

Abstract
The interaction between Lymphocyte function-associated antigen 1 (LFA-1) and intercellular-adhesion molecule-1 (ICAM-1) plays important roles in the cell-mediated immune response and inflammation associated with dry eye disease. LFA-1/ICAM-1 antagonists can be used for the treatment of dry eye disease, such as Lifitegrast which has been approved by the FDA in 2016 as a new drug for the treatment of dry eye disease. In this study, we designed and synthesized some new structure compounds that are analogues to Lifitegrast, and their biological activities were evaluated by in vitro cell-based assay and also by in vivo mouse dry eye model. Our results demonstrated that one of these analogues of Lifitegrast (compound 1b) showed good LFA-1/ICAM-1 antagonist activity in in vitro assay; meanwhile, it also significantly reduced ocular surface epithelial cells damage, increased goblet cell density in dry eye mouse and highly improved the symptoms of dry eye mouse.
Graphical abstract
Keywords: Lifitegrast, Analogues, LFA-1/ICAM-1antagoinst, Dry eye, Corneal epithelial in jury
Introduction
Dry eye disease (DED) is a complex, multifactorial condition characterized by inflammation of the ocular surface and lacrimal glands, as well as the reduction in the quality and/or quantity of tears [1]. DED can be categorized as “dry eye with reduced tear production (aqueous deficient) and dry eye with increased evaporation of the tear film known as the hyperevaporative type” [2]. Dry eye patients suffer from ocular-related symptoms, such as stinging, burning, itching, light sensitivity, and blurry vision which limit the quality of their life, as well as their ability to work [3]. The prevalence of DED is between 5% and 50% in the different countries, and this prevalence can be up to 75% in adults over age 40, with mostly female patients. In adults aged 18–40, 2.7% of these people have had an experience of DED [4].
Lymphocyte function-associated antigen 1 (LFA-1) is a leukocyte cell surface glycoprotein widely expressed on cells of a hematopoietic lineage [5]. Previous functional studies indicated that intercellular-adhesion molecule-1 functions as a ligand for LFA-1-dependent adhesion by a variety of leukocytes [6]. LFA-1 functions are extremely varied but play a critical role in facilitating effective immune responses. It has been reported that LFA-1 was implicated in numerous autoimmune and inflammatory conditions including inflammatory bowel disease, psoriasis, diabetes, and arthritis [7]. The previous study suggested that LFA-1/ICAM-1 interaction may play important roles in the cell-mediated immune response and inflammation associated with DED. Notably, inhibition of LFA-1/ICAM-1 binding offers a novel and possible approach to reducing ocular surface inflammation in this condition [8].
The immunosuppressive mechanism of Lifitegrast is different from the first-generation immunosuppressor (cyclosporine A) for dry eye treatment. Lifitegrast (Fig. 1) is a small molecule that inhibits T-cell-mediated inflammation by blocking the binding of LFA-1 and ICAM-1, thus lessening overall inflammatory responses [9]. In vitro, Lifitegrast potently inhibited the attachment of LFA-1 to ICAM-1 in a concentration-dependent manner, with a half-maximal inhibitory concentration (IC50) of 2.98 nmol/L [10]. The results from the phase 3 clinic trial of Lifitegrast have shown its ability to improve ocular surface epithelial health and associated symptoms of dry eye patients, thereby demonstrating its potential as a relatively fast-acting second-generation agent for the treatment of DED [11]. Lifitegrast became the second topical anti-inflammatory agent which has been approved by the FDA in 2016 for the treatment of DED [12].
Fig. 1.
Chemical structure of Lifitegrast
Lifitegrast ophthalmic solution (Xiidra) is labeled for the treatment of DED in concentrations as high as 5.0%. Such high concentrations result in many side effects, such as ocular irritation, dysgeusia and reduced visual acuity, especially an unusual taste sensation in the mouth. The LFA-1/ICAM-1 antagonist activity of Lifitegrast is not high enough to overcome these side effects. Therefore, it is very interesting to find novel higher LFA-1/ICAM-1 antagonist activity compounds. The discovery process and structure–activity relationships (SARs) of Lifitegrast were reported in a previous report [13]. So far, no other medicinal chemistry studies about Lifitegrast analogues have been reported. In this study, a novel series of Lifitegrast analogues with systematic modifications were further designed, synthesized, and their activities as LFA-1/ICAM-1 antagonists were evaluated by in vitro and in vivo assays.
Results and discussion
Design of Lifitegrast analogues
Lifitegrast is used as a lead compound for designing analogues. It has been established that the structure of central tetrahydroisoquinoline (THIQ) scaffold (Fig. 2) is a pharmacophore that is essential for LFA-1/ICAM-1 antagonist activity [14]. Previous SAR studies have shown that an aromatic group in the “left-wing” residue is necessary for LFA-1/ICAM-1 antagonist activity [15]. Therefore, we attempted to link tetrahydroisoquinoline and benzofuran with different groups (Fig. 2, 1a–1c) to investigate the LFA-1/ICAM-1 antagonist activity. In order to investigate the effect of X groups with different electron-absorbing properties and different sizes on the activity, we selected sulfone group, N-methylene carbamoyl and methylene carbonyl as substituents. Meanwhile, the effects of substitution of benzofuran with different aromatic groups (Fig. 2, 1d–1e) on the activity were investigated. Although benzotriazole and benzofuran have different atomic compositions, they are both benzo five-membered rings. From the activity results of benzotriazole, it can be seen whether there is a more suitable Ar1 group. Ethynylbenzene was selected to investigate whether Ar1 ring opening would have a significant effect on activity. At the same time, we incorporated different aromatic groups into “right-wing” amino acids (Fig. 2, 1f–1j) to investigate the LFA-1/ICAM-1 antagonist activity. We cyclized methylsulfonyl with phenyl and converted methylsulfonylphenyl to benzene six- membered rings such as quinoline and 2-oxo-1,2,3,4-tetrahydroquinolin to investigate the effect of cyclization and different group size on the activity. According to the results of previous studies, Lifitegrast is located in the I-domain allosteric site of LFA-1/ICAM-1. Ar2 group of Lifitegrast interact with the protein mainly through hydrogen-bonds with residues such as Tyr257 and Lys287 [16]. Therefore, methylsulfonyl group was converted to acetyl group and hydroxyl group to change the interaction mode of the hydrogen bond between Ar2 with the protein to investigate the effect on the activity.
Fig. 2.
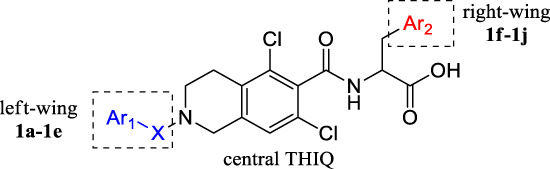
Design rationale of the target LFA-1/ICAM-1 antagonists
Chemistry
Amide intermediate (4) was synthesized by condensation of carboxylic acid (2) with primary amine (3) in DMF in the presence of condensating agent HATU, which was deprotected Boc group to yield amide intermediate (5) (Scheme 1) [17]. The yield of condensation reaction is low due to steric size of compound 2 and 3.
Scheme 1.

Reagents and conditions: a HATU, TEA, DMF, 20 °C, 16 h, 25.2%; b HCl, EtOAc, 20 °C, 3 h
Analogues 1a–1e were generated through the synthetic route outlined in Scheme 2. Amide intermediate (5) was treated with benzofuran-6-sulfonyl chloride (10), benzofuran-6-ylmethanamine (14), 2-(benzofuran-5-yl)acetic acid (16) [18], [1,2,4]triazolo[1,5-a]pyridine-6-carboxylic acid (18) [19] and 3-ethynylbenzoic acid 20 [20] in DCM in the presence of condensating agent HATU or CDI to yield benzyl group analogues 11, 15, 17, 19 and 21, which provided analogues 1a–1e in the presence of debenzylation agent H2 or TMSOK in MeOH or THF through removing the benzyl group [21]. The condensation activity of carboxyl with amino is stronger than that of amine with amino group, so the yield of compound 11, 17, 19, 21 was higher than that of compound 15.
Scheme 2.

Reagents and conditions: a TEA, HATU, DCM, 20 °C, 3 h, 69.8– 87.2%; b H2, Raney-Ni, MeOH, 25 °C, 3 h, 9.42–33.4%; c CDI, DIEA, DCM, 0–25 °C, 1 h, 9.42%; d TMSOK, THF, 70 °C, 1 h, 22.0–22.2%
Williamson reaction of 2-bromo-1,1-diethoxyethane (6) with 3-bromophenol (7) yielded 1-bromo-3-(2,2-diethoxyethoxy)benzene (8), which was cyclized to the corresponding 6-bromobenzofuran (9) by heating in toluene containing cyclizing agent PPA. Sulfonylation of intermediate 9 by treatment with sulfuryl chloride in THF catalyzed by halogen-lithium exchange reagent n-BuLi gave the respective benzofuran-6-sulfonyl chloride (10) (Scheme 3) [22]. The sulfonation of intermediate 9 first involved the exchange of halogen atom to lithium atom, and then lithium atom was replaced by sulfonyl group. The complexity of the reaction mechanism leaded to the low yield of this reaction.
Scheme 3.

Reagents and conditions: a K2CO3, DMF, 100 °C, 10 h, 47.8%; b PPA, Toluene, 80 °C, 4 h, 40.3%; c n-BuLi, SO2, Sulfuryl Chloride, THF, −78 °C, 12 h, 22.7%
6-bromobenzofuran (12) was treated with zinc cyanide and cyanation catalyst tetrakis(triphenylphosphine)palladium to give benzofuran-6-carbonitrile (13), which was reduced by reducing agent lithium aluminum hydride yield benzofuran-6-ylmethanamine (14) (Scheme 4) [23]. The cyanidation reaction (13) and reduction reaction (14) were both classical reactions, the yields of this two reaction were normal.
Scheme 4.
Reagents and conditions: a Zn(CN)2, Pd(PPh3)4, DMF, 80 °C, 16 h, 68.8%; b LiAlH4, THF, 20 °C, 1 h, 48.6%
Condensation reaction of benzofuran-6-carboxylic acid (22) with methyl 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxylate (23) by condensating agent EDCI yielded carboxylic acid methyl ester intermediate (24), which was hydrolyzed to the corresponding carboxylic acid intermediate (25) by heating in pyridine containing hydrolysis reagent lithium iodide (Scheme 5) [24]. The hydrolysis of methyl carboxylate (24) involved the formation of methyl iodide and carboxylic acid lithium, then the reaction yield was slightly lower.
Scheme 5.

Reagents and conditions: a EDCI, DMAP, TEA, DMF, 25 °C, 15 h, 58.6%; b LiI, Pyridine, 100 °C, 3 h, 48.1%
Analogues 1f–1h were generated through the synthetic route outlined in Scheme 6. Carboxylic acid intermediate (25) was treated with ethyl 2-amino-3-(quinolin-6-yl)propanoate (30), ethyl 2-amino-3-(1,1-dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)propanoate (41), ethyl 2-amino-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)propanoate (50) in DCM in the presence of condensating agent HATU to yield carboxylic acid ethyl ester analogues 31, 42, 51 which provided analogues 1f–1h in the presence of hydrolysis reagent sodium hydroxide in EtOH, H2O and THF mixed solvent. The hydrolysis of ethyl carboxylate (31, 42, 51) is more difficult than that of methyl carboxylate (24), the yield is lower than that of methyl carboxylate.
Scheme 6.

Reagents and conditions: a HATU, TEA, DMAP, DCM, 20–30 °C, 20.5 h, 37.7–38.9%; b NaOH, EtOH/H2O/THF = 2/1/1, 20 °C, 2 h, 12.0–12.8%
Quinoline-6-carbaldehyde (26) was reduced in methanol in the presence of reducing agent sodium borohydride to yield 6-Hydroxymethyl-quinoline (27), which provided 6-(bromomethyl)quinoline (28) in acetic acid in the presence of hydrogen bromide. The activity of hydrogen bromide as a halogenated agent generally resulted in a slightly lower yield of halogenated reaction (28). The intermediate (28) was treated with ethyl N-(diphenylmethylene)glycinate in THF in the presence of alkylation catalyst sodium hydride to yield ethyl 2-((diphenylmethylene)amino)-3-(quinolin-6-yl)propanoate (29), which was hydrolyzed in ethyl acetate in the presence of hydrolysis reagent hydrochloric acid to yield ethyl 2-amino-3-(quinolin-6-yl)propanoate (30) (Scheme 7) [25]. Imine (29) was hydrolyzed to primary amine (30) under hydrochloric acid with normal yield.
Scheme 7.

Reagents and conditions: a NaBH4, MeOH, 0–15 °C, 3 h, 65.8%; b HBr, AcOH, 20 °C, 3 h, 37.5%; c Ethyl N-(diphenylmethylene)glycinate, NaH, THF, 5–20 °C, 20.5 h, 70.4%; d HCl, EtOAc, 20 °C, 0.5 h, 66.8%
2-amino-3-(1,1-dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)ethyl propanoate (41) was generated from benzo[b]thiophene 1,1-dioxide (32) in Scheme 8. 6-nitrobenzo[b]thiophene 1,1-dioxide (33) was obtained by treating compound (32) with nitric and sulfuric acid. This nitrification reaction (33) was a classic reaction, and the yield is in normal range. The nitro group is reduced to an amine group to give 6-amino-2,3-dihydrobenzo[b]thiophene 1,1-dioxide (34), which is replaced by the bromine atom to form 6-bromo-2,3-dihydrobenzo[b]thiophene 1,1-dioxide (35). The activity of substituting amino group (34) with hydrogen bromide as halogenated reagent was higher than that of substituting hydroxyl group (27). Zinc cyanide reacted with intermediate (35) to produce 2,3-dihydrobenzo[b]thiophene-6-carbonitrile 1,1-dioxide (36), which was reduced to give 2,3-dihydrobenzo[b]thiophene-6-carbaldehyde 1,1-dioxide (37). 2-amino-3-(1,1-dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)ethyl propanoate (41) was synthesized from intermediate (37) through reduction reaction, halogenating reaction, hydrocarbylation reaction and hydrolysis reaction [26]. These reactions belonged to the classical reactions, the yields were in the normal range.
Scheme 8.

Reagents and conditions: a HNO3/H2SO4 (1/1), 0 °C, 1 h, 70.8%; b Pd/C, H2, MeOH, 50 °C, 12 h, 64.0%; c NaNO2, CuBr, HBr, 0–70 °C, 1 h, 59.1%; d Zn(CN)2, Pd(PPh3)4, DMF, 100 °C, 16 h, 63.9%; e Ni, HCOOH, 100 °C, 2 h, 88.6%; f NaBH4, MeOH, 0–15 °C, 3 h, 49.4%; g HBr, AcOH, 20 °C, 2 h, 60.7%; h Ethyl N-(diphenylmethylene)glycinate, NaH, THF, 0–20 °C, 16 h, 59.5%; i HCl, EtOAc, 20 °C, 1 h, 94.7%
2-amino-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)ethyl propanoate (50) was generated from 7-hydroxy-3,4-dihydroquinolin-2(1H)-one (43) in Scheme 9. The hydroxyl group of compound (43) was esterified by trifluoroacetic anhydride to give 2-oxo-1,2,3,4-tetrahydroquinolin-7-yl trifluoromethanesulfonate (44). Zinc cyanide reacted with intermediate (44) to produce 2-oxo-1,2,3,4-tetrahydroquinoline-7-carbonitrile (45), which was reduced to give 2-oxo-1,2,3,4-tetrahydroquinoline-7-carbaldehyde (46). Since trifluoroacetate was an excellent leaving group, the cyanidation reaction yield was over 50%. 2-amino-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)ethyl propanoate (50) was synthesized from intermediate (46) through reduction reaction, halogenating reaction, hydrocarbylation reaction and hydrolysis reaction [27]. These reactions belonged to the classical reactions, the yields were in the normal range.
Scheme 9.

Reagents and conditions: a (Tf)2O, Py, CHCl3, 0–25 °C, 40 min, 55.2%; b Zn(CN)2, Pd(PPh3)4, DMF, 80 °C, 12 h, 55.2%; c Ni, HCOOH, 100 °C, 2 h, 58.9%; d NaBH4, MeOH, 15 °C, 3 h, 49.4%; e HBr, AcOH, 25 °C, 0.5 h, 33.9%; f Ethyl N-(diphenylmethylene)glycinate, NaH, THF, 0–25 °C, 5 h, 61.9%; g HCl, EtOAc, 0–25 °C, 0.5 h, 88.6%
Analogues 1i–1j were generated through the synthetic route outlined in Scheme 10. Acyl chloride intermediate (52) was obtained by treating carboxylic acid intermediate (25) with dichlorosulfoxide. Compound 52 was directly used in the next reaction without purification. Acyl chloride intermediate (52) was acylated with compound 3-(3-acetyl-4-hydroxyphenyl)-2-aminopropanoic acid (53) and 2-amino-3-(3,4-dihydroxyphenyl)propanoic acid (54) in the presence of TEA in DMF to yield analogues 1i and 1j, respectively. The yields of condensations (1i, 1j) were low because the carboxyl group at the α position reduces the nucleophilicity of the amino group (53, 54).
Scheme 10.

Reagents and conditions: a SOCl2, 70 °C, 2 h; b TEA, DMF, 70 °C, 2 h, 5.19–5.43%
A total of 5 target “left-wing” analogues, 5 target “right-wing” analogues and more than 30 intermediates were successfully synthesized. All the designed target analogues were confirmed correct structure by 1H NMR, 13C NMR, LCMS and HRMS, and the LCMS content reached more than 95%. The structures of analogues are shown in Table 1 and the detailed synthesis process of each compound can be found in Supplementary Materials of this article.
Table 1.
LFA-1/ICAM-1 antagonist activities of Lifitegrast analogues

n.a. no activity, Ar aromatic groups
aThe IC50 value is an average of three titrations with six concentration points
Cell attachment assay
The IC50 value activity test results were shown in Fig. 3 and Table 1. The IC50 value of Lifitegrast was about 10.5 nM (Fig. 3) measured by our assay, this IC50 value was very close to the previously reported IC50 value of Lifitegrast (IC50 = 2.98 nM and 9 nM in Jurkat or in HUT 78T cells, respectively) [9]. The activity test results showed that the IC50 value of compound 1b reached the level of 262 nM, and had a good LFA-1 inhibitory activity. The IC50 of compounds 1c, 1f and 1h also reached several micromol levels. Among them, 1f and 1h are racemes, and the activity of their S configuration will be higher.
Fig. 3.
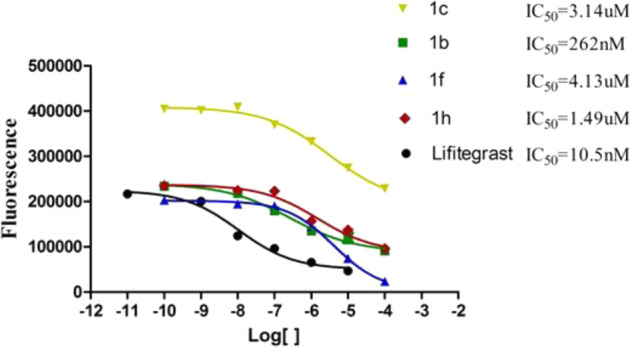
Titration curves of compounds 1c, 1b, 1f, 1h and Lifitegrast in the Jurkat T-cell adhesion assay. Compounds 1c, 1b, 1f or 1h inhibit the attachment of Jurkat cells to intercellular-adhesion molecule (ICAM)-1 in vitro, 1/10 serial dilutions of the compounds, and labeled Jurkat cells were incubated with either the compounds or Lifitegrast on plates containing captured ICAM-1. The solid lines are the fits of the data. The IC50 values are provided in the legends
From the results of Jurkat T-cell adhesion assay, we may summarize some rules about the relationship between the structures of the designed target compounds and LFA-1/ICAM-1 antagonist activities. With Lifitegrast as a lead compound, in the “left-wing” residue, changing carbonyl group in Lifitegrast to sulfone group (compound 1a), or changing furyl to triazole (compound 1d) or acetenyl (compound 1e) decreased LFA-1/ICAM-1 antagonist activity obviously when compared with Lifitegrast. The activity of compound 1a showed that the carbonyl group could not be replaced by the more electron-absorbing sulfone group between benzofuran and tetrahydroisoquinoline, probably because the charge distribution of the whole molecule was affected. The charge distribution of triazole is quite different from that of furyl, which may contribute to the poor activity of compound 1d. The ring structure of benzofuran was opened is the reason for the poor activity of compound 1e, indicating that the ring structure plays an important role in the activity. While the carbonyl was changed to the N-methylene carbamoyl (compound 1b) and methylene carbonyl (compound 1c), the analogues show certain LFA-1/ICAM-1 antagonist activity. This result indicated that the length of the carbonyl between benzofuran and tetrahydroisoquinoline can be appropriately extended without adversely affecting the binding of the molecule to the target protein. The IC50 value of compound 1b reached the level of 262 nM, this result suggested that the LFA-1/ICAM-1 antagonist activity can be maintained or even improved by only changing the X group to the other groups. In the “right-wing” Ar2 residue of the Lifitegrast, replacing methylsulfonyl benzene with quinoline (compound 1f), 1-dioxido-2,3-dihydrobenzothiophen (compound 1g), 2-oxo-1,2,3,4-tetrahydroquinolin (compound 1h), 3-acetyl-4-hydroxyphenyl (compound 1i) or 3,4-dihydroxyphenyl (compound 1j) respectively decreased LFA-1/ICAM-1 antagonist activity. The cyclization of the sulfone group with benzene (compound 1g) resulted in a loss of activity because of the influence on the binding mode of the sulfone group to the target protein. Meanwhile, the IC50 value of compounds 1f and 1h also reached several micromol levels. These results showed that replacing the Ar2 residue with benzene six-membered ring could not improve the LFA-1/ICAM-1 antagonist activity, this may be related to increasing the size of the AR2 group. Conversion of methylsulfonyl group to acetyl or hydroxyl groups on benzene ring also could not produce good activity. This result indicated that adding hydroxyl groups to the benzene ring did not increase the hydrogen bond interaction between the analogues and the target protein.
Animal experiments
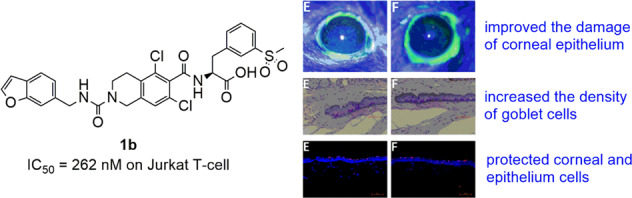
Compound 1b reduced the damage of corneal epithelium in dry eye mice model
After the 14 days of desiccating stress and drug-induction, we established a dry eye model that causes dry eye symptoms closer to the symptoms in patients with chronic DED [28]. Then we evaluated if compound 1b could improve the permeability barrier integrity of the ocular surface in this animal model.
Fluorescein staining in Fig. 4 showed punctate staining of green on the corneal surfaces in the experimental and control groups at day 14. We can see that the fluorescence dyeing areas on the ocular surface were significantly increased in the dry eye model group (B, EDE group) compared with the blank group (A, control group). Treatment with 5% or 7% compound 1b eye drops (E, F groups) improved the damage of corneal epithelium with less fluorescence dyeing areas, and treatment with 5% Lifitegrast (C group) was used as the positive control, which reveals the prominent therapeutically effects on DED.
Fig. 4.

Effect of compound 1b on ocular surface damage in dry eye mice. After 14 days treatment on dry eye mice model, ocular surface of mice were stained with fluorescein sodium, the green fluorescence staining points represent the damaged area of ocular surface epithelium (A–F), and the corneal fluorescence staining score in graph A–F were calculated and shown in a bar graph (G). Data were shown as mean ± SEM (n = 10, eyes/group). *P < 0.05, **P < 0.01, ***P < 0.001. Control: non-treated normal mice group; EDE: experimental dry eye mice group; EDE + Lifitgrast: experimental dry eye mice treated with Lifitgrast; EDE + Vehicle: experimental dry eye mice treated with control solvent; EDE + 5%1b: experimental dry eye mice treated with 5% compound 1b; EDE + 7%1b: experimental dry eye mice treated with 7% compound 1b
Compound 1b enhanced the down-regulated goblet cells in dry eye mice model
The conjunctival goblet cells were distributed mainly among the fornix epithelial cells. Goblet cells could secrete the mucin and keep the tear film stable. The symptoms of dry eye are usually accompanied by the loss of goblet cells [29]. Therefore, we assessed the effects of compound 1b on goblet cells in dry eye mice model. As shown in Fig. 5, The number of goblet cells from the superior and inferior conjunctiva were markedly decreased in dry eye model group (B) compared with blank group (A, control group). Treatment with 5% or 7% compound 1b (E and F groups) significantly increased the density of goblet cells compared with the dry eye model group (B). It is obvious that compound 1b prevented the missing of the goblet cells in dry eye mice.
Fig. 5.

Compound 1b enhanced the density of goblet cells on conjunctival epithelium in dry eye mice after 14 days treatment. The results of histopathological evaluation of the goblet cells staining by periodic acid-schiff were shown in A–F images (size, ×400). The deep purple dyeing in conjunctiva represents goblet cells. The quantification of goblet cells densities in graph A–F was shown in G graph, data was shown as mean ± SEM (n = 10 eyes/group). *P < 0.05, **P < 0.01, ***P < 0.001. Control: non-treated normal mice group; EDE: experimental dry eye mice group; EDE + Lifitgrast: experimental dry eye mice treated with Lifitgrast; EDE + Vehicle: experimental dry eye mice treated with control solvent; EDE + 5%1b: experimental dry eye mice treated with 5% compound 1b; EDE + 7%1b: experimental dry eye mice treated with 7% compound 1b
Compound 1b inhibited the apoptosis of corneal epithelial cells in dry eye mice model
In order to investigate if compound 1b has anti-apoptotic effects to protect the ocular surface damage in dry eye mice, we detected the signals of apoptosis on corneal epithelium by using TUNEL staining method in dry eye mice model with or without compound 1b treatment. TUNEL immunofluorescence staining is a method to analyze apoptosis at a late stage by detecting DNA fragmentation through labeling the terminal end of nucleic acids in the cells. The results of TUNEL immunofluorescence staining in Fig. 6 demonstrated that compared to control group, mean numbers of corneal epithelium cells which had the signals of TUNEL staining with Cy3 were significantly increased in the dry eye model groups (EDE, and EDE + Vehicle groups). In compound 1b treatment group (EDE + 5% 1b, or EDE + 7% 1b), the amount of cells labeled with Cy3 were markedly decreased in cornea tissues when compared with that of dry eye model group, and the quantitative statistical analysis data of TUNEL positive cells in each group was shown in Fig. 6G. But the anti-apoptotic effect of compound 1b was weaker than that of Lifitegrast treatment group. These results in Fig. 6 suggested that treatment with compound 1b could protect corneal and conjunctival epithelium cells from apoptosis, maintained the integrity of ocular epithelium in dry eye mice.
Fig. 6.

Compound 1b attenuated the apoptosis of corneal epithelium in dry eye mice model. The red fluorescence stain (TUNEL) represents apoptotic cells and the blue stain (DAPI) represents cell nucleus. The quantification of TUNEL positive staining cells in corneal epithelium were shown in G graphs, respectively. Data were shown as mean ± SEM (n = 10 eyes/group).*P < 0.05, **P < 0.01, ***P < 0.001. Control: non-treated normal mice group; EDE: experimental dry eye mice group; EDE + Lifitgrast: experimental dry eye mice treated with Lifitgrast; EDE + Vehicle: experimental dry eye mice treated with control solvent; EDE + 5%1b: experimental dry eye mice treated with 5% compound 1b; EDE + 7%1b: experimental dry eye mice treated with 7% compound 1b
The Lifitegrast eye drops showed a statistically significant reduction in ocular surface injury compared to the model control group, and the effect was stronger than compound 1b. As shown in Fig. 4, 5% and 7% compound 1b significantly reduced ocular surface epithelial cell damage in dry eye mice. 5% and 7% compound 1b reduced the damage of corneal epithelium in dry eye mice model. The conjunctiva goblet cells secrete the mucin to maintain the stability of the tear film. The symptoms of dry eye are usually accompanied by a decreasing of the number or density of goblet cells. We evaluated the effect of 5% and 7% compound 1b on goblet cells in dry eye mice model. A statistical histogram of goblet cell density is shown in Fig. 5. 5% and 7% compound 1b significantly increased goblet cell density and improved dry eye symptoms in mice with dry eye. As shown in Fig. 6, 5% and 7% compound 1b has a protective effect on apoptosis of ocular surface cells in dry-eye mice, but the effect is weaker than that of Lifitegrast. The results from our in vivo animal model experiments showed that topical administration of 5% and 7% (W/V) compound 1b improved the dry eye symptoms through reducing the ocular surface injuries, increasing the density of goblet cells on conjunctival epithelium and decreasing the apoptosis of cornea and conjunctiva. There was no statistical difference in the effect between 5% and 7% compound 1b. There was no statistical difference in the effect between 5% and 7% compound 1b. The in vitro assay showed the IC50 value of compound 1b reached the level of 262 nM which is lower than that of Lifitegrast (IC50 value is about 11 nM). This result suggested that the LFA-1/ICAM-1 antagonist activity of compound 1b may be weaker than that of Lifitegrast also in vivo, therefore, the anti-dry eye effects of compound 1b is less as effective as Lifitegrast.
Although the effect of compound 1b is weaker than Lifitegrast in vitro and in vivo, the structure of compound 1b is different from Lifitegrast and circumvented the scope of Lifitegrast original patent. These results may serve as important milestone references for future structure optimization.
Conclusions
In summary, in this study, 10 analogues with different groups in the “left-wing” or “right-wing” residues in Lifitegrast were designed according to molecular dynamics simulations and were synthesized through a variety of chemical methods. The Jurkat cell attachment assay was used to assess the LFA-1/ICAM-1 antagonist activity of these 10 analogues of Lifitegrast. Among them, the IC50 value of compound 1b reached 262 nM. To further verify the biological activity of compound 1b in vivo, dry eye mice model was developed to investigate if compound 1b could improve the symptoms of dry eye. The results of animal model demonstrated that 5%(W/V) compound 1b significantly improved the symptoms of dry eye mice through reducing ocular surface epithelial cells damage, increasing goblet cell density and decreasing the apoptosis of cornea and conjunctiva in dry eye mice. The results of this study encourage us to further optimize the structure of compound 1b in our future work, looking for a compound with a better LFA-1/ICAM-1 antagonist activity.
Experimental
Chemistry
All solvents and reagents were purchased from WuXi AppTec (Shanghai, China), Accela ChemBio Co., Ltd. (Shanghai, China), Titan Scientific Co.,Ltd. (Shanghai, China), and Hao Hong Pharma (Shanghai, China). Solvents and reagents were generally the best quality commercial-grade products and were used without further purification. 1H and 13C NMR spectra were taken on Bruker Avance III 500 or 400 NMR spectrometers operating at 400 MHz for 1H NMR, and 100 MHz for 13C NMR, with tetramethylsilane (TMS) as the internal standard and CDCl3, MeOD and DMSO-d6 as the solvent. Chemical shift values (δ) are reported in ppm relative to that of the internal TMS. The following abbreviations were used: s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet, and b = broad. 13C NMR spectra were recorded with complete proton decoupling. ESIMS data were recorded on Finnigan LCQ/DECA instruments. HRMS data were collected on Micromass QTOF Ultima (ESI) spectrometers. Silica gel F254 was used in analytical thin-layer chromatography (TLC), and silica gel was used in column chromatography; visualizations were accomplished with UV light (254 nm).
(S)-2-(2-(tert-Butoxycarbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl) benzyl propanoate (4)
A mixture of compound 2 (5.61 g, 16.2 mmol), compound 3 (4.50 g, 13.5 mmol), HATU (7.70 g, 20.2 mmol) and TEA (2.05 g, 20.25 mmol) in DMF (30 mL) was stirred at 20 °C for 16 h. TLC showed new spot was formed. H2O (30 mL) was added and the mixture was extracted with EtOAc (30 mL × 2). The organic layer was isolated and dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash silica gel chromatography. Compound 4 (2.20 g, 2.83 mmol, 25.2% yield) was obtained as light yellow gum. 1H NMR (CDCl3, 400 MHz) δ 7.84 (s, 1H, CONH), 7.82–7.81 (m, 2H, Ar–H), 7.47–7.31 (m, 5H, Ar–H), 7.25–7.24 (s, 1H, Ar–H), 7.10–7.07 (m, 1H, Ar–H), 6.43–6.35 (m, 1H, Ar–H), 5.33–5.23 (m, 1H, CHCO), 5.22–5.16 (m, 2H, CH2N), 4.57 (s, 2H, CH2, Bn), 3.64 (t, J = 6.0 Hz, 2H, CH2N), 3.40 (m, 1H, *CH2CH2N), 3.34–3.24 (m, 1H, *CH2CH2N), 2.99 (s, 3H, CH3SO2), 2.81–2.75 (m, 2H, CH2CH), 1.48 (s, 9H, CH3, Boc).
(S)-2-(5,7-Dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl) benzyl propanoate (5)
To a solution of compound 4 (2.20 g, 3.33 mmol) in EtOAc (10.0 mL), HCl/EtOAc (4 M, 15 mL) was added. The mixture was stirred at 20 °C for 3 h. TLC showed new spot was formed. The residue was concentrated under reduced pressure. Compound 5 (1.80 g, crude, HCl) was obtained as white solid. 1H NMR (MeOD, 400 MHz) δ 7.91 (s, 1H, Ar–H), 7.82 (d, J = 7.8 Hz, 1H, Ar–H), 7.64 (d, J = 7.8 Hz, 1H, Ar–H), 7.58–7.51 (m, 1H, H-4, Ar–H), 7.42–7.29 (m, 5H, Ar–H), 7.31 (s, 1H, Ar–H), 5.22 (d, J = 4.4 Hz, 2H, CH2N), 5.16 (m, 1H, CHCO), 4.36 (s, 2H, CH2, Bn), 3.56–3.50 (m, 2H, CH2N), 3.49–3.41 (m, 1H, *CH2CH2N), 3.21–3.12 (m, 1H, *CH2CH2N), 3.07 (s, 3H, CH3SO2), 3.01 (t, J = 6.4 Hz, 2H, CH2CH), 2.01 (s, 1H, CH2NHCH2).
(S)-2-(2-(Benzofuran-6-ylsulfonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)benzyl propanoate (11)
To a solution of compound 5 (0.300 g, 501 µmol, HCl) in DCM (3.00 mL), TEA (152 mg, 1.51 mmol) and HATU (286 mg, 752 µmol) were added. The solution was stirred at 20 °C for 3 h, then compound 10 (108 mg, 501 µmol) was added via several portions. The reaction mixture was stirred at 20 °C for 2 h under N2 atmosphere. LCMS showed starting material was consumed completely. The reaction was quenched with water (3.00 mL), and the organic was extracted with DCM (3.00 mL × 3), then the organic layer was concentrated under reduced pressure. Compound 11 (0.260 g, 350 µmol, 69.8% yield) was obtained as a brown solid. 1H NMR (CDCl3, 400 MHz) δ 8.01 (s, 1H, CONH), 7.81–7.85 (m, 1H, O*CH=CH), 7.81–7.85 (m, 1H, Ar–H), 7.77–7.81 (m, 1H, Ar–H), 7.75 (s, 1H, Ar–H), 7.67–7.71 (m, 1H, Ar–H), 7.40–7.43 (m, 2H, Ar–H), 7.36–7.27 (m, 5H, Ar–H), 7.02 (s, 1H, Ar–H), 6.88 (d, J = 1.6 Hz, 1H, *CH=CHO), 6.39 (d, J = 7.6 Hz, 1H, Ar–H), 5.22–5.29 (m, 1H, CHCO), 5.19 (d, J = 6.0 Hz, 2H, CH2N), 4.20 (s, 2H, CH2, Bn), 3.35–3.39 (m, 2H, CH2N), 3.24–3.31 (m, 1H, *CH2CH2N), 2.93 (s, 3H, CH3SO2), 2.86 (m, 1H, *CH2CH2N), 2.18 (m, 2H, CH2CH); LCMS m/z 741.0 [M + H]+.
(S)-2-(2-(Benzofuran-6-ylsulfonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)propanoic acid (1a)
To a solution of compound 11 (0.260 g, 350 µmol) in MeOH (1.00 mL), Raney-Ni (30.0 mg, 350 µmol) was added under N2. The suspension was degassed under vacuum and purged with H2 several times. Then the mixture was stirred under H2 (15 psi) at 25 °C for 3 h. TLC indicated starting material was consumed completely. After that, the suspension was filtered through a pad of Celite and the pad was washed with EtOH (2.00 mL × 3). The combined filtrates were concentrated to dryness to give product, and the crude product was purified by prep-HPLC (HCl condition). Compound 1a (0.022 g, 33.0 µmol, 9.42% yield, 97.7% purity) was obtained as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 12.88 (s, 1H, COOH), 9.02 (d, J = 8.4 Hz, 1H, CONH), 8.29 (d, J = 2.4 Hz, 1H, O*CH=CH), 8.09 (s, 1H, Ar–H), 7.93 (d, J = 8.0 Hz, 1H, Ar–H), 7.85 (s, 1H, Ar–H), 7.76 (d, J = 7.6 Hz, 1H, Ar–H), 7.72 (m, 1H, Ar–H), 7.63 (m, 1H, Ar–H), 7.53–7.59 (m, 1H, Ar–H), 7.33 (s, 1H, Ar–H), 7.15 (d, J = 1.2 Hz, 1H, *CH=CHO), 4.72–4.81 (m, 1H, CHCO), 4.22 (s, 2H, CH2N), 3.75 (m, 2H, CH2N), 3.28 (dd, J = 14.4, 4.4 Hz, 1H, *CH2CH2N), 3.14 (s, 3H, CH3SO2), 3.00 (dd, J = 14.0, 10.6 Hz, 1H, *CH2CH2N), 2.73 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.514 (1C, COOH), 163.885 (1C, CONH), 153.768 (1C, Ar), 150.431 (1C, O*CH=CH) 141.065 (1C, Ar), 139.534 (1C, Ar), 136.104 (1C, Ar), 135.129 (1C, Ar), 134.905 (1C, Ar), 132.240 (1C, Ar), 131.886 (1C, Ar), 131.497 (1C, Ar), 130.991 (1C, Ar), 129.720 (1C, Ar), 128.731 (1C, Ar), 128.211 (1C, Ar), 126.218 (1C, Ar), 125.503 (1C, Ar), 122.824 (1C, Ar), 122.412 (1C, Ar), 111.710 (1C, Ar), 107.608 (1C, *CH=CHO), 53.490 (1C, *CHCO), 47.395 (1C, CH2N), 44.066 (1C, CH2N), 43.474 (1C, CH3SO2), 36.765 (1C, *CH2CH), 26.951 (1C, *CH2CH2N). LCMS m/z 651.0 [M + H]+; HR-MS m/z calcd. for C28H24Cl2N2O8S2 (650.04) 651.0 [M + H]+, found: 651.0436 [M + H]+.
(S)-2-(2-((Benzofuran-6-ylmethyl)carbamoyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)benzyl propanoate (15)
CDI (220 mg, 1.36 mmol) was dissolved in DCM (3.00 mL) and cooled to 0 °C. A solution of compound 14 (0.20 g, 1.36 mmol) and DIEA (193 mg, 1.49 mmol) in DCM (3.00 mL) was added, then the reaction mixture was warmed to 25 °C for 0.5 h. A solution of compound 5 (763 mg, 1.28 mmol) and DIEA (176 mg, 1.36 mmol) in DCM (5.00 mL) was added and the reaction mixture was stirred for 0.5 h at 25 °C. LCMS showed the desired MS was detected. The reaction mixture was diluted with DCM (10.0 mL), washed with 1 M aq. Na2CO3 (2 × 25.0 mL), dried MgSO4 and concentrated in vacuum. Compound 15 (0.022 g, 33.0 µmol, 9.42% yield) was obtained as a light yellow solid. 1H NMR (MeOD, 400 MHz) δ 9.01 (s, 1H, CONH), 7.93 (s, 1H, Ar–H), 7.84 (d, J = 7.8 Hz, 1H, Ar–H), 7.72 (d, J = 2.0 Hz, 1H, O*CH=CH), 7.67–7.63 (m, 1H, Ar–H), 7.58–7.52 (m, 2H, Ar–H), 7.44 (m, 1H, Ar–H), 7.42–7.34 (m, 5H, Ar–H), 7.23 (s, 1H, Ar–H), 7.18 (s, 1H, Ar–H), 6.81 (d, J = 1.4 Hz, 1H, *CH=CHO), 5.23 (d, J = 3.4 Hz, 2H, CH2N), 5.20–5.12 (m, 1H, CHCO), 4.58 (s, 2H, CH2, Bn), 4.50 (s, 2H, CH2, CH2NH), 3.70 (br t, J = 5.8 Hz, 2H, CH2NH), 3.45 (dd, J = 5.4, 14.2 Hz, 1H, *CH2CH2N), 3.40–3.36 (m, 1H, *CH2CH2N), 3.22–3.11 (m, 1H, NHCH2), 3.08 (s, 3H, CH3SO2), 2.80 (t, J = 5.8 Hz, 2H, CH2CH); LCMS m/z 734.0[M + H]+.
(S)-2-(2-((Benzofuran-6-ylmethyl)carbamoyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)propanoic acid (1b)
To a solution of compound 15 (0.200 g, 272 µ mol) in MeOH (5.00 mL), Raney-Ni (46.6 mg, 544 µ mol) was added under N2. The suspension was degassed under vacuum and purged with H2 several times. The mixture was stirred under H2 (15 psi) at 25 °C for 1 h. LCMS indicated starting material was consumed completely and one major new peak with desired mass was detected. The suspension was filtered through a pad of Celite and the pad was washed with EtOH (2.00 mL × 3). The combined filtrates were concentrated to dryness to give product. The crude product was purified by prep-HPLC (HCl condition). Compound 1b (0.025 g, 37.8 µmol, 13.8% yield) was obtained as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.02 (d, J = 8.4 Hz, 1H, CONH), 7.93 (d, J = 2.0 Hz, 1H, O*CH=CH), 7.87 (s, 1H, Ar–H), 7.77 (d, J = 7.6 Hz, 1H, Ar–H), 7.67 (d, J = 7.6 Hz, 1H, Ar–H), 7.59 (m, 1H, Ar–H), 7.54 (m, 1H, Ar–H), 7.45 (s, 1H, Ar–H), 7.35 (s, 1H, NHCH2), 7.26 (s, 1H, Ar–H), 7.17 (d, J = 8.0 Hz, 1H, Ar–H), 6.90 (d, J = 1.2 Hz, 1H, *CH=CHO), 4.74–4.83 (m, 1H, CHCO), 4.53 (s, 2H, CH2N), 4.35 (d, J = 4.0 Hz, 2H, CH2NH), 3.63 (m, 2H, CH2N), 3.29 (dd, J = 14.0, 4.2 Hz, 1H, *CH2CH2N), 3.15 (s, 3H, CH3SO2), 3.01 (dd, J = 13.6, 10.8 Hz, 1H, *CH2CH2N), 2.66 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.518 (1C, COOH), 164.076 (1C, CONH), 157.670 (1C, NHCON), 154.940 (1C, Ar), 146.260 (1C, O*CH=CH), 141.103 (1C, Ar), 139.558 (1C, Ar), 138.770 (1C, Ar), 138.106 (1C, Ar), 134.914 (1C, Ar), 134.770 (1C, Ar), 132.394 (1C, Ar), 131.614 (1C, Ar), 129.721 (1C, Ar), 128.595 (1C, Ar), 128.219 (1C, Ar), 126.183 (1C, Ar), 125.937 (1C, Ar), 125.518 (1C, Ar), 122.832 (1C, Ar), 121.243 (1C, Ar), 110.223 (1C, Ar), 106.987 (1C, *CH=CHO), 53.531 (1C, *CHCO), 49.075 (1C, CH2N), 45.739 (1C, CH2N), 44.092 (1C, CH3SO2), 41.225 (1C, CH2NH), 36.820 (1C, *CH2CH), 26.420 (1C, *CH2CH2N). LCMS m/z 644.1 [M + H]+; HR-MS m/z calcd. for C30H27Cl2N3O7S (643.09) 644.0 [M + H]+, found: 644.1075 [M + H]+.
(S)-2-(2-(2-(Benzofuran-5-yl)acetyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)benzyl propanoate (17)
To a solution of compound 5 (0.200 g, 334 µmol) in DCM (2.00 mL), TEA (101 mg, 1.00 mmol, 139 µL) and HATU (191 mg, 502 µmol) were added, the solution was stirred at 20 °C for 1 h, then compound 16 (58.9 mg, 334 µmol) was added via several portions. The reaction mixture was stirred at 20 °C for 2 h under N2 atmosphere. LCMS showed starting material was consumed completely and one major new peak with desired mass was detected. The solution was quenched with water (2.00 mL) and the organic was extracted with DCM (2.00 mL × 3), then organic was concentrated under reduced pressure. Compound 17 (0.210 g, 292 µmol, 87.2% yield) was obtained as a brown solid; LCMS m/z 719.1 [M + H]+.
(S)-2-(2-(2-(Benzofuran-5-yl)acetyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)propanoic acid (1c)
To a solution of compound 17 (97.9 mg, 136 µmol) in MeOH (1.00 mL), Raney-Ni (11.6 mg, 136 µmol) was added under N2. The suspension was degassed under vacuum and purged with H2 several times. The mixture was stirred under H2 (15 psi) at 25 °C for 3 h. TLC indicated starting material was consumed completely. The suspension was filtered through a pad of Celite and the filter cake was washed with MeOH (2.00 mL × 3). The combined filtrates were concentrated to dryness to give product. The crude product was purified by prep-HPLC (HCl condition). Compound 1c (0.030 g, 45.5 µmol, 33.4% yield) was obtained as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.79 (d, J = 7.6 Hz, 1H, CONH), 7.90 (d, J = 2.1 Hz, 1H, O*CH=CH), 7.86 (s, 1H, Ar–H), 7.77 (d, J = 7.6 Hz, 1H, Ar–H), 7.65 (d, J = 7.7 Hz, 1H, Ar–H), 7.57 (d, J = 7.55 Hz, 1H, Ar–H), 7.51 (s, 1H, Ar–H), 7.47 (d, J = 8.4 Hz, 1H, Ar–H), 7.31 (s, 1H, Ar–H), 7.19 (d, J = 8.4 Hz, 1H, Ar–H), 6.88 (d, J = 2.2 Hz, 1H, *CH=CHO), 4.85–4.75 (m, 1H, CHCO), 4.62 (s, 2H, CH2N), 3.88 (s, 2H, CH2N), 3.77 (s, 2H, CH2CO), 3.30 (dd, J = 14.0, 4.7 Hz, 1H, *CH2CH2N), 3.12 (s, 3H, CH3SO2), 3.05 (dd, J = 14.3, 10.0 Hz, 1H, *CH2CH2N), 2.67 (s, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ ppm 172.512 (1C, COOH), 170.113 (1C, CONH), 169.982 (1C, CH2CON), 164.002 (1C, Ar), 153.663 (1C, O*CH=CH), 146.721 (1C, Ar), 141.099 (1C, Ar), 139.546 (1C, Ar), 137.643 (1C, Ar), 134.909 (1C, Ar), 134.843 (1C, Ar), 130.680 (1C, Ar), 129.724 (1C, Ar), 128.798 (1C, Ar), 127.814 (1C, Ar), 126.013 (1C, Ar), 125.889 (1C, Ar), 125.524 (1C, Ar), 122.003 (1C, Ar), 121.857 (1C, Ar), 111.473 (1C, Ar), 111.379 (1C, Ar), 107.062 (1C, *CH=CHO), 53.512 (1C, CHCO), 44.077 (1C, CH2N), 43.756 (1C, CH3SO2), 42.779 (1C, CH2N), 38.973 (1C, CH2CON), 36.807 (1C, *CH2CH), 27.262 (1C, *CH2CH2N). LCMS m/z 629.1 [M + H]+; HR-MS m/z calcd. for C30H26Cl2N2O7S (628.08) 629.0 [M + H]+, found: 629.0922 [M + H]+.
(S)-2-(2-([1,2,4]Triazolo[1,5-a]pyridine-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)benzyl propanoate (19)
To a solution of compound 5 (0.200 g, 334 µmol) in DCM (2.00 mL), TEA (101 mg, 1.00 mmol) and HATU (190 mg, 502 µmol) were added, the solution was stirred at 20 °C for 1 h, then compound 18 (54.6 mg, 334 µmol) was added via several portions. The reaction mixture was stirred at 20 °C for 2 h under N2 atmosphere. LCMS showed starting material was consumed completely and one major new peak with desired mass was detected. The solution was quenched with water (2.00 mL) and the organic was extracted with DCM (2.00 mL × 3), then the organic was concentrated under reduced pressure. Compound 19 (0.220 g, crude) was obtained as a brown solid; LCMS m/z 706.1 [M + H]+.
(S)-2-(2-([1,2,4]Triazolo[1,5-a]pyridine-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)propanoic acid (1d)
To a solution of compound 19 (0.200 g, 283 µmol) in THF (2.00 mL), TMSOK (109 mg, 849 µmol) was added, then the mixture was stirred at 70 °C for 1 h. LCMS indicated starting material was consumed completely and one major new peak with desired mass was detected. The mixture was quenched with water (2.00 mL) and acidified with 1 N HCl to pH = 3, the mixture was extracted with EtOAc (2.00 mL × 3), then the organic layer was concentrated under reduced pressure. The crude product was purified by prep-HPLC (HCl condition). Compound 1d (0.04 g, 62.4 µmol, 22.0% yield) was obtained as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.20 (s, 1H, Ar–H), 9.03 (d, J = 8.0 Hz, 1H, CONH), 8.62 (s, 1H, CH=N), 7.93 (d, J = 9.2 Hz, 1H, Ar–H), 7.86 (s, 1H, Ar–H), 7.77 (d, J = 7.6 Hz, 1H, Ar–H), 7.72 (d, J = 7.6 Hz, 1H, Ar–H), 7.67 (d, J = 7.6 Hz, 1H, Ar–H), 7.55 (s, 1H, Ar–H), 7.54–7.60 (m, 1H, Ar–H), 4.77–4.82 (m, 1H, CHCO), 4.74 (s, 2H, CH2N), 3.74 (m, 2H, CH2N), 3.30 (dd, J = 14.0, 4.3 Hz, 1H, *CH2CH2N), 3.15 (s, 3H, CH3SO2), 3.02 (dd, J = 14.0, 10.6 Hz, 1H, *CH2CH2N), 2.80 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.526 (1C, COOH), 164.002 (1C, CONH), 155.184 (1C, CON), 155.034 (1C, CH=N), 150.221 (1C, Ar), 141.099 (1C, Ar), 139.561 (1C, Ar), 137.220 (1C, Ar), 135.018 (1C, Ar), 134.931 (1C, Ar), 132.109 (1C, Ar), 131.525 (1C, Ar), 130.133 (1C, Ar), 129.724 (1C, Ar), 129.061 (1C, Ar), 128.820 (1C, Ar), 128.193 (1C, Ar), 126.217 (1C, Ar), 125.524 (1C, Ar), 123.337 (1C, Ar), 116.634 (1C, Ar), 53.52 (1C, *CHCO), 44.08 (1C, CH2N), 40.65 (1C, CH3SO2), 40.44 (1C, CH2N), 40.02 (1C, *CH2CH), 36.80 (1C, *CH2CH2N). LCMS m/z 616.0 [M + H]+; HR-MS m/z calcd. for C27H23Cl2N5O6S (615.07) 616.0 [M + H]+, found: 616.0825 [M + H]+.
(S)-2-(2-(3-Ethynylbenzoyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)benzyl propanoate (21)
To a solution of compound 5 (0.200 g, 334 µmol) in DCM (2.00 mL), TEA (101 mg, 1.00 mmol) and HATU (190 mg, 501 µmol) were added, the solution was stirred at 20 °C for 1 h, then compound 20 (48.9 mg, 334 µmol) was added via several portions. The reaction mixture was stirred at 20 °C for 2 h under N2 atmosphere. LCMS showed starting material was consumed completely and one major new peak with desired mass was detected. The reaction was quenched with water (2.00 mL) and the organic was extracted DCM (2.00 mL × 3), then the organic was concentrated under reduced pressure. Compound 21 (0.200 g, 290 µmol, 86.7% yield) was obtained as a brown solid; LCMS m/z 689.1 [M + H]+.
(S)-2-(2-(3-Ethynylbenzoyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3-(methylsulfonyl)phenyl)propanoic acid (1e)
To a solution of compound 21 (0.200 g, 290 µmol) in THF (2.00 mL), TMSOK (111 mg, 870 µmol) was added, then the mixture was stirred at 70 °C for 1 h. LCMS indicated starting material was consumed completely, and new peak with desired mass was detected. The mixture was quenched with water (2.00 mL) and acidified with 1 N HCl to pH = 3, the mixture was extracted with EtOAc (2.00 mL × 3), then the organic layer was concentrated under reduced pressure. The crude product was purified by prep-HPLC (HCl condition). Compound 1e (0.040 g, 64.4 µmol, 22.2% yield) was obtained as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.04 (d, J = 7.6 Hz, 1H, CONH), 7.86 (s, 1H, Ar–H), 7.77 (d, J = 7.6 Hz, 1H, Ar–H), 7.67 (d, J = 7.6 Hz, 1H, Ar–H), 7.54–7.61 (m, 2H, Ar–H), 7.49 (s, 1H, Ar–H), 7.45–7.54 (m, 1H, Ar–H), 7.40–7.45 (m, 1H, Ar–H), 7.33 (s, 1H, Ar–H), 4.73–4.82 (m, 1H, CHCO), 4.68 (s, 2H, CH2N), 4.12 (s, 1H, CH≡C), 3.69 (m, 2H, CH2N), 3.29 (dd, J = 14.0, 4.5 Hz, 1H, *CH2CH2N), 3.15 (s, 3H, CH3SO2), 3.02 (dd, J = 14.0, 10.6 Hz, 1H, *CH2CH2N), 2.75 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.534 (1C, COOH), 164.010 (1C, CONH), 141.114 (1C, CON), 139.554 (1C, Ar), 137.300 (1C, Ar), 136.659 (1C, Ar), 135.00 (1C, Ar), 134.923 (1C, Ar), 133.458 (1C, Ar), 132.072 (1C, Ar), 131.576 (1C, Ar), 130.50 (1C, Ar), 130.410 (1C, Ar), 129.732 (1C, Ar), 129.564 (1C, Ar), 128.879 (1C, Ar), 128.193 (1C, Ar), 127.821 (1C, Ar), 125.786 (1C, Ar), 125.532 (1C, Ar), 122.549 (1C, Ar), 83.196 (1C, *C≡CH), 82.183 (1C, *CH≡C), 53.512 (1C, *CHCO), 44.084 (1C, CH2N), 40.671 (1C, CH3SO2), 40.462 (1C, CH2N), 40.253 (1C, *CH2CH), 36.814 (1C, *CH2CH2N). LCMS m/z 599.1 [M + H]+; HR-MS m/z calcd. for C29H24Cl2N2O6S (598.07) 599.0 [M + H]+, found: 599.0801 [M + H]+.
1-Bromo-3-(2,2-diethoxyethoxy)benzene (8)
To a solution of compound 6 (10.0 g, 57.8 mmol) and 2-bromo-1,1-diethoxy-ethane 7 (22.8 g, 115 mmol) in DMF (60.0 mL), K2CO3 (7.99 g, 57.8 mmol) was added, then air in the reaction system was replaced with nitrogen three times, and the mixture was stirred at 100 °C for 10 h. TLC indicated starting material was consumed completely. After that, the reaction solution was diluted with water (200 mL) and then extracted with ethyl acetate (60 mL × 3). The extract was dried over anhydrous sodium sulfate and concentrated. Then the oil was purified by silica gel column chromatography. Compound 8 (8.00 g, 27.6 mmol, 47.8% yield) was obtained as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ 7.07–7.17 (m, 3H, Ar–H), 6.83–6.90 (m, 1H, Ar–H), 4.82 (t, J = 5.2 Hz, 1H, CHO2), 3.99 (d, J = 5.2 Hz, 2H, CH2O), 3.71–3.83 (m, 2H, CH2CH3), 3.55–3.70 (m, 2H, CH2CH3), 1.22–1.29 (m, 6H, CH3CH2).
6-Bromobenzofuran (9)
PPA (2.27 g, 83.0 mmol) was added to a solution of compound 8 (8.00 g, 27.6 mmol) in toluene (40.0 mL), the mixture was stirred at 80 °C for 4 h. TLC indicated starting material was consumed completely. The solution was cooled to room temperature, quenched with water (60.0 mL), extracted with EtOAc (40 mL × 3), the organic layer was washed with brine (40.0 mL), dried (Na2SO4), filter. Then the organic layer was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography. Compound 9 (4.40 g, 11.1 mmol, 40.3% yield, ~50% purity) was obtained as brown oil. 1H NMR (CDCl3, 400 MHz) δ 7.70 (s, 1H, H-7), 7.61 (d, J = 2.4 Hz, 1H, H-2), 7.47–7.50 (m, 1H, H-4), 7.37 (dd, J = 8.4, 1.8 Hz, 1H, H-5), 6.76 (dd, J = 2.4, 0.8 Hz, 1H, H-3).
Benzofuran-6-sulfonyl chloride (10)
To a solution of compound 9 (0.400 g, 2.03 mmol) in THF (4.00 mL), n-BuLi (0.130 g, 812 µL) was added at −78 °C with atmosphere of nitrogen, the solution was stirred at −78 °C for 0.5 h. Then to the solution was added SO2 (1.30 g, 20.3 mmol) in THF (2.00 mL), the solution was stirred at 20 °C for another 1.5 h. The solution was concentrated under vacuum. The residue was dissolved in hexane, then the mixture was added sulfuryl chloride (548 mg, 4.06 mmol) at 0 °C. The resulting solution was stirred for 10 h at 20 °C. TLC indicated starting material was consumed completely. After that, the mixture reaction was filter, and the organic layer was concentrated under reduced pressure to left a residue. The residue was purified by prep-TLC. Compound 10 (0.200 g, 923 µmol, 22.7% yield) was obtained as a white solid. 1H NMR (CDCl3, 400 MHz) δ 8.24 (s, 1H, H-7), 7.92–7.99 (m, 2H, H-2 H-4), 7.83 (d, J = 8.4 Hz, 1H, H-5), 6.95 (s, 1H, H-3).
Benzofuran-6-carbonitrile (13)
To a solution of compound 12 (2.00 g, 10.2 mmol) in DMF (12.0 mL), Zn(CN)2 (19.6 g, 167 mmol) and Pd(PPh3)4 (1.41 g, 1.22 mmol) were added under a nitrogen atmosphere, the reaction mixture was stirred at 80 °C for 16 h. TLC indicated starting material was consumed completely, and two new spots were detected. The reaction mixture was diluted with toluene (10.0 mL) and the phases were separated. The aqueous phase was extracted twice with toluene (10.0 mL × 2). The combined organic was washed with brine (100 mL) and saturated aqueous ammonium hydroxide (20 mL), then dried (Na2SO4) and concentrated to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 100/1 to 10/1). Compound 13 (1.00 g, 6.99 mmol 68.8% yield) was obtained as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ = 7.84 (s, 1H, H-7), 7.82 (d, J = 2.0 Hz, 1H, H-2), 7.70 (d, J = 8.0 Hz, 1H, H-4), 7.52 (d, J = 8.0 Hz, 1H, H-5), 6.87 (d, J = 1.3 Hz, 1H, H-3).
Benzofuran-6-ylmethanamine (14)
To a mixture of LiAlH4 (265 mg, 6.99 mmol) in THF (10.0 mL), compound 13 (1.00 g, 6.99 mmol) was added in one portion at 20 °C. The mixture was stirred at 20 °C for 1 h. TLC indicated one new spot was detected. 1 mL of saturated Na2SO4 was added to the reaction mixture carefully, and then filtered. The filtration was concentrated to give compound 14 (0.5 g, 3.40 mmol, 48.60% yield) as a light yellow oil. 1H NMR (MeOD, 400 MHz) δ 7.71 (d, J = 2.0 Hz, 1H, H-2), 7.55 (d, J = 8.0 Hz, 1H, H-4), 7.49 (s, 1H, H-7), 7.21 (d, J = 8.0 Hz, 1H, H-5), 6.79–6.80 (m, 1H, H-3), 3.88 (s, 2H, CH2, CH2NH2); LCMS m/z 148.1 [M + H]+.
2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-methyl carboxylate (24)
To a mixture of compound 23 (5.00 g, 16.8 mmol), ECDI (3.56 g, 18.5 mmol), DMAP (164 mg, 1.35 mmol) and TEA (5.12 g, 50.58 mmol) in DMF (30 mL), compound 22 (2.73 g, 16.8 mmol) was added. Then the mixture was stirred at 25 °C for 16 h. TLC indicated reactant 23 was consumed completely, and the reaction was clean according to TLC. The reaction mixture was poured into H2O (120 mL). The mixture was diluted with DCM 10.0 mL and washed with H2O (50.0 mL), aq.citric acid (50.0 mL), aq. NaHCO3 (50.0 mL). The combined organic layers were washed with brine 100 mL dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. Compound 24 (4.00 g, 9.90 mmol, 58.6% yield) was obtained as yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.12 (d, J = 2.4 Hz, 1H, O*CH=CH), 7.78–7.71 (m, 1H, Ar–H), 7.74 (s, 1H, Ar–H), 7.68–7.47 (m, 1H, Ar–H), 7.35 (br d, J = 7.6 Hz, 1H, Ar–H), 7.05 (d, J = 1.6 Hz, 1H, *CH=CHO), 4.79 (s, 2H, CH2N), 3.91 (s, 3H, CH3, CH3O), 3.79–3.55 (m, 2H, CH2N), 2.93–2.79 (m, 2H, CH2CH2N).
2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxylic acid (25)
To a solution of compound 24 (4.00 g, 9.90 mmol) in pyridine (24.0 mL), LiI (5.30 g, 39.5 mmol) was added. Then the mixture was stirred at 100 °C for 16 h. TLC indicated reactant 24 was consumed completely and the reaction was clean according to TLC. The reaction mixture was concentrated under reduced pressure to remove pyridine. The reaction mixture was adjusted the pH to 2 by adding HCl (3 M, 40.0 mL), solid was precipitate out, then the mixture was filtered and the filter cake was washed with H2O (50.0 mL), dried in vacuum to give residue. The crude product was purified by re-crystallization from Petroleum ether: Ethyl acetate = 1: 1 (20.0 mL) at 15 °C. Compound 25 (2.00 g, 4.77 mmol, 48.1% yield) was obtained as brown solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.12 (d, J = 2.4 Hz, 1H, O*CH=CH), 7.78 (s, 1H, Ar–H), 7.70 (s, 1H, Ar–H), 7.64–7.45 (m, 1H, Ar–H), 7.35 (d, J = 7.2 Hz, 1H, Ar–H), 7.05 (d, J = 1.6 Hz, 1H, *CH=CHO), 4.78 (d, J = 1.2 Hz, 2H, CH2N), 4.01–3.71 (m, 2H, CH2N), 2.85 (t, J = 5.6 Hz, 2H, CH2CH2N); LCMS m/z 390.1 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(quinolin-6-yl)ethyl propanoate (31)
To a solution of compound 25 (0.300 g, 768 µmol) in DCM (3.00 mL), TEA (23.3 mg, 2.31 mmol) and HATU (438 mg, 1.15 mmol) were added, the mixture was stirred at 20 °C for 0.5 h, then compound 30 (187 mg, 768 µmol) and DMAP (75.1 mg, 615 µmol) were added, the mixture was stirred for 2 h at 20 °C. TLC indicated reactant 25 was consumed completely. The mixture poured into H2O (10 mL), extracted with EtOAc (10 mL × 2), dried over Na2SO4, concentrated to give crude product. The residue was purified by column chromatography. Compound 31 (0.200 g, 291 µmol, 37.9% yield, 90.0% purity) was obtained as a white solid. 1HNMR (CDCl3, 400 MHz) δ 8.88 (d, J = 3.2 Hz, 1H, CONH), 8.10 (d, J = 8.0 Hz, 1H, Ar–H), 8.02 (d, J = 8.8 Hz, 1H, Ar–H), 7.73 (d, J = 1.6 Hz, 1H, O*CH=CH), 7.72 (s, 1H, Ar–H), 7.65 (d, J = 8.0 Hz, 1H, Ar–H), 7.62–7.56 (m, 3H, Ar–H), 7.40 (dd, J = 4.0, 8.3 Hz, 1H, Ar–H), 7.31 (d, J = 7.6 Hz, 1H, Ar–H), 6.83 (d, J = 1.2 Hz, 1H, *CH=CHO), 6.59 (s, 1H, Ar–H), 5.34–5.19 (m, 1H, CHCO), 4.75 (s, 2H, CH2N), 4.22 (q, J = 7.2 Hz, 2H, CH2CH3), 3.89–3.58 (m, 2H, CH2N), 3.49 (d, J = 5.8 Hz, 2H, CH2CH2N), 2.98–2.83 (m, 2H, CH2CH), 1.31–1.17 (m, 3H, CH3, CH3CH2); LCMS m/z 616.3 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(quinolin-6-yl)propanoic acid (1f)
To a solution of compound 31 (200 mg, 324 µmol) in EtOH (2.00 mL) and THF (1.00 mL), NaOH (38.9 mg, 973 µmol) in H2O (1.00 mL) were added, the mixture was stirred at 20 °C for 2 h. TLC indicated reactant 31 was consumed completely. EtOH and THF was removed, the mixture was adjusted pH = 4, then solid was formed, filtered and the filter cake concentrated to give crude product. The crude product was triturated with Petroleum ether: Ethyl acetate = 10: 1 (20 mL) at 30 °C for 30 min. Compound 1f (25.0 mg, 41.6 µmol, 12.8% yield) was obtained as a white solid. 1HNMR (DMSO-d6, 400 MHz) δ 8.89 (s, 1H, CONH), 8.82 (d, J = 8.8 Hz, 1H, Ar–H), 8.32 (d, J = 8.8 Hz, 1H, Ar–H), 8.06 (s, 1H, Ar–H), 7.97 (d, J = 9.2 Hz, 1H, Ar–H), 7.89 (s, 1H, O*CH=CH), 7.75 (d, J = 8.8 Hz, 1H, Ar–H), 7.74 (d, J = 8.8 Hz, 1H, Ar–H), 7.66 (s, 1H, Ar–H), 7.54 (dd, J = 4.8, 8.2 Hz, 1H, Ar–H), 7.31 (d, J = 10.0 Hz, 1H, Ar–H), 7.30 (d, J = 10.0 Hz, 1H, Ar–H), 7.02 (s, 1H, *CH=CHO), 4.88 (m, 1H, CHCO), 4.72 (s, 2H, CH2N), 3.74 (m, 2H, CH2N), 3.46–3.33 (m, 1H, *CH2CH2N), 3.27–3.20 (m, 1H, *CH2CH2N), 2.78 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.698 (1C, COOH), 169.912 (1C, CONH), 164.022 (1C, CON), 154.114 (1C, Ar), 149.783 (1C, Ar), 148.209 (1C, Ar), 145.927 (1C, O*CH=CH), 137.477 (1C, Ar), 137.456 (1C, Ar), 136.806 (1C, Ar), 135.027 (1C, Ar), 132.461 (1C, Ar), 132.133 (1C, Ar), 132.046 (1C, Ar), 131.623 (1C, Ar), 129.151 (1C, Ar), 128.852 (1C, Ar), 128.495 (1C, Ar), 128.262 (1C, Ar), 128.028 (1C, Ar), 126.206 (1C, Ar), 122.480 (1C, Ar), 121.992 (1C, Ar), 121.897 (1C, Ar), 110.801 (1C, Ar), 107.316 (1C, *CH=CHO), 53.701 (1C, *CHCO), 49.072 (1C, CH2N), 40.654 (1C, CH2N), 40.246 (1C, *CH2CH), 37.137 (1C, *CH2CH2N). LCMS m/z 588.1 [M + H]+; HR-MS m/z calcd. for C31H23Cl2N3O5 (587.10) 588.1 [M + H]+; found: 588.1070 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(1,1-dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)ethyl propanoate (42)
To a solution of compound 25 (300 mg, 768 μmol) in DCM (5.00 mL), TEA (233 mg, 2.31 mmol) and HATU (438 mg, 1.15 mmol) were added. The mixture was stirred for 30 min, then compound 41 (217 mg, 768 μmol), and DMAP (75.1 mg, 615 μmol) were added. The mixture was stirred for 20 h at 30 °C. TLC indicated reactant 25 was consumed completely. The mixture was poured into H2O (10 mL), extracted with EtOAc (20 mL), washed with 0.5 M HCl (10 mL), NaHCO3 (10 mL), brine (10 mL), dried over Na2SO4, concentrated to give crude product. The residue was purified by column chromatography. Compound 42 (0.200 g, 289 µmol, 37.7% yield) was obtained as a white solid. 1HNMR (CDCl3, 400 MHz) δ 7.73 (d, J = 2.4 Hz, 1H, O*CH=CH), 7.69–7.59 (m, 3H, Ar–H), 7.59–7.49 (m, 1H, Ar–H), 7.32 (d, J = 8.0 Hz, 2H, Ar–H), 6.83 (s, 1H, Ar–H), 6.57–6.44 (m, 1H, *CH=CHO), 5.18–5.09 (m, 1H, CHCO), 4.89–4.62 (s, 2H, CH2N), 4.29–4.16 (m, 2H, CH2, CH2CH3), 3.88–3.62 (m, 2H, CH2N), 3.52–3.39 (m, 2H, *CH2CH2N), 3.39–3.28 (m, 4H, CH2CH2), 2.90 (m, 2H, CH2CH), 1.31–1.27 (m, 3H, CH3CH2); LCMS m/z 655.3 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(1,1-dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)propanoic acid (1g)
To a solution of compound 42 (0.200 g, 305 µmol) in EtOH (2.00 mL) and THF (1.00 mL), NaOH (36.6 mg, 915 µmol) in H2O (1.00 mL) was added. The mixture was stirred at 20 °C for 20 h. TLC indicated reactant 42 was consumed completely. EtOH and THF was removed, the mixture was adjusted pH = 4, then solid was formed, filtered and the filter cake concentrated to give crude product. The crude product was triturated with Petroleum ether: Ethyl acetate = 10: 1 (20 mL) at 30 °C for 30 min. Compound 1g (24.0 mg, 36.6 µmol, 12.0% yield) was obtained as a white solid. 1HNMR (DMSO-d6, 400 MHz) δ 12.85 (s, 1H, COOH), 9.02 (d, J = 8.8 Hz, 1H, CONH), 8.11 (d, J = 2.0 Hz, 1H, O*CH=CH), 7.81–7.66 (m, 2H, Ar–H), 7.62 (s, 1H, Ar–H) 7.64–7.55 (m, 1H, Ar–H), 7.43 (d, J = 7.6 Hz, 2H, Ar–H), 7.32 (s, 1H, Ar–H), 7.03 (s, 1H, *CH=CHO), 4.69–4.70 (m, 1H, CHCO), 4.69 (s, 2H, CH2N), 3.87–3.59 (m, 2H, CH2N), 3.54 (t, J = 6.8 Hz, 2H, CH2CH2), 3.30–3.16 (m, 2H, CH2CH2), 3.25–3.16 (m, 1H, *CH2CH2N), 3.07–2.94 (m, 1H, *CH2CH2N), 2.77 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.522 (1C, COOH), 170.011 (1C, CON), 164.116 (1C, CONH), 154.114 (1C, Ar), 148.221 (1C, O*CH=CH), 139.209 (1C, Ar), 139.029 (1C, Ar), 137.505 (1C, Ar), 136.682 (1C, Ar), 134.999 (1C, Ar), 134.892 (1C, Ar), 132.146 (1C, Ar), 132.081 (1C, Ar), 131.735 (1C, Ar), 129.142 (1C, Ar), 128.940 (1C, Ar), 127.857 (1C, Ar), 126.225 (1C, Ar), 122.484 (1C, Ar), 121.906 (1C, Ar), 121.668 (1C, Ar), 110.800 (1C, Ar), 107.319 (1C, *CH=CHO), 53.750 (1C, *CHCO), 51.056 (1C, *CH2CH2), 44.440 (1C, CH2N), 40.711 (1C, CH2N), 40.385 (1C, *CH2CH), 36.512 (1C, *CH2CH2), 25.081 (1C, *CH2CH2N). LCMS m/z 627.0 [M + H]+; HR-MS m/z calcd. for C30H24Cl2N2O7S (626.07) 627.0 [M + H]+; found: 627.0760 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)ethyl propanoate (51)
To a solution of compound 25 (0.300 g, 768 µmol) in DCM (5.00 mL), TEA (233 mg, 2.31 mmol) and HATU (438 mg, 1.15 mmol) were added. The mixture was stirred at 20 °C for 0.5 h, then compound 50 (201 mg, 768 µmol) and DMAP (75.1 mg, 615 µmol) were added. The mixture was stirred at 30 °C for 20 h. TLC indicated reactant 25 was consumed completely. The mixture was poured into H2O (10 mL), extracted with EtOAc (20 mL), washed with 0.5 M HCl (10 mL), NaHCO3 (10 mL), brine (10 mL), dried over Na2SO4, concentrated to give crude product. The residue was purified by column chromatography. Compound 51 (0.200 g, 299 µmol, 38.9% yield) was obtained as a white solid. 1HNMR (CDCl3, 400 MHz) δ 8.13 (s, 1H, CONH), 7.74 (d, J = 2.0 Hz, 1H, O*CH=CH), 7.67 (d, J = 8.0 Hz, 1H, Ar–H), 7.63 (s, 1H, Ar–H), 7.33 (d, J = 8.0 Hz, 1H, Ar–H), 7.08 (d, J = 7.6 Hz, 1H, Ar–H), 6.89–6.82 (m, 2H, Ar–H), 6.65 (s, 1H, *CH=CHO), 6.38 (d, J = 8.0 Hz, 1H, Ar–H), 5.23–5.09 (m, 1H, CHCO), 4.80 (s, 2H, CH2N), 4.23 (q, J = 7.2 Hz, 2H, CH2CH3), 3.94–3.69 (m, 2H, CH2N), 3.29–3.14 (m, 1H, *CH2CH2N), 3.29–3.14 (m, 1H, *CH2CH2N), 3.00–2.95 (m, 2H, CH2CH2), 2.95–2.85 (m, 2H, CH2CH2), 2.69–2.55 (m, 2H, CH2CH), 1.36–1.20 (m, 3H, CH3, CH3CH2); LCMS m/z 634.4 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)propanoic acid (1h)
To a solution of compound 51 (0.200 g, 315 µmol) in MeOH (2.00 mL) and THF (1.00 mL), NaOH (37.8 mg, 945 µmol) in H2O (1.00 mL) were added, the mixture was stirred at 20 °C for 2 h. TLC indicated reactant 51 was consumed completely. EtOH and THF was removed, the mixture was adjusted pH = 4, then solid was formed, filtered and the filter cake concentrated to give crude product. The crude product was triturated with Petroleum ether: Ethyl acetate = 10: 1 (20 mL) at 30 °C for 30 min. Compound 1h (0.024 g, 37.9 µmol, 12.0% yield) was obtained as a white solid. 1HNMR (DMSO-d6, 400 MHz) δ 12.76 (s, 1H, COOH), 10.05 (s, 1H, CH2CONH), 9.00 (d, J = 8.4 Hz, 1H, CONHCH), 8.13 (d, J = 2.0 Hz, 1H, O*CH=CH), 7.83–7.70 (m, 1H, Ar–H), 7.70–7.65 (m, 1H, Ar–H), 7.50–7.36 (m, 1H, Ar–H), 7.36–7.26 (m, 1H, Ar–H), 7.14–7.10 (m, 1H, Ar–H), 7.10–7.00 (m, 1H, Ar–H), 6.83 (d, J = 7.6 Hz, 1H, Ar–H), 6.75 (s, 1H, *CH=CHO), 4.75 (s, 2H, CH2N), 4.66–4.57 (m, 1H, CHCO), 3.65 (m, 2H, CH2N), 3.04 (dd, J = 5.2, 13.6 Hz, 1H, *CH2CH2N), 2.89–2.82 (m, 2H, CH2CH), 2.80 (m, 1H, *CH2CH2N), 2.82–2.74 (m, 2H, CH2CH2), 2.39 (t, J = 7.6 Hz, 2H, CH2CH2). 13C NMR (DMSO-d6, 101 MHz) δ 172.731 (1C, COOH), 170.724 (1C, CONH), 170.322 (1C, CH2*CONH), 163.957 (1C, CONH), 154.121 (1C, Ar), 148.214 (1C, O*CH=CH), 138.574 (1C, Ar), 137.447 (1C, Ar), 136.739 (1C, Ar), 135.114 (1C, Ar), 132.110 (1C, Ar), 132.074 (1C, Ar), 131.713 (1C, Ar), 129.157 (1C, Ar), 128.962 (1C, Ar), 127.907 (1C, Ar), 126.196 (1C, Ar), 123.358 (1C, Ar), 122.499 (1C, Ar), 122.159 (1C, Ar), 121.906 (1C, Ar), 116.274 (1C, Ar), 110.807 (1C, Ar), 107.319 (1C, *CH=CHO), 59.719 (1C, *CHCO), 54.017 (1C, CH2N), 44.550 (1C, CH2N), 37.155 (1C, *CH2CH), 31.067 (1C, *CH2CH2), 27.123 (1C, CH2*CH2), 24.973 (1C, *CH2CH2N). LCMS m/z 606.0 [M + H]+; HR-MS m/z calcd. for C31H25Cl2N3O6 (605.11) 606.0 [M + H]+; found: 606.1202 [M + H]+.
6-Hydroxymethyl-quinoline (27)
To a solution of compound 26 (3.00 g, 19.0 mmol) in MeOH (15.0 mL), NaBH4 (2.20 g, 58.1 mmol) was added in portions at 0 °C. Then the mixture was stirred at 15 °C for 3 h. TLC showed the starting material was consumed completely. The reaction mixture was partitioned between H2O (100 mL) and EtOAc (60.0 mL, 40.0 mL, 20.0 mL). The organic phase was separated, washed with brine 50.0 mL, dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. Compound 27 (2.00 g, 12.5 mmol, 65.8% yield) was obtained as yellow oil. 1H NMR (CDCl3, 400 MHz) δ 8.80 (dd, J = 1.6, 4.0 Hz, 1H, H-2), 8.08 (d, J = 8.0 Hz, 1H, H-4), 8.02 (d, J = 8.4 Hz, 1H, H-8), 7.77 (s, 1H, H-5), 7.65 (dd, J = 1.6, 8.8 Hz, 1H, H-7), 7.34 (dd, J = 4.4, 8.4 Hz, 1H, H-3), 4.89 (s, 2H, CH2).
6-(Bromomethyl)quinoline (28)
To a solution of compound 27 (0.700 g, 4.40 mmol) in HBr/AcOH (5.00 mL), the mixture was stirred at 20 °C for 3 h. TLC indicated reactant 27 was consumed completely. The mixture was concentrated to give product. The crude product was triturated with Petroleum ether: Ethyl acetate = 5: 1 (10 mL) at 20 °C for 30 min. Compound 28 (0.500 g, 1.65 mmol, 37.5% yield) was obtained as a yellow solid. 1HNMR (DMSO-d6, 400 MHz) δ 9.39–9.28 (m, 1H, H-2), 9.12 (d, J = 8.4 Hz, 1H, H-4), 8.41 (s, 1H, H-5), 8.34–8.25 (m, 1H, H-8), 8.24–8.14 (m, 1H, H-7), 8.08 (dd, J = 5.2, 8.3 Hz, 1H, H-3), 4.99 (s, 2H, CH2); LCMS m/z 22.2 [M + H]+.
2-((Diphenylmethylene)amino)-3-(quinolin-6-yl)ethyl propanoate (29)
To a solution of ethyl N-(diphenylmethylene)glycinate (882 mg, 3.30 mmol) in THF (10 mL), NaH (132 mg, 3.30 mmol) was added at 5 °C, the mixture was stirred for 0.5 h, then compound 28 (0.500 g, 1.65 mmol) was added, the mixture was stirred at 20 °C for 2 h. TLC indicated reactant 28 was consumed completely. The mixture was poured into NH4Cl (10 mL), extracted with EtOAc (20 mL), brine (10 mL), dried over Na2SO4, concentrated to give crude product. The residue was purified by column chromatography. Compound 29 (0.500 g, 1.16 mmol, 70.4% yield, 95.0% purity) was obtained as yellow oil. 1HNMR (CDCl3, 400 MHz) δ 8.87 (dd, J = 1.6, 4.3 Hz, 1H, Ar–H), 8.01 (d, J = 8.0 Hz, 1H, Ar–H), 7.94 (d, J = 8.8 Hz, 1H, Ar–H), 7.59–7.53 (m, 2H, Ar–H), 7.51 (s, 1H, Ar–H), 7.45–6.56 (m, 10H, Ar–H), 4.38 (dd, J = 4.4, 9.3 Hz, 1H, CHCO), 4.29–4.12 (m, 2H, CH2CH3), 3.54–3.44 (m, 1H, *CH2CHCO), 3.43–3.32 (m, 1H, *CH2CHCO), 1.27 (t, J = 7.2 Hz, 3H, CH3CH2); LCMS m/z 409.4 [M + H]+.
2-Amino-3-(quinolin-6-yl)ethyl propanoate (30)
To a solution of compound 29 (0.500 g, 1.22 mmol) in EtOAc (3 mL) and HCl (1.00 M, 2 mL), the mixture was stirred at 20 °C for 0.5 h. TLC indicated reactant 29 was consumed completely. The mixture was extracted with EtOAc (10 mL), the aqueous phase adjusted pH = 8 with aq.NaCO3, and extracted with EtOAc (10 mL × 2), brine (10 mL), dried over Na2SO4, concentrated to give product. Compound 30 (0.200 g, 818 µmol, 66.8% yield) was obtained as yellow oil. 1HNMR (CDCl3, 400 MHz) δ 8.89 (dd, J = 1.6, 4.2 Hz, 1H, H-2), 8.11 (d, J = 8.4 Hz, 1H, H-4), 8.06 (d, J = 8.8 Hz, 1H, H-8), 7.65 (s, 1H, H-5), 7.59 (dd, J = 1.6, 8.7 Hz, 1H, H-7), 7.39 (dd, J = 4.4, 8.3 Hz, 1H, H-3), 4.18 (q, J = 7.2 Hz, 2H, CH2CH3), 3.82 (dd, J = 5.2, 7.8 Hz, 1H, CHCO), 3.28 (dd, J = 5.2, 13.6 Hz, 1H, *CH2CHCO), 3.06 (dd, J = 8.0, 13.6 Hz, 1H, *CH2CHCO), 1.23 (t, J = 7.2 Hz, 3H, CH3CH2).
6-Nitrobenzo[b]thiophene 1,1-dioxide (33)
To a solution of HNO3 (35.0 g, 333 mmol) in H2SO4 (46.0 g, 460 mmol) at 0 °C, then compound 32 (10.0 g, 60.2 mmol) was added at 0 °C for 1 h. TLC showed the starting material was consumed completely. The mixture was poured into ice water (300 mL) and the mixture was partitioned with EtOAc (200 mL) and the organic layer was wished with aq. NaHCO3 (200 mL), dried over Na2SO4 and evaporated to dryness. The residue was purified by column chromatography. Compound 33 (9.00 g, 42.6 mmol, 70.8% yield) was obtained as light yellow solid. 1HNMR (CDCl3, 400 MHz) δ 8.57 (d, J = 1.6 Hz, 1H, H-7), 8.48 (dd, J = 2.0, 8.3 Hz, 1H, H-5), 7.59 (d, J = 8.0 Hz, 1H, H-4), 7.34 (dd, J = 0.8, 7.0 Hz, 1H, H-3), 7.01 (d, J = 6.8 Hz, 1H, H-2).
6-Amino-2,3-dihydrobenzo[b]thiophene 1,1-dioxide (34)
To a solution of compound 33 (9.00 g, 42.6 mmol) in MeOH (90.0 mL), Pd/C (0.90 g, 10% purity) was added under N2. The suspension was degassed under vacuum and purged with H2 several times. The mixture was stirred under H2 (50 Psi) at 50 °C for 12 h. TLC showed the starting material was consumed completely. The suspension was filtered through a pad of Celite and the filter cake was washed with MeOH (100 mL × 3). The combined filtrates were concentrated to dryness. The crude product was triturated with EtOAc (20 mL) at 25 °C for 30 min. Compound 34 (5.00 g, 27.3 mmol, 64.0% yield) was obtained as light yellow solid. 1HNMR (DMSO-d6, 400 MHz,) δ 7.13 (d, J = 8.4 Hz, 1H, H-4), 6.83 (dd, J = 2.4, 8.3 Hz, 1H, H-5), 6.75 (d, J = 2.0 Hz, 1H, H-7), 5.58 (s, 2H, NH2), 3.52–3.40 (m, 2H, H-2), 3.12 (t, J = 6.8 Hz, 2H, H-3); LCMS m/z 184.1 [M + H]+.
6-Bromo-2,3-dihydrobenzo[b]thiophene 1,1-dioxide (35)
To a solution of compound 34 (5.00 g, 27.3 mmol) in HBr (50.0 mL, 40 % purity) at 0 °C, NaNO2 (2.45 g, 35.5 mmol) was added portion wise. The temperature was kept BELOW 8 °C during this addition. The resulting mixture was then added to a suspension of CuBr (2.74 g, 19.1 mmol) in HBr (10.0 mL) at 70 °C. The resulting mixture was stirred at 70 °C for 1 h. TLC showed the starting material was consumed completely. The mixture was poured into ice water (200 mL) and then was partitioned with EtOAc (100 ml × 3), the organic layer was washed with H2O (150 mL) dried over Na2SO4 and evaporated to dryness. The residue was purified by column chromatography. Compound 35 (4.00 g, 16.2 mmol, 59.1% yield) was obtained as light yellow solid. 1HNMR (CDCl3, 400 MHz) δ 7.85 (d, J = 1.2 Hz, 1H, H-7), 7.67 (dd, J = 1.6, 8.2 Hz, 1H, H-5), 7.27 (s, 1H, H-4), 3.55–3.47 (m, 2H, H-2), 3.36–3.30 (m, 2H, H-3); LCMS m/z 246.9 [M + H]+.
2,3-Dihydrobenzo[b]thiophene-6-carbonitrile 1,1-dioxide (36)
To a solution of compound 35 (4.00 g, 16.2 mmol) in DMF (24.0 mL), Zn(CN)2 (3.04 g, 25.9 mmol) and Pd(PPh3)4 (2.24 g, 1.95 mmol) were added under a nitrogen atmosphere, the reaction mixture was stirred at 80 °C for 16 h. TLC showed the starting material was consumed completely. The mixture was prateritioned between EtOAc (100 mL × 3) and H2O (100 mL), the organic layer was dried over Na2SO4 and evaporated to dryness. The crued product was triturated with EtOAc (10.0 mL) for 1 h at 25 °C. Compound 36 (2.00 g, 10.4 mmol, 63.9% yield) was obtained as light yellow solid. 1HNMR (DMSO-d6, 400 MHz,) δ 8.41 (d, J = 0.8 Hz, 1H, H-7), 8.11 (dd, J = 1.2, 8.0 Hz, 1H, H-5), 7.76 (dd, J = 0.8, 8.0 Hz, 1H, H-4), 3.71–3.63 (m, 2H, H-2), 3.49–3.41 (m, 2H, H-3).
2,3-Dihydrobenzo[b]thiophene-6-carbaldehyde 1,1-dioxide (37)
Compound 36 (0.500 g, 2.59 mmol) was added into a solution of Raney-Ni (221.70 mg, 2.59 mmol) in HCOOH (5.00 mL) in one portion at 25 °C under N2. The mixture was stirred at 100 °C for 2 h. Two reactions were combined for work up. The mixture was cooled to 20 °C and filter by Celite. The filtration was concentrated in reduced pressure to give compound 37 (0.900 g, 4.59 mmol, 88.6% yield) as a light yellow solid. 1H NMR (CDCl3, 400 MHz) δ 10.10 (s, 1H, CHO), 8.28 (s, 1H, H-7), 8.16 (dd, J = 1.4, 7.8 Hz, 1H, H-5), 7.61 (d, J = 7.8 Hz, 1H, H-4), 3.65 - 3.59 (m, 2H, H-2), 3.56–3.50 (m, 2H, H-3).
6-(Hydroxymethyl)-2,3-dihydrobenzo[b]thiophene 1,1-dioxide (38)
To a solution of compound 37 (2.00 g, 10.1 mmol) in MeOH (10.0 mL), NaBH4 (1.16 g, 30.5 mmol)was added in portions at 0 °C. Then the mixture was stirred at 15 °C for 3 h. TLC showed the starting material was consumed completely. The reaction mixture was partitioned between H2O (100 mL) and EtOAc (60.0 mL, 40.0 mL, 20.0 mL). The organic phase was separated, washed with brine 50.0 mL, dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. Compound 38 (1.00 g, 5.04 mmol, 49.4% yield) was obtained as yellow solid. 1H NMR (CDCl3, 400 MHz) δ 7.74 (s, 1H, H-7), 7.62–7.56 (m, 1H, H-5), 7.38 (d, J = 7.8 Hz, 1H, H-4), 4.77 (s, 2H, CH2, CH2OH), 3.56–3.46 (m, 2H, H-2), 3.43–3.34 (m, 2H, H-3), 2.01 (s, 1H, OH).
6-(Bromomethyl)-2,3-dihydrobenzo[b]thiophene 1,1-dioxide (39)
A mixture of compound 38 (500 mg, 2.52 mmol) in HBr (3.00 mL) was stirred at 20 °C for 2 h. TLC showed new spots were formed. The residue was poured into H2O (10 mL) and extracted with EtOAc (10 mL × 2). The organic layer was isolated, dried (MgSO4) and concentrated in vacuo. Compound 39 (500 mg, 1.53 mmol, 60.7% yield) was obtained as yellow oil. 1H NMR (CDCl3, 400 MHz,) δ 7.76 (s, 1H, H-7), 7.71 (d, J = 8.0 Hz, 1H, H-5), 7.48 (d, J = 8.0 Hz, 1H, H-4), 4.66 (s, 2H, CH2, CH2Br), 3.61–3.50 (m, 2H, H-2), 3.40 (d, J = 7.2 Hz, 2H, H-3); LCMS m/z 261.1 [M + H]+.
3-(1,1-Dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)-2-((diphenylmethylene)amino)ethyl propanoate (40)
A mixture of compound 39 (500 mg, 1.91 mmol), ethyl N-(diphenylmethylene)glycinate (767 mg, 2.87 mmol), NaH (114 mg, 2.87 mmol, 60% purity) in THF (3.00 mL) was stirred at 0–20 °C for 16 h. TLC showed new spots were formed. The residue was poured into H2O (15 mL) was the mixture was extracted with EtOAc (15 mL × 2). The organic layer was isolated, dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash silica gel chromatography. Compound 40 (600 mg, 1.14 mmol, 59.5% yield) was obtained as yellow gum. 1H NMR (CDCl3, 400 MHz) δ 7.58 (d, J = 7.6 Hz, 1H, Ar–H), 7.45–29 (m, 10H, Ar–H), 7.22 (d, J = 8.0 Hz, 1H, Ar–H), 6.75 (d, J = 4.8 Hz, 1H, Ar–H), 4.33–4.26 (m, 1H, CHCO), 4.13 (q, J = 7.2 Hz, 2H, CH2CH3), 3.52–3.43 (m, 2H, CH2CH2), 3.40–3.36 (m, 2H, CH2CHCO), 3.36–3.26 (m, 2H, CH2CH2), 1.27 (t, J = 7.2 Hz, 3H, CH3CH2); LCMS m/z 448.3 [M + H]+.
2-Amino-3-(1,1-dioxido-2,3-dihydrobenzo[b]thiophen-6-yl)ethyl propanoate (41)
To a solution of compound 40 (500 mg, 1.12 mmol) in EtOAc (1.00 mL), HCl (1 M, 2.50 mL) was added. The mixture was stirred at 20 °C for 1 h. TLC showed new spots were formed. The residue was poured into H2O (5 mL) and extracted with EtOAc (5 mL). The aqueous phase was basified by NaHCO3 to pH = ~8 and extracted with DCM (5 mL × 3). The organic phase was concentrated under reduced pressure. Compound 41 (300 mg, 1.06 mmol, 94.7% yield) was obtained as yellow oil. 1H NMR (CDCl3, 400 MHz,) δ 7.60 (s, 1H, H-7), 7.45 (dd, J = 1.2, 7.9 Hz, 1H, H-5), 7.32 (d, J = 8.0 Hz, 1H, H-4), 4.18 (q, J = 7.2 Hz, 2H, CH2CH3), 3.75–3.66 (m, 1H, CHCO), 3.55–3.44 (m, 2H, CH2CH2), 3.41–3.32 (m, 2H, CH2CH2), 3.15 (dd, J = 5.2, 13.7 Hz, 1H, *CH2CHCO), 2.93 (dd, J = 8.0, 13.7 Hz, 1H, *CH2CHCO), 1.26 (t, J = 7.2 Hz, 3H, CH3CH2).
2-Oxo-1,2,3,4-tetrahydroquinolin-7-yl trifluoromethanesulfonate (44)
To a solution of compound 43 (5.00 g, 30.6 mmol) in CHCl3 (50.0 mL), pyridine (5.28 g, 66.8 mmol, 5.39 mL) was added. The mixture was stirred at 25 °C for 10 min and then cooled to 0 °C. (Tf)2O (8.60 g, 30.6 mmol) was slowly added drop wise under 0 °C followed by stirring for 10 min. The reaction was stirred at 25 °C for 20 min. TLC showed the reaction was complete. The solution was poured into water (20.0 mL) and DCM (10.0 mL) was added. The organic phase was separated and the aqueous was extracted with DCM (2 × 10.0 mL). The organic phase were dried over magnesium sulfate, filtered and concentrated in vacuum. The crude product was purified by column chromatography. Compound 44 (5.00 g, 16.9 mmol, 55.2% yield) was obtained as white solid. 1H NMR (CDCl3, 400 MHz,) δ 9.44 (s, 1H, CONH), 7.23 (d, J = 8.4 Hz, 1H, H-8), 6.90 (dd, J = 2.4, 8.3 Hz, 1H, H-5), 6.80 (d, J = 2.4 Hz, 1H, H-6), 3.01 (t, J = 7.6 Hz, 2H, H-4), 2.80–2.57 (m, 2H, H-3); LCMS m/z 296.0 [M + H]+.
2-Oxo-1,2,3,4-tetrahydroquinoline-7-carbonitrile (45)
To a solution of compound 44 (5.00 g, 16.9 mmol) in DMF (30.0 mL), Zn(CN)2 (1.59 g, 13.5 mmol) and Pd(PPh3)4 (1.96 g, 1.69 mmol) were added, then the reaction was stirred at 80 °C for 12 h. TLC showed the reaction was complete. The solution was poured into water (20.0 mL) and DCM (10.0 mL) was added. The organic phase was separated and the aqueous was extracted with DCM (2 × 10.0 mL). The organic phase were dried over magnesium sulfate, filtered and concentrated in vacuum. Compound 45 (2.00 g, 9.35 mmol, 55.2% yield) was obtained as off-white solid. 1HNMR (DMSO-d6, 400 MHz) δ 10.34 (s, 1H, CONH), 7.39–7.36 (m, 2H, H-8 H-5), 7.15 (s, 1H, H-6), 2.96 (t, J = 7.6 Hz, 2H, H-4), 2.49–2.46 (m, 2H, H-3); LCMS m/z 173.1 [M + H]+.
2-Oxo-1,2,3,4-tetrahydroquinoline-7-carbaldehyde (46)
To a solution of compound 45 (2.00 g, 11.6 mmol) in HCOOH (20.0 mL), Ni (2.00 g, 34.0 mmol) was added, then the reaction was stirred at 100 °C for 2 h. TLC showed the reaction was complete. The mixture was filtered, the filtrate was concentrated under reduced pressure. The crude product was purified by column chromatography. Compound 46 (1.20 g, 6.85 mmol, 58.9% yield) was obtained as brown solid. 1HNMR (DMSO-d6, 400 MHz) δ 10.34 (s, 1H, CHO), 9.90 (s, 1H, CONH), 7.49 (dd, J = 1.6, 7.6 Hz, 1H, H-8), 7.44–7.39 (m, 1H, H-5), 7.33 (d, J = 1.6 Hz, 1H, H-6), 2.97 (t, J = 7.6 Hz, 2H, H-4), 2.49–2.45 (m, 2H, H-3); LCMS m/z 176.1 [M + H]+.
7-(Hydroxymethyl)-3,4-dihydroquinolin-2(1H)-one (47)
To a solution of compound 46 (2.00 g, 11.4 mmol) in MeOH (10.0 mL), NaBH4 (0.86 g, 22.0 mmol) was added in portions at 0 °C. Then the mixture was stirred at 15 °C for 3 h. TLC showed the starting material was consumed completely. The reaction mixture was partitioned between H2O (100 mL) and EtOAc (60 mL, 40 mL, 20 mL). The organic phase was separated, washed with brine 50.0 mL, dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. Compound 47 (1.00 g, 5.64 mmol, 49.4% yield) was obtained as yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 10.05 (s, 1H, CONH), 7.08 (d, J = 8.2 Hz, 1H, H-8), 6.85–6.80 (m, 2H, H-5 H-6), 4.40 (d, J = 4.6 Hz, 2H, CH2, CH2OH), 2.83 (t, J = 7.4 Hz, 2H, H-4), 2.45–2.37 (m, 2H, H-3); LCMS m/z 178.0 [M + H]+.
7-(Bromomethyl)-3,4-dihydroquinolin-2(1H)-one (48)
A solution of compound 47 (1.00 g, 6.13 mmol) in HBr (6.00 mL), then the mixture was stirred at 25 °C for 0.5 h. TLC showed the reaction was complete. The solution was poured into water (10.0 mL) and DCM (10.0 mL) was added. The organic phase was separated and the aqueous was extracted with DCM (2 × 10.0 mL). The organic phase were dried over magnesium sulfate, filtered and concentrated in vacuum. The crude product was triturated with EtOAc (10.0 mL) at 25 °C for 10 min. Compound 48 (0.500 g, 2.08 mmol, 33.9% yield) was obtained as a white solid. 1HNMR (DMSO-d6, 400 MHz,) δ 10.15 (s, 1H, CONH), 7.14 (d, J = 7.6 Hz, 1H, H-8), 6.98 (dd, J = 1.6, 7.6 Hz, 1H, H-5), 6.90 (s, 1H, H-6), 4.64 (s, 2H, CH2, CH2Br), 2.85 (t, J = 7.6 Hz, 2H, H-4), 2.46–2.40 (m, 2H, H-3); LCMS m/z 240.0 [M + H]+.
2-((Diphenylmethylene)amino)-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)ethyl propanoate (49)
To a solution of compound 48 (0.500 g, 2.08 mmol) and ethyl N-(diphenylmethylene)glycinate (1.11 g, 4.16 mmol) in THF (3.00 mL), NaH (166 mg, 4.16 mmol, 60% purity) was added at 0 °C, then the mixture was strried at 25 °C for 5 h. TLC showed the reaction was complete. The solution was poured into water (5.00 mL) and DCM (5.00 mL) was added. The organic phase was separated and the aqueous was extracted with DCM (2 × 5.00 mL). The organic phase were dried over magnesium sulfate, filtered and concentrated in vacuum. The crude product was purified by column chromatography. Compound 49 (0.550 g, 1.29 mmol, 61.9% yield. 1HNMR (DMSO-d6, 400 MHz) δ 7.74–6.67 (m, 10H, Ar–H), 6.99 (d, J = 7.6 Hz, 1H, Ar–H), 6.75–6.67 (m, 1H, Ar–H), 6.39 (d, J = 1.2 Hz, 1H, Ar–H), 4.27–4.22 (m, 1H, CHCO), 4.22–4.12 (m, 2H, CH2CH3), 3.28–3.18 (m, 1H, *CH2CHCO), 3.17–3.07 (m, 1H, *CH2CHCO), 2.91 (t, J = 7.6 Hz, CH2CH2), 2.64–2.55 (m, 2H, CH2CH2), 1.27 (t, J = 7.2 Hz, 3H, CH3CH2); LCMS m/z 427.2 [M + H]+.
2-Amino-3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)ehyl propanoate (50)
To a solution of 49 (0.550 g, 1.29 mmol) in EtOAc (3.00 mL), HCl (2 M, 2.58 mL) was added at 0 °C, then the mixture was stirred at 25 °C for 0.5 h. TLC showed the reaction was complete. The mixture extracted with EtOAc (5.00 mL). The organic phase was adjusted to pH = 7–8 with NaHCO3, extracted with DCM (10 × 5.00 mL). The combined organic phase was washed with brine (20.0 mL), dried over Na2SO4, filtered and concentrated in vacuum. Compound 50 (0.300 g, 1.14 mmol, 88.6% yield) was obtained as colorless oil. 1HNMR (DMSO-d6, 400 MHz) δ 8.28 (s, 1H, CONH), 7.10 (d, J = 7.6 Hz, 1H, Ar–H), 6.83 (d, J = 7.6 Hz, 1H, Ar–H), 6.63 (s, 1H, Ar–H), 4.20 (q, J = 7.2 Hz, 2H, CH2CH3), 3.70 (dd, J = 5.2, 7.9 Hz, 1H, CHCO), 3.05 (dd, J = 5.2, 13.6 Hz, 1H, *CH2CHCO), 2.94 (t, J = 7.6 Hz, 2H, CH2CH2), 2.81 (dd, J = 8.0, 13.6 Hz, 1H, *CH2CHCO), 2.63 (t, J = 7.6 Hz, 2H, CH2CH2), 1.29–1.25 (m, 3H, CH3CH2); LCMS m/z 263.1 [M + H]+.
3-(3-Acetyl-4-hydroxyphenyl)-2-(2-(benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)propanoic acid (1i)
To a solution of compound 25 (0.300 g, 768 µmol) in SOCl2 (3.00 mL), was stirred at 70 °C for 2 h, until the mixture was concentrated. A solution of 3-(3-acetyl-4-hydroxyphenyl)-2-aminopropanoic acid 53 (188 mg, 845 µmol) in DMF (3 mL) and TEA (233 mg, 2.31 mmol) were added, and the reaction mixture was stirred at 20 °C for 2 h. TLC indicated reactant 25 was consumed completely. The mixture was poured into citric acid (10 mL), extracted with EtOAc (20 mL), concentrated to give crude product. The residue was purified by prep-HPLC. Compound 1i (0.025 g, 39.8 µmol, 5.19% yield) was obtained as a yellow solid. 1HNMR (DMSO-d6, 400 MHz) δ 11.73 (s, 1H, COOH), 8.77 (d, J = 7.6 Hz, 1H, CONH), 8.07 (d, J = 2.0 Hz, 1H, O*CH=CH), 7.81 (s, 1H, Ar–H), 7.74 (d, J = 7.9 Hz, 1H, Ar–H), 7.67 (s, 1H, Ar–H), 7.50–7.42 (m, 1H, Ar–H), 7.37–7.28 (m, 1H, Ar–H), 7.35 (s, 1H, Ar–H), 7.05–7.00 (m, 1H, *CH=CHO), 6.87 (d, J = 8.4 Hz, 1H, Ar–H), 4.80–4.65 (m, 1H, CHCO), 4.75 (s, 2H, CH2N), 4.28 (s, 1H, Ar–OH), 3.76 (s, 2H, CH2N), 3.16 (d, J = 4.8 Hz, 1H, *CH2CH2N), 2.95 (dd, J = 9.6, 13.8 Hz, 1H, *CH2CH2N), 2.81 (m, 2H, CH2CH), 2.61 (s, 3H, CH3CO). 13C NMR (DMSO-d6, 101 MHz,) δ 205.022 (1C, COCH3), 172.752 (1C, COOH), 167.452 (1C, CON), 164.025 (1C, CONH), 160.030 (1C, Ar), 154.124 (1C, Ar), 148.211 (1C, O*CH=CH), 137.843 (1C, Ar), 137.501 (1C, Ar), 135.109 (1C, Ar), 132.630 (1C, Ar), 132.193 (1C, Ar), 132.157 (1C, Ar), 132.084 (1C, Ar), 131.646 (1C, Ar), 129.153 (1C, Ar), 128.883 (1C, Ar), 128.373 (1C, Ar), 122.482 (1C, Ar), 121.906 (1C, Ar), 120.331 (1C, Ar), 117.779 (1C, Ar), 110.794 (1C, Ar), 107.316 (1C, *CH=CHO), 53.838 (1C, CHCO), 36.208 (1C, CH2N), 30.273 (1C, CH2N), 28.830 (1C, *CH2CH), 27.933 (1C, *CH3CO), 22.858 (1C, *CH2CH2N). LCMS m/z 595.0 [M + H]+; HR-MS m/z calcd. for C30H24Cl2N2O7 (594.10) 595.0 [M + H]+; found: 595.0844 [M + H]+.
2-(2-(Benzofuran-6-carbonyl)-5,7-dichloro-1,2,3,4-tetrahydroisoquinoline-6-carboxamido)-3-(3,4-dihydroxyphenyl)propanoic acid (1j)
To a solution of compound 25 (0.300 g, 768 μmol) in SOCl2 (3 mL), was stirred at 70 °C for 2 h, until the mixture was concentrated. A solution of 2-amino-3-(3,4-dihydroxyphenyl)propanoic acid 54 (166 mg, 845 μmol) in DMF (3 mL) and TEA (233 mg, 2.31 mmol) were added, and the reaction mixture was stirred at 20 °C for 2 h. TLC indicated reactant 25 was consumed completely. The mixture was poured into citric acid (10 mL), extracted with EtOAc (20 mL), concentrated to give crude product. The residue was purified by prep-HPLC. Compound 1j (0.025 g, 41.7 µmol, 5.43% yield) was obtained as a off-white solid. 1HNMR (DMSO-d6, 400 MHz) δ 8.64 (s, 1H, CONH), 8.38 (s, 2H, Ar–OH), 8.07 (d, J = 1.6 Hz, 1H, O*CH=CH), 7.75 (d, J = 8.0 Hz, 1H, Ar–H), 7.68 (s, 1H, Ar–H), 7.40–7.29 (m, 1H, Ar–H), 7.34(s, 1H, Ar–H) 7.02 (s, 1H, Ar–H), 6.68 (s, 1H, *CH=CHO), 6.62 (d, J = 7.6 Hz, 1H, Ar–H), 6.54 (d, J = 7.6 Hz, 1H, Ar–H), 4.74 (s, 2H, CH2N), 4.65–4.55 (m, 1H, CHCO), 3.76 (s, 2H, CH2N), 2.95 (dd, J = 5.6, 14.0 Hz, 1H, *CH2CH2N), 2.89–2.82 (m, 1H, *CH2CH2N), 2.82–2.78 (m, 2H, CH2CH). 13C NMR (DMSO-d6, 101 MHz) δ 172.935 (1C, COOH), 163.974 (1C, CONH), 154.131 (1C, CON), 148.211 (1C, Ar), 145.412 (1C, O*CH=CH), 145.339 (1C, Ar), 144.252 (1C, Ar), 137.391 (1C, Ar), 135.201 (1C, Ar), 132.110 (1C, Ar), 132.084 (1C, Ar), 131.756 (1C, Ar), 129.145 (1C, Ar), 129.001 (1C, Ar), 128.489 (1C, Ar), 126.200 (1C, Ar), 122.496 (1C, Ar), 121.913 (1C, Ar), 120.425 (1C, Ar), 116.999 (1C, Ar), 115.701 (1C, Ar), 110.801 (1C, Ar), 107.316 (1C, *CH=CHO), 54.421 (1C, *CHCO), 40.687 (1C, CH2N), 40.475 (1C, CH2N), 40.311 (1C, *CH2CH), 36.872 (1C, *CH2CH2N). LCMS m/z 569.2 [M + H]+; HR-MS m/z calcd. for C28H22Cl2N2O7 (568.08) 569.2 [M + H]+; found: 569.0673 [M + H]+.
Jurkat T-cell adhesion assay
Cells and Cell Culture: Lifitegrast (Selleck, S3714, Shanghai, China), Human leukemia T lymphocytes (Jurkat, Clone E6-1, the catalog number is SPSC-513, was obtained from the cell bank of Chinese Academy of Sciences, Shanghai, China) were cultured in RPMI 1640 Medium (Invitrogen, 11875-093) supplemented with 5% fetal bovine serum at 37 °C with 5% CO2 in a humidifified atmosphere.
To evaluate the ability of compounds 1a–1j to inhibit the attachment of Jurkat cells to intercellular-adhesion molecule ICAM-1 in vitro, 100 mM stock solutions of the compounds and a positive control (Lifitegrast) were prepared in an aqueous solution of dimethylsulfoxide (1:1 (v/v)), respectively, and then diluted by adding assay medium to achieve and maintain the desired concentration throughout the assay. The Jurkat cells were labeled with an 8 μM solution of 2′, 7′-bis-(2-carboxyethyl)-5-(and-6)- carboxyfluorescein in growth medium at room temperature for 15 min. Labeled cells were incubated in 70 μL of assay medium in each well of a 96-well plate at 5 × 105 cells per well with 70 μL of compound or the positive control in assay medium at 37 °C for 30 min. A 100 μL aliquot of this fluorescence-labeled Jurkat cell suspension was allowed to settle in the presence of compound or the positive control in wells of a 96-well plate coated with recombinant human ICAM-1 expressed as an Fc chimera (R&D Systems, Minneapolis, MN) at 37 °C for 1 h. A dose titration of each test compound was run (10−10–10−4 M) in three separate lanes on a single plate, non-adherent cells were removed by washing with the medium. The adherent cells were quantitated by measuring the intensity of fluorescence from adherent cells, and the IC50 values were calculated by using a standard four-parameter logistic nonlinear regression analysis of the data (GraphPad Software Inc., San Diego, CA) [13].
Animal experiments
Dry eye mice model
Drugs and instruments: Scopolamine (0.5 mg/0.2 mL, Sigma-Aldrich, S-098-1g, St. Louis, MO); 5% (W/V) or 7% (W/V) compound 1b eye drops (containing 2.6% glycerinum and 0.01% benzalkonium chloride 0.01%, pH range from 8.0 to 8.5). Lifitegrast (5%, Shire, 07548, Columbia, USA); OCT (VWR, 8551-00, Suwanee, USA); TUNEL(Beyotime Institute of Biotechnology, C1090, Haimen, China); (Nikon Eclipse Ni-U, Japan); Stereographic fluorescence microscope (Nikon SMZ1500, Japan); Frozen slicing machine (LEIKA CM1860UV, Germany); Intelligent drying oven (ZD-890C; Ouyi Company, Hangzhou, China). In this study, the animal experimental procedures were approved by the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006), and adhered to the standards in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
All experimental procedures related to animals’ welfare in this research were performed according to the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006), and the standards in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All animal experimental protocols in this study were also approved by the animal ethics committee of He University. Thirty female C57/BL6 mice (age range, 6–8 weeks) were purchased from Liaoning Changsheng Biotechnology Co., LTD. The animals were divided into 6 groups (each group, n = 5): blank control group, model control group, two compound 1b treatment groups (concentrations: 5% and 7% W/V), vehicle control group treated with the vehicle of compound 1b solution and positive control group treated with 5% Lifitegrast. The dry eye model of mice was created by intelligent environment control system combined with drug induction. Two-step dehumidification method was used to control the environmental humidity, The relative humidity in the room was reduced to 40 ± 5% using the temperature dehumidifier, and the intelligent dehumidifier (dehumidification range RH 10–80%) is used to further reduce the humidity of the cabinet. At the same time, the air flow in the cabinet were maintained at 2.1 ± 0.2 m/s by the ventilator located 20 cm from the mouse cages in the cabinet. Through the above measures, humidity, temperature and air flow are effectively regulated. C57/BL6 mice were exposed to dry conditions (RH = 15; WV = 2 m/s; T = 21–23 °C) for 14 days, subcutaneously were injected to hindquarters alternately with scopolamine hydrobrominde (0.5 mg/0.2 ml) three times a day (9:00 am, 1:00 PM, 4:00 pm) for 14 days to induce dry eye symptoms [30]. At the same time of dry eye development, the treatment groups mice were administrated on the surface of the both eyeball with 5 µL compound 1b eye drops, or treated with Lifitegrast (5%, USA) individually, and treatment with the vehicle of compound 1b solution was used as vehicle control group. There were 5 mice in each group, and twice a day for topical administration of drugs (at 9:00 am and 3:00 pm, respectively).
Frozen section preparation
Periodic acid-Schiff (PAS) and TUNEL staining were performed on frozen section in this study. The animals were sacrificed, and the eyeballs and appendages (n = 10 eyes/group) were surgically excised and then embedded by OCT (VWR, Suwanee, GA, USA). The central sagittal plane of the eyeball was sectioned with 6 µm thickness and stored at −80 °C [31].
Corneal fluorescein staining
The mice were in an unanesthetized state, 2 µL of 1% fluorescein sodium solution was dropped into the conjunctival sac of mice using a micropipette. The cornea was examined using slit-lamp microscopy in cobalt blue light 2 min after fluorescein instillation. The damaged areas of corneal epithelium were stained in green, the stained areas were assessed according to a standardized grading system ranging from 0 to 3, with the corneal surface divided into five regions, 0 score: no dyeing; 1 score: few punctate dyeing; 2 score: diffuse punctate dyeing; 3 score: flake dyeing [32]. Then the total score from the five regions was analyzed.
Measurement of goblet cell density
Ocular sections were cut at the center of the eye, where the lens has its maximum diameter. Sections (6 µm) were stained by periodic-Schiff (PAS) reagent for measuring goblet cell density. The sections were observed and taken for counting with a digital camera (Eclipse Ni-U; Nikon). Goblet cell density was measured in the superior and inferior conjunctival of the eyes of each group. And using image-analysis software (NIS Elements Software, Nikon) and expressed as number of goblet cells per mm [33].
TUNEL staining
Frozen slices of 6 μm were used for the terminal deoxynucleotidyl transferase-mediated FITC-dUTP nick end labeling (TUNEL) apoptosis Assay kit (C1089, Beyotime Institute of Biotechnology, Haimen, China) detection. According to the manufacture’s protocol, sections were fixed in 4% paraformaldehyde for 15 min at room temperature, permeabilized in 0.3% Triton X-100 for 3 min at 4 °C. Then the frozen slices were incubated with TUNEL assay at room temperature for 60 min, and the slices were then stained with DAPI (0.5 μg/ml, staining the nucleus) for 5 min. After washing with PBS, the sections were visualized, and the images were captured on an inverted fluorescent microscope (×400 amplification, Nikon, ECLIPSE Ni, Japan). The central corneal epithelial field of 200 µm was selected, and the number of TUNEL positive cells was counted and recorded [34].
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. The data were analyzed by one-way analysis of variance (ANOVA) with SPSS 20.0 (IBM). The histograms were plotted with Graphpad prism 8. Image J was used to show the image and calculate the gray value of protein bands in western blot.
Supplementary Information
Acknowledgements
This research was funded by Technology Program of Liaoning, grant number 2019JH2/10300011.
Abbreviations
- DED
dry eye disease
- LFA-1
lymphocyte function-associated antigen 1
- ICAM-1
intercellular-adhesion molecule-1
- THIQ
Tetrahydroisoquinoline
- SAR
structure–activity relationship
- DMF
Dimethylformamide
- HATU
2-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- Boc
tert-Butyldicarbonate
- TEA
Triethylamine
- DCM
Dichloromethane
- CDI
Carbonyldiimidazole
- TMSOK
Potassium trimethylsilanolate
- THF
Tetrahydrofuran
- DIEA
Diisopropylethylamine
- n-BuLi
n-Butyllithium
- PPA
Polyphosphoric acid
- EDCI
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- DMAP
Dimethylaminopyridine
- (Tf)2O
Trifluoroacetic anhydride
- Ar
Aromatic groups
- NMR
Nuclear Magnetic Resonance
- TMS
Tetramethylsilane
- ppm
parts per million
- MS
mass spectrum
- ESI
electrospray ionization
- HRMS
high resolution mass spectrum
- LCMS
liquid chromatography mass spectrometry
- TLC
thin-layer chromatography
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Consent for publication
All authors have seen the manuscript and agree to its publication.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mingyue Zheng, Email: myzheng@simm.ac.cn.
Donglei Zhang, Email: zhangdonglei@huh.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1007/s00044-022-02851-9.
References
- 1.Bartlett J, Keith M, Sudharshan L, Snedecor S. Associations between signs and symptoms of dry eye disease: a systematic review. Clin Ophthalmol. 2015;9:1719–30. doi: 10.2147/OPTH.S89700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rouen PA, White ML. Dry eye disease: prevalence, assessment, and management. Home Healthc Now. 2018;36:74–83. doi: 10.1097/NHH.0000000000000652. [DOI] [PubMed] [Google Scholar]
- 3.Pult H, Purslow C, Murphy P. The relationship between clinical signs and dry eye symptoms. Eye. 2011;25:502–10. doi: 10.1038/eye.2010.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potvin R, Makari S, Rapuano CJ. Tear film osmolarity and dry eye disease: a review of the literature. Clin Ophthalmol. 2015;9:2039–47. doi: 10.2147/OPTH.S95242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–9. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 6.Makgoba MW, Sanders ME, Luce GEG, Dustin ML, Springer TA, Clark EA, et al. ICAM-1 a ligand for LFA-1-dependent adhesion of B, T and myeloid cells. Nature. 1988;331:86–88. doi: 10.1038/331086a0. [DOI] [PubMed] [Google Scholar]
- 7.Walling BL, Kim M. LFA-1 in T Cell Migration and Differentiation. Front Immunol. 2018;9:952.. doi: 10.3389/fimmu.2018.00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pflugfelder SC, Stern M, Zhang S, Shojaei A. LFA-1/ICAM-1 Interaction as a Therapeutic Target in Dry Eye Disease. J Ocul Pharmacol Therap. 2017;33:5–12. doi: 10.1089/jop.2016.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez VL, Pflugfelder SC, Zhang S, Shojaei A, Haque R. Lifitegrast, a Novel Integrin Antagonist for Treatment of Dry Eye Disease. Ocul Surf. 2016;14:207–15. doi: 10.1016/j.jtos.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Keating GM. Lifitegrast Ophthalmic Solution 5%: A Review in Dry Eye Disease. Drugs. 2017;77:201–8. doi: 10.1007/s40265-016-0681-1. [DOI] [PubMed] [Google Scholar]
- 11.Sheppard JD, Torkildsen GL, Lonsdale JD, D’Ambrosio FA, McLaurin EB, Eiferman RA, et al. Lifitegrast Ophthalmic Solution 5.0% for Treatment of Dry Eye Disease: Results of the OPUS-1 Phase 3 Study. Ophthalmology. 2014;121:475–83. doi: 10.1016/j.ophtha.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 12.Lollett IV, Galor A. Dry eye syndrome: developments and Lifitegrast in perspective. Clin Ophthalmol. 2018;12:125–39. doi: 10.2147/OPTH.S126668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong M, Gadek TR, Bui M, Shen W, Burnier J, Barr KJ, et al. Discovery and Development of Potent LFA-1/ICAM-1 Antagonist SAR 1118 as an Ophthalmic Solution for Treating Dry Eye. ACS Med Chem Lett. 2012;3:203–6. doi: 10.1021/ml2002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong M, Shen W, Barr KJ, Arbitrario JP, Arkin MR, Bui M, et al. Discovery of tetrahydroisoquinoline (THIQ) derivatives as potent and orally bioavailable LFA-1/ICAM-1 antagonists. Bioorg Med Chem Lett. 2020;20:5269–73. doi: 10.1016/j.bmcl.2010.06.145. [DOI] [PubMed] [Google Scholar]
- 15.Zhong M, Hanan EJ, Shen W, Bui M, Arkin MR, Barr KJ, et al. Structure–activity relationship (SAR) of the α-amino acid residue of potent tetrahydroisoquinoline (THIQ)-derived LFA-1/ICAM-1 antagonists. Bioorg Med Chem Lett. 2011;21:307–10. doi: 10.1016/j.bmcl.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Abdullahi M, Olotu FA, Soliman ME. Allosteric inhibition abrogates dysregulated LFA-1 activation: Structural insight into mechanisms of diminished immunologic disease. Comput Biol Chem. 2018;73:49–56. doi: 10.1016/j.compbiolchem.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Vuković G, Marinković A, Obradović M, Radmilović V, Čolić M, Aleksić R, et al. Synthesis, characterization and cytotoxicity of surface amino-functionalized water-dispersible multi-walled carbon nanotubes. Appl Surf Sci. 2009;255:8057–75. doi: 10.1016/j.apsusc.2009.05.016. [DOI] [Google Scholar]
- 18.Michael DM, Arthur AH, Karin T, Kevin BS, Rajnandan P, David MS, et al. Structure−Activity Studies for a Novel Series of N-(Arylethyl)-N-(1,2,3,4-tetrahydronaphthalen-1-ylmethyl)-N-methylamines Possessing Dual 5-HT Uptake Inhibiting and α2-Antagonistic Activities. J Med Chem. 1997;40:1049–62. doi: 10.1021/jm960723m. [DOI] [PubMed] [Google Scholar]
- 19.Wolfgang G, Matthias N, Bernd P, Claus R, Sébastien S. Comparison of inhibitory activity of isomeric triazolopyridine derivatives towards adenosine receptor subtypes or do similar structures reveal similar bioactivities? Bioorg Med Chem Lett. 2004;14:3307–12. doi: 10.1016/j.bmcl.2004.03.104. [DOI] [PubMed] [Google Scholar]
- 20.Takayoshi S, Yosuke O, Masaki R, Masashige B, Aogu G, Yukihiro I, et al. Rapid Discovery of Highly Potent and Selective Inhibitors of Histone Deacetylase 8 Using Click Chemistry to Generate Candidate Libraries. J Med Chem. 2012;55:9562–75. doi: 10.1021/jm300837y. [DOI] [PubMed] [Google Scholar]
- 21.Bajwa JS. Chemoselective deprotection of benzyl esters in the presence of benzyl ethers, benzyloxymethyl ethers and n-benzyl groups by catalytic transfer hydrogenation. Tetrahedron Lett. 1992;33:2299–302. doi: 10.1002/chin.199243125. [DOI] [Google Scholar]
- 22.Wang Y, Chen F, Di H, Xu Y, Xiao Q, Wang X, et al. Discovery of Potent Benzofuran Derived Diapophytoene Desaturase (CrtN) Inhibitors with Enhanced Oral Bioavailability for the Treatment of Methicillin-Resistant S. aureus (MRSA) Infections. J Med Chem. 2016;59:3215–30. doi: 10.1021/acs.jmedchem.5b01984. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Huang W, Xin M, Chen P, Gui L, Zhao X, et al. Identification of 4-(2-furanyl)pyrimidin-2-amines as Janus kinase 2 inhibitors. Bioorg Med Chem. 2017;25:75–83. doi: 10.1016/j.bmc.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Ozaki Y, Hosoya A, Okamura K, Kim SW. A Convenient Synthesis of 2-Alkylated 1,4-Benzenediols. Synlett. 1997;4:365–6. doi: 10.1055/s-1997-806. [DOI] [Google Scholar]
- 25.Kilbourn MR, Dischino DD, Welch MJ. Synthesis of DL-[3-11C]phenylalanine. Int J Appl Radiat Isotopes. 1984;35:603–5. 10.1016/0020-708X(84)90103-0. [DOI] [PubMed]
- 26.Wang XF, Tian XT, Ohkoshi E, Qin B, Liu YN, Wu PC, et al. Design and synthesis of diarylamines and diarylethers as cytotoxic antitumor agents. Bioorg Med Chem Lett. 2012;22:6224–8. doi: 10.1016/j.bmcl.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 27.Kubota H, Rice KC. Palladium-catalyzed cyanation of hindered, electron-rich aryl triflates by zinc cyanide. Tetrahedron Lett. 1998;39:2907–10. doi: 10.1002/chin.199830049. [DOI] [Google Scholar]
- 28.Zhang Y, An Y, He X, Zhang D, He W. Esculetin protects human corneal epithelial cells from oxidative stress through Nrf-2 signaling pathway. Exp Eye Res. 2020;202:108360.. doi: 10.1016/j.exer.2020.108360. [DOI] [PubMed] [Google Scholar]
- 29.Gipson IK. Goblet cells of the conjunctiva: A review of recent findings. Prog Retinal Eye Res. 2016;54:49–63. doi: 10.1016/j.preteyeres.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Chen W, De Paiva CS, Corrales RM, Volpe EA, Mcclellan AJ, et al. Interferon-γ Exacerbates Dry Eye–Induced Apoptosis in Conjunctiva through Dual Apoptotic Pathways. Investig Ophthalmol Vis Sci. 2011;52:6279–85. doi: 10.1167/iovs.10-7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma N, Siegfried C, Kubota M, Huang J, Liu Y, Liu M, et al. Expression profiling of ascorbic Acid-Related transporters in human and mouse eyes. Investig Ophthalmol Vis Sci. 2016;57:3440–50. doi: 10.1167/iovs.16-19162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemp MA. Report of the National Eye Institute/Industry workshop on Clinical Trials in Dry Eyes. Clao J. 1995;21:221–32. doi: 10.1016/j.susc.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Chen W, Zhang X, Zhang J, Chen J, Wang S, Wang Q, et al. A Murine Model of Dry Eye Induced by an Intelligently Controlled Environmental System. Investig Ophthalmol Vis Sci. 2008;49:1386–91. doi: 10.1167/iovs.07-0744. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Fang Q, Zhong P, Chen L, Wang L, Zhang Y, et al. EGFR Inhibition Blocks Palmitic Acid-induced inflammation in cardiomyocytes and Prevents Hyperlipidemia-induced Cardiac Injury in Mice. Sci Rep. 2016;6:24580.. doi: 10.1038/srep24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.