

Abstract
Emerging viral diseases can substantially threaten national and global public health. Central to our ability to successfully tackle these diseases is the need to quickly detect the causative virus and neutralize it efficiently. Here we present the rational design of DNA nanostructures to inhibit dengue virus infection. The designer DNA nanostructure (DDN) can bind to complementary epitopes on antigens dispersed across the surface of a viral particle. Since these antigens are arranged in a defined geometric pattern that is unique to each virus, the structure of the DDN is designed to mirror the spatial arrangement of antigens on the viral particle, providing very high viral binding avidity. We describe how available structural data can be used to identify unique spatial patterns of antigens on the surface of a viral particle. We then present a procedure for synthesizing DDNs using a combination of in silico design principles, self-assembly, and characterization using gel electrophoresis, atomic force microscopy and surface plasmon resonance spectroscopy. Finally, we evaluate the efficacy of a DDN in inhibiting dengue virus infection via plaque-forming assays. We expect this protocol to take 2–3 d to complete virus antigen pattern identification from existing cryogenic electron microscopy data, ~2 weeks for DDN design, synthesis, and virus binding characterization, and ~2 weeks for DDN cytotoxicity and antiviral efficacy assays.
Introduction
The challenges underlying current coronavirus disease 2019 treatment and rapid diagnostic development are already well known from previous encounters with newly emerging pathogens (e.g., the 2009 H1N1 pandemic1,2). Inhibition and treatment of virus infections typically relies on neutralizing antibodies (NAbs) that target virus surface-specific epitopes mainly in a one-to-one fashion3. Production of NAbs can be triggered by vaccination or active virus infection in the host. However, safe and effective vaccines normally take years to develop for an emerging virus. Therapeutic antibodies can be administered in response to viral infections. However, producing antibodies for treatment is very costly and time consuming. Importantly, NAbs may induce unwanted antibody-dependent enhancement of infection4,5 (for example, with dengue virus (DENV) vaccine), where antibodies induce increased viral infectivity in vivo. Viruses present unique spatial patterns of antigens on their surfaces6. Such patterns facilitate multivalent binding of the virus to host cells for enhanced pathogenic infectivity. Based on this naturally occurring multivalent virus-cell binding mechanism, creating polyvalent virus entry blockers is a promising and practical approach to producing potent inhibitors of virus infections.
Development of the DNA star antiviral platform
Our group recently designed and synthesized a ~43 nm diameter star-shaped DNA architecture, called a ‘DNA star’ to multivalently bind to viral epitopes to efficiently inhibit virus infection. We designed the antiviral DNA star to specifically target complex epitopes on the DENV envelope protein domain 3 (ED3). The five-point DNA star provides structural rigidity to display ten dengue ED3 targeting aptamers in a 2D pattern precisely mirroring the complex spatial arrangement of DENV ED3s7. Each DENV (~50 nm diameter) can bind up to two DNA stars, one on each hemisphere. The DNA star demonstrated high virus-binding avidity and specificity to DENV and was a highly potent DENV inhibitor in human blood with a half-maximal effective concentration (EC50) of 2 nM (~7,500-fold more effective than the monovalent aptamer). We anticipate that our strategy can be tailored to target any viral epitope pattern to combat emerging and re-emerging viruses by generating the requisite ligand patterns on customized designer DNA nanostructures (DDNs).
The DNA star was designed to afford local structural flexibility through a single-stranded DNA (ssDNA) region on each of the five internal edges and unpaired thymine bases (Ts) at the five internal junctions. The ssDNA regions offer limited but sufficient local structural flexibility to ensure effective binding to DENV under a range of physiological conditions and temperatures8, in which the pattern of DENV envelope proteins may change slightly. Additionally, the unpaired Ts introduced at the junctions allow bendability of the DNA star, enhancing its ability to wrap around and inhibit a virus particle. We found that more closely matching the geometry of the antigen pattern, by adding more triangles into the structure to create five- or six-point DNA stars whose vertices precisely match the interspatial pattern of ED3 clusters, results in stronger virus binding avidity. In theory, more points can be added to the current five- or six-point DNA star scaffold to increase avidity. However, larger structures assemble in a lower yield and require more DNA oligonucleotides, thus increasing cost. For a new virus target, an optimal DDN size can be determined by testing the virus binding avidity and antiviral activity of various DDN sizes. An optimal DDN construct can then be selected by collectively considering cost, assembly yield and antiviral performance of the candidate DDNs. The cost of making a DDN platform can be largely reduced if the DNA oligos are synthesized at a larger scale (>1 μmol from existing commercial sources such as IDTDNA or Sangon) or produced in a microbiome using molecular cloning9–12.
The DNA star platform is a complex tile-based DDN that consists of multiple branched DNA junctions assembled from short DNA oligonucleotides13–15. Despite growing interest in DNA origami-based biological applications16–19, to date there are no comprehensive protocols to guide researchers through the process of creating tile-based DDNs for antiviral applications. Here we describe how to design tile-based DDNs, specifically antiviral DNA stars. The process begins by identifying antigen spatial patterns on virus particles using available cryogenic electron microscopy (cryo-EM) data. A DNA star with antigen-targeting ligands is then designed to mirror this spatial pattern. The binding avidity between the DNA star and intact viral particles is investigated using surface plasmon resonance (SPR) spectroscopy, and antiviral efficacy of DNA stars is evaluated using plaque-forming assays. While sample preparation strategies for atomic force microscopy (AFM) imaging of DNA origami nanostructures16,17,20 are well established, we discuss the adjustments required to image small tile-based DDNs like the DNA stars. Furthermore, generic guidelines for employing SPR in studying individual ligand–protein interactions have been discussed in recent review articles21–24. However, these scenarios do not recapitulate the polyvalent interactions between DNA stars and intact virus particles that we outline here. In addition, plaque-forming assays are commonly used to screen small molecule-, peptide- and protein-based antiviral drugs25,26, but we have adapted the standard experimental conditions here to evaluate the potency of DNA star-based virus inhibitors. In our procedure, we provide a detailed step-by-step protocol for virus surface antigen pattern analysis, DNA star inhibitor design, synthesis, characterization and evaluation of DNA star cytotoxicity and antiviral efficacy.
Comparison with other methods
One strategy in the development of antivirals is to create materials designed to bind to a virus to prevent it from interacting with and entering host cells. Multivalent virus entry blockers have been previously constructed by linking epitope-targeting ligands to a synthetic scaffold to improve binding avidity27–29. We recently designed dendrimer conjugates and demonstrated that matching average ligand spacing with the distance of viral epitopes is a key determinant for effectively inhibiting influenza viral infection in mice27. However, synthetic scaffolds (polymers, dendrimers, nanofibers, inorganic nanoparticles, lipid nano-emulsions, etc.) exhibit substantial drawbacks as part of an antiviral ‘drug’, such as toxicity and limited control over the scaffold shape, ligand spacing and ligand valency30–33.
Nucleic acid-based scaffolds are able to overcome some limitations of synthetic scaffolds because they can be designed into biologically stable and biocompatible 2D and 3D platforms while controlling ligand spacing, valency and spatial arrangements with nanometer precision7,34–45. The DDN platform acts as an ideal template to display multiple binding motifs. For instance, a DDN platform has been used to arrange ligands with well-defined patterns to direct and regulate receptor activation, and thus invasiveness, of breast cancer cells46. A DDN platform was also used to precisely display antigens to study optimal distances for bivalent antibody binding47. Recently, virus-like DDN nanoparticles were created to display clinical vaccine immunogens to determine the impact of DDN particle size and rigidity, and immunogen spacing on B-cell receptor activation, and thus elucidate DDN-based vaccine design principles48.
In terms of stability and biocompatibility, DDNs have exhibited blood circulation lifetime of up to 24 h (ref.38) and are eventually removed by the liver38 and kidney40,49. Certain DNA sequences can drive functional immune responses and thus can be used as vaccine adjuvants50,51. Three-dimensional DNA origami structures can be designed to mimic virus-like particles and thus can exhibit relatively high immunogenicity37,52. Coating DNA origami with biocompatible ligands (i.e., PEGylated lipids37, BSA52, PEGylated oligolysines53,54) increases the DDNs’ in vivo stability by reducing nuclease degradation and/or low salt denaturation, and also can be used to attenuate the immune response activated by these structures. This molecular coating strategy can be used to tune the in vivo lifetime and immune activity of a DDN for specific biomedical applications. Please refer to recent review articles for extended discussion on DDNs’ biostability and biocompatibility55–57.
The building blocks of DDNs, DNA oligonucleotides, can be affordably produced at scale9–11,58,59. They are thermally stable for hours or days in solution7,60 and can be stored for months as a dried powder10. In response to emerging viruses and mutants, new high-affinity virus-targeting aptamer sequences can be readily and economically produced and evolved through the in vitro selection process, called systematic evolution of ligands by exponential enrichment (SELEX)61–63. DDNs and aptamers have already found applications in biomedicine and biosensing7,38,64–79. Additionally, other antigen-binding ligands such as antibodies or nanobodies can be decorated on customized DDN scaffolds to target viruses or to serve as coating molecules to enhance DDN in vivo stability and compatibility.
Application of the DNA star platform
The threat of emerging and re-emerging infectious diseases has emphasized the need to develop robust surveillance technologies and novel therapeutics. The DNA star platform presented here has been effectively used to both detect and inhibit DENV. We anticipate that our platform can be tailored and tuned to target antigens displayed on other viruses. The DNA star can be used as a multivalent sensor for diagnostic testing, or as a therapeutic candidate potentially capable of inhibiting in vivo viral entry into host cells (Fig. 1a). The DNA star is designed to interact with the intact/infectious virions with partially or fully preserved surface antigens that interact with host cell receptors. Thus, it may be possible to use a DNA star sensor to distinguish between infectious and noninfectious forms of a virus and minimize false positive test results that can be generated by nucleic acid-based tests such as PCR. Additionally, DNA structures customized to target different viruses can be designed to display distinct fluorescent readouts for multiplexed diagnostics.
Fig. 1 |. Schematic of applications of the DNA star strategy.
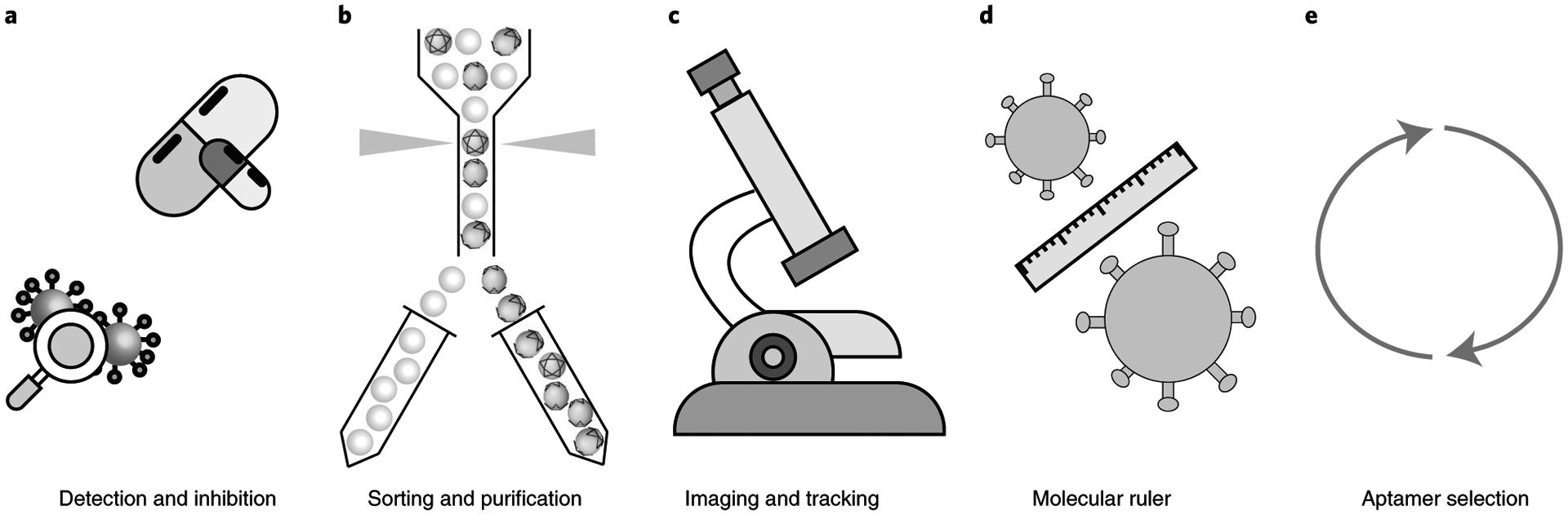
Schematic of use of DNA star strategy for a range of applications. a, Detection and inhibition of viral infections. b, Sorting and purification of viral particles for vaccination and drug delivery. c, Serving as a molecular tag for virus imaging and tracking. d, Serving as a molecular ruler for the elucidation of virus surface antigen arrangement. e, Evolution and selection of specific viral antigen binding aptamers.
Beyond the potential clinical applications in diagnostics and antiviral therapeutics, it may also be possible to use the DNA star strategy for the following five purposes. (1) Virus titer determination: a fluorophore-labeled DNA star may be used to quantify virus titers of stocks, clinical samples or environmental samples, for instance, by sorting the DNA star–virus mixture using flow virometry technique80,81, a derivative of flow cytometry. (2) Purification: DNA star may be incorporated into resins to improve purification of viral particles manufactured in cell culture (e.g., to purify virus particles that are used in gene therapy) (Fig. 1b). (3) Imaging and tracking: fluorescently labeled DNA stars may be used as a molecular tag to track how the intact/infectious virions behave in biological fluids and environments (Fig. 1c). Furthermore, the specificity provided by newly customized DDNs can be tailored to other scenarios such as single-cell imaging. The architecture of cells and distribution of cell surface receptors is typically altered as cells transition between different developmental stages and physiological states. We can use the availability of these cell surface receptors as targeting moieties to perform single-cell analysis on multiple populations of cells. (4) A molecular ruler: for an emerging virus that has one or more previously known ‘cousins’ from the same virus family/subfamily, a candidate DDN can first be designed to match the antigen spatial pattern of a cousin virus to get an approximate structure for the emerging virus before the cryo-EM data are obtained. The candidate DDN can be turned into a ‘molecular ruler’ by presenting affinity ligands at varied distances compared with the initial design to elucidate the optimal antigen pattern on the outer surface of the emerging virus (Fig. 1d). Identifying the distribution and arrangements of viral antigens may assist in vaccine and antiviral drug development. (5) Binder selection: a newly customized DDN can serve as a synergetic platform to template multiple DNA libraries for the simultaneous evolution and selection of specific viral antigen-binding aptamers in a multivalent and pattern-matching interaction with the virus particles (Fig. 1e).
Overall, we envision that this protocol can be used to develop customized DDNs that can be tailored to address a broad range of biological and virological questions. Implementation of this protocol requires a multidisciplinary team of structural biologists, chemists and virologists, and may be useful to researchers in the fields of drug design, biology, materials science, bioengineering, nanotechnology and chemistry.
Challenges of applying the ‘DNA star strategy’ to an emerging virus
Designing an antiviral DNA star is dependent on our ability to identify unique patterns of antigens displayed on the surface of a viral particle. When using our DNA star strategy to tackle an emerging virus, two key pieces of information are needed: knowledge of antigen arrangement on the outer surface of the virus (e.g., from cryo-EM data) and availability of antigen-binding ligands (e.g., antibodies, nanobodies or aptamers). Most emerging viruses have one or more previously known ‘cousins’ from the same virus family/subfamily (i.e., severe acute respiratory syndrome coronavirus and severe acute respiratory syndrome coronavirus 2). They differ in antigen sequences but have the same or similar antigen arrangement. Thus, the DDN design can start with a known antigen pattern for a similar virus. The DDN design can then be further optimized based on the confirmed patterns from cryo-EM data. It normally takes months to obtain antibodies from recovered patients or to engineer nanobodies that can specifically bind to the antigen of an emerging virus. In contrast, it is much easier and faster to derive and produce aptamers that target an emerging virus antigen and its mutants with at least submicromolar avidity. Our DNA star-based virus capture probe allows for polyvalent and spatial pattern-matching interactions, dramatically improving virus-binding avidity compared with individual monomeric aptamers7. As a result, a modest affinity aptamer can be turned into an excellent binder when presented on a multivalent DDN structure.
As described in this protocol, live DENV is used under biosafety level (BSL)-2 containment to measure DNA star antiviral efficacy. However, highly virulent and pathogenic viruses such as severe acute respiratory syndrome-associated viruses must be handled under BSL-3+ containment. Therefore, when targeting these highly virulent viruses, we suggest initially screening candidate antiviral DNA star constructs using pseudoviral assays (i.e., pseudomodels of severe acute respiratory syndrome coronavirus 2 (ref.82–84)) before working with the live virus.
Limitations of the DNA star platform
The in vitro DENV inhibition performance of the DNA star platform shows promising therapeutic potential. However, biostability of the DNA star, which is composed of unmodified DNA oligos, must be improved before moving into in vivo assays and potential future clinical use. Chemically modified component DNA oligos can be used to improve the DNA star’s in vivo nuclease resistance and biostability57. Unpaired thymine nucleotides introduced at each vertex can be UV crosslinked42 to improve the DNA star’s lifetime in vivo. Additionally, the aptamer targeting ligand can be substituted with antibodies or peptides to improve virus-binding affinity. Protein or peptide coatings on the DNA star can also improve its biostability and repress potential immunogenicity, as observed for other DNA nanostructures37,52.
When applying the DNA star platform to biosensing (which is not outlined in the Procedure here), the sensor relies on the strong interaction between the patterned aptamers on the DNA star and antigens on the surface of the virus7. For DENV, the envelope proteins on the virus surface act as steady anchors that, when bound to the DNA star, can effectively stretch the hairpin loops in the molecular beacon-like motifs on the DNA star, which can trigger the release of a quencher dye and restore a fluorescent detection signal. However, unlike DENV antigens that are immobile, spike glycoproteins on viruses such as coronaviruses, influenza and human immunodeficiency virus have flexible stems and mobile roots, which cannot serve as steady anchors to stretch the hairpin loops of DNA star sensors to report viral binding. Thus, the mechanism by which the detection signal is generated with the current DNA star strategy cannot be applied to viruses or cells whose surface proteins are flexible and mobile. For these targets, it may be possible to integrate different signal generation and detection technologies (e.g., lateral flow assay85, light scattering imaging86, aptamer switching87) with the DNA star platform to enable rapid, ultrasensitive and inexpensive disease detection.
Overview of the procedure
Here we describe how to develop DDNs for inhibition of DENV. This protocol comprises five main stages as illustrated in Fig. 2. The procedure starts with identifying unique spatial patterns of antigens on a viral particle surface using available cryo-EM data of dengue virions (Fig. 2a). After the virus antigen spatial pattern is determined, the next step is to design a DNA nanostructure mirroring this pattern using a combination of in silico and our in-house DNA design principles (Fig. 2b). Component DNA oligos are purified by denaturing gel electrophoresis, and then the DDN is self-assembled via thermal annealing. The structural formation of a DDN is characterized first by nondenaturing gel electrophoresis and then by AFM (Fig. 2c). Interactions between DDNs and intact virus particles are analyzed using SPR spectroscopy to determine the binding avidity (Fig. 2d). For DENV, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and plaque-forming assays are used to evaluate DDN’s cytotoxicity and antiviral efficacy, respectively (Fig. 2e). While not specifically outlined in this Procedure, Fig. 3 illustrates the ‘mix-and-read’ strategy for DDN-based virus sensing.
Fig. 2 |. Schematic overview of the design, assembly, and characterization of DDNs for inhibition of virus infection.
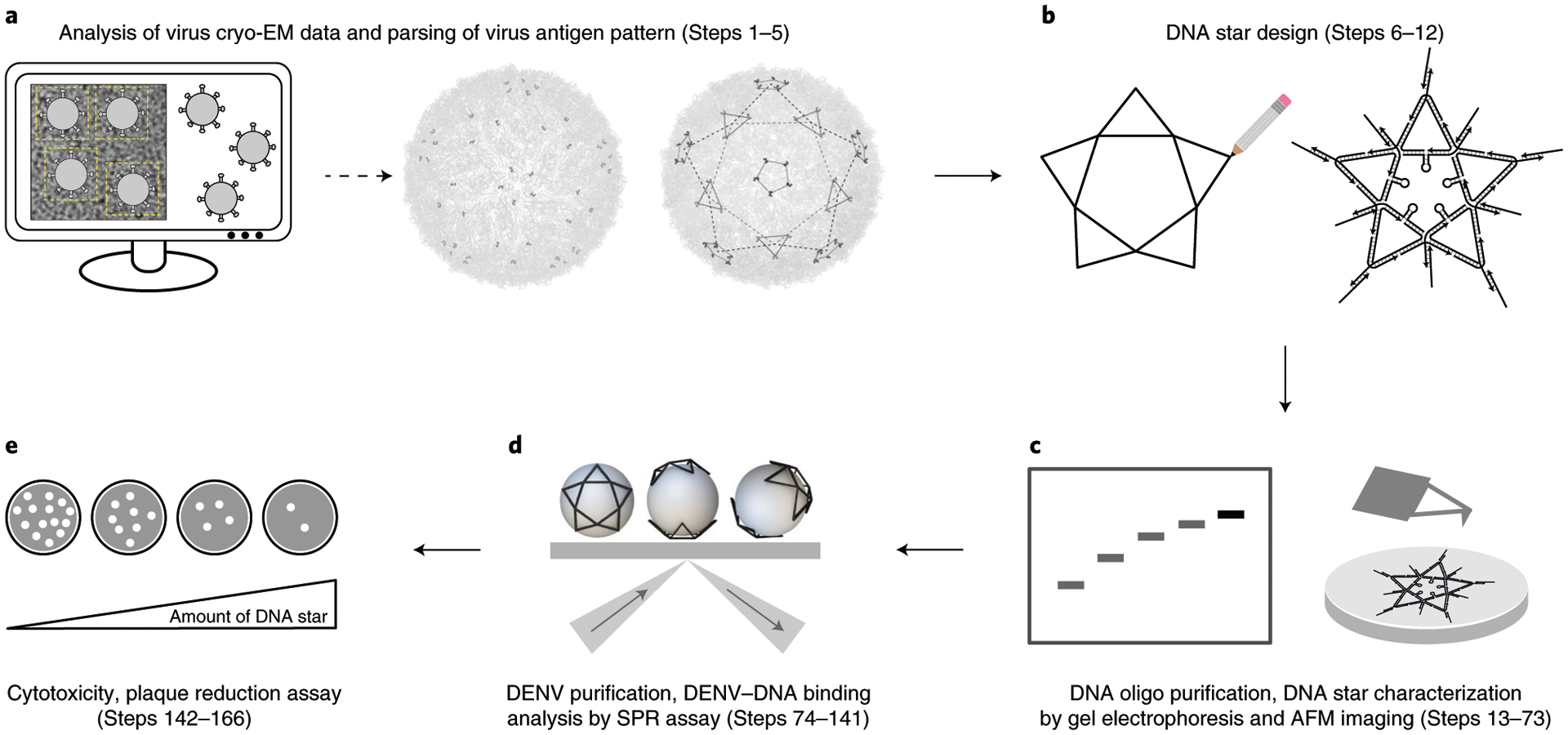
a, Identification of unique spatial patterns of antigens on a viral particle surface using available cryo-EM data. b, Design of a DNA nanostructure mirroring the antigen patterns identified in a. c, Characterization of the DNA structure formation using nondenaturing gel electrophoresis and AFM. d, Characterization of the interaction between virus and DNA structure using SPR spectroscopy. e, Evaluation of DNA structure in virus inhibition using plaque-forming assays. Part of a and b adapted with permission from ref.7, Springer Nature Ltd.
Fig. 3 |. Schematic overview of DENV detection by DNA star sensor.
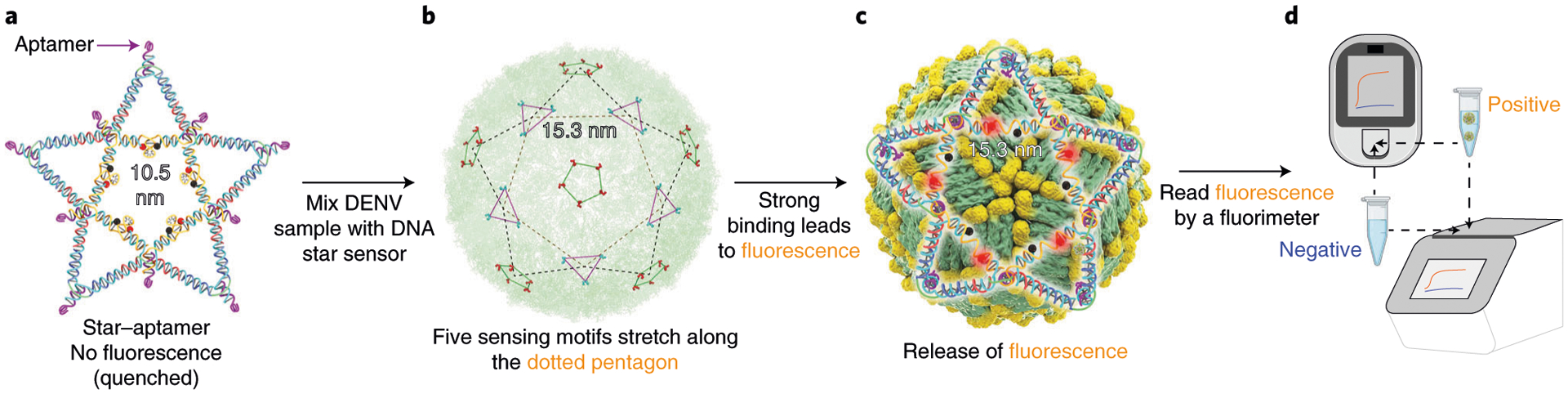
a, The ten aptamers placed on the DNA star scaffold to match the pattern and spacing of ED3 clusters on outer surface of the DENV. Five fluorophore-quencher pairs flanking the five molecular beacon-like motifs along the inner pentagon of the DNA star remain in a quenching fluorescence resonance energy transfer. b, Mixing DENV with the DNA star will trigger the release of fluorescence signal. c, When DENV is present, the polyvalent and pattern-matching-based interactions between the DNA star-templated aptamers and ED3 on DENV unzip the hairpins into ssDNA, resulting in separation of the fluorophores from the quenchers to afford a fluorescence signal. d, The signal can be read by a portable fluorimeter in point-of-care settings, or by an RT-PCR system in laboratory and high-throughout settings. b adapted with permission from ref.7, Springer Nature Ltd.
Experimental design
This protocol should be accessible to researchers with training in structural biology, DNA nanotechnology or biochemistry. The experiments must be conducted under suitable biosafety containment based on the virus of interest. The DNA star protocol is broken down into subsections, which should be adaptable to other DNA nanostructures: (a) purification of DNA oligos, (b) DDN self-assembly and gel/AFM characterization, (c) purification of the DDN complex, (d) SPR assays and (e) plaque-forming assays. Preparation of control samples for experiments throughout the Procedure are outlined in ‘Reagent setup’. DDN stability can be monitored by gel electrophoresis of the DDN samples after incubating in physiological environments up to 24 h (ref.57).
Identification of virus antigen spatial patterns (Steps 1–5)
Advances in the field of structural biology have made it possible to develop methods such as cryo-EM that provide us with the ability to visualize proteins and protein complexes at up to atomic resolution88,89. Cryo-EM has been used to determine the arrangement of proteins in macromolecular assemblies such as the dengue viral particle, which has enabled an understanding of the mechanism of viral infection48,49. The structure of the dengue viral particle (PDB: 1p58) determined by Zhang et al.90, was used to investigate the distribution of the ‘QHGTI’ amino acid sequence motif in the ED3 across the entire viral particle (Fig. 4a). Considering that the 1p58 biological assembly contains 12 five-point stars, with each point on a star representative of three monomers of the envelope protein (Supplementary Fig. 1), we first needed to identify individual monomers containing neighboring epitopes that were surface accessible before determining the distance separating them. The ‘QHGTI’ motif was previously identified as part of a conserved region in B- and T-cell epitopes of dengue envelope glycoprotein that could be used to inhibit viral infection91. Once these individual epitopes were identified, we next determined unique patterns of the distribution of epitopes accessible on the surface of the viral particle (i.e., not buried) (Fig. 4b,c). To accomplish this, we measured the distance between the Cα of the middle residue ‘G’ in neighboring ‘QHGTI’ motifs. The arc length was then measured to determine the surface distance between individual epitopes in each ED3 cluster and in between ED3 clusters (Fig. 4d and Fig. 5a).
Fig. 4 |. Identification of DENV antigen spatial patterns.
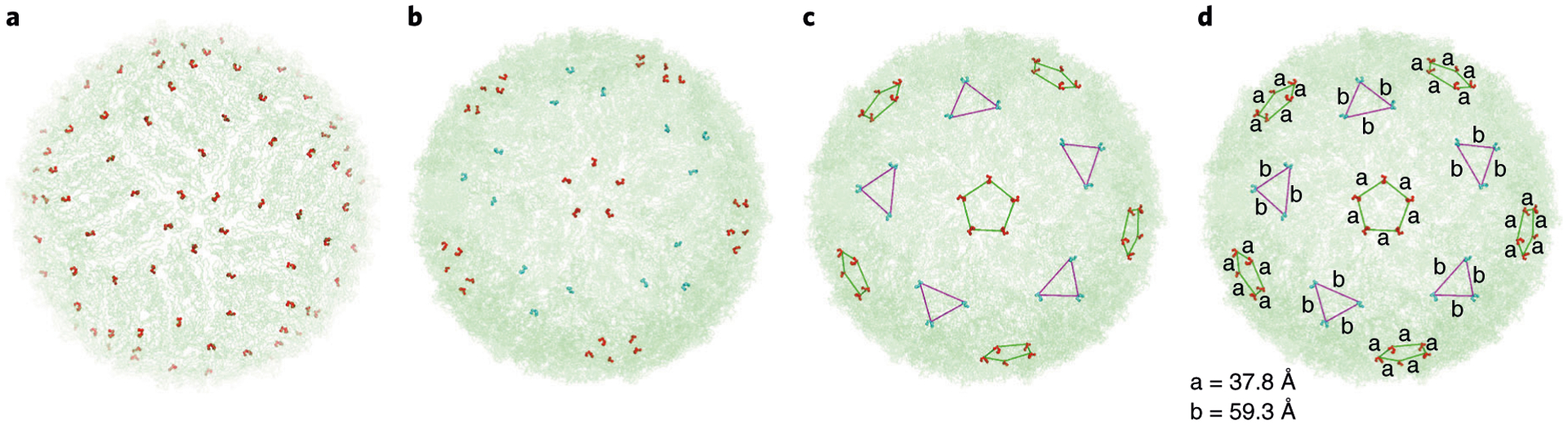
a, The QHGTI epitope was selected and colored red to illustrate the distribution of these epitopes on the dengue viral particle. b, Surface accessible epitopes on one face of the viral particle were selected and colored based on the specific patterns that were observed to cluster into trimers or pentamers7. c, The specific cluster patterns of epitope arrangement were determined by measuring the distance between the Cα of G in the QHGTI motif on neighboring epitopes (triangular clusters were colored in cyan and pentagonal clusters colored in red). d, The atomic distances that defines the arrangement of epitopes as measured in PyMol. a and b adapted with permission from ref.7, Springer Nature Ltd.
Fig. 5 |. DNA star design.
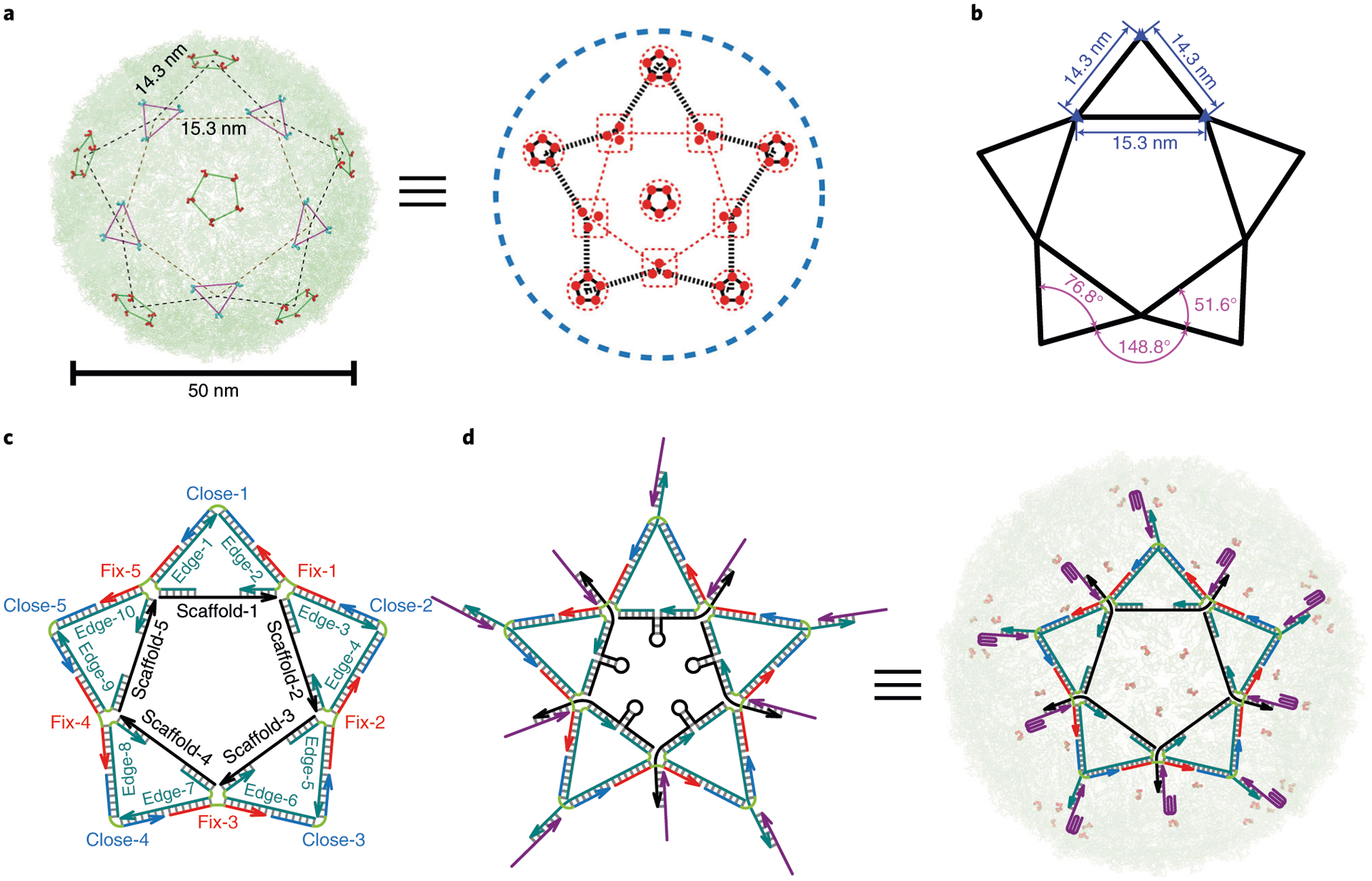
a, Distribution of DENV ED3 trivalent and pentavalent clusters presents a shape of five-pointed star7. b, Orthodromic distance between trivalent–trivalent or trivalent–pentavalent clusters is 15.3 nm or 14.3 nm. The angle of external edge–external edge (inside of an isosceles triangle), external edge–external edge (between two adjacent isosceles triangles) or external edge–internal edge is 76.8°, 148.8° or 51.6°. c, The DNA star structure consists of five ‘scaffold’ strands (S-1 to S-5) that form the pentagon inner edges, ten ‘edge’ strands that connect internal and external edges, five ‘fix’ strands that connect the external edges, and ‘close’ strands that cap the external edges of the triangle. A single-stranded region of each scaffold strand forms a hairpin loop. The sequence for each DNA strand was programmed in SEQUIN. d, 3′ overhangs on the DNA star allow aptamer incorporation7. The ten incorporated aptamers match the pattern and spacing of ED3 clusters. When bound to a virion, each of the five hairpins is stretched to a distance that fits the spacing between adjacent trivalent ED3 clusters. The switchable DNA star can be turned into a virus inhibitor or sensor (when fluorescence components are added to report virus binding). a and d adapted with permission from ref. 7, Springer Nature Ltd.
Design of the DNA star (Steps 6–12)
Our analysis of the DENV cryo-EM data90 has focused on the ED3 viral epitope because it is a major interaction point for DENV NAbs that strongly inhibit DENV infection92–97. This motivated us to develop a DNA-based viral inhibition strategy targeting the ED3 epitopes. QHGTI sequence motifs in ED3 were found to organize into a repeating pattern on the DENV surface with alternating trivalent or pentavalent clusters (Fig. 5a). By connecting the ED3 trivalent and pentavalent cluster sites linearly (centered on a pentavalent cluster), we identified a star shape, consisting of an interior pentagon connected to five exterior triangles, which we hypothesized would provide an optimal multivalent scaffold for high DENV binding avidity (Fig. 5a). The orthodromic center to center distances between trivalent–trivalent and trivalent–pentavalent ED3 clusters are respectively 15.3 nm and 14.3 nm.
Based on structural information, we designed a DNA star scaffold, with vertices matching the spatial pattern of the ED3 clusters on DENV surface. Since the size of the DNA star was smaller (~43 nm diameter) and simpler than a typical DNA origami structure43, we used DNA tile nanostructure design principles (e.g., minimizing sequence symmetry) and assembly strategies7,13,98–102. Specifically, the five-point DNA star consists of five geometrically identical isosceles triangles. The orthodromic center-to-center distances between trivalent–trivalent and trivalent–pentavalent ED3 (or other antigen) clusters determine the lengths of the isosceles triangle internal and external edges as 52 bp and 42 bp long, respectively (Fig. 5b). As the design steps are further illustrated in Fig. 6 and elaborated in Steps 6–12 of the Procedure, the DNA star is constructed using 25 ssDNA oligonucleotides: five ‘scaffold’ strands, ten ‘edge’ strands, five ‘close’ strands and five ‘fix’ strands (Fig. 5c). The specific sequences we used were obtained in SEQUIN program13,103, in which we were able to input the structural information and generate the DNA sequences that assemble into a structure to match the DENV surface antigen geometric arrangement. The ‘scaffold’ and ‘edge’ DNA constitute internal edges of the isosceles triangles. The ‘close’ and ‘edge’ DNA form external edges of the isosceles triangles. The five ‘fix’ strands connect the five isosceles triangles together to assemble a full star structure. The DNA star is assembled of five rigid four-way crossover structures at the five pentagon corners. Linking them together without providing flexible ssDNA linkers may cause torsion in the whole structure, thus deforming canonical B-DNA duplex to make the DNA star structure unstable. In this regard, measures must be taken to provide the crossovers in the DNA star with more flexibility to relieve such torsional stress. For this purpose, several noncomplementary thymine bases (free Ts) can be added at the corner of internal pentagon crossovers and at the five tips of the star (indicated by the green lines in Fig. 5c,d). Different numbers of free Ts at the crossover define different angles between its two-flanking duplex: adding more free Ts results in a smaller crossover angle104,105. Thus, the optimal number of free Ts can be determined based on the crossover angles required to form a DNA nanostructure. For our (five-point) DNA star, the three respective angles of scaffold/edge–edge/fix, edge/fix–edge/fix, and edge/close–edge/close duplexes, in theory, are 51.6°, 148.8° and 76.8°. These angles dictate the number of free Ts used at the respective corners, which are 7, 3 and 7 nt. After adding the free Ts at the crossovers, the DNA star structure becomes more flexible, allowing it to better wrap around a virus particle. If applying the DNA star strategy to target a different virus, we can repeat the entire DNA nanostructure design procedure, starting with identifying unique spatial patterns of antigens on a viral particle surface using available cryo-EM data of the virion of interest, then design a DNA nanostructure mirroring this pattern, and automatically program the DNA sequences for such a DNA nanostructure using the SEQUIN program.
Fig. 6 |. Process flow diagram of the DNA star design.
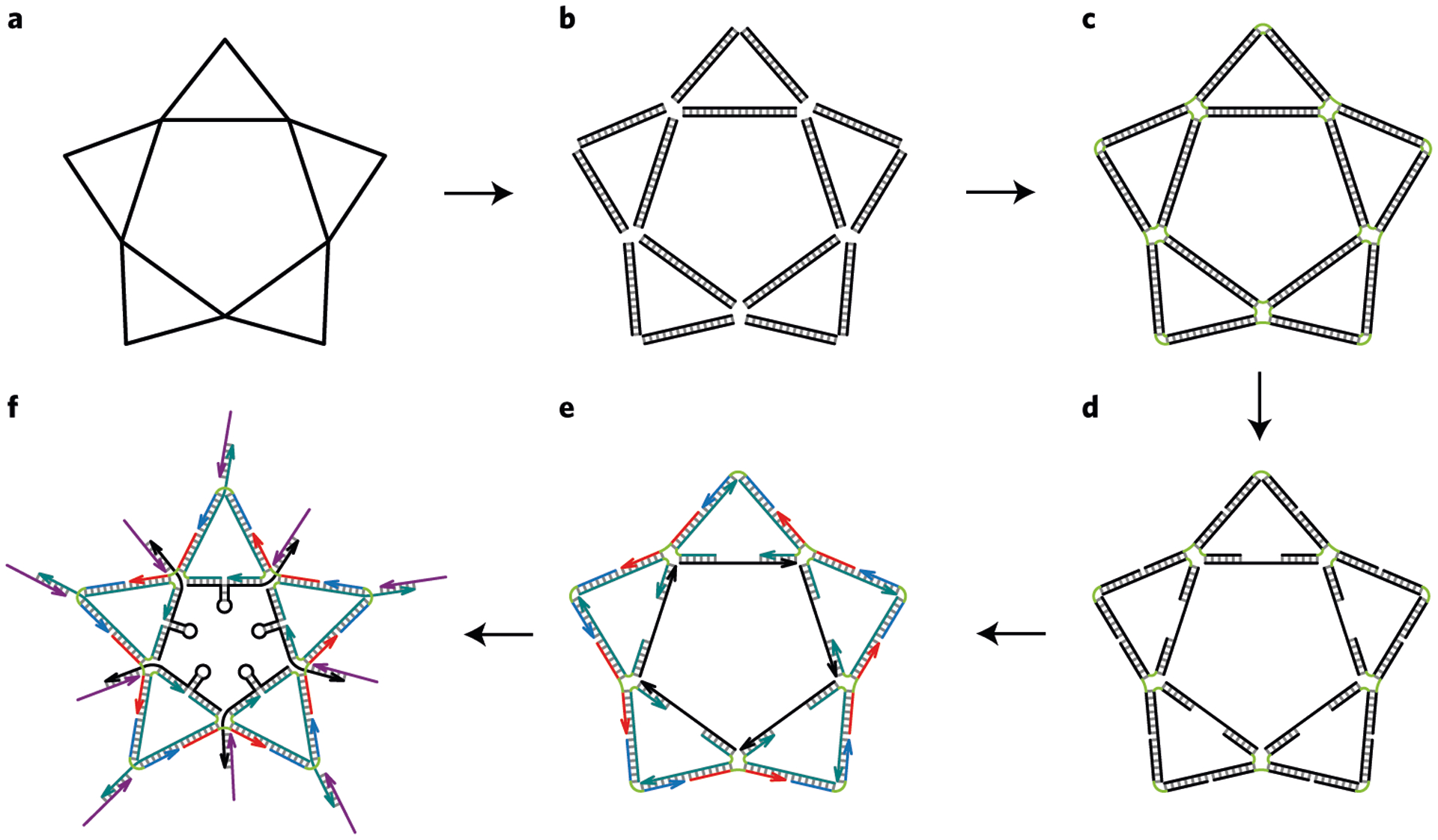
a, A single-line skeleton of the star shape with the angle degrees and DNA base pair equivalent edge lengths indicated. b, A double-line skeleton of the star shape with DNA hydrogen bonds indicated by the short perpendicular lines. c, All gaps on the lines are sealed with short green lines that indicate unpaired Ts. d, Generation of nicks and five ssDNA regions on the inner edges of the star. e, 3′-end of each strand is indicated with an arrow. Strands of the same type are colored with the same color. f, Schematic of the native DNA star–aptamer complex that contains five hairpin loops and ten aptamers displayed at ten vertices to mirror the DENV ED3 cluster spatial pattern7. Note that the five hairpin loops are drawn not to scale. e and f adapted with permission from ref.7, Springer Nature Ltd.
The DNA star is functionalized for inhibition and detection of DENV by conjugating an ED3-targeting aptamer at each of the ten vertices of the DNA star scaffold to form a DNA star–aptamer complex that superimposes the spatial pattern of ED3 clusters on DENV surface (Fig. 5d). To turn the star–aptamer complex into an effective DENV inhibitor and sensor, we introduced a ssDNA region into each of the five internal star edges to form a hairpin structure with 7 bp stem and 6 nt loop. The hairpins unzipping into ssDNAs stretches each internal edge of the DNA star to a distance that fits the spacing between adjacent trivalent ED3 clusters. These hairpins play a dual function. First, for DENV inhibition, they provide the overall rigid DNA star scaffold with local structural flexibility to ensure binding and inhibition of DENV under various physiological environments and temperatures8. DENV typically enters cells first through electrostatic interactions with the host cell plasma membrane and glycosaminoglycans. After binding to the DENV surface with high avidity, the DNA star physically and electrostatically (through interactions between DENV surface and negatively charged DNA scaffold) traps and prevents virions from interacting with and entering host cells. Second, for DENV detection (which is not a focus of this protocol), hairpins provide the DNA star with structurally switchable motifs to report the presence of DENV in a sample. Specifically, fluorophore and quencher carrying strands (e.g., FAM-BHQ1 pair or Cy3-BHQ2 pair) are hybridized to each inner edge of the star so they are brought together to flank the hairpin by canonical Watson–Crick base pairing, much like a molecular beacon motif. However, unlike a molecular beacon that generates a fluorescence signal for target ssRNA or ssDNA hybridization, on the DNA star hairpins are pulled apart as a result of strong aptamer–ED3 binding. Such potent interactions can separate fluorophores from quenchers to restore fluorescence as a signal readout.
Denaturing PAGE for oligonucleotide purification (Steps 13–41)
DNA oligo synthesis using phosphoramidite chemistry106–108 typically generates truncated products that can interfere with formation of DNA nanostructures. A critical prerequisite for ensuring the success of DNA star assembly is to obtain purified full-length products of each of the component DNA oligos. Denaturing polyacrylamide gel electrophoresis (PAGE) can separate full-length products from shorter species (including n-1 species) with high efficiency. PAGE purification is effective for separating unmodified oligos from unwanted truncated products. The component DNA oligos for the self-assembly of the DNA star range from 40 to 80 nt. Different lengths of DNA strands can be resolved by altering the concentration of polyacrylamide gel, where a higher gel percentage is used to purify shorter DNA oligos. After electrophoresis, the desired DNA band is identified using UV visualization, excised, and eluted from the gel using elution buffer (Buffer 3) and concentrated using ethanol precipitation. Precipitates of the DNA oligos are suspended in nuclease-free water, and DNA concentration is quantified using an absorbance measurement at 260 nm. In comparison with other purification methods, like HPLC, denaturing PAGE offers high-resolution separation of full-length products from shorter side products with length differences as small as a single nucleotide. Several samples can be run simultaneously without requiring any special equipment.
Assembly of the DNA star (Steps 42–46)
Forming a DNA star with high yield relies on three factors: a close to 1:1 stoichiometric ratio among all component DNA strands, a suitable annealing buffer and an appropriate thermal annealing protocol. To construct the DNA star (100 μL in each PCR tube at a final concentration ranging from 0.1 to 0.4 μM final concentration of DNA star complexes for different applications), stoichiometrically equivalent quantities (scaffold strands: edge strands: fix strands: close strands = 1:1:1:1) of all component strands are thoroughly mixed in 1× TAE-Mg2+ (Buffer 5) annealing buffer. The mixture is then heated in a standard thermal cycler to 95 °C and incubated for 5 min before it is slowly cooled down from 95 °C to 20 °C at a rate of 0.15 °C per min over 8 h (an overnight protocol). Samples are incubated for 5 min at a high temperature (80–95 °C) to break all previously formed Watson–Crick base pairs to ensure that all component DNAs can hybridize with their correctly designed partners. A shorter annealing protocol may work as well, but we have not tested it for the formation of DENV-targeting DNA stars7.
Nondenaturing gel electrophoretic characterization and purification of the DNA star (Steps 47–67)
Nondenaturing (native) gel electrophoresis is a reliable method for quickly characterizing the structural formation and yield of a DNA nanostructure assembly. There are two gel matrices to choose from based on the size and complexity of DNA nanostructures. Nondenaturing agarose gel electrophoresis (AGE) is normally used to characterize DNA assemblies with very high molecular weight or complex nanostructures such as 2D and 3D DNA origami. Nondenaturing PAGE is normally used for DNA nanostructures such as DNA tile motifs with a molecular weight <1,000 base pairs (bp). Since certain 3D nanostructures are too big to travel through the polyacrylamide gel matrix, they may not enter the PAGE. Thus, higher-percentage AGE (e.g., 3%) can also be used to characterize the formation of tile-based DNA motifs, but with lower-resolution molecular weight separation (20–30 bp) among different DNA species. The size of the full DENV-targeting five-point DNA star is 615 bp (or ~470.80 kDa). Its length and width in 2D space are ~39.26 nm and ~37.34 nm, respectively. Because the DNA star has a relatively low molecular weight and small size, and the molecular weight difference between the partial and full DNA star is small, we use a 4% nondenaturing PAGE gel to characterize the yield of the structure formation presented here. The electrophoresis is run in 1× TAE-Mg2+ buffer (Buffer 5) at 60 V for 90 min. The gel running buffer contains magnesium ions, which can generate heat during electrophoresis. Therefore, we put the gel box either in a 4 °C cold room or in an ice bath to make sure the gel box is not overheated, so it does not denature the DNA star complex. Post electrophoresis, gels can be stained with DNA intercalating reagents such as GelRed, ethidium bromide or Sybr dyes. The resultant PAGE of the partial or full DNA star typically shows a distinct and dominant band corresponding to each of the DNA complexes, confirming that DNA star structures are assembled in high yield. It is worth noting that byproducts with lower molecular weight may form as a result of imperfect stoichiometric ratios among the component DNA oligos. After confirming the yield by PAGE gel, the desired DNA stars may be purified as intact complexes using a high percentage (i.e., 3%) nondenaturing AGE gel and dialysis for DNA star elution.
AFM characterization of the DNA star (Steps 68–73)
AFM imaging can show the entire morphology of a DNA assembly at <5 nm resolution. AFM is particularly useful for visualizing large DNA 2D arrays and DNA origami nanostructures. For more fragile tile-based DNA star nanostructures, special care is needed during AFM sample preparation. This includes modifying the mica substrate surface with positively charged (3-aminopropyl)triethoxysilane (APTES) to facilitate sample deposition of negatively charged DNA109, and using ‘soft tapping in fluid’ mode on the AFM. The AFM images of the DNA stars in this protocol are obtained on a Bruker MultiMode VIII microscope in 1× TAE-Mg2+ buffer (Buffer 5) with SNL-10 probe.
Purification of the DENV (Steps 74–132)
Purified virus is required for SPR analysis of the interaction between viral particles and the DNA star complexes. We provide a DENV purification protocol that is streamlined for laboratories that do not have access to a gradient maker. This protocol is optimized for small-scale virus preparation for up to 60 mL supernatant of DENV-infected cells110,111, typically from four T-75 cell culture flasks (called T-75 flask herein). DENV is a human pathogen and can cause serious illness. Thus, proper precautions should be taken to ensure the safety of everyone in a BSL-2 laboratory. Researchers need to consult with their institution’s safety office with regard to appropriate personal protective equipment (PPE) and handling precautions. Also, we highly recommend consulting the latest edition of Biosafety in Microbiological and Biomedical Laboratories (BMBL)112 by the Centers for Disease Control and Prevention (CDC) and the National Institutes of Health (NIH) about regulations on handling biohazardous materials.
SPR analysis of the interaction between viral particles and the DNA star complexes (Steps 133–141)
SPR spectroscopy is based on an optical biosensor technique that measures molecular binding events at a metal surface by detecting changes in the local refractive index113. It is a powerful tool to measure biomolecular interactions in real time and in a label-free environment. SPR assays can typically answer the following questions in regard to ligand–ligand interaction: (i) How specific is an interaction? (ii) How strong is an interaction, and what is the binding affinity (KD)? (iii) How fast is an interaction, and what are the association and dissociation rate constants (ka and kd)? (iv) What are the thermodynamic parameters (enthalpy (ΔH) and entropy (ΔS)) for an interaction? (v) What is the biologically active concentration of a specific molecule in a sample? In this protocol, intact virions are immobilized on the SPR chip surface to characterize their interaction with DNA star nanostructures for their relative and absolute binding affinities. This setup recapitulates the scenario for DNA stars interacting with intact virions for virus inhibition. The interactions between viral particles and DNA stars are analyzed using the SPR instrument BIAcore 3000.
MTT assay to measure DNA star cytotoxicity (Steps 142–155)
To assess the cytotoxicity of DNA star nanostructures, MTT assay is used to measure cell viability and proliferation. In the MTT assay, cellular oxidoreductase/dehydrogenase enzymes in living cells reduce the yellow tetrazolium dye MTT to an insoluble formazan, which has a purple color, which can be easily quantified by measuring absorbance at 570 nm on a spectrophotometer or plate reader. The absorbance is directly proportional to the number of viable cells. In this assay, cytotoxicity is evaluated by measuring the inhibition of cell proliferation114,115. This measurement of cell proliferation and viability in terms of reductive activity is widely accepted as a reliable way to examine the cytotoxicity effect of drugs or compounds116,117. Serially diluted DNA star samples as well as controls with only the dilution buffer are added to the Vero cell monolayer in triplicates. Average of the triplicate reading for each sample can be used to calculate the percentage of cytotoxicity with the following equation: % cytotoxicity = (100 × (control − sample) / control).
Plaque reduction test (antiviral assay) (Steps 156–166)
The plaque reduction neutralization test is considered the ‘gold standard’ in quantifying the titer of neutralization antibody for a virus. Here we use a modified plaque reduction neutralization test assay, called the plaque reduction test, to measure the inhibition of plaque formation efficiency by an antiviral drug or compound118–121. The solution of antiviral compound is diluted serially and mixed with a known concentration of virus. After a period of incubation allowing the drug to act on the virus, the mixture is added to cell monolayers for assessment. The assay conditions can be further modified to assess the possible mode or timing of action. The assay is typically performed in triplicate, and the arithmetic mean is used to calculate the percentage reduction in plaque numbers by statistical analysis. A dilution buffer without any test compound should always be included to measure the background of the assay conditions. The concentration of test compound to reduce the number of plaques by 50% (EC50) or 90% (EC90) on a dose–response curve, when compared with the medium or buffer controls gives a measurement of the compound’s antiviral efficiency.
Materials
Biological materials
Cell line of interest. We used a Vero cell line (ATCC, cat. no. CCL-81; RRID: CVCL_0059) ! CAUTION The cell lines should be regularly checked to ensure they are authentic and are not infected with mycoplasma. Morphology of cultures should be checked prior to each usage. Healthy Vero cells grow as an adherent monolayer and exhibit a slightly elongated fibroblast morphology. Find more information on ATCC website (https://www.atcc.org/products/ccl-81).
Virus of interest. We used a DENV sample with known titer from previous preparations (BEI Resources, cat. no. NR-84) ! CAUTION DENV is a human pathogen and can cause serious illness. Tubes and any containers should only be opened and handled in a biosafety cabinet within a BSL-2 laboratory. Follow the biosafety guidelines for working with infectious agents in the laboratory, which can be found in the most recent version of the Arbovirus Laboratory Manual, and in the BMBL112.
Reagents
Reagents used in more than one section
Ultrapure water (from Milli-Q direct water purification system)
Tris(hydroxymethyl)aminomethane (Tris base; Sigma-Aldrich, cat. no. 252859; CAS no. 77-86-1)
Acetic acid (CH3COOH; Sigma-Aldrich, cat. no. 695092; CAS no. 64-19-7)
EDTA disodium salt dihydrate 99.0–101.0% (VWR BDH Chemicals, cat. no. BDH4616–500G; CAS no. 6381-92-6)
Sodium chloride (NaCl; VWR Life Science, cat. no. 97061–270; CAS no. 7647-14-5)
N,N,N′,N′-tetramethylethylenediamine (TEMED), molecular biology grade (C6H16N2; MP Biomedicals, cat. no. 0219401925; CAS no. 110-18-9) ! CAUTION TEMED is highly flammable and toxic liquid and can cause severe skin burns and eye damage. When handling TEMED, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential hazard exposure to the reagent. Collect any liquid waste in a bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute. If exposed to TEMED spills, take necessary washing and cleaning steps as trained and seek help from the corresponding health professionals.
Ammonium persulfate (APS, (NH4)2S2O8; VWR Life Science, cat. no. 97064–594; CAS no. 7727-54-0) ! CAUTION APS is highly flammable and toxic liquid and can cause severe skin burns and eye damage. When handling APS, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential hazard exposure to the reagent. Collect any liquid waste in a bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute. If exposed to APS spills, take necessary washing and cleaning steps as trained and seek help from the corresponding health professionals.
40% acrylamide:bisacrylamide, 19:1 (VWR Life Science, cat. no. 97064–870; CAS no. 79-06-1) ! CAUTION Unpolymerized acrylamide monomer has been reported to affect peripheral nervous system and cause sensory impairment. When handling acrylamide monomer, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential hazard exposure to the reagent. Collect any liquid waste in a bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute. If exposed to acrylamide monomer spills, take necessary washing and cleaning steps as trained and seek help from the corresponding health professionals.
Sodium hydroxide (NaOH, 3.0 N; VWR BDH Chemicals, cat. no. BDH7472–1; CAS no. 1310-73-2)
Magnesium acetate tetrahydrate (Mg (CH3COO)2 · 4H2O; VWR Life Science, cat. no. 97061–060; CAS no. 16674-78-5)
Minimal essential medium (MEM; Sigma-Aldrich, cat. no. 56416C-1L) ▲ CRITICAL Follow the manufacturer’s suggested storage condition and shelf life.
Fetal bovine serum (FBS; Sigma-Aldrich, cat. no. F2442–1L)
Penicillin G sodium salt (C16H17N2NaO4S; Sigma-Aldrich, cat. no. P3032–100MU; CAS no. 69-57-8)
Streptomycin solution, 1 mg/mL (C21H39N7O12 · 1.5 H2SO4; Sigma-Aldrich, cat. no. 85886–10ML; CAS no. 3810-74-0)
1 M Tris-HCl buffer, pH 7.5 (Thermo Fisher Scientific, cat. no. 15567027; CAS no. 1185-53-1)
Sodium bicarbonate (NaHCO3; Sigma-Aldrich, cat. no. S5761–500G; CAS no. 144-55-8)
Gentamicin sulfate salt (Sigma-Aldrich, cat. no. G1264–5G; CAS no. 1405-41-0)
Fungizone (C47H73NO17; Sigma-Aldrich, cat. no. A2411–1G; CAS no. 1397-89-3)
Coverage Plus NPD Disinfectant (Steris Life Sciences, cat. no. 638708)
70% ethanol (Fisher Scientific, cat. no. BP8201500; CAS no. 64-17-5)
10× M199-Hank’s medium with Hanks’ salts (Sigma-Aldrich, cat. no. M9163) ▲ CRITICAL Follow the manufacturer’s suggested storage condition and shelf life.
Potassium chloride (KCl; VWR Life Science, cat. no. 97061–566; CAS no. 7447-40-7)
Oxoid Agar Bacteriological (Thermo Fisher Scientific, cat. no. LP0011B)
Neutral Red Solution (C15H17CIN4 in DPBS, 3.3 g/L, 0.33% wt/vol; Sigma-Aldrich, cat. no. N2889; CAS no. 553-24-2)
Design of the DNA star (Steps 6–12)
The DNA oligonucleotide sequences for making a five-point DNA star are shown in Supplementary Table 1 ▲ CRITICAL 2 μmol DNA oligo synthetic scale is required for the assays in this protocol.
The DNA oligonucleotide sequences for making a six-point DNA star are shown in Supplementary Table 2 ▲ CRITICAL 2 μmol DNA oligo synthetic scale is required for the assays in this protocol.
The DNA oligonucleotide sequences for making a seven-point DNA star are shown in Supplementary Table 3 ▲ CRITICAL 2 μmol DNA oligo synthetic scale is required for the assays in this protocol.
Denaturing PAGE for oligonucleotide purification (Steps 13–41)
Boric acid (H3BO3; Sigma-Aldrich, cat. no. B6768–1KG; CAS no. 10043-35-3)
n-Butanol (Sigma-Aldrich, cat. no. 1.01988.1000; CAS no. 71-36-3) ! CAUTION n-Butanol is highly flammable and causes eye irritation. Prolonged exposure to skin causes drying, redness and possible blistering. When handling butanol, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential hazard exposure to the reagent. Collect any liquid waste in a glass bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute. If exposed to butanol spills, take necessary washing and cleaning steps as trained and seek help from the corresponding health professionals.
Ethidium bromide (VWR Life Science, cat. no. 97062–734; CAS no. 1239-45-8) ! CAUTION Ethidium bromide is a powerful mutagen. When handling ethidium-bromide-containing buffer solutions, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential hazard exposure to the reagent. Collect any liquid waste in a bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute. If exposed to ethidium bromide spills, take necessary washing and cleaning steps as trained and seek help from the corresponding health professionals.
Ethanol, 200-proof (C2H5OH; Fisher Scientific, cat. no. A4094; CAS no. 64-17-5)
Urea (H2NCONH2; VWR Life Science, cat. no. 97061–920; CAS no. 57-13-6)
Bromophenol blue (C19H10Br4O5S; Sigma-Aldrich, cat. no. B0126–25G; CAS no. 115-39-9)
Xylene cyanol FF (C25H27N2NaO6S2; Sigma-Aldrich, cat. no. X4126–10G; CAS no. 2650-17-1)
(Optional) Orange G (C16H10N2Na2O7S2; Sigma-Aldrich, cat. no. O3756–25G; CAS no. 1936-15-8)
Glycerol (HOCH2CH(OH)CH2OH; Sigma-Aldrich, cat. no. G5516–100ML; CAS no. 56-81-5)
Ammonium acetate (C2H7NO2; VWR Life Science, cat. no. 97061–014; CAS no. 631-61-8)
Assembly of the DNA star (Steps 42–46)
DNA strands (Integrated DNA Technologies or Sangon Biotech) (the DNA star sequences used in this protocol are included in Supplementary Tables 1–3)
Nondenaturing gel electrophoretic characterization and purification of the DNA star (Steps 47–67)
6× loading buffer (Thermo Fisher Scientific, cat. no. R0611)
4S GelRed, 10,000× in water (Sangon Biotech, cat. no. A616697) ! CAUTION GelRed is a potential carcinogen. When handling GelRed-containing buffer solutions, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential hazard exposure to the reagent. Collect any liquid waste in a bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute. If exposed to GelRed spills, take necessary washing and cleaning steps as trained and seek help from the corresponding health professionals.
Agarose RA, Biotechnology Grade (VWR Life Science, cat. no. 97064–258; CAS no. 9012-36-6)
SYBR Green I nucleic acid gel stain, 10,000× (Thermo Fisher Scientific, cat. no. S7585)
AFM characterization of the DNA star (Steps 68–73)
APTES (Sigma-Aldrich, cat. no. 706493; CAS no. 919-30-2)
Purification of the DENV (Steps 74–132)
DENV sample with known titer from previous preparations (BEI Resources, cat. no. NR-84) ! CAUTION DENV is a human pathogen and can cause serious illness. Tubes and any containers should only be opened and handled in a biosafety cabinet within a BSL-2 laboratory. Follow the biosafety guidelines for working with infectious agents in the laboratory, which can be found in the most recent version of the Arbovirus Laboratory Manual and in the BMBL112.
Sodium phosphate dibasic, anhydrous (Na2HPO4; VWR Life Science, cat. no. 97061–588; CAS no. 7558-79-4)
Potassium dihydrogen phosphate, anhydrous (KH2PO4; VWR Life Science, cat. no. 97062–350; CAS no. 7778-77-0)
Sucrose (C12H22O11; VWR Life Science, cat. no. 97063–790; CAS no. 57-50-1)
l-glutamine (C5H10N2O3; VWR Life Science, cat. no. 97061–506; CAS no. 56-85-9)
100× Penicillin–streptomycin, 10,000 U/mL (Thermo Fisher Scientific, cat. no. 15140122)
Eagle’s MEM (EMEM, ATCC, cat. no. 30–2003)
SPR analysis on the interaction between viral particles and the DNA star complexes (Steps 133–141)
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC; from amine coupling kit, Cytiva, cat. no. BR100050; CAS no. 25952-53-8)
N-hydroxysuccinimide (NHS; from amine coupling kit, Cytiva, cat. no. BR100050; CAS no. 6066-82-6)
Ethanolamine (from amine coupling kit, Cytiva, cat. no. BR100050; CAS no. 141-43-5)
Sodium acetate anhydrous (Na (CH3COO); VWR Life Science, cat. no. 97061–998; CAS no. 127-09-3)
MTT assay to measure DNA star cytotoxicity (Steps 142–155)
MTT cell proliferation assay kit (ATCC, cat. no. 30–1010K) ▲ CRITICAL Follow the manufacturer’s suggested storage condition and shelf life.
Trypan Blue Solution, 0.4% (Thermo Fisher Scientific, cat. no. 15250061; CAS no. 72-57-1)
Plaque reduction test (antiviral assay) (Steps 156–166)
BSA (Sigma-Aldrich, cat. no. A7030–50G; CAS no. 9048-46-8)
Equipment
Equipment used in more than one section
Pipettes (Eppendorf Research Plus package, cat. no. 2231000773)
Pipette tips (USA Scientific, cat. nos. 1160–3700, 1163–1700, 1161–1720)
Milli-Q direct water purification system (Millipore, cat. no. ZR0Q008WW)
Benchtop centrifuge for 1.5 mL or 2.0 mL Eppendorf tubes (Eppendorf Centrifuge 5425, cat. no. 5405000247, or Eppendorf Centrifuge 5427R, cat. no. 022620702)
0.2 mL PCR tube (USA Scientific, cat. no. 1402–1800)
Nonstick, RNase-free microfuge tubes, 1.5 mL (Thermo Fisher Scientific, cat. no. AM12450)
Vortex mixer (VWR, cat. no. 97043–562)
Razor blades, single edge, individually wrapped (VWR, cat. no. 55411–055) ! CAUTION Take care when using a razor to avoid cuts, and wear thick gloves. Properly dispose of the razor blades in a biohazard sharps container.
PAGE gel system (e.g., Bio-Rad Mini Protean II system (Bio-Rad))
Power supply (Thermo Scientific EC1000XL, cat. no. FBEC1000XL)
UV transilluminator (VWR, cat. no. 76407–432)
UV-Vis spectrophotometer (Eppendorf BioSpectrometer, cat. no. 6135000923) ! CAUTION Protect yourself from exposure to UV by wearing face and eye protection as well as a laboratory coat.
5% CO2 incubator (Thermo Fisher Scientific, cat. no. Water Jacketed Incubator model 3110)
500 mL and 1 L 0.22 μm vacuum filtration system (VWR, cat. no. 76010–402 and 76010–416)
Centrifuge with aerosol tight lids for 50 mL conical tubes (Sorvall, cat. no. Legend XTR with Fiberlite F13–14 × 50cy Fixed Angle Rotor)
Balance, analytical and precision, 10 kg capacity (VWR, cat. no. 10204–998)
Balance, top loading, 120 g capacity (VWR, cat. no. 75802–860)
Glass medium bottles, graduated, various sizes (Corning, cat. nos. 1395–50, 1395–100, 1395–150, 1395–250, 1395–500, 1395–1L, 1395–2L)
pH meter (Sartorius, cat. no. PB-10)
Water bath, temperature range: ambient + 5 to 100 °C (VWR, cat. no. 76308–896, or Fisher Scientific Isotemp General Purpose Deluxe, cat. no. FSGPD10)
Biosafety cabinet (Baker Company, cat. no. Model SG603 Sterilgard III Advance, or Thermo Scientific, 1300 Series Class II or equivalent) ▲ CRITICAL Biosafety cabinet needs to be used when preparing samples containing intact virus particles. PPE, including gloves, laboratory coat, closed-toe shoes, face mask and laboratory safety goggles, is used to mitigate potential hazard exposure to the reagents.
Inverted microscope (Nikon Eclipse, cat. no. TS100)
Pipette controller (Pipet-Aid, cat no. 4-000-101)
Sterile serological pipettes (Corning, cat. nos. CLS4010, CLS4100, CLS4250)
Microcentrifuge tubes, 1.5 mL (Eppendorf Safe-Lock Tubes, cat. no. 022363204)
Multichannel pipette (Rainin, cat. no. Pipet-Lite Multi Pipette L8–200XLS+)
Cell culture flask T-75 cm2 rectangular canted neck with vented cap (Corning, cat. no. 353136)
Identification of virus antigen spatial patterns (Steps 1–5)
Personal computer running Windows/MacOS/Linux operating system
Installation of PyMol (https://pymol.org/2/) or similar structure visualization software such as Molecular Operating Environment (MOE) (https://www.chemcomp.com/), Visual Molecular Dynamics (VMD) (https://www.ks.uiuc.edu/Research/vmd/) or UCSF Chimera (https://www.cgl.ucsf.edu/chimera/)
Protein Data Bank (PDB) file of the structure coordinates of the dengue viral particle as determined by cryo-EM (PDB: 1p58)
Design of the DNA star (Steps 6–12)
Assembly of the DNA star (Steps 42–46)
Thermal cycler (Thermo Fisher Scientific, cat. no. 4452299, or Biometra TRIO Thermal Cycler, cat. no. 846-4-070-723)
Nondenaturing gel electrophoretic characterization and purification of the DNA star (Steps 47–67)
Ultrasonic cleaner (Kunshan Ultrasonic Instruments, cat. no. KQ2200E)
Ice maker (Changshu Xueke Electric, cat. no. IMS-50)
Mini-PROTEAN Tetra electrophoresis tank (Bio-Rad, cat. no. 1658001)
Ten-well, 1.0 mm gel casting module (Bio-Rad, cat. no. 1658015)
Gel imager (Syngene, cat. no. GBOX-F3)
Owl EasyCast B1A mini gel electrophoresis system (Thermo Fisher Scientific, cat. no. B1A)
Dialysis tubing, 3.5K MWCO (Thermo Fisher Scientific, cat. no. 88242)
Dialysis tubing clips (Thermo Fisher Scientific, cat. no. 68011)
Erlenmeyer flask, 250 mL (VWR, cat. no. 10536–914)
Erlenmeyer flask, 500 mL (VWR, cat. no. 10536–926)
Microwave oven, 1.2 ft3 (Toshiba, cat. no. EM131A5C-SS)
Hot hand protector mitt (Bel-Art, cat. no. F38000–0001)
AFM characterization of the DNA star (Steps 68–73)
Plastic tweezers (Agar Scientific, cat. no. AGT5811)
Atomic force microscope (Bruker, Multimode 8)
AFM tip (Bruker, SNL-10)
Mica sheet (Ted Pella, cat. no. 50)
NanoScope Analysis v1.50
Purification of the DENV (Steps 74–132)
Nunc cryogenic tubes, sterile 1.8 mL of screw-cap tubes (Sigma Aldrich, cat. no. TMO375418)
Mechanical beam balance (Ohaus, cat. no. 1550-SD)
Ultracentrifuge (Beckman, cat. no. XL-100K)
Beckman SW41 Ti rotor (Beckman, cat. no. 331336)
Ultraclear SW41 ultracentrifuge tubes (Beckman, cat. no. 344059)
Pointed stainless forceps (Fisher Scientific, cat. no. 12-000-157)
Costar six-well flat-bottom cell culture plates (Corning, cat. no. 3506)
SPR analysis on the interaction between viral particles and the DNA star complexes (Steps 133–141)
Biacore SPR system (Biacore 3000, GE Healthcare) ▲ CRITICAL To comply with biosafety regulations, access to the SPR laboratory should be highly restricted to authorized researchers who have passed intensive biosafety training for handling samples containing virus particles. Virus-containing samples should only be handled within a suitable biosafety cabinet.
CM5 sensor chip (Cytiva, cat. no. BR100012 for Biacore 3000)
BiaEvaluation software 4.0.1. (GE Healthcare, Uppsala, Sweden)
MTT assay to measure DNA star cytotoxicity (Steps 142–155)
Hemocytometer (Hausser Scientific, cat. no. 3100)
Microtiter plate reader with a filter in the wavelength range of 550–600 nm (Synergy, cat. no. H1 Hybrid Multi-Mode Reader)
96-well flat-bottomed microtiter plate (Falcon, cat. no. 353072)
Reagent reservoirs (Corning Costar Reagent Reservoirs, cat. no. 4870)
Plaque reduction test (antiviral assay) (Steps 156–166)
Light Box for visualization of plaques (Fisher Scientific, cat. no. 50-949-588)
Six-well tissue culture plates (Corning, cat. no. 3506)
Aerosol tight rotor (Eppendorf, cat. no. FA-45-30-11)
Sterile tubes, 5 mL (Falcon, cat. no. 3–352063)
Reagent setup
Buffer 1
Buffer 1 is 10× TBE buffer (1.3 M Tris, 0.45 M boric acid and 25 mM EDTA). Add 108 g of Tris base, 55 g of boric acid and 7.5 g of EDTA to 800 mL of Milli-Q water, and mix to dissolve. Use HCl or NaOH solution to adjust the pH of the buffer to 8.0. Then, add Milli-Q water to a final volume of 1 L, and store the buffer in a 1.0 L or 2.0 L glass bottle. If precipitation is present, warm to 37 °C and mix using a magnetic stir bar and plate until completely dissolved prior to dilution. ▲ CRITICAL The buffer should be stored at room temperature (RT, 22–25 °C) for <3 months before a fresh buffer is prepared again for optimal buffer performance. Discard buffer if it becomes cloudy during the 3-month storage period.
Buffer 2
Buffer 2 is 1× TBE buffer (0.13 M tris, 45 mM boric acid and 2.5 mM EDTA). Add 100 mL of 10× TBE buffer (Buffer 1) to 900 mL of Milli-Q water, and use a magnetic stir bar and plate to mix the solution to prepare 1 L of 1× TBE buffer. Filter the 1× working solutions through a 0.22 μm filter before use. 1× working solutions can be used at RT for 3 months. 1 L 1× TBE buffer will be used immediately before gel electrophoresis, which is sufficient for running four denaturing gel using Bio-Rad Mini Protean II system. ▲ CRITICAL The buffer should be stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance. Discard buffer if it becomes cloudy during the 3-month storage period.
Buffer 3
Buffer 3 is elution buffer (0.5 M ammonium acetate, 10 mM magnesium acetate). Add 3.85 g of ammonium acetate and 0.14 g of magnesium acetate in 100 mL of Milli-Q water, and mix to dissolve using a magnetic stir bar and plate. Store the buffer in a 250 mL or 500 mL glass bottle. 100 mL elution buffer is sufficient for eluting ~100 ssDNA after PAGE purification. ▲ CRITICAL The buffer should be stored at RT for <6 months before a fresh buffer is prepared again for optimal buffer performance. Discard buffer if it becomes cloudy during the 6-month storage period.
Buffer 4
Buffer 4 is 10× TAE-Mg2+ buffer (400 mM Tris base, 200 mM acetic acid, 20 mM EDTA, 125 mM Mg (CH3COO)2). Add 48.5 g of Tris base, 11.4 mL of acetic acid, 7.5 g of EDTA and 26.8 g of Mg (CH3COO)2 to 800 mL of Milli-Q water, and mix using a magnetic stir bar and plate to dissolve. Use acetic acid or NaOH solution to adjust the pH of the buffer to 7.5. Then, add Milli-Q water to a final volume of 1 L. Store the buffer in a 1.0 L or 2.0 L glass bottle. ▲ CRITICAL The buffer should be stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance. Discard buffer if it becomes cloudy during the 3-month storage period.
Buffer 5
Buffer 5 is 1× TAE-Mg2+ buffer (40 mM Tris base, 20 mM acetic acid, 2 mM EDTA and 12.5 mM Mg (CH3COO)2). Add 100 mL of 10× TAE-Mg2+ buffer (Buffer 4) to 900 mL of Milli-Q water, and use a magnetic stir bar and plate to mix the solution to prepare 1 L of 1× TAE-Mg2+ buffer immediately before the gel electrophoresis. 1 L 1× TAE-Mg2+ buffer is sufficient for running four PAGE gels using Bio-Rad Mini Protean II system, or for running one agarose gel using an Owl EasyCast B1A mini gel electrophoresis system. ▲ CRITICAL The buffer should be stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance. Discard buffer if it becomes cloudy during the 3-month storage period.
Buffer 6
Buffer 6 is 10× TAE-Mg2+-K+ buffer (400 mM Tris base, 200 mM acetic acid, 20 mM EDTA, 125 mM Mg (CH3COO)2, 100 mM KCl). Add 48.5 g of Tris base, 11.4 mL of acetic acid, 7.5 g of EDTA, 26.8 g of Mg (CH3COO)2 and 7.5 g KCl to 800 mL of Milli-Q water, and mix using a magnetic stir bar and plate to dissolve. Use acetic acid or NaOH solution to adjust the pH of the buffer to 7.5. Then, add Milli-Q water to make a final volume of 1 L. Store the buffer in a 1.0 L or 2.0 L glass bottle. ▲ CRITICAL The buffer should be stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance.
Buffer 7
Buffer 7 is 1× TAE-Mg2+-K+ buffer (40 mM Tris base, 20 mM acetic acid, 2 mM EDTA and 12.5 mM Mg (CH3COO)2, 10 mM KCl). Add 100 mL of 10× TAE-Mg2+-K+ buffer (Buffer 6) to 900 mL of Milli-Q water, and use a magnetic stir bar and plate to mix the solution to prepare 1 L of 1× TAE-Mg2+-K+ buffer. 1 L 1× TAE-Mg2+-K+ buffer is sufficient for running four PAGE gels using Bio-Rad Mini Protean II system, or for running one agarose gel using an Owl EasyCast B1A mini gel electrophoresis system, or for running three replicates of SPR assays. ▲ CRITICAL The buffer should be stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance.
Buffer 8
Buffer 8 is 10× PBS (1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 18 mM KH2PO4). Add 80 g of NaCl, 2 g of KCl, 14.4 g of Na2HPO4 and 2.4 g of KH2PO4 to 800 mL of Milli-Q water, and mix using a magnetic stir bar and plate to dissolve. Use HCl to adjust the pH of the buffer to 7.4. Then, add Milli-Q water to make a final volume of 1 L. Store the buffer in a 1.0 L or 2.0 L glass bottle. ▲CRITICAL The buffer should be stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance.
Buffer 9
Buffer 9 is 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). Add 100 mL of 10× PBS (Buffer 8) to 900 mL of Milli-Q water, and use a magnetic stir bar and plate to mix the solution to prepare 1 L of 1× PBS buffer. Sterilize PBS by either autoclave in liquid cycle or filtration through a 0.22 μm membrane. PBS buffer is aliquoted in 200 mL volumes in a 250 mL glass bottle and sufficient for multiple procedures in the assays in this protocol. ▲ CRITICAL The buffer is stored at RT for <3 months before a fresh buffer is prepared again for optimal buffer performance.
20% denaturing gel solution (20% acrylamide, 1× TBE buffer, 7 M urea)
Mix 500 mL 40% acrylamide, 100 mL 10× TBE buffer (Buffer 1) and 420 g urea. Use a magnetic stir bar and plate to mix the mixture overnight on a hot plate heated to ~30 °C to dissolve. Add Milli-Q water to 1 L and mix. Store the gel solution in a 1.0 L or 2.0 L brown glass bottle. 1 L 20% denaturing gel solution is sufficient for making ~15 20% gels, ~30 10% gels or ~60 5% gels. ▲ CRITICAL The gel solution should be stored at RT for <3 months before a fresh gel solution is prepared again for optimal performance. Discard if the gel solution becomes cloudy during the 3-month storage period. ▲ CRITICAL Do not store denaturing gel solution in 4 °C fridge or cold room because urea can precipitate out of the solution over time. ! CAUTION Acrylamide monomer has been reported to affect the peripheral nervous system and cause sensory impairment. When handling the gel solution, PPE, including laboratory coat, gloves and goggles, should be worn throughout.
0% denaturing gel solution (0% acrylamide, 1× TBE buffer, 7 M urea)
Mix 500 mL Milli-Q water, 100 mL 10× TBE buffer (Buffer 1) and 420 g urea. Use a magnetic stir bar and plate to mix the mixture overnight on a hot plate set to ~30 °C to dissolve. Add Milli-Q water to 1 L and mix. Store the gel solution in a 1.0 L or 2.0 L brown glass bottle. 1 L 20% denaturing gel solution is sufficient for making ~15 20% gels, ~30 10% gels or ~60 5% gels. ▲ CRITICAL The gel solution should be stored at RT for <3 months before a fresh gel solution is prepared again for optimal performance. Discard if the gel solution becomes cloudy during the 3-month storage period. ▲ CRITICAL Do not store denaturing gel solution in 4 °C fridge or cold room because urea can precipitate out of the solution over time.
2x denaturing loading dye (10 mM NaOH, 1 mM EDTA, 30% (vol/vol) glycerol, 0.25% (wt/vol) bromophenol blue, 0.25% (wt/vol) xylene cyanol FF)
Add 0.33 mL of NaOH (3 M), 0.2 mL of EDTA (0.5 M), 30 mL glycerol, 0.25 g of bromophenol blue, and 0.25 g of xylene cyanol FF to 50 mL Milli-Q water, and use a magnetic stir bar and plate to mix to dissolve. Add Milli-Q water to a final volume of 100 mL loading dye solution. Store the loading dye in a 250 mL or 500 mL glass bottle. If bromophenol blue and/or xylene cyanol FF migrate at the same rate as the interested DNA oligos, they can be replaced with orange G at a concentration of 0.15% (wt/vol). 100 mL denaturing loading dye can be used to load 100 mL DNA samples onto denaturing gels. ▲ CRITICAL The solution should be stored at RT for <1 year before a fresh denaturing loading dye is prepared again for optimal performance.
Sodium chloride, NaCl (5 M)
Dissolve 2.92 g of sodium chloride in 8 mL of water. Use a magnetic stir bar and plate to mix to dissolve, and add water to get a final volume of 10 mL. Store the solution in a 50 mL glass bottle. ▲ CRITICAL The solution should be stored at RT for <6 months before a fresh solution is prepared again for optimal performance. Discard solution if it becomes cloudy during the 6-month storage period.
70% ethanol
Mix and manually shake 70 mL 200-proof ethanol with 30 mL water. Store it in a 250 mL glass bottle. ▲ CRITICAL 70% ethanol should be stored at −20 °C for <2 months before a fresh solution is prepared again for optimal performance.
10% (wt/vol) APS
Add 0.5 g of APS to 5 mL of Milli-Q water and vortex to dissolve. Aliquot the solution into 0.5 mL each, and store in 1.5 mL Eppendorf tubes. ▲ CRITICAL APS solution should be stored at 4 °C for <2 weeks before a fresh solution is prepared again for optimal performance.
Gel staining solution
Add 5 μL of GelRed nucleic acid stain (10,000× aqueous solution) to 50 mL of Milli-Q water, and vortex to dissolve. ▲ CRITICAL The solution should be stored at RT in a dark place for <2 weeks before a fresh solution is prepared again for optimal performance.
Sodium acetate, Na (CH3COO) (100 mM)
Add 8.2 g sodium acetate to 900 mL Milli-Q water, and use a magnetic stir bar and plate to mix to dissolve. Use HCl solution to adjust the pH of the solution to 4.5. Then, add Milli-Q water to make a final volume of 1 L. Store the solution in a 1.0 or 2.0 L glass bottle. ▲ CRITICAL The solution should be stored at 2–8 °C for <2 months before a fresh solution is prepared again for optimal performance.
Sodium chloride, NaCl (2 M)
Add 11.7 g of NaCl in 70 mL Milli-Q water, and vortex to dissolve. Then, add Milli-Q water to make a final volume of 100 mL. Store the solution in a 250 mL glass bottle. ▲ CRITICAL The solution should be stored at RT for <6 months before a fresh solution is prepared again for optimal performance.
Growth medium for Vero cells
EMEM with 10% (vol/vol) heat-inactivated FBS. Heat inactivate FBS at 56 °C in a water bath for 30 min. Optional: addition of 100 units/mL penicillin and 100 μg/mL streptomycin sulfate. Add antibiotics if opting for absolutely no bacterial contamination in cell medium. ▲ CRITICAL The solution should be stored at 2–8 °C and prepared freshly monthly for optimal performance.
Maintenance medium for Vero cells
EMEM with 2% (vol/vol) heat-inactivated FBS. Heat inactivate FBS at 56 °C in a water bath for 30 min. Optional: addition of 100 units/mL penicillin and 100 μg/mL streptomycin sulfate. Add antibiotics if opting for absolutely no bacterial contamination in cell medium. ▲ CRITICAL The solution should be stored at 2–8 °C and prepared freshly monthly for optimal performance.
Sucrose gradient solutions (10%, 20%, 40%, 60% wt/vol)
Add 10 g, 20 g, 40 g or 60 g of sucrose to 10 mL 10× PBS buffer (Buffer 8) and 40 mL of Milli-Q water, and use a magnetic stir bar and plate to mix to dissolve. Add Milli-Q water to 100 mL final volume. Store each solution in a 250 mL glass bottle at 4 °C.
BA-1 diluent
BA-1 diluent contains M199-Hank’s medium, 1% (wt/vol) BSA, 0.05 M Tris buffer, 0.35 g/L sodium bicarbonate, 100 units/mL penicillin, 100 μg/mL streptomycin and 1 μg/mL fungizone. Add 100 mL 10× M199 with Hank’s salts (without l-glutamine), 10 g BSA, 50 mL 1M Tris-HCl pH 7.5, 10 mL l-glutamine (100×, 200 mM ready to use solution), 0.35 g sodium bicarbonate, 10 mL 100× penicillin–streptomycin, 1 g fungizone and Milli-Q water to 1 L. Use a magnetic stir bar and plate to mix the mixture to dissolve. Store the solution in a 1.5 L glass bottle. 100 mL BA-1 diluent should be sufficient for the assays in this protocol. ▲ CRITICAL The solution should be stored at 2–8 °C for up to 6 weeks before a fresh solution is prepared again for optimal performance.
First overlay medium
2× EMEM with 10% (vol/vol) FBS, 200 units/mL penicillin and 100 μg/mL streptomycin. Dissolve two packages of 1 L MEM powder in Milli-Q water into 1 L. Add 100 mL heat-inactivated FBS, 20 mL 100× penicillin–streptomycin. Use a magnetic stir bar and plate to mix the mixture to dissolve. Use 0.22 μm filter to sterilize the solution. Store the solution in a 1 L glass bottle. Prepare at least 10 mL for each six-well plate used in the assay. ▲ CRITICAL The solution should be stored at 2–8 °C for up to 4 weeks before a fresh solution is prepared again for optimal performance.
Second overlay medium
2× EMEM with 2% (vol/vol) FBS, 200 units/mL penicillin and 100 μg/mL streptomycin. Dissolve two packages of 1 L MEM powder in Milli-Q water into 1 L. Add 20 mL heat-inactivated FBS and 20 mL 100× penicillin–streptomycin.
EMEM supplemented with 20% FBS and penicillin–streptomycin
Add 10 mL heat-inactivated FBS and 10 mL 100× penicillin–streptomycin to 500 mL EMEM. Use a magnetic stir bar and plate to mix the mixture to dissolve. Use 0.22 μm filter to sterilize the solution. Store the solution in a 1.0 L glass bottle. ▲ CRITICAL The solution should be stored at 2–8 °C for up to 4 weeks before a fresh solution is prepared again for optimal performance.
Oxoid agar for overlay
Add 1.2 g Oxoid agar to 100 mL Milli-Q water, and sterilize by autoclaving in liquid cycle and store at RT in a 250 mL glass bottle. Prepare 20 mL for each six-well plate used in the assay. Use Oxoid agar in plaque reduction or titration assays.
Controls used in the procedures
Step 50: ssDNA and partial DNA star controls prepared for nondenaturing gel electrophoretic characterization of the DNA star
Fix-1 control: add 1 μL of 6× loading buffer to 5 μL of 2 μM fix-1 strand post thermal annealing
Close-1 control: add 1 μL of 6× loading buffer to 5 μL of 2 μM close-1 strand post thermal annealing
Scaffold-1 control: add 1 μL of 6× loading buffer to 5 μL of 2 μM scaffold-1 strand post thermal annealing
Edge-1 control: add 1 μL of 6× loading buffer to 5 μL of 2 μM edge-1 strand post thermal annealing
Triangle control: add 1 μL of 6× loading buffer to 5 μL of 0.5 μM one-triangle complex (scaffold-1, edge-1, edge-2, close-1, fix-1, fix-5) post thermal annealing
Triangle control: add 1 μL of 6× loading buffer to 5 μL of 0.25 μM two-triangle complex (scaffold-1, scaffold-2, edge-1, edge-2, edge-3, edge-4, close-1, close-2, fix-1, fix-2, fix-5) post thermal annealing
Triangle control: add 1 μL of 6× loading buffer to 5 μL of 0.2 μM three-triangle complex (scaffold-1, scaffold-2, scaffold-3, edge-1, edge-2, edge-3, edge-4, edge-5, edge-6, close-1, close-2, close-3, fix-1, fix-2, fix-3, fix-5) post thermal annealing
Triangle control: add 1 μL of 6× loading buffer to 5 μL of 0.15 μM four-triangle complex (scaffold-1, scaffold-2, scaffold-3, scaffold-4, edge-1, edge-2, edge-3, edge-4, edge-5, edge-6, edge-7, edge-8, close-1, close-2, close-3, close-4, fix-1, fix-2, fix-3, fix-4, fix-5) post thermal annealing
Unclosed star control: add 1 μL of 6× loading buffer to 5 μL of 0.1 μM unclosed five-triangle complex (scaffold-1, scaffold-2, scaffold-3, scaffold-4, scaffold-5, edge-1, edge-2, edge-3, edge-4, edge-5, edge-6, edge-7, edge-8, edge-9, edge-10, fix-1, fix-2, fix-3, fix-4, fix-5) post thermal annealing
Step 79: mock infection control
Mock-infection control flask: add 5 mL of BA-1 diluent or maintenance medium to a T-75 flask.
Step 112: mock infection control
Mock-infection control six-well plate: add 3 mL per well of BA-1 diluent to a six-well plate.
Step 137: control flow cell preparation for SPR assays
Control flow cell: activate carboxymethyl groups on CM5 chip surface using an injection pulse of 35 μL (flow rate 5 μL/min) of an equimolar mix of EDC and NHS in water (final concentration of 50 μM, manually mix immediately before injection). Following the activation, inject 1 mL 100 mM sodium acetate pH 5.0 buffer over the activated biosensor surface. Then, deactivate excess unreacted carboxymethyl groups on the sensor surface with a 35 μL injection of 1 M ethanolamine.
Step 140: partial and full stars prepared for relative binding affinity comparison using SPR assays
One-triangle, two-triangle, three-triangle, four-triangle and full star scaffolds were used to carry three, five, seven, nine and ten aptamers, respectively. All scaffolds were prepared with a normalized aptamer concentration of 1.2 μM.
Step 152: cytotoxicity assay of the DNA star complex
Background control: medium-only (no cells)
Untreated cell control: cells in medium without DNA star complexes. This gives the optimal cell viability reading
Dilution buffer control: if the dilution buffer for DNA star complexes is not the cell growth medium, include a buffer control with no DNA star complexes to assess the impact of dilution buffer on the cells. Some cell types may be more sensitive to dilution buffers, so it is preferable to add test samples in as small a volume as possible to reduce the buffer’s impact on cell growth
Step 160: antiviral assay of the DNA star complex
Medium-only control: consists of virus suspensions mixed with an equal volume of BA-1. This serves as the untreated control and is the considered the maximal number of PFU under the assay condition. For each well of a six-well plate, mix 100 μL of virus suspension with 100 μL of BA-1 diluent
Buffer-only control: consists of DNA star dilution buffer mixed with an equal volume of virus suspension. This serves to measure the impact from DNA star dilution buffer. For each well of a six-well plate, mix 100 μL of virus suspension with 100 μL of DNA star dilution buffer
Positive control: consists of a NAb (or other positive control when it is appropriate and available). For each well of a six-well plate, mix 100 μL of virus suspension with 100 μL of NAb or positive control
Cell control or mock-infection control with no virus or treatment: This control should yield no virus infection, hence no plaques, and serves as the internal control of no cross-contamination of virus. For each well of a six-well plate, prepare 200 μL of BA-1 diluent
Procedure
Identification of virus antigen spatial patterns ● Timing ~2–3 d
▲ CRITICAL The hands-on time to use PyMol to identify a viral antigen pattern based on existing cryo-EM data is ~2–3 d. This does not include time to collect or generate the cryo-EM structure of the virus as this is assumed to be available. Here we use the PDB file 1p58 that was solved by Zhang et al90. Visualization of the arrangement of the QHGTI motif on the dengue viral particle was achieved using the commands in Steps 1–4 below. Identification of the arrangement of the QHGTI motif can also be achieved by using the 3J27 PDB structure previously solved by Zhang et al.90.
Open 1p58 PDB structure file on PyMol.
-
View the structure of each of the individual subunits in the biological assembly (viral particle) simultaneously in PyMol to determine their arrangement. Use the following commands to visualize the biological assembly:
fetch 1p58, Type=pdb1 split_states 1p58
▲ CRITICAL STEP If it is easier to work with a white background, this can be adjusted using the command: bg_color white.
- Use the PyMol sequence viewer to select the QHGTI sequence motif on the dengue E protein (see a PyMol window in Supplementary Fig. 2) following the instructions below. Then, color the sequence to highlight its exact location on the viral particle. Repeat for each of the 60 subunits of the biological assembly to identify the location of all antigenic epitopes on the virus.
-
Use command ‘color red, resi 316–320’ to color all of the QHGTI motifs red on the viral particle (Fig. 4a). This command colors the QHGTI motif throughout the entire viral particle structure and allows for differentiation from other amino acids
-
Distinguish three-member and five-member antigen clusters using the color command included above to differentially color the amino acids in the QHGTI motif that make up each of the two clusters (Fig. 4b)
-
-
Measure the distance between neighboring epitopes visible on the surface of the viral particle to identify epitopes present in repeating clusters (Fig. 4c). We recommend measuring the distance between the Cα of the glycine residue ‘G’ in the QHGTI motif of neighboring epitopes, since epitopes are present on domains from neighboring subunits in the biological assembly (Fig. 4d). Use the following command to measure the distance between two atoms on different domains on two different subunits to generate a pentagon cluster of epitopes:
dist /1p58_0001//C/318/CA, /1p58_0002//C/318/CA dist /1p58_0002//C/318/CA, /1p58_0003//C/318/CA dist /1p58_0003//C/318/CA, /1p58_0004//C/318/CA dist /1p58_0004//C/318/CA, /1p58_0005//C/318/CA dist /1p58_0001//C/318/CA, /1p58_0004//C/318/CA
In the commands above: 000* represents the specific subunit in the biological assembly, C is the domain where the epitope is located, ‘318’ is the amino acid position in the domain, and CA is the Cα carbon that will be used to measure the distance between individual epitopes. When using another PDB structure file, for example the 3J27 structure instead of 1p58, replace ‘1p58’ in the commands above with ‘3j27’ (or the relevant PDB file name of choice).
▲ CRITICAL STEP Identify repeating clusters by groups of antigenic epitopes that are separated by the same distance. It is important to only use epitopes visible on the surface of the viral particle as these are the only ones that the aptamer on the DDN can bind to.
▲ CRITICAL STEP DNA_Star_Viral_Protocol_Dengue_Model.pse. PyMol session file (available on 10.6084/m9.figshare.c.5409411) was used for generating Fig. 3 (second panel), Fig. 4, Fig. 5a,b and Supplementary Figs. 1 and 2. This file is loaded with the structure of the dengue viral particle (PDB: 1p58) as solved by Zhang et al. Once the file is loaded, the user will notice that the biological assembly is loaded using 60 subunits labeled 1p58_0001–1p58_0060. Additionally, users will be able to identify antigen binding sites that are part of a triangular (cyan) and pentagonal cluster. Lines used to determine the distance between individual binding sites within each cluster are displayed in magenta for the triangular clusters and green for the pentagonal clusters. Note that the commands necessary to generate this analysis are available in the above procedure steps.
Determine the distance between individual clusters by measuring the arc length from the midpoint of each triangular or pentagonal cluster (Fig. 5a). PyMol allows for direct distance measurements between two atoms in the rendered object.
Design of the DNA star ● Timing ~4 h
-
3
Use a software such as Microsoft Powerpoint or Adobe Illustrator to draw a single-line skeleton of the star shape based on the known center to center distances between ED3 clusters on the viral outer surface (from Step 5) (Fig. 6a).
-
4
Convert the single-line edge skeleton into a diagram of double-line edges, representing complementary DNA duplexes. Use short perpendicular lines between the two parallel lines to represent hydrogen bonds (Fig. 6b).
-
5
Use a software such as Microsoft Powerpoint or Adobe Illustrator to draw green lines (indicating unpaired Ts) to connect all the nicks from the above double-line skeleton diagram to make a star shape drawing with continuous lines (Fig. 6c).
-
6
Make nicks on the closed lines to generate line segments indicating individual component DNA oligos (‘scaffold’, ‘edge’, ‘fix’, ‘close’). Use single-line regions to indicate ssDNA form of the hairpins after they are fully stretched (Fig. 6d).
-
7Place arrows at the end of the lines to indicate the 3′-ends of the DNA. Color all the strands of a same kind (‘scaffold’, ‘edge’, ‘fix’, ‘close’) using the same color (Fig. 6e). Use the following key commands (functions), as clearly elaborated in the SEQUIN_User_Instruction.PDF, to assign the DNA sequences in SEQUIN, which uses a sequence-symmetry minimization algorithm to maximize control over the secondary structure of branched nucleic acids13,103.
- NEWARM: define each edge of the DNA star as a new ‘arm’ of a given length in SEQUIN
- LOOPS: establish a ssDNA loop connection (indicated as a green line at each of the triangle corners) between two adjacent DNA star edges
- SET: set the sequence of all the green-line-indicated loops with base T
- CRUNCH: after the sequence programming preparation using the above key commands, this CRUNCH command automatically sets the sequence of a short stretch of DNA segment. We suggest CRUNCH-ing segments three bases long one at a time when programming the DNA star sequences, including the 3′ extended sequences on the five ‘scaffold’ strands (scaffold-1, 2, 3, 4, 5) and five ‘edge’ strands (edge-1, 3, 5, 7, 9) to arrange ten aptamer molecules in Step 11
-
8
Extend the 3′-end of all the ‘scaffold’ strands and five ‘edge’ strands at ten specific vertices to arrange ten aptamer molecules into a star shape (Fig. 6f).
-
9Order DNA sequences from your preferred vendor. We order DNA oligonucleotides from IDTDNA or Sangon Biotech. The turnaround time for DNA oligos from IDTDNA is ~2 d for 250 nmol scale or ~7 d for 1–2 μmol scale, and the oligos are desalted by the manufacturer but not PAGE purified. Turnaround time for DNA oligos from Sangon Biotech is 3–4 d for 250 nmol scale or above, and oligos are PAGE purified by the manufacturer. If your oligos are PAGE purified by the manufacturer, proceed to Step 42. If your oligos are not PAGE purified by the manufacturer, continue to Step 13. The sequences we used to form a DENV-targeting antiviral DNA star are listed in Supplementary Tables 1–3. For the following procedure, we recommend ordering 2 μmol scale of each sequence. For a five-point DNA star you will need to order 26 sequences including:
- Five scaffold sequences (2 μmol scale)
- Ten edge sequences (2 μmol scale)
- Five fix sequences (2 μmol scale)
- Five close sequences (2 μmol scale)
-
One aptamer sequence (2 μmol scale). Use ED3-targeting aptamer to target ED3 on DENV virus surface.▲ CRITICAL STEP After receiving the samples, centrifuge all the DNA-containing tubes at 13,600g at RT for 3 min to spin down the lyophilized DNA product before use. Do not open the tube cap before centrifugation to prevent the loss of the lyophilized DNA product.
Denaturing PAGE for oligonucleotide purification ● Timing ~20 h Gel preparation
-
10For optimal resolution of DNA fragments using denaturing PAGE, select the appropriate concentration of polyacrylamide gel based on the length of your DNA oligo product using the following table:
Acrylamide (%) Fragment sizes separated (bases) 20 8–25 10 25–35 8 35–45 6 45–70 5 70–300 4 100–500 -
11Prepare the acrylamide concentration for the optimal resolution based on the length of your DNA following the recipe in the table below for a 20 mL denaturing gel solution. You will need 5 mL of gel solution for a 10.1 cm × 8.2 cm × 1 mm gel, or 60 mL of gel solution for a larger, 20 cm × 22 cm × 1.5 mm gel. Scale the below recipe accordingly. Prepare gel solution in a 50 mL conical tube.
Acrylamide concentration 20% 10% 8% 6% 5% 4% 20% denaturing gel solution (see ‘Reagent setup’) 20 mL 10 mL 8 mL 6 mL 5 mL 4 mL 0% denaturing gel solution (see ‘Reagent setup’) 0 mL 10 mL 12 mL 14 mL 15 mL 16 mL Total gel volume 20 mL 20 mL 20 mL 20 mL 20 mL 20 mL ▲ CRITICAL STEP Use longer gels to resolve single base pair differences in products.
-
12
Assemble the gel box and fix the gel in the gel-casting chamber according to the manufacturers’ description.
-
13
Add 50 μL of 10% (wt/vol) APS to the gel solution prepared in a 50 mL conical tube (from Step 14), and vortex thoroughly.
-
14
Add 8 μL TEMED solution to the same 50 mL conical tube, and vortex again to ensure thorough mixing.
▲ CRITICAL STEP Polymerization begins as soon as APS is added to the mixture, so all subsequent actions must be performed quickly, especially for high-concentration gels, which polymerize faster than low-concentration gels.
! CAUTION Polyacrylamide might contain small quantities of unpolymerized acrylamide, which has been reported to affect peripheral nervous system and cause sensory impairment. Handle carefully with appropriate PPE.
! CAUTION TEMED and APS are highly flammable and toxic liquids. They can cause severe skin burns and eye damage.
! CAUTION When handling these reagents, PPE, including laboratory coat, gloves and goggles, should be worn throughout and when handling the gel.
-
15
Immediately pour the acrylamide gel solution between the gel plates (Fig. 7a), and then insert the comb (Fig. 7b).
-
16
Clamp the comb in place at the top of the gel to avoid gel leakage and gel separation from the plates during polymerization. Let the gel polymerize for 30 min to solidify.
-
17
Once solidified, remove the comb from the gel.
? TROUBLESHOOTING
-
18
Place the gel into the electrophoresis tank with the shorter plate facing the inner reservoir.
-
19
Add 1× TBE buffer (Buffer 2) to the fill line, and rinse out the wells with buffer to remove urea or bubbles.
-
20
Prerun the gel for at least 30 min at 5 V/cm (constant voltage) before adding your samples.
-
21
Prepare the DNA oligo samples (from Step 12) using a maximum DNA concentration of 2 OD per gel. Add an equal volume of 2× denaturing loading dye/buffer. Do not use the wells on the edges/end of the gel (Fig. 7c).
▲ CRITICAL STEP Do not load >10 μL of DNA sample per well. If the loading dye and the target DNA migrate at the same rate, load the samples without the tracking dye, and instead load 5 μL of tracking dye mixture into an unused well to prevent interference with UV detection. Alternatively, use orange-G-containing loading dye.
-
22Run the gel at 450 V for 60 min or until the dye has migrated two-thirds down the gel. The migration of tracking dye (equivalent to DNA oligo size in bases), bromophenol blue and xylene cyanol FF in different acrylamide gel concentrations is provided in the following table, and can be used to track the gel progress (Fig. 7d).
Acrylamide (%) Migration of bromophenol blue (bases) Migration of xylene cyanol FF (bases) 20 8 28 10 12 55 8 19 75 6 26 105 5 35 130 4 ~50 ~230 ? TROUBLESHOOTING
-
23
Carefully remove the gel from the tank. Stain the gel in ~50 mL of H2O with 5 μL of 10 mg/mL ethidium bromide aqueous solution for ~10–30 min without rocking. Alternatively, use GelRed nucleic acid gel stain (Biotium, cat. no. 41003), which is less toxic than ethidium bromide, to stain the gel for DNA oligo purification.
▲ CRITICAL STEP Gently pick the gel up from the plate or staining box as it is so thin that it can be easily torn.
! CAUTION Ethidium bromide is a powerful mutagen. When handling ethidium-bromide-containing buffer solutions, use proper PPE, including double gloves, laboratory coat, closed-toe shoes and laboratory safety goggles, to mitigate potential exposure to the reagent. Collect any liquid waste in a bottle with a clear label. Manage waste pickup with the Division of Research Safety or a similar office in the institute.
Fig. 7 |. Process flow schematic of the purification of DNA oligos using denaturing gel electrophoresis.
a,b, Preparing acrylamide solution with comb for gel electrophoresis. c, Loading DNA oligo samples into the wells of the gel. d, Gel electrophoresis of DNA oligo samples. e, Examining the gel using a UV transilluminator, cut and collect gel pieces into an Eppendorf tube for DNA sample elution.
Isolation of target band from polyacrylamide gels
-
24
Examine the gel using a UV transilluminator. In a 4 °C cold room, use a clean and sharp scalpel or razor blade to excise the band of interest (corresponding to the molecular weight of your purified DNA oligo) from the gel. Cut as closely as possible around the band to avoid contamination.
! CAUTION Protect yourself from UV exposure by wearing face and eye protection as well as a laboratory coat.
! CAUTION Wear thick gloves and take care to avoid injuring yourself when using the scalpel.
? TROUBLESHOOTING
-
25
Cut the polyacrylamide gel fragments into small pieces (~2–3 mm squares), and collect them into a 1.5 mL Eppendorf tube (Fig. 7e).
-
26
Add 400 μL of elution buffer (Buffer 3) to the tube, and elute the DNA by shaking the tube gently in a 4 °C cold room overnight.
-
27
Bring the tube to RT, and centrifuge the DNA solution in the Eppendorf tube at 16,000g for 2 min. Transfer the elution buffer (Buffer 3) into a new Eppendorf tube by leaving all the gel debris in the original tube.
-
28
Add 100 μL of elution buffer (Buffer 3) to the remaining gel debris in the original Eppendorf tube. Vortex and centrifuge the tube again at 16,000g for 2 min. Collect the extra elution buffer (Buffer 3), and mix it with the previous supernatant in the collection tube. Repeat this step again. You should ultimately have ~600 uL of elution buffer (Buffer 3) containing your purified DNA oligo.
-
29
Add 800 μL of n-butanol to this ~600 μL of elution buffer (Buffer 3), and vortex briefly. At this point, two separate layers are formed, with n-butanol on the top of the elution buffer (Buffer 3). ! CAUTION n-Butanol is highly flammable. It causes eye irritation. Prolonged exposure to skin causes drying, redness and possible blistering. Handle with care and appropriate PPE.
-
30
Remove the top layer carefully using a pipette, and discard it into a chemical waste bottle.
-
31
Add 800 μL of n-butanol to the same tube, vortex, and repeat Step 33.
-
32
Add 800 μL of n-butanol to the same tube, vortex, and repeat Step 33. The final solution remaining in the Eppendorf tube should be reduced to ~200 μL (this is your purified DNA oligo).
Ethanol precipitation of DNA oligos
-
33
Add 20 μL of 5M NaCl and 1 mL of 100% ethanol to the ~200 μL DNA oligo solution.
-
34
Briefly vortex and incubate the sample on dry ice for ~30 min in a 4 °C cold room.
-
35
Quickly transfer the samples to a centrifuge, and spin down at 16,000g for 30 min at 4 °C.
-
36
Discard the supernatant, and wash the pellet with 1 mL of cold 70% ethanol (precooled in −20 °C freezer).
-
37
Centrifuge again immediately after adding 70% ethanol at 16,000g for 5 min, and discard the supernatant.
-
38
Dry the DNA pellet by leaving the cap of the Eppendorf tube open at RT overnight.
▲ CRITICAL STEP Use a piece of Kimwipe paper to cover the open tubes to protect the sample from contamination.
? TROUBLESHOOTING
Assembly of the DNA star ● Timing ~12 h
-
39
Dissolve each of the purified DNA oligonucleotides (from Step 12 or Step 41) in nuclease-free water. We typically add ~100 μL of nuclease-free water to the DNA pellet from Step 41. If your DNA is PAGE-purified by the manufacturer from Step 12 (e.g., from Sangon Biotech), add nuclease-free water to make an initial ~100 μM stock solution based on the manufacturer-reported amounts. Vortex the tube until the DNA powder is completely dissolved (~15 s).
■ PAUSE POINT The dissolved DNA oligonucleotides should be stored at −20 °C for <1 year for optimal performance.
-
40
Use a UV-visible spectrophotometer to measure the absorbance of each DNA oligo stock at 260 nm. Calculate the precise concentration of DNA using Beer–Lambert law: A = ɛbc (A is the absorbance at 260 nm; b is the path length of the cuvette, in cm; ɛ is the extinction coefficient provided by the DNA manufacturer, in L/(mole·cm)). After measuring the stock concentrations, dilute each DNA oligo to 10 μM in nuclease-free water to a final volume of 200 μL. Of note, aptamer DNA oligo will be used as is without PAGE purification. Dilute aptamer oligo to 100 μM in nuclease-free water to a final volume of 200 μL.
▲ CRITICAL STEP Accurate DNA concentrations are critical for successfully producing DNA stars as oligos must be added in precise stoichiometric ratios for efficient self-assembly.
-
41Add stoichiometrically equivalent amounts of each of the component DNA strands to a PCR tube containing nuclease-free water and annealing buffer. For a five-point star, this includes adding stoichiometric amounts of the 25 components (all DNA sequences from Supplementary Table 1, excluding the virus-targeting aptamer). An ideal recipe for making 100 μL of the five-point DNA star scaffold is shown in the following table:
Component Amount (μL) Final concentration 25 purified DNA strands (10 μM, from Step 42) 2 (of each strand; 50 total) 100 nM 10× TAE-Mg2+ buffer (Buffer 4) 10 1× Nuclease-free water 40 Total 100 -
42
Put the PCR tube into a thermocycler and incubate the mixture at 95 °C for 5 min, and then slowly cool it down to 20 °C at a rate of 0.15 °C per min over a period of 8 h.
-
43
Mix 2 μL of the aptamer (100 μM, from Step 42) with 100 μL of the annealed five-point DNA star scaffold (0.2 μM). Incubate in the thermocycler at 30 °C for 1 h and then 25 °C for 1 h to produce five-point DNA star complexes functionalized with the virus-targeting aptamers. For six-point star, mix 1.2 μL of the aptamer (100 μM, from Step 42) with 100 μL of the annealed six-point DNA star scaffold (0.2 μM) before thermal annealing. For seven-point star, mix 1.4 μL of the aptamer (100 μM, from Step 42) with 100 μL of the annealed seven-point DNA star scaffold (0.2 μM) before thermal annealing. ■
PAUSE POINT Store the DNA star complex at 4 °C for short-term storage (<4 weeks) before downstream use. Store DNA star complex at −20 °C for long-term storage.
Nondenaturing gel electrophoretic characterization and purification of the DNA star ● Timing ~6 h
-
44
Wash the gel-casting module (a casting stand, a casting frame, a comb, a short plate and a spacer plate) before casting the gel.
-
45Follow the recipe in the table below to prepare 15 mL of a 4% (wt/vol) nondenaturing PAGE gel. Mix the reagents below in a 50 mL conical tube or a 100 mL glass beaker, and vortex or stir the mixture well before pouring into a gel casting module.
Component Amount (mL) 40% acrylamide/bis-acrylamide (19:1) 1.5 10× TAE-Mg2+ buffer (Buffer 4) 1.5 Ultrapure water 11.9 10% APS 0.11 TEMED 0.01 Total 15 ! CAUTION Polyacrylamide might contain small quantities of unpolymerized acrylamide which has been reported to affect peripheral nervous system and cause sensory impairment. Handle with care and wear appropriate PPE.
! CAUTION TEMED and APS are highly flammable and toxic liquids. They can cause severe skin burns and eye damage.
! CAUTION When handling these reagents, PPE, including laboratory coat, gloves and goggles, should be worn throughout and when handling the gel.
-
46
Insert a comb after the gel casting module is fully filled with the gel solution. Ensure there are no bubbles in the module. Wait 30 min for the gel to solidify. Pull the comb out of solidified gel, and put the casting module with gel into the electrophoresis tank. Fill the electrophoresis tank with the gel running buffer that contains 1× TAE-Mg2+ (Buffer 5).
? TROUBLESHOOTING
-
47Prepare DNA samples for PAGE characterization. Add 1 μL of 6× loading buffer to 5 μL of DNA samples and controls (ssDNA controls, partial DNA star controls and full DNA star). Preparation of the controls is described in ‘Reagent setup’. The concentration of DNA in each of the samples should be prepared according to the following table:
Sample DNA complex concentration before adding loading buffer (μM) Fix strand 2 Close strand 2 Scaffold strand 2 Edge strand 2 One-triangle 0.5 Two-triangle 0.25 Three-triangle 0.2 Four-triangle 0.15 Star 0.1 Star without five ‘close’ strands 0.1 -
48
Load the DNA samples into the wells, put the lid on the electrophoresis tank and run electrophoresis at 60 V for 90 min.
▲ CRITICAL STEP Put the gel box in a 4 °C cold room or in an ice bath to prevent thermal denaturation of the DNA nanostructures.
? TROUBLESHOOTING
-
49
Carefully remove the gel from the glass plates and place the gel into GelRed staining solution at RT for 10 min. ! CAUTION GelRed is a potential carcinogen. Handle the solution carefully in a fume hood with PPE, including gloves, laboratory coat and goggles.
▲ CRITICAL STEP Handle the gel gently as it is thin so can tear easily.
-
50
Use a gel imaging doc/system to visualize and analyze the gel image. Adjust the imaging parameters, such as brightness and contrast, to clearly capture all DNA bands/species without overexposing. An example gel image for the DNA star is shown in Fig. 8. Of note, this step is for gel imaging and DNA star complex formation yield measurement. It is not for the purification of the DNA star complex, so DNA extraction is not needed.
! CAUTION UV radiation is harmful to eyes and skin, so be sure to wear UV light protection goggles and protective laboratory coat and gloves.
▲ CRITICAL STEP Avoid overexposing the gel image so that the intensities of the DNA species/bands on the gel can be used to accurately estimate DNA nanostructure yield.
? TROUBLESHOOTING
Fig. 8 |. Gel electrophoretic characterization of the DNA star nanostructure.
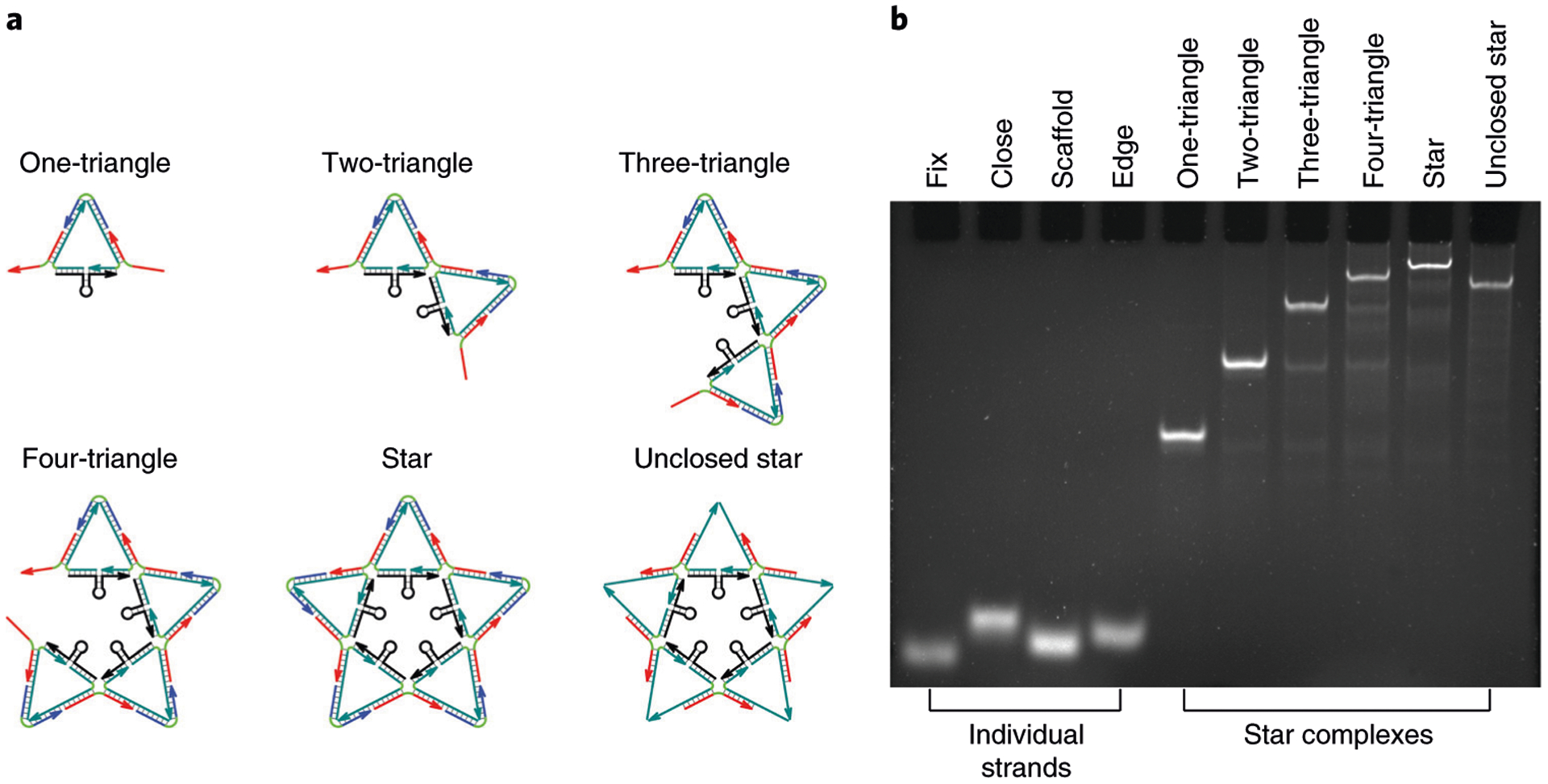
a, Schematic of the partial or complete star complexes. b, Characterization of the partial or complete DNA star structures using nondenaturing PAGE. Figure adapted with permission from ref.7, Springer Nature Ltd.
Purification of the DNA star complex
-
51
Prepare an agarose gel. Add 40 mL 1× TAE-Mg2+ buffer (Buffer 5) to a 250 mL Erlenmeyer flask.
▲ CRITICAL STEP Use a 250 or 500 mL flask that is large enough to avoid spilling the solution when melting the agarose.
-
52
Use an analytical balance to weigh 1.2 g of agarose powder, and add this to Buffer 5 in the flask. Record the total weight of agarose powder, buffer and flask.
-
53
Melt the agarose in a microwave oven (~2 min in total) to prepare a 3% (wt/vol) agarose gel.
▲ CRITICAL STEP Use a microwave oven to melt high-percentage agarose gels stepwise by heating the solution for 30 s and swirling the flask to fully mix the solution. Repeat the heating and swirling steps several times until the agarose powder is fully dissolved.
! CAUTION Use heatproof gloves when handling the hot flask for agarose gel preparation.
-
54
Weigh the flask after fully melting the agarose powder, and add Milli-Q water to recover the total weight recorded in Step 55 to ensure you maintain the correct percentage gel.
-
55
Wait for the agarose gel solution to cool to ~45–50 °C (but still above the agarose gelling temperature of 34–38 °C).
■ PAUSE POINT Keep the flask in a 45–50 °C water bath if not casting the gel immediately.
-
56
Add 4 μL SYBR Green I dye (10,000×) to the agarose solution, swirl to mix, pour into a casting tray containing a ten-tooth comb (1.5 mm thick) and wait for ~30 min until the prestained gel is cooled and fully solidified.
-
57
Slowly pull the comb out of the gel. Fit the casting tray into the gel box and cover the gel with 1× TAE-Mg2+ (Buffer 5) running buffer.
▲ CRITICAL STEP Do not add excess running buffer above the manufacturer-suggested ‘fill line’ marked on the gel box.
-
58
Load each well with 30 μL DNA star complex and dye mixture (5 μL of 6× loading buffer and 25 μL of 0.2 μM DNA star samples (from Step 46). Perform electrophoresis at 70 V for 120–150 min.
▲ CRITICAL STEP To minimize the volume of buffer used to elute the DNA star complex from the gel in Step 62, use a minimum number of wells by loading each well with a maximum amount of the DNA star sample. The maximum amount of DNA that can be loaded into each well is 30 μL.
▲ CRITICAL STEP Put the gel box in a 4 °C cold room or in an ice bath during electrophoresis to prevent thermal denaturation of the DNA star complex.
-
59
Examine the agarose gel using a UV transilluminator. Use a clean, sharp scalpel or razor blade to excise the band of interest from the gel.
▲ CRITICAL STEP Keep the size of the agarose gel slice as small as possible by removing all the non-DNA-containing parts of a gel piece. This will help to minimize the amount of buffer used to elute the DNA star in Step 60.
! CAUTION Protect yourself from UV exposure by wearing face and eye protection as well as a laboratory coat.
! CAUTION Wear thick gloves and take care to avoid injuring yourself when using the scalpel.
-
60
Insert the excised gel pieces into 5-cm-long 3.5 kDa dialysis tubing. Clip one end of the dialysis tubing, and add a small amount of 1× TAE-Mg2+ buffer (Buffer 5) to the tubing to immerse all the gel pieces within the tubing in buffer. Then, clip the other end of the dialysis tubing. Of note, use 3.5 kDa dialysis tubing for any DNA complex with a molecular weight >3.5 kDa.
-
61Insert the dialysis tubing containing gel pieces into the gel electrophoresis box, and fill the surrounding box with ~500 μL 1× TAE-Mg2+ buffer (Buffer 5) as illustrated in Fig. 9.
- Perform electrophoresis at 70 V for 30 min to run the SYBR Green-dye-labeled DNA star out of the gel piece and into the surrounding buffer within the dialysis tubing.
- Examine the gel pieces when they are still in dialysis tubing for green fluorescence signal under a UV transilluminator; the gel pieces should have no remaining fluorescence signal if the DNA stars have successfully run out of the gel pieces and entered the surrounding buffer in the dialysis tubing.
- If the gel pieces have residual fluorescence, run electrophoresis again at 70 V for additional 10 min. Examine the gel pieces and run electrophoresis again at 70 V for an additional 10 min until all DNA oligos have run into the dialysis buffer.
-
62
Perform electrophoresis for 1 min (while the gel pieces are still in the tubing) by reversing the current polarity applied to the gel box (swapping anode and cathode on the power supply) so the DNA star molecules that may have adhered to the tubing inner surface can run into the buffer to improve sample recovery efficiency.
-
63
Remove the clip from one end of the dialysis tubing and recover the DNA star sample by transferring the buffer from dialysis tubing to an Eppendorf tube using a 100 μL pipette. Measure DNA concentration by UV-visible. The purified DNA star sample can be stored at 4 °C or −20 °C. After freeze–thaw cycles, the DNA star complex shows the same mobility as that of the DNA star complex prior to a freeze–thaw cycle (Supplementary Fig. 3).
▲ CRITICAL STEP Avoid touching the gel pieces with the pipette tip so the DNA star sample will not be contaminated with small gel pieces that may interfere with the downstream assays.
■ PAUSE POINT The purified DNA star sample can be stored at 4 °C for at least 4 weeks. Store the DNA star sample in −20 °C freezer for long-term storage.
-
64
Repeat Steps 54–62 to run 10 μL amount of the purified and unpurified DNA star complex on a 3% AGE gel to confirm that they have the same mobility (Supplementary Fig. 4). Of note, this step is only for gel electrophoresis characterization. No DNA needs to be recovered.
Fig. 9 |. Purification of the DNA star.
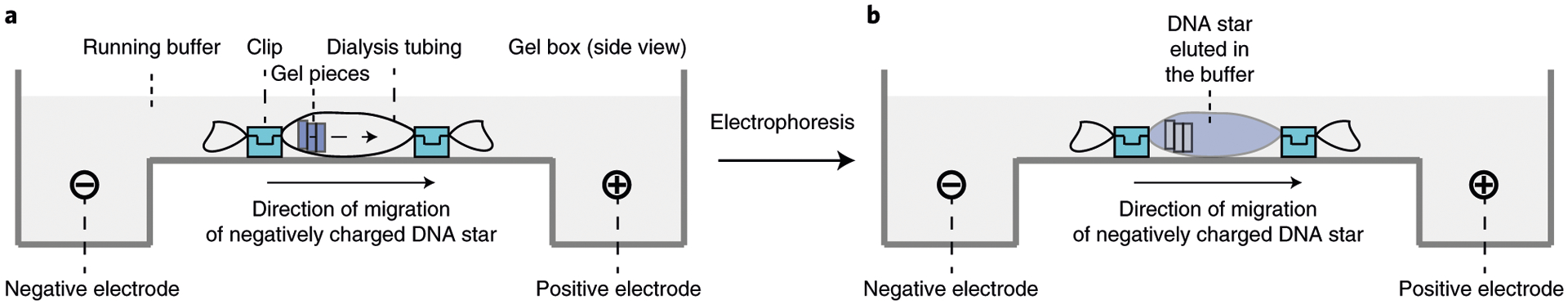
a, The DNA star (indicated by blue color) is retained in the excised gel pieces after an initial AGE, and the gel pieces are inserted into dialysis tubing with buffer and clipped. b, The DNA star molecules migrate into the buffer held by dialysis tubing through secondary electrophoresis. The DNA star sample can then be recovered by transferring the buffer to an Eppendorf tube.
AFM characterization of the DNA star ● Timing ~6 h
-
65
Stick the round-shape mica substrate (10 mm diameter) onto the surface of the round metal plate (12 mm diameter) to support mica substrate using double-sided tape (cut off the edges of the mica with a scissor, if necessary, to fit the size of the metal plate). Place transparent tape on top of the mica disk, and then peel away the tape gently to obtain a freshly cleaved mica surface for adsorption of sample.
▲ CRITICAL STEP Obtaining a smooth surface after mica cleavage is critical to minimize or avoid mica background interference to achieve high-resolution imaging.
-
66
Pretreat mica surface with 2 μL of 0.5% (vol/vol) APTES solution for 1 min. Positively charged APTES is used to increase the electrostatic interaction of small negatively charged DNA star with the mica surface. Rinse the surface with 200 μL nuclease-free water to remove free APTES, and softly dry the surface with compressed air.
▲ CRITICAL STEP The incubation time for the pretreatment of mica surface by APTES needs to be limited to 1 min. Otherwise, excessive APTES molecules on the mica surface will have a negative impact on subsequent AFM imaging.
-
67
Dilute the DNA star solution (from Step 46 or Step 66) to 25 nM using 0.22 μm filtered 1× TAE-Mg2+ buffer (Buffer 5). Place 5 μL diluted DNA star onto APTES-pretreated mica surface, and incubate for 2 min before loading 30 μL 1× TAE-Mg2+ buffer (Buffer 5) on top of the DNA star sample.
-
68
Turn the AFM instrument on, open the Nanoscope software, select ‘Soft tapping in fluid’ mode and assemble a SNL-10 AFM tip in the fluid cell holder. Put the round metal plate that carries a droplet of DNA star sample onto the AFM magnetic stand.
▲ CRITICAL STEP Make sure that the scanner stage is low enough to prevent possible damage to the AFM probe by touching the mica surface.
-
69
Move the AFM probe close to the mica surface before engaging the AFM probe. For high-resolution AFM imaging, perform with scanning rate of 1.5 Hz, scanning range of 500 nm2 and samples per line of 512.
? TROUBLESHOOTING
-
70
After the AFM scan, open raw AFM files with NanoScope Analysis software (v1.50). Use the ‘Flatten’ function to adjust the images to reflect the actual height of the sample. If needed, adjust the image color bar scale to fine tune the display contrast. The images can be exported as a JPG or BMP file. Example AFM images of the five-, six- and seven-point DNA stars are shown in Fig. 10.
? TROUBLESHOOTING
Fig. 10 |. AFM images of the DNA stars.
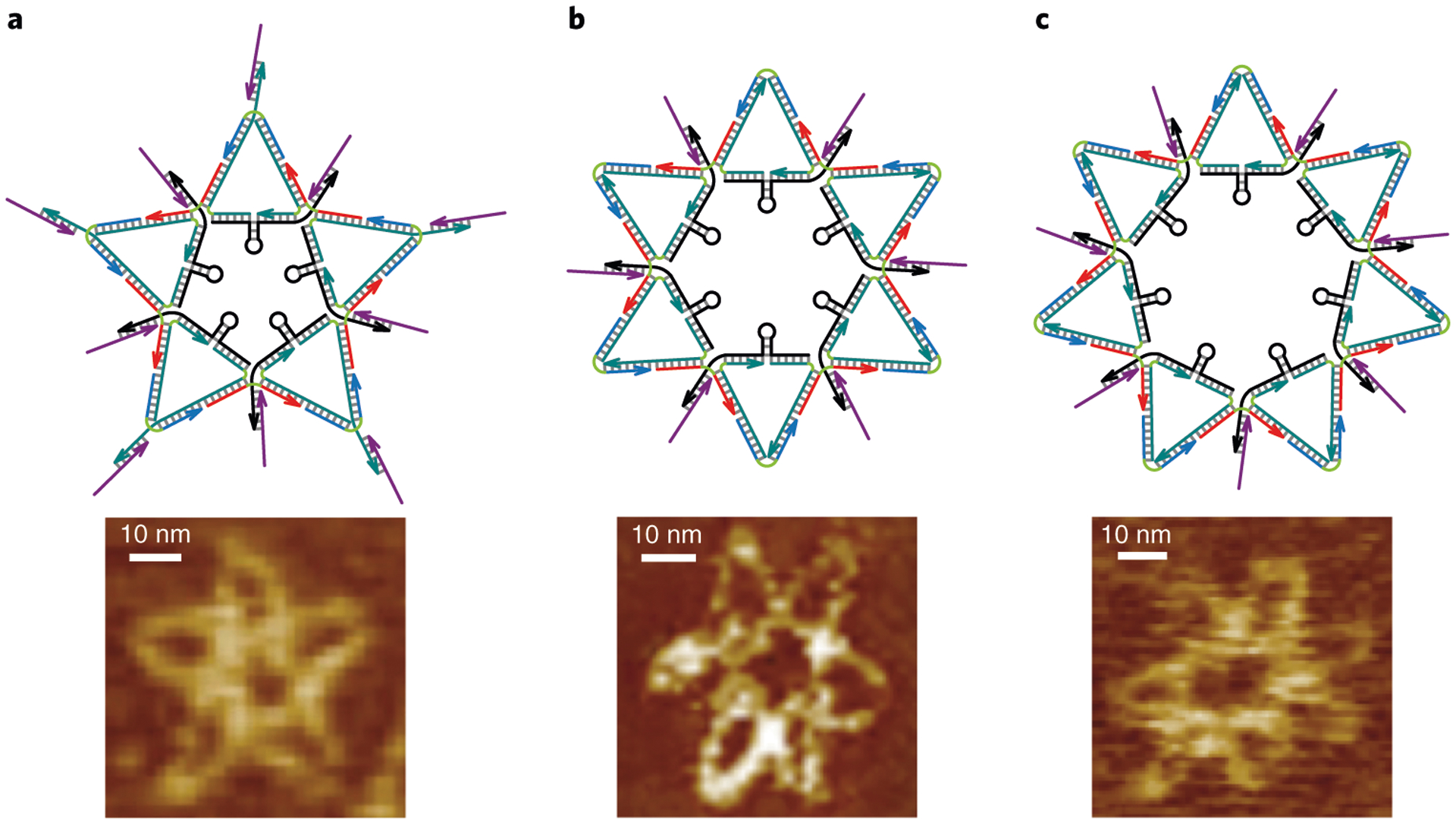
a, Five-point star. b, Six-point star. c, Seven-point star. Scale bars, 10 nm. Figure adapted with permission from ref.7, Springer Nature Ltd.
Purification of the DENV ● Timing ~10 d
! CAUTION DENV is a human pathogen and can cause serious illness. Tubes and any containers should only be opened and handled in a biosafety cabinet within a BSL-2 laboratory. Follow the biosafety guidelines for working with infectious agents in the laboratory, which can be found in the most recent version of the Arbovirus Laboratory Manual and in the BMBL112.
! CAUTION All waste should be considered biohazardous and be disinfected or autoclaved before disposal.
Propagation and storage of DENV
-
71
Seed Vero cells with a concentration of 2 × 105 cells per mL at least 48–72 h before the planned time of infection. We typically add 15 mL of 2 × 105 cells per mL suspension into a T-75 flask. We recommend preparing five T-75 flasks for each DENV propagation, with four flasks to grow the virus and the fifth for a mock infection control. Incubate the Vero cells at 37 °C in a 5% CO2 incubator.
-
72
Inspect cells daily to monitor the growth and to make sure there is no sign of contamination. Monolayers should be adherent to flasks and have few dead or floating cells. Make sure that cultures are not contaminated before use. Signs of contamination include cloudy medium, rounded cells, acidity (yellow medium) and presence of fungal mycelium. A careful examination under an inverted light microscope can often reveal contamination by yeast, fungus and bacteria.
▲ CRITICAL STEP Make sure the cells are 85–100% confluent but not overgrown at the time of infection.
-
73
On the day of infection, thaw the DENV (~2.8 × 107 TCID50/mL), if frozen, quickly at 37 °C, refrigerate or store on ice until you’re ready to use it for infection of the cells.
-
7477 Set up biological safety hood. Disinfect the biosafety cabinet surface with 70% ethanol.
- Place absorbent pad on floor of hood. Line a stainless steel or plastic container with plastic bag to collect solid waste such as pipette tips, pipettes and tubes.
- Prepare stainless steel or plastic container with disinfectant for all liquid waste. We use Coverage Plus NPD disinfectant for a minimum of 15 min contact time
- Assemble pipettes, pipettor, pipette tips, BA-1, tubes, racks and spray bottle with 70% ethanol in the hood
-
75
Dilute the virus stock in a total volume of 5 mL in BA-1 to achieve a multiplicity of infection (MOI, number of virions that are added per cell during infection) of 0.01–0.1. Based on the cell seeding density in Step 74, we estimate there to be ~7 × 106 cells per T-75 flask when 85–100% confluent. To calculate the plaque-forming unit (PFU) needed to infect a flask, multiply the MOI by the total number of cells per flask. For example, to infect a T-75 flask at MOI of 0.01, 7 × 104 PFU is needed. Similarly, an inoculum of 7 × 105 PFU will be required to reach MOI of 0.1.
! CAUTION DENV is a human pathogen and can cause serious disease. All precautions, including proper PPE, should be taken to ensure the safety of yourself and others working in the laboratory. All waste should be autoclaved or disinfected before disposal. Consult the safety officer in your institute on proper disposal procedures of biohazardous waste.
▲ CRITICAL STEP When propagating a mutant virus with less stable genotype, it may be necessary to use a higher MOI (e.g., 1–5) to limit multiple rounds of replication to prevent the occurrence and selection of revertant or new variants.
-
76
Remove growth medium from the T-75 Vero cell-containing flask (from Step 74) with a serological pipet and discard into the liquid waste container. Add the 5 mL of diluted virus (from Step 78) to the flask. For the mock-infection control flask, add 5 mL of BA-1 diluent to a T-75 flask (also described in ‘Reagent setup’).
-
77
Return both infection and mock-infection flasks back to incubator at 37 °C in a 5% CO2 incubator for 60 min. Gently rock flasks side to side a few times every 15–20 min for virus absorption to cells.
-
78
At the end of the virus absorption period, add 10 mL of appropriate cell maintenance medium (we used EMEM supplemented with 2% FBS and penicillin–streptomycin) to each T-75 flask. There is no need to remove the inoculum.
-
79
Return the flasks to incubate at 37°C in a 5% CO2 incubator for 4–6 d based on the progression of infection.
-
80
Examine the flasks each day under an inverted microscope with phase contrast. The infection of DENV often gives rise to morphological changes in the host cell known as cytopathogenic effect (CPE). Detectable changes in the host cell due to infection may include cell rounding, fusion, swelling or shrinking, death, or detachment from the surface. Use the following scale, rate the CPE: 1+ (0–25% cells effected by CPE), 2+ (26–50% CPE), 3+ (51–75% CPE) and 4+ (76–100% CPE).
-
81
Perform the virus harvest at about 3+ or 4 CPE (>75% of the monolayer affected). Different DENV strains or isolates grow at different rates, so the time to harvest can differ substantially depending on the virus. Typically, we harvest a wild-type DENV ~4–6 d post infection.
-
82
Set up the biological safety hood as described in Step 77. Set up a sterile 50 mL conical tube to collect harvested virus.
-
83
Use a sterile serological pipet to remove supernatant from the T-75 flask and transfer into the 50 mL conical tube. If infecting multiple flasks, pool the supernatant and aliquot it into 50 mL conical tubes.
-
84
Balance the conical tubes and centrifuge in a benchtop centrifuge with aerosol-tight lid rotor at 4 °C at 850g for 20 min to precipitate any cell debris to clarify the supernatant.
-
85
Transfer 40 mL supernatant to a new 50 mL conical tube. For virus purification, take an aliquot of 0.4 mL and mix with 100 μL FBS for titration (Step 112); proceed to Step 92 with the clarified supernatant for purification.
-
86
For storing virus stock, add 10 mL of heat inactivated FBS for a final FBS concentration of 20% (vol/vol). Thoroughly mix the solution by pipetting up and down for a few times. To store unpurified virus as stocks for future infections, prepare 0.5–1.0 mL aliquots of the virus stock in 1.8 mL screw-cap tubes with O-rings.
-
87
Label each tube—’name of virus’, ‘date of harvest’, ‘passage number’, ‘cell type used’ and ‘passage history’—and record in your laboratory inventory.
■ PAUSE POINT The harvested and clarified virus can be stored at −80 °C until downstream procedures.
Gradient purification and concentration of DENV
▲ CRITICAL Gradient purification and concentrating virus stock should be performed before running the SPR assay (Step 133). We recommend performing the virus titer (Steps 112–132) on an aliquot of the unpurified virus reserved from Step 88 before performing gradient purification and concentration (Steps 91–111).
-
88
On day 1 of virus purification, set up biological safety hood as described in Step 77.
-
89
Thaw the harvested supernatant in a 37 °C water bath, if frozen. In the biosafety cabinet, place the supernatant (from Step 88) on ice while preparing SW41 centrifuge tubes for the first centrifugation.
-
90
Take out 5 mL of 60% (wt/vol) sucrose gradient solution (see ‘Reagent setup’) and set on ice to chill. This will be used to concentrate the virus solution so that the concentration is suitable for downstream SPR assays.
-
91
Pipette 500 μL of ice cold 60% (wt/vol) sucrose gradient solution into the bottom of each SW41 centrifugation tube. Prepare six tubes each for 10 mL of clarified supernatant. Set the tubes on ice to keep cold.
-
92
Use a Pasteur pipette to slowly overlay 10 mL of the thawed supernatant (from Step 92) onto the 60% sucrose gradient cushion.
▲ CRITICAL STEP Overlay slowly and gently to avoid disturbing the sucrose–medium interface. It is recommended to start with small volumes of 0.5–1 mL at a time until at least 5 mL solution is loaded so disturbance is less likely to occur.
-
93
Put all the filled tubes into the SW41 buckets. Do not cap the buckets until each pair of tubes to be placed opposite each other on the rotor are balanced on a beam-balance scale (Step 97). Match each bucket’s position on the rotor by numbers, by placing bucket #1 in position #1 on the rotor.
▲ CRITICAL STEP There are six positions on a SW41 rotor, bucket #1 is on the opposite side of #4, and they are considered to be a pair of buckets. Each pair needs to be balanced before centrifugation. If not using all the buckets, they still need to be placed on the rotor for balance during the spin.
-
94
Balance the tubes. Position a benchtop beam balance in the biosafety hood, and make sure it is on a level surface. Then, place one 250 mL beaker on each side of the beam balance and balance them on the scale. Place one filled SW41 bucket in one beaker and another on the one on the other side to make sure they are accurately balanced. If needed, add 1× PBS buffer (Buffer 9) to the tubes to balance them.
▲ CRITICAL STEP Make sure to include the bucket caps on the beam balance during weighing. Failure to balance the tubes on the rotor can result in catastrophic damage to the rotor or the centrifuge.
-
95
Once balanced, screw the caps tightly onto the bucket. Then, place the whole set of six buckets on a bucket rack and transfer them out of the biosafety hood.
▲ CRITICAL STEP While transporting the bucket to the centrifuge, make sure to hold the buckets upright and in a level position to minimize disturbance to the sucrose cushion.
-
96
Place each bucket onto its position on the rotor according to the number. Load the rotor into the centrifuge chamber. Make sure the rotor is correctly and securely positioned in the chamber.
▲ CRITICAL STEP SW41 rotor is a swinging bucket style rotor. Make sure that each bucket is hooked on correctly and the rotor is positioned correctly. Failure to do so can cause serious damage to both the bucket and the centrifuge during high-speed centrifugation.
-
97
Set the ultracentrifuge to a speed of 151,000g for SW41 rotor, temperature to 4 °C and time to 2.5 h. Press the VACUUM first to make sure the vacuum is functioning properly, then press the START button to start the run.
-
98
During the first ultracentrifugation step, prepare 10–20–40–60% (wt/vol) sucrose step gradient solutions (see ‘Reagent setup’) in a new set of six SW41 tubes. These will be used to further purify the DENV by discontinuous step gradients. For each SW41 centrifugation tube, sequentially, from bottom to top, layer 0.5 mL of 60% sucrose gradient solution, 3 mL of 40% sucrose gradient solution, and 2 mL of 20% sucrose gradient solution, and finally top with 1 mL of 10% sucrose gradient solution. Allow the gradients to layer, and position each tube upright in ice until ready to centrifuge.
▲ CRITICAL STEP Make sure to keep each gradient solution on ice to facilitate gradient layering. Take care to layer each solution slowly and gently onto the previous one to minimize disturbance at the interface. The interfaces between each sucrose gradient solution should be distinctively visible.
-
99
After the first centrifugation is complete, release the vacuum in the ultracentrifuge chamber, take the rotor out of the chamber and set up on the stand. Gently take the buckets off the rotor and place them on the rack. Transfer the buckets to the biosafety hood, and unscrew the caps in the hood. Use a pair of pointed forceps to carefully take the centrifuge tubes out of the buckets and set the tubes securely on ice.
-
100
In the biosafety hood, prepare a new 50 mL conical tube for 20 mL of 1× PBS buffer (Buffer 9) on ice for pelleted virus. Prepare a beaker or plastic container with disinfectant for waste.
-
101
Set up a tube rack to hold the centrifuge tubes steady during virus collection. Position one tube upright on the rack, and use a sterile Pasteur pipette to progressively take 9–9.5 mL of the supernatant from the top of the tube until ~0.5 mL sucrose above the interface of the 60% sucrose cushion. Discard the supernatant liquid into the beaker with disinfectant for later disposal. The pelleted virus may be visible as a thin opaque layer of white band at the interface when illuminating the tube from above against a dark background.
▲ CRITICAL STEP When pipetting, make sure not to get too close to or disturb the gradient interface.
-
102
Use a new Pasteur pipette to resuspend the virus and the 60% sucrose cushion. Transfer the mixture to a new 50 mL conical tube on ice. Wash the tube twice with 1 mL of ice-cold 1× PBS buffer (Buffer 9) (Step 103) each time, and pool with the collected virus. This will dilute the sucrose concentration of the collected virus to ~10%.
-
103
Repeat Steps 104–105 for all other tubes, and pool the collected virus in the 50 mL conical tube.
▲ CRITICAL STEP Keep in mind that the virus is still infectious, so handle only one tube at a time and take all necessary precaution throughout the process.
-
104
Carefully overlay 3–3.5 mL of resuspended pooled virus on top of each prepared step gradient (from Step 102). Load the tubes into the SW41 buckets, and balance each pair following Steps 96–101.
-
105
Centrifuge at 151,000g at 4 °C for 2.5 h. Alternatively, centrifuge at 77,100g at 4 °C overnight.
-
106
Post spin, collect the virus in the biosafety cabinet by repeating the procedures as described in Steps 102–106. The virus ‘band’ now resides at the 20%–40% sucrose interface after the ultrahigh-speed centrifugation and may be visible as an opaque thin layer, depending on the concentration of the virus particles.
▲ CRITICAL STEP Take care not to disturb the gradient while transporting the centrifuge buckets to the biosafety cabinet or retrieving the SW41 centrifuge tubes.
-
107
A faint white band of virus (visible at the 20%–40% interface) may be visible when examined against a black background. Use a Pasteur pipette to slowly and gradually remove the top 4.5–5 mL gradients solution without disturbing the interface where the virus lies. Stop at ~1 mL above the 20%–40% interface. Discard the liquid waste into a waste beaker with disinfectant. To collect the virus, change to a new Pasteur pipette and collect the virus band.
▲ CRITICAL STEP If the virus band is not visible, collect ~1 mL above and below the 20%–40% interface.
-
108
Pool and aliquot the concentrated virus for storage at −80 °C. Proceed to Step 133 for SPR analysis.
Plaque titration of unpurified DENV stock using Vero cells
-
109
Seed cells in six-well plates 72 h prior to the assay. Prepare confluent cell monolayers by seeding cells in six-well plates at a density that will produce at least 90% confluency on the day of infection. Typically, we dilute Vero cells to 2 × 105 cells per mL in cell growth medium (EMEM + 10% FBS) and seed into six-well plates at 3 mL per well (= 6 × 105 cells per well). Prepare an extra six-well plate for mock infection control.
▲ CRITICAL STEP To decide the number of plates needed for the assay, assess how many dilutions of each virus sample and replicates you need. It is standard to run each dilution in triplicate. For example, if you are planning six tenfold dilutions in triplicate (18 wells total), you will need to seed three six-well plates of cells for each sample.
? TROUBLESHOOTING
-
110
Culture cells for 72 h or until they reach 90% confluency.
-
111
On day 1 of the plaque titration, set up the biological safety hood following Step 77.
-
112
Examine cell monolayers for density and appearance under the microscope; note any abnormalities.
▲ CRITICAL STEP Make sure the monolayers are at least 90% confluent, but not overgrown. Make sure that cultures are not contaminated. Signs of contamination include cloudy medium, rounded cells, acidity (yellow medium) and presence of fungal threads.
-
113
Label cell culture plates and tubes for the virus dilutions.
-
114
Thaw viral samples (from Step 89) in a 37 °C water bath, then store on ice until use.
-
115
Fill 1.5 mL snap cap tubes with appropriate amount of BA-1 diluent for tenfold serial dilutions. More specifically, prepare enough of each dilution to add 100 μL of sample to each well. Prepare at least 450 μL BA-1 per tube in order to have sufficient sample volume for triplicate wells of each dilution.
-
116
Prepare viral dilutions (usually tenfold serial dilutions) in BA-1 in 1.5 mL snap cap tubes prepared in Step 118. Achieve serial dilutions by mixing 50 μL virus with 450 μL of BA-1 diluent for the first dilution, by vortexing briefly or pipetting up and down. Continue the serial dilutions by diluting into the remaining tubes stepwise.
▲ CRITICAL STEP Make sure to change pipette tips for each dilution tube to avoid carryover errors, which will result in inaccurate titer measurement.
-
117
For infection of cell monolayers, remove medium from uninfected cell monolayers with a serological pipette. Take care to leave ~200 μL of medium over the cell monolayer to prevent cells from drying.
-
118
Add 100 μL of appropriate viral inoculum to each well. Ensure you inoculate three wells for each dilution. Prepare a mock-infection control six-well plate: add 100 μL per well of BA-1 diluent to a six-well plate (also described in ‘Reagent setup’).
-
119
Incubate plates at 37 °C for 60 min to allow adsorption of virus to cells. Rock plates gently by tilting at ~45° angles every 15 min to keep monolayer moist and to evenly distribute viral inoculum.
-
120
Prepare first agar overlay solution. Calculate the total amount of Oxoid agar overlay needed to cover your plates. You will need to add 3 mL of Oxoid agar–EMEM mixture to each well, so a minimum of 18 mL is required per six-well plate. Always prepare a few extra milliliters of overlay solution. The overlay mixture is a 1:1 mixture of 1.2% Oxoid agar and 2× EMEM + 10% FBS. Prepare 20 mL of mixture (10 mL of each component), for every six-well plate in order to have sufficient Oxoid agar–EMEM mixture for overlay following the steps below. Melt 10 mL of 1.2% Oxoid agar for every six-well plate following the steps below.
? TROUBLESHOOTING
-
121
Calculate the amount of 2× EMEM + 10% FBS needed, based on the number of six-well plates in the assay. The volume of 2× EMEM + 10% FBS is the number of six-well plates multiplied by 10 mL. For example, for ten six-well plates, you will need 100 mL of 2× EMEM + 10% FBS. In the biological safety hood, transfer the needed amount of 2× EMEM + 10% FBS to a clean sterile glass bottle, place it in a 37 °C water bath and allow it to equilibrate to the temperature.
-
122
Melt the required amount of 1.2% (wt/vol) Oxoid agar in a microwave oven. When fully melted, place it at 56 °C in a water bath and allow to equilibrate.
▲ CRITICAL STEP Make sure to loosen caps before melting the Oxoid agar in the microwave oven. During the melting process, inspect the bottle from time to time and swirl it gently to ensure all agar has dissolved. The agar will become very hot during the melting process; take precaution when handling to prevent burns.
▲ CRITICAL STEP Prewarming the 2× EMEM + 10% FBS medium and preparing the Oxoid agar solution can also be done in advance before the virus infection (Step 122).
-
123
In the biological safety cabinet, combine equal amounts of 2× EMEM + 10% FBS and 1.2% (wt/vol) Oxoid agar that have been equilibrated to 37 °C and 56 °C, respectively. Place the mixture in 45 °C water bath until needed.
▲ CRITICAL STEP When handling a large number of plates, we recommend preparing the overlay mixture in multiple bottles of 200–250 mL. Keep the bottles at 45 °C to avoid the agar mixture solidifying prematurely during overlaying.
-
124
At the end of the 1 h virus–cell absorption (Step 122), gently add 3 mL EMEM+agar mixture (Step 126) per well to each six-well plate. To avoid cross-contamination, make sure not to touch the pipette tip to any part of the well. Allow the agar overlay to solidify for ~15 min at RT before returning to 5% CO2 incubator at 37 °C. Incubate samples for 5 d (days 1–6).
▲ CRITICAL STEP Before adding the overlay mixture onto the cell monolayer, make sure the mixture is not too hot, which can result in cell death. This can be done by feeling the outside of the bottle—it should be warm, not hot, to touch.
? TROUBLESHOOTING
-
125
Grow the virus until day 6. On day 6 of the plaque titration assay, prepare a second overlay containing neutral red stain. Neutral red stains viable cells and leaves dead or virally infected cells opaque or clear. You will need to prepare the same volume of second overlay as you did for the first overlay (Steps 125–126). The second overlay is a mixture of equal volume of 1.2% Oxoid agar and 2× EMEM + 2% FBS, with 2.0 mL of neutral red stock solution (0.33%, wt/vol) added to every 100 mL of the overlay mixture. To avoid crystal formation of neutral red during staining, add neutral red to hot agar once it is melted and place in 56 °C until time to mix with 2× EMEM + 2% FBS.
▲ CRITICAL STEP Keep stained cells in dark when possible since the stain is sensitive to light.
▲ CRITICAL STEP Depending on the strains or isolates of DENV, the best timing for the second overlay to stain the monolayer may vary and can only be determined empirically. We suggest incubating samples with the first overlay for 5 d before adding the second overlay.
? TROUBLESHOOTING
-
126
Add 3 mL of the second overlay agar mixture to each well on top of the first overlay.
-
127
Allow the overlay to solidify at RT for 15 min, and then incubate at 37 °C overnight in a 5% CO2 incubator to allow for diffusion of the stain into the monolayer of cells.
? TROUBLESHOOTING
-
128
Count plaques. On day 7, count plaques to determine the viral titer. Invert the plates on a light box, and count the number of plaques per well by marking them with a marker on the bottom of the plate. Neutral red stains viable cells, so the general background will be dark red; plaques resulting from cell death due to virus infection will not be stained and remain clear or colorless. Plaques are generally round with a diameter of 1–4 mm. Record the number of plaques counted per well.
? TROUBLESHOOTING
-
129Calculate the viral titer in PFU/mL. Use the wells containing 10–80 plaques per well to determine the titer in PFU/mL. Do not use plaque counts from wells in which there are too many plaques because plaques may be fused or be too crowded to count accurately.
- Average the triplicate values for plaque counts from wells with ~10–80 plaques
- Use the following equation to calculate viral titer: PFU/mL = average of triplicate (number of plaques) × dilution factor from virus stock × 10
- For example, for a sample with an average of 25 plaques at 1:10,000 (104) dilution, with 100 μL inoculum added per well, the viral titer = 25 × 104 × 10 = 2.5 × 106 PFU/mL
SPR analysis of the interaction between viral particles and the DNA star complexes ● Timing ~5 h
-
130
Insert CM5 chip into the SPR instrument Biacore 3000.
-
131
Prepare 1 mL of an equimolar mix of EDC and NHS at a final concentration of 50 μM. Mix immediately before injection into the SPR instrument.
-
132
Activate carboxymethyl groups on the CM5 chip surface using an injection pulse of 35 μL (flow rate 5 μL/min) of the EDC/NHS solution.
-
133
Following the activation, inject 1 mL purified and concentrated virus solution from Step 111 (~1010 PFU/mL in a 100 mM sodium acetate pH 5.0 buffer) over the activated biosensor surface.
▲ CRITICAL STEP The successful immobilization of the virus particles is confirmed by the observation of a 1,000–2,000 resonance unit (RU) increase above baseline signal.
? TROUBLESHOOTING
-
134
Deactivate excess unreacted carboxymethyl groups on the sensor surface with a 35 μL injection of 1 M ethanolamine. Prepare a reference or control flow cell as described in ‘Reagent setup’.
! CAUTION Use a biosafety cabinet to prepare and handle samples containing intact virus particles. To comply with the biosafety regulation, access to the SPR laboratory should be restricted to authorized researchers who have passed intensive biosafety training. PPE, including gloves, laboratory coat, closed-toe shoes, face mask and laboratory safety goggles, are used to mitigate potential exposure to the reagents.
■ PAUSE POINT The virus particle immobilized SPR chip can be stored at 4 °C for 7 d.
-
135
To measure the binding kinetics, equilibrium dissociation constant of the full DNA star, inject DNA star samples from Step 66 (five different concentrations equivalent to aptamer concentrations from 0 to 10 μM in 1:2 serial dilution for each sample) over the virus chip at a flow rate of 50 μL/min with 1× TAE-Mg2+-K+ buffer (Buffer 7) as running buffer (Fig. 11a). Prepare partial and full stars for this comparison assay with a normalized aptamer concentration of 1.2 μM (also described in ‘Reagent setup’).
-
136
At the end of the sample injection, flow 1× TAE-Mg2+-K+ buffer (Buffer 7) over the sensor surface to facilitate dissociation. After a 3 min dissociation, regenerate the sensor surface by injecting 50 μL of 2 M NaCl.
-
137
Monitor and record the SPR response (sensorgram) as a function of time at RT. Use the sensorgram to compare relative binding avidity between each of the partial/full DNA stars and dengue virions (Fig. 11b,c).
? TROUBLESHOOTING
-
138
Determine the binding kinetics and affinity using the resulting sensorgrams (signal from reference flow cell should be subtracted from each dataset) including association rate constant (ka), dissociation rate constant (kd) and binding equilibrium dissociation constant (KD, KD = kd/ka) by globally fitting the entire association and dissociation phases using 1:1 Langmuir binding model using the BiaEvaluation software 4.0.1 (Cytiva Life Sciences) (Fig. 11d).
▲ CRITICAL STEP The SPR assay for each DNA star complex should be performed at least in triplicate to obtain the binding affinity (KD) to the virus particles. Each of the KD values obtained from triplicate measurements of each DNA star complex should be statistically indistinguishable (P > 0.05; no significant difference; otherwise, more trials will be carried out until P-value measurement is satisfied).
▲ CRITICAL STEP After each SPR experiment, the laboratory bench, SPR instrument and biosafety cabinet surface should be thoroughly cleaned and disinfected using 10% fresh bleach and 70% ethanol for a minimum of 30 min contact time. SPR chips (if not reused), pipet tips, plastic tubes and microscope slides or coverslips should be properly disposed as biohazardous waste in a biohazard waste container located inside the biosafety cabinet.
Fig. 11 |. SPR binding assays.
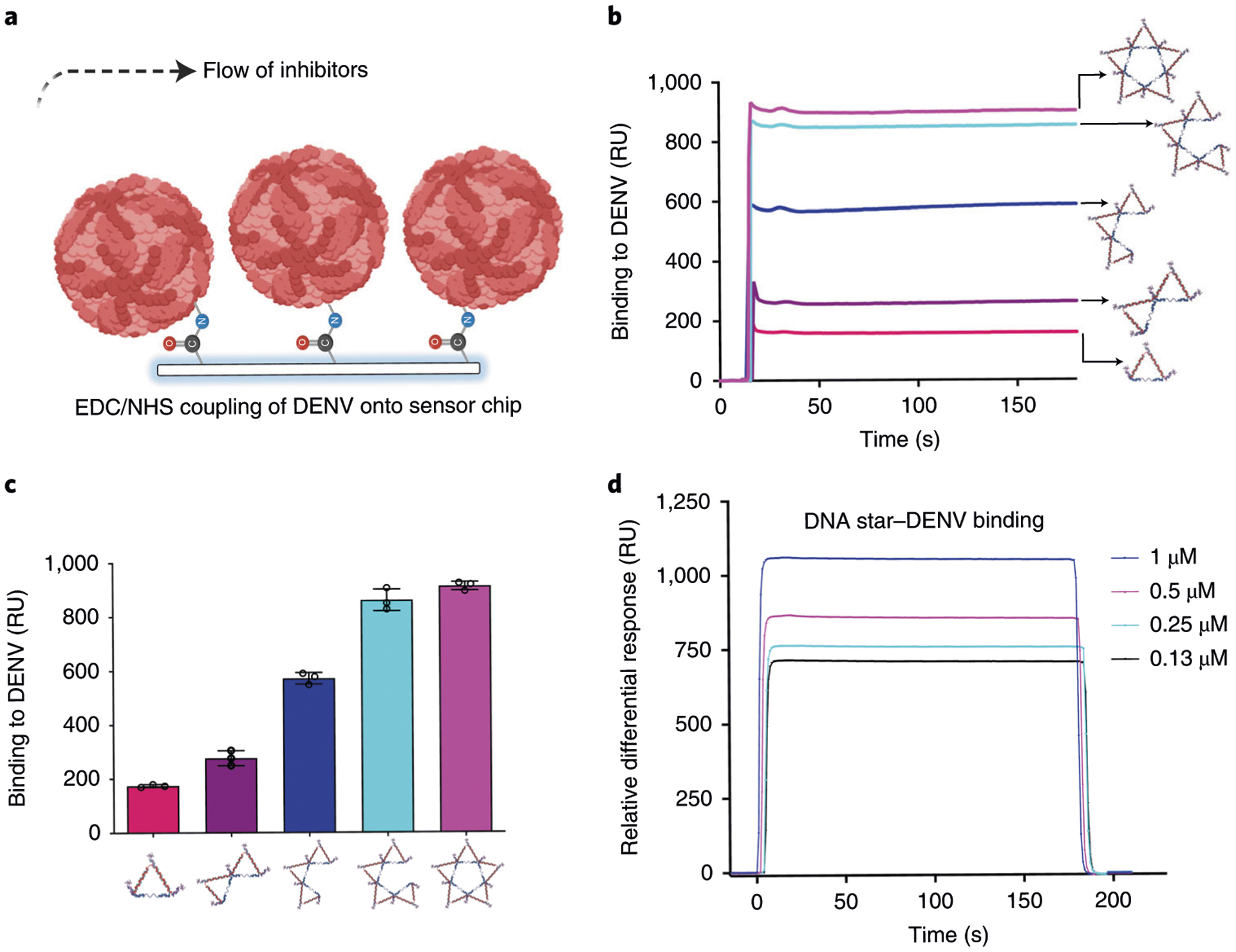
a, Schematic of the SPR binding assay used to determine the binding avidity of the DNA star and DENV particles. b,c, Binding avidity comparison of one-triangle, two-triangle, three-triangle, four-triangle or full star–aptamer complex and DENV particles. Higher-degree pattern matching (by increasing the number of points of the star to better match the ED3 geometry) leads to stronger binding avidity. b shows exemplar response curves. Error bars are mean ± s.d., n = 3 biologically independent samples in c. d, Dissociation constant (KD) measurement of the DNA star when binding to DENV particles is calculated by globally fitting the entire association and dissociation phases using 1:1 Langmuir binding model from BiaEvaluation software 4.0.1. (GE Healthcare, Uppsala, Sweden). d shows exemplar response curves. Figure adapted with permission from ref.7, Springer Nature Ltd.
MTT assay to measure DNA star cytotoxicity ● Timing ~2 d
▲ CRITICAL Many commercial MTT assay kits are available to make the assay more streamlined in a 96-well plate format. Refer to the manufacturer’s protocol for specific variations and details. Use sterile techniques throughout the entire assay as contamination will result in high background.
Determine cell density for specific assay conditions
-
139
Prepare cell suspension. Harvest Vero cells by trypsinization, and resuspend in fresh growth medium at a concentration of 1 × 106 cells per mL. Use a hemocytometer and Trypan Blue staining to determine the cell concentration and viability.
▲ CRITICAL STEP For cells grown in suspension, take an aliquot of the suspension to determine the cell concentration and viability. Harvest the cells by centrifugation, then resuspend in growth medium.
-
140
Prepare dilutions of cells from 1 × 104 to 1 × 106 cells per mL. Pipette 100 μL per well of cell suspension in triplicate into a 96-well cell culture plate. This will result in 103 –105 cells per well. Seed cells at several densities to determine the conditions that will result in final absorbance readings at 570 nm in a linear range, usually between 0.1 and 1.25.
▲ CRITICAL STEP The number of cells per well required for optimal results may vary depending upon cell types, culture conditions, doubling time of cells, and experimental conditions. The condition needs to be determined empirically for specific experimental designs.
▲ CRITICAL STEP Include three wells with growth medium (no cells). These wells serve as the background controls for the absorbance readings. This is important especially if the growth medium contains ingredients that generate higher background or interfere with absorbance measurement.
-
141
Incubate the plate for 12–24 h at 37 °C to allow cells to recover and start to grow. Observe the cells on an inverted microscope to check on the cell conditions before proceeding to the next step.
▲ CRITICAL STEP The temperatures and incubation time needed for cell recovery may vary depending on specific cell types.
-
142
Using a multichannel pipette, add 10 μL of MTT reagent (i.e., 1/10 of the medium volume) per well.
-
143
Return the plate back to the incubator for 2–4 h until purple precipitate is visible. During the incubation, observe the cells periodically under an inverted microscope for the formation of intracellular punctate purple precipitates.
▲ CRITICAL STEP Longer incubation times may be required for some cell types.
-
144
Using a multichannel pipette, add 110 μL (equal volume of growth medium) of detergent reagent to all wells, when the purple precipitate is clearly visible.
-
145
Leave plate at RT in the dark for 2–4 h or overnight. Measure the absorbance at 570 nm in a microplate reader. Absorbances can be read with any filter in the wavelength range of 550–600 nm. If required, a reference wavelength should be set at >650 nm. The medium-only control typically gives a near-zero reading.
▲ CRITICAL STEP Keep the plate in the dark during incubation. The plate can be covered with foil to protect it from the light.
▲ CRITICAL STEP Return the plate to the dark and incubate longer if the absorbance readings are low.
-
146
Take the average values of the triplicate readings and subtract the background reading, which is the average of the medium-only controls. Plot the absorbance reading against cell number per mL, and select the seeding density of cells that yields an absorbance ~0.75–1.2 for the cytotoxicity assay. This will represent the maximal cell viability if there is no detrimental impact on the cells in the cytotoxicity assay.
Cytotoxicity assay of the DNA star complexes
-
147
Seed Vero cells at the optimal density (from Step 149) in a 96-well plate, and allow the cells to recover for 4–8 h at 37°C in a 5% CO2 incubator.
-
148
Serially dilute the DNA star complexes (from Step 66) in dilution buffer or growth medium over a range of at least 5 logs in microcentrifuge tubes. The starting concentration of each DNA star complex should be 10 μM aptamer equivalent. Serially dilute over a range of 5 logs for cytotoxicity assay.
-
149
Add 10 μL of each serially diluted DNA star complex in triplicate to cell monolayers, and return the plate back to the incubator. Include background, untreated cell and dilution buffer controls as described in ‘Reagent setup’. Incubate overnight.
-
150
Use a multichannel pipette to add 11 μL of MTT reagent, and return the plate back to the incubator and incubate for 2–4 h. Observe the cells periodically under an inverted microscope to evaluate the formation of purple precipitate.
-
151
Use a multichannel pipette to add 110 μL of detergent reagent to all wells when the purple color is visible. Leave the plate at RT in the dark for 2–4 h. Measure the absorbance at 570 nm in a microplate plate reader.
-
152
Average the triplicate measurements for each sample and subtract the average from medium-only (no cells) background control, which gives the background-adjusted absorbance value. The % cytotoxicity can be calculated using the following equation: % cytotoxicity = 100 × (corrected untreated control average − corrected sample average) / corrected medium-control average.
Plaque reduction test (antiviral assay) ● Timing ~8 d
▲ CRITICAL Sterile technique should be practiced throughout the entire process to avoid contamination.
! CAUTION DENV is a human pathogen and can cause serious disease; the assay needs to be performed under BSL-2 conditions by referring to the requirements listed in the CDC’s Biosafety Microbiological and Biomedical Laboratories, 6th edition112. All precautions, including proper PPE, should be taken to ensure the safety of yourself and others working in the laboratory.
▲ CRITICAL All waste should be thoroughly disinfected or autoclaved before discarding. Consult the safety offices of your institution about correct biohazardous waste disposal procedures and guidelines
-
153
Prepare and seed cells for the plaque reduction assay 3–4 d before the assay. Prepare Vero cell suspension at 2.0 × 105 cells per mL in EMEM + 10% FBS (growth medium as in ‘Reagent setup’). Dispense 3 mL per well of cell suspension in six-well plates, and incubate the plates in 5% CO2 incubator at 37 °C. Vero cells are usually at least 90% confluent and ready for use after 72–96 h of culture. Prepare one six-well plate for each DNA star complex dilution or control. For example, to assess a five-point DNA star complex over eight serial dilutions in triplicate (starting with 2.5 μM aptamer equivalent concentration with fourfold dilutions), one should prepare eight six-well plates of cells per series of dilutions. Prepare extra plates for all controls.
▲ CRITICAL STEP Before using the cells, examine them for appearance and density under an inverted microscope to ensure the health of the cells and ensure they are free of contamination.
▲ CRITICAL STEP Discard the plates if there is sign of abnormalities, cell death/detachment or bacterial/fungal contamination. Signs of contamination include cloudy medium, rounded cells, acidity (yellow medium) and presence of fungal threads. Cells that are overgrown or cultured for >7 d should not be used.
? TROUBLESHOOTING
-
154
Set up biological safety hood following Step 77.
-
155
Day 1: dilute virus stock with determined titer (from Step 132) to 200 PFU per 0.1 mL (2 × 103 PFU/mL) in fresh BA-1 diluent. Inoculum per well is 100 μL per well. At 200 PFU per 0.1 mL, the inoculum will contain 100 PFU per 100 μL after mixing 1:1 with test samples. For convenience and consistency between assays, virus stock (from Step 132) can be diluted in EMEM + 20% FBS to 105 PFU/mL and frozen in 0.5 mL aliquots at −80 °C for storage. To reach 2,000 PFU/mL needed for plaque reduction assay, one 0.5 mL aliquot is diluted into 25 mL BA-1.
▲ CRITICAL STEP Once thawed and diluted, leftover virus should be discarded and not used for a different assay.
-
156
Serially dilute each DNA star complex in 1× TAE-Mg2+-K+ buffer (Buffer 7) (starting with 2.5 μM aptamer equivalent concentration in 1:4 from the stock solution over the range of 8 logs). Other dilution factors may be preferred depending on the specific range of concentrations to be assayed. To prepare enough amount of diluted DNA star complex to be used in Step 160, the dilutions are prepared in 1 mL aliquots. Aliquot 750 μL of 1× TAE-Mg2+-K+ (Buffer 7) to seven sterile 1.5 mL snap-cap or screw-cap tubes. Prepare the first dilution by adding 250 μL of DNA star complex to the first tube of 750 μL of 1× TAE-Mg2+-K+ buffer (Buffer 7), mixing by briefly pipetting up and down, then continue with serial dilutions.
-
157
Mix 250 μL of diluted virus (from Step 158) with 250 μL of each serially diluted DNA star complex (from Step 159) in a sterile 1.5 mL microcentrifuge tube, and incubate in 5% CO2 incubator at 37 °C for 1 h. Prepare at least duplicate sets for each test sample. Include medium-only, buffer-only, positive, and cell controls as described in ‘Reagent setup’.
▲ CRITICAL STEP The incubation parameters such as length of incubation period or temperature can be modified, depending on the interaction dynamics and stability of the complexes in the virus diluent.
-
158
For virus infection in cell culture, prepare a new set of sterile microcentrifuge tubes—one for each DNA star complex mixture from Step 160—and fill with 400 μL of BA-1 diluent for inoculum dilution.
-
159
Label each plate before the inoculation. Work with one cell culture plate (per sample) at a time to avoid drying the cell monolayer. Aspirate or use a pipette to remove culture medium from wells; keep the plate covered until inoculation. Take care to leave ~200 μL of medium on the cell monolayer to avoid dehydrating the cells during next steps.
-
160
Take 100 μL of each virus–DNA star complex mixture (from Step 160), and mix well with the prepared 400 μL of BA-1 (fivefold diluted mixture) in microcentrifuge tubes from Step 161. Add 100 μL per well of either the fivefold diluted mixture or the undiluted mixture (from Step 160) in triplicate to inoculate the cell monolayer along the side of the wells. Gently rock the plate to evenly distribute the inoculum over the monolayer. For each six-well plate, there should be three wells of cells inoculated with fivefold diluted virus/DNA star mixture and the other three with undiluted virus/DNA star mixture.
? TROUBLESHOOTING
-
161
Incubate the inoculated six-well plate at 37 °C for 1 h to allow virus adsorption. Rock the plates gently every 15–20 min throughout the incubation.
▲ CRITICAL STEP Infecting the cell monolayer at two different dilutions, undiluted and fivefold diluted, ensures the numbers of plaques per well fall in a reliable range, usually between 20 and 100, for enumeration. Plaque numbers >100 or <10 per well may not be considered sufficiently reliable for calculation.
? TROUBLESHOOTING
-
162
Follow Steps 123–130 for the first and second agar overlay.
-
163
Day 7 or 8: examination and enumeration of the plaques (1–2 d after staining). Stained plaques become visible as early as 24 h after staining; however, some may take longer to appear. Plaques should be observed and counted daily until there is no substantial increase in numbers (Fig. 12a). Ideally, the plaques should be evenly distributed over the well and the numbers should fall between 30 and 100 to be reliably counted. The plaque numbers collected from each of the serially diluted DNA star complexes can be used to generate an inhibition curve (Fig. 12b). The concentration of DNA star complexes that is able to reduce the plaque number by 50% (compared with untreated control) is designated as EC50 (Fig. 12c).
Fig. 12 |. Evaluation of the inhibitors.
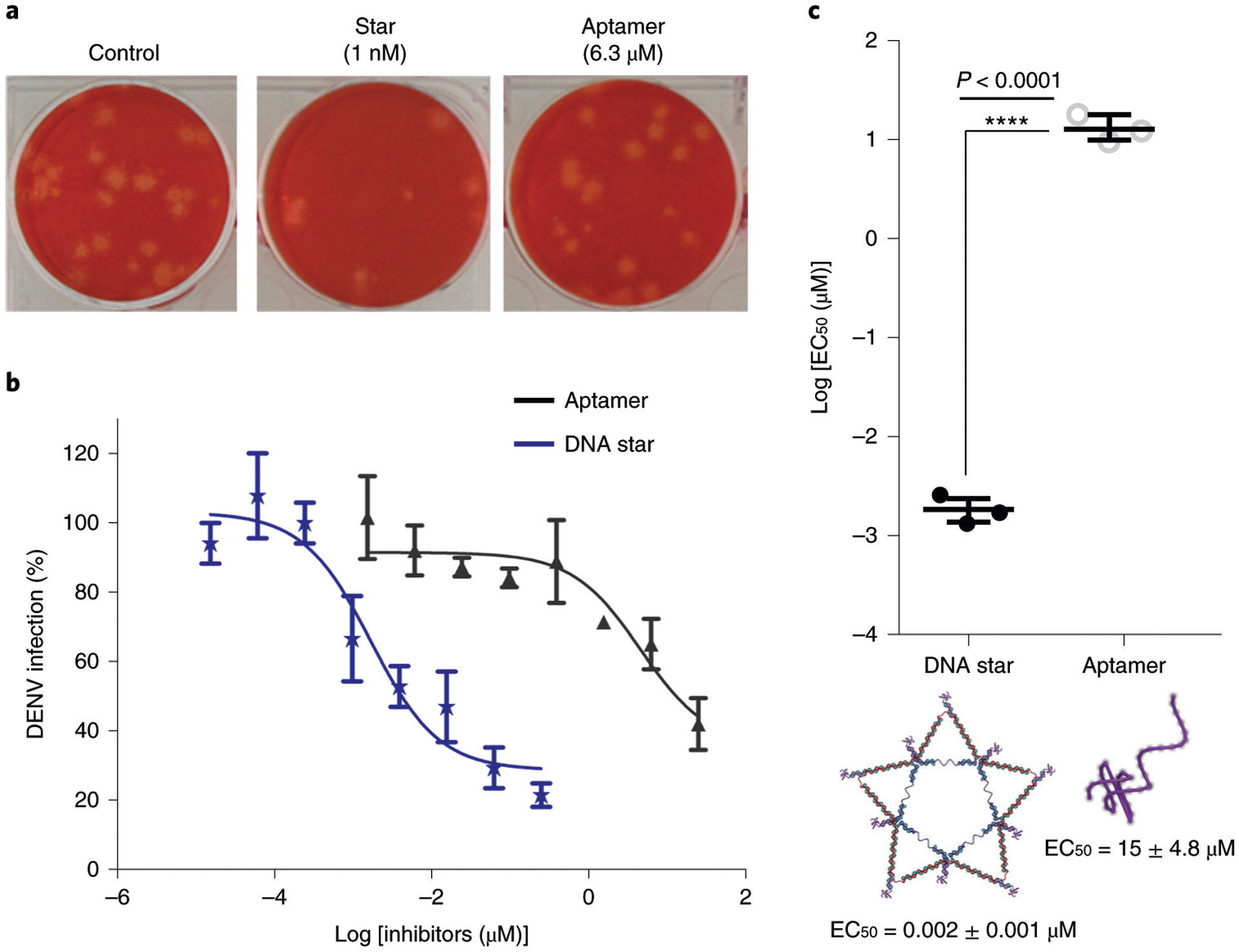
a, Representative plaque-reduction assays corresponding to the monovalent aptamer and DNA star complex concentrations. The no-inhibitor treated control well is present as a no-drug control. b, Dose-dependent, plaque-reducing inhibition curves for the monovalent aptamer (abbreviated as aptamer) and DNA star inhibitors. Inhibitor concentration was standardized through aptamer concentration. Data are presented as mean ± s.d., n = 3 biologically independent experiments. c, EC50 comparison of the DNA star and monovalent aptamer. Data are presented as mean ± s.d., n = 3 biologically independent experiments. A t-test was performed to test significance against the DNA star (****P < 0.0001). Figure adapted with permission from ref. 7, Springer Nature Ltd.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 20, 49 | Gel does not solidify at all or solidifies very slowly | TEMED and/or APS solution is not fresh | Order new TEMED and/or make fresh APS solution |
| 25, 51 | Bromophenol blue and xylene cyanol FF dyes migrate together at the gel front | The gel running buffer may be too old to maintain the pH and ionic strength | Make and use fresh gel running buffer |
| 27 | DNA oligos run as a smear spanning the partial or the entire lane | The DNA manufacturer sometimes could underestimate the actual amount of oligo provided in the tube. The gel could be overloaded with too much crude DNA | Measure the DNA concentration yourself using a UV-Vis spectrophotometer at 260 nm to correct the amount of unpurified DNA |
| 41 | No DNA pellet or a very small DNA pellet is formed | Too little NaCl (Step 36) is added, and/or the 70% ethanol added to the second centrifugation (Step 39) is not cold enough | Make sure to add enough NaCl before centrifugation, and precool 70% ethanol in −20 °C for at least 3 h before using |
| 53 | Gel electrophoresis and AFM imaging may show that a specific DNA star structure does not form at all or at a low yield | The number of unpaired Ts used at the inner corners is not sufficient (if the downstream experimental conditions were checked and optimized) | Change the number of the unpaired Ts by adding or removing one T at a time until the formation yield is ~90% or above |
| Nondenaturing gel shows partial DNA star products | The component DNA oligos are not pure or start to breakdown, probably due to DNA nuclease contamination | Check the DNA oligos purity and integrity by running them on a denaturing gel as described in Step 25 | |
| 72 | Mica starts moving during an AFM scan | Thin glue from double-sided tape is displaced by buffer used in tapping in buffer AFM mode | Consider using stronger glue/tape to get the mica adhered to the metal plate |
| 73 | Dirty AFM image background | Too much APTES residues are left on the mica surface (Step 69) | Stick to the mica activation procedures of using 0.5% (vol/vol) APTES for 1 min |
| AFM imaging cannot resolve the hollow structures on DNA star | Too much DNA is loaded on mica surface and the structures are overlaid on each other (Step 70) | Lower the amount of DNA loaded on the mica. Titrate to find the best working concentration | |
| 112, 123, 127, 156,163, 164 | Wells exhibit mold or bacterial growth | Cells became contaminated upon initial seeding or during infection | Check diluent, virus stocks and overlay medium for any signs of contamination |
| Field virus samples were not collected in a sterile manner | Supplement extra antibiotics such as gentamicin or antimicrobial fungizone in the first agar overlay | ||
| 127, 130 | Agar overlay solidifies before use or during use | Agar solution temperature is too low before preparing the overlay | Check if water baths are set to and measuring the correct temperatures |
| If large volumes of overlay are required, aliquot the mixture into smaller volumes and keep at 45 °C until immediately before use | |||
| 128 | A large area of cells does not show neutral red stain | This results from a large area of cell death usually because of monolayer drying during infection or incubation. The cell death could also occur if the temperature of agar overlay, especially the first overlay, is too high | Handle a small number of plates at a time and minimize the time when the cell monolayers are without medium. Additionally, make sure to check the temperature of the overlay mixture before adding onto the cell monolayer |
| 130, 131 | Cells show very light staining and plaques do not form clear borders | Incorrect amount of neutral red stain was added to the overlay medium | A third overlay containing the correct amount of neutral red can be added; wait for another day for plaques to appear |
| Neutral red might precipitate out of the solution | Add the neutral red to freshly made agar before combining the agar with 2× EMEM | ||
| Unhealthy or older cells also take up less stain | Seed fresh cells | ||
| 131 | There are no apparent or visible plaques shown in the wells | Cells may be lysed | Evaluate cells with microscopy to look for neutral red uptake |
| Viral stock may have been diluted too much | Test a more concentrated viral stock from Step 90 | ||
| Plaques are very small | Increase incubation time (in Step 130) | ||
| 136 | 1,000 to 2,000 RU increase is not seen after SPR chip activation by virus particles | Mix of EDC and NHS are not fresh, and/or virus particles are degraded | Make a fresh EDC/NHS solution and thaw a new aliquot of the virus sample to repeat the SPR chip activation process |
| 140 | The SPR response of different DNA nanostructures or of the same structure at different concentrations does not correlate | The DNA complexes do not form well (see Troubleshooting advice for Step 53) | Prepare freshly activated SPR chips, and remake DNA complexes (Steps 42–46) |
Timing
Steps 1–5, identification of virus antigen spatial patterns: ~2–3 d
Steps 6–12, design of the DNA star: ~4 h
Steps 13–41, denaturing PAGE for oligonucleotide purification: ~20 h
Steps 42–46, assembly of the DNA star: ~12 h
Steps 47–67, nondenaturing gel electrophoretic characterization and purification of the DNA star: ~6 h
Steps 68–73, AFM characterization of the DNA star: ~6 h
Steps 74–132, purification and concentration of the DENV: ~10 d
Steps 133–141, SPR analysis of the interaction between viral particles and the DNA star complexes: ~5 h
Steps 142–155, MTT assay to measure DNA star cytotoxicity: ~2 d
Steps 156–166, plaque reduction test (antiviral assay): ~8 d
Anticipated results
Spatial arrangements, patterns and spacing of the envelope proteins on the outer surface of the virus can be obtained using the described computational analysis of the structure of the dengue viral particle48. One of the patterns can be used as a mirroring template for the design of a (five-point) DNA star as displayed in Figs. 4 and 5a. The DNA stars can be created using the described ‘DNA tile’ nanostructure design procedures illustrated in Figs. 5 and 6. The resulting component DNA sequences predicted to self-assemble into the desired DNA star structure can be obtained from the SEQUIN program and have been listed in Supplementary Tables 1–3. An example of nondenaturing gel electrophoretic characterization of the individual component DNA oligos and the partial/full DNA star complexes is shown in Fig. 8b. The gel shows a distinct and dominant band corresponding to each of the DNA complexes, confirming that DNA star structures are successfully self-assembled in high yield. Figure 10 shows the exemplar AFM images of the DNA stars, confirming that the DNA species has an expected DNA star structure. An example of nondenaturing AGE shows a distinct and dominant band corresponding to the DNA before and after freeze–thaw cycles, confirming that the DNA star sample can tolerate storage in a −20 °C freezer and several freeze–thaw cycles (Supplementary Fig. 3). When a pure DNA star sample is needed for certain assays or applications (e.g., SPR), it can be obtained using a high-percentage (i.e., 3%) nondenaturing AGE purification procedure. An example of nondenaturing AGE shows a distinct and dominant band corresponding to the DNA before and after the gel purification procedures, validating the method (Supplementary Fig. 4). Figure 11 shows the DNA star–DENV binding curves obtained using the SPR binding assay and data analysis procedures, which confirms that the polyvalent, pattern-matching-based DNA star–virus interaction can offer high virus-binding avidity. We hypothesize that the binding avidities of the four-triangle and five-triangle (full star) do not differ much for the following two reasons: (1) the four-triangle and five-triangle (full star) complexes used in the SPR assays carry nine and ten aptamers, respectively, so there is only one aptamer difference carried by two ‘scaffolds’; (2) four-triangle complex is able to interact similarly with DENV compared with the five-triangle star, with just one tip shy of a full star. Therefore, the two complexes should behave similarly when interacting with a dengue virion. Figure 12 shows the plaque-reducing inhibition curves for the monovalent aptamer and DNA star inhibitors obtained using the described plaque reduction assay procedures for inhibitory evaluation. The ~7,500-fold improvement of antiviral efficacy confirms that the high virus-binding avidity offered by the DNA star results in a highly potent virus inhibitor.
Supplementary Material
Acknowledgements
Preparation of this paper was partially supported by NIH NIAAA (UO1 AA029348) and NIH NIAID (RO1 AI159454) to X.W. The authors thank A. F. Payne for helping with the list of equipment and supplies used in assays to measure DNA star cytotoxicity and plaque reduction test.
Footnotes
Competing interests
A Patent Cooperation Treaty application (PCT/US2020/033398) has been filed on DNA stars by X.W., R.J.L. and P.S.K.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-021-00641-y.
Code availability
The SEQUIN program package used in this study runs on a Windows 10 operating system. It is deposited at figshare and available for download without any access restrictions at https://doi.org/10.6084/m9.figshare.c.5409411. The files include SEQUIN_Program_Package.zip (software package) and SEQUIN_User_Instruction.pdf (software command instruction).
Data availability
All data supporting the findings of this work are available within this paper (figures and description) and the Supplementary Information. All the raw and source data have been deposited at figshare. They can be accessed at https://doi.org/10.6084/m9.figshare.c.5409411. These files include: DNA_Star_Viral_Protocol_Dengue_Model.pse. PyMol session file used for generating Fig. 3 (second panel), Fig. 4, Fig. 5a,b and Supplementary Figs. 1 and 2. Source data are included for Fig. 8 (DNA_Star_Viral_Protocol_Figure_8_Gel_Image.pdf), Fig. 11c (DNA_Star_Viral_Protocol_Figure_11c.xlsx), Fig. 12c (DNA_Star_Viral_Protocol_Figure_12c.xlsx), Supplementary Fig. 3 (DNA_Star_Viral_Protocol_Supplementary_Figure_3_Gel_Image.pdf) and Supplementary Fig. 4 (DNA_Star_Viral_Protocol_Supplementary_Figure_4_Gel_Image.pdf).
References
- 1.Dawood FS et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect. Dis 12, 687–695 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Shrestha SS et al. Estimating the burden of 2009 pandemic influenza A (H1N1) in the United States (April 2009–April 2010). Clin. Infect. Dis 52, S75–S82 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Ali MG et al. Recent advances in therapeutic applications of neutralizing antibodies for virus infections: an overview. Immunol. Res 68, 325–339 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tirado SM & Yoon KJ Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 16, 69–86 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Whitehead SS, Blaney JE, Durbin AP & Murphy BR Prospects for a dengue virus vaccine. Nat. Rev. Microbiol 5, 518–528 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Prasad BV & Schmid MF Principles of virus structural organization. Adv. Exp. Med. Biol 726, 17–47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon PS et al. Designer DNA architecture offers precise and multivalent spatial pattern-recognition for viral sensing and inhibition. Nat. Chem 12, 26–35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fibriansah G et al. Structural changes in dengue virus when exposed to a temperature of 37 degrees C. J. Virol 87, 7585–7592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosuri S & Church GM Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Praetorius F et al. Biotechnological mass production of DNA origami. Nature 552, 84–87 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ducani C, Kaul C, Moche M, Shih WM & Hogberg B Enzymatic production of ‘monoclonal stoichiometric’ single-stranded DNA oligonucleotides. Nat. Methods 10, 647–652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin C et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl Acad. Sci. USA 105, 17626–17631 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seeman NC Nucleic acid junctions and lattices. J. Theor. Biol 99, 237–247 (1982). [DOI] [PubMed] [Google Scholar]
- 14.Chandrasekaran AR & Zhuo RA ‘tile’ tale: hierarchical self-assembly of DNA lattices. Appl. Mater. Today 2, 7–16 (2016). [Google Scholar]
- 15.Duan J, Wang X & Kizer ME Biotechnological and therapeutic applications of natural nucleic acid structural motifs. Top. Curr. Chem 378, 26 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Chao J et al. Programming DNA origami assembly for shape-resolved nanomechanical imaging labels. Nat. Protoc 13, 1569–1585 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Lanphere C et al. Design, assembly, and characterization of membrane-spanning DNA nanopores. Nat. Protoc 16, 86–130 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Sigl C et al. Programmable icosahedral shell system for virus trapping. Nat. Mater 20, 1281–1289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chauhan N & Wang X Nanocages for virus inhibition. Nat. Mater 20, 1176–1177 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Kuzuya A, Sakai Y, Yamazaki T, Xu Y & Komiyama M Nanomechanical DNA origami ‘single-molecule beacons’ directly imaged by atomic force microscopy. Nat. Commun 2, 449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikolovska-Coleska Z Studying protein–protein interactions using surface plasmon resonance. Methods Mol. Biol 1278, 109–138 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Drescher DG, Selvakumar D & Drescher MJ Analysis of protein interactions by surface plasmon resonance. Adv. Protein Chem. Struct. Biol 110, 1–30 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Douzi B Protein–protein interactions: surface plasmon resonance. Methods Mol. Biol 1615, 257–275 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Rath PP, Anand G & Agarwal S Surface plasmon resonance analysis of the protein–protein binding specificity using Autolab ESPIRIT. Bio Protoc. 10, e3519 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baer A & Kehn-Hall K Viral concentration determination through plaque assays: using traditional and novel overlay systems. J. Vis. Exp e52065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendoza EJ, Manguiat K, Wood H & Drebot M Two detailed plaque assay protocols for the quantification of infectious SARS-CoV-2. Curr. Protoc. Microbiol 57, ecpmc105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon SJ et al. Nanostructured glycan architecture is important in the inhibition of influenza A virus infection. Nat. Nanotechnol 12, 48–54 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Nie C et al. Spiky nanostructures with geometry-matching topography for virus inhibition. Nano Lett. 20, 5367–5375 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Lauster D et al. Phage capsid nanoparticles with defined ligand arrangement block influenza virus entry. Nat. Nanotechnol 15, 373–379 (2020). [DOI] [PubMed] [Google Scholar]
- 30.King DJ & Noss RR Toxicity of polyacrylamide and acrylamide monomer. Rev. Environ. Health 8, 3–16 (1989). [DOI] [PubMed] [Google Scholar]
- 31.Malik N et al. Dendrimers: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J. Control Release 65, 133–148 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Ahmad KM, Xiao Y & Soh HT Selection is more intelligent than design: improving the affinity of a bivalent ligand through directed evolution. Nucleic Acids Res 40, 11777–11783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauch EM et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol 35, 667–671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mei QA et al. Stability of DNA origami nanoarrays in cell lysate. Nano Lett. 11, 1477–1482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hahn J, Wickham SFJ, Shih WM & Perrault SD Addressing the instability of DNA nanostructures in tissue culture. ACS Nano 8, 8765–8775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal NP, Matthies M, Gur FN, Osada K & Schmidt TL Block copolymer micellization as a protection strategy for DNA origami. Angew. Chem. Int. Ed. Engl 56, 5460–5464 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Perrault SD & Shih WM Virus-inspired membrane encapsulation of DNA nanostructures to achieve in vivo stability. ACS Nano 8, 5132–5140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat. Biotechnol 36, 258–264 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Kizer ME et al. Hydroporator: a hydrodynamic cell membrane perforator for high-throughput vector-free nanomaterial intracellular delivery and DNA origami biostability evaluation. Lab Chip 19, 1747–1754 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Jiang D et al. DNA origami nanostructures can exhibit preferential renal uptake and alleviate acute kidney injury. Nat. Biomed. Eng 2, 865–877 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y & Yin P Enhancing biocompatible stability of DNA nanostructures using dendritic oligonucleotides and brick motifs. Angew. Chem. Int. Ed. Engl 59, 700–703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerling T, Kube M, Kick B & Dietz H Sequence-programmable covalent bonding of designed DNA assemblies. Sci. Adv 4, eaau1157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothemund PW Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Chandrasekaran AR, Anderson N, Kizer M, Halvorsen K & Wang X Beyond the fold: emerging biological applications of DNA origami. Chembiochem 17, 1081–1089 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Wilner OI & Willner I Functionalized DNA nanostructures. Chem. Rev 112, 2528–2556 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Shaw A et al. Spatial control of membrane receptor function using ligand nanocalipers. Nat. Methods 11, 841–846 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Shaw A et al. Binding to nanopatterned antigens is dominated by the spatial tolerance of antibodies. Nat. Nanotechnol 14, 184–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veneziano R et al. Role of nanoscale antigen organization on B-cell activation probed using DNA origami. Nat. Nanotechnol 15, 716–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee H et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol 7, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klinman DM Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol 4, 249–258 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Liu X et al. A DNA nanostructure platform for directed assembly of synthetic vaccines. Nano Lett. 12, 4254–4259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Auvinen H et al. Protein coating of DNA nanostructures for enhanced stability and immunocompatibility. Adv. Healthc. Mater 6 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Ponnuswamy N et al. Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation. Nat. Commun 8, 15654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anastassacos FM, Zhao Z, Zeng Y & Shih WM Glutaraldehyde cross-linking of oligolysines coating DNA origami greatly reduces susceptibility to nuclease degradation. J. Am. Chem. Soc 142, 3311–3315 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Ramakrishnan S, Ijas H, Linko V & Keller A Structural stability of DNA origami nanostructures under application-specific conditions. Comput. Struct. Biotechnol. J 16, 342–349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bila H, Kurisinkal EE & Bastings MMC Engineering a stable future for DNA-origami as a biomaterial. Biomater. Sci 7, 532–541 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Chandrasekaran AR Nuclease resistance of DNA nanostructures. Nat. Rev. Chem 5, 225–239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kick B, Praetorius F, Dietz H & Weuster-Botz D Efficient production of single-stranded phage DNA as scaffolds for DNA origami. Nano Lett. 15, 4672–4676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palluk S et al. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol 36, 645–650 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Deng Y et al. Intracellular delivery of nanomaterials via an inertial microfluidic cell hydroporator. Nano Lett. 18, 2705–2710 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Ellington AD & Szostak JW In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990). [DOI] [PubMed] [Google Scholar]
- 62.Tuerk C & Gold L Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
- 63.Robertson DL & Joyce GF Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 344, 467–468 (1990). [DOI] [PubMed] [Google Scholar]
- 64.Kizer ME, Linhardt RJ, Chandrasekaran AR & Wang X A molecular hero suit for in vitro and in vivo DNA nanostructures. Small 15, e1805386 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Ke Y, Castro C & Choi JH Structural DNA nanotechnology: artificial nanostructures for biomedical research. Annu. Rev. Biomed. Eng 20, 375–401 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Bujold KE, Lacroix A & Sleiman HF DNA nanostructures at the interface with biology. Chem 4, 495–521 (2018). [Google Scholar]
- 67.Jorge AF & Eritja R Overview of DNA self-assembling: progresses in biomedical applications. Pharmaceutics 10, 268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Udomprasert A & Kangsamaksin T DNA origami applications in cancer therapy. Cancer Sci. 108, 1535–1543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu W et al. Functional nucleic acid nanomaterials: development, properties, and applications. Angew. Chem. Int. Ed. Engl (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weng Y et al. Improved nucleic acid therapy with advanced nanoscale biotechnology. Mol. Ther. Nucleic Acids 19, 581–601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaur H, Bruno JG, Kumar A & Sharma TK Aptamers in the therapeutics and diagnostics pipelines. Theranostics 8, 4016–4032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shum KT, Zhou J & Rossi JJ Aptamer-based therapeutics: new approaches to combat human viral diseases. Pharmaceuticals 6, 1507–1542 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keller A & Linko V Challenges and perspectives of DNA nanostructures in biomedicine. Angew. Chem. Int. Ed. Engl 59, 15818–15833 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang S, Ge Z, Mou S, Yan H & Fan C Designer DNA nanostructures for therapeutics. Chem 7, 1156–1179 (2021). [Google Scholar]
- 75.Zeng Y, Nixon RL, Liu W & Wang R The applications of functionalized DNA nanostructures in bioimaging and cancer therapy. Biomaterials 268, 120560 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Wang H, Luo D, Wang H, Wang F & Liu X Construction of smart stimuli-responsive DNA nanostructures for biomedical applications. Chemistry 27, 3929–3943 (2021). [DOI] [PubMed] [Google Scholar]
- 77.He L, Mu J, Gang O & Chen X Rationally programming nanomaterials with DNA for biomedical applications. Adv. Sci 8, 2003775 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang Z, Qiu L, Zhang T & Tan W Integrating DNA nanotechnology with aptamers for biological and biomedical applications. Matter 4, 461–489 (2021). [Google Scholar]
- 79.Smith DM & Keller A DNA nanostructures in the fight against infectious diseases. Adv. Nanobiomed. Res 2000049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lippe R Flow virometry: a powerful tool to functionally characterize viruses. J. Virol 92, e01765–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zamora JLR & Aguilar HC Flow virometry as a tool to study viruses. Methods 134–135, 87–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xia S et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 30, 343–355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nie J et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect 9, 680–686 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shang J et al. Cell entry mechanisms of SARS-CoV-2. Proc. Natl Acad. Sci. USA 117, 11727–11734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koczula KM & Gallotta A Lateral flow assays. Essays Biochem 60, 111–120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li N et al. Photonic resonator interferometric scattering microscopy. Nat. Commun 12, 1744 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang Z et al. Aptamer switch probe based on intramolecular displacement. J. Am. Chem. Soc 130, 11268–11269 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Yip KM, Fischer N, Paknia E, Chari A & Stark H Atomic-resolution protein structure determination by cryo-EM. Nature 587, 157–161 (2020). [DOI] [PubMed] [Google Scholar]
- 89.Nakane T et al. Single-particle cryo-EM at atomic resolution. Nature 587, 152–156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang X et al. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat. Struct. Mol. Biol 20, 105–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wadood A et al. Epitopes based drug design for dengue virus envelope protein: a computational approach. Comput. Bio. Chem 71, 152–160 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Lin B, Parrish CR, Murray JM & Wright PJ Localization of a neutralizing epitope on the envelope protein of dengue virus type 2. Virology 202, 885–890 (1994). [DOI] [PubMed] [Google Scholar]
- 93.Crill WD & Roehrig JT Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J. Virol 75, 7769–7773 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sukupolvi-Petty S et al. Type- and subcomplex-specific neutralizing antibodies against domain III of dengue virus type 2 envelope protein recognize adjacent epitopes. J. Virol 81, 12816–12826 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gromowski GD & Barrett AD Characterization of an antigenic site that contains a dominant, type-specific neutralization determinant on the envelope protein domain III (ED3) of dengue 2 virus. Virology 366, 349–360 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Lok SM et al. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat. Struct. Mol. Biol 15, 312–317 (2008). [DOI] [PubMed] [Google Scholar]
- 97.Williams KL, Wahala WM, Orozco S, de Silva AM & Harris E Antibodies targeting dengue virus envelope domain III are not required for serotype-specific protection or prevention of enhancement in vivo. Virology 429, 12–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dong Y et al. DNA functional materials assembled from branched DNA: design, synthesis, and applications. Chem. Rev 120, 9420–9481 (2020). [DOI] [PubMed] [Google Scholar]
- 99.Wang YL, Mueller JE, Kemper B & Seeman NC Assembly and characterization of five-arm and six-arm DNA branched junctions. Biochemistry 30, 5667–5674 (1991). [DOI] [PubMed] [Google Scholar]
- 100.Fu TJ & Seeman NC DNA double-crossover molecules. Biochemistry 32, 3211–3220 (1993). [DOI] [PubMed] [Google Scholar]
- 101.Wang X & Seeman NC Assembly and characterization of 8-arm and 12-arm DNA branched junctions. J. Am. Chem. Soc 129, 8169–8176 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang X et al. Paranemic crossover DNA: there and back again. Chem. Rev 119, 6273–6289 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Seeman NC De novo design of sequences for nucleic acid structural engineering. J. Biomol. Struct. Dyn 8, 573–581 (1990). [DOI] [PubMed] [Google Scholar]
- 104.He Y et al. Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature 452, 198–201 (2008). [DOI] [PubMed] [Google Scholar]
- 105.Wang P et al. Retrosynthetic analysis-guided breaking tile symmetry for the assembly of complex DNA nanostructures. J. Am. Chem. Soc 138, 13579–13585 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Caruthers MH A brief review of DNA and RNA chemical synthesis. Biochem. Soc. Trans 39, 575–580 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Roy S & Caruthers M Synthesis of DNA/RNA and their analogs via phosphoramidite and H-phosphonate chemistries. Molecules 18, 14268–14284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hughes RA & Ellington AD Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol 9, a023812 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shlyakhtenko LS, Gall AA & Lyubchenko YL Mica functionalization for imaging of DNA and protein–DNA complexes with atomic force microscopy. Methods Mol. Biol 931, 295–312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fosmire JA, Hwang K & Makino S Identification and characterization of a coronavirus packaging signal. J. Virol 66, 3522–3530 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kuo L, Koetzner CA & Masters PS A key role for the carboxy-terminal tail of the murine coronavirus nucleocapsid protein in coordination of genome packaging. Virology 494, 100–107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Biosafety in Microbiological and Biomedical Laboratories 6th edn (US Department of Health and Human Services, Centers for Disease Control and Prevention, National Institutes of Health, 2020). [Google Scholar]
- 113.Jonsson U et al. Real-time biospecific interaction analysis using surface plasmon resonance and a sensor chip technology. Biotechniques 11, 620–627 (1991). [PubMed] [Google Scholar]
- 114.van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG & Langenhuijsen MM A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 174, 311–320 (1994). [DOI] [PubMed] [Google Scholar]
- 115.Mosmann T Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983). [DOI] [PubMed] [Google Scholar]
- 116.Chacon E, Acosta D & Lemasters JJ Primary cultures of cardiac myocytes as in vitro models for pharmacological and toxicological assessments. in In Vitro Methods in Pharmaceutical Research 209–223 (Elsevier, 1997). [Google Scholar]
- 117.Patravale V, Dandekar P & Jain R Nanoparticulate Drug Delivery: Perspectives on the Transition from Laboratory to Market 123–155 (Elsevier, 2012). [Google Scholar]
- 118.Calisher CH, Monath TP, Karabatsos N & Trent DW Arbovirus subtyping: applications to epidemiologic studies, availability of reagents, and testing services. Am. J. Epidemiol 114, 619–631 (1981). [DOI] [PubMed] [Google Scholar]
- 119.Lindsey HS, Calisher CH & Mathews JH Serum dilution neutralization test for California group virus identification and serology. J. Clin. Microbiol 4, 503–510 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Russell PK, Nisalak A, Sukhavachana P & Vivona S A plaque reduction test for dengue virus neutralizing antibodies. J. Immunol 99, 285–290 (1967). [PubMed] [Google Scholar]
- 121.Abou-Karam M & Shier WT A simplified plaque reduction assay for antiviral agents from plants. Demonstration of frequent occurrence of antiviral activity in higher plants. J. Nat. Prod 53, 340–344 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this work are available within this paper (figures and description) and the Supplementary Information. All the raw and source data have been deposited at figshare. They can be accessed at https://doi.org/10.6084/m9.figshare.c.5409411. These files include: DNA_Star_Viral_Protocol_Dengue_Model.pse. PyMol session file used for generating Fig. 3 (second panel), Fig. 4, Fig. 5a,b and Supplementary Figs. 1 and 2. Source data are included for Fig. 8 (DNA_Star_Viral_Protocol_Figure_8_Gel_Image.pdf), Fig. 11c (DNA_Star_Viral_Protocol_Figure_11c.xlsx), Fig. 12c (DNA_Star_Viral_Protocol_Figure_12c.xlsx), Supplementary Fig. 3 (DNA_Star_Viral_Protocol_Supplementary_Figure_3_Gel_Image.pdf) and Supplementary Fig. 4 (DNA_Star_Viral_Protocol_Supplementary_Figure_4_Gel_Image.pdf).