

Abstract
Background
Waldenström macroglobulinemia (WM) is an incurable disease and, while treatable, can develop resistance to available therapies and be fatal. Chimeric antigen receptor (CAR) T cell therapy directed against the CD19 antigen has demonstrated efficacy in relapsed or refractory B lymphoid malignancies, and is now approved for B cell acute lymphoblastic leukemia and certain B cell lymphomas. However, CAR T therapy has not been evaluated for use in WM.
Methods and results
We performed preclinical studies demonstrating CAR T cell activity against WM cells in vitro, and developed an in vivo murine model of WM which demonstrated prolonged survival with use of CAR T therapy. We then report the first three patients with multiply relapsed and refractory WM treated for their disease with CD19-directed CAR T cells on clinical trials. Treatment was well tolerated, and observed toxicities were consistent with those seen in CAR T treatment for other diseases, and no grade 3 or higher cytokine release syndrome or neurotoxicity events occurred. All three patients attained at least a clinical response to treatment, including one minimal residual disease-negative complete response, though all three eventually developed recurrent disease between 3 and 26 months after initial treatment.
Conclusions
This report summarizes preclinical and clinical activity of CD19-directed CAR T therapy in WM, demonstrating early tolerability and efficacy in patients with WM, and representing a possible treatment option in patients with heavily pretreated and relapsed or refractory WM. Larger studies evaluating CAR T therapy in WM are warranted, along with further evaluation into mechanisms of resistance to CAR T therapy.
Keywords: hematologic neoplasms, translational medical research, t-lymphocytes, receptors, chimeric antigen, cell engineering
Introduction
Waldenström macroglobulinemia (WM) is a rare lymphoma characterized by neoplastic proliferation of lymphoplasmacytic lymphoma (LPL) cells that infiltrate the bone marrow and other organs and secrete a monoclonal IgM paraprotein.1 Many available therapies are effective in attaining disease control, including Bruton tyrosine kinase (BTK) inhibitors, proteasome inhibitors, chemotherapy, and anti-CD20 monoclonal antibodies.2 However, these therapies are generally not curative, and a subset of patients will develop treatment-refractory WM and die of their disease.3 Therefore, novel therapeutic strategies for relapsed/refractory WM are required.
Anti-CD19-directed chimeric antigen receptor (CAR) T cell therapy is now an established treatment modality in several B cell leukemias and lymphomas.4–9 Given these encouraging results and usually strong CD19 positivity of WM clones, CAR T cell therapy is an intuitive option in patients with WM who have exhausted other treatment options.
In this report we describe the development and implementation of in vitro and in vivo models demonstrating promising anti-WM activity of CD19-directed CAR T cells. Following these positive preclinical studies, we opened an investigation into CD19-targeted CAR T cell therapies for WM. Three patients with WM were treated with anti-CD19 CAR T cell therapy in phase I clinical trials. We report on the safety and clinical activity of anti-CD19 CAR T cell therapy in these patients with multiply relapsed or refractory WM.
Materials and methods
In vitro assessment
A human MYD88 L265P-positive WM cell line, BCWM.1 (generously provided by Steven Treon, Dana-Farber Cancer Institute), was modified to express firefly luciferase (BCWM.1-Luc) and co-cultured with donor lymphocytes which were genetically modified to express either a 19-28z CAR, an irrelevantly targeted MUC16-28z CAR, or were left untransduced. CAR transduction was via gammaretrovirus as previously reported.10 Co-cultures were performed at effector:target ratios of 1:1 and 1:10 for each CAR T cell variant. In vitro cytotoxicity was assessed via adenosine-triphosphate (ATP)-dependent bioluminescence after 4-hour co-culture. Supernatant interferon (IFN)-γ and interleukin (IL)-2 levels were measured by xMAP Luminex 200 (Luminex Corp, Austin, Texas) at each time point. All in vitro studies were performed twice with independent donors to ensure reproducibility.
Preclinical in vivo studies
In vivo studies were conducted using sublethally irradiated SCID/beige mice to generate a systemic model of WM.11 Tail vein injection of 1×106 luciferase transduced BCWM.1 cells was performed. This model is characterized by widespread tumor growth in the bone marrow, liver, lungs, and kidney. 19-28ζ CAR modified T cell therapy was administered 7 days after WM infusion. Mice were monitored daily and assessed by weekly bioluminescent imaging and ultimately were sacrificed when they developed hind leg paralysis. All in vivo experiments were performed twice with independent donors.
Clinical protocols
Patients with relapsed or refractory WM were considered for one of two phase I clinical trials, NCT00466531 or NCT03085173, each investigating the safety of autologous T cells genetically modified to express a CAR targeted against CD19 antigen. In NCT00466531, patients with WM were eligible if they had relapsed or chemotherapy-refractory disease; in NCT03085173, patients were eligible if they had disease refractory to or relapsed following at least two lines of chemoimmunotherapy (including at least one course of anti-CD20 antibody) or one prior biologic agent. In both studies patients were required to have measurable disease via demonstrable monoclonal paraprotein and bone marrow involvement.
Bridging therapy was permitted and patients received investigator’s choice of conditioning chemotherapy followed by CAR T cell infusion. Cytokine and CAR T cell persistence were monitored following CAR T infusion per online supplemental methods.
jitc-2021-004128supp001.pdf (92.6KB, pdf)
Protocol NCT00466531
Two patients with WM (Patients 1 and 2) were enrolled in stage 3 of clinical trial NCT00466531, a phase I, single-center study designed to evaluate the safety of autologous T cells genetically modified to express a CAR targeting CD19 antigen (19-28ζ) with or without conditioning chemotherapy in patients with chronic lymphocytic leukemia (CLL) or indolent B cell lymphoma (online supplemental file 6). Data on the CLL cohort has previously been published.12 Bridging therapy was permitted. For patients on ibrutinib therapy prior to CAR T cell infusion, ongoing ibrutinib therapy was permitted. Ibrutinib was held immediately prior to CAR T infusion, and resumed after resolution of acute toxicities. Patients were treated with the investigator’s choice of conditioning chemotherapy, followed by administration of modified T cells. Cells were administered per a dose escalation scheme with 1×106 CAR T cells/kg at level 1, 3×106 at level 2, and 1×107 at level 3. Toxicity, including cytokine release syndrome and neurotoxicity, was graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), V.4.0.
jitc-2021-004128supp006.pdf (76.8KB, pdf)
Protocol NCT03085173
One patient with WM (Patient 3) was enrolled in a separate Phase 1 clinical trial (NCT03085173) investigating CD19-targeted EGFRt/19-28ζ/4-1BBL ‘armored’ CAR-modified T cells in patients with relapsed or refractory CD19 positive hematologic malignancies (online supplemental file 7). The data for this study were previously reported in abstract form.13 After clinician’s choice of conditioning therapy, patients received treatment per a dose escalation scheme with 1×105 CAR T cells/kg at level 1, 3×105 at level 2, and 1×106 at level 3. Toxicity, including neurotoxicity, was graded according to the NCI CTCAE, V.4.0; cytokine release syndrome (CRS) toxicity was graded according to Memorial Sloan Kettering Cancer Center criteria, as previously published.14
jitc-2021-004128supp007.pdf (62.2KB, pdf)
Results
19-28ζ CAR T cells have antitumor function in vitro
We first assessed whether CD19-targeted CAR T cells could lyse the BCWM.1 human WM line in vitro. We co-cultured BCWM.1 human WM cell line expressing firefly luciferase (BCWM.1-Luc) with donor lymphocytes genetically modified to express either a 19-28ζ CAR, an irrelevantly targeted MUC16-28ζ CAR, or left untransduced. After a 4-hour co-culture, we observed significant cytotoxicity by bioluminescence, even at low effector:target ratios (52% lysis at 1:1; 92% lysis at 10:1; figure 1A). IFN-γ and IL-2 were significantly higher in the supernatant from the 19-28ζ co-cultures at 24 hours compared with the controls (figure 1B).
Figure 1.
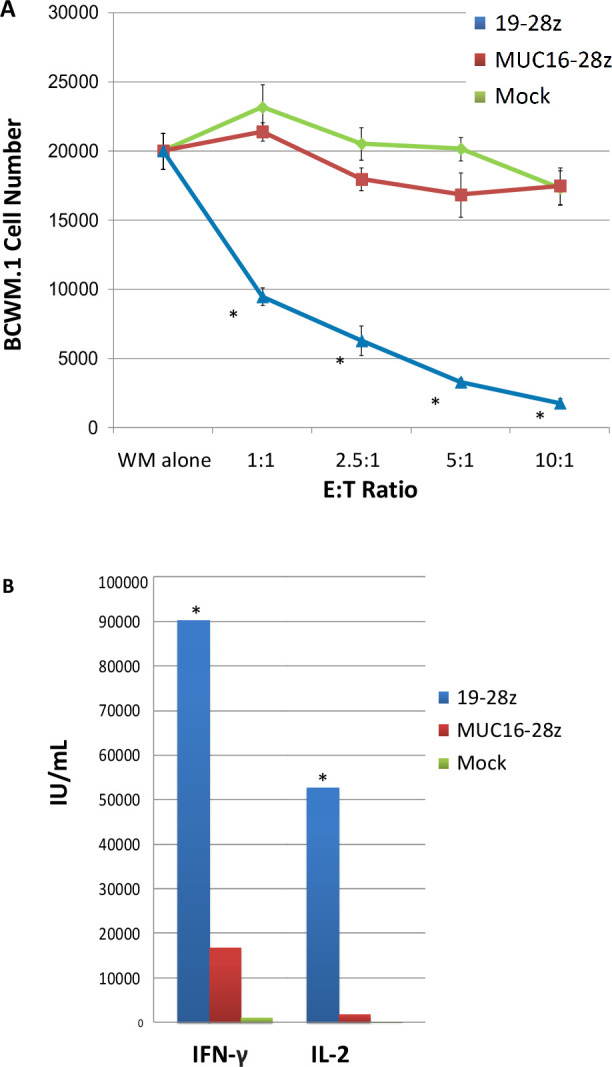
CD19-targeted CAR T cells have anti-WM activity in vitro. (A) Lysis of BCWM.1 human WM cell line after 4-hour co-culture with CAR T cells at indicated E:T ratio. (B) Effector cytokines detected in supernatant after 24 hours co-culture of CAR T cells with BCWM.1 WM cells; 1:1 CAR+:WM ratio. All in vitro studies were performed twice with independent donors. *p<0.05. CAR, chimeric antigen receptor; IFN, interferon; IL, interleukin; WM, Waldenström macroglobulinemia.
19-28ζ CAR T cells have antitumor function in vivo
To test whether CD19-targeted CAR T cell therapy would be effective in vivo, we generated a systemic model of WM by injecting BCMW.1-Luc cells intravenously into sublethally irradiated SCID/beige mice. Treatment with 19-28ζ CAR modified T cells significantly extended the survival of these mice (figure 2A, B). Of note, although BCWM.1 is a CD19-positive cell line, in two out of three 19-28ζ CAR treated mice assessed at relapse by immunohistochemistry (IHC), we found human-CD19 negative antigen loss, while none of the control treated mice evaluated by IHC had evidence of hCD19 antigen loss (figure 2C).
Figure 2.

CD19-targeted CAR T cells in a systemic WM in vivo model demonstrate initial efficacy followed by antigen escape mediated relapse. (A) Survival curve of SCID/beige mice after sublethal irradiation and engraftment of GFP/Luc+BCWM0.1 cells (day 0) and CAR +T cell treatment (day 7). Pooled data from two separate experiments. (B) Representative serial bioluminescent imaging over time. (C) hCD19 immunohistochemistry status of relapsed/residual tumor at time of humane endpoint. CAR, chimeric antigen receptor; WM, Waldenström macroglobulinemia.
Clinical trial results
Patients
Three patients with WM were treated with CAR T therapy, two in protocol NCT00466531 and one in protocol NCT03085173. Baseline characteristics are summarized in table 1. All patients were female, and ages ranged from 64 to 75 at time of CAR T cell infusion. All patients had relapsed or refractory WM, had been diagnosed at least 10 years before CAR T therapy (range 10–12 years), and had received at least five prior lines of treatment (range 5–7), including chemoimmunotherapy with an anti-CD20 monoclonal antibody, ibrutinib, and bortezomib. Manifestations of WM requiring treatment at time of
Table 1.
Baseline demographics, disease characteristics, treatment, clinical outcomes, and toxicities for three patients with relapsed/refractory Waldenström macroglobulinemia treated with CAR Tcell therapy
| Baseline patient characteristics | Patient 1 | Patient 2 | Patient 3 |
| Age, gender | 73, female | 75, female | 64, female |
| Genotype | MYD88 p.L265P CXCR4 p.S342fs TP53 p.R175H/R196Q |
MYD88 p.L265P Loss of MYB and ATM by FISH |
MYD88 p.L265P DNMT3A p.R823K |
| Time from diagnosis to CAR T | 12.1 years | 10.4 years | 10.2 years |
| Prior lines of therapy (best response by IWWM criteria) | Rituximab (PR) Rituximab+Fludarabine (PR) Rituximab+Bendamustine (VGPR) Duvelisib (NE) Ibrutinib (PD) BDR ->R-Bortezomib maint (MR) IMO-8400 (TLR antagonist) (NE) Ofatumumab+Bendamustine (MR) |
Rituximab (PR) Rituximab+Bendamustine (PR) BDR (PR) Venetoclax (MR) Ibrutinib (PR) Ibrutinib/Ofatumumab (PD) |
Rituximab+Bortezomib (PR) Rituximab+Bendamustine (PR) Ibrutinib (PD) Ofatumumab+Cyclophosphamide + Fludarabine (CR, MRD +on BM) Ofatumumab (PD) |
| Marrow involvement | Hypercellular marrow (80%) LPL involvement (50%) |
Hypercellular marrow (70%) LPL involvement (40%–60%) |
Hypercellular marrow (95%) LPL involvement (70%–80%) |
| Transfusion dependence | Pancytopenic, requiring PRBC and platelet transfusions multiple times per week | Anemia requiring transfusions | No |
| Pretreatment IgM | 2570 mg/dL | 3789 mg/dL | 7520 mg/dL |
| Treatment | |||
| CAR-T product | CD19-CD28-CD3ζ (19-28z) | CD19-CD28-CD3ζ (19-28z) | CD19-CD28-CD3ζ (19-28z) plus 4-1BBL (armored CAR product) |
| Bridging therapy | Bendamustine | Ibrutinib | Ofatumumab |
| Conditioning | Cyclophosphamide 1 g/m2 (day −3), Fludarabine 25 mg/m2 (day −3 to −1) |
Cyclophosphamide 1 g/m2 (day −3) Fludarabine 25 mg/m2 (day −3 to −1) |
Cyclophosphamide 300 mg/m2 (day −3 to −1) Fludarabine 30 mg/m2 (day −3 to −1) |
| CAR T cell dose | 1×106 CAR T cells/kg | 1×106 CAR T cells/kg | 1×105 CAR T cells/kg |
| Response | |||
| Best response | Stable disease by IgM Hematologic response |
PR | VGPR |
| Vital status | Deceased | Deceased | Alive |
| Cause of death | Disease | Sepsis 2/2 PsA bacteremia | N/A |
| PFS (months) | 6.8 | 4.0 | 26.5 |
| OS (months) | 12.4 | 4.0 | Not reached |
| Toxicity | |||
| CRS | Grade 1 | Grade 2 | Grade 2 |
| Neurotoxicity | None | None | Grade 2 |
| Infections | None | None | None |
| Other | Grade 2 peripheral edema Grade 2 nasal congestion Grade 2 anemia (baseline) Grade 1 rash (pityriasis rosacea) |
Grade 1 edema Grade 1 alopecia Grade 1 pleural effusion Grade 2 hypothyroidism |
Grade 2 dyspnea/wheezing Grade 2 elevated bicarbonate Grade 2 anemia Grade 1 diarrhea Grade 1 nausea |
BDR, bortezomib, dexamethasone, rituximab; CAR, chimeric antigen receptor; CR, complete response; CRS, cytokine release syndrome; IHC, immunohistochemistry; IWWM, International Workshop on Waldenström Macroglobulinemia; LPL, lymphoplasmacytic lymphoma; MRD, minimal residual disease; NE, response not evaluable; OS, overall survival; PFS, progression-free survival; PR, partial response; PRBC, packed red blood cells; PsA, pseudomonas aeruginosa; TLR, toll-like receptor; VGPR, very good partial response.
CAR T therapy included transfusion-dependent anemia and/or thrombocytopenia in two patients, and symptomatic hyperviscosity requiring plasmapheresis in the third. Patients 1 and 2 had lymphadenopathy on baseline CT scans prior to treatment, while Patient 3 had no evidence of extramedullary disease. All three patients had baseline marrow evaluations prior to CAR T therapy demonstrating hypercellular marrow and at least 50% marrow involvement by LPL. All patients had molecular analysis demonstrating MYD88L265P mutations. Genetic analysis demonstrated additional genetic alterations, including, for Patient 1, CXCR4S342fs and TP53R175H/R196Q; and for Patient 2, FISH demonstrating loss of MYB, ATM, and del13q.
Treatment
All patients received bridging therapy with bendamustine (Patient 1), ibrutinib (Patient 2), or ofatumumab (Patient 3). All received cyclophosphamide/fludarabine lymphodepleting therapy. Two were treated with second-generation anti-CD19 CAR T cells with a CD28ζ costimulatory domain (Patients 1 and 2, NCT00466531). The third (Patient 3, NCT03085173) was treated with ‘armored’ 19-28ζ/4-1BBL CAR T cells expressing both CD28ζ and 4-1BB ligand (4-1BBL) costimulatory domains. Respective doses of CAR T cells are reflected in table 1. Patient 1 was re-treated with a second dose of the same CAR T cell product after developing clinical progression of disease, 7 months after first CAR T infusion. Of note, Patient 2 had been on ibrutinib for 3 years prior to CAR T therapy, and had documented serologic and radiologic progression of disease on full dose ibrutinib which prompted CAR T therapy. However, it was felt that ibrutinib may be slowing disease progression, so it was continued as bridging therapy prior to CAR T therapy, and was resumed on day 20 after CAR T infusion, which was permitted per protocol.
Safety
No grade 3 or higher events were recorded in any patients. Grade 1–2 CRS was reported in all patients; none required treatment with steroids or tocilizumab for CRS. Onset of CRS was 11 hours – 11 days after CAR T infusion. Patient 3 had grade 2 neurotoxicity on day 7 after CAR T, characterized by lethargy and tremor, for which she received one dose of 10 mg intravenous dexamethasone and was transferred to an intensive care unit for monitoring. Neurotoxicity completely resolved within 24 hours of onset, and the patient did not require intubation. The other two patients had no neurotoxicity. Other toxicities are shown in table 1. Peak levels of C reactive protein, IL-2, IL-6, IL-10, tumor necrosis factor-α, and IFN-γ occurred 2–17 days after therapy, and were similar to with regards to timing and peak levels to similar CAR T studies performed in B cell lymphoma, CLL, and multiple myeloma (figure 3, online supplemental figure 1).12 15 16
Figure 3.

Clinical responses and inflammatory marker profiles following CAR T therapy. (A) Serum IgM, hemoglobin, and platelet measurements over time. (B) Levels of CRP, IL-6, and IL-10 after CAR T infusion on day 0. CAR, chimeric antigen receptor; CRP, C reactive protein; IL, interleukin.
jitc-2021-004128supp002.pdf (62.9KB, pdf)
Clinical responses
Two patients had a response to treatment by International Workshop on Waldenström Macroglobulinemia criteria, including one with complete response and minimal residual disease (MRD)-negative marrow by molecular analysis, one partial response, and one stable disease by IgM with prolonged hematologic response. Responses and duration are summarized in table 21.
Patient 1 had stable disease based on IgM; however, she had a 7-month-long hematologic response based on increased hemoglobin and platelet values (figure 3), allowing for transfusion independence and an associated increase in performance status during this time. Platelets increased from median 32
(range, 14–56) to median 93 (range, 73–125) in the 6 months after discharge. Hemoglobin increased from median 80 g/L (range, 60.0–90.9) pre-CAR T to 10.6 g/dL (range, 8.6–12.6) after discharge. Repeat marrow at day 11 post-CAR T therapy showed improvement in marrow involvement by LPL cells from 50% to 5%–10% of cellularity. She had a radiologic complete response with resolution of lymphadenopathy on repeat CT scan 6 months after CAR T therapy. She had progression of disease 7 months after CAR T therapy, as evidenced by recurrence of anemia and thrombocytopenia, and an associated increase in marrow involvement by LPL from 20% to 60%. She was re-treated with 1×106 CAR T cells/kg shortly after her progression. While an interval improvement in LPL involvement in marrow was noted from 60% to 25%, she did not have significant improvement in cytopenias or IgM level. She died shortly thereafter due to progression of disease.
Patient 2 had a partial response to treatment based on 77% reduction in serum IgM levels and improvement in serum platelet and hemoglobin levels (figure 3). Bone marrow evaluation 22 days post-CAR T therapy showed improvement from 70% LPL involvement by cellularity to less than 5%. However, a CT scan after 3 months showed worsening lymphadenopathy suggesting extramedullary progression of disease. She ultimately died from a documented Influenza A and Pseudomonas aeruginosa bacteremia 4 months after CAR T cell therapy. Prior to infection, her cell counts, including absolute neutrophil count, had recovered to normal levels, though she had ongoing hypogammaglobulinemia post-CAR T.
Patient 3 achieved a complete response with MRD-negative marrow by flow and cytogenetics, and immunofixation was negative following CAR T therapy. Notably while very good partial response (VGPR) was achieved within 6 weeks of treatment, she did not achieve molecular MRD negative status until day 350 post-CAR T. She had recurrence 26 months after CAR T cell infusion based on an increase in serum IgM, new splenomegaly on CT scan, and bone marrow demonstrating hypercellularity with 80% involvement by LPL (online supplemental figure 2). She was subsequently treated with bortezomib-based therapy followed by acalabrutinib, and is alive with ongoing VGPR as of August 13, 2021.
jitc-2021-004128supp003.pdf (646.9KB, pdf)
CAR T cell expansion and persistence
Peak expansion of CAR T cells occurred 12 and 30 days following initial CAR T cell infusion for Patients 1 and 2 receiving 19-28ζ CAR T cells, and 11 days post-infusion for Patient 3 who received 19-28ζ/4-1BBL CAR T cells (online supplemental figure 3). Different assays were used between patients, precluding direct comparison of CAR T expansion data. CAR T cells were detectable by either PCR or flow cytometry up to 210, 79, and 343 days following infusion in Patients 1, 2, and 3, respectively. Patient 1 received a second infusion 215 days after first infusion; CAR T cells were detectable 14 days after this infusion but not at day 21 or at later time points.
jitc-2021-004128supp004.pdf (91KB, pdf)
Disease characteristics at relapse
We retrospectively assessed bone marrow B cell and plasma cell content, along with CD19 expression, before CAR T therapy, within 1–4 weeks after CAR T administration, and at time of disease progression (online supplemental table 1). In all patients CD19 expression by malignant B cells was appreciated prior to therapy. CD19 expression on malignant B cells was also present in all three patients at time of disease progression. In Patient 1, there was a decrease in B cell CD19 expression by mean fluorescence intensity (MFI) at time of progression after CAR T therapy, which may suggest that antigen escape contributed to resistance. Direct quantification of changes in CD19 expression for Patient 3 is limited due to differences in fluorochromes used for the assays between samples.
jitc-2021-004128supp005.pdf (43KB, pdf)
Discussion
In this report, we demonstrate in vitro and in vivo evidence of anti-WM activity in second-generation anti-CD19 CAR T cells, along with the first series of three patients treated with CAR T products, demonstrating early signs of safety and clinical activity. Clinically, treatment was well-tolerated with only grade 1–2 toxicities. Responses were seen in all three patients, ranging from stable disease with hematologic response to a prolonged complete response. However, subsequent progression of disease was seen in all patients. Similarly encouraging results have been reported in one other patient who was treated with CAR T therapy for a large cell transformation of WM, and had an ongoing CR from both large cell transformation and indolent WM at 1 year post-CAR T infusion.17
Our preclinical and clinical findings suggest that CAR T resistance by may be due in part to decreased CD19 expression, though this does not account for treatment failure in all cases. Our preclinical models demonstrated selective proliferation of CD19-negative, plasma cell-like LPL clones as a mechanism of resistance to CAR T therapy. One of three patients had a decline in CD19 expression at time of disease progression, which may suggest that antigen escape contributed to CAR T resistance; however, no significant change was noted in the other two patients, so other mechanisms likely also contribute. Further studies in larger cohorts evaluating antigen escape and other mechanisms, such as CAR T exhaustion, will be necessary to fully understand CAR T resistance in WM. There is little data on CD19 antigen density on WM as compared with other B cell malignancies; one study suggests a possible decrease in expression compared with marginal zone lymphoma.18 Should CD19 loss or plasmacytic differentiation arise as a significant escape mechanism of resistance in WM, a potential solution would be the employment of dual-targeted CAR T cells directed against both CD19 and a plasma cell antigen such as CD38, CD138 or BCMA.19–22
Of note, all patients in this study were heavily pretreated with at least five lines of prior therapy, including immunochemotherapy with rituximab–bendamustine. Research in other malignancies suggests that heavy pretreatment may negatively impact the patients’ T cells and the resulting CAR T product, raising questions over whether earlier treatment may allow for greater CAR T efficacy.23 However, given the availability of effective and less toxic therapies such as BTK inhibitors and proteasome inhibitors, the role for CAR T therapy in the early phases of treatment is likely limited until further studies in the relapsed/refractory setting evaluating efficacy and safety in larger numbers can be performed.2
One patient received ibrutinib before and after treatment with CAR T therapy, as permitted in protocol. Recent preclinical studies suggest ibrutinib may improve the antitumor activity of CAR T cells, and the combination of ibrutinib with CAR T therapy in CLL was well tolerated in one study.24 25 While sample sizes are too small to draw conclusions regarding efficacy of combination ibrutinib and CAR T therapy in WM, this would be a reasonable approach in future studies, particularly if patients are still deriving some degree of benefit from BTK inhibitors.
In all three patients, treatments were on clinical trial protocols and using CAR T products which are not commercially available. Patients 1 and 2 were treated with a second-generation CAR T product with CD28ζ costimulatory domain, similar to that seen in axicabtagene ciloleucel.14 Patient 3 was treated with an ‘armored’ CAR T product using a CD28ζ costimulatory domain, along with constitutive 4-1BBL expression, providing an additional costimulatory element. Results with this product were reported previously, though no such product is commercially available at this time.13
Overall, this study demonstrates feasibility, safety, and efficacy of anti-CD19-directed CAR T in a small population of patients with heavily pretreated WM. Further studies in larger cohorts of highly selected patients with WM should be pursued.
Footnotes
Contributors: MLP was an investigator on the described clinical trials, oversaw care of all described patients, and collected and interpreted data. DQ collected and interpreted data, and wrote manuscript. SM, SS, AD and MR performed the pathologic analysis of marrow and biopsy samples and assessment of mechanisms of resistance to therapy. BS, XW, IR, MS, RJB, and ELS designed and developed the chimeric antigen receptor T cells used in the two described clinical trials. BS, MS, RJB, and ELS performed the in vitro and in vivo studies. JHP designed and was principal investigator for both described clinical trials. All authors participated in the drafting and review of the manuscript.
Funding: ELS acknowledges NIH (K08CA241400-01) support. RJB acknowledges NIH (R01 CA138738-05), the Annual Terry Fox Run for Cancer Research organized by the Canada Club of New York, Kate’s Team, the Carson Family Charitable Trust, the William Lawrence and Blanche Hughes Foundation, the Emerald Foundation, and the Experimental Therapeutics Center of MSKCC. All investigators acknowledge the MSKCC Core Facilities Grants (P30 CA008748 and U54 OD020355-01).
Competing interests: MLP has received consulting fees from BeiGene, Novartis and Synthekine. An immediate family member of MLP is a Board Member with DKMS, has IP rights with Juno Therapeutics and Seres Therapeutics, and receives consulting fees from Rheos Medicines, Ceramedix, GKS, Priotera, and WindMIL Therapeutics. MR has received consulting fees from Celgene and Physicians’ Education Resource, and consulting fees plus equity interest in Auron Therapeutics. IR has received consulting fees and funding support from Juno Therapeutics, in which she had an equity interest. RJB has licensed intellectual property to and collect royalties from BMS, Caribou and Sanofi. RJB received research funding from BMS. RJB is a consultant to BMS and was a consultant for Gracell Biotechnologies but ended employment in the past 24 months. JHP received consulting fees from Amgen, Allogene, Artiva Biotherapeutics, Autolus, BMS, Curocell, GSK, Incyte, Innate Pharma, Intellia Therapeutics, Kite Pharma, Kura Oncology, Minerva Biotechnologies, Novartis, Pfizer and Servier, and serves as a DSMB member for Affyimmune and Bright Pharmaceuticals. ELS received IP or royalties from BMS, Sanofi, and is a consultant to BMS, Novartis, and Chimeric Therapeutics. DQ, SM, SS, AD, BS, XW, and MS have no conflicts of interest to disclose.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request. Please contact the corresponding author regarding any information requests.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study is a retrospective review including three patients who were treated on clinical trials. These studies were approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board, ID numbers 06-138 and 16-1570. Participants gave informed consent to participate in the study before taking part.
References
- 1. Swerdlow SH, Campo E, Harris NL. WHO classification of tumours of haematopoietic and lymphoid tissues. revised 4t. Lyon, France: IARC Press, International Agency for Research on Cancer, 2017. [Google Scholar]
- 2. Castillo JJ, Advani RH, Branagan AR, et al. Consensus treatment recommendations from the tenth International workshop for Waldenström macroglobulinaemia. Lancet Haematol 2020;7:e827–37. 10.1016/S2352-3026(20)30224-6 [DOI] [PubMed] [Google Scholar]
- 3. Dalal NH, Dores GM, Curtis RE, et al. Cause-specific mortality in individuals with lymphoplasmacytic lymphoma/Waldenström macroglobulinaemia, 2000-2016. Br J Haematol 2020;189:1107–18. 10.1111/bjh.16492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439–48. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2020;382:1331–42. 10.1056/NEJMoa1914347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020;396:839–52. 10.1016/S0140-6736(20)31366-0 [DOI] [PubMed] [Google Scholar]
- 7. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol 2019;20:31–42. 10.1016/S1470-2045(18)30864-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siddiqi T, Soumerai JD, Dorritie KA, et al. Updated follow-up of patients with relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma treated with lisocabtagene maraleucel in the phase 1 monotherapy cohort of TRANSCEND CLL 004, including high-risk and ibrutinib-treated patients. Blood 2020;136:40–1. 10.1182/blood-2020-140491 [DOI] [Google Scholar]
- 9. Jacobson C, Chavez JC, Sehgal AR, et al. Primary analysis of Zuma-5: a phase 2 study of Axicabtagene Ciloleucel (Axi-Cel) in patients with relapsed/refractory (R/R) indolent non-Hodgkin lymphoma (iNHL). Blood 2020;136:40–1. 10.1182/blood-2020-136834 [DOI] [Google Scholar]
- 10. Smith EL, Staehr M, Masakayan R, et al. Development and evaluation of an optimal human single-chain variable fragment-derived BCMA-Targeted CAR T cell vector. Mol Ther 2018;26:1447–56. 10.1016/j.ymthe.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ditzel Santos D, Ho AW, Tournilhac O, et al. Establishment of BCWM.1 cell line for Waldenström's macroglobulinemia with productive in vivo engraftment in SCID-hu mice. Exp Hematol 2007;35:1366–75. 10.1016/j.exphem.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 12. Geyer MB, Rivière I, Sénéchal B, et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI Insight 2019;4. 10.1172/jci.insight.122627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park JH, Palomba ML, Batlevi CL, et al. A Phase I First-in-Human Clinical Trial of CD19-Targeted 19-28z/4-1BBL "Armored" CAR T Cells in Patients with Relapsed or Refractory NHL and CLL Including Richter’s Transformation. Blood 2018;132:224. 10.1182/blood-2018-99-117737 [DOI] [Google Scholar]
- 14. Park JH, Rivière I, Gonen M, et al. Long-Term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med 2018;378:449–59. 10.1056/NEJMoa1709919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther 2017;25:285–95. 10.1016/j.ymthe.2016.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med 2019;380:1726–37. 10.1056/NEJMoa1817226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bansal R, Jurcic JG, Sawas A, et al. Chimeric antigen receptor T cells for treatment of transformed Waldenström macroglobulinemia. Leuk Lymphoma 2020;61:465–8. 10.1080/10428194.2019.1665668 [DOI] [PubMed] [Google Scholar]
- 18. Ocio EM, Hernández JM, Mateo G, et al. Immunophenotypic and cytogenetic comparison of Waldenstrom's macroglobulinemia with splenic marginal zone lymphoma. Clin Lymphoma 2005;5:241–5. 10.3816/CLM.2005.n.007 [DOI] [PubMed] [Google Scholar]
- 19. Munshi NC, Anderson LD, Shah N, Larry Anderson D D, et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 2021;384:705–16. 10.1056/NEJMoa2024850 [DOI] [PubMed] [Google Scholar]
- 20. Fernández de Larrea C, Staehr M, Lopez AV, et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA Escape-Driven relapse in multiple myeloma. Blood Cancer Discov 2020;1:146–54. 10.1158/2643-3230.BCD-20-0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah NN, Johnson BD, Schneider D, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med 2020;26:1569–75. 10.1038/s41591-020-1081-3 [DOI] [PubMed] [Google Scholar]
- 22. Osborne W, Marzolini M, Tholouli E, et al. Phase I Alexander study of AUTO3, the first CD19/22 dual targeting CAR T cell therapy, with pembrolizumab in patients with relapsed/refractory (r/r) DLBCL. JCO 2020;38:8001. 10.1200/JCO.2020.38.15_suppl.8001 [DOI] [Google Scholar]
- 23. Das RK, Vernau L, Grupp SA, et al. Naïve T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discov 2019;9:492–9. 10.1158/2159-8290.CD-18-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruella M, Kenderian SS, Shestova O, et al. The addition of the Btk inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res 2016;22:2684–96. 10.1158/1078-0432.CCR-15-1527 [DOI] [PubMed] [Google Scholar]
- 25. Gauthier J, Hirayama AV, Purushe J, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020;135:1650–60. 10.1182/blood.2019002936 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-004128supp001.pdf (92.6KB, pdf)
jitc-2021-004128supp006.pdf (76.8KB, pdf)
jitc-2021-004128supp007.pdf (62.2KB, pdf)
jitc-2021-004128supp002.pdf (62.9KB, pdf)
jitc-2021-004128supp003.pdf (646.9KB, pdf)
jitc-2021-004128supp004.pdf (91KB, pdf)
jitc-2021-004128supp005.pdf (43KB, pdf)
Data Availability Statement
Data are available upon reasonable request. Please contact the corresponding author regarding any information requests.