

ABSTRACT
The rare capacity for heat shock protein 90 (Hsp90) chaperones to support almost the entire cellular signaling network was viewed as a potential breakthrough to combat tumor resistance to single-oncogene-based therapeutics. Over 2 decades, several generations of Hsp90 ATP binding inhibitors have entered numerous cancer clinical trials, but few have advanced to FDA approval for treatment of human cancers. Herein, we report that Hsp90 expression varies dramatically, especially among different types of noncancer cells and organs. The highly variable levels of Hsp90, from as low as 1.7% to as high as 9% of their total cellular proteins, were responsible for either an extreme sensitivity or an extreme resistance to a classical Hsp90 ATP-binding inhibitor. Among randomly selected cancer cell lines, the same client proteins for regulation of cell growth exhibited unexpectedly heterogenous reactions in response to an Hsp90 ATP-binding inhibitor, inconsistent with the current understanding. Finally, a minimum amount (<10%) of Hsp90β was still required for client protein stability and cell survival even in the presence of full Hsp90α. These new findings of Hsp90 expression in host and isoform compensation in tumor cells could complicate biomarker selection, toxicity readout, and clinical efficacy of Hsp90-ATP-binding inhibitors in cancer clinical trials.
KEYWORDS: Hsp90α, Hsp90β, chaperones, ATP-binding inhibitors, druggable window, cancer therapy, clients, geldanamycin, heat shock protein 90, isoforms
INTRODUCTION
The heat shock protein 90 (Hsp90) family proteins serve as ATP-dependent critical chaperones for stability and functionality of signaling proteins distributed across almost the entire cellular signaling networks in most cell types. The reported higher expression or higher affinity of Hsp90 for cellular oncogene products in tumor cells has been targeted by many molecule inhibitors that bind the N-terminal ATP/ADP binding site of Hsp90 in at least 60 cancer clinical trials over the past 2 decades (1–8). To date, few have received FDA approval for clinical treatment of human cancers due to various cited reasons (5–7). In humans, the constitutively expressed Hsp90β and the stress-inducible Hsp90α differ at a total of 100 amino acid residues, most significantly in the highly charged linker region, along their 724 (Hsp90β)- and 732 (Hsp90α)-amino-acid sequences. In cultured cells, complete Hsp90α knockout did not affect cell morphology, survival, or growth rates, whereas a similar attempt to knock out Hsp90β led to cell death during drug selection (9) or the single-cell cloning period (10) in both cancer and noncancer cells. The seemingly noninterchangeable roles of Hsp90α and Hsp90β in cultured cells have further been substantiated by mouse genetic studies. Mice with either chaperone-defective mutations in Hsp90α (10, 11) or complete Hsp90α knockout (10) showed little phenotypic difference from their wild-type counterparts other than defective spermatogenesis in male mice (11), blockade of extracellular antigen translocation (12), and hydrocephalus-like syndrome in approximately 20% of homogenous mice (10). In contrast, mice with Hsp90β deficiency disrupted the placental labyrinth formation and died on E10.5 (13). The straightforward interpretations of the above findings seem to be that (i) Hsp90β is more critical than Hsp90α for cell survival and during mouse development and (ii) Hsp90α function could be replaced by Hsp90β alone in the Hsp90α-knockout cells and mice.
A large number of previous publications provided in vitro and in vivo support for the notion of higher accumulation or higher sensitivity of tumor cells or tumor-bearing mice to the N-terminal ATP-binding inhibitors of Hsp90 than normal cell and mice. Kamal et al. reported a 100-fold difference in binding affinity of cell-free Hsp90 protein complex between tumor cells and normal cells to 17-AAG (17-N-allylamino-17-demethoxygeldanamycin) (14). Solit et al. reported that the maximally tolerated dose of 17-AAG was higher in control mice than in tumor-bearing mice (15). Chiosis’ group showed that normal cells could reach up to 700- to 3,000-fold more resistance than tumor cells to purine-scaffold inhibitors (16–20). The higher inhibitor-binding affinity by tumor cells was thought to be due to posttranslational modifications in Hsp90 (21–23), mutations (24), higher drug retention by tumor cells (25), formation of oncogenic complexes such as the “epichaperome” (26), or all of the above. In contrast to these cell-free protein binding results, data obtained with intact cells in response to 17-AAG, 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin), and purine-based inhibitors were less unclear. For instance, Premkumar et al. reported moderate difference in cellular toxicity between 20% in normal cells and 50% in cancer cells to 17-AAG (27). Similarly, Lukasiewicz and colleagues showed 30 to 50% normal cell death versus 55 to 80% cancer cell death upon treatment with 17-AAG or 17-DMAG (28). Instead of a 1,000-fold difference in cell-free binding assays, two groups showed a modest 20-fold difference in cell growth inhibition between tumor cells and a normal cell line (17, 18). Furthermore, Vilenchik et al. reported less than a 10-fold difference in the 50% inhibitory concentrations (IC50s) for the inhibitor PU24FCI in cell growth inhibition between two normal and 15 cancer cell lines (19). Therefore, results of the above studies showed significant data variation and inconsistency.
In the current study, we first used a well-characterized tumor cell model to explore the individual roles for Hsp90α and Hsp90β in their protection of client proteins and their support of cell survival in the absence or presence of 17-DMAG. Then, we expanded the investigations to eight randomly selected cancer and four noncancer cell lines. We measured the Hsp90 expression (as a percentage of the total soluble proteins) in these cells and correlated it to the cells’ ability to survive after exposure to 17-DMAG. Results of this new study raise previously unrecognized complexities and hurdles for Hsp90 inhibitors to become clinical therapeutics, in either an isoform-specific or global targeting strategy, and thus provide valuable information for ongoing and future cancer clinical trials with the class of Hsp90 inhibitors.
RESULTS
Only Hsp90α/Hsp90β double depletions cause collapse of cellular client proteins.
Prompted by previous reports on “distinct roles” for Hsp90α and Hsp90β during mouse development (10–13), we were interested in determining the biochemical basis of the distinctions by studying the individual roles of Hsp90α and Hsp90β in protecting client protein stability in the same cells. To begin with, we chose the highly malignant human breast cancer cell line, MDA-MB-231, which has an elevated level of Hsp90 protein, up to 3.66% of its total cellular proteins, in comparison to an average of 2.3% in four selected nontransformed cell lines (29). Using CRISPR-Cas9 technology, we obtained Hsp90α knockout (Hsp90α-KO) cell clones. However, similar attempts to obtain Hsp90β-KO cell clones resulted in cell death during drug selection and cell cloning (9, 10). Therefore, using a lentivirus infection system to deliver short-hairpin RNA (shRNA) against the Hsp90β gene, we obtained (90 to 95%) Hsp90β knockdown (Hsp90β-KD) cells, as well as Hsp90α-KO/Hsp90β-KD cells (Fig. 1A, panels a and b). The Hsp90β-KD cells exhibited normal morphology, survived, and proliferated with only a slightly lower growth rate than their parental counterpart. Why the presence of even a small fraction of Hsp90β is needed, together with Hsp90α, for preventing client protein degradation and cell death remains to be further studied. However, the Hsp90α- and Hsp90β-depleted cells survived and expanded for a period of 10 to 14 days. Similar observations were previously reported in Saccharomyces cerevisiae (30) and in a human oral cancer cell line (31).
FIG 1.

Single and double depletions of Hsp90α and Hsp90β on client protein stability. (A) The CRISPR-Cas9 gene editing technique was utilized to create Hsp90α-KO, and a lentiviral infection-mediated shRNA delivery system, FG-12, was used to downregulate Hsp90β or Hsp90α or both in MDA-MB-231 cells. The efficiency of knockout and knockdown of Hsp90β and Hsp90α is shown (panels a and b). (B and C) Parental MDA-MB-231 cells cultured in medium with 10% FBS to 80% confluence were utilized to establish a standard duration and dosage of 17-DMAG treatment. Total lysates of the untreated cells (vehicle alone) and cells treated with 100 nM 17-DMAG for indicated time points up to 72 h (B) and of untreated cells and cells treated with 0 to 1,000 nM 17-DMAG for 48 h (C). All samples were subjected to Western immunoblotting analysis with antibodies against Hsp90α and Hsp90β or five signaling proteins of the mitogenic signaling pathway. Equal loadings of the cell lysates were confirmed with β-actin. Similar results were obtained from three independent experiments, in which the reduced level of the bands in (A, asterisks) was reproducible.
With these four lines of MDA-MB-231 cells, i.e., the (i) parental, (ii) Hsp90α-KO, (iii) Hsp90β-KD, and (iv) Hsp90α-KO/Hsp90β-KD cells, cultured in complete medium with 10% fetal bovine serum (FBS), we examined the steady-state protein or phosphorylation levels of five common signaling molecules in the so-called mitogenic pathway, from the cell surface via the cytosol to the nucleus, including epidermal growth factor receptor (EGFR), Akt1, Akt2, phospho-Erk1/2, and cyclin D1 (Fig. 1A, panels c to g, lanes 1 to 4), with β-actin as the sample loading control (Fig. 1A, panel h). All five client proteins or phosphoprotein were clearly detected in the parental cells (Fig. 1A, lanes 1). In either Hsp90β-KD cells (Fig. 1A, lanes 2) or Hsp90α-KO (lanes 3), all five protein or phosphorylation levels were largely indistinguishable from those in the parental cells, except a modest decrease in Akt2, phospho-Erk1/2 and cyclin D1 levels (marked with asterisks) in Hsp90β-KD cells. In contrast, all five signaling proteins, but not the β-actin control (Fig. 1A, panel h), showed a dramatic decline in Hsp90α-KO/Hsp90β-KD cells (Fig. 1A, lanes 4, as indicated by a dashed rectangle). The variations among the different molecules were not due to their different half-lives, since these gene-manipulated cells were all cultured for 4 to 5 days prior to the analyses. We concluded that Hsp90α and Hsp90β have strong compensatory functions for each other.
Increasing sensitivities in cells with single and double Hsp90 depletions to a classical ATP-binding inhibitor.
The pioneering ATP-binding inhibitor of Hsp90, geldanamycin (GA), and several of its derivatives have been extensively studied as benchmarks for inhibiting Hsp90 chaperone functions and entered numerous clinical trials as potential antitumor therapeutics (2–4). For this study, we chose the semisynthetic and water-soluble derivative of G,A 17-DMAG, as a classical representative for all ATP-binding inhibitors of Hsp90 chaperones. First, we set out to identify the minimum time and dosage of 17-DMAG that start to cause dramatic destabilization of the five client proteins using MDA-MB-231 cells. Consistent with a previous report (10), we found that treatment of the cells with a fixed 100 nM concentration of 17-DMAG at 48 h significantly decreased the cellular levels of EGFR, Akt1, Akt2, phospho-Erk1/2, and cyclin D1 (Fig. 1B). In contrast, upon treatment with increasing dosages of 17-DMAG for 48 h, 100 nM 17-DMAG started to cause significant declines of the five client proteins (Fig. 1C).
Next, we used the treatments to study the individual roles for Hsp90α and Hsp90β in protecting client proteins using Hsp90α-KO, Hsp90β-KD, and Hsp90α/Hsp90β-depleted MDA-MB-231 cells in response to 17-DMAG. In the parental cells, significant degradation of the client proteins was detected upon treatment with 100 nM 17-DMAG (Fig. 2A, lanes 4), as previously shown. Interestingly, a similar degree of client protein degradation shifted to 30 nM 17-DMAG in cells with single depletion of either Hsp90α (Fig. 2B, lanes 9) or Hsp90β (Fig. 2C, lanes 15). Moreover, significant client protein degradation was already detected in cells with Hsp90α and Hsp90β double depletions even in the absence of 17-DMGA treatment (Fig. 2D, lanes 19, versus Fig. 2A, lane 1), and, as expected, a catastrophic degradation of client proteins occurred at the lowest dosage (10 nM) of 17-DMAG (lanes 20). When three of the four cell lines were subjected to a 48-h cell survival and growth analysis, as shown in Fig. 2E, the Hsp90α-KO and Hsp90β-KD cells showed a weaker survival, slower growth, and more death than the parental cells. Since a majority of the Hsp90α/Hsp90β-depleted cells died within 10 to 14 days, they were not included in this assay. Taken together, these findings indicate that (i) Hsp90α and Hsp90β make up a threshold level of chaperoning activity to maintain the maximum stability of client proteins and cell viability and (ii) even a fraction of the endogenous level of Hsp90β was sufficient to work with Hsp90α. It should be pointed out that, in Fig. 2A to D, equalized amounts of total proteins were used for the Western immunoblotting analyses, but the total cell numbers were unequal after 48 h of treatment with increasing dosages of 17-DMAG. For instance, the cells in medium with 300 nM and 1,000 nM 17-DMAG were unable to expand, in comparison to their numbers on day 0, as indicated in Fig. 2E.
FIG 2.

Only a small fraction of Hsp90β is needed for protecting cells from ATP-binding inhibitor. (A to D) The parental, Hsp90α-KO, Hsp90β-KD, and Hsp90β/Hsp90α-depleted MDA-MB-231 cells (Fig. 1) were treated with increasing concentrations of 17-DMAG for 48 h. Total lysates of the cells were subjected to Western immunoblotting analysis with indicated antibodies (A to D). (E) The parental, Hsp90α-KO and Hsp90β-KD MDA-MB-231 cells were subjected to a cell growth assay in the absence and presence of increasing dosages of 17-DMAG treatment. The percentage of attached cells was determined on days 0 and 2 and plotted against various dosages of 17-DMAG. This experiment was repeated three times. Data are means and SD. P < 0.05, as significant.
Dramatic variations among the same client proteins in different cancer cells in response to 17-DMAG.
However, in contrast to the current understanding, the client protein response from 17-DMAG-treated MDA-MB-231 cells does not represent any common theme among other cancer cells. When we compared the responses of the same five client proteins in seven additional cancer cell lines (with total cellular proteins equalized) to that in the standardized treatment with 17-DMAG, as shown in Fig. 3, we found a dramatically heterogenous range of variations. Specifically, (i) EGFR was the most sensitive client protein among the five tested (Fig. 3a, lanes 2, 6, 10, 12, and 14), consistent with a previous report (23), except in HeLa cells, where the EGFR level remained unchanged (lane 8, band 4); (ii) similarly, the Akt1 level changed in some cancer cell lines (Fig. 3b, lanes 2, 4, 6, 8, and 10), while it remained unchanged in other cancer cell lines (lanes 12, 14, and 16); (iii) to our surprise, some client proteins were downregulated, unchanged, or even upregulated even in the same cells, such as Akt1, Akt2, and cyclin D in B16 cells (Fig. 3, lanes 4, bands 1, 2, and 3) and Akt1 versus Akt2 in in HeLa cells (lanes 8, bands 5 and 6), following 17-DMAG treatment. Finally, these biochemical differences correlate with neither their total Hsp90 expression nor viability (see Fig. 5 and 6). The cause of these unexpected variations could be due to such reported mechanisms as posttranslational modifications of Hsp90 proteins (23) or oncogenic mutations in client molecules (24) or both.
FIG 3.

Highly heterogenous responses of the same client proteins in different tumor cells in response to ATP-binding inhibitor. Eight cancer cell lines were treated without (vehicle alone) or with 100 nM 17-DMAG for 48 h. Total cell lysates were equalized (within each cell line itself) for total cellular proteins and subjected to Western immunoblotting analysis with the indicated antibodies. Measurements of 17-DMAG-caused changes in client protein levels were restricted within each of the cell lines without or with 17-DMAG treatment. The results were confirmed in three independent repetitions of the same experiment.
FIG 5.

Comparison of the relative levels (percentages) of Hsp90 in selected normal and tumor cells. The total cellular proteins of 13 cell lines were equalized and subjected to Western immunoblotting analysis with a pan-anti-Hsp90 antibody. The relative amount of the total cellular Hsp90 protein (percentage of total cellular proteins) of the tested cell lines was quantitated via NIH ImageJ analysis against their own β-actin and then compared to the previously reported 3.66% of total Hsp90 protein in MDA-MB-231 cells (23) to calculate the percent Hsp90. (A) Two separate Western anti-Hsp90 immunoblots of equalized total cell lysates with β-actin as a loading control; (B) method of calculation; (C) plot of the results of the NIH ImageJ analyses. Three independent experiments were carried out, and data are means and SD. P < 0.05, as significant.
FIG 6.

Lack of a druggable window between normal and tumor cells for a Hsp90 ATP-binding inhibitor. The eight cancer cell lines and four nontransformed cell lines were seeded in culture plates for the first 48 h to settle, and the cell number per well was designated the day 0 number. The cells were incubated for an additional 48 h in media with increasing concentrations of 17-DMAG. Following three washes with PBS, attached cells were lifted by trypsin and counted in triplicate, designated day 2 (see Materials and Methods). Relative cell number (as a percentage of that of day 0) for each cell line was plotted against the various dosages of 17-DMAG. The experiment was repeated multiple times. Data are means and SD. P < 0.05, as significant.
Highly variable sensitivities of client proteins among different noncancer cells to 17-DMAG.
One of the key theories to support both previous and current cancer clinical trials with Hsp90 inhibitors is that the corresponding noncancer counterpart cells are less sensitive than the cancer cells to the inhibitors. First, we repeated the same experiments to examine the client protein response to 17-DMAG using two randomly selected human and two randomly selected mouse nontransformed primary or cell lines. As shown in Fig. 4, under conditions in which a vast majority of the cells were still alive for generating sufficient cell lysates, we found dramatic variations in sensitivity of the same signaling molecules in response to increasing dosages of 17-DMAG treatment. Interestingly, unlike the eight cancer cell lines shown in Fig. 3, we found less heterogenous responses of the client proteins in the same cells to 17-DMAG. In terms of cell viability at the various dosages of 17-DMAG, the sensitivities were highest for mouse embryo fibroblasts (MEF) (Fig. 4B), followed by normal dermal fibroblasts (NDF) (Fig. 4A), NIH 3T3 cells (Fig. 4C), and 293T cells (Fig. 4D), showing lack of a consensus to support the theory of clinical trials. While the number of cell lines was small, these data suggest that a given dosage of an Hsp90 inhibitor in a clinical trial may be safe for that specific cell marker but toxic to other cell types and organs in the same patient.
FIG 4.

Highly heterogenous sensitivities of client proteins in different normal cells in response to ATP-binding inhibitor. Four normal cell lines were treated without (vehicle alone) or with 100 nM 17-DMAG for 48 h. Total cell lysates were equalized (within each cell line itself) for total cellular proteins and subjected to Western immunoblotting analysis with the indicated antibodies. Measurements of 17-DMAG-caused changes in client protein levels were restricted within each of the cell lines without or with 17-DMAG treatment. The results were reproducible in multiple independent repetitions of the same experiment.
Heterogenous expression and variations in protection, especially in normal cells and organs, could complicate the design of cancer clinical trials with Hsp90 inhibitors.
In an attempt to understand the reason for these observations, we decided to measure the relative percentage of the total Hsp90 protein in the four noncancer and the eight tumor cell lines and to study if there was any correlation between Hsp90 expression and sensitivity of the cells to 17-DMAG. Using our previously published synchronized Western blotting protocol and the 3.66% Hsp90 (of the total cellular soluble proteins) in MDA-MB-231 cells (28) as the reference, we obtained the relative Hsp90 levels (as percentages) for rest of the 11 cell lines. As shown in Fig. 5A, lysates of all 12 cell lines were equalized for total cellular proteins and subjected to “synchronized” steps of gel electrophoresis (corun), immunoblotting (in the same containers all the time), enhanced chemiluminescence (ECL) reaction (together), and film exposure (in same cassette) with a pan-anti-Hsp90 antibody, with β-actin as the loading control. Then, following the equation shown in Fig. 5B, ImageJ scanning data of Hsp90 were divided by the scanning data of its own β-actin band from each cell line, followed by calibration in reference to the same set of ImageJ scanning data from MDA-MB-231. Under these calculations, as summarized shown in Fig. 5C, most tumor cells showed compatible levels of Hsp90, except B16 cells. In contrast, among the four noncancer cell lines, NDF showed as little as 1.7% and 293T cells showed as much as 9% of their corresponding total cellular proteins. Consistently, we also observed such heterogenous patterns of Hsp90 expression in different mouse organs. The total Hsp90 proteins varied greatly among the organ tissues tested, of which brain and testis (11) showed the highest levels and lung and kidney the lowest (Fig. 5D, panel a). More interestingly, the variations were largely due to the differences in Hsp90α expression (Fig. 5D, panel b), since Hsp90β remained relatively constant (panel c). Grad and colleagues previously showed extremely high expression in brain and testis and extremely low expression in heart, muscle, and pancreas (11).
When all the cancer and noncancer cells were subjected to cell survival assay in medium with increasing dosages of 17-DMAG. As shown in Fig. 6, the overall trend of the cell survival profiles was similar: i.e., all cells showed a decline in the number of the viable cells in a dose-dependent fashion in response to 17-DMAG. Among the four noncancer cell lines (NDF, 293T, MEF, and NIH 3T3), the human NDF, which showed the lowest level of Hsp90 expression (1.74%), exhibited the highest sensitivity, with 60% live cells in response to the lowest concentration of 10 nM 17-DMAG in comparison to the control (100%) point (Fig. 6A); human 293T cells, which showed the highest level of Hsp90 expression (∼9%), exhibited the lowest sensitivity with 60% live cells even after treatment with 300 nM 17-DMAG (Fig. 6D), followed by MEF and NIH 3T3 cells (Fig. 6B and C). The eight tumor cell lines also showed heterogenous cell survival profiles, largely according to their total Hsp90 levels, in which 30 nM to 1 μM was the effective dosage range in comparison to their control points (Fig. 6E to L). These results suggest that (i) there is a lack of a clear window in sensitivity between noncancer and cancer cells for the ATP-binding inhibitor and (ii) there is a lack of a clear cutoff dosage among normal cells for the inhibitor.
Hsp90 level- and ATPase-dependent protection of client proteins and cell survival.
To confirm the dose- and ATPase-dependent mechanism for Hsp90 in protection of client proteins, we carried out rescue experiments using the Hsp90α-KO MDA-MB-231 cells, where only endogenous Hsp90β is left and 17-DMAG treatment (100 nM, 48 h) of the cells causes a massive decrease in client proteins. We used the pRRLsinh-CMV lentiviral system to exogenously overexpress Hsp90α, the most variable isoform in organs (Fig. 5), in comparison to the parental control cells. To distinguish it from endogenous Hsp90, we utilized green fluorescent protein (GFP)-tagged wild-type or ATP-binding mutant (D93N) human Hsp90α. The lysates of the cells were immunoblotted with a pan-anti-Hsp90 antibody that recognizes both Hsp90α and Hsp90β. The endogenous Hsp90 from the parental cells represented the total Hsp90 (Hsp90α and Hsp90β) proteins (Fig. 7A, panel a, lanes 1 and 2), whereas the endogenous Hsp90 from the Hsp90α-KO cells represented only Hsp90β protein (panel a, lanes 3 to 8, lower bands). It should be pointed out that Hsp90β level is significantly elevated under Hsp90α-KO stress (10–12), making it appear similar to the total Hsp90 level in the control cells. The expression of GFP-Hsp90α (Fig. 7A, panel a, lanes 5 to 8, upper bands) was 3- to 5-fold higher than the total endogenous Hsp90 (α and β combined) in the parental cells (panel a, lanes 1 and 2). 17-DMAG treatment caused degradation of all five client proteins in the parental (Fig. 7A, panels b to f, lanes 2) and stronger degradation in Hsp90α-KO (panels b to f, lanes 4) cells, as previously shown (Fig. 2). However, the 17-DMAG-caused client protein degradation was completely reversed in cells with overexpressed GFP-Hsp90α-wt (Fig. 7A, panels b to f, lanes 6) but not the GFP-Hsp90α-D93N mutant (panels b to f, lanes 8), which was best represented by the EGFR (indicated by numbered circles). Accordingly, the cells with overexpressed GFP-Hsp90α-wt, but not the GFP-Hsp90α-D93N mutant, showed increased resistance to 17-DMAG in cell survival assays. As shown in Fig. 7B, with increasing dosages of 17-DMAG in the culture medium, the cells with overexpressed GFP-Hsp90α-wt showed significantly stronger resistance than the cells with GFP alone or the GFP-Hsp90α-D93 mutant. Under our experimental conditions, these observations suggested that high levels of Hsp90 could make tumor cells more, rather than less, resistant to ATP-binding inhibitors, although it is unclear if the overexpressed GFP-Hsp90α-wt protein underwent posttranslational modifications similar to those of the endogenous Hsp90α.
FIG 7.

Exogenously elevated Hsp90 confers Hsp90-KO tumor cells full resistance to an ATP-binding inhibitor. Hsp90α-KO MDA-MB-231 cells were exogenously re-expressed at higher levels with GFP-labeled wild-type (GFP-90α-wt) or ATPase-defective mutant (GFP-90α-D93N) Hsp90α by the pRRLsinh-CMV lentiviral infection system (see Materials and Methods). These cells were subjected to both client protein protection (Western) assay and cell growth assay in response to 17-DMAG, in comparison to the parental and GFP-only Hsp90α-KO cells. (A) Western immunoblotting analysis of the total cell lysates with the indicated antibodies, in which comparison of the data (panels a to f) is highlighted by rectangles. β-Actin was used as the sample loading control (panel g). (B) Cell growth/death analysis in complete medium in the absence or presence of increasing dosages of 17-DMAG, in which the number of viable cells was counted on day 0 and day 2. This experiment was repeated three times, with results in excellent agreement. Data are means and SD. P < 0.05, as significant.
To further correlate Hsp90 level and cell sensitivity to the ATP-binding inhibitor, we turned to noncancer cell lines by choosing the highest-Hsp90-expressing HEK 293T cells and the lowest-Hsp90-expressing human normal dermal fibroblasts (NDF). Since Hsp90α (not Hsp90β) represents the variations of the total Hsp90 levels in cells and organs (Fig. 5), our idea was to downregulate Hsp90α in 293T cells, to upregulate Hsp90α in NDF by lentiviral infections, and to subject the cells to a cell survival assay in the presence of increasing dosages of 17-DMAG. We were able to express a GFP-Hsp90α gene at levels severalfold higher than its endogenous counterpart (Fig. 8A, inset, lane 2 versus lane 1) in NDF. Under these conditions, we found that the GFP-Hsp90α-overexpressing NDF cells exhibited significantly higher resistance than the vector control NDF cells to the treatment of 10 nM to 30 nM 17-DMAG but were still not able to resist 100 nM or 300 nM 17-DMAG. In contrast, we were able to downregulate the endogenous Hsp90α in 293T cells to approximately 30% of the vector alone-infected cells (Fig. 8B, inset, lane 2 versus lane 1). The 293T cells with reduced Hsp90α showed a higher degree of sensitivity to 17-DMAG at all tested dosages. Taken together, these in vitro findings further support the main point of this study, that the heterogeneity in Hsp90 expression and responses to Hsp90 inhibitors in different normal cell types and organs could greatly complicate the design and critical readouts of a clinical trial.
FIG 8.
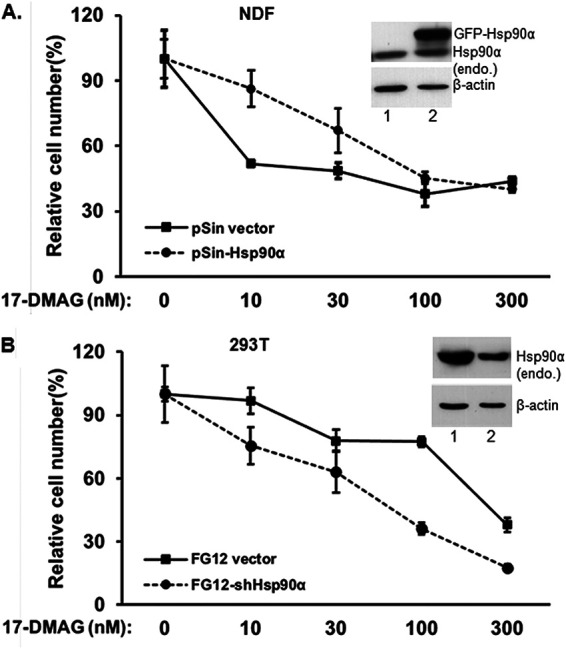
Hsp90 levels determine cell sensitivity or resistance to ATP-binding inhibitor. (A) The lowest Hsp90-expressing NDF cells were exogenously overexpressed with GFP-Hsp90α, and (B) the highest Hsp90-expressing 293T cells had an shRNA introduced to downregulate the endogenous Hsp90α. Both cell lines were then subjected to a cell survival assay with increasing dosages of 17-DMAG and compared to their wild-type counterparts. The results shown represent one of three repeated experiments with data in excellent agreement. Data are means and SD. P < 0.05, as significant.
DISCUSSION
The first Hsp90 inhibitor, 17-AAG, entered cancer clinical trials in 1999, followed by its water-soluble derivative, 17-DMAG, and subsequently by a dozen more inhibitors that all target the N-terminal ATP-binding site of the Hsp90 family proteins (5–7). A key scientific support for launching the initial clinical trials came from the study showing that 17-AAG directly binds to Hsp90, instead of its client protein v-src tyrosine kinase, resulting in dissociation of Hsp90 from v-src and subsequent v-src protein degradation, in v-src-transformed mouse NIH 3T3 and human PC3 prostate cancer cell lines (32). Apart from this early landmark study and related studies in yeast during the same period (33, 34), there had been limited in vitro and in vivo studies that support existence of a druggable window prior to the clinical trials. Only several years later, a number of studies reported that (i) Hsp90 complexes from tumor cells had higher binding affinity for the inhibitors than those from normal cells (14), (ii) the proliferation of normal cells has higher resistant to the inhibitors than tumor cells (16), and (iii) tumor-bearing nu/nu mice were more sensitive to 17-AAG than control mice (15).
The overall interpretations of these early studies have been less definitive, if not controversial. For instance, in the study by Solit and colleagues, not only was the maximally tolerated dose of 17-AAG drug administration schedule dependent, but the reported higher sensitivity in the tumor-bearing mice could simply be due to the fact that these mice had poorer health than the tumor-free control mice (15). In the current study, we found a large degree of heterogeneity in Hsp90, specifically Hsp90α, expression in different types of normal cells in culture and different organs in mice. The cells with lower Hsp90 expression are far more sensitive than cells with higher Hsp90 expression to ATP-binding inhibitors. While previous clinical trials often chose a few and easily accessible host cell types or organs as the biomarkers, such as peripheral blood mononuclear cells (PBMCs) for drug pharmacokinetics and liver for drug toxicity, our current findings suggest that a potentially wide range of pharmacokinetics and toxicity profiles of Hsp90 ATP-binding inhibitors from normal cells and organs in the host may significantly complicate the design of a clinical trial. In addition, the highly compensatory functions between Hsp90α and Hsp90β can dim the hope for isotope-specific drugs.
The reverse correlation between Hsp90 expression and degree of cellular toxicity of 17-DMAG was substantiated by overexpressing the wild-type Hsp90α in NDF cells and by downregulating Hsp90α in 293T cells, granting the former cells increased resistance and the latter cells increased sensitivity to 17-DMAG. These findings have several clinically relevant implications to currently ongoing and future cancer clinical trials with the class of Hsp90 ATP-binding inhibitors. First, the toxicity-tolerable dosages of an inhibitor may greatly vary among different types of normal cells, tissues, and organs in the same human patient. It would be technically difficult to monitor potentially dangerous, if left unnoticed, side effects of the inhibitor during clinical trials, which often include a single biomarker. The interpretation of a trial clinical data from a single or selected few cell biomarkers as the “overall” pharmacokinetic and toxicity readouts could lead to unexpected long-term harm to the patient. On the other hand, it may not be technically and economically feasible to measure drug toxicity in all organs of the patient. Second, it is known that different cancer cells, even among cells of the same type of cancer, show either variable degrees of elevated Hsp90 or remain unchanged in comparison to their corresponding normal cell counterparts (29, 35). There has been little evidence that even the same clinically defined tumor exhibits similar Hsp90 expression among different patients, posing a potential difficulty of choosing the right dosage range, especially during the late stage of clinical trials with a large number of patients. Therefore, prior to drug treatment, it may be necessary to measure the tumor Hsp90 levels in cancer patients, grouping patients with similar Hsp90 levels and treating different groups with different dosages of the inhibitor. If these concerns prove valid, they will greatly complicate the design of a cancer clinical trial. In retrospect, the wide range of Hsp90 expression in normal organs and even in the same tumors in different patients could have been among the factors that caused the failure of the previous clinical trials.
Our finding that Hsp90α and Hsp90β compensate for each other’s absence to protect client protein stability and cell survival does not explain either the distinct phenotypes of Hsp90α- and Hsp90β- gene knockout mice (10–13) or the opposite outcomes of CRISPR-Cas9 knockouts of Hsp90α and Hsp90β in cultured cells (9, 10). We speculate that the complete absence of a gene isoform, such as by various gene-editing technologies, does not reflect naturally occurring scenarios. As far as Hsp90α and Hsp90β are concerned, the difference between the Hsp90 isoforms in reality is limited to lower or higher levels, instead of total absence, between the two gene products (Fig. 5D). Therefore, interpretation of a phenotype of a complete gene knockout should be done with caution in terms of its physiological relevance and its actual cause, i.e., whether it is directly due to the knocked-out gene or something else that never naturally takes place. Such an irrelevant incident that has actually caused cell death bears little biological significance.
Instead, we believe that partial gene knockdown more closely reflects the changes under physiological conditions. Our definition of “compensation” is based on this understanding, in which Hsp90α and Hsp90β under the condition of a partial gene knockdown highly compensate each other’s shortage both in protecting client proteins and in supporting cell survival in response to a drug insult. While Hsp90β gene knockout by CRISPR-Cas9 technology led to death of cultured tumor and normal cells (9, 10), the cells with shRNA-mediated Hsp90β knockdown showed client protection, cell growth profiles, and resistance comparable to those seen with 17-DMAG. It is possible that the remaining (small) portion of Hsp90β in Hsp90β-knockdown cells is sufficient to support the yet-to-be identified distinct and critical client protein(s), which is destabilized in Hsp90β-knockout cells and mice. On the other hand, the Picard laboratory was able to obtain Hsp90β-knockout cell lines, albeit with a higher degree of difficulty and longer selection process (D. Picard, personal communication). It is of great interest to identify the putative factor(s) that is specifically chaperoned by Hsp90β and essential for cell survival, which could provide valuable new drug targets for the design of effective anti-Hsp90 cancer drugs.
MATERIALS AND METHODS
Cell culture and treatments.
The cancer cell lines MDA-MB-231, MDA-MB-468, MCF-7, HeLa, HT-29, H460, A549, and B16 and the normal cell lines NIH 3T3, 293T, NDF, and MEF were cultured in Dulbecco’s modified Eagle medium (DMEM) with penicillin-streptomycin (100 U/mL-0.1 mg/mL) and 10% fetal bovine serum (FBS) (Thermo Scientific, MA, USA). All cells were tested to ensure that they were mycoplasma free every 2 months at USC Tissue Culture Core. The third or fourth passages of the primary human cells, such as NDF, were used for this study. CRISPR-Cas9 knockout of Hsp90α (gene ID, 3320) and human Hsp90β (gene ID, 3326) in MDA-MB-231 cells was performed as previously described, in which attempts to knock out the human Hsp90β gene led to cell death after the second round of double drug selection (24). Similarly, cell death occurred after attempts to knock out mouse Hsp90β in MEF (25). 17-DMAG hydrochloride (1 mg) (no. 1776-1; BioVision, Milpitas, CA) was dissolved in 100 μl dimethyl sulfoxide (DMSO) to make a 15.3 mM stock solution.
Antibodies.
The antibodies used in this study include anti-Hsp90α antibody (NB120-2928) from Novus Biologicals (Littleton, CO); anti-Hsp90β (H9010; StressMarq Biosciences Inc, Victoria, BC, Canada); anti-EGFR (D38B1; no. 4267), anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (D13.14.4E; no. 4370), anti-Akt1 (C73H10; no. 2938), and anti-Akt2 (D6G4; no. 3063) antibodies from Cell Signaling Technology (Beverly, MA); anti-cyclin D1 antibody (EPR2241; GTX61845; GeneTex, Irvine, CA); and anti-β-actin antibody (AC038; Transduction Laboratories, San Jose, CA). ECL Western blotting detection reagent (no. RPN2106) was from Amersham, Inc. (Marlborough, MA).
Gene knockout and knockdown cell lines.
The lentiviral infection system pRRLsinh-CMV was used to overexpress exogenous Hsp90 genes, and FG-12 delivery was used to deliver shRNAs against human Hsp90 genes, as previously described (9, 10). The shRNAs GGAAAGAGCTGCATATTAA (sense) and GCATCTATCGCATGATCAA (sense) were delivered to downregulate human Hsp90α and human Hsp90β, respectively, in cells. pRRLsinh-CMV was utilized to overexpress GFP-tagged wild type and ATP-binding mutants of the human Hsp90α gene.
Western immunoblotting analysis and quantitation.
Cellular lysates were equalized using a BCA protein assay kit (Thermo Scientific) and were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Ponceau S solution was used to stain the membrane to confirm efficient and even protein transfer. The primary antibodies against the indicated signaling proteins were as described above. Secondary anti-rabbit IgG (1:10,000) and anti-mouse IgG (1:10,000) were used as instructed by the manufacturers. The intensity of protein bands was quantitated using the NIH ImageJ software via the following procedure. Digital images of radiograph films were opened and converted to grayscale. Using a rectangular selection tool, a rectangle was drawn to cover all the protein bands incubated with the same primary antibody. “Plot lanes” was selected from the “Analyze” menu to create a profile plot of all bands. Lines were drawn between the peaks that represented darker bands. All measurements were recorded for the highlighted peaks. The peak of the control band (such as Hsp90 from MDA-MB-231) was selected as the standard, and the relative density of the other bands was calculated in reference to the control band.
Cell survival and growth assay.
Cells in exponential growth phase were used for this assay. Cells were reseeded at 8 × 104 cells/well in 12-well plates and allowed to grow for 48 h. Cells in triplicate were lifted with trypsin, and cells were counted as the day 0 starting point (without treatments). The rest of the cells (triplicate cells per condition) were incubated without or with the indicated concentrations of 17-DMAG for 48 h. The viable cells in each well were counted, and the averaged numbers from triplicates were plotted as number of cells versus increasing concentrations of 17-DMAG treatments (P < 0.05, as significant).
Statistical analysis.
All numerical results are reported as means and standard deviations (SD). The band intensity in Western blotting was quantified with ImageJ software (National Institutes of Health). Cell numbers were calculated as a percentage of the number on day 0 (100%). Statistical significance was determined using a two-tailed Student's t test and one-way analysis of variance (ANOVA). Final presentation as means and SD was based on at least three independent and corroborating experiments. Confirmation of a difference as statistically significant requires rejection of the null hypothesis of no difference between means obtained from replicate sets. A P value equal to or less than 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM067100 (to W.L.) and grant W81XWH-1810558 from the Congressionally Directed Medical Research Program (to M.C.).
We have neither financial nor nonfinancial conflict of interest. We have no commercial conflict of interest.
REFERENCES
- 1.Kim YS, Alarcon SV, Lee S, Lee M-J, Giaccone G, Neckers L, Trepel JB. 2009. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem 9:1479–1492. 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trepel J, Mollapour M, Giaccone G, Neckers L. 2010. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10:537–549. 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jhaveri K, Taldone T, Modi S, Chiosis G. 2012. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 1823:742–755. 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neckers L, Workman P. 2012. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18:64–76. 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuno A, Lee MJ, Lee S, Tomita Y, Rekhtman D, Moore B, Trepel JB. 2018. Clinical evaluation and biomarker profiling of Hsp90 inhibitors. Methods Mol Biol 1709:423–441. 10.1007/978-1-4939-7477-1_29. [DOI] [PubMed] [Google Scholar]
- 6.Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, Picard D. 2018. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones 23:467–482. 10.1007/s12192-018-0877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez J, Carter TR, Cohen MS, Blagg BS. 2020. Old and new approaches to target the Hsp90 chaperone. Curr Cancer Drug Targets 20:253–270. 10.2174/1568009619666191202101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koren J, Blagg BS. 2020. The right tool for the job: an overview of Hsp90 inhibitors. Adv Exp Med Biol 1243:135–146. 10.1007/978-3-030-40204-4_9. [DOI] [PubMed] [Google Scholar]
- 9.Zou M, Bhatia A, Dong H, Jayaprakash P, Guo J, Sahu D, Hou Y, Tsen F, Tong C, O'Brien K, Situ AJ, Schmidt T, Chen M, Ying Q, Ulmer TS, Woodley DT, Li W. 2017. Evolutionarily conserved dual lysine motif determines the non-chaperone function of secreted Hsp90alpha in tumour progression. Oncogene 36:2160–2171. 10.1038/onc.2016.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang X, Chang C, Hao M, Chen M, Woodley DT, Schönthal AH, Li W. 2021. Heat shock protein-90alpha (Hsp90α) stabilizes hypoxia-inducible factor-1α (HIF-1α) in support of spermatogenesis and tumorigenesis. Cancer Gene Ther 28:1058–1070. 10.1038/s41417-021-00316-6. [DOI] [PubMed] [Google Scholar]
- 11.Grad I, Cederroth CR, Walicki J, Grey C, Barluenga S, Winssinger N, De Massy B, Nef S, Picard D. 2010. The molecular chaperone Hsp90α is required for meiotic progression of spermatocytes beyond pachytene in the mouse. PLoS One 5:e15770. 10.1371/journal.pone.0015770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, Ichiyanagi T, Hikida M, Wang J-Y, Udono H. 2011. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci USA 108:16363–16368. 10.1073/pnas.1108372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voss AK, Thomas T, Gruss P. 2000. Mice lacking HSP90beta fail to develop a placental labyrinth. Development 127:1–1. 10.1242/dev.127.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. 2003. A high-affinity conformation of Hsp90 confers tumor selectivity on Hsp90 inhibitors. Nature 425:407–410. 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 15.Solit DB, Zheng FF, Drobnjak M, Münster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, Scher HI, Rosen N. 2002. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 8:986–993. [PubMed] [Google Scholar]
- 16.Chiosis G, Lucas B, Huezo H, Solit D, Basso A, Rosen N. 2003. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr Cancer Drug Targets 3:371–376. 10.2174/1568009033481778. [DOI] [PubMed] [Google Scholar]
- 17.Llauger L, He H, Kim J, Aguirre J, Rosen N, Peters U, Davies P, Chiosis G. 2005. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. J Med Chem 48:2892–2905. 10.1021/jm049012b. [DOI] [PubMed] [Google Scholar]
- 18.He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, Wu N, Immormino RM, Gewirth DT, Chiosis G. 2006. Identification of potent water-soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem 49:381–390. 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 19.Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G. 2004. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol 11:787–797. 10.1016/j.chembiol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Chiosis G, Vilenchik M, Kim J, Solit D. 2004. Hsp90: the vulnerable chaperone. Drug Discov Today 9:881–888. 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 21.Woodford MR, Truman AW, Dunn DM, Jensen SM, Cotran R, Bullard R, Abouelleil M, Beebe K, Wolfgeher D, Wierzbicki S, Post DE, Caza T, Tsutsumi S, Panaretou B, Kron SJ, Trepel JB, Landas S, Prodromou C, Shapiro O, Stetler-Stevenson WG, Bourboulia D, Neckers L, Bratslavsky G, Mollapour M. 2016. Mps1 mediated phosphorylation of Hsp90 confers renal cell carcinoma sensitivity and selectivity to Hsp90 inhibitors. Cell Rep 14:872–884. 10.1016/j.celrep.2015.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mollapour M, Bourboulia D, Beebe K, Woodford MR, Polier S, Hoang A, Chelluri R, Li Y, Guo A, Lee M-J, Fotooh-Abadi E, Khan S, Prince T, Miyajima N, Yoshida S, Tsutsumi S, Xu W, Panaretou B, Stetler-Stevenson WG, Bratslavsky G, Trepel JB, Prodromou C, Neckers L. 2014. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol Cell 53:317–329. 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Backe SJ, Sager RA, Woodford MR, Makedon AM, Mollapour M. 2020. Post-translational modifications of Hsp90 and translating the chaperone code. J Biol Chem 295:11099–11117. 10.1074/jbc.REV120.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zurawska A, Urbanski J, Matuliene J, Baraniak J, Klejman MP, Filipek S, Matulis D, Bieganowski P. 2010. Mutations that increase both Hsp90 ATPase activity in vitro and Hsp90 drug resistance in vivo. Biochim Biophys Acta 1803:575–583. 10.1016/j.bbamcr.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Ohkubo S, Kodama Y, Muraoka H, Hitotsumachi H, Yoshimura C, Kitade M, Hashimoto A, Ito K, Gomori A, Takahashi K, Shibata Y, Kanoh A, Yonekura K. 2015. TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol Cancer Ther 14:14–22. 10.1158/1535-7163.MCT-14-0219. [DOI] [PubMed] [Google Scholar]
- 26.Rodina A, Wang T, Yan P, Gomes ED, Dunphy MPS, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H, Caldas-Lopes E, Alpaugh M, Corben A, Riolo M, Beattie B, Pressl C, Peter RI, Xu C, Trondl R, Patel HJ, Shimizu F, Bolaender A, Yang C, Panchal P, Farooq MF, Kishinevsky S, Modi S, Lin O, Chu F, Patil S, Erdjument-Bromage H, Zanzonico P, Hudis C, Studer L, Roboz GJ, Cesarman E, Cerchietti L, Levine R, Melnick A, Larson SM, Lewis JS, Guzman ML, Chiosis G. 2016. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538:397–401. 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Premkumar DR, Arnold B, Pollack IF. 2006. Cooperative inhibitory effect of ZD1839 (Iressa) in combination with 17-AAG on glioma cell growth. Mol Carcinog 45:288–301. 10.1002/mc.20141. [DOI] [PubMed] [Google Scholar]
- 28.Lukasiewicz E, Miekus K, Kijowski J, Gozdzik J, Wilusz M, Bobis-Wozowicz S, Wiecha O, Majka M. 2009. High anti tumor activity against rhabdomyosarcoma cells and low normal cells cytotoxicity of heat shock protein 90 inhibitors, with special emphasis on 17-[2-(pyrrolidin-1-yl)ethyl]-aminno-17-demethoxygeldanamycin. J Physiol Pharmacol 12:161–166. [PubMed] [Google Scholar]
- 29.Sahu D, Zhao Z, Tsen F, Cheng C-F, Park R, Situ AJ, Dai J, Eginli A, Shams S, Chen M, Ulmer TS, Conti P, Woodley DT, Li W. 2012. A potentially common peptide target in secreted heat shock protein-90α for hypoxia-inducible factor-1α–positive tumors. Mol Biol Cell 23:602–613. 10.1091/mbc.E11-06-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S. 1989. hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol 9:3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ono K, Eguchi T, Sogawa C, Calderwood SK, Futagawa J, Kasai T, Seno M, Okamoto K, Sasaki A, Kozaki K-I. 2018. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J Cell Biochem 119:7350–7362. 10.1002/jcb.27039. [DOI] [PubMed] [Google Scholar]
- 32.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. 1994. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA 91:8324–8328. 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y, Lindquist S. 1993. Heat-shock protein hsp90 governs the activity of pp60v-src kinase. Proc Natl Acad Sci USA 90:7074–7078. 10.1073/pnas.90.15.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Y, Singer MA, Lindquist S. 1999. Maturation of the tyrosine kinase c-src as a kinase and as a substrate depends on the molecular chaperone Hsp90. Proc Natl Acad Sci USA 96:109–114. 10.1073/pnas.96.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong H, Zou M, Bhatia A, Jayaprakash P, Hofman F, Ying Q, Chen M, Woodley DT, Li W. 2016. Breast cancer MDA-MB-231 cells use secreted heat shock protein-90alpha (Hsp90α) to survive a hostile hypoxic environment. Sci Rep 6:20605. 10.1038/srep20605. [DOI] [PMC free article] [PubMed] [Google Scholar]