

Abstract
Eosinophils are potent innate effector cells associated mainly with type 2 immune responses elicited by helminths and allergens. Their activity needs to be tightly controlled in order to prevent severe inflammation and tissue damage. Eosinophil degranulation and secretion of inflammatory effector molecules including cytokines, chemokines and lipid mediators can be regulated by activating and inhibitory receptors on the cell surface. Here, we investigated the modulation of proliferation, apoptosis, gene expression and cytokine/chemokine secretion from IL-33-activated Mus musculus eosinophils upon cross-linking of the transmembrane receptor Sialic acid-binding immunoglobulin-like lectin F (Siglec-F). Siglec-F contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) plus an ITIM-like motif in its intracellular tail and is mainly regarded as an inhibitory and apoptosis-inducing receptor. In vitro co-stimulation of bone marrow-derived eosinophils with anti-Siglec-F and IL-33 compared to treatment with either alone lead to enhanced STAT6 phosphorylation, stronger induction of hypoxia/glycolysis related pro-inflammatory genes, and elevated secretion of type 2 cytokines (IL-4, IL-13) and chemokines (CCL3, CCL4) with only minor effects on proliferation and apoptosis. Using a competitive mixed bone marrow chimera approach with wild-type and Siglec-F-deficient eosinophils we observed no evidence for Siglec-F-regulated inhibition of Aspergillus fumigatus-elicited lung eosinophilia. Truncation of the Siglec-F cytoplasmic tail but not mutation of the ITIM and ITIM like motifs ablated the effect of enhanced cytokine/chemokine secretion. This provides evidence for an ITIM phosphorylation independent signaling pathway from the cytoplasmic tail of the Siglec-F receptor that enhances effector molecule release from activated eosinophils.
Introduction
Sialic acid-binding immunoglobulin-like lectin F (Siglec-F) is a surface receptor that belongs to the CD33 family of immunoglobulin-like lectins and is expressed on murine eosinophils, alveolar macrophages and a few other cell types (1–3). Siglec-F and its human paralog Siglec-8 contain an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an ITIM-like motif in their cytoplasmic tail region (1, 2). Both receptors are able to induce apoptosis in eosinophils (4–6), while mechanistic differences were reported (7, 8). Studies with Siglec mutants revealed that the ITIMs play a critical role in the recruitment of the tyrosine phosphatases SHP-1 and SHP-2 that are able to inhibit activating signaling events (9–12). Siglec-F and Siglec-8 both bind to 6’-sulfo sialyl LewisX which contains terminal sialic acids bound via α2,3 linkage to galactose-6-sulfate in glycoproteins (1). Although the mucins Muc5b and Muc4 were described to bind to Siglec-F (13) the identification and characterization of in vivo sialic acid ligands is still ongoing (13–15). It has been described that after ligand binding Siglec-F limits eosinophil-mediated tissue damage during allergic inflammation by induction of apoptosis (4). However, this initial finding seems to be dependent on the experimental model. For example, Siglec-F-deficient mice showed enhanced lung eosinophilia in ovalbumin-induced lung inflammation when the dissolved antigen was given intranasally, while this effect was not observed with aerosolized ovalbumin, despite similar expression of Siglec-F ligands in both models (16). The mechanism of Siglec-F-induced apoptosis appears to be caspase-dependent but relatively inefficient (8). Recent studies even describe Siglec-F-dependent enhanced effector molecule production. As such, Siglec-F knock-down on bone marrow derived macrophages, that start to express Siglec-F after GM-CSF stimulation, leads to a reduced IL-4-induced phosphorylation of STAT6 and arginase-1 expression (17). In addition, combined stimulation of eosinophils with the alarmin IL-33 and anti-Siglec-F antibody caused a synergistically enhanced release of IL-4 and IL-13 compared to stimulation by either IL-33 or anti-Siglec-F treatment alone (18). This indicates that Siglec-F can act as an activating receptor besides its described pro-apoptotic function.
In this study, we analyzed the Siglec-F-mediated modulation of IL-33-induced chemokine and cytokine secretion from eosinophils. In vitro stimulation of bone-marrow-derived eosinophils with IL-33 induced Siglec-F upregulation and progressive secretion of IL-4, IL-13, MIP1-α/CCL3 and MIP1-β/CCL4 over several hours. IL-33 further impaired the proliferation of developing eosinophils in culture and inhibited the accumulation of eosinophils in the lung during Aspergillus fumigatus-induced allergic lung inflammation. Receptor cross-linking with anti-Siglec-F during IL-33 stimulation in vitro had little effect on viability, proliferation and expression of activation markers but strongly enhanced cytokine/chemokine secretion. Interestingly, the IL-4/IL-13-activated transcription factor STAT6 was required for IL-33-induced secretion of IL-4, IL-13, CCL3 and CCL4 but anti-Siglec-F treatment could partially compensate for the STAT6 deficiency. Using retroviral reconstitution experiments, we demonstrate that the cytoplasmic tail but not the tyrosine residues in the ITIM and ITIM-like motifs is critical for the enhanced effector release upon anti-Siglec-F cross-linking.
Material and Methods
Mice and bone marrow chimeras
The following strains of mice were used on C57BL/6 background: IL-4eGFP reporter (4get_B6) (19), 4get_IL-5tg (20), IL-4/IL-13−/− (21), ST2−/− (22), MyD88−/− (23), and CD45.1_B6-congenic mice; or on BALB/c background: 4get_BALB/c, 4get_STAT6−/− (24), and CD45.1_BALB/c-congenic mice. To generate mixed bone marrow chimeras (MBMC), mice were irradiated with 1100 rad and 50:50 mixtures of either ST2−/− and CD45.1-congenic or Siglec-F−/− and CD45.1-congenic bone marrow cells were transferred by tail vein injection. MBMC were kept with antibiotics in drinking water (2 g/L neomycin sulfate and 100 mg/L polymyxin B sulfate in drinking water; MilliporeSigma, Burlington, MA) for 8 weeks after reconstitution and were analyzed after 8 to 10 weeks. Mice were kept under specific pathogen free conditions and maintained in the Franz-Penzoldt Center in Erlangen. All experiments were performed in accordance with German animal protection law and European Union guidelines 86/809 and were approved by the Federal Government of Lower Franconia.
Cell culture
Bone marrow derived eosinophils (BMDEs) were generated as described before (25). In short, single cell suspensions of bone marrow from femur and tibia were cultured in eosinophil culture medium (ECM; RPMI complemented with 20% (v/v) fetal bovine serum, 25 mM Hepes, 55 μM β-mercaptoethanol, 2 mM L-glutamine, 10 mM non-essential amino acids, 1 mM sodium pyruvate, 100 μg/mL streptomycin, 100 U/mL penicillin) at a density of 1×106 cell/mL. For the first four days, 100 ng/mL SCF and 100 ng/mL FLT3-L (both Peprotech) were added to the culture and then changed to 10 ng/mL IL-5 (R&D Systems) for further ten days with media change every other day. Eosinophils were cultured in the presence of IL-5 to support their survival. At day 14 bone marrow derived eosinophils were analyzed by flow cytometry (Siglec-F+, high side scatter, purity usually above 90%). For ELISA and flow cytometry analysis if not differently indicated, cells were seeded at a density of 0.2×106 cells/mL and stimulated with 5 μg/mL purified rat anti-mouse Siglec-F antibody (clone E50–2440) or purified rat IgG2a isotype control (BD Pharmingen or Invitrogen) and/or 10 ng/mL recombinant murine IL-33 (R&D Systems) for 24 hrs. For Western blot, cells were seeded at a density of 1×106 cells/mL and stimulated with 1 μg/mL of anti-Siglec-F or isotype control antibody and 10 ng/mL IL-33.
Isolation and stimulation of human eosinophils from peripheral blood
Isolation of human eosinophils was performed on freshly isolated peripheral blood of healthy donors with informed consent and approval by the Ethics committee of the Faculty of Medicine at the Friedrich-Alexander Universität Erlangen-Nürnberg (FAU) (no. 224_14B). About 50 mL whole blood was collected in an EDTA containing tube, diluted 1:1 with PBS and subjected to density gradient centrifugation with BioColl (1.077 g/mL; Bio&SELL) at 1000g for 30 min at room temperature. Supernatant was discarded and the granulocyte-erythrocyte pellet was subjected twice to red blood cell lysis (15 min and 10 min) in hypotonic lysis buffer (155 mM NH4CL, 10 mM KHCO3 in H2O) on ice. Eosinophils were further separated from other granulocytes by magnet bead negative separation using the human Eosinophil isolation Kit (Miltenyi Biotec) according to manufacturer’s instructions. Purity of the isolated eosinophils was determined directly after purification by flow cytometry defining eosinophils as CCR3+Siglec-8+ singlets pre-gated on viable cells. Purity was at least above 88% and on average 93% (Suppl. Fig. 2A). Stimulation experiments for activation marker flow cytometry and qRT-PCR cells were performed in ECM medium supplemented with 10 ng/mL recombinant human IL-5 (R&D Systems). 0.2×106 cells per well in 200 μL were stimulated for 24 hrs with 5 μg/ml anti-Siglec-8 or isotype control antibody. 10 ng/mL recombinant human IL-33 were optionally added.
Luminex assay
Supernatants of IL-33 stimulated or unstimulated control BMDEs were collected over a kinetic from 30 min to 16 hrs and directly subjected to the ProcartaPlex Mouse Cytokine and Chemokine Panel 1A (36-plex) kit (eBioscience/ Thermo Fisher Scientific). The assay was performed according to the manufacturer’s protocol and measured on a Bio-Rad Bio-Plex MAGPIX Multiplex Reader instrument.
Retroviral transduction
Retroviral supernatants were generated in Phoenix or Plat-E cultures transfected with Siglec-F mutant sequences in a MSCV-IRES-Thy1.1 backbone and packaging helper plasmid pCLEco (Biocarta Europe GmbH). Siglec-F−/− cells were transduced with retroviral supernatant in six well plates at day 2 and 3 of BMDE generation. At day 13 Thy1.1+ cells were sorted. At day 14 0.85×106 cells were seeded in 100 μL ECM medium with 10 ng/mL IL-5 and stimulated with 5 μg/mL anti-Siglec-F (clone E50–2440), Rat IgG2a kappa Isotype control (Becton Dickinson) and 10 ng/mL IL-33 (R&D Systems) as indicated in figures for 24 hrs. Supernatants were used for ELISA.
Aspergillus fumigatus infection
2×106 conidia of the ATCC 46645 strain of A. fumigatus were intranasally administered 5 times every 3–4 days as previously described (26). Mice were sacrificed on day 17 after initial application.
Flow cytometry
Either cultured cells or single cell suspensions that were prepared using 70 μm or 100 μm cell strainers were analyzed. If necessary, erythrocytes were lysed with ACK-buffer (0.15 M NH4Cl, 1 mM KHO3, 0.1 mM Na2EDTA). If not indicated differently stainings of up to 4 million cells were performed in 100 μl FACS buffer (PBS with 2 % FCS and 0.1 % NaN3) for 25 min at 4°C. Cells were pre-treated with Fc-receptor blocking antibody (anti-CD16/32, clone 2.4G2, Bio X Cell, West Lebanon, NH) and stained with fluorophore-coupled antibodies. The following antibodies for murine eosinophils were used: from Becton Dickinson: anti-Siglec-F BV421, Af647, PerCP-Cy5.5, PE or unlabeled clone: E50–2440, anti-B220 BV711 clone: RA3–6B2, anti-PD-L1 APC clone: MIH5, anti-CD125 Af488 clone: T21, anti-Ly6G BUV395 clone: 1A8, anti-CD45.1 Af700 clone: A20, anti-CD11b BV785 clone: M1/70, anti-ST2 BUV421 clone:U29–93, anti-CD11c BUV737 clone: N418; from BioLegend: anti-Siglec-F unlabeled clone S17007L, anti-CD11c APC-Cy7 clone: N418, anti-CD45.2 PE-Cy7 clone: 104, anti-CD115 PerCP-Cy5.5 clone: AF598, anti-CD193/CCR3 FITC or Af647 clone: J073E5, anti-CD11b A700 clone: M1/70; from eBioscience: anti-Thy1.1 (anti-CD90.1) FITC or PerCP-Cy5.5 clone: HIS51, anti-CD62L PE-Cy7 clone: MEL-14, anti-CD11b e780 clone: M1/70; from Invitrogen: anti-CD101 PE clone: Moushi101, fixable viability dye in AmCyan. For human eosinophils the following antibodies were used: from Biolegend: anti-CD11b e780 clone: M1/70, anti-CD62L e450 clone: DREG-56, anti-CD193/CCR3 FITC clone: 5E8, from R&D Systems: anti-Siglec-8 PE clone: FAB7975P.
Proliferation and apoptosis assays
For proliferation assay, BMDEs of culture day 8 were seeded at 0.5×106 cells/mL in 500 μL BM-medium in a 48 well plate and stimulated with 5 μg/mL rat IgG2a, к isotype control antibody or purified rat anti-mouse Siglec-F antibody (clone E50–2440) and optional 10 ng/mL recombinant murine IL-33 (R&D Systems) for 24 hrs. Additionally, 10 μM EdU (Invitrogen) per well were added and cells were incubated for further 24 hrs at 37°C 5% CO2. Then cells were stained for surface antigens followed by EdU staining with the Click-iT Plus EdU Alexa Fluor 647 Kit (Invitrogen) according to manufacturer’s instructions. For apoptosis assay, mature BMDEs were seeded at 0.2×106 cells/mL in 200 μL BM-medium in a 96 well plate and stimulated as in the proliferation assay described above for 24 hrs. For flow cytometry staining, Annexin binding buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2) was used instead of FACS buffer. AnnexinV in FITC or APC (ImmunoTools) was used and DAPI (Sigma/Merck) was added to each sample directly before measuring in the flow cytometer.
Enzyme-linked immunosorbent assay (ELISA)
For IL-4 ELISA anti-IL-4 clones 11B11 (BioXcell) and biotinylated clone BVD6–24G2 (Southern Biotech, Birmingham, AL) were used as coating or detection antibody respectively. For detection, phosphatase-coupled streptavidin and para-Nitrophenylphosphate substrate (Southern Biotech) were used. For IL-13 detection an ELISA Development Kit (Peprotech) was used. For CCL3 and CCL4 ELISA were also performed with commercial kits (CCL3/MIP-1 alpha DuoSet ELISA and Mouse CCL4/MIP-1 beta DuoSet ELISA, R&D Systems).
Western blot
Cells were lysed in RIPA-lysis buffer (1% NP-40, 50 mM Tris (pH7.4), 0.15 M NaCl, 1 mM EDTA (pH 8.0), 0.25% deoxycholic acid) containing PhosSTOP and cOmplete Proteinase Inhibitor Cocktail (both Roche). 5 μg protein were run on a SDS-PAGE and blotted to PVDF membrane (Bio-RAD). Binding of primary antibodies (all from Cell Signaling Technology) against phosho-STAT6 (Y641), STAT6 (either from Santa Cruz or Cell Signaling Technology), phospho-AKT (D9E), AKT (pan, C67E7) and beta-actin (13E5) was detected by HRP-coupled donkey-anti-rabbit (Rockland Immunochemicals) and the SignalFire™ Plus ECL reagent (Cell Signaling Technology) using the ChemiDoc™ imager (Bio-Rad). For incubation with a second primary antibody, membranes were stripped with the Restore PLUS Western Blot Stripping Buffer (Thermo Fischer Scientific).
qRT-PCR of human eosinophils
Freshly isolated human eosinophils were stimulated with 5 μg/mL anti-human Siglec-8 antibody (clone 7C9, Biolegend) or Purified Mouse IgG1 κ isotype control antibody (clone MOPC-21, Biolegend) and optionally 10 ng/mL recombinant human IL-33 (Gibco) in ECM including 10 ng/mL recombinant human IL-5. After 4 hrs, cells were harvested in RLT buffer (Qiagen, Hilden, Germany) containing 0.1M DTT and frozen overnight at −80°C. RNA was isolated with the RNeasy kit (Qiagen) according to the manufacturer’s protocol and cDNA was synthesized with the SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). The SYBR Select Master Mix premix (Thermo Fisher Scientific) was used for qPCRs and Ct values were measured on an Applied Biosystems ViiA 7 Real‐Time PCR System (Thermo Fisher Scientific) and normalized to GAPDH as a housekeeper gene. The following primer sequences were used: GAPDH-fw 5’- GGAGAAGGCTGGGGCTCATTTG-3’, GAPDH-rv 5’- GCATGGACTGTGGTCATGAGTCC-3’, CCL3-fw 5’- CCAGTTCTCTGCATCACTTGCTGC-3’, CCL3-rv 5’- CGCTTGGTTAGGAAGATGACACCG-3’, CCL4-fw 5’- CGCTCTCAGCACCAATGGGC-3’, CCL4-rv 5’- GGAATACCACAGCTGGCTGGG-3’.
Transcriptome analysis
Eosinophils were derived from mice with systemic eosinophilia due to overexpression of IL-5 (4get_IL-5tg). Splenic single cell suspension from male mice was treated with ACK buffer to lyse erythrocytes (0.15 M NH4Cl, 1 mM KHO3, 0.1 mM Na2EDTA). Untouched eosinophils were sorted as SSChigh4get+ cells. Purity (Fixable Viability Dye−Siglec-F+CCR3+SSChigh) was >95%. Cells were seeded into 24 well plates (3×106/mL) in 1 mL ECM with 10 ng/mL IL-5. Cells were stimulated with 5 μg/mL anti-Siglec-F (clone E50–2440) or isotype control (BD) and/or 10 ng/mL recombinant murine IL-33 (R&D Systems) for 4 hrs. RNA was isolated by phenol/chloroform extraction (SIGMA-ALDRICH 83913) according to manufacturer’s instructions except that 1/6 of recommended volumes were used, samples were sheared through 27G cannulas during initial lysis and precipitation was performed at −80°C instead of −20°C. RNA was dissolved in DEPC H2O. It was challenging to extract RNA from the RNAse rich eosinophils especially after stimulation and generated RNA had RNA Integrity Numbers between 5.7 and 7.8 (average 6.9; Agilent Bioanalyzer 2100). RNA was sent to Novogene Europe (Cambridge, UK) for sequencing as a service. There, library preparation was performed with NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB) following manufacturer’s recommendations and paired-end sequencing was performed on an Illumina NovaSeq6000. Filtered clean reads were aligned to reference genome mm10 using STAR (v2.6.1d) (27). FeatureCounts (v1.5.0-p3) was used to generate counts used for downstream analysis in the lab (28). Data were loaded into R (3.5.3; The R Foundation for Statistical Computing, Vienna, Austria) and genes with zero counts were removed. Deseq2 (1.20.0) (29) was used to normalize counts and perform differential expression analysis without further shrinkage. Heatmaps of log2 normalized counts were drawn with the gplots package. For gene set enrichment Bubble GUM software (1.3.19) (30) based on GSEA (31) with normalized count values as input and recommended settings (default values but permutation type set to “gene_set”) was used. For the analysis HALLMARK gene sets (32) of MSigDB database version 6.2 were combined with custom added gene sets of IL-33 or IL-4 stimulated eosinophils (GSE43660; significantly altered and log2 fold change >0.25) (33). GSEA built in conversion of mouse to human gene identifiers was used if necessary (31). Data is available via the GEO database (https://www.ncbi.nlm.nih.gov/geo/ accession no. GSE166968).
Generation of Siglec-F mutants
Siglec-F cDNA was derived from splenic cells of an IL-5tg mouse (identical with NCBI CCDS ID 21167.1). Primer: 5’G GCGGCCGC GCCACC ATGCGGTGGGCATG 3’ (SigF-for; features in order separated by space: guanine, NotI, Kozak, complementary sequence); 5’ G GTCGAC TCAGCACTTGTGGATCTTGATCTCTGTG 3’ (SigF-rev; guanine; SalI, complementary sequence). Overlap PCR with the cloned-Siglec-F as a template generated a Siglec-F version with mutated ITIM and ITIM like motif. Primer for overlap PCR 1 (mutated triplets highlighted by extra spaces): 5’ GAGCCTGAACTCCAC TTT GCCTCCCTCTCCTTCC 3’ (SigF-for-ITIMmut), 5’ TCAGCACTTGTGGATCTTGATCTCTGT GAA TACAG 3’ (SigF-rev-ITIM-like-mut). In parallel, another PCR with the SigF-for primer and a complementary reverse version of the SigF-for-ITIMmut primer was performed to generate the second template. Both templates were used in an overlap PCR with the SigF-for and the SigF-rev primer to finalize the Siglec-F sequence with mutated ITIM and ITIM like motifs. To generate the Siglec-F version with a truncated intracellular tail SigF-for primer combined with 5’ G GTCGAC TCA CTTCACTGTGAAAAAGATG 3’ (guanine, SalI, stop codon, complementary sequence) were used. The sequential tail truncation variants were generated analogously to the outlined tail truncation variant using corresponding reverse primers. Siglec-F variants were cloned into an MSCV-IRES-Thy1.1 via NotI and SalI restriction sites.
Statistics
Statistical analysis was performed with GraphPad PRISM 5 and Sigmaplot 12.3 except for RNA sequencing analysis. Statistical tests were used as indicated in figure legends.
Results
IL-33 elicits chemokine and cytokine secretion and Siglec-F upregulation in eosinophils
The alarmin IL-33 is known to activate eosinophils although the spectrum of secreted cytokines and chemokines has not been analyzed in detail. To address this point we analyzed supernatants of IL-33-stimulated, bone marrow-derived eosinophils (BMDE; culture exemplary visualized in Suppl. Fig. 1A) by a Luminex bead array. The secretion of the pro-inflammatory cytokine IL-6 and the IL-1 family member IL-18 as well as the type 2 immunity-associated cytokines IL-4 and IL-13 was strongly induced upon IL-33 stimulation (Fig. 1A). The chemokines MCP-1/CCL2, MIP-1α/CCL3, MIP-1β/CCL4 and MIP-2/CCL8 that contribute to the recruitment of immune cells were also elevated (Fig. 1A). Kinetic ELISA experiments further revealed that secretion of IL-4, IL-13, CCL3 and CCL4 was already detectable at 2 hrs after stimulation and the concentrations significantly increased after 8 and 16 hrs compared to unstimulated controls (Fig. 1B). Simultaneously, IL-33 induced the up-regulation of Siglec-F on the cell surface (Fig. 1C). As Siglec-F has been described as an inhibitory and pro-apoptotic receptor (4, 5) these results raise the question how the potentially opposing processes of enhanced activation/cytokine production by IL-33 and inhibitory effects from Siglec-F engagement are balanced and integrated to mediate an appropriate immune response.
Figure 1: IL-33 activates eosinophils to release cytokines and chemokines and increase their surface expression of Siglec-F.
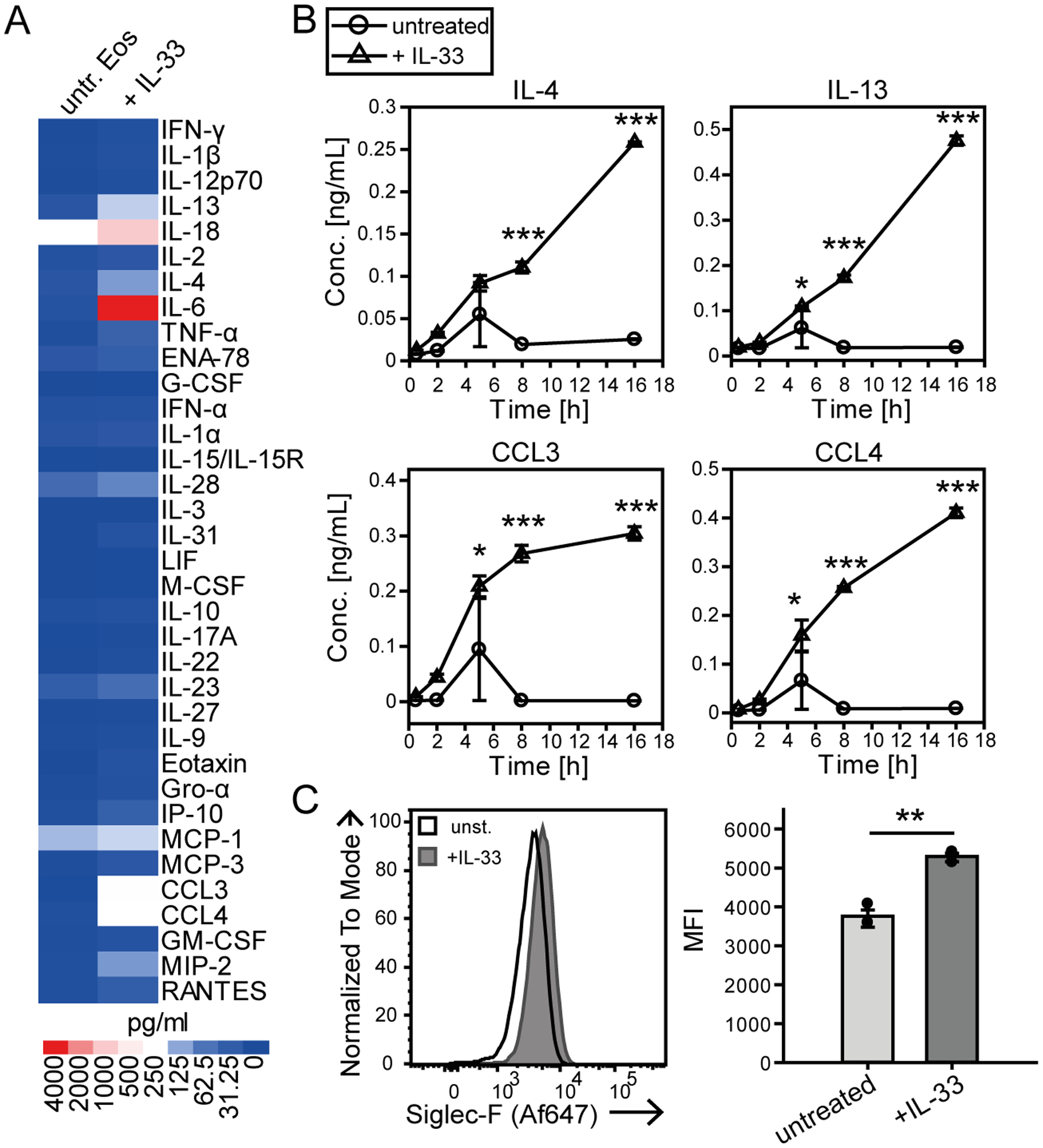
A, B) Multiplex cytokine assay screen from the supernatants of d14 BMDE cultures stimulated with IL-33 and their unstimulated controls. Data were obtained from one BMDE culture in technical duplicates. A) An overview of the 35 analyzed cytokines is displayed as a heatmap for 8 hrs of stimulation. B) Time course analysis for the release of selected cytokines over the course of 0.5, 2, 5, 8 and 16 hrs of stimulation with IL-33 or of unstimulated cultures. C) Siglec-F surface expression on d14 BMDE cultures. Displayed are representative histograms of one culture and the mean fluorescence intensity (MFI) of pooled data from three biological distinct cultures after 6 hrs of IL-33 stimulation or for unstimulated controls. For B) Significance was determined by Two-way ANOVA with Holm-Sidak post-hoc test and is indicated for significant differences between unstimulated and IL-33 stimulated BMDEs of the same time point. For quantification in C) data are displayed as the mean ± SEM and a t-test was performed with significance levels of *p < 0.05; **p < 0.01; ***p < 0.001.
Siglec-F engagement minimally affects survival and proliferation of eosinophils while IL-33 inhibits proliferation
Next, we investigated the combined effect of IL-33 and Siglec-F stimulation on eosinophil survival. BMDE were stimulated with IL-33 or anti-Siglec-F alone or with a combination of both. After 24 hours the viability (percent of DAPI−Annexin− cells, gating exemplary visualized in Suppl. Fig. 1B) was only mildly affected by anti-Siglec-F treatment (Fig. 2A and B). This is in accordance with published in vitro data that used the same anti-Siglec-F antibody clone (4, 7, 8). Related, we determined if Siglec-F mediates inhibition of eosinophil proliferation. We measured proliferation by the uptake of EdU (gating exemplary visualized in Suppl. Fig. 1B) starting at day eight of the eosinophil culture, when cells still proliferate. Cells were stimulated for 48 hrs and EdU was added for the last 24 hrs of stimulation. While IL-33 treatment caused profound inhibition of BMDE proliferation, there was only a mild inhibitory effect caused by additional anti-Siglec-F stimulation (Fig. 2C and D).
Figure 2: Siglec-F signaling barely affects survival and proliferation of IL-33-stimulated eosinophils.
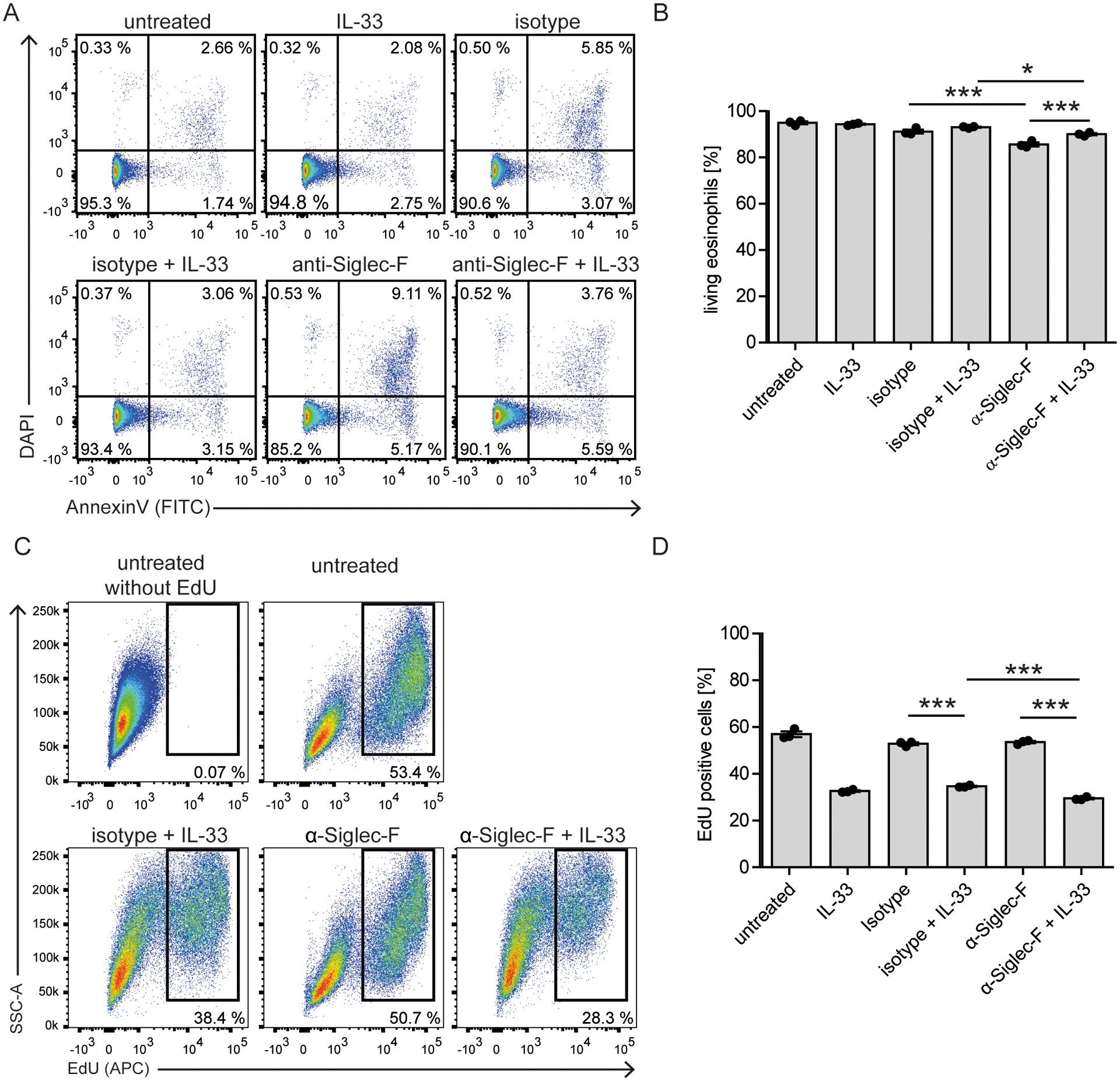
A, B) Survival analysis. BMDE of culture d14 were stimulated with anti-Siglec-F or isotype control and optional IL-33 for 24 hrs and stained with AnnexinV and DAPI. Cells were pregated on eosinophils defined as CCR3+ cells (as in Suppl. Fig. 1B). A) Representative flow cytometry plots and B) Quantification of AnnexinV and DAPI double negative eosinophils. Shown are technical triplicates from one representative BMDE culture representative for three biologically distinct cultures. C, D) Proliferation analysis. BMDEs of culture d8 were stimulated with indicated conditions for 48 hrs and EdU was added for the last 24 hrs. C) Representative flow cytometry plots for incorporated EdU pregated on eosinophils and expected eosinophil precursors based on Siglec-F and CCR3 expression as in Suppl. Fig 1B. D) Quantification of the percentage of EdU+ cells from technical triplicates of one BMDE culture representative for two biologically distinct cultures. For B) and D) Mean ± SEM is shown. Two-way ANOVA with Bonferroni posttests. Only selected significances that highlight IL-33 and Siglec-F mediated effects are indicated; *p <0.05, ***p <0.001.
To further analyze the regulation of eosinophilia by IL-33 and Siglec-F in vivo, we generated mixed bone marrow chimeras (MBMC) and subjected them to intranasal A. fumigatus infection to elicit allergic inflammation in the lung. First, we analyzed MBMC generated with 50% CD45.2_ST2−/− (IL-33-receptor deficient) and 50% congenic CD45.1_wild-type donors. While the ratio of eosinophils in the lung remained about 50/50 in naïve MBMC, the ratio changed to a higher percentage of ST2−/− cells after A. fumigatus infection (70/30) (Fig. 3A). Representative flow cytometry plots for naïve and A. fumigatus infected mice highlight A. fumigatus-induced eosinophil (SiglecF+Ly6G− gate) recruitment to the lung (Fig. 3B). Further sub-gating of eosinophils by congenic markers reveals the fraction of eosinophils derived from ST2−/− or wild type bone marrow (Fig. 3B).
Figure 3: IL-33- rather than Siglec-F signaling dampens eosinophil expansion in A. fumigatus-elicited lung inflammation.
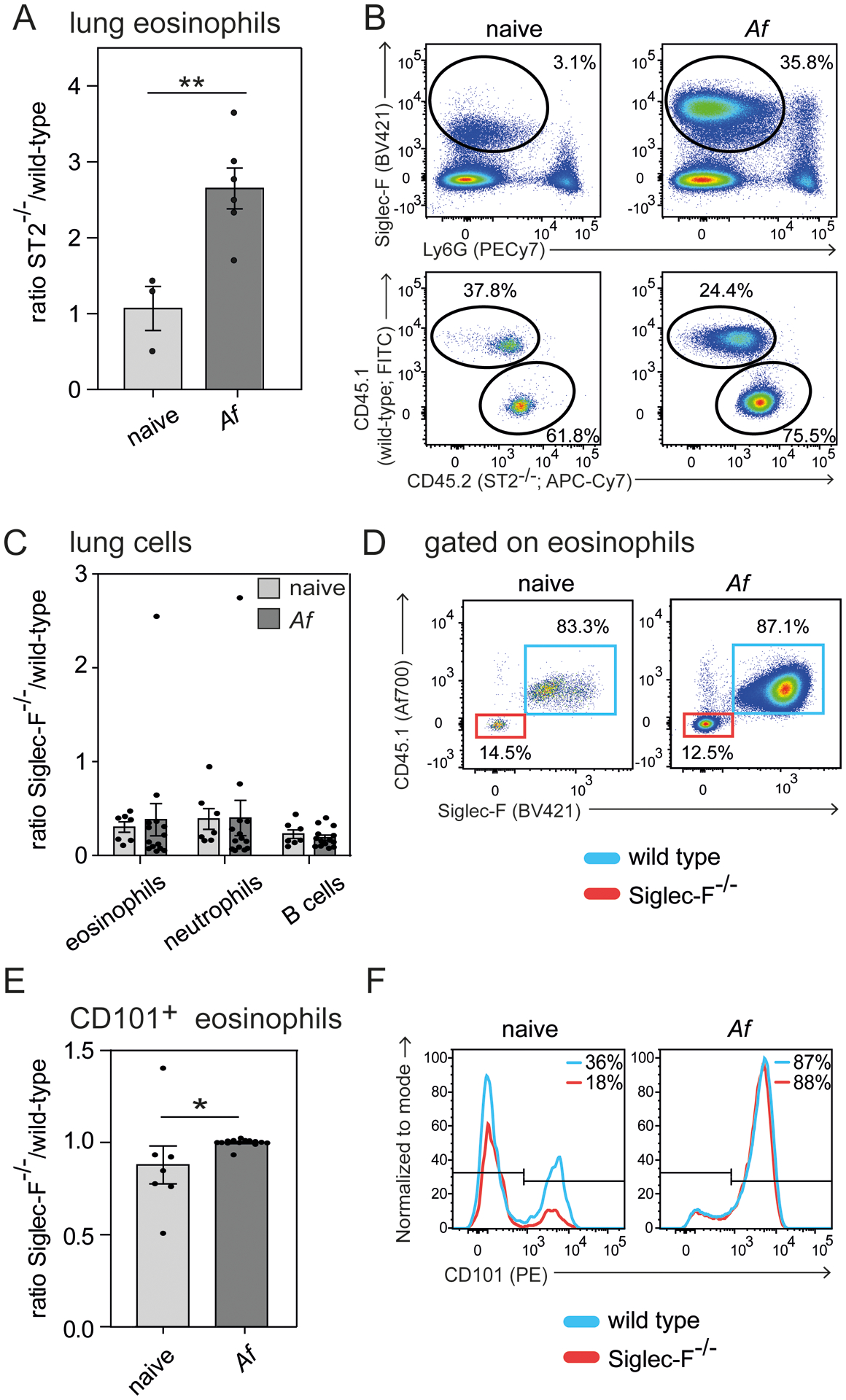
Competitive mixed bone marrow chimeras for ST2−/− (A and B) or Siglec-F−/− (C-F) (both CD45.2) generated in a 50:50 mix with CD45.1 congenic wild-type bone marrow were subjected to intranasal A. fumigatus (Af) infection and analyzed on day 17 by flow cytometry. A) Ratio of ST2−/− knock-out against wild-type eosinophils of the lung. B) Representative flow cytometric plots highlight eosinophil gating (Siglec-F+Ly6G−) and further subgating of eosinophils based on congenic markers. C) Ratio of Siglec-F−/− against wild-type cells for indicated populations. D) Representative flow cytometric plots display pre-gated eosinophils (Suppl. Fig. 1C) subdivided into Siglec-F−/− (Siglec-F−CD45.1−, red) or wild-type derived eosinophils (Siglec-F+CD45.1+, blue). E) Ratio of Siglec-F−/− against wild-type CD101+ eosinophils in the lung. F) Representative histograms for CD101 expression on eosinophils. n=2 independent experiments with a total of 3–6 mice (A and B) or 7–14 mice (C-F) per group. Two-way-ANOVA with Bonferroni posttests was performed to determine significance for C). Students t-test was performed for eosinophil ratio in ST2−/− chimeras (A) and frequency of CD101+ eosinophils (E). Bars show the mean ± SEM; *p < 0.05; **p < 0.01.
Next, we analyzed eosinophils in MBMC generated with 50% CD45.2_Siglec-F−/− and 50% CD45.1_wild-type donors. We used a gating strategy to identify eosinophils as CD11b+SSChi cells without using Siglec-F as an actual marker (Suppl. Fig. 1C). We observed an expected increase of lung eosinophils from 8±5% SD in naïve to 46±14% SD in A. fumigatus infected chimeras (data not shown). While we expected a 50/50 ratio of bone marrow derived cells after reconstitution we observed a ~20/80 (Siglec-F−/−/wild-type) ratio of eosinophils, neutrophils and B cells in the lung of naïve and A. fumigatus-infected MBMC (Fig. 3C and D). This indicates that Siglec-F may play a general role for bone marrow reconstitution but does not significantly reduce the number of eosinophils in the inflamed lung, which does not fit with a pro-apoptotic function. The subset of CD101+ inflammatory eosinophils (34) was slightly increased within the Siglec-F−/− fraction in uninfected MBMC but this difference disappeared after A. fumigatus infection (Fig. 3E and F).
In conclusion, IL-33 inhibits eosinophil proliferation in vitro and IL-33-responsive eosinophils have a competitive disadvantage to establish lung eosinophilia after A. fumigatus infection which we have previously shown to promote eosinophilopoiesis in the bone marrow (35). In contrast, anti-Siglec-F caused only minor effects on eosinophil survival and proliferation in vitro. In accordance, Siglec-F-deficient eosinophils did not outcompete wild-type eosinophils in A. fumigatus-induced lung eosinophilia of MBMC. Therefore, the results do not support a strong pro-apoptotic role of Siglec-F under these conditions.
Siglec-F engagement promotes synergistic cytokine/chemokine secretion of IL-33-stimulated eosinophils
As we observe increased Siglec-F expression after IL-33 stimulation and in inflamed lungs that was accompanied by enhanced cytokine/chemokine secretion upon IL-33 stimulation, we further investigated how anti-Siglec-F regulates the IL-33-induced cytokine/chemokine secretion and expression of activation markers on the cell surface. Therefore, BMDEs were stimulated with IL-33, anti-Siglec-F or a combination of both. Concentrations of the type 2 immunity-associated effector molecules IL-4, IL-13, CCL3 and CCL4 were then measured in supernatants by ELISA. Double stimulation of eosinophils with IL-33 and anti-Siglec-F antibody (either clone E50–2440 or clone S17007L) had a strong synergistic effect compared to stimulation with IL-33 or anti-Siglec-F alone (Fig. 4A, 4B, and data not shown). In contrast, anti-Siglec-F treatment did not significantly alter the IL-33-induced up-regulation of CD11b, CD11c, CD101 or ST2 but slightly enhanced the down-regulation of CD62L (Fig. 4C). To further investigate whether the IL-33-induced change of activation marker expression are a cell-intrinsic effect we analyzed IL-33-stimulated BMDE cultures generated with 50% CD45.2_ST2−/− and 50% congenic CD45.1_wild-type cells derived from MBMC mice. The activation markers CD11b and CD101 were indeed expressed at lower levels on ST2−/− eosinophils as compared to wild-type eosinophils after IL-33 stimulation while expression of CD62L that is downregulated after IL-33 stimulation and CCR3 remained higher on ST2−/− eosinophils (Fig. 5). This result proves that cell-intrinsic ST2-mediated signaling events are directly required for IL-33-induced changes of the activation marker profile and the effect cannot be caused in a paracrine manner by soluble factors released from bystander wild-type eosinophils. In addition, we found that activation marker expression as well as mediator secretion after IL-33 stimulation require the ST2-associated signaling adaptor molecule MyD88 (Suppl. Fig. 3A–C). In summary, anti-Siglec-F treatment promotes IL-33-elicited and MyD88-dependent mediator secretion but anti-Siglec-F antibody by itself does not affect expression of activation markers.
Figure 4: Siglec-F signaling promotes synergistic cytokine and chemokine secretion of IL-33-stimulated eosinophils.
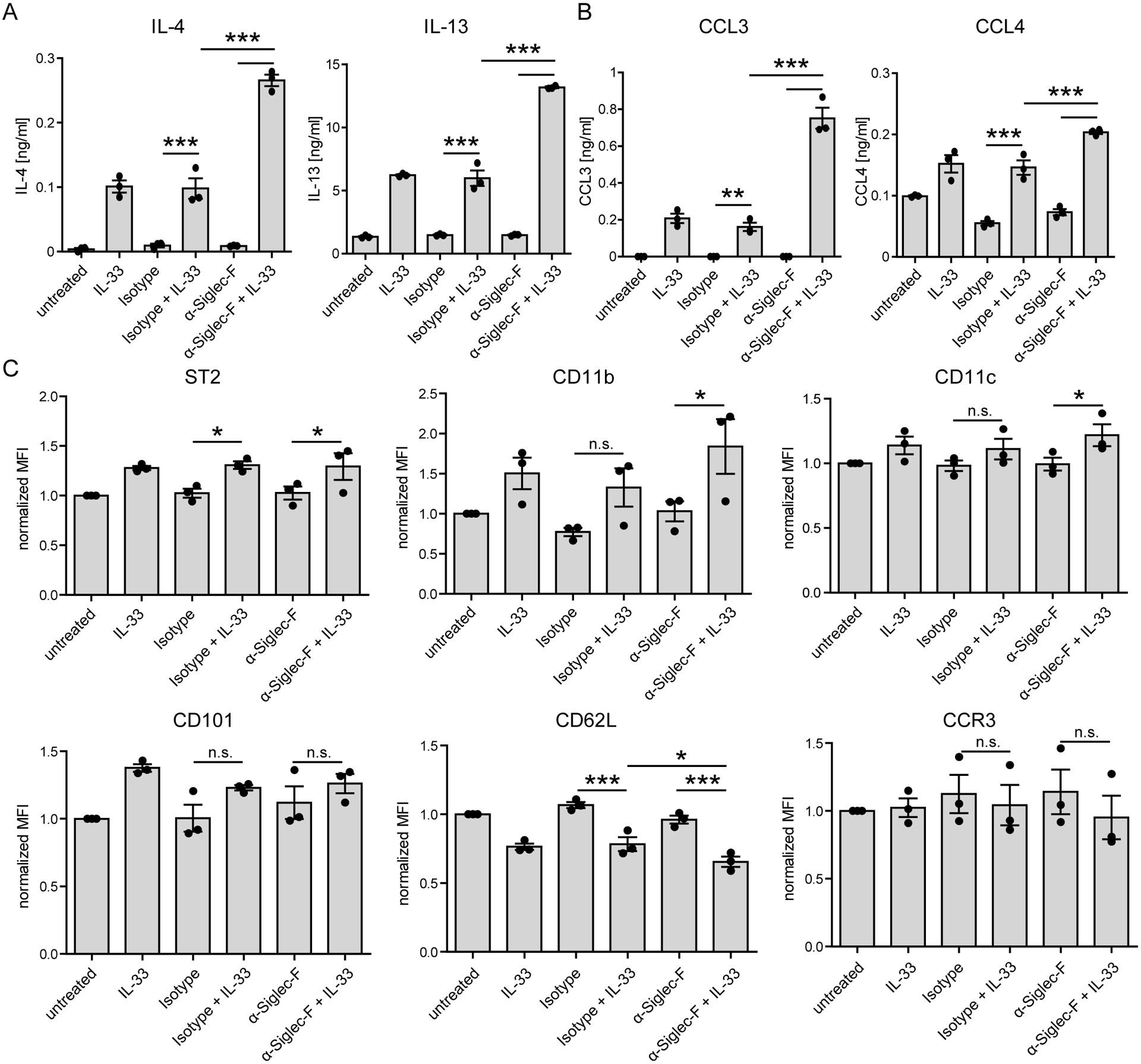
BMDE were generated from BALB/c mice and stimulated on d14 with anti-Siglec-F or isotype antibody and optional IL-33 for 24 hrs. Bars show mean ± SEM of A) Cytokine and B) Chemokine secretion for triplicates of one BMDE culture (representative for three biologically distinct cultures) measured by ELISA. C) Bars show mean of MFI ± SEM of surface markers measured by flow cytometry pregated on living cells. Means of pooled triplicates of three biologically distinct culture (n=3) normalized to the unstimulated control of the respective culture are shown. Two-way ANOVA with Holm-Sidak post-hoc test was performed. Only selected significances that highlight IL-33 and Siglec-F mediated effector release are indicated; ns=not significant, *p <0.05, **p <0.01, ***p <0.001.
Figure 5: IL-33-induced activation does not modulate activation marker expression on ST2-deficient bystander eosinophils.
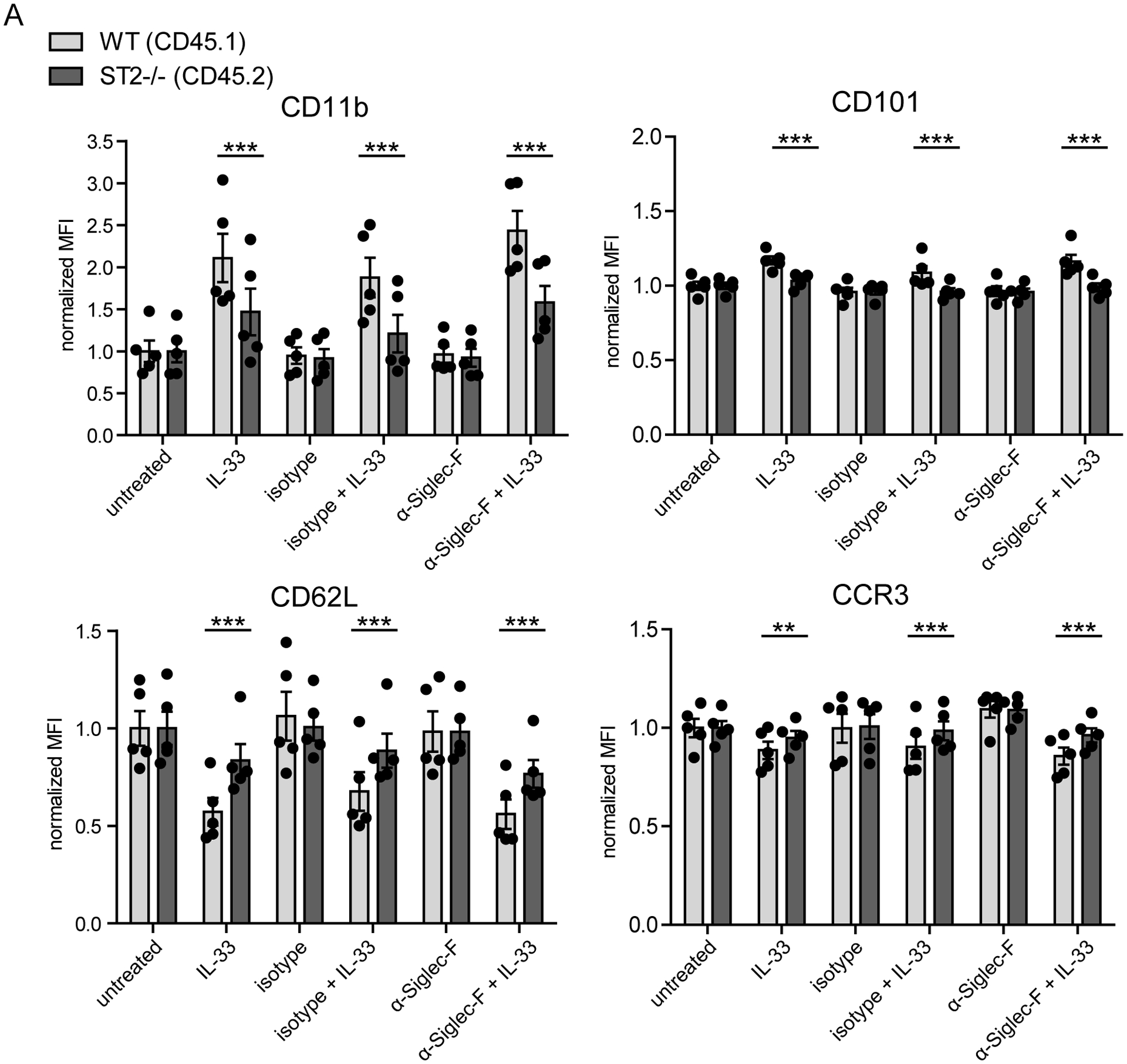
BMDE were generated from five biologically distinct ST2−/−/WT (50/50) mixed bone marrow chimeras. Cells were stimulated on culture d14 with anti-Siglec-F or isotype antibody and optional IL-33 for 24 hrs and were analyzed for expression of indicated surface markers by flow cytometry. Bars show mean fluorescence ± SEM pregated on living cells and the distinct genotype portion and normalized to the mean of all unstimulated controls of the respective genotype (n=5 of 2 independent experiments; Two-way ANOVA with Bonferroni posttests; **p <0.01, ***p <0.001).
To determine if Siglec-F mediated enhanced effector release of IL-33 stimulated eosinophils is transferrable to the human system we targeted Siglec-8, a described functional paralog of Siglec-F in humans, on eosinophils isolated from the peripheral blood (Suppl. Fig. 2A). We confirmed published Siglec-8 induced cell death by treatment with a commonly used anti-Siglec-8 antibody (clone 7C9) (Suppl. Fig. 2B) (5) but also upregulation of the activation marker CD11b (Suppl. Fig. 2C) (7, 36). As a further sign of activation, we observed CD62L downregulation on human eosinophils, while CCR3 expression was not altered (Suppl. Fig. 2C). CCL3 and CCL4 gene expression was also upregulated after anti-Siglec-8 antibody stimulation (Suppl. Fig. 2D). However, combined anti-Siglec-8 antibody and IL-33 treatment at indicated time points did not induce a synergistic upregulation of activation markers or chemokines beyond single stimulations (Suppl. Fig. 2). Furthermore, IL-4 or IL-13 were not detected upon combined anti-Siglec-8 antibody and IL-33 treatment or treatment with one or the other alone (data not shown). These findings indicate that Siglec-8 does not promote IL-33-elicited eosinophil activation, but, in contrast to Siglec-F, can activate eosinophils directly as reported before (36).
Siglec-F promotes STAT6 but not AKT signaling in IL-33-stimulated eosinophils
Next, we extended our investigation of the synergistic IL-33 and anti-Siglec-F effect on potentially involved signaling pathways. We analyzed phosphorylation of the IL-4/IL-13-activated transcription factor STAT6 and the serine/threonine kinase AKT that is involved in regulation of cell metabolism and survival. As expected, AKT phosphorylation was induced by IL-33 but anti-Siglec-F treatment did not modulate this response (Fig. 6A). However, anti-Siglec-F treatment enhanced and prolonged the IL-33-induced phosphorylation of STAT6 (Fig. 6A). To further investigate whether IL-4/IL-13-mediated activation of STAT6 was indeed required for mediator secretion or expression of activation markers, we used BMDE from STAT6−/− mice. The induction of activation markers in the absence of STAT6 was still functional with subtle STAT6 dependent alterations and only moderate (for CD11b, CD11c) or minor (for ST2, CD101, CD62L, CCR3) alterations were observed compared to wild-type (Suppl. Fig. 3D). However, the IL-33-induced secretion of IL-4, IL-13, CCL3 and CCL4 was almost completely abrogated in STAT6−/− BMDE (Fig. 6B). Only low levels of IL-13 were detected in supernatants of IL-33-stimulated STAT6−/− eosinophils. This demonstrates that IL-33 signaling does not directly result in pronounced secretion of IL-4, IL-13, CCL3 and CCL4, which suggests that IL-33-elicited autocrine IL-4/IL-13 signaling through STAT6 is required for substantial secretion of all four mediators. Consistently, CCL3 and CCL4 concentrations were also reduced in IL-33-stimulated IL-4/IL-13−/− BMDE cultures (Fig. 6C), whereas only minor changes of activation marker expression could be observed (Suppl. Fig. 3E).
Figure 6: Siglec-F promotes STAT6 phosphorylation but also enhances effector release from IL-33-stimulated eosinophils of STAT6-deficient mice.
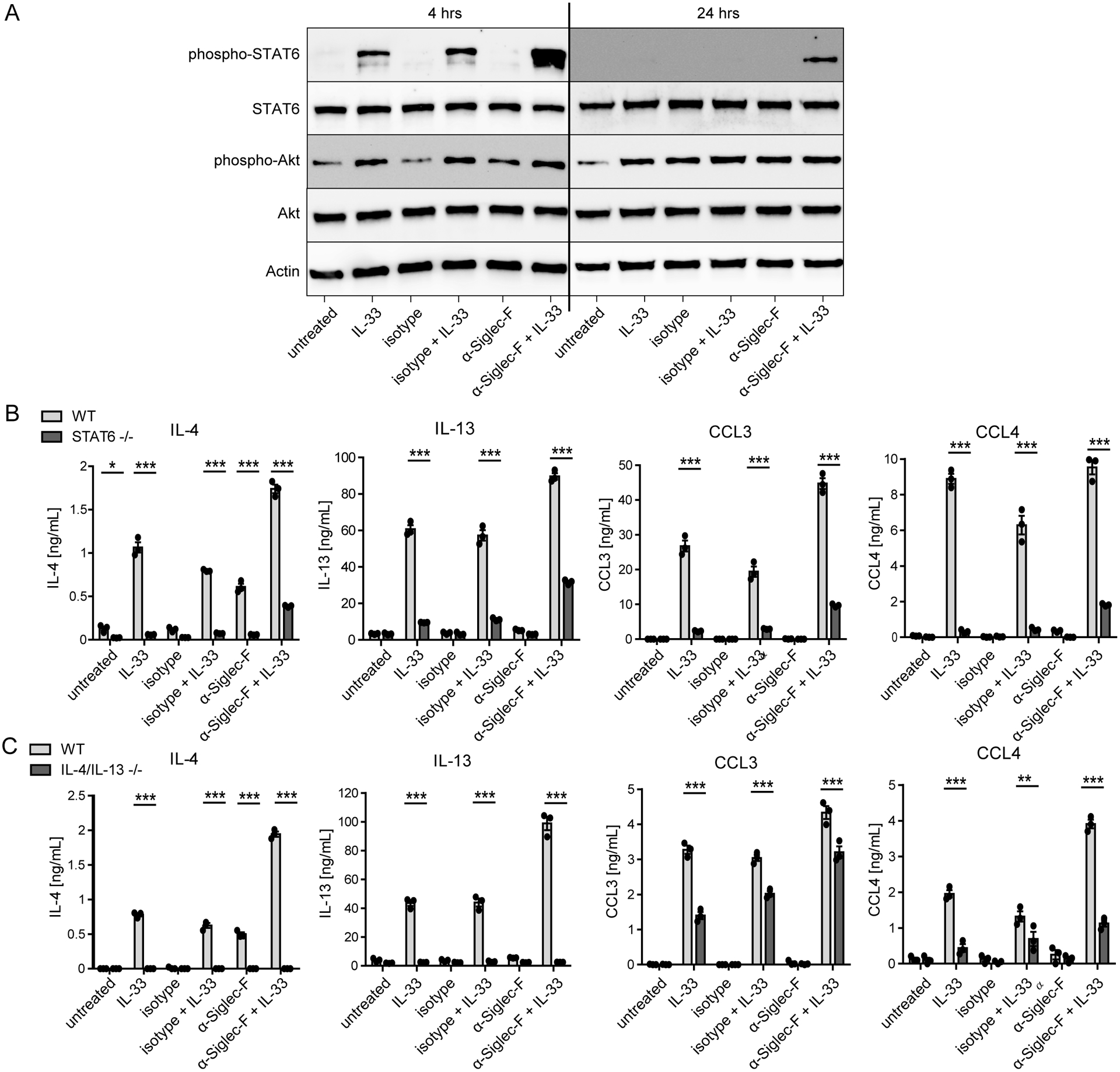
A) Western blots for indicated targets from wild-type BMDE stimulated for 4 hrs or 24 hrs with anti-Siglec-F or isotype control antibody and optionally IL-33. B) and C) ELISA results from stimulated BMDE cultures of 4get_STAT6−/− and 4get_BALB/c mice (B) or IL-4/IL-13−/− and CD45.1_BALB/c mice (C). Results show mean ± SEM of technical triplicates of one BMDE culture representative for two biologically distinct cultures per genotype. Two-way ANOVA with Bonferroni posttests; *p <0.05, ***p <0.001.
Interestingly, while anti-Siglec-F alone does not induce effector release, anti-Siglec-F and IL-33 co-treatment was able to elicit low-level secretion of all four mediators in STAT6−/− BMDE cultures indicating that the STAT6-dependency for IL-33-induced effector secretion could be partially restored by Siglec-F signaling (Fig. 6B). This finding led us to further investigate Siglec-F-mediated transcriptional changes to understand which pathways might promote enhanced effector release from IL-33-stimulated eosinophils.
Anti-Siglec-F alone is a weak modulator of gene expression but it elevates gene expression induced by IL-33
In addition to signaling analysis on protein level, we analyzed how IL-33 and anti-Siglec-F double stimulation impacts transcription by RNA sequencing. While anti-Siglec-F alone barely changed transcription (only Etv5, Peg10, Spred1, Spred2, Tnf, Tram1, Trem14 were weak but significantly upregulated), IL-33 induced the expression of 224 genes more than two fold compared to the isotype control. These included the cytokines/chemokines Il6, Il13, Ccl3, Ccl4 and Cxcl2. Combined IL-33 and anti-Siglec-F treatment lead to an even more pronounced upregulation of the highly induced genes. In addition, a higher total number of genes was induced more than two fold compared to IL-33 treatment alone (454) but changes were moderate for most of them (Fig. 7A). To determine differentially expressed genes specific for the IL-33 and anti-Siglec-F double treatment, genes that were significantly different versus all of the single/control stimulations (isotype, isotype + IL-33 or anti-Siglec-F) were determined (Fig. 7B). Amongst the upregulated genes were hypoxia-associated genes (Fam162a, Egln2, Ankrd37, Hilpda, Ftl1, Prdx1) and likely hypoxia-induced metabolism/glycolysis associated genes (Pfkl, Tpi1, Pgk1, Pgm2) as well as a gene encoding for the hypoxia-induced, pro-inflammatory cytokine migration inhibitory factor (MIF). How exactly Siglec-F signaling confers a hypoxia-associated gene expression profile are beyond the scope of this manuscript and remains to be determined in future studies. We further noticed an enhanced expression of Tarm1 which encodes for a co-stimulator of cytokine secretion (37). This could at least partially explain the anti-Siglec-F-mediated increase of cytokine/chemokine concentrations in supernatants of IL-33-stimulated eosinophils. Other upregulated genes upon anti-Siglec-F and IL-33 co-stimulation included pro- and anti-apoptotic genes (Bcl2a1b, Bcl2a1d, Mien1, Bnip3), integrin-related/adhesion-related Cd53, the gene for the surface receptor CD52 involved in cell activation and proliferation, genes encoding for transcription factors and transcription factor binding proteins (Eef1e1, Batf3, Scand1), the protease inhibitor coding gene Cstb, the G-protein signaling related gene Rgs10 and ribosomal proteins (Nhp2, Rps27l, Rps14) (Fig. 7C). The downregulated genes included repressors of NFκB (Mturn) and TGFβ (Ldlrad4) pathways. To analyze the data beyond the top regulated genes we performed a gene set enrichment analysis on HALLMARK gene sets (32) where we also compared IL-33 and anti-Siglec-F treatment to the other conditions. Myc target genes, and gene sets associated with Mtorc signaling, oxidative phosphorylation, generation of reactive oxygen species, glycolysis and adipogenesis were all significantly upregulated after double stimulation compared to any of the single treatments (Fig. 7D). As expected a huge proportion of gene sets was enriched upon stimulation with IL-33 alone. Of note, most sets enriched by IL-33 showed at least a tendency to further enrichment by additional anti-Siglec-F treatment (i.e. the HYPOXIA gene set).
Figure 7: Anti-Siglec-F alone is a weak modulator of gene expression but it elevates gene expression induced by IL-33.
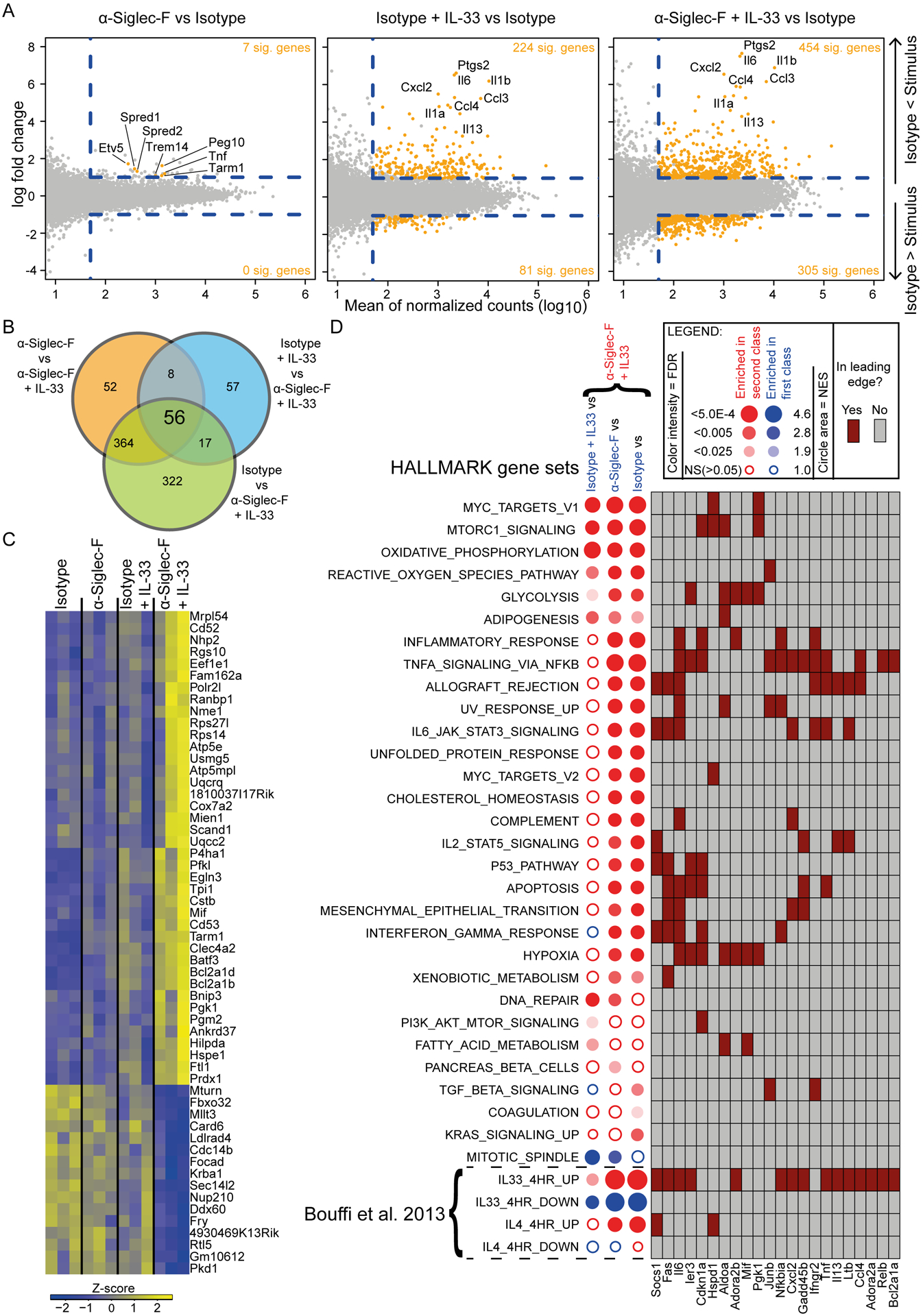
RNAseq for ex vivo sorted eosinophils of 4get_IL-5tg mice. Eosinophils were stimulated with combinations of IL-33, anti-Siglec-F and isotype control antibody as indicated for 4 hrs. A) MA plots of different stimulations versus isotype control as indicated. Genes with a fold change >2, an adjusted p-value <0.05 and a mean expression >50 are highlighted in orange. Dashed lines highlight the cut-offs. A few selected genes are labeled. B) Venn diagram of genes that passed criteria as in A for indicated contrasts. The number of genes significantly altered in the anti-Siglec-F + IL-33 condition versus all other conditions is shown in the center. C) Genes altered in anti-Siglec-F + IL-33 versus all other conditions are visualized in a heatmap. Every condition column contains three sub columns of biological replicates. D) Gene set enrichment analysis of contrasts that compare Siglec-F + IL-33 to indicated conditions. Contrasts were analyzed for enrichment in HALLMARK, IL-4 and IL-33 related gene sets. Circles indicate for the direction of regulation (color), the normalized enrichment score (size) and the false discovery rate (FDR>0.05 = filled). A contingency table with selected genes found in the leading edge of one or more gene sets and in all analyzed contrasts of at least one gene set. Three biological replicates were performed per stimulation condition.
In line with HALLMARK analysis results, effects on published IL-33 target genes (33) were also elevated when eosinophils were treated with anti-Siglec-F + IL-33 compared to isotype + IL-33 treatment. In contrast, IL-4 signature genes were enriched when stimulation contained IL-33 but additional significant effects upon combined anti-Siglec-F treatment were not observed (Fig. 7D). Of note, IL-4 expression itself was also not significantly altered by any of the stimulations applied in the experiment. Therefore, increased IL-4 secretion observed upon anti-Siglec-F + IL-33 treatment did not result in a stronger IL-4-mediated transcriptional response compared to IL-33 alone. In contrast, IL-13 transcription is upregulated by IL-33 stimulation but there is no significant further upregulation upon additional anti-Siglec-F treatment.
In summary, anti-Siglec-F treatment generally does very little by itself, but elevates gene expression induced by IL-33 but together with IL-33 stimulation also induces expression of additional i.e. hypoxia/glycolysis associated genes.
Siglec-F ITIM and ITIM-like motifs are dispensable but the tail is required for enhanced effector release
The anti-Siglec-F-mediated enhanced effector release from IL-33-stimulated eosinophils suggested a critical role for the cytoplasmic tail of Siglec-F in this process. The tail of Siglec-F contains one ITIM and one ITIM-like motif that mediate downstream signaling. To analyze if these motifs or other parts of the tail are required for enhanced effector release in the context of IL-33 and anti-Siglec-F double stimulation we generated BMDE from Siglec-F−/− bone marrow cells that were retrovirally complemented with wild-type or mutant Siglec-F. Either, both the ITIM and ITIM-like motif were mutated by exchange of their tyrosine residues to phenylalanine (ITIMmut) or the cytoplasmic tail was completely deleted (Stop codon behind K463; Taildel) (Fig. 8A). The transduced Siglec-F−/− BMDE successfully expressed all constructs on the cell surface demonstrating that the tail is not required for surface expression of Siglec-F (Fig. 8B). Reconstitution with wild-type Siglec-F did not spontaneously modulate IL-33-induced secretion of IL-4, IL-13, CCL3 and CCL4 (Fig. 8C, Suppl. Fig. 4A). However, enhanced secretion occurred after additional anti-Siglec-F treatment of wild-type and ITIMmut samples (with the exception of CCL4) but not with Taildel samples (Fig. 8C–E, Suppl. Fig. 4A–B). In an attempt to further specify which region of the cytoplasmic tail enhances effector release we generated two additional truncation mutants. The first lacks the terminal part of the tail that includes the ITIM-like motif and the ITIM motif (Stop codon behind D528; ITIMdel) and the second one retains only a short part of the cytoplasmic tail (Stop codon behind S493; Shorttail). In both mutants, the synergistic effect of IL-33 and anti-Siglec-F treatment was lost (Suppl. Fig. 4C). This indicates that the last forty-two amino acids deleted in the ITIMdel mutant are required to mediate the synergistic effect. In conclusion, the terminal part of the Siglec-F tail is critical for the elevated production of IL-4, IL-13 and CCL3 in activated eosinophils, but tyrosines in its ITIM and ITIM-like motifs are dispensable. Our study shows an important activating function of the Siglec-F tail independently of known cytoplasmic signaling motifs generally associated with inhibitory functions. Therefore, Siglec-F should not simply be regarded as pro-apoptotic receptor but also as an activating modulator of cytokine and chemokine secretion from eosinophils.
Figure 8: The cytoplasmic tail of Siglec-F promotes the enhanced cytokine/chemokine secretion from IL-33-stimulated eosinophils but the ITIM motifs are dispensable.
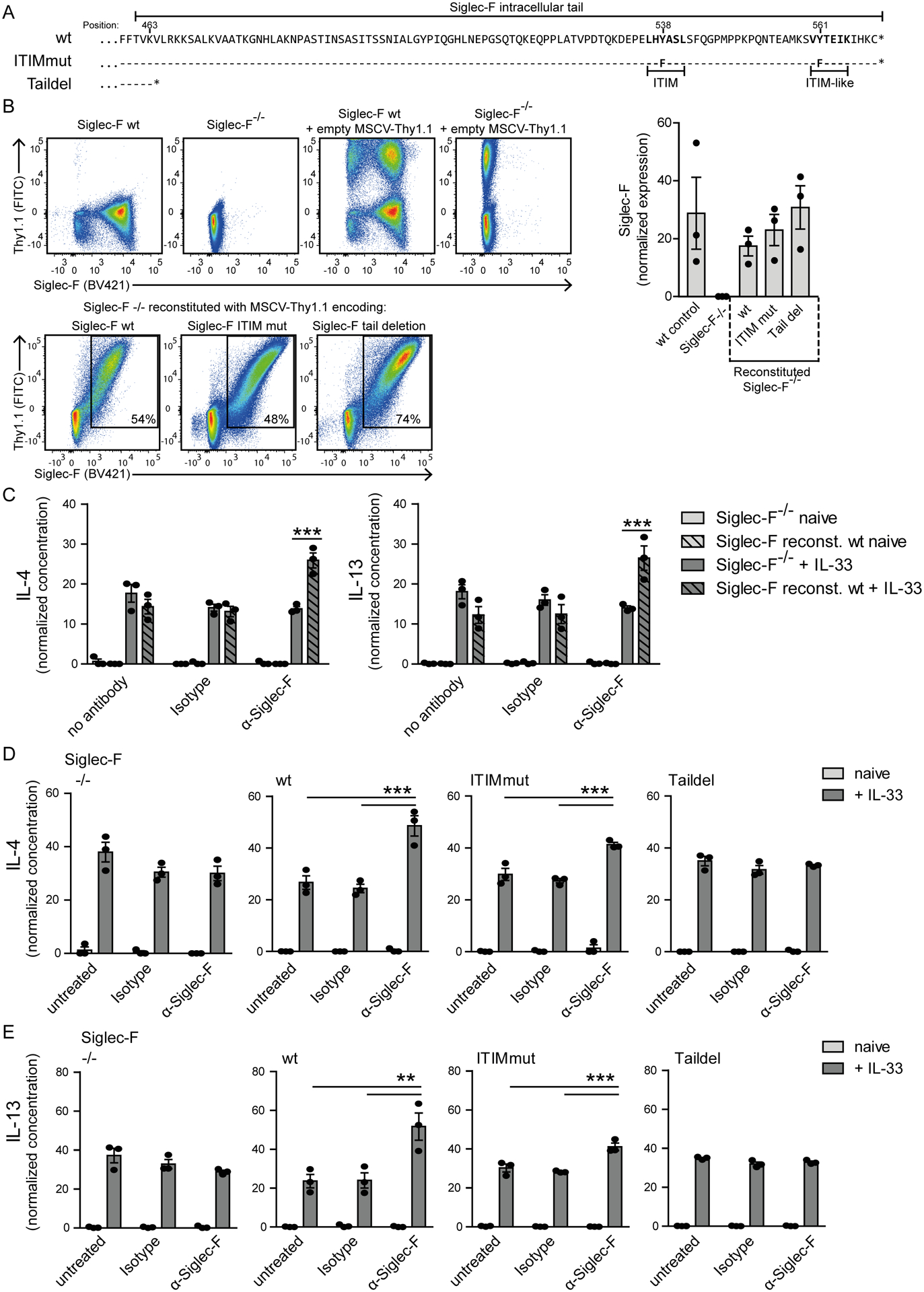
Siglec-F−/− bone marrow cells were transduced with Siglec-F variants-encoding MSCV-IRES-Thy1.1 retroviral vectors. A) Illustration of cytoplasmic tail sequences for Siglec-F variants. Dashes indicate matched amino acids and stars position of stop codons. B) Surface expression of Siglec-F and Thy1.1 from indicated populations. The bar graph shows the mean Siglec-F expression levels between wild-type BMDE cultures and retrovirally expressed Siglec-F variants. C) IL-4 and IL-13 concentrations in supernatants for MSCV-IRES-Thy1.1 transduced Siglec-F−/− and wild-type reconstituted Siglec-F−/− BMDE. Cells were unstimulated (light grey) or IL-33-stimulated (dark grey) and optionally treated with anti-Siglec-F or isotype antibody. D) and E) IL-4 (D) and IL-13 (E) normalized concentrations in supernatants of BMDE reconstituted with indicated Siglec-F mutants upon indicated treatment. In D) and E) culture conditions are compared individually for each variant. Normalization between experiments was performed with sum of replicate normalization. Bars show the mean ± SEM from n = 3 different cultures. One-way ANOVA with Bonferroni posttests was performed to determine significance. Only selected significances that highlight IL-33 and Siglec-F mediated effector release are indicated; *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
Eosinophils are involved in allergic responses as tissue damaging effector cells. Therefore, various treatments have been developed that target eosinophils to ameliorate diseases like allergy and asthma. These include eosinophil depletion or the activation of inhibitory receptors like Siglec-8 on human and as a model Siglec-F on murine eosinophils (4–6, 8, 38–44). Based on their ITIM and ITIM-like motifs both receptors are thought to mediate inhibition and apoptosis of eosinophils (4, 5). However, at least in the murine system it was shown that the induction of apoptosis by Siglec-F was not always effective (4, 7, 8) and strongly dependent on the chosen disease model (16). Such context-dependent differences are currently not well understood. A recent study with bone marrow-derived macrophages, that are able to express Siglec-F after GM-CSF stimulation, described enhanced Siglec-F-dependent STAT6 phosphorylation and in the presence of IL-4 also arginase 1 induction (17). In contrast to the described functions of Siglec-F this might argue for promotion of type 2 immunity by Siglec-F, rather than functional inhibition of the macrophages. Along the same line our group found that IL-33 and Siglec-F co-stimulation lead to elevated IL-4 and IL-13 secretion by eosinophils in vitro (18). We expand these initial findings now to increased secretion of CCL3 and CCL4 chemokines. Both chemokines can also be sensed by murine eosinophils (45) and mediate effector cell recruitment to sites of inflammation where they can contribute to tissue damage. We further describe that the apoptosis induction by Siglec-F is only a moderate effect as also seen by others (4, 7, 8) and largely independent of the IL-33 costimulation. Therefore, the elevated effector release after IL-33 and Siglec-F costimulation cannot simply be explained by passive release caused by Siglec-F-induced apoptosis. Of note, IL-33 inhibited proliferation measured at day 7–8 of our BMDE cultures while others reported an IL-33-dependent expansion of bone marrow eosinophil precursors and in turn mature eosinophils (46). However, BMDE cells probably develop uniformly in culture and early precursor stages that likely proliferate and drive an IL-33-mediated eosinophil expansion might not be present in the middle of the BMDE culture. Therefore, IL-33 might modulate eosinophil proliferation dependent on the eosinophil developmental stage. We also find no obvious pro-apoptotic function of Siglec-F in vivo by analyzing A. fumigatus-induced lung eosinophilia in competitive MBMC. In combination, our findings in context of the current literature point to a more diversified role of Siglec-F with inhibitory and activating aspects that are regulated in a context-dependent way. It might be that Siglec-F elevates the immune response until a signal, maybe via a feedback mechanism, induces inhibition/apoptosis to prevent overwhelming immune responses as suggested in the literature (4). How these findings relate to the human system remains incompletely understood as cell death induction by Siglec-8 is described to be more pronounced and mechanistically different to the induction by Siglec-F (8). This might indicate a higher inhibitory capacity of Siglec-8, despite the described analogy of Siglec-8 and Siglec-F. It has even been described that cytokines like IL-5 and IL-33 that enhance effector secretion in combination with Siglec-F in the murine system, in contrast enhance Siglec-8 dependent cell death induction of human eosinophils (36, 47, 48). As induction of cell death by Siglec-8 includes activation of signaling pathways and ROS effector production, Siglec-8 was already reported as an activating receptor in this regard (36). We also observed activation-associated surface receptor regulation but no enhancement of MIP1-α, MIP1-β, IL-4 or IL-13 chemokine/cytokine release upon anti-Siglec-8 antibody treatment of IL-33 stimulated human eosinophils that would point to synergistic activating functions of IL-33 and Siglec-8 in the chosen setup (47).
The effector release after IL-33 or combined IL-33 and anti-Siglec-F treatment is almost exclusively dependent on MyD88 as a main mediator of IL-33 signaling (23, 49, 50) which we show in MyD88−/− BMDE cultures. Downstream we find IL-33 to induce STAT6 phosphorylation. This has been observed before and was linked to an IL-33 mediated IL-4 release that induces STAT6 phosphorylation via an autocrine IL-4 loop (33). Compatible with this model the further enhanced IL-4 release observed upon combined IL-33 and anti-Siglec-F treatment probably also induces the elevated and prolonged STAT6 phosphorylation. Using BMDE from STAT6-deficient mice we confirm that STAT6-signaling enhances effector release. However, a Siglec-F-induced effector release is also observed in STAT6-deficient BMDE, albeit at a lower level leaving the possibility of additional STAT6-independent pathways of Siglec-F-induced secretion of effectors.
The transcriptome analysis revealed hypoxia- and glycolysis-related genes such as the cytokine MIF to be highly upregulated by Siglec-F co-stimulation. MIF promotes inflammation, eosinophil maturation, chemotaxis and activation in type 2 immunity-related diseases (51), and may contribute to enhanced IL-13 secretion as suggested by others for murine asthma models (52, 53). Tarm1, another gene upregulated by Siglec-F costimulation, has been shown to enhance inflammatory cytokine secretion by macrophages and neutrophils upon LPS stimulation (37) suggesting that it may also promote Siglec-F-mediated enhanced effector release in eosinophils. On a broad perspective, a higher number of genes is differentially expressed upon Siglec-F co-stimulation and gene set enrichment analysis additionally suggests that Siglec-F primarily elevates enrichment of gene sets induced by IL-33. Therefore, Siglec-F is able to function as a co-stimulatory molecule for IL-33 induced expression changes.
Our mutation analysis shows that tyrosine phosphorylation of the inhibitory ITIM and ITIM-like motifs of the cytoplasmic tail are not mandatory for enhanced effector release. We mutated their tyrosine residues to phenylalanine, which has been described as effective to also prevent phosphorylation-independent SHP-1 and SHP-2 binding for another Siglec (12) and to mediate loss of inhibitory capacity of Siglec-8 on mast cells (54). Therefore, we likely suppress most of the described signaling capacity of the tail in the ITIM-mutant. Nevertheless, enhanced effector release of IL-33 and anti-Siglec-F treatment was only absent when the whole tail of Siglec-F was deleted also excluding that the remaining domains act as coreceptors for other proteins to mediate effector release. However, enhanced effector release was still seen in the ITIM-mutant. This again suggests ITIM-independent signaling capacity that induces activating functions of Siglec-F. However, we cannot exclude structural changes of the tail deletion mutant that might lead to an altered localization or interaction behavior.
In summary, Siglec-F is able to enhance IL-33-dependent secretion of the cytokines IL-4, IL-13 and the chemokines CCL3, CCL4 mediated by its cytoplasmic tail but independent of tyrosine phosphorylation in the ITIM and ITIM-like motifs, likely related to increased expression of hypoxia-induced pro-inflammatory molecules and enhanced/prolonged STAT6 phosphorylation.
Supplementary Material
Key points.
Siglec-F does not dampen A. fumigatus-elicited lung eosinophilia.
Siglec-F cross-linking enhances mediator release from IL-33-stimulated eosinophils.
This effect depends on the cytoplasmic tail but not the ITIM/ITIM-like motifs.
Acknowledgments
We thank Roland Lang for providing MyD88−/− bone marrow, Sven Krappmann, Sebastian Schrüfer and Michaela Dümig for providing A. fumigatus conidia, Christian Schwartz for providing human IL-33, Mark Gresnigt for advice on human eosinophil isolation, and the sequencing core unit at the human genetics of the University Hospital Erlangen, especially Arif Ekici for help to determine RNA quality.
The Deutsche Forschungsgemeinschaft (grants VO944/9-1 and CRC1181_A02 to D.V.) and a grant from the National Institute of Allergy and Infectious Diseases (U19 AI136443 to B.S.B.) supported this work.
Footnotes
Disclosures
B.S.B. receives remuneration for serving on the scientific advisory board of Allakos, Inc. and owns stock in Allakos. B.S.B. is a co-inventor on existing Siglec-8–related patents and thus may be entitled to a share of royalties received by Johns Hopkins University during development and potential sales of such products. B.S.B is also a co-founders of Allakos, Inc. which makes him subject to certain restrictions under University policy. The terms of this arrangement are being managed by Johns Hopkins University and Northwestern University in accordance with their conflict of interest policies. The remaining authors have no financial conflicts of interest.
References
- 1.Tateno H, Crocker PR, and Paulson JC. 2005. Mouse Siglec-F and human Siglec-8 are functionally convergent paralogs that are selectively expressed on eosinophils and recognize 6’-sulfo-sialyl Lewis X as a preferred glycan ligand. Glycobiology 15: 1125–1135. [DOI] [PubMed] [Google Scholar]
- 2.Angata T, Hingorani R, Varki NM, and Varki A. 2001. Cloning and characterization of a novel mouse Siglec, mSiglec-F: differential evolution of the mouse and human (CD33) Siglec-3-related gene clusters. J Biol Chem 276: 45128–45136. [DOI] [PubMed] [Google Scholar]
- 3.Zhang JQ, Biedermann B, Nitschke L, and Crocker PR. 2004. The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur J Immunol 34: 1175–1184. [DOI] [PubMed] [Google Scholar]
- 4.Zhang M, Angata T, Cho JY, Miller M, Broide DH, and Varki A. 2007. Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood 109: 4280–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nutku E, Aizawa H, Hudson SA, and Bochner BS. 2003. Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood 101: 5014–5020. [DOI] [PubMed] [Google Scholar]
- 6.Zimmermann N, McBride ML, Yamada Y, Hudson SA, Jones C, Cromie KD, Crocker PR, Rothenberg ME, and Bochner BS. 2008. Siglec-F antibody administration to mice selectively reduces blood and tissue eosinophils. Allergy 63: 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knuplez E, Krier-Burris R, Cao Y, Marsche G, O’Sullivan J, and Bochner BS. 2020. Frontline Science: Superior mouse eosinophil depletion in vivo targeting transgenic Siglec-8 instead of endogenous Siglec-F: Mechanisms and pitfalls. J Leukoc Biol 108: 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mao H, Kano G, Hudson SA, Brummet M, Zimmermann N, Zhu Z, and Bochner BS. 2013. Mechanisms of Siglec-F-induced eosinophil apoptosis: a role for caspases but not for SHP-1, Src kinases, NADPH oxidase or reactive oxygen. PLoS One 8: e68143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor VC, Buckley CD, Douglas M, Cody AJ, Simmons DL, and Freeman SD. 1999. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. J Biol Chem 274: 11505–11512. [DOI] [PubMed] [Google Scholar]
- 10.Yu Z, Maoui M, Wu L, Banville D, and Shen S. 2001. mSiglec-E, a novel mouse CD33-related siglec (sialic acid-binding immunoglobulin-like lectin) that recruits Src homology 2 (SH2)-domain-containing protein tyrosine phosphatases SHP-1 and SHP-2. Biochem J 353: 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avril T, Floyd H, Lopez F, Vivier E, and Crocker PR. 2004. The membrane-proximal immunoreceptor tyrosine-based inhibitory motif is critical for the inhibitory signaling mediated by Siglecs-7 and −9, CD33-related Siglecs expressed on human monocytes and NK cells. J Immunol 173: 6841–6849. [DOI] [PubMed] [Google Scholar]
- 12.Avril T, Freeman SD, Attrill H, Clarke RG, and Crocker PR. 2005. Siglec-5 (CD170) can mediate inhibitory signaling in the absence of immunoreceptor tyrosine-based inhibitory motif phosphorylation. J Biol Chem 280: 19843–19851. [DOI] [PubMed] [Google Scholar]
- 13.Kiwamoto T, Katoh T, Evans CM, Janssen WJ, Brummet ME, Hudson SA, Zhu Z, Tiemeyer M, and Bochner BS. 2015. Endogenous airway mucins carry glycans that bind Siglec-F and induce eosinophil apoptosis. J Allergy Clin Immunol 135: 1329–1340 e1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patnode ML, Cheng CW, Chou CC, Singer MS, Elin MS, Uchimura K, Crocker PR, Khoo KH, and Rosen SD. 2013. Galactose 6-O-sulfotransferases are not required for the generation of Siglec-F ligands in leukocytes or lung tissue. J Biol Chem 288: 26533–26545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiwamoto T, Brummet ME, Wu F, Motari MG, Smith DF, Schnaar RL, Zhu Z, and Bochner BS. 2014. Mice deficient in the St3gal3 gene product alpha2,3 sialyltransferase (ST3Gal-III) exhibit enhanced allergic eosinophilic airway inflammation. J Allergy Clin Immunol 133: 240–247 e241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMillan SJ, Richards HE, and Crocker PR. 2014. Siglec-F-dependent negative regulation of allergen-induced eosinophilia depends critically on the experimental model. Immunol Lett 160: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tateyama H, Murase Y, Higuchi H, Inasaka Y, Kaneoka H, Iijima S, and Nishijima KI. 2019. Siglec-F is induced by granulocyte-macrophage colony-stimulating factor and enhances interleukin-4-induced expression of arginase-1 in mouse macrophages. Immunology 158: 340–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willebrand R, and Voehringer D. 2016. IL-33-Induced Cytokine Secretion and Survival of Mouse Eosinophils Is Promoted by Autocrine GM-CSF. PLoS One 11: e0163751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohrs M, Shinkai K, Mohrs K, and Locksley RM. 2001. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity 15: 303–311. [DOI] [PubMed] [Google Scholar]
- 20.Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, and Lee JJ. 1997. Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol 158: 1332–1344. [PubMed] [Google Scholar]
- 21.McKenzie GJ, Fallon PG, Emson CL, Grencis RK, and McKenzie AN. 1999. Simultaneous disruption of interleukin (IL)-4 and IL-13 defines individual roles in T helper cell type 2-mediated responses. J Exp Med 189: 1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshino K, Kashiwamura S, Kuribayashi K, Kodama T, Tsujimura T, Nakanishi K, Matsuyama T, Takeda K, and Akira S. 1999. The absence of interleukin 1 receptor-related T1/ST2 does not affect T helper cell type 2 development and its effector function. J Exp Med 190: 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, and Akira S. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9: 143–150. [DOI] [PubMed] [Google Scholar]
- 24.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, Doherty PC, Grosveld G, Paul WE, and Ihle JN. 1996. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 380: 630–633. [DOI] [PubMed] [Google Scholar]
- 25.Dyer KD, Moser JM, Czapiga M, Siegel SJ, Percopo CM, and Rosenberg HF. 2008. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol 181: 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willebrand R, Dietschmann A, Nitschke L, Krappmann S, and Voehringer D. 2018. Murine eosinophil development and allergic lung eosinophilia are largely dependent on the signaling adaptor GRB2. Eur J Immunol 48: 1786–1795. [DOI] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao Y, Smyth GK, and Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spinelli L, Carpentier S, Montanana Sanchis F, Dalod M, and Vu Manh TP. 2015. BubbleGUM: automatic extraction of phenotype molecular signatures and comprehensive visualization of multiple Gene Set Enrichment Analyses. BMC Genomics 16: 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P. 2015. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouffi C, Rochman M, Zust CB, Stucke EM, Kartashov A, Fulkerson PC, Barski A, and Rothenberg ME. 2013. IL-33 markedly activates murine eosinophils by an NF-kappaB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J Immunol 191: 4317–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D, Janss T, Starkl P, Ramery E, Henket M, Schleich FN, Radermecker M, Thielemans K, Gillet L, Thiry M, Belvisi MG, Louis R, Desmet C, Marichal T, and Bureau F. 2016. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest 126: 3279–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dietschmann A, Schruefer S, Krappmann S, and Voehringer D. 2020. Th2 cells promote eosinophil-independent pathology in a murine model of allergic bronchopulmonary aspergillosis. Eur J Immunol 50: 1044–1056. [DOI] [PubMed] [Google Scholar]
- 36.Carroll DJ, O’Sullivan JA, Nix DB, Cao Y, Tiemeyer M, and Bochner BS. 2018. Sialic acid-binding immunoglobulin-like lectin 8 (Siglec-8) is an activating receptor mediating beta2-integrin-dependent function in human eosinophils. J Allergy Clin Immunol 141: 2196–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radjabova V, Mastroeni P, Skjodt K, Zaccone P, de Bono B, Goodall JC, Chilvers ER, Juss JK, Jones DC, Trowsdale J, and Barrow AD. 2015. TARM1 Is a Novel Leukocyte Receptor Complex-Encoded ITAM Receptor That Costimulates Proinflammatory Cytokine Secretion by Macrophages and Neutrophils. J Immunol 195: 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, and Yancopoulos GD. 2016. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov 15: 35–50. [DOI] [PubMed] [Google Scholar]
- 39.Song DJ, Cho JY, Lee SY, Miller M, Rosenthal P, Soroosh P, Croft M, Zhang M, Varki A, and Broide DH. 2009. Anti-Siglec-F antibody reduces allergen-induced eosinophilic inflammation and airway remodeling. J Immunol 183: 5333–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song DJ, Cho JY, Miller M, Strangman W, Zhang M, Varki A, and Broide DH. 2009. Anti-Siglec-F antibody inhibits oral egg allergen induced intestinal eosinophilic inflammation in a mouse model. Clin Immunol 131: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdala Valencia H, Loffredo LF, Misharin AV, and Berdnikovs S. 2016. Phenotypic plasticity and targeting of Siglec-F(high) CD11c(low) eosinophils to the airway in a murine model of asthma. Allergy 71: 267–271. [DOI] [PubMed] [Google Scholar]
- 42.Schanin J, Gebremeskel S, Korver W, Falahati R, Butuci M, Haw TJ, Nair PM, Liu G, Hansbro NG, Hansbro PM, Evensen E, Brock EC, Xu A, Wong A, Leung J, Bebbington C, Tomasevic N, and Youngblood BA. 2020. A monoclonal antibody to Siglec-8 suppresses non-allergic airway inflammation and inhibits IgE-independent mast cell activation. Mucosal Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dellon ES, Peterson KA, Murray JA, Falk GW, Gonsalves N, Chehade M, Genta RM, Leung J, Khoury P, Klion AD, Hazan S, Vaezi M, Bledsoe AC, Durrani SR, Wang C, Shaw C, Chang AT, Singh B, Kamboj AP, Rasmussen HS, Rothenberg ME, and Hirano I. 2020. Anti-Siglec-8 Antibody for Eosinophilic Gastritis and Duodenitis. N Engl J Med 383: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youngblood BA, Leung J, Falahati R, Williams J, Schanin J, Brock EC, Singh B, Chang AT, O’Sullivan JA, Schleimer RP, Tomasevic N, Bebbington CR, and Bochner BS. 2020. Discovery, Function, and Therapeutic Targeting of Siglec-8. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Post TW, Bozic CR, Rothenberg ME, Luster AD, Gerard N, and Gerard C. 1995. Molecular characterization of two murine eosinophil beta chemokine receptors. J Immunol 155: 5299–5305. [PubMed] [Google Scholar]
- 46.Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, Berdnikovs S, and Bryce PJ. 2016. IL-33 Precedes IL-5 in Regulating Eosinophil Commitment and Is Required for Eosinophil Homeostasis. J Immunol 197: 3445–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Na HJ, Hudson SA, and Bochner BS. 2012. IL-33 enhances Siglec-8 mediated apoptosis of human eosinophils. Cytokine 57: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nutku-Bilir E, Hudson SA, and Bochner BS. 2008. Interleukin-5 priming of human eosinophils alters siglec-8 mediated apoptosis pathways. Am J Respir Cell Mol Biol 38: 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroeger KM, Sullivan BM, and Locksley RM. 2009. IL-18 and IL-33 elicit Th2 cytokines from basophils via a MyD88- and p38alpha-dependent pathway. J Leukoc Biol 86: 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, and Janeway CA Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2: 253–258. [DOI] [PubMed] [Google Scholar]
- 51.Bozza MT, Lintomen L, Kitoko JZ, Paiva CN, and Olsen PC. 2020. The Role of MIF on Eosinophil Biology and Eosinophilic Inflammation. Clin Rev Allergy Immunol 58: 15–24. [DOI] [PubMed] [Google Scholar]
- 52.Torii M, Wang L, Ma N, Saito K, Hori T, Sato-Ueshima M, Koyama Y, Nishikawa H, Katayama N, Mizoguchi A, Shiku H, Yodoi J, Kuribayashi K, and Kato T. 2010. Thioredoxin suppresses airway inflammation independently of systemic Th1/Th2 immune modulation. Eur J Immunol 40: 787–796. [DOI] [PubMed] [Google Scholar]
- 53.Magalhaes ES, Mourao-Sa DS, Vieira-de-Abreu A, Figueiredo RT, Pires AL, Farias-Filho FA, Fonseca BP, Viola JP, Metz C, Martins MA, Castro-Faria-Neto HC, Bozza PT, and Bozza MT. 2007. Macrophage migration inhibitory factor is essential for allergic asthma but not for Th2 differentiation. Eur J Immunol 37: 1097–1106. [DOI] [PubMed] [Google Scholar]
- 54.Yokoi H, Choi OH, Hubbard W, Lee HS, Canning BJ, Lee HH, Ryu SD, von Gunten S, Bickel CA, Hudson SA, Macglashan DW Jr., and Bochner BS. 2008. Inhibition of FcepsilonRI-dependent mediator release and calcium flux from human mast cells by sialic acid-binding immunoglobulin-like lectin 8 engagement. J Allergy Clin Immunol 121: 499–505 e491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.