

SUMMARY
Skeletal muscle atrophy is a debilitating condition that occurs with aging and disease, but the underlying mechanisms are incompletely understood. Previous work determined that common transcriptional changes occur in muscle during atrophy induced by different stimuli. However, whether this holds true at the proteome level remains largely unexplored. Here, we find that, contrary to this earlier model, distinct atrophic stimuli (corticosteroids, cancer cachexia, and aging) induce largely different mRNA and protein changes during muscle atrophy in mice. Moreover, there is widespread transcriptome-proteome disconnect. Consequently, atrophy markers (atrogenes) identified in earlier microarray-based studies do not emerge from proteomics as generally induced by atrophy. Rather, we identify proteins that are distinctly modulated by different types of atrophy (herein defined as “atroproteins”) such as the myokine CCN1/Cyr61, which regulates myofiber type switching during sarcopenia. Altogether, these integrated analyses indicate that different catabolic stimuli induce muscle atrophy via largely distinct mechanisms.
Graphical Abstract
In brief
Skeletal muscle wasting is caused by many catabolic stimuli, which were thought to act via shared mechanisms. Hunt et al. now show that distinct catabolic stimuli induce muscle wasting via largely different molecular changes. The authors identify atrophy-associated proteins (“atroproteins”) that may represent diagnostic biomarkers and/or therapeutic targets.
INTRODUCTION
Skeletal muscle atrophy is a debilitating condition associated with aging and with many human diseases, with the main outcome being reduced muscle mass and strength. Several studies have demonstrated that muscle atrophy worsens disease outcome and increases mortality, whereas preserving skeletal muscle mass and function is protective (Johnston et al., 2015; Shavlakadze and Grounds, 2006; Tisdale, 2010; Zhou et al., 2010). However, the mechanisms responsible for muscle atrophy are only in part understood, and no therapies are available in the clinic.
At the cellular level, it is well established that muscle atrophy is primarily determined by atrophy of muscle cells (myofibers), whose size is determined by the balance between protein synthesis and degradation (Bonaldo and Sandri, 2013; Shavlakadze and Grounds, 2006). Previous pioneering work has found that multiple types of skeletal muscle atrophy involve a common program of gene expression. In particular, the transcriptional induction of a few ubiquitin ligases has been considered a standard response that occurs in multiple types of skeletal muscle atrophy induced by diverse stimuli, such as cancer-associated cachexia, starvation-induced atrophy, denervation, diabetes, kidney failure, and infections (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007). In particular, these studies have highlighted that myofiber atrophy primarily results from protein degradation via the autophagy-lysosome and ubiquitin-proteasome systems (Bonaldo and Sandri, 2013; Demontis et al., 2013; Piccirillo et al., 2014; Reid et al., 2014; Shavlakadze and Grounds, 2006). Specifically, transcriptional upregulation of several E3 ubiquitin ligases, including Fbxo32 (atrogin-1/MAFbx), Trim63 (MuRF1), Fbxo30 (MUSA), and Fbxo21 (SMART), occurs in atrophy and is responsible for poly-ubiquitin tagging and proteasomal degradation of target proteins during muscle atrophy (Bonaldo and Sandri, 2013; Demontis et al., 2013; Piccirillo et al., 2014; Reid et al., 2014; Shavlakadze and Grounds, 2006). The definition of a common transcriptional program that is activated in many types of muscle atrophy has greatly impacted this field of research and has led to the hypothesis that anti–atrophy interventions may find general application to treat muscle atrophy induced by diverse stimuli.
However, these meritorious early studies were based on gene expression analysis via microarrays (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007), which have technical limitations compared to current RNA sequencing (RNA-seq) technologies, such a decreased coverage and a lower quantitative range of detection of gene expression changes (Rao et al., 2019; Wang et al., 2014a). In recent years, application of RNA-seq to the analysis of muscle atrophy has indeed provided novel insight into the transcriptional changes associated with atrophy and muscle homeostasis (Chapman et al., 2020; Kunkel et al., 2011; Llano-Diez et al., 2019; Mahmassani et al., 2019; Terry et al., 2018).
In addition to gene expression changes, muscle atrophy is characterized by profound remodeling of the proteome due to changes in protein synthesis and degradation (Bonaldo and Sandri, 2013; Demontis et al., 2013; Piccirillo et al., 2014; Reid et al., 2014). However, the application of proteomics to study muscle atrophy is an approach that has been employed by only a few studies (Fuller et al., 2016; Lang et al., 2017; Lang et al., 2018; Mugahid et al., 2019; Shum et al., 2018; Ubaida-Mohien et al., 2019), some of which have limited proteome coverage due to technical constrains and/or focus on a single type of muscle atrophy (Ibebunjo et al., 2013). Therefore, although these studies have provided new insight into the proteomic changes associated with certain types of muscle atrophy, an integrated cross-comparison of different types of muscle atrophy is missing.
Here, we have used RNA-seq and quantitative mass spectrometry to determine the molecular changes that occur in mouse skeletal muscle during atrophy induced by dexamethasone, cancer cachexia, and aging. This deep multi-omics approach detected >15,000 unique mRNAs for each mode (with an overlap of for ~13,000 mRNAs) and ~5,000 unique proteins each mode (with approximately ~3,000 overlapping).
Contrary to the model that posits that a common molecular signature underlies atrophy induced by different stimuli, we find a remarkable diversity in the mRNA and protein changes induced by distinct atrophic stimuli, in particular at the proteome level, and disconnect between transcriptional and proteomic changes. Moreover, most of the common markers initially identified in microarrays studies and commonly referred to as biomarkers of atrophy (“atrogenes”; Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007) do not emerge from these proteomic analyses as the most relevantly associated with all modes of atrophy. On this basis, these integrated analyses indicate that different catabolic stimuli can induce muscle atrophy via a diversity of protein changes. Altogether, these integrated multi-omics provide a framework for understanding the specificity of muscle atrophy induced by distinct stimuli and provide potential leads that could be targeted to prevent muscle atrophy. As proof of concept, we demonstrate that Cyr61, which emerged from this analysis as an atroprotein associated with aging, regulates myofiber type switching that occurs alongside muscle atrophy with aging. We propose that these integrated analyses and datasets are a resource that may help develop therapies tailored for specific types of muscle atrophy.
RESULTS
Analysis of skeletal muscle atrophy induced by dexamethasone, cancer cachexia, and aging
Dexamethasone is a glucocorticoid analog that is widely prescribed for its anti-inflammatory functions and, similar to endogenous glucocorticoids, induces skeletal muscle atrophy (Braun and Marks, 2015; Hasselgren et al., 2010; Schakman et al., 2013). As expected based on previous studies (Hong et al., 2019; Jesinkey et al., 2014; Li et al., 2017), dexamethasone led to body mass loss and skeletal muscle atrophy, as indicated by the reduction in the weight of the tibialis anterior (TA) muscle (Figure 1A). At the cellular level, dexamethasone-induced muscle atrophy was characterized by a decrease in the size of type 2B myofibers, whereas the size of type 2A and 2X myofibers was only marginally affected, consistent with previous reports (Hong et al., 2019; Jesinkey et al., 2014; Li et al., 2017). No changes in the number of myofibers present in the TA muscle and in their relative proportion was seen in response to treatment with dexamethasone.
Figure 1. Dexamethasone, LLC cancer cachexia, and aging all induce myofiber atrophy.
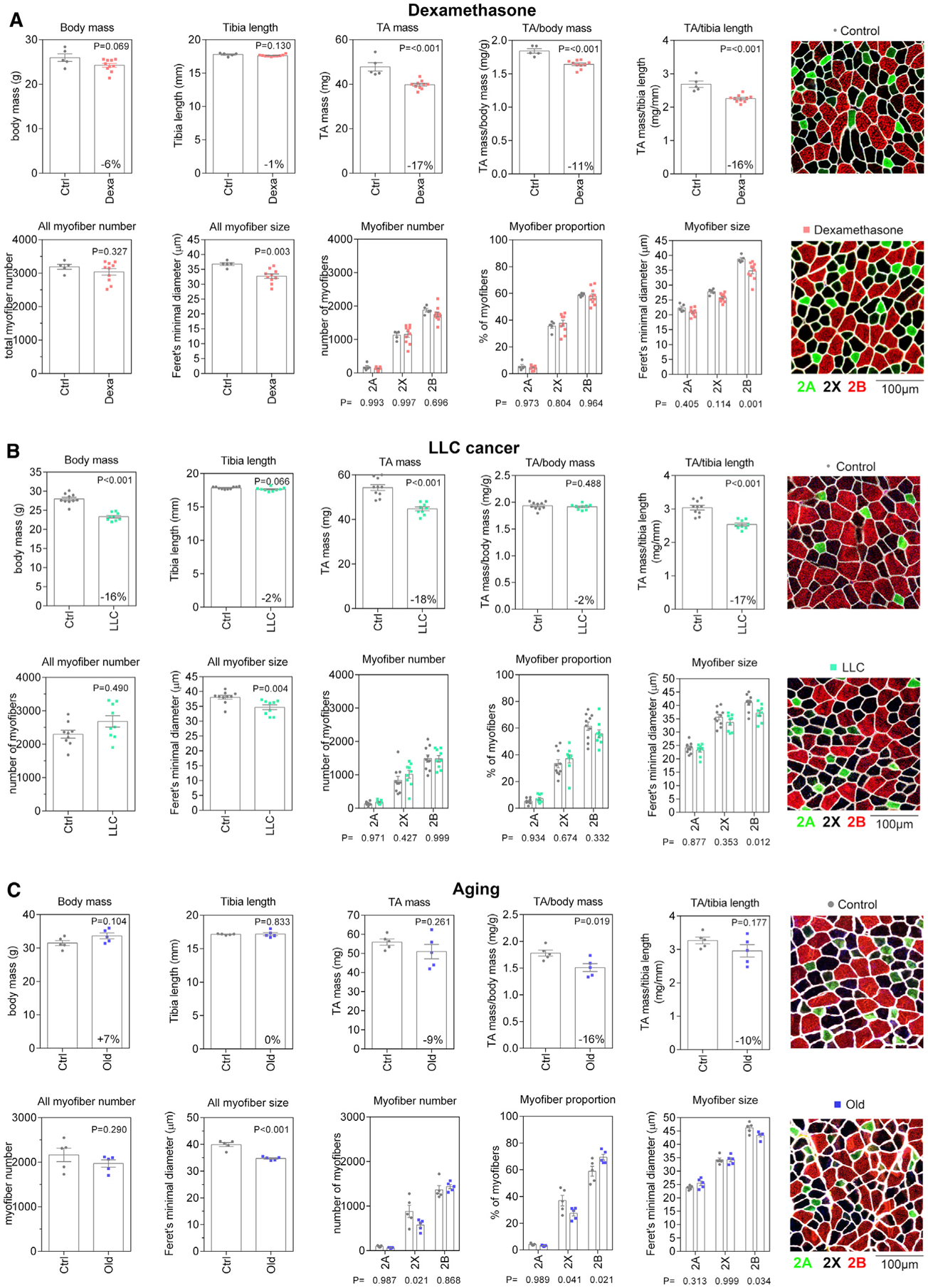
(A) Dexamethasone-treated mice display a trend toward decreased body weight with decreased mass of the tibialis anterior (TA) muscle. Dexamethasone induces a significant decrease in the mean myofiber size, which is significant for type 2B myofibers.
(B) Also, tumor-bearing mice display a significant decrease in the size of type 2B myofibers.
(C) The total number of type 2X myofibers significantly decreases and is indicative of a shift in proportion from type 2X toward type 2B myofibers during aging. Mean myofiber size is significantly reduced and, as observed for dexamethasone treatment and cancer cachexia, the size of type 2B myofibers is the most affected. Representative images of TA muscle cross-sections are shown (right) with myofiber types determined by immunostaining for myosin heavy chain (MHC) isoforms, i.e., for type 2A (green) and 2B (red) MHC; type 2X (black) myofibers are myofibers unstained for type 2A and type 2B isoforms. Myofiber boundaries are shown in white based on laminin immunostaining.
Changes similar to those found in response to dexamethasone were also observed in response to body wasting (cachexia) induced by cancer cells (Figure 1B), as previously observed (Hunt et al., 2019b; Puppa et al., 2014). Specifically, atrophy of the TA muscle induced by Lewis lung carcinoma (LLC) cancer cells was primarily due to atrophy of type 2B myofibers, whereas the number of myofibers and the relative proportion of myofiber types did not change.
Lastly, we compared TA muscles from 6- versus 24-month-old mice (Figure 1C). There was an age-related increase in body mass, presumably due to age-associated obesity (Houtkooper et al., 2011), with a considerably smaller reduction in TA mass compared to other modes of atrophy (9% decrease compared to LLC 18% and Dexa 17%). However, age-induced decrease in TA muscle mass is significantly higher (~16%) when TA mass is normalized to body mass. Age-induced muscle atrophy (sarcopenia) was also characterized by a decline in the size of type 2B myofibers in the TA, as observed for atrophy induced by dexamethasone and LLC cancers. However, different from atrophy induced by these stimuli, aging resulted in a decline in the number of type 2X myofibers, presumably due to a differentiation shift from type 2X to type 2B myofibers, as previously observed (Crupi et al., 2018). Altogether, these histological analyses reveal overall similar changes in response to different atrophic stimuli: atrophy impacts predominantly type 2B myofibers, whereas myofiber type shifts occur only with aging and lead to an increased proportion of type 2B myofibers in aged mouse TA muscle, similar to what previously reported (Giacomello et al., 2020; Sakellariou et al., 2016).
Gene expression analyses reveal largely distinct transcriptional responses of skeletal muscle to corticosteroids, cancer cachexia, and aging
We next used RNA-seq of TA muscles to determine the transcriptional changes associated with atrophy induced by dexamethasone, LLC cancer cells, and aging. For each of these RNA-seq datasets, >15,000 mRNAs/condition were quantified. Dexamethasone and aging induced an overall increase in gene expression, whereas there was a similar number of downregulated and upregulated genes in response to LLC cancer cells (Figures 2A–2C; Table S1).
Figure 2. RNA sequencing (RNA-seq) of skeletal muscles undergoing atrophy induced by dexamethasone, LLC cancer cachexia, and aging reveals stimulus-specific transcriptional signatures of atrophy.
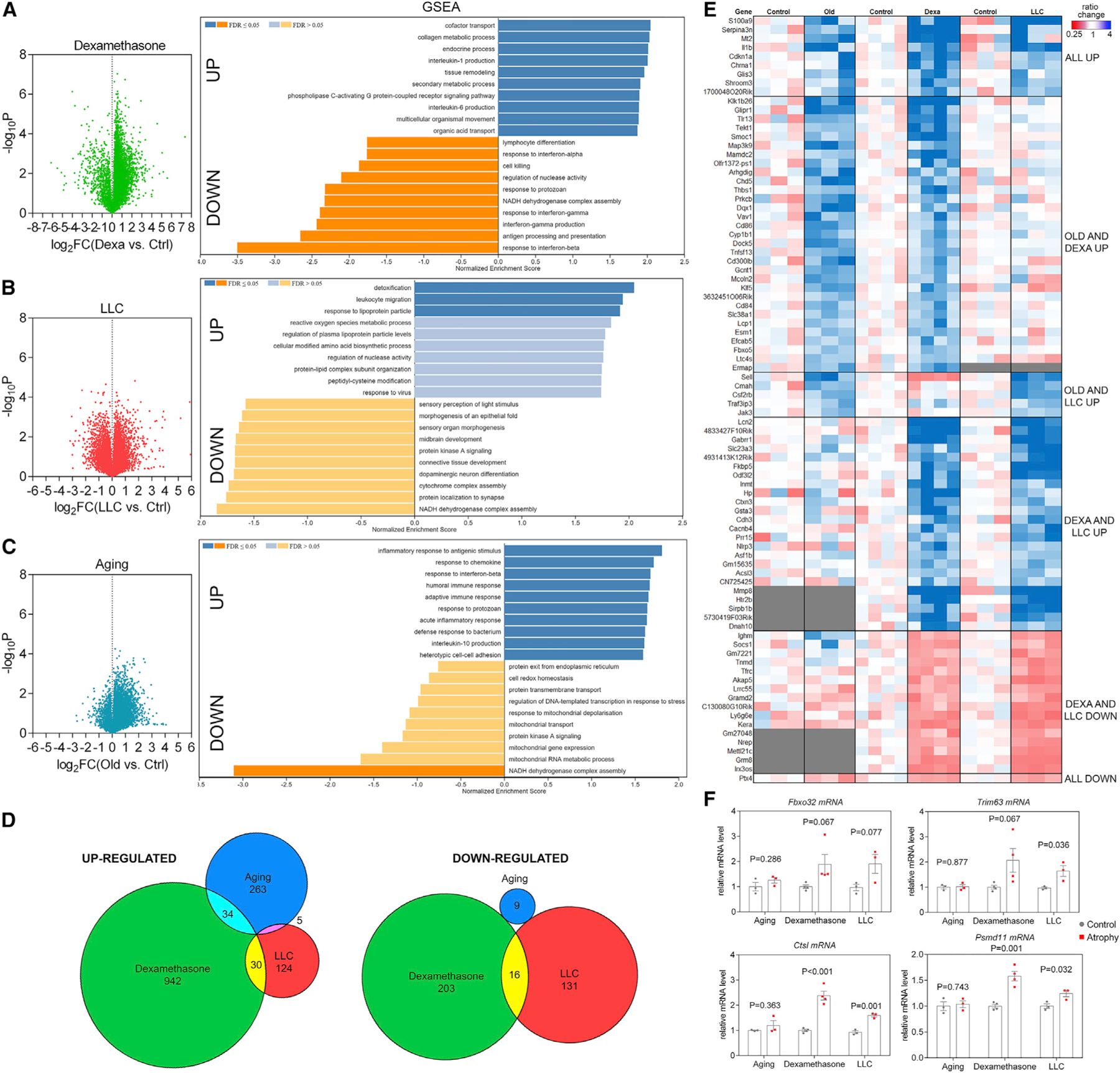
(A–C) Volcano plots of RNA-seq data and GSEA from TA muscles from (A) dexamethasone, (B) LLC cancer cachexia, and (C) aging.
(D) Comparison of significantly regulated mRNAs (p < 0.05 and log2FC > 1 for upregulated and log2FC < −1 for downregulated mRNAs) in distinct modes of myofiber atrophy.
(E) Heatmap of the genes that met the criteria above and shared overlap with at least 2 modes of atrophy. Genes up- and downregulated in all modes of atrophy (denoted as “all up” or “all down”) met changes in magnitude criteria but not p value criteria for all 3 modes.
(F) Atrogenes previously described as mediators of atrophy are not regulated in all modes of atrophy.
Gene set enrichment analysis (GSEA) revealed the upregulation of many gene categories involved in immunity and inflammation, such as “interleukin 1 (IL1) and IL6 production” (dexamethasone), “leukocyte migration” and “response to virus” (LLC cancer), and “acute inflammatory response” and “humoral and adaptive immune response” (aging). In addition to an overall involvement of inflammation in muscle atrophy induced by distinct stimuli, there were gene categories that were upregulated in a stimulus-specific manner. For example, “heterotypic cell-cell adhesion” was upregulated with aging, “collagen metabolic process” with dexamethasone, and “response to lipoprotein particle” with LLC cancer (Figures 2A–2C and 2E; Table S2), which may sustain the excessive fatty oxidation that is known to occur in muscle atrophy associated with cancer cachexia (Fukawa et al., 2016).
There were also several gene categories that were downregulated during skeletal muscle atrophy. “NADH dehydrogenase complex assembly” was commonly downregulated in response to dexamethasone, LLC cancer, and aging, consistent with a decline in mitochondrial function with muscle atrophy (Ibebunjo et al., 2013; Liu et al., 2016; Powers et al., 2012; Romanello and Sandri, 2016. In addition, “cytochrome complex assembly” was downregulated in response to LLC cancer cells, whereas “mitochondrial transport,” “mitochondrial gene expression,” and “mitochondrial RNA metabolic process” were downregulated in aging. Moreover, “protein kinase A signaling” was downregulated with LLC cancer and aging but not by dexamethasone (Figures 2A–2C and 2E).
We also cross-compared the significantly regulated genes by dexamethasone, LLC cancer, and aging and found that there is very limited overlap in the gene expression induced. Specifically, 263 genes were uniquely induced by aging, 942 by dexamethasone, and 124 by LLC cancers. Only 34 genes were commonly induced by aging and dexamethasone, 30 by LLC cancer and dexamethasone, and only 5 were shared between aging and LLC cancer. Analysis of downregulated genes led to similar conclusions, i.e., most gene expression changes were uniquely induced in response to aging (9), dexamethasone (203), and LLC cancer (131), whereas only 16 were commonly downregulated by dexamethasone and LLC cancer (Figure 2D). Altogether, gene expression analyses indicate that largely distinct transcriptional changes characterize muscle atrophy induced by dexamethasone, LLC cancer cells, and aging (Figures 2E, S1A, and S1C).
Despite overall differences in transcriptional responses, there was a small number of genes, including S100a9 and Il1b, which are associated with inflammatory cells, and Serpina3n, the expression of which has been suggested to correlate with and predict muscle atrophy (Gueugneau et al., 2018; Hulmi et al., 2020), that trended toward upregulation for all modes of atrophy. This suggests that, despite overall differences, few atrogenes are consistently modulated by distinct types of atrophy. The expression of previously described atrogenes such as Fbxo32 (atrogin-1/MAFbx), Trim63 (MuRF1), Ctsl, and Psmd11 was increased in response to dexamethasone and LLC cancer-induced cachexia, as expected based on previous studies (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007), but the expression of these genes was not induced by aging (Figure 2F). Therefore, atrogenes previously described as upregulated and mediating acute modes of muscle atrophy may not be as relevant for the progressive muscle atrophy that occurs with aging.
Proteomic analyses reveal that different atrophic inducers shape the proteome in distinct manners
We next used the tandem mass tag (TMT) mass spectrometry method to define protein changes associated with muscle atrophy. For each of these TMT datasets, ~5,000 proteins/condition were quantified. Dexamethasone and LLC cancer led to an overall similar number of downregulated and upregulated proteins, whereas aging showed a directional bias toward protein upregulation. Similar to the transcriptional changes (Figure 2), the proteomic changes induced by distinct atrophic stimuli were largely divergent (Figures 3, S1B, and S1D; Table S3).
Figure 3. TMT-mass spectrometry (MS) of skeletal muscles undergoing atrophy induced by dexamethasone, LLC cancer cachexia, and aging reveals distinct proteomic signatures of stimulus-specific atrophy.
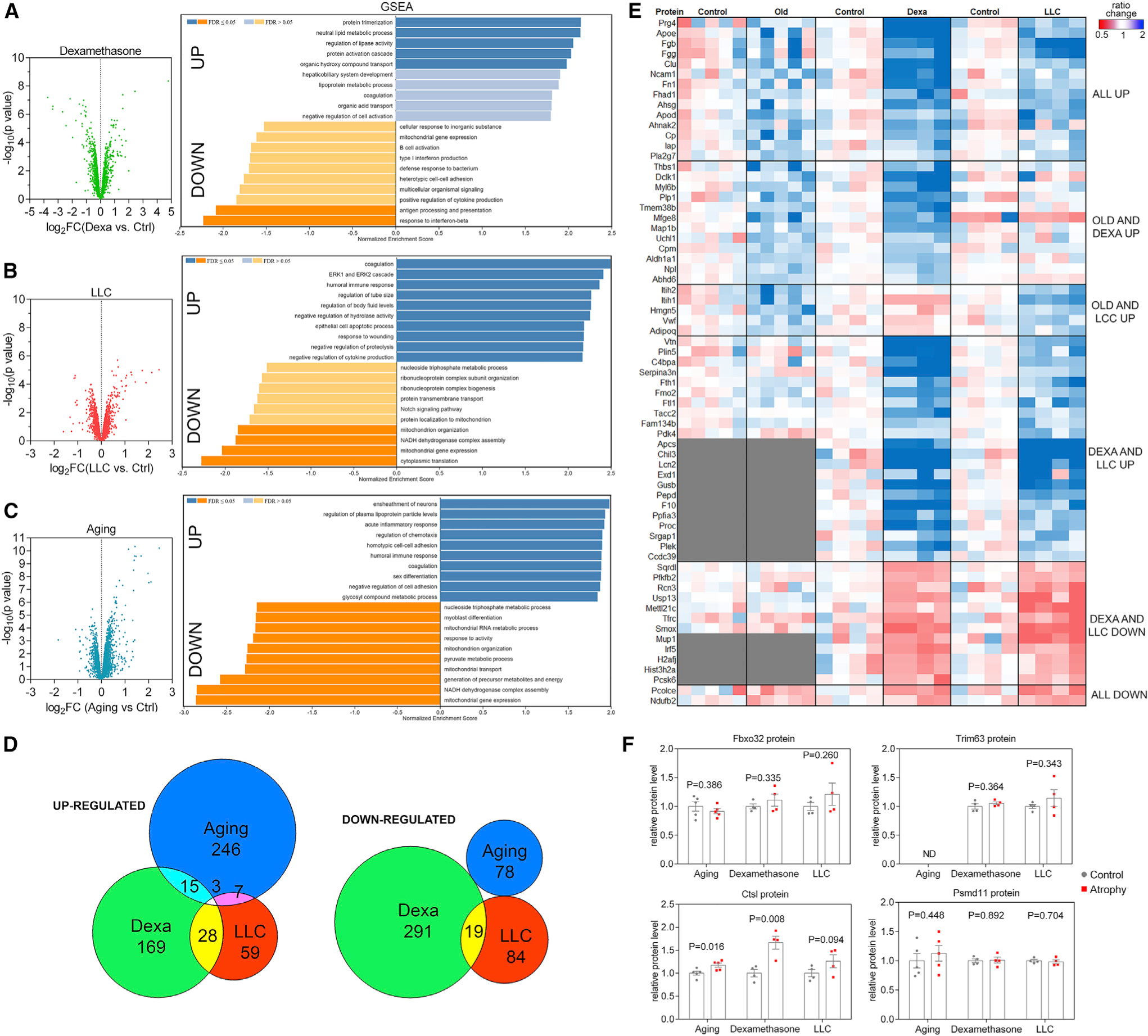
(A–C) Volcano plots of TMT data and GSEA from TA muscles from (A) dexamethasone, (B) LLC cancer cachexia, and (C) aging.
(D) Comparison of significantly regulated proteins (p < 0.05 and log2FC > 0.3 for upregulated and log2FC < −0.3 for downregulated proteins) in distinct modes of myofiber atrophy. Relatively few of these up- and downregulated proteins showed overlap between dexamethasone, LLC cancer cachexia, and aging.
(E) Heatmap of the proteins which met the criteria above and shared overlap with at least 2 modes of atrophy.
(F) Atrogenes previously described as mediators of atrophy are not consistently regulated at the protein level.
GSEA of protein changes revealed some categories that were detected also from the transcript analyses whereas other protein categories were not. For example, “neutral lipid metabolic process,” “regulation of lipase activity,” and “lipoprotein metabolic process” were upregulated protein categories in response to dexamethasone whereas they did not emerge from the analysis of the corresponding RNA-seq data obtained from the same muscle samples. Downregulated protein categories included “antigen processing and presentation” (dexamethasone), “myoblast differentiation” (aging), and “mitochondrial gene expression” (all 3 atrophic stimuli) (Figures 3A–3C; Table S2), further indicating that the dysregulation of mitochondrial proteins might be common to all modes of muscle atrophy and could indicate decline in mitochondrial function with atrophy.
We also cross-compared the proteins significantly regulated by dexamethasone, LLC cancer, and aging and found that, similar to transcriptional changes, there is very limited overlap in the protein changes induced by different atrophic stimuli. Specifically, 246 proteins were uniquely induced by aging, 169 by dexamethasone, and 59 by LLC cancers. Only 15 proteins were commonly induced by aging and dexamethasone, 28 by LLC cancer and dexamethasone, 7 by aging and LLC cancer, and only 3 proteins were shared between all 3 atrophic stimuli. Likewise, downregulated proteins were uniquely modulated by aging (78), dexamethasone (291), and LLC cancer cells (84), whereas only 19 were commonly downregulated by dexamethasone and LLC (Figure 3D). Altogether, the proteome is reshaped in a largely distinct manner by dexamethasone, LLC cancer cells, and aging (Figure 3E).
Once again, despite overall differences between proteomic changes induced by different modes of atrophy, there were few commonly regulated proteins that included Prg4, Fgb, Fgg, Apod, and Apoe, which are all extracellular secreted proteins. Interestingly, although the mRNA levels of the atrogenes Fbxo32 (atrogin-1/MAFbx), Trim63 (MuRF1), and Psmd11 increase in response to dexamethasone and LLC-cancer-induced cachexia (Figure 2F), their protein levels did not significantly change (Figure 3F), suggesting that these proteins are not robust markers of atrophy as originally assumed based on their transcriptional modulation (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007). However, the protein levels of the lysosomal protease Cathepsin L (Ctsl) increased in all modes of atrophy, indicating that at least some of the originally identified atrogenes are regulated at the protein level and may be responsible for muscle protein degradation associated with atrophy.
Another overarching conclusion from these genomic and proteomic surveys is that atrophic muscles are characterized by an inflammatory state. For example, there was an increase in the expression of several mRNAs and proteins, which are normally considered distinctive markers of neutrophils. Specifically, levels of S100a8 and S100a9 are increased consistently by dexamethasone, cancer cachexia, and aging (Figure S2A). There was also upregulation by dexamethasone and LLC of Lcn2 (a lipocalin secreted in high amount by neutrophils), Il1b (an interleukin largely produced due to inflammasome-mediated signaling), and genes associated with the complement cascade. Because these genes are markers of inflammatory cells (Wang et al., 2018), their increased levels in muscles undergoing atrophy may be indicative of increased recruitment of inflammatory cells to atrophic muscle. However, immunostaining of muscle cryosections with antibodies for neutrophil markers Ly6g and macrophage marker F4/80 did not reveal any significant recruitment of inflammatory cells to atrophic muscles compared to controls, especially in comparison to the extreme inflammatory cell recruitment that occurs with cardiotoxin-induced muscle injury (Figures S2B and S2C). Together, these findings suggest that increased levels of neutrophil and inflammatory markers arise from heightened expression independent of the total number of inflammatory cells present.
Although we have examined the transcriptional and proteomic changes associated with atrophy induced by aging, cancer, and glucocorticoids, there are many other catabolic stimuli that can induce muscle atrophy. Importantly, denervation is a common cause of muscle atrophy in neuromuscular disorders (Bonaldo and Sandri, 2013; Demontis et al., 2013; Piccirillo et al., 2014; Reid et al., 2014; Shavlakadze and Grounds, 2006). Microarrays studies have previously suggested that denervation-induced atrophy occurs via the induction of the same atrogenes triggered by other atrophic stimuli (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007), but this model has not been tested with proteomics. On this basis, we have compared our proteomic surveys of muscle atrophy induced by dexamethasone, cancer, and aging with that of denervation-induced muscle atrophy, for which proteomic data are publicly available (Lang et al., 2017). Similar to our analyses that have demonstrated remarkable divergence in the proteomic changes induced by aging, dexamethasone, and cancer cachexia, we find that denervation induces muscle atrophy via protein changes largely different from those induced by other atrophic stimuli (Figure S3). Interestingly, aging and denervation (particularly at the later stages examined: day 14), although somewhat weakly correlated, appear to share the greatest correlation (r2 = 0.120) among all comparisons, suggesting that denervation may play a role in age-associated muscle atrophy, consistent with previous reports (Chai et al., 2011). Thus, our proteomic dataset can be used both as a resource to identify potential mediators of muscle atrophy and for cross-comparison of different atrophic states.
Transcriptome-proteome disconnect occurs in muscle atrophy induced by diverse atrophic stimuli
We have previously found a disconnect of the skeletal muscle transcriptome and proteome during aging in Drosophila melanogaster (Hunt et al., 2019a), in line with other studies that similarly have found an unanticipated limited correspondence between mRNA and protein levels in other organisms (Ghazalpour et al., 2011; Gygi et al., 1999; Yeung, 2011).
On this basis, we have examined the correlation between the transcriptome and proteome and have found a relatively low similarity between mRNA and protein changes in skeletal muscles undergoing atrophy in response to dexamethasone, cancer, and aging (Figure 4A; r2 values of 0.164, 0.082, and 0.202, respectively). As expected, there was an overall tendency for modulation of mRNA levels leading to changes in the abundance of the corresponding proteins (Figure 4B). That is, when mRNA levels are increased (log2FC > 0.5), the average corresponding protein levels are typically increased, and when mRNA levels are decreased (log2FC < −0.5), the corresponding proteins are on average decreased. However, a striking disconnect was found for mRNAs that were unchanged (−0.5 > log2FC < 0.5). Strikingly, there was an overall increase (aging) and decrease (dexamethasone and cancer) in the levels of the proteins corresponding to these mRNAs that were unaffected by atrophic stimuli (Figure 4B). GSEAs revealed several categories that were overrepresented in dexamethasone-regulated proteins without mRNA changes (Figure 4C), cancer-regulated proteins without mRNA changes (Figure 4D), and age-regulated proteins without mRNA changes (Figure 4E; Table S2). Interestingly, several mitochondrial proteins were downregulated without a corresponding large magnitude decline in their expression in response to dexamethasone, cancer, and aging (“mitochondrion organization,” “mitochondrial gene expression,” and “NADH dehydrogenase complex assembly”). Other categories of proteins that were upregulated via mechanisms independent from transcription were “regulation of plasma lipoprotein particle levels” (cancer and aging) and “response to endoplasmic reticulum stress” (aging), which is consistent with the activation of endoplasmic reticulum (ER) stress during aging in skeletal muscle (Demontis et al., 2013; Iwawaki et al., 2004).
Figure 4. Disconnect of mRNA and protein changes during muscle atrophy.
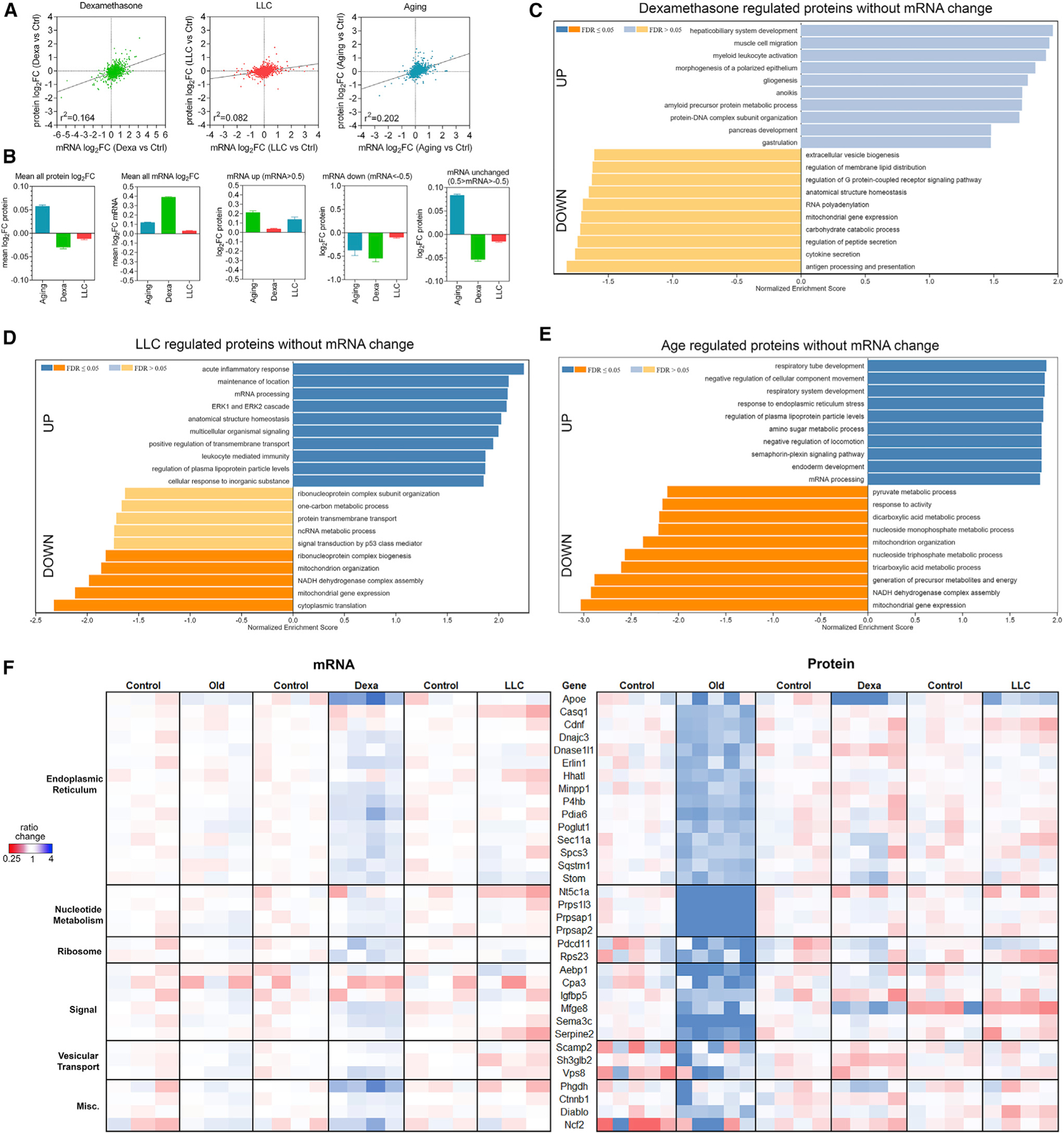
(A) Overall low correlation between mRNA and protein changes occurs with dexamethasone, LLC cancer, and aging.
(B) The average change for all proteins is skewed toward upregulation for aging but downregulation to neutral for dexamethasone and LLC. In contrast, the average of all mRNA changes is skewed up.
(C and D) GSEA for proteins not regulated at the mRNA level shows that for (C) dexamethasone and (D) LLC, many of the same protein categories are enriched and de-enriched compared to the whole proteome.
(E) Aging shows enrichment for unique groups of proteins, including those related to endoplasmic reticulum (ER) stress.
(F) Heatmap of proteins upregulated post-transcriptionally with age and categorized based on their function.
The post-transcriptional regulation of proteins involved in ER stress suggests that protein quality control diminishes with aging and that this response is not substantially modulated by dexamethasone and LLC. Protein levels of proteostasis regulators (Casq1, Dnajc3/Hsp40, Pdia6, and Sqstm1) are examples of a large group of proteins associated with ER function that were significantly induced with aging but not in response to dexamethasone and cancer (Figure S4), suggesting that they are part of an adaptive response to declining protein quality control in the aging muscle (Demontis et al., 2013; Jiao and Demontis, 2017). Furthermore, SQSTM1/p62 protein levels were found to increase in detergent insoluble protein fractions during aging together with poly-ubiquitinated proteins, suggesting that there is an accumulation during aging of ubiquitinated proteins that are likely misfolded and aggregation-prone (Figures S5A and S5B). Interestingly, there were no significant changes in the caspase-like, chymotrypsin-like, and trypsin-like proteolytic activities of the proteasome in TA muscles from 24- versus 6-month-old mice (Figure S5C), suggesting that decline in the proteasome proteolytic capacity may not be the original cause of protein quality control decline with aging (Jiao and Demontis, 2017). Altogether, these analyses demonstrate a disconnect between the transcriptome and the proteome during muscle atrophy induced by diverse atrophic stimuli and highlight protein quality control as uniquely impacted by aging (Figures 4C–4F).
Transcriptomic and proteomic surveys from whole muscles primarily reflect changes that occur in myofibers rather than in other muscle-associated cell types
Although myofibers constitute the bulk of skeletal muscle, this also includes nerves and muscle progenitor cells (satellite cells) as well as infiltrating non-muscle cells, such as immune cells, endothelial cells, and fibro-adipogenic progenitors (FAPs) (Mukund and Subramaniam, 2020). On this basis, we examined the relative contribution of these cells to the overall transcriptome and proteome data that we have obtained from muscles undergoing atrophy in response to distinct stimuli (Figures 2 and 3; Tables S1 and S2).
Although we found an enrichment of immune markers in atrophy (Figures 2 and 3), immunostaining suggests that this is not due to any enrichment in the number of immune cells infiltrating the muscle (Figure S2). Rather, these changes may indicate altered activity of immune cells and/or transcripts/proteins derived from other cell sources within the muscle, such as myofibers.
Moreover, the abundance of markers for other cell types is considerably lower than those for myofibers (Table S5). For example, the satellite cell marker Pax7 (Relaix et al., 2005) shows TPM values between 0.3 and 2 in RNA-seq from the whole muscles, whereas sarcomeric proteins from differentiated myofibers, such as MYH1 and MYH4 (representing MYHC2A and 2B, respectively, the most abundant myosin types in the TA; Schiaffino and Reggiani, 2011), display TPMs in the thousands (Table S4). Likewise, TMT did not detect Pax7 in muscle samples, whereas spectral counts for myosin heavy chains are in the millions (Table S4). Similarly, abundance of mRNA and protein markers of FAPs such as Pdgfra (Heredia et al., 2013) is low and does not change with atrophy (Table S4).
Overall, the abundance of mRNA and protein markers for cell types other than myofibers appears to be low in our muscle datasets. Moreover, these markers do not substantially change with atrophy (Table S4). However, there are a few exceptions. Specifically, the mRNA levels of the muscle progenitor cell markers Myod1, Myog, and Myf6 (Bentzinger et al., 2012) increase in muscles in response to dexamethasone (Table S4).
Collectively, these analyses indicate that the vast majority of mRNAs and proteins detected from whole muscles primarily reflect changes that occur in myofibers rather than in other cell types associated with the muscle.
Integrated analyses reveal atrophy-regulated proteins (“atroproteins”) that are biomarkers and that may regulate skeletal muscle atrophy
We have found an unexpected uncoupling between the transcriptome and the proteome and that such disconnect is a pervasive feature of muscle atrophy irrespective of the underlying atrophic stimulus (Figure 4). Moreover, our integrated analyses have revealed that many atrophy-induced genes previously identified via microarray analyses, such as Fbxo32 (atrogin-1/MAFbx), Trim63 (MuRF1), and Psmd11, are not modulated at the protein level during atrophy induced by diverse atrophic stimuli and that other biomarkers may provide more robust information on the level of atrophy of skeletal muscle (Figures 2 and 3).
Here, we have examined in more detail the protein changes associated with distinct atrophic stimuli, herein defined as “atroproteins,” and propose that they might be used as type-specific markers of muscle atrophy (Figures 5A and 5B). Of such atroproteins, only a few appear to be significantly regulated in a consistent manner by all atrophic stimuli. Specifically, protein levels of Apod, Clu, Fhad1, and Pla2g7 increase in response to aging, dexamethasone, and cancer, whereas protein levels of the mitochondrial complex I component Ndufb2 are reduced (Figure 5A).
Figure 5. Atroproteins, i.e., robust protein biomarkers of general and type-specific skeletal muscle atrophy.
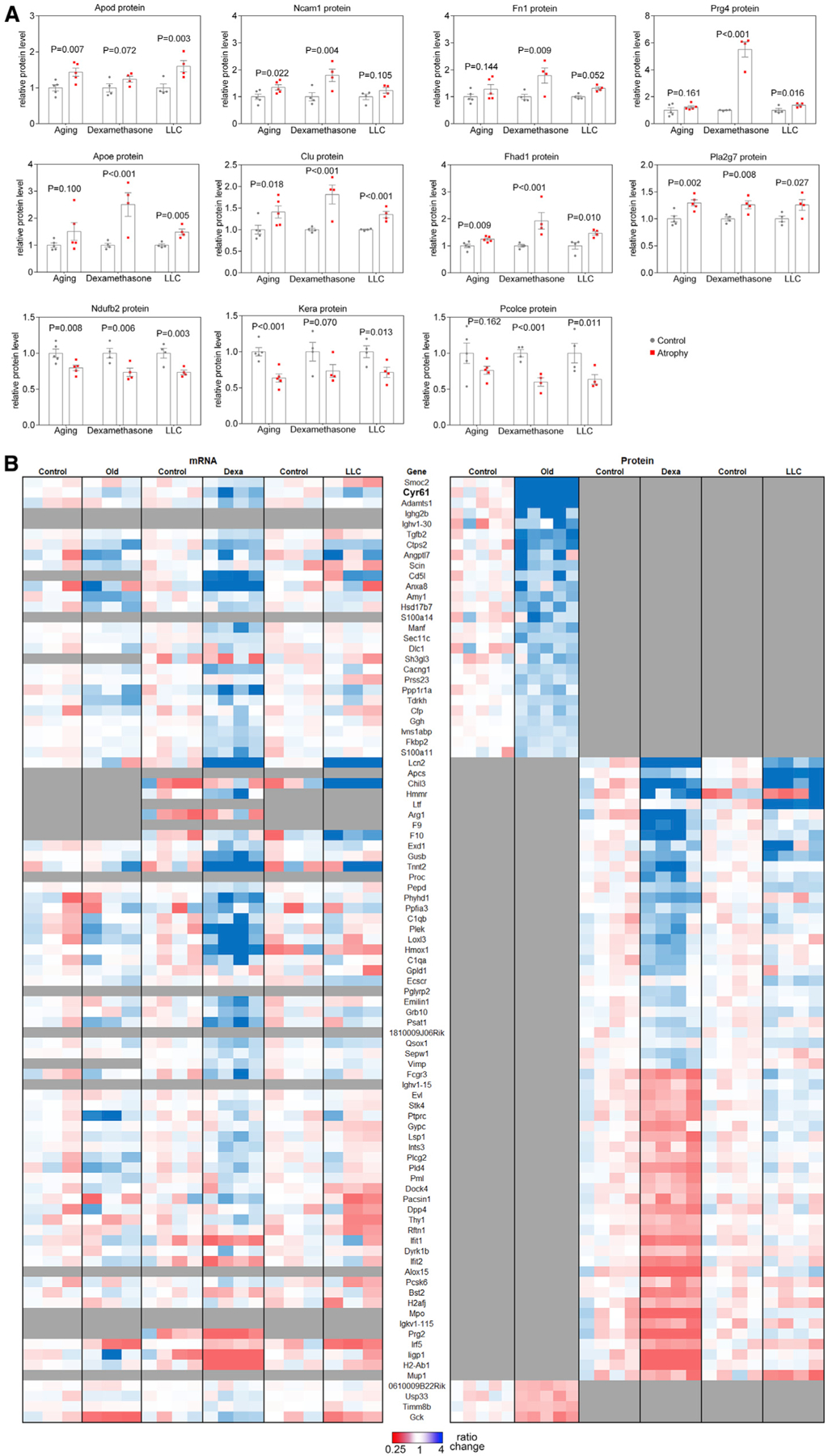
(A) Proteins that are detected in all datasets and most consistently regulated by all three modes of atrophy.
(B) Heatmap of atroproteins significantly regulated by at least one mode of atrophy but not detected in all datasets.
In order to ensure that potential atroproteins were not excluded on the basis of non-detection, we also considered proteins that were upregulated in at least one mode of atrophy but for which data were missing from one of the other modes of wasting (Figure 5B). Comparison of their transcriptional data indicates that upregulation of Cyr61 mRNA can be detected in dexamethasone, cachexia, and aging, but increased Cyr61 protein levels is found only with aging (Cyr61 protein was not detected in proteomic analyses with dexamethasone and cancer cachexia). Therefore, Cyr61 may be an atroprotein that is upregulated also in other modes of atrophy. Remarkably, we find that many proteins that change in response to atrophy consist of secreted proteins, including extracellular matrix proteins (Smoc2, Cyr61, Prg4, Fn1), secreted proteins with metabolic functions (Apod and Apoe), an extracellular chaperone (Clu), and signaling factors such as Tgfb2, whose protein levels increase with aging and may contribute to age-associated myofiber atrophy (Figures 5A and 5B).
These proteins may associate with and/or regulate muscle mass and homeostasis also in physiologic and pathologic contexts different from those examined here. For example, Pla2g7 mutations have been associated with a greater increase in lean muscle mass following training (Wootton et al., 2007). Clu expression has also been linked with declining muscle mass in osteoporosis and found to negatively regulate muscle cell viability (Pucci et al., 2019). Altogether, these integrated analyses provide robust protein biomarkers associated with atrophy (atroproteins), some of which could causally determine atrophy-associated changes of skeletal muscle in response to dexamethasone, cancer cachexia, and aging. The identification of these atroproteins will allow further studies to determine whether they are solely biomarkers of muscle atrophy or whether they also causally contribute to muscle wasting and homeostasis.
The myokine CCN1/Cyr61 is upregulated by aging and regulates myofiber switching during sarcopenia
Because the myokine CCN1/Cyr61 is among the most age-upregulated proteins (Figure 5), we next wanted to test if Cyr61 regulates any sarcopenia-associated phenotype. CCN1/Cyr61 (cysteine-rich angiogenic protein 61) is a matricellular protein that has been shown to regulate a variety of processes in different target tissues, such as cell survival, proliferation, differentiation, migration, and adhesion (Chaqour and Goppelt-Struebe, 2006; Malik et al., 2015). In skeletal muscle, Cyr61 has been shown to induce senescence and to decrease proliferation of muscle progenitor cells during aging (Du et al., 2014) and to be required for myoblast migration, differentiation, and myofiber formation during muscle regeneration (O’Connor et al., 2007). Interestingly, in addition to being a target of Wnt and Hippo/YAP signaling (Du et al., 2014; Malik et al., 2015), Cyr61 is an exercise-induced myokine, as shown by its rapid induction by muscle contraction and mechanical tension (Chaqour and Goppelt-Struebe, 2006; Kivela et al., 2007).
Via genomic surveys (Figures 2, 3, 4, 5, and 6), we have found that Cyr61 mRNA levels significantly increase in skeletal muscle in response to aging, dexamethasone, and cancer-induced cachexia (Figure 6A). Moreover, an age-related increase is also detected for Cyr61 protein in muscle (Figure 6B). Altogether, Cyr61 appears to be a robust biomarker of muscle atrophy. On this basis, we next tested whether, in addition to being a biomarker, Cyr61 functionally regulates any aspects of sarcopenia. For these experiments, we electroporated Cyr61 siRNAs into the TA muscles of 30-month-old mice and, after 7 days, compared its effects to control non-targeting (NT) siRNAs electroporated into the contralateral leg. As expected, Cyr61 siRNAs led to an ~70% knock down in Cyr61 mRNA and protein levels (Figure 6C).
Figure 6. Cyr61 regulates myofiber type shifting during aging.
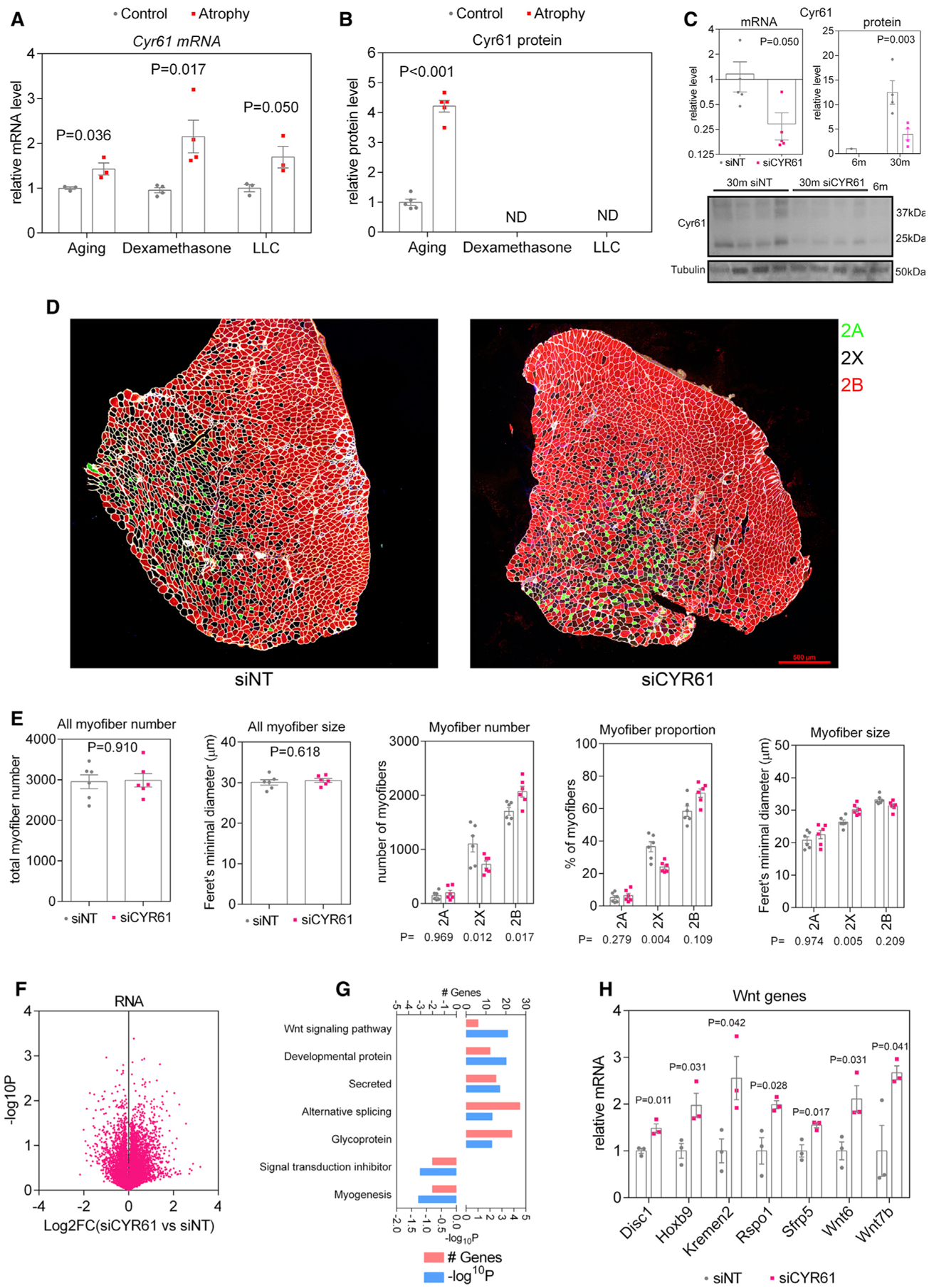
(A) Cyr61 expression increases in all modes of atrophy.
(B) By TMT-MS, Cyr61 protein was significantly upregulated with aging (but not detected with LLC and dexamethasone).
(C) Cyr61 mRNA and protein levels were significantly reduced in TA muscles of 30-month-old mice after electroporation of siRNAs targeting Cyr61 compared to control non-targeting (NT) siRNAs.
(D) Representative images of cross-sections from control (siNT) and Cyr61 siRNA (siCYR61)-treated TA muscles and immunostained for myosin heavy chain isotypes, as explained for Figure 1.
(E) There are no changes in the number of myofibers following Cyr61 knock down. However, the proportion of myofiber types shifts from 2X toward 2B, simultaneously causing a significant increase in the size of type 2X myofibers.
(F–H) Volcano plot of RNA-seq from TA muscles with Cyr61 knock down shows substantial transcriptional changes (F), including significant regulation of Wnt signaling signaling pathway components (G), which are shown in (H).
Because protein quality control contributes to sarcopenia (Figure S5; Demontis et al., 2013; Jiao and Demontis, 2017), we examined whether Cyr61 regulates proteostasis, which declines during skeletal muscle aging (Demontis and Perrimon, 2010; Hunt et al., 2021; Jiao and Demontis, 2017). However, analysis of ubiquitinated proteins found in detergent-insoluble fractions revealed that loss of Cyr61 does not modulate this process (Figure S6).
We next examined whether Cyr61 modulates age-associated myofiber atrophy. Analysis of myofiber size (irrespective of myofiber type) revealed that Cyr61 siRNAs does not alter the overall size or number of myofibers. However, Cyr61 siRNAs impacted the relative proportion of myofiber types (Figures 6D and 6E). Specifically, the number and relative proportion of type 2X myofibers decreased whereas that of 2B myofibers increased in response to Cyr61 siRNAs, indicating a shift from type 2X toward type 2B myofibers upon loss of Cyr61. When examining the size of different myofiber types, Cyr61 siRNA increased the size of type 2X myofibers compared to control NT siRNAs (Figures 6D and 6E), and this most likely stems from detection of type 2X myofibers that are en route to differentiate into type 2B myofibers, which are on average bigger than type 2X myofibers.
Because aging leads to a type 2X to 2B switch in the TA muscle (Figure 1; Crupi et al., 2018), which is similar to what is seen here with Cyr61 siRNAs (Figures 6D and 6E), we hypothesize that the age-associated increase in Cyr61 constitutes an adaptive mechanism to counteract or limit age-associated myofiber type-switching from 2X to 2B.
To determine the mechanisms of action of Cyr61 in sarcopenia, we next examined the transcriptional changes induced by Cyr61 siRNAs (Figure 6F). Compared to NT siRNAs, several gene categories were modulated by Cyr61, including splicing, myogenesis, and expression of components of the Wnt signaling pathway, such as Wnt6, Wnt7b, Sfrp5, Rspo1, and Kremen2 (Figures 6F–6H; Table S5). Whereas decreased expression of myogenic genes is consistent with previous reports on the requirement for Cyr61 in muscle regeneration (O’Connor et al., 2007), the increase in the expression of components of the Wnt signaling pathway provides a possible explanation for how Cyr61 may regulate myofiber type switching. Specifically, it has been previously shown that Wnt signaling plays a role in myofiber type determination (Anakwe et al., 2003; von Maltzahn et al., 2012) and that, for example, Wnt4 overexpression increases differentiation toward type 2B myofibers compared to control conditions (Takata et al., 2007). Altogether, these findings indicate that Cyr61 modulates myofiber type-switching during aging, and this may possibly occur via its capacity to modulate Wnt signaling.
In summary, muscle Cyr61 expression increases with aging most likely due to higher circulating levels of Wnt ligands (Brack et al., 2007; Du et al., 2014), which are known inducers of Cyr61 expression in multiple systems (Du et al., 2014; Malik et al., 2015; Si et al., 2006). Interestingly, age-associated increase in Wnt signaling may contribute to the shift in type 2X to 2B myofibers that occurs in the TA muscle with aging (Figure 1; Crupi et al., 2018). By limiting the expression of Wnt pathway components, age-upregulation of Cyr61 may constitute a negative feedback that restrains myofiber type switching during aging. However, it is currently unknown whether Cyr61 indeed relies on Wnt signaling to regulate age-dependent myofiber shifts.
Proteomic analyses and individual variability within an isogenic population reveal biomarkers and putative regulators of sarcopenia
Although Cyr61 is upregulated with aging and regulates age-associated myofiber type switching, we did not find a role for Cyr61 in the regulation of age-associated muscle atrophy. Therefore, we utilized the aging proteomic dataset to identify candidate proteins that may correlate with and regulate muscle mass. We have found that the loss of muscle mass is variable even in isogenic animals housed in the same cage (Figure 1). On this basis, we interrogated this individual variability to determine whether aged muscles that were more prone to atrophy demonstrated a different proteomic signature. For these studies, we have examined the correlation between muscle mass and protein levels in individual mice, at 6 and 24 months of age (Figures 7A and 7B).
Figure 7. Proteomic analyses of the individual variability in sarcopenia within an isogenic population.
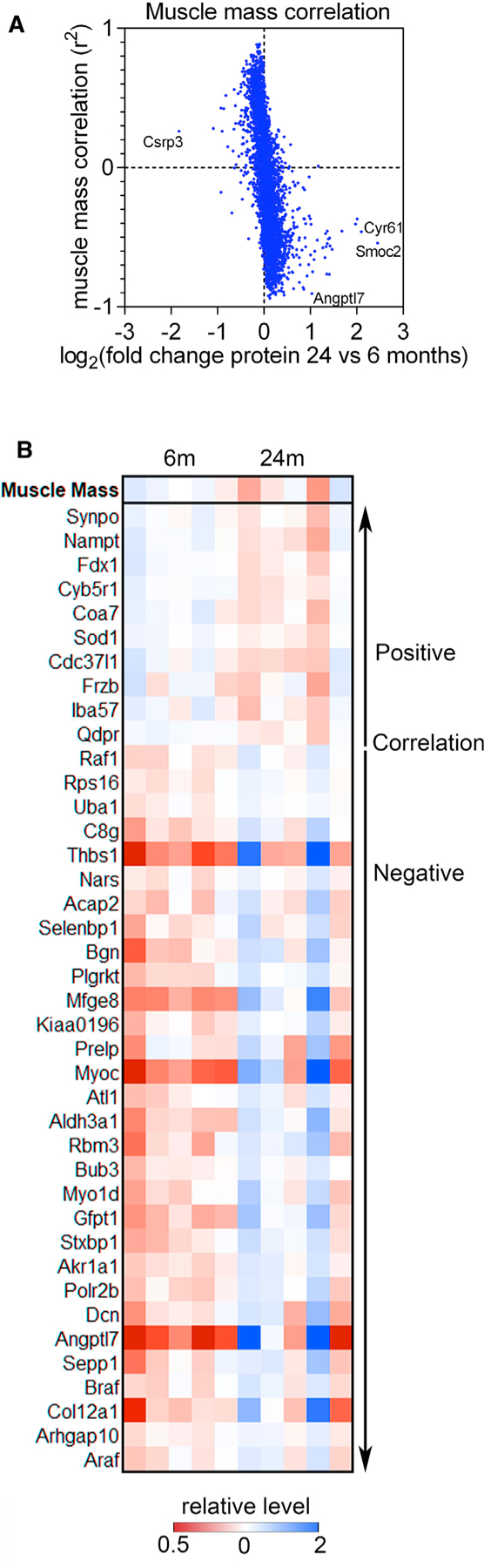
(A) The correlation across individual samples for each protein to muscle mass was determined and is plotted versus the log2FC for that protein. In general, the proteins with the highest correlation with muscle mass tend to have lower log2FC with aging whereas those with large log2FC tend to have a lower correlation.
(B) Heatmap of proteins with the best positive and negative correlations with muscle mass. The relative change in protein levels compared to relative change in muscle mass are shown for that sample on top.
This analysis has revealed several protein biomarkers of muscle mass loss with aging. Unexpectedly, the proteins with the strongest association to muscle mass loss are not those with the strongest age-related changes (Figure 7A), which may contribute to other muscle parameters that are modulated by aging but distinct from muscle mass loss. For example, the myokine Cyr61 is among the most age-upregulated proteins, and although its levels in individual mice are negatively correlated with muscle mass (r2 = −0.459), many other proteins have a greater degree of negative correlation. This suggests that this myokine is consistently induced by aging regardless of the degree of atrophy. Our functional analysis with Cyr61 siRNAs (Figure 6) indeed indicates that Cyr61 does not cause age-related muscle mass loss.
On the other hand, the protein levels of the myokine angiopoietin-like 7 (Angptl7) increase with aging and have a strong negative correlation (r2 = −0.903) to skeletal muscle mass, which could indicate that Angptl7 is a myokine that promotes sarcopenia (Figure 7B). Conversely, the protein levels of superoxide dismutase 1 (Sod1) and nicotinamide phosphoribosyltransferase (Nampt), an enzyme essential for NAD biosynthesis and sirtuin function (Imai, 2011), positively correlate with muscle mass (r2 = 0.887 and 0.885, respectively). Specifically, Sod1 and Nampt protein levels decline during aging, but muscle mass is better preserved in the mice that retain higher Sod1 and Nampt protein levels (Figure 7B). In support of this hypothesis, Nampt-deficient skeletal muscles display myofiber atrophy and reduced mass (Frederick et al., 2016).
Altogether, by examining variability in muscle mass within an isogenic population, age-regulated candidate proteins with possible distinct roles in sarcopenia can be identified. These are likely involved in age-related muscle mass loss and in other features of sarcopenia that are independent from muscle size determination.
Comparison of protein changes associated with sarcopenia in the TA versus soleus muscle
Skeletal muscles throughout the body have different characteristics based on the anatomical location and function (Ciciliot et al., 2013; Schiaffino and Reggiani, 2011). For example, the TA muscle here used for transcriptomics and proteomics is a relatively fast glycolytic muscle (comprised mostly of type 2X and 2B myofibers and a small population of type 2A myofibers), whereas the soleus is a slower oxidative muscle (comprised mostly of type 1 and 2A myofibers).
These muscles are known to respond differently to atrophic stimuli with the slower and more oxidative muscles generally being less susceptible to atrophy, which also translates to the individual myofiber types: the faster type 2B and 2X myofibers are more susceptible to atrophy than type 1 and 2A (Bonaldo and Sandri, 2013; Ciciliot et al., 2013; Schiaffino and Reggiani, 2011). Thus, although the TA is a useful muscle to study atrophy due to its susceptibility to atrophic stimuli, it is also valuable to understand how other muscles that are relatively resistant to atrophy, such as the soleus, respond at the transcriptome and proteome level to atrophic stimuli. In this respect, we have conducted a brief survey of candidate proteins that were identified from TA proteomics to determine how they are modulated in the soleus muscle with aging (Figure S7). Consistent with TMT mass spectrometry, these western blots confirmed that Cyr61, clusterin, ApoD, and Casq1 trend toward or are significantly upregulated in TA muscles in old age. However, only Cyr61 and clusterin showed increased levels in the soleus. This suggests that certain proteins, such as Cyr61 and clusterin, may be generally upregulated with age and not necessarily correlate with the susceptibility to or degree of atrophy. Conversely, because ApoD and Casq1 protein levels are induced only in TA but not soleus with aging, these findings suggest that they may reflect susceptibility to atrophy.
The mouse TA and EDL are fast-twitch muscles with relatively similar proportions of different myofiber types, although EDLs contain a higher proportion (15% versus 5%) of type 2A myofibers (Bloemberg and Quadrilatero, 2012). Western blot analysis also indicates that Cyr61, clusterin, and ApoD are higher in EDL muscles from 24- versus 6-month-old mice, as found for the TA. However, Casq1 levels are not significantly modulated (Figure S7). Altogether, these analyses indicate that there is overall similar induction of atroproteins across different muscles with aging, although some differences also exist, such as the case of Casq1 that is upregulated with aging in the TA but not in the soleus and EDL.
DISCUSSION
Skeletal muscle atrophy is a debilitating feature of many human diseases (Bonaldo and Sandri, 2013; Piccirillo et al., 2014; Shavlakadze and Grounds, 2006). Previous studies have defined a common set of genes (atrogenes) that are modulated during muscle atrophy in diverse disease settings, such as cancer-associated cachexia, starvation-induced atrophy, denervation, diabetes, kidney failure, and infections (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007). The definition of such molecular signature of muscle atrophy has profoundly impacted this field of research and enabled the discovery of many mediators of muscle atrophy. However, age-induced muscle atrophy (sarcopenia) was not included in these earlier studies, and the ablation of genetic targets that were discovered to mediate muscle atrophy, such as Fbxo32 (MAFbx/atrogin-1) and Trim63 (MuRF1), does not successfully prevent age-associated muscle atrophy and actually compromise muscle function (Sandri et al., 2013). Moreover, the analysis of proteomic changes has remained largely unexplored.
Here, we have applied integrated deep genomic and proteomic analyses and quantified the abundance of >15,000 mRNAs and ~5,000 proteins for each condition (Figures 2 and 3). On the basis of this deep genomic and proteomic profiling, we have established that distinct muscle atrophic stimuli, i.e., glucocorticoids (dexamethasone), cancer cachexia, and aging, induce largely distinct molecular changes in muscle. In general, acute modes of muscle atrophy induced by dexamethasone and LLC cancer cachexia overlap more with each other than compared to aging. For example, the aforementioned Fbxo32 and Trim63 mRNAs are upregulated in muscle in response to dexamethasone and LLC but not with aging (Figures 2, 3, and 4). We also present evidence for declining protein quality control that is unique to aging (Figures 5, S4, and S5). Altogether, this indicates a high degree of diversity in the etiology and response to atrophic stimuli and that especially sarcopenia shall be considered differently with regards to developing treatments.
Furthermore, many atrogenes previously identified from gene expression profiling are not modulated at the protein level. For example, we have found that Fbxo32 (MAFbx/atrogin-1) and Trim63 (MuRF1), which are among the most widely known atrogenes (>2,000 PubMed entries), are induced by dexamethasone and LLC cancer at the mRNA level (Figure 2F), as previously reported (Bodine and Baehr, 2014; Bodine et al., 2001; Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007). However, no increase in the levels of these proteins was found in atrophic muscles (Figure 3F).
Interestingly, despite a prominent role for proteolysis and for inhibition of protein synthesis during skeletal muscle atrophy (Bonaldo and Sandri, 2013; Demontis et al., 2013; Piccirillo et al., 2014; Reid et al., 2014), we have found few proteomic changes that could provide mechanistic understanding for how these processes are modulated during atrophy. Specifically, there was downregulation of ribosomal proteins in LLC-cancer-induced muscle atrophy (Figure 3), which may impact protein synthesis. More importantly, the lysosomal protease Ctsl was a notable exception, as its protein levels were increased in all modes of atrophy (Figures 2 and 3). However, ubiquitin ligases did not emerge as prominently regulated at the protein level (Figure 3).
Because atrophy is characterized by an overall increase in muscle protein ubiquitination (Bonaldo and Sandri, 2013; Demontis et al., 2013; Lecker et al., 1999; Piccirillo et al., 2014; Reid et al., 2014; Solomon et al., 1998), much attention has been devoted to the analysis of ubiquitin ligases as they could promote such atrophy-associated ubiquitination and proteasome-mediated degradation. Interestingly, an increase in target protein ubiquitination could be similarly achieved via reduction in the levels of deubiquitinating enzymes (DUBs) (Komander et al., 2009). However, the role of the >100 DUBs in muscle atrophy is largely unexplored (Bédard et al., 2015). Here, we have found that the levels of some DUBs decrease with atrophy, including USP33, whose protein levels decline during aging (Figure 5A), and USP13 (Figure 2E), whose protein levels decline with dexamethasone and LLC cancer. On this basis, we propose that these DUBs may contribute to changes in muscle protein ubiquitination during atrophy, together with post-translational modulation of ubiquitin ligases.
As exemplified by Fbxo32 and Trim63 and in line with surveys in other systems (Ghazalpour et al., 2011; Gygi et al., 1999; Hunt et al., 2019a; Yeung, 2011), we have found a lower-than-expected correlation between the transcriptome and proteome during muscle atrophy (Figure 4) and that several protein categories are modulated by dexamethasone, cancer, and aging independently from mRNA changes.
Because protein changes with limited mRNA changes trend downward for dexamethasone and LLC, whereas they trend upward for aging, this suggests that dexamethasone and LLC may be characterized by a bias toward either decreased protein translation and/or increased degradation, whereas aging has a bias toward decreased degradation. In agreement with this model, we have found increased detergent-insoluble levels of ubiquitinated proteins and SQSTM1 in aging muscles and age-associated increase in ER chaperones (Figures 5, S4, and S5), which indicates that proteostasis is compromised in sarcopenia, consistent with previous reports (Ayyadevara et al., 2016; Demontis and Perrimon, 2010; Fernando et al., 2019; Hunt et al., 2021; Piccirillo et al., 2014; Sakuma et al., 2016; Wohlgemuth et al., 2010; Yamaguchi et al., 2007). Therefore, we suggest that aging may differ from acute modes of muscle atrophy because of dysfunction in protein degradation and protein quality control pathways.
By surveying the proteomic changes that characterize different modes of atrophy, this study has identified many proteins that are robustly modulated by atrophy and herein propose to define these protein biomarkers of atrophy as “atroproteins,” as similarly done for atrophy-modulated genes, i.e., atrogenes (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007). However, whereas it was proposed that atrogenes are commonly modulated by and may mediate muscle atrophy in response to diverse atrophic stimuli (Gomes et al., 2001; Jagoe et al., 2002; Lecker et al., 2004; Sacheck et al., 2007), our analyses suggest that distinct sets of atroproteins define and may contribute to stimulus-specific atrophy.
Overall, this study provides an integrated framework for understanding stimulus-specific muscle atrophy and indicates that, contrary to a previous model, stimulus-specific protein changes molecularly differentiate myofiber atrophies that otherwise have remarkable phenotypic similarities (Figure 1). On this basis, we envision that the datasets of this study may help direct therapeutic efforts toward the development of interventions for stimulus-specific atrophies. In particular, the proteomic surveys here generated provide a resource for shortlisting candidate protein biomarkers of atrophy that could be experimentally tested to determine their role in myofiber atrophy induced by dexamethasone, cancer cachexia, and aging. We envision that therapeutically targeting some of these atroproteins may eventually provide suitable interventions to resolve atrophy in distinct disease settings.
Limitations of the study
A limitation of this study is that it does not examine wasting in human samples. Nonetheless, the mouse proteomic dataset reported here provides a starting point for ascertaining the modulation of atroproteins in humans and their potential use as therapeutic targets.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fabio Demontis (Fabio.Demontis@stjude.org).
Materials availability
There are no restrictions to the availability of tools generated in this study.
Data and code availability
All data supporting the findings of this study are available within the paper and the Supplemental information, including Figures S1–S7 and Tables S1, S2, S3, S4, S5, and S6. The RNA-seq data discussed in this publication has also been deposited in the NCBI’s Gene Expression Omnibus and is accessible through GEO series accession number GSE159952. The raw proteomics data generated in this study has been deposited to the PRIDE database and is available via ProteomeXchange with identifiers PXD027464 and PXD027490.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
LLC cells were obtained from the ATCC and screened regularly to ensure the absence of mycoplasma. Cells were maintained at 37°C with 5% CO2 in DMEM containing 10% fetal bovine serum, Glutamax and penicillin/streptomycin.
Mouse models of skeletal muscle atrophy
All mice were housed and handled in accordance with approved St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee protocols and fed standard chow with 12-hour light/dark cycles. To generate dexamethasone-induced muscle atrophy, 6-month-old male C57BL/6J mice (The Jackson Laboratory, JAX#000664) were treated with dexamethasone (#D2915, Sigma-Aldrich, MO) for 14 days via intraperitoneal injections of 20 mg/kg/day, as previously done (Hong et al., 2019; Jesinkey et al., 2014; Li et al., 2017). For the analysis of sarcopenia, 6-month versus 24-month-old male C57BL/6J mice were compared. For cancer-induced atrophy, 5×105 LLC cells were each injected into the right and left flank of 6-month-old male C57BL/6J mice, as previously done (Hunt et al., 2019b; Puppa et al., 2014; Talbert et al., 2019), and tumors were allowed to grow for ~3 weeks before analysis of muscle atrophy.
METHOD DETAILS
Immunostaining and laser scanning confocal microscopy
Tibialis anterior muscles were bisected at the mid-belly, mounted onto tragacanth gum, and frozen in isopentane cooled by liquid nitrogen. 10 μm sections were cut on a cryostat and immunostained as previously described (Hunt et al., 2019b). Unfixed slides holding the sections were incubated with blocking buffer (PBS with 2% BSA) for 1 hour before incubation with primary antibodies against type 2A (DSHB, SC-71) and 2B myosin heavy chain (DSHB, BF-F3) and laminin α2 (Santa Cruz, 4H8–2) overnight at 4°C. The sections were then washed and incubated with secondary antibodies for type 2A (anti-mouse IgG1 Alexa488), type 2B (anti-mouse IgM Alexa555) and laminin (anti-rat IgG Alexa633). The fiber types and sizes were analyzed with the Nikon Elements software using the inverse threshold of laminin α2 staining to determine myofiber boundaries. The myosin heavy chain staining was used to classify type 2B fibers (red), type 2A (green) and presumed 2X fibers (black) that were not stained for 2B or 2A myosin heavy chain. After myofibers were classified and parameters measured, the Feret’s minimal diameter was used as the measurement of myofiber size due its accuracy in estimating the size of unevenly shaped or cut objects (Bloemberg and Quadrilatero, 2012). For the quantification of the number of myofibers, all fibers in the cross sections of entire tibialis anterior muscles were counted based on the myofiber borders identified by laminin immunostaining. The size and number of myofibers was measured from the inverse images of laminin immunostaining (for identifying myofiber borders), excluding myofibers with diameters < 5 and > 100 μm. To categorize myofiber types, the intersections of the inverse images of laminin and myosin heavy chain-specific staining was used. These analyses were performed in an automated manner by using the Nikon Elements software and the “Object count” function.
For immunostaining of infiltrating immune cells, similar procedures as above were used together with anti-Ly6G (127601, BioLegend) and anti-F4/80 (123101, BioLegend) primary antibodies. Schemes were drawn with BioRender.
Mouse siRNA electroporation procedures
The tibialis anterior (TA) muscles of 30-month-old male C57BL6/J mice were electroporated with either Cyr61 siRNAs (Dharmacon #L-043717-01) or control non-targeting (NT) siRNAs (Dharmacon #D-001810-10) by first anaesthetizing mice with isoflurane, removing hair from the hind legs, and then injecting the tibialis anterior with 30 μL of 0.4U/L Hyaluronidase using a 29G1/2 insulin syringe. After recovering for two hours, the mice were anesthetized again and 500 pmol of siRNAs in 50 μL of Dharmacon’s recommended siRNA resuspension buffer (60 mM KCl, 6 mM HEPES-pH 7.5, and 0.2 mM MgCl2) were injected into the tibialis anterior muscle (one leg injected with NT siRNAs and the contralateral leg with Cyr61 siRNAs) followed by electroporation by using a BTX ECM 830 apparatus with metal electrodes placed parallel to the tibia orientation. Specifically, 4 pulses at 200V/cm with 20 msec length at 1Hz were delivered, followed by another 4 pulses after the orientation of the electrodes was switched to perpendicular to the tibia. After 7 days from electroporation, mice were sacrificed, and TA muscles dissected and immediately snap frozen for subsequent analyses.
RNA-seq and qRT-PCR
Tissues were homogenized in TRIzol and RNA extracted by isopropanol precipitation from the aqueous phase. RNA sequencing was performed as previously described (Hunt et al., 2019b). RNA sequencing libraries for each sample were prepared with 1 μg total RNA using the Illumina TruSeq RNA Sample Prep v2 Kit per the manufacturer’s instructions, and sequencing was completed on the Illumina HiSeq 4000. Approximately 100 million reads were obtained for each sample. The 100 bp paired-end reads were trimmed, filtered against quality (Phred-like Q20 or greater) and length (50 bp or longer), and aligned to a mouse reference sequence GRCm38 (UCSC mm10), using CLC Genomics Workbench v12.0.1 (QIAGEN). For gene expression comparisons, we obtained the TPM (transcript per million) counts from the RNA-Seq Analysis tool assigned at the gene level (individual transcript isoforms were not considered). The differential gene expression analysis was performed using the non-parametric ANOVA using the Kruskal-Wallis and Dunn’s tests on log-transformed TPM between three replicates of experimental groups, implemented in Partek Genomics Suite v7.0 software (Partek Inc.). For each of these RNA-seq datasets, > 15,000 mRNAs/condition were quantified. The data discussed in this publication have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE159952.
For qRT-PCR, cDNAs were reverse transcribed from 500 ng total RNA. qRT-PCR was performed as previously described by using GAPDH, HPRT, and PPIA as normalization (Hunt et al., 2015). The following oligonucleotides were used to detect expression of these specific genes:
Cyr61: 5′-AGAGGCTTCCTGTCTTTGGC-3′ and 5′-CCAAGACGTGGTCTGAACGA-3′
GAPDH: 5′-CCAGAACATCATCCCTGCATCC-3′ and 5′-ATACTTGGCAGGTTTCTCCAGG-3′
HPRT: 5′-GATTAGCGATGATGAACCAGGTT-3′ and 5′-TCCAAATCCTCGGCATAATGAT-3′
PPIA: 5′-GGCCGATGACGAGCCC-3′ and 5′-TGTCTTTGGAACTTTGTCTGCAA-3′
Protein digestion and peptide isobaric labeling by tandem mass tags
For preparation of tibialis anterior muscles for TMT-MS, muscle samples were ground into a fine powder by using a mortar and pestle cooled by liquid nitrogen, as previously done (Hunt et al., 2019b). Similar volumes of the powdered samples were used for subsequent analyses, which were performed with slight modifications to a previously published protocol (Bai et al., 2017). Specifically, tissue samples were extracted in lysis buffer (50 mM HEPES, pH 8.5, 8 M urea and 0.5% sodium deoxycholate), and protein concentration of the lysates was determined by Coomassie-stained short gels using bovine serum albumin (BSA) as standard (Xu et al., 2009). 100 μg of protein for each sample was digested with LysC (Wako) at an enzyme-to-substrate ratio of 1:100 (w/w) for 2h in the presence of 1 mM DTT. Following this, the samples were diluted to a final 2 M Urea concentration with 50 mM HEPES (pH 8.5), and further digested with trypsin (Promega) at an enzyme-to-substrate ratio of 1:50 (w/w) for at least 3h. The peptides were reduced by adding 1 mM DTT for 30 min at room temperature (RT) followed by alkylation with 10 mM iodoacetamide (IAA) for 30 min in the dark at RT. The unreacted IAA was quenched with 30 mM DTT for 30 min. Finally, the digestion was terminated and acidified by adding trifluoroacetic acid (TFA) to 1%, desalted using C18 cartridges (Harvard Apparatus), and dried by speed vac. The purified peptides were resuspended in 50 mM HEPES (pH 8.5) and labeled with 16-plex Tandem Mass Tag (TMT) reagents (Thermo Scientific) following manufacturer’s recommendations.
Two-dimensional HPLC and mass spectrometry
TMT-labeled samples were mixed equally, desalted, and fractionated on an offline HPLC (Agilent 1220) using basic pH reverse phase liquid chromatography (pH 8.0, XBridge C18 column, 4.6 mm × 25 cm, 3.5 μm particle size, Waters). The fractions were dried and resuspended in 5% formic acid and analyzed by acidic pH reverse phase LC-MS/MS analysis. The peptide samples were loaded on a nanoscale capillary reverse phase C18 column (New objective, 75 um ID × ~25 cm, 1.9 μm C18 resin from Dr. Maisch GmbH) by a HPLC system (Thermo Ultimate 3000) and eluted by a 60-min gradient. The eluted peptides were ionized by electrospray ionization and detected by an inline Orbitrap Fusion mass spectrometer (Thermo Scientific). The mass spectrometer is operated in data-dependent mode with a survey scan in Orbitrap (60,000 resolution, 1 × 106 AGC target and 50 ms maximal ion time) and MS/MS high resolution scans (60,000 resolution, 2 × 105 AGC target, 120 ms maximal ion time, 32 HCD normalized collision energy, 1 m/z isolation window, and 15 s dynamic exclusion).
MS data analysis
The MS/MS raw files were processed by the tag-based hybrid search engine, JUMP, which showed better sensitivity and specificity than commercial packages (e.g., Proteome Discoverer), (Wang et al., 2014b). The raw data were searched against the UniProt mouse database concatenated with a reversed decoy database for evaluating false discovery rate. Searches were performed using a 15-ppm mass tolerance both precursor and product ions, fully tryptic restriction with two maximal missed cleavages, three maximal modification sites, and the assignment of a, b, and y ions. TMT tags on Lys and N-termini (+304.20715 Da) were used for static modifications and Met oxidation (+15.99492 Da) was considered as a dynamic modification. Matched MS/MS spectra were filtered by mass accuracy and matching scores to reduce protein false discovery rate to ~1%. Proteins were quantified by summing reporter ion intensities across all matched PSMs using the JUMP software suite (Pagala et al., 2015). For each of these TMT datasets, ~5,000 proteins/condition were quantified, specifically 5229 were detected in the batch for aging, while 4848 were detected in the batch containing LLC and dexamethasone comparisons. Raw proteomics data has been deposited to the PRIDE database and is available via ProteomeXchange with identifiers PXD027464 and PXD027490.
Western blots analysis of detergent-insoluble protein fractions
Detergent-soluble protein fractions were extracted by homogenizing tissues in NP40 cell lysis buffer (Invitrogen #FNN0021; 100 μL per ~5 mg of mouse muscle tissue) by using a bullet blender (NextAdvance) with zirconium 0.5-mm beads, and then centrifuging at 15,000 rpm for 10 minutes to pellet insoluble material (Hunt et al., 2021; Rai et al., 2021b). The supernatant (detergent-soluble protein fraction) was removed and the protein concentration determined by using the Bio-Rad protein assay. Subsequently, equal protein quantities of detergent-soluble fractions were loaded into 4%–20% gradient polyacrylamide gels (Bio-Rad) with SDS blue loading buffer. Insoluble pellets were washed three times with NP40 cell lysis buffer. Subsequently, 8M urea and 1% SDS in PBS was added to solubilize the insoluble pellet (the specific volume added was determined based on the concentration of the corresponding detergent-soluble fraction), combined with SDS blue loading buffer, and loaded into polyacrylamide gels. The gels were transferred to PVDF membranes and blocked with either 10% skim milk powder or BSA in Tris buffered saline with 0.1% Tween-20 (TBST) for 1 hour. The membranes were then incubated with primary antibodies in TBST overnight at 4°C, washed and incubated with either anti-mouse or anti-rabbit HRP-linked secondary antibodies (Cell Signaling Technologies #7076 and #7074, respectively) for 2 hours. Membranes were washed and incubated with ECL (GE Healthcare) and film used to measure the signal. Adobe Photoshop was used to quantify band density and proteins were normalized to tubulin band density and presented as a ratio relative to controls. The primary antibodies used are listed in the Key resource table.
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Mouse IgG1 anti-Myosin heavy chain type IIA | DSHB | SC-71; RRID:AB_2147165 |
| Mouse IgM anti-Myosin heavy chain type IIB | DSHB | BF-F3; RRID:AB_2266724 |
| Rat anti-laminin α−2 (4H8-2) | Santa Cruz | Sc-59854; RRID:AB_784266 |
| Mouse anti-ubiquitin (P4D1) | Santa Cruz | Sc-8017; RRID:AB_2762364 |
| Rabbit anti-p62/SQSTM1 | Cell Signaling Technologies | 5114S; RRID:AB_10624872 |
| Rabbit anti-alpha- tubulin (11H10) | Cell Signaling Technologies | 2125S; RRID:AB_2619646 |
| Rabbit anti-GAPDH (D16H11) | Cell Signaling Technologies | 5174S; RRID:AB_10622025 |
| Rat anti-F4/80 | BioLegend | 123101; RRID:AB_893504 |
| Rat anti-Ly6G (1A8) | BioLegend | 127601; RRID:AB_1089179 |
| Mouse anti-Cyr61 (A-10) | Santa Cruz | sc-374129; RRID:AB_10947399 |
| Mouse anti-Clusterin/ApoJ | R&D | AF2747-SP; RRID:AB_2083314 |
| Rabbit anti-ApoD | Novus Bio | NBP2-92650; RRID:N/A |
| Rabbit anti-Calsequestrin1 | Novus Bio | NBP1-88180; RRID:AB_11010381 |
| AlexaFluor488 anti-mouse IgG1 | Life Technologies | A21121; RRID:AB_2535764 |
| AlexaFluor555 anti-mouse IgM | Life Technologies | A21426; RRID:AB_1500929 |
| AlexaFluor647 anti-rat | Life Technologies | A21247; RRID:AB_141778 |
| Anti-mouse IgG, HRP-linked | Cell Signaling Technologies | 7076S; RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked | Cell Signaling Technologies | 7074S; RRID:AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| AlexaFluor635 Phalloidin | Life Technologies | A22284 |
| Dexamethasone | Sigma-Aldrich | D2915 |
| DAPI | Roche | 10236276001 |
| TRIzol | Ambion | 15596018 |
| MG132 | Sigma-Aldrich | M8699 |
| IQ Sybr Green supermix | Bio-Rad | 170-8885 |
| 6-well plates | Corning | REF3516 |
| 96-well PCR plates | Biorad | HSP9601 |
| Transparent 96-well plates | Corning | REF3599 |
| High glucose DMEM, with glutamax | GIBCO | 10566016 |
| Fetal bovine serum | GIBCO | 10437-028 |
| Penicillin streptomycin | GIBCO | 15140122 |
| PBS | GIBCO | 10010023 |
| Blue loading buffer pack | Cell Signaling | 7722 |
| Precision Plus protein standard | Bio-Rad | 1610374 |
| 4-20% Mini-PROTEAN TGX pre-cast gels | Bio-Rad | 4561096 |
| Immobilon-P PVDF membrane | Millipore | IPVH00010 |
| Ponceau S | Sigma-Aldrich | P7170-1L |
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 |
| Critical commercial assays | ||
| Pierce BCA protein assay kit | Thermo Scientific | 23225 |
| Proteasome-Glo 3-substrate cell-based assay system | Promega | G1180 |
| iScript reverse transcriptase | Bio-Rad | 1708840 |
| Deposited data | ||
| Reagent or resource | Source | Identifier |
| RNA-seq data from mouse muscles with different types of atrophy | This paper | Gene Expression Omnibus, GSE159952 |
| RNA-seq data from mouse muscles with Cyr61 siRNAs and control NT siRNAs | This paper | Gene Expression Omnibus, GSE159952 |
| TMT mass spectrometry data from mouse muscles with different types of atrophy | This paper | PRIDE database, ProteomeXchange identifiers PXD027464 and PXD027490 |
| Experimental models: Cell lines | ||
| Mouse: LLC cells | ATCC | CRL-1642 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | The Jackson Laboratory | JAX#000664 |
| Oligonucleotides | ||
| ON-TARGET Plus Non-targeting siRNA pool | Dharmacon | D-001810-10-05 |
| ON-TARGET Cyr61 siRNA pool | Dharmacon | L-043717-01 |
| Software and algorithms | ||
| GraphPad Prism | Graphpad | https://www.graphpad.com/ |
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/ products/software |
| Photoshop CSX | Adobe | https://www.adobe.com/products/photoshop.html |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| WebGestalt | Wang et al., 2017 | https://www.webgestalt.org |
Other western blot analyses
Western blots of Cyr61, Clusterin/ApoJ, ApoD, and Calsequestrin1 levels were done with muscle samples homogenized in NP40 cell lysis buffer (Invitrogen #FNN0021) and subsequently processed according to standard procedures (Hunt et al., 2015).
Proteasome assays
Muscles were homogenized in NP40 cell lysis buffer (Invitrogen #FNN0021) and protein quantitation determined by using the Bio-Rad protein assay. Subsequently, muscle extracts were then all normalized to the same final protein concentration (1 mg/mL) by dilution with additional NP40 cell lysis buffer. 10 μL of the normalized NP40 muscle extracts were then used for the measurement of proteasome activity with the Proteasome-Glo 3-substrate cell-based assay system (Promega #G1180), as previously done (Hunt et al., 2021; Rai et al., 2021a). Specifically, 100 μL of the three different substrate reagents (to measure caspase-like, trypsin-like, and chymotrypsin-like proteolytic activities) were added individually to plates with 10 μL of the normalized NP40 muscle extracts. The plates were then incubated for 30 min at room temperature and the luminescence read on a Tecan Infinite 200 PRO plate reader. To ensure that the observed proteolytic activities indeed depend on the proteasome, some samples were treated with MG132 at a final concentration of 50 μM: this was added to the muscle extracts just before addition of the Proteasome-Glo3 substrates.
Dataset analysis
Bonferonni’s adjustment was used to adjust for multiple comparisons within transcriptomic data whereas Benjamini-Hochberg was used for p value adjustment of proteomics data. For Gene Set Enrichment Analysis (GSEA; Table S2), the WebGestalt online tool (Wang et al., 2017) was used to determine enrichment of Gene Ontology (GO) terms by biological process (BP) by using the totality of detected mRNAs/proteins in the dataset as background. For determining the significant overlapping mRNA and protein changes for Venn diagrams, the criteria used were p < 0.05 and −1 > log2FC > 1 for mRNA and −0.3 > log2FC > 0.3 for protein. This lower criterion was used for protein changes partly because of ratio compression, a common issue of TMT-based quantification due to interference of co-isolated ions from complex whole proteome samples (Rauniyar and Yates, 2014); thus the measured fold change is generally smaller than the bona fide fold change in the real sample (Niu et al., 2017). When considering categorization and organization of heatmaps, only the magnitude was considered as long as at least one atrophic condition met the above criteria. Comparisons on absolute values between sample groups were not considered due to different animal cohorts and batch processing between these. PCA scores (Figures S1C and S1D) were calculated by using Spotfire Decision.
QUANTIFICATION AND STATISTICAL ANALYSES
The unpaired two-tailed Student’s t test was used to compare the means of two independent groups to each other. One-way ANOVA with Tukey’s post hoc test was used for multiple comparisons of more than two groups of normally distributed data. The “n” for each experiment can be found in the figures and legends and represents independently generated samples (i.e., biological replicates) for all experiments. Bar graphs represent the mean ± SEM. Throughout the figures, the exact P values are annotated. Statistical analyses were done with Excel and GraphPad Prism.
Supplementary Material
Highlights.
Deep-coverage proteomics reveal molecular changes responsible for muscle wasting
Different catabolic stimuli induce muscle atrophy via largely distinct mechanisms
Transcriptome-proteome disconnect occurs during muscle wasting
Atroproteins are proteins modulated by atrophic stimuli
ACKNOWLEDGMENTS
This work was supported by research grants to F.D. from The Hartwell Foundation (Individual Biomedical Research award), the National Cancer Institute (P30CA021765-developmental funds), and the National Institute on Aging (R01AG055532 and R56AG63806). L.C.H. was supported by a Glenn/AFAR Postdoctoral Fellowship. The mass spectrometry analysis was performed at the St. Jude Children’s Research Hospital Center for Proteomics and Metabolomics, partially supported by NIH Cancer Center Support grant P30CA021765 and by NIH grant R01AG053987 (J.P.). M.L. is supported by the National Cancer Institute (R01CA245301). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research at St. Jude Children’s Research Hospital is supported by ALSAC.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109971.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anakwe K, Robson L, Hadley J, Buxton P, Church V, Allen S, Hart-mann C, Harfe B, Nohno T, Brown AM, et al. (2003). Wnt signalling regulates myogenic differentiation in the developing avian wing. Development 130, 3503–3514. [DOI] [PubMed] [Google Scholar]
- Ayyadevara S, Balasubramaniam M, Suri P, Mackintosh SG, Tackett AJ, Sullivan DH, Shmookler Reis RJ, and Dennis RA (2016). Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging (Albany NY) 8, 3486–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Tan H, Pagala VR, High AA, Ichhaporia VP, Hendershot L, and Peng J (2017). Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol. 585, 377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bédard N, Jammoul S, Moore T, Wykes L, Hallauer PL, Hastings KE, Stretch C, Baracos V, Chevalier S, Plourde M, et al. (2015). Inactivation of the ubiquitin-specific protease 19 deubiquitinating enzyme protects against muscle wasting. FASEB J. 29, 3889–3898. [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX, and Rudnicki MA (2012). Building muscle: molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol 4, a008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D, and Quadrilatero J (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multi-color immunofluorescence analysis. PLoS ONE 7, e35273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, and Baehr LM (2014). Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab 307, E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. [DOI] [PubMed] [Google Scholar]
- Bonaldo P, and Sandri M (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech 6, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, and Rando TA (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810. [DOI] [PubMed] [Google Scholar]
- Braun TP, and Marks DL (2015). The regulation of muscle mass by endogenous glucocorticoids. Front. Physiol 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai RJ, Vukovic J, Dunlop S, Grounds MD, and Shavlakadze T (2011). Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS ONE 6, e28090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Arif M, Emanuelsson EB, Reitzner SM, Lindholm ME, Mardinoglu A, and Sundberg CJ (2020). Skeletal Muscle Transcriptomic Comparison between Long-Term Trained and Untrained Men and Women. Cell Rep. 31, 107808. [DOI] [PubMed] [Google Scholar]
- Chaqour B, and Goppelt-Struebe M (2006). Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J. 273, 3639–3649. [DOI] [PubMed] [Google Scholar]
- Ciciliot S, Rossi AC, Dyar KA, Blaauw B, and Schiaffino S (2013). Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol 45, 2191–2199. [DOI] [PubMed] [Google Scholar]
- Crupi AN, Nunnelee JS, Taylor DJ, Thomas A, Vit JP, Riera CE, Gottlieb RA, and Goodridge HS (2018). Oxidative muscles have better mitochondrial homeostasis than glycolytic muscles throughout life and maintain mitochondrial function during aging. Aging (Albany NY) 10, 3327–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F, and Perrimon N (2010). FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F, Piccirillo R, Goldberg AL, and Perrimon N (2013). Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis. Model. Mech 6, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Klein JD, Hassounah F, Zhang J, Zhang C, and Wang XH (2014). Aging increases CCN1 expression leading to muscle senescence. Am. J. Physiol. Cell Physiol 306, C28–C36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando R, Drescher C, Nowotny K, Grune T, and Castro JP (2019). Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med 132, 58–66. [DOI] [PubMed] [Google Scholar]
- Frederick DW, Loro E, Liu L, Davila A Jr., Chellappa K, Silverman IM, Quinn WJ 3rd, Gosai SJ, Tichy ED, Davis JG, et al. (2016). Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 24, 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian CN, Ong P, Li Z, Chen S, Mak SY, et al. (2016). Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med 22, 666–671. [DOI] [PubMed] [Google Scholar]
- Fuller HR, Gillingwater TH, and Wishart TM (2016). Commonality amid diversity: Multi-study proteomic identification of conserved disease mechanisms in spinal muscular atrophy. Neuromuscul. Disord 26, 560–569. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N, et al. (2011). Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 7, e1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello E, Crea E, Torelli L, Bergamo A, Reggiani C, Sava G, and Toniolo L (2020). Age Dependent Modification of the Metabolic Profile of the Tibialis Anterior Muscle Fibers in C57BL/6J Mice. Int. J. Mol. Sci 21, 3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, and Goldberg AL (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 98, 14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueugneau M, d’Hose D, Barbé C, de Barsy M, Lause P, Maiter D, Bindels LB, Delzenne NM, Schaeffer L, Gangloff Y-G, et al. (2018). Increased Serpina3n release into circulation during glucocorticoid-mediated muscle atrophy. J. Cachexia Sarcopenia Muscle 9, 929–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, and Aebersold R (1999). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselgren PO, Alamdari N, Aversa Z, Gonnella P, Smith IJ, and Tizio S (2010). Corticosteroids and muscle wasting: role of transcription factors, nuclear cofactors, and hyperacetylation. Curr. Opin. Clin. Nutr. Metab. Care 13, 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, and Chawla A (2013). Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Lee JH, Jeong KW, Choi CS, and Jun HS (2019). Amelioration of muscle wasting by glucagon-like peptide-1 receptor agonist in muscle atrophy. J. Cachexia Sarcopenia Muscle 10, 903–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Cantó C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, and Auwerx J (2011). The metabolic footprint of aging in mice. Sci. Rep 1, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulmi JJ, Penna F, Pöllänen N, Nissinen TA, Hentilä J, Euro L, Lautaoja JH, Ballarò R, Soliymani R, Baumann M, et al. (2020). Muscle NAD+ depletion and Serpina3n as molecular determinants of murine cancer cachexia-the effects of blocking myostatin and activins. Mol. Metab 41, 101046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt LC, Xu B, Finkelstein D, Fan Y, Carroll PA, Cheng PF, Eisenman RN, and Demontis F (2015). The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev. 29, 2475–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt LC, Jiao J, Wang YD, Finkelstein D, Rao D, Curley M, Robles-Murguia M, Shirinifard A, Pagala VR, Peng J, et al. (2019a). Circadian gene variants and the skeletal muscle circadian clock contribute to the evolutionary divergence in longevity across Drosophila populations. Genome Res. 29, 1262–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt LC, Stover J, Haugen B, Shaw TI, Li Y, Pagala VR, Finkelstein D, Barton ER, Fan Y, Labelle M, et al. (2019b). A Key Role for the Ubiquitin Ligase UBR4 in Myofiber Hypertrophy in Drosophila and Mice. Cell Rep. 28, 1268–1281.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt LC, Schadeberg B, Stover J, Haugen B, Pagala V, Wang YD, Puglise J, Barton ER, Peng J, and Demontis F (2021). Antagonistic control of myofiber size and muscle protein quality control by the ubiquitin ligase UBR4 during aging. Nat. Commun 12, 1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibebunjo C, Chick JM, Kendall T, Eash JK, Li C, Zhang Y, Vickers C, Wu Z, Clarke BA, Shi J, et al. (2013). Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol 33, 194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S (2011). Dissecting systemic control of metabolism and aging in the NAD World: the importance of SIRT1 and NAMPT-mediated NAD biosynthesis. FEBS Lett. 585, 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K, and Miura M (2004). A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med 10, 98–102. [DOI] [PubMed] [Google Scholar]
- Jagoe RT, Lecker SH, Gomes M, and Goldberg AL (2002). Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J. 16, 1697–1712. [DOI] [PubMed] [Google Scholar]
- Jesinkey SR, Korrapati MC, Rasbach KA, Beeson CC, and Schnell-mann RG (2014). Atomoxetine prevents dexamethasone-induced skeletal muscle atrophy in mice. J. Pharmacol. Exp. Ther 351, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, and Demontis F (2017). Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol 34, 1–6. [DOI] [PubMed] [Google Scholar]
- Johnston AJ, Murphy KT, Jenkinson L, Laine D, Emmrich K, Faou P, Weston R, Jayatilleke KM, Schloegel J, Talbo G, et al. (2015). Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 162, 1365–1378. [DOI] [PubMed] [Google Scholar]
- Kivela R, Kyrolainen H, Selanne H, Komi PV, Kainulainen H, and Vihko V (2007). A single bout of exercise with high mechanical loading induces the expression of Cyr61/CCN1 and CTGF/CCN2 in human skeletal muscle. J. Appl. Physiol 103, 1395–1401. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, and Urbé S (2009). Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol 10, 550–563. [DOI] [PubMed] [Google Scholar]
- Kunkel SD, Suneja M, Ebert SM, Bongers KS, Fox DK, Malmberg SE, Alipour F, Shields RK, and Adams CM (2011). mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab. 13, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Aravamudhan S, Nolte H, Türk C, Hölper S, Müller S, Günther S, Blaauw B, Braun T, and Krüger M (2017). Dynamic changes in the mouse skeletal muscle proteome during denervation-induced atrophy. Dis. Model. Mech 10, 881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Khaghani S, Türk C, Wiederstein JL, Hölper S, Piller T, Nogara L, Blaauw B, Günther S, Müller S, et al. (2018). Single Muscle Fiber Proteomics Reveals Distinct Protein Changes in Slow and Fast Fibers during Muscle Atrophy. J. Proteome Res 17, 3333–3347. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, and Goldberg AL (1999). Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J. Clin. Invest 104, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, and Goldberg AL (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 18, 39–51. [DOI] [PubMed] [Google Scholar]
- Li J, Chan MC, Yu Y, Bei Y, Chen P, Zhou Q, Cheng L, Chen L, Ziegler O, Rowe GC, et al. (2017). miR-29b contributes to multiple types of muscle atrophy. Nat. Commun 8, 15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Peng Y, Wang X, Fan Y, Qin C, Shi L, Tang Y, Cao K, Li H, Long J, and Liu J (2016). Mitochondrial Dysfunction Launches Dexamethasone-Induced Skeletal Muscle Atrophy via AMPK/FOXO3 Signaling. Mol. Pharm 13, 73–84. [DOI] [PubMed] [Google Scholar]
- Llano-Diez M, Fury W, Okamoto H, Bai Y, Gromada J, and Larsson L (2019). RNA-sequencing reveals altered skeletal muscle contraction, E3 ligases, autophagy, apoptosis, and chaperone expression in patients with critical illness myopathy. Skelet. Muscle 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmassani ZS, Reidy PT, McKenzie AI, Stubben C, Howard MT, and Drummond MJ (2019). Age-dependent skeletal muscle transcriptome response to bed rest-induced atrophy. J. Appl. Physiol 126, 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AR, Liszewska E, and Jaworski J (2015). Matricellular proteins of the Cyr61/CTGF/NOV (CCN) family and the nervous system. Front. Cell. Neurosci 9, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugahid DA, Sengul TG, You X, Wang Y, Steil L, Bergmann N, Radke MH, Ofenbauer A, Gesell-Salazar M, Balogh A, et al. (2019). Proteomic and Transcriptomic Changes in Hibernating Grizzly Bears Reveal Metabolic and Signaling Pathways that Protect against Muscle Atrophy. Sci. Rep 9, 19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukund K, and Subramaniam S (2020). Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med 12, e1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu M, Cho JH, Kodali K, Pagala V, High AA, Wang H, Wu Z, Li Y, Bi W, Zhang H, et al. (2017). Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal. Chem 89, 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor RS, Mills ST, Jones KA, Ho SN, and Pavlath GK (2007). A combinatorial role for NFAT5 in both myoblast migration and differentiation during skeletal muscle myogenesis. J. Cell Sci 120, 149–159. [DOI] [PubMed] [Google Scholar]
- Pagala VR, High AA, Wang X, Tan H, Kodali K, Mishra A, Kavdia K, Xu Y, Wu Z, and Peng J (2015). Quantitative protein analysis by mass spectrometry. Methods Mol. Biol 1278, 281–305. [DOI] [PubMed] [Google Scholar]
- Piccirillo R, Demontis F, Perrimon N, and Goldberg AL (2014). Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn 243, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Wiggs MP, Duarte JA, Zergeroglu AM, and Demirel HA (2012). Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab 303, E31–E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci S, Greggi C, Polidoro C, Piro MC, Celi M, Feola M, Gasbarra E, Iundusi R, Mastrangeli F, Novelli G, et al. (2019). Clusterin silencing restores myoblasts viability and down modulates the inflammatory process in osteoporotic disease. J. Transl. Med 17, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa MJ, Gao S, Narsale AA, and Carson JA (2014). Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 28, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Coleman Z, Curley M, Nityanandam A, Platt A, Robles-Murguia M, Jiao J, Finkelstein D, Wang YD, Xu B, et al. (2021a). Proteasome stress in skeletal muscle mounts a long-range protective response that delays retinal and brain aging. Cell Metab. 33, 1137–1154.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Curley M, Coleman Z, Nityanandam A, Jiao J, Graca FA, Hunt LC, and Demontis F (2021b). Analysis of proteostasis during aging with western blot of detergent-soluble and insoluble protein fractions. STAR Protoc 2, 100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Van Vleet TR, Ciurlionis R, Buck WR, Mittelstadt SW, Blomme EAG, and Liguori MJ (2019). Comparison of RNA-Seq and Micro-array Gene Expression Platforms for the Toxicogenomic Evaluation of Liver From Short-Term Rat Toxicity Studies. Front. Genet 9, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N, and Yates JR 3rd. (2014). Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MB, Judge AR, and Bodine SC (2014). CrossTalk opposing view: The dominant mechanism causing disuse muscle atrophy is proteolysis. J. Physiol 592, 5345–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Rocancourt D, Mansouri A, and Buckingham M (2005). A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 435, 948–953. [DOI] [PubMed] [Google Scholar]
- Romanello V, and Sandri M (2016). Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, and Goldberg AL (2007). Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 21, 140–155. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, Pearson T, Lightfoot AP, Nye GA, Wells N, Giakoumaki II, Vasilaki A, Griffiths RD, Jackson MJ, and McArdle A (2016). Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci. Rep 6, 33944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma K, Kinoshita M, Ito Y, Aizawa M, Aoi W, and Yamaguchi A (2016). p62/SQSTM1 but not LC3 is accumulated in sarcopenic muscle of mice. J. Cachexia Sarcopenia Muscle 7, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, Mammucari C, Meskers CGM, Pallafacchina G, Paoli A, et al. (2013). Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 14, 303–323. [DOI] [PubMed] [Google Scholar]
- Schakman O, Kalista S, Barbé C, Loumaye A, and Thissen JP (2013). Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol 45, 2163–2172. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, and Reggiani C (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev 91, 1447–1531. [DOI] [PubMed] [Google Scholar]
- Shavlakadze T, and Grounds M (2006). Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. BioEssays 28, 994–1009. [DOI] [PubMed] [Google Scholar]
- Shum AMY, Poljak A, Bentley NL, Turner N, Tan TC, and Polly P (2018). Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: alterations in sarcomeric and mitochondrial protein expression. Oncotarget 9, 22001–22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si W, Kang Q, Luu HH, Park JK, Luo Q, Song WX, Jiang W, Luo X, Li X, Yin H, et al. (2006). CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol. Cell. Biol 26, 2955–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon V, Lecker SH, and Goldberg AL (1998). The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J. Biol. Chem 273, 25216–25222. [DOI] [PubMed] [Google Scholar]
- Takata H, Terada K, Oka H, Sunada Y, Moriguchi T, and Nohno T (2007). Involvement of Wnt4 signaling during myogenic proliferation and differentiation of skeletal muscle. Dev. Dyn 236, 2800–2807. [DOI] [PubMed] [Google Scholar]
- Talbert EE, Cuitino MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, Leone GW, Ostrowski MC, Paleo B, Weisleder N, et al. (2019). Modeling Human Cancer-induced Cachexia. Cell Rep. 28, 1612–1622.e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry EE, Zhang X, Hoffmann C, Hughes LD, Lewis SA, Li J, Wallace MJ, Riley LA, Douglas CM, Gutierrez-Monreal MA, et al. (2018). Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. eLife 7, e34613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ (2010). Reversing cachexia. Cell 142, 511–512. [DOI] [PubMed] [Google Scholar]
- Ubaida-Mohien C, Lyashkov A, Gonzalez-Freire M, Tharakan R, Shardell M, Moaddel R, Semba RD, Chia CW, Gorospe M, Sen R, and Ferrucci L (2019). Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. eLife 8, e49874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Maltzahn J, Chang NC, Bentzinger CF, and Rudnicki MA (2012). Wnt signaling in myogenesis. Trends Cell Biol. 22, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Gong B, Bushel PR, Thierry-Mieg J, Thierry-Mieg D, Xu J, Fang H, Hong H, Shen J, Su Z, et al. (2014a). The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol 32, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J (2014b). JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 13, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, and Zhang B (2017). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45 (W1), W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Song R, Wang Z, Jing Z, Wang S, and Ma J (2018). S100A8/A9 in Inflammation. Front. Immunol 9, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, and Leeuwenburgh C (2010). Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp. Gerontol 45, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton PTE, Flavell DM, Montgomery HE, World M, Humphries SE, and Talmud PJ (2007). Lipoprotein-associated phospholipase A2 A379V variant is associated with body composition changes in response to exercise training. Nutr. Metab. Cardiovasc. Dis 17, 24–31. [DOI] [PubMed] [Google Scholar]
- Xu P, Duong DM, and Peng J (2009). Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res 8, 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Arai H, Katayama N, Ishikawa T, Kikumoto K, and Atomi Y (2007). Age-Related Increase of Insoluble, Phosphorylated Small Heat Shock Proteins in Human Skeletal Muscle. J. Gerontol. A. Biol. Sci. Med. Sci 62, 481–489. [DOI] [PubMed] [Google Scholar]
- Yeung ES (2011). Genome-wide correlation between mRNA and protein in a single cell. Angew. Chem. Int. Ed. Engl 50, 583–585. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. (2010). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and the Supplemental information, including Figures S1–S7 and Tables S1, S2, S3, S4, S5, and S6. The RNA-seq data discussed in this publication has also been deposited in the NCBI’s Gene Expression Omnibus and is accessible through GEO series accession number GSE159952. The raw proteomics data generated in this study has been deposited to the PRIDE database and is available via ProteomeXchange with identifiers PXD027464 and PXD027490.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.