressed by cDC1. EBI2 function in activated cDC2 depends on Hsd3b7 expression in both stromal cells and cDC1. When Hsd3b7 is lacking from DC, activated cDC2 fail to move to the outer T zone and instead distribute throughout the T zone, in accord with cDC1 metabolizing EBI2 ligands and thereby helping maintain the gradients needed for cDC2 positioning (10). Naïve CD4 T cells show an EBI2-dependent bias in their T zone distribution towards the follicle-T zone interface and this bias is rapidly strengthened during CD4 T cell activation due to upregulation of EBI2 (112, 152). Colocalization of cDC2 and early activated CD4 T cells at the follicle-T zone interface is thought to favor the repeated DC interactions needed for CD4 T cell activation while also positioning them for interaction with activated B cells. While less characterized, cDC2 can contribute to B cell activation by displaying or releasing antigen and producing cytokines (153).
Although cDC2 favor the follicle-T zone interface in several activation conditions, following immunization with the dsRNA mimetic polyinosinic:polycytidylic acid (polyI:C) they instead localize uniformly in the T zone (12). This altered distribution reflects polyI:C induction of type I IFN and thereby causing upregulation of Ch25h within the T zone and increased production of 7α,25-HC, likely disrupting its organizing gradient. In accord with these findings, although EBI2 was important for effective Tfh cell induction in response to missing self-CD47 cells, it was not required for induction of Tfh cells following poly I:C and missing self-CD47 cell coinjection (unpubl. obs.). A recent study on tumor-associated cDC showed that type I IFN promotes MHC class I cross-dressing of cDC2 (154). Comingling of cDC2 and cDC1 in splenic T zones after type I IFN exposure may enable cross-dressing and enhanced CD8 T cell responses.
During some viral and bacterial infections, CCL21 expression in the T zone can become strongly downregulated (155–157). Given that Ch25h is IFN-inducible and 7α,25-HC abundance generally increases during inflammation (45), EBI2 may help ensure that cDC2 and helper T cells continue to interact under conditions of reduced CCR7 ligand abundance.
Initiation of splenic Tfh cell responses
As well as positioning to efficiently present antigen to CD4 T cells, activated cDC2 have properties that can favor induction of Tfh cell responses (158). Depending on the activation stimulus, this can include strong expression of ICOSL and OX40L, costimulatory molecules that favor Tfh cell responses (112, 159, 160). Activated cDC2 strongly upregulate expression of CD25 (IL2Rα) and this may contribute to reducing the availability of IL2, a cytokine that can antagonize Tfh cell induction (112). However, the actions of IL2 during early T cell activation are complex (161) and cDC2 expression of CD25 was not linked with augmentation of Tfh cell induction in another study (34). DC expression of IL6 can also contribute to favoring Tfh cell induction under some conditions (162, 163). In addition to DC-derived factors, there is evidence that TCR affinity can sometimes be a more import determinant than cDC2 versus cDC1 properties in Tfh cell fate decision making (164). The relative contributions of cDC2A and cDC2B to the induction of Tfh cell responses requires more investigation though the finding of DC Notch2 dependence in promoting Tfh cells in response to foreign RBCs implicates cDC2A in this setting (34).
Conclusions
The DC compartment is remarkably complex, but a core requirement of cDC is an ability to position in a sentinel location in tissue and then, upon activation, to travel to a lymphoid organ T zone to present antigen and secrete cytokines. The requirements for cDC migration, positioning and activation in the spleen discussed in this review provide a conceptual framework for understanding related requirements in other tissues, especially in cases where cells are exposed to fluid flow. This review highlights the ability of cDC2 to sense RBC using both CD97 and SIRPα and to engulf altered RBC using CD11c/CD18. It seems unlikely to be a coincidence that the SIRPα+ subset of cDC that responds very rapidly to circulating missing self-CD47 cells is also the subset that depends on CD97 engagement with CD55 on RBC to be retained in the spleen. We suggest that cDC2 use the open circulation of the spleen to continually monitor the health of RBC. Since RBC are non-migratory, and passively flow through the spleen, this monitoring requires cDC2 to be situated in areas with significant blood flow. Expression of the mechanosensing aGPCR CD97 allows cDC2 to measure their exposure to blood flow and to withdraw if their movement is leading to a region with sufficient shear forces to cause their dislodgement and loss from the spleen. While only some BC cDC2 are blood exposed at any given time, it is possible that these cDC2 are motile and continually exchanging between blood-exposed and protected areas. Real time intravital imaging studies have provided valuable initial insight into cDC movements in subcapsular regions, but a full understanding of the biology awaits technical advances that permit visualization of cDC migration and interaction dynamics deep within the spleen.
This review has focused almost entirely on findings in the mouse. While understanding of human cDC has advanced greatly in recent years due to the application of scRNA-seq, more work is needed to understand human cDC function in the physiological context of the tissue. Looking through the therapeutic lens, it should be explored whether blocking CD47 or SIRPα to augment phagocytosis of cancer cells also leads to cDC2 activation, providing a possible second anti-tumor action of such blocking reagents but possibly also causing reduced cDC2 function over time. Finally, as the efforts to engineer immune cells for disease therapy become more advanced, the mechanosensing property of CD97 might offer new opportunities for controlling cell position or behavior based on sensing cells in flow.
Acknowledgements
We thank past lab members Sanjiv Luther, Kenji Kabashima, Tangsheng Yi, Erick Lu, Jagan Muppidi and Lauren Rodda for their contributions to work discussed in this review and Marco De Giovanni for comments on the manuscript. DL was supported by a CRI Irvington postdoctoral fellowship. JGC is an Investigator of the Howard Hughes Medical Institute. Work discussed in this review was supported by NIH grants AI40098 and AI45073.
Footnotes
Conflict of Interest
J.G.C. is a Scientific Advisory Board member of MiroBio and BeBio and owns stock in ALX Oncology. The other authors have no financial or personal relationships that could be viewed as a conflict of interest.
References:
- 1.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol.2005;5:606–616. [DOI] [PubMed] [Google Scholar]
- 2.Lewis SM, Williams A, Eisenbarth SC. Structure and function of the immune system in the spleen. Sci Immunol.2019;4:eaau6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto K, Kobayashi T, Murakami T. Arterial terminals in the rat spleen as demonstrated by scanning electron microscopy of vascular casts. Scan Electron Microsc.1982;Pt 1:455–458. [PubMed] [Google Scholar]
- 4.Schmidt EE, MacDonald IC, Groom AC. Comparative aspects of splenic microcirculatory pathways in mammals: the region bordering the white pulp. Scanning Microsc.1993;7:613–628. [PubMed] [Google Scholar]
- 5.Bajenoff M, Glaichenhaus N, Germain RN. Fibroblastic reticular cells guide T lymphocyte entry into and migration within the splenic T cell zone. J Immunol.2008;181:3947–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chauveau A, Pirgova G, Cheng HW, et al. Visualization of T Cell Migration in the Spleen Reveals a Network of Perivascular Pathways that Guide Entry into T Zones. Immunity.2020;52:794-807 e797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol.2019;19:89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson DA 3rd, Dutertre CA, Ginhoux F, Murphy KM. Genetic models of human and mouse dendritic cell development and function. Nat Rev Immunol.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis ESC. Dendritic Cells Revisited. Annu Rev Immunol.2021;39:131–166. [DOI] [PubMed] [Google Scholar]
- 10.Yi T, Cyster JG. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. Elife.2013;2:e00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calabro S, Liu D, Gallman A, et al. Differential Intrasplenic Migration of Dendritic Cell Subsets Tailors Adaptive Immunity. Cell Rep.2016;16:2472–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu E, Dang EV, Cyster JG. Distinct oxysterol requirements for positioning naive and activated dendritic cells in the spleen. Science Immunology.2017;2:eaal5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu D, Wu J, An J, Cyster JG. Requirements for cDC2 positioning in blood-exposed regions of the neonatal and adult spleen. J Exp Med.2020;217:e20192300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnon TI, Horton RM, Grigorova IL, Cyster JG. Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature.2013;493:684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi T, Li J, Chen H, et al. Splenic Dendritic Cells Survey Red Blood Cells for Missing Self-CD47 to Trigger Adaptive Immune Responses. Immunity.2015;43:764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudziak D, Kamphorst AO, Heidkamp GF, et al. Differential antigen processing by dendritic cell subsets in vivo. Science.2007;315:107–111. [DOI] [PubMed] [Google Scholar]
- 17.Liu D, Duan L, Rodda LB, et al. CD97 promotes spleen dendritic cell positioning and homeostasis through sensing of red blood cells. Science.2021;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uto T, Fukaya T, Takagi H, et al. Clec4A4 is a regulatory receptor for dendritic cells that impairs inflammation and T-cell immunity. Nature communications.2016;7:11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chappell CP, Draves KE, Giltiay NV, Clark EA. Extrafollicular B cell activation by marginal zone dendritic cells drives T cell-dependent antibody responses. J Exp Med.2012;209:1825–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durai V, Murphy KM. Functions of Murine Dendritic Cells. Immunity.2016;45:719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waskow C, Liu K, Darrasse-Jeze G, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol.2008;9:676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito Y, Boddupalli CS, Borsotti C, Manz MG. Dendritic cell homeostasis is maintained by nonhematopoietic and T-cell-produced Flt3-ligand in steady state and during immune responses. Eur J Immunol.2013;43:1651–1658. [DOI] [PubMed] [Google Scholar]
- 23.Miloud T, Fiegler N, Suffner J, Hammerling GJ, Garbi N. Organ-specific cellular requirements for in vivo dendritic cell generation. J Immunol.2012;188:1125–1135. [DOI] [PubMed] [Google Scholar]
- 24.Nobs SP, Schneider C, Dietrich MG, et al. PI3-Kinase-gamma Has a Distinct and Essential Role in Lung-Specific Dendritic Cell Development. Immunity.2015;43:674–689. [DOI] [PubMed] [Google Scholar]
- 25.Birnberg T, Bar-On L, Sapoznikov A, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity.2008;29:986–997. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Dislich B, Brakebusch CH, Lichtenthaler SF, Brocker T. Control of Homeostasis and Dendritic Cell Survival by the GTPase RhoA. J Immunol.2015;195:4244–4256. [DOI] [PubMed] [Google Scholar]
- 27.Kabashima K, Banks TA, Ansel KM, et al. Intrinsic Lymphotoxin-beta Receptor Requirement for Homeostasis of Lymphoid Tissue Dendritic Cells. Immunity.2005;22:439–450. [DOI] [PubMed] [Google Scholar]
- 28.Vanderkerken M, Baptista AP, De Giovanni M, et al. ILC3s control splenic cDC homeostasis via lymphotoxin signaling. J Exp Med.2021;218:e20190835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satpathy AT, Briseno CG, Lee JS, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol.2013;14:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu L, D’Amico A, Winkel KD, et al. RelB is essential for the development of myeloid-related CD8alpha- dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity.1998;9:839–847. [DOI] [PubMed] [Google Scholar]
- 31.Briseno CG, Gargaro M, Durai V, et al. Deficiency of transcription factor RelB perturbs myeloid and DC development by hematopoietic-extrinsic mechanisms. Proc Natl Acad Sci U S A.2017;114:3957–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seki T, Yamamoto M, Taguchi Y, et al. Visualization of RelB expression and activation at the single-cell level during dendritic cell maturation in Relb-Venus knock-in mice. J Biochem.2015;158:485–495. [DOI] [PubMed] [Google Scholar]
- 33.Lewis KL, Caton ML, Bogunovic M, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity.2011;35:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Briseno CG, Satpathy AT, Davidson JTt, et al. Notch2-dependent DC2s mediate splenic germinal center responses. Proc Natl Acad Sci U S A.2018;115:10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diener N, Fontaine JF, Klein M, et al. Posttranslational modifications by ADAM10 shape myeloid antigen-presenting cell homeostasis in the splenic marginal zone. Proc Natl Acad Sci U S A.2021;118:e2111234118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan JB, Xu K, Cretegny K, et al. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity.2009;30:254–263. [DOI] [PubMed] [Google Scholar]
- 37.Fasnacht N, Huang HY, Koch U, et al. Specific fibroblastic niches in secondary lymphoid organs orchestrate distinct Notch-regulated immune responses. J Exp Med.2014;211:2265–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng HW, Onder L, Novkovic M, et al. Origin and differentiation trajectories of fibroblastic reticular cells in the splenic white pulp. Nature communications.2019;10:1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujita K, Chakarov S, Kobayashi T, et al. Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen. Proc Natl Acad Sci U S A.2019;116:14714–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanderkerken M, Maes B, Vandersarren L, et al. TAO-kinase 3 governs the terminal differentiation of NOTCH2-dependent splenic conventional dendritic cells. Proc Natl Acad Sci U S A.2020;117:31331–31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown CC, Gudjonson H, Pritykin Y, et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell.2019;179:846-863 e824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanderkerken M, Baptista AP, De GIovanni M, et al. Type 3 innate lymphoid cells control splenic conventional dendritic cell homeostasis via lymphotoxin signaling. J Exp Med.2020;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinman RM, Pack M, Inaba K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol Rev.1997;156:25–37. [DOI] [PubMed] [Google Scholar]
- 44.Gatto D, Wood K, Caminschi I, et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nat Immunol.2013;14:446–453. [DOI] [PubMed] [Google Scholar]
- 45.Cyster JG, Dang EV, Reboldi A, Yi T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol.2014;14:731–743. [DOI] [PubMed] [Google Scholar]
- 46.Hannedouche S, Zhang J, Yi T, et al. Oxysterols direct immune cell migration via EBI2. Nature.2011;475:524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang YG, Kim KD, Wang J, Yu P, Fu YX. Stimulating lymphotoxin beta receptor on the dendritic cells is critical for their homeostasis and expansion. J Immunol.2005;175:6997–7002. [DOI] [PubMed] [Google Scholar]
- 48.Iwama T, Kano K, Saigusa D, et al. Development of an On-Tissue Derivatization Method for MALDI Mass Spectrometry Imaging of Bioactive Lipids Containing Phosphate Monoester Using Phos-tag. Anal Chem.2021;93:3867–3875. [DOI] [PubMed] [Google Scholar]
- 49.Zhang B, Vogelzang A, Miyajima M, et al. B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol.2018;19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Czeloth N, Schippers A, Wagner N, et al. Sphingosine-1 phosphate signaling regulates positioning of dendritic cells within the spleen. J Immunol.2007;179:5855–5863. [DOI] [PubMed] [Google Scholar]
- 52.Cinamon G, Matloubian M, Lesneski MJ, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol.2004;5:713–720. [DOI] [PubMed] [Google Scholar]
- 53.Cinamon G, Zachariah M, Lam O, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol.2008;9:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arnon TI, Xu Y, Lo C, et al. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science.2011;333:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muppidi JR, Arnon TI, Bronevetsky Y, et al. Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J Exp Med.2011;208:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pirgova G, Chauveau A, MacLean AJ, Cyster JG, Arnon TI. Marginal zone SIGN-R1(+) macrophages are essential for the maturation of germinal center B cells in the spleen. Proc Natl Acad Sci U S A.2020;117:12295–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aoshi T, Zinselmeyer BH, Konjufca V, et al. Bacterial entry to the splenic white pulp initiates antigen presentation to CD8+ T cells. Immunity.2008;29:476–486. [DOI] [PubMed] [Google Scholar]
- 58.Edelson BT, Bradstreet TR, Hildner K, et al. CD8alpha(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity.2011;35:236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu D, Yin X, Olyha SJ, et al. IL-10-Dependent Crosstalk between Murine Marginal Zone B Cells, Macrophages, and CD8alpha(+) Dendritic Cells Promotes Listeria monocytogenes Infection. Immunity.2019;51:64-76 e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghilas S, Ambrosini M, Cancel JC, et al. Natural killer cells and dendritic epidermal gammadelta T cells orchestrate type 1 conventional DC spatiotemporal repositioning toward CD8(+) T cells. iScience.2021;24:103059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyake Y, Asano K, Kaise H, et al. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest.2007;117:2268–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravishankar B, Liu H, Shinde R, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A.2012;109:3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jenkins MM, Bachus H, Botta D, et al. Lung dendritic cells migrate to the spleen to prime long-lived TCF1(hi) memory CD8(+) T cell precursors after influenza infection. Sci Immunol.2021;6:eabg6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larsen CP, Morris PJ, Austyn JM. Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J Exp Med.1990;171:307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Z, Castellaneta A, De Creus A, et al. Heart, but not skin, allografts from donors lacking Flt3 ligand exhibit markedly prolonged survival time. J Immunol.2004;172:5924–5930. [DOI] [PubMed] [Google Scholar]
- 66.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science.2002;297:409–412. [DOI] [PubMed] [Google Scholar]
- 67.Lammermann T, Bader BL, Monkley SJ, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature.2008;453:51–55. [DOI] [PubMed] [Google Scholar]
- 68.Alon R, Ley K. Cells on the run: shear-regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr Opin Cell Biol.2008;20:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tedford K, Steiner M, Koshutin S, et al. The opposing forces of shear flow and sphingosine-1-phosphate control marginal zone B cell shuttling. Nature communications.2017;8:2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langenhan T, Aust G, Hamann J. Sticky signaling--adhesion class G protein-coupled receptors take the stage. Sci Signal.2013;6:re3. [DOI] [PubMed] [Google Scholar]
- 71.Purcell RH, Hall RA. Adhesion G Protein-Coupled Receptors as Drug Targets. Annu Rev Pharmacol Toxicol.2018;58:429–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vizurraga A, Adhikari R, Yeung J, Yu M, Tall GG. Mechanisms of adhesion G protein-coupled receptor activation. J Biol Chem.2020;295:14065–14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scholz N, Monk KR, Kittel RJ, Langenhan T. Adhesion GPCRs as a Putative Class of Metabotropic Mechanosensors. Handb Exp Pharmacol.2016;234:221–247. [DOI] [PubMed] [Google Scholar]
- 74.Petersen SC, Luo R, Liebscher I, et al. The adhesion GPCR GPR126 has distinct, domain-dependent functions in Schwann cell development mediated by interaction with laminin-211. Neuron.2015;85:755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boyden SE, Desai A, Cruse G, et al. Vibratory Urticaria Associated with a Missense Variant in ADGRE2. N Engl J Med.2016;374:656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang T, Tian L, Haino M, et al. Improved antibacterial host defense and altered peripheral granulocyte homeostasis in mice lacking the adhesion class G protein receptor CD97. Infect Immun.2007;75:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Veninga H, Becker S, Hoek RM, et al. Analysis of CD97 expression and manipulation: antibody treatment but not gene targeting curtails granulocyte migration. J Immunol.2008;181:6574–6583. [DOI] [PubMed] [Google Scholar]
- 78.Heidkamp GF, Sander J, Lehmann CHK, et al. Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Sci Immunol.2016;1. [DOI] [PubMed] [Google Scholar]
- 79.Lin HH, Hsiao CC, Pabst C, et al. Adhesion GPCRs in Regulating Immune Responses and Inflammation. Adv Immunol.2017;136:163–201. [DOI] [PubMed] [Google Scholar]
- 80.Hamann J, Stortelers C, Kiss-Toth E, et al. Characterization of the CD55 (DAF)-binding site on the seven-span transmembrane receptor CD97. Eur J Immunol.1998;28:1701–1707. [DOI] [PubMed] [Google Scholar]
- 81.Hamann J, Vogel B, van Schijndel GM, van Lier RA. The seven-span transmembrane receptor CD97 has a cellular ligand (CD55, DAF). J Exp Med.1996;184:1185–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kwakkenbos MJ, Pouwels W, Matmati M, et al. Expression of the largest CD97 and EMR2 isoforms on leukocytes facilitates a specific interaction with chondroitin sulfate on B cells. J Leukoc Biol.2005;77:112–119. [DOI] [PubMed] [Google Scholar]
- 83.Wandel E, Saalbach A, Sittig D, Gebhardt C, Aust G. Thy-1 (CD90) is an interacting partner for CD97 on activated endothelial cells. J Immunol.2012;188:1442–1450. [DOI] [PubMed] [Google Scholar]
- 84.Wang T, Ward Y, Tian L, et al. CD97, an adhesion receptor on inflammatory cells, stimulates angiogenesis through binding integrin counterreceptors on endothelial cells. Blood.2005;105:2836–2844. [DOI] [PubMed] [Google Scholar]
- 85.Karpus ON, Veninga H, Hoek RM, et al. Shear stress-dependent downregulation of the adhesion-G protein-coupled receptor CD97 on circulating leukocytes upon contact with its ligand CD55. J Immunol.2013;190:3740–3748. [DOI] [PubMed] [Google Scholar]
- 86.Ward Y, Lake R, Yin JJ, et al. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res.2011;71:7301–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hilbig D, Sittig D, Hoffmann F, et al. Mechano-Dependent Phosphorylation of the PDZ-Binding Motif of CD97/ADGRE5 Modulates Cellular Detachment. Cell Rep.2018;24:1986–1995. [DOI] [PubMed] [Google Scholar]
- 88.Bhudia N, Desai S, King N, et al. G Protein-Coupling of Adhesion GPCRs ADGRE2/EMR2 and ADGRE5/CD97, and Activation of G Protein Signalling by an Anti-EMR2 Antibody. Sci Rep.2020;10:1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lu E, Cyster JG. G-protein coupled receptors and ligands that organize humoral immune responses. Immunol Rev.2019;289:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dho SH, Lim JC, Kim LK. Beyond the Role of CD55 as a Complement Component. Immune Netw.2018;18:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ozen A, Comrie WA, Ardy RC, et al. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy, and Thrombosis. N Engl J Med.2017;377:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Niu M, Xu S, Yang J, et al. Structural basis for CD97 recognition of the decay-accelerating factor CD55 suggests mechanosensitive activation of adhesion GPCRs. J Biol Chem.2021;296:100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsiao CC, Wang WC, Kuo WL, et al. CD97 inhibits cell migration in human fibrosarcoma cells by modulating TIMP-2/MT1- MMP/MMP-2 activity--role of GPS autoproteolysis and functional cooperation between the N- and C-terminal fragments. FEBS J.2014;281:4878–4891. [DOI] [PubMed] [Google Scholar]
- 94.Liebscher I, Schon J, Petersen SC, et al. A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep.2014;9:2018–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sun Y, Zhang D, Ma ML, et al. Optimization of a peptide ligand for the adhesion GPCR ADGRG2 provides a potent tool to explore receptor biology. J Biol Chem.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aittaleb M, Boguth CA, Tesmer JJ. Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol Pharmacol.2010;77:111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carter AM, Gutowski S, Sternweis PC. Regulated localization is sufficient for hormonal control of regulator of G protein signaling homology Rho guanine nucleotide exchange factors (RH-RhoGEFs). J Biol Chem.2014;289:19737–19746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Waite JC, Leiner I, Lauer P, et al. Dynamic imaging of the effector immune response to listeria infection in vivo. PLoS Pathog.2011;7:e1001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Siehler S Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol.2009;158:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takuwa N, Du W, Kaneko E, et al. Tumor-suppressive sphingosine-1-phosphate receptor-2 counteracting tumor-promoting sphingosine-1-phosphate receptor-1 and sphingosine kinase 1 - Jekyll Hidden behind Hyde. American journal of cancer research.2011;1:460–481. [PMC free article] [PubMed] [Google Scholar]
- 101.Green JA, Cyster JG. S1PR2 links germinal center confinement and growth regulation. Immunol Rev.2012;247:36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kranenburg O, Poland M, van Horck FP, et al. Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Mol Biol Cell.1999;10:1851–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu H, Marton TF, Goodman CS. Plexin B mediates axon guidance in Drosophila by simultaneously inhibiting active Rac and enhancing RhoA signaling. Neuron.2001;32:39–51. [DOI] [PubMed] [Google Scholar]
- 104.Obara Y, Ueno S, Yanagihata Y, Nakahata N. Lysophosphatidylinositol causes neurite retraction via GPR55, G13 and RhoA in PC12 cells. PLoS One.2011;6:e24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Batson J, Maccarthy-Morrogh L, Archer A, Tanton H, Nobes CD. EphA receptors regulate prostate cancer cell dissemination through Vav2-RhoA mediated cell-cell repulsion. Biol Open.2014;3:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schill Y, Bijata M, Kopach O, et al. Serotonin 5-HT4 receptor boosts functional maturation of dendritic spines via RhoA-dependent control of F-actin. Commun Biol.2020;3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Laudanna C, Campbell JJ, Butcher EC. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science.1996;271:981–983. [DOI] [PubMed] [Google Scholar]
- 108.Yeung J, Adili R, Stringham EN, et al. GPR56/ADGRG1 is a platelet collagen-responsive GPCR and hemostatic sensor of shear force. Proc Natl Acad Sci U S A.2020;117:28275–28286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iguchi T, Sakata K, Yoshizaki K, et al. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J Biol Chem.2008;283:14469–14478. [DOI] [PubMed] [Google Scholar]
- 110.Ackerman SD, Garcia C, Piao X, Gutmann DH, Monk KR. The adhesion GPCR Gpr56 regulates oligodendrocyte development via interactions with Galpha12/13 and RhoA. Nature communications.2015;6:6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Persson EK, Uronen-Hansson H, Semmrich M, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity.2013;38:958–969. [DOI] [PubMed] [Google Scholar]
- 112.Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature.2016;533:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature.2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Felce JH, Parolini L, Sezgin E, et al. Single-Molecule, Super-Resolution, and Functional Analysis of G Protein-Coupled Receptor Behavior Within the T Cell Immunological Synapse. Front Cell Dev Biol.2020;8:608484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Abbott RJ, Spendlove I, Roversi P, et al. Structural and functional characterization of a novel T cell receptor co-regulatory protein complex, CD97-CD55. J Biol Chem.2007;282:22023–22032. [DOI] [PubMed] [Google Scholar]
- 116.Cerny O, Godlee C, Tocci R, et al. CD97 stabilises the immunological synapse between dendritic cells and T cells and is targeted for degradation by the Salmonella effector SteD. PLoS Pathog.2021;17:e1009771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Capasso M, Durrant LG, Stacey M, et al. Costimulation via CD55 on human CD4+ T cells mediated by CD97. J Immunol.2006;177:1070–1077. [DOI] [PubMed] [Google Scholar]
- 118.Sutavani RV, Bradley RG, Ramage JM, et al. CD55 costimulation induces differentiation of a discrete T regulatory type 1 cell population with a stable phenotype. J Immunol.2013;191:5895–5903. [DOI] [PubMed] [Google Scholar]
- 119.Liu J, Miwa T, Hilliard B, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med.2005;201:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Veninga H, Hoek RM, de Vos AF, et al. A novel role for CD55 in granulocyte homeostasis and anti-bacterial host defense. PLoS One.2011;6:e24431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martin GH, Roy N, Chakraborty S, et al. CD97 is a critical regulator of acute myeloid leukemia stem cell function. J Exp Med.2019;216:2362–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hoek RM, de Launay D, Kop EN, et al. Deletion of either CD55 or CD97 ameliorates arthritis in mouse models. Arthritis Rheum.2010;62:1036–1042. [DOI] [PubMed] [Google Scholar]
- 123.Ward Y, Lake R, Martin PL, et al. CD97 amplifies LPA receptor signaling and promotes thyroid cancer progression in a mouse model. Oncogene.2013;32:2726–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ward Y, Lake R, Faraji F, et al. Platelets Promote Metastasis via Binding Tumor CD97 Leading to Bidirectional Signaling that Coordinates Transendothelial Migration. Cell Rep.2018;23:808–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hsiao CC, Keysselt K, Chen HY, et al. The Adhesion GPCR CD97/ADGRE5 inhibits apoptosis. Int J Biochem Cell Biol.2015;65:197–208. [DOI] [PubMed] [Google Scholar]
- 126.Hsiao CC, Chen HY, Chang GW, Lin HH. GPS autoproteolysis is required for CD97 to up-regulate the expression of N-cadherin that promotes homotypic cell-cell aggregation. FEBS Lett.2011;585:313–318. [DOI] [PubMed] [Google Scholar]
- 127.Oldenborg PA. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN hematology.2013;2013:614619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Feng M, Jiang W, Kim BYS, et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer.2019;19:568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol.2014;32:25–50. [DOI] [PubMed] [Google Scholar]
- 130.Wu J, Wu H, An J, Ballantyne CM, Cyster JG. Critical role of integrin CD11c in splenic dendritic cell capture of missing-self CD47 cells to induce adaptive immunity. Proc Natl Acad Sci U S A.2018;115:6786–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Latour S, Tanaka H, Demeure C, et al. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J Immunol.2001;167:2547–2554. [DOI] [PubMed] [Google Scholar]
- 132.Schakel K, von Kietzell M, Hansel A, et al. Human 6-sulfo LacNAc-expressing dendritic cells are principal producers of early interleukin-12 and are controlled by erythrocytes. Immunity.2006;24:767–777. [DOI] [PubMed] [Google Scholar]
- 133.Morrissey MA, Kern N, Vale RD. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity.2020;53:290-302 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vorup-Jensen T, Carman CV, Shimaoka M, et al. Exposure of acidic residues as a danger signal for recognition of fibrinogen and other macromolecules by integrin alphaXbeta2. Proc Natl Acad Sci U S A.2005;102:1614–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sadhu C, Ting HJ, Lipsky B, et al. CD11c/CD18: novel ligands and a role in delayed-type hypersensitivity. J Leukoc Biol.2007;81:1395–1403. [DOI] [PubMed] [Google Scholar]
- 136.Vorup-Jensen T, Jensen RK. Structural Immunology of Complement Receptors 3 and 4. Frontiers in Immunology.2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Astrof NS, Salas A, Shimaoka M, Chen J, Springer TA. Importance of force linkage in mechanochemistry of adhesion receptors. Biochemistry (Mosc).2006;45:15020–15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hagnerud S, Manna PP, Cella M, et al. Deficit of CD47 results in a defect of marginal zone dendritic cells, blunted immune response to particulate antigen and impairment of skin dendritic cell migration. J Immunol.2006;176:5772–5778. [DOI] [PubMed] [Google Scholar]
- 139.Van VQ, Lesage S, Bouguermouh S, et al. Expression of the self-marker CD47 on dendritic cells governs their trafficking to secondary lymphoid organs. EMBO J.2006;25:5560–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saito Y, Iwamura H, Kaneko T, et al. Regulation by SIRPalpha of dendritic cell homeostasis in lymphoid tissues. Blood.2010;116:3517–3525. [DOI] [PubMed] [Google Scholar]
- 141.Saito Y, Respatika D, Komori S, et al. SIRPalpha(+) dendritic cells regulate homeostasis of fibroblastic reticular cells via TNF receptor ligands in the adult spleen. Proc Natl Acad Sci U S A.2017;114:E10151–E10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Loetsch C, Warren J, Laskowski A, et al. Cytosolic Recognition of RNA Drives the Immune Response to Heterologous Erythrocytes. Cell Rep.2017;21:1624–1638. [DOI] [PubMed] [Google Scholar]
- 143.Lam LKM, Murphy S, Kokkinaki D, et al. DNA binding to TLR9 expressed by red blood cells promotes innate immune activation and anemia. Science translational medicine.2021;13:eabj1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sosale NG, Rouhiparkouhi T, Bradshaw AM, et al. Cell rigidity and shape override CD47’s “self”-signaling in phagocytosis by hyperactivating myosin-II. Blood.2015;125:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ayi K, Lu Z, Serghides L, et al. CD47-SIRPalpha Interactions Regulate Macrophage Uptake of Plasmodium falciparum-Infected Erythrocytes and Clearance of Malaria In Vivo. Infect Immun.2016;84:2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen GY, Brown NK, Zheng P, Liu Y. Siglec-G/10 in self-nonself discrimination of innate and adaptive immunity. Glycobiology.2014;24:800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Smith BAH, Bertozzi CR. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat Rev Drug Discov.2021;20:217–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gunn MD, Kyuwa S, Tam C, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med.1999;189:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schumann K, Lammermann T, Bruckner M, et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity.2010;32:703–713. [DOI] [PubMed] [Google Scholar]
- 150.Yanagawa Y, Onoe K. CCL19 induces rapid dendritic extension of murine dendritic cells. Blood.2002;100:1948–1956. [DOI] [PubMed] [Google Scholar]
- 151.Kiermaier E, Moussion C, Veldkamp CT, et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science.2016;351:186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Baptista AP, Gola A, Huang Y, et al. The Chemoattractant Receptor Ebi2 Drives Intranodal Naive CD4(+) T Cell Peripheralization to Promote Effective Adaptive Immunity. Immunity.2019;50:1188-1201 e1186. [DOI] [PubMed] [Google Scholar]
- 153.Heath WR, Kato Y, Steiner TM, Caminschi I. Antigen presentation by dendritic cells for B cell activation. Curr Opin Immunol.2019;58:44–52. [DOI] [PubMed] [Google Scholar]
- 154.Duong E, Fessenden TB, Lutz E, et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Benedict CA, De Trez C, Schneider K, et al. Specific remodeling of splenic architecture by cytomegalovirus. PLoS Pathog.2006;2:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mueller SN, Hosiawa-Meagher KA, Konieczny BT, et al. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science.2007;317:670–674. [DOI] [PubMed] [Google Scholar]
- 157.St John AL, Abraham SN. Salmonella disrupts lymph node architecture by TLR4-mediated suppression of homeostatic chemokines. Nat Med.2009;15:1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Krishnaswamy JK, Alsen S, Yrlid U, Eisenbarth SC, Williams A. Determination of T Follicular Helper Cell Fate by Dendritic Cells. Front Immunol.2018;9:2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shin C, Han JA, Koh H, et al. CD8alpha(−) Dendritic Cells Induce Antigen-Specific T Follicular Helper Cells Generating Efficient Humoral Immune Responses. Cell Rep.2015;11:1929–1940. [DOI] [PubMed] [Google Scholar]
- 160.Brocker T, Gulbranson-Judge A, Flynn S, et al. CD4 T cell traffic control: in vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B cell follicles. Eur J Immunol.1999;29:1610–1616. [DOI] [PubMed] [Google Scholar]
- 161.DiToro D, Winstead CJ, Pham D, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science.2018;361:eaao2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity.2009;31:491–501. [DOI] [PubMed] [Google Scholar]
- 163.De Giovanni M, Cutillo V, Giladi A, et al. Spatiotemporal regulation of type I interferon expression determines the antiviral polarization of CD4(+) T cells. Nat Immunol.2020;21:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kotov DI, Mitchell JS, Pengo T, et al. TCR Affinity Biases Th Cell Differentiation by Regulating CD25, Eef1e1, and Gbp2. J Immunol.2019;202:2535–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]