

Abstract
Antibiotic tolerance is an understudied potential contributor to antibiotic treatment failure and the emergence of multidrug-resistant bacteria. The molecular mechanisms governing tolerance remain poorly understood. A prominent type of β-lactam tolerance relies on the formation of cell wall-deficient spheroplasts, which maintain structural integrity via their outer membrane (OM), an asymmetric lipid bilayer consisting of phospholipids on the inner leaflet and a lipid-linked polysaccharide (lipopolysaccharide, LPS) enriched in the outer monolayer on the cell surface. How a membrane structure like LPS, with its reliance on mere electrostatic interactions to maintain stability, is capable of countering internal turgor pressure is unknown. Here, we have uncovered a novel role for the PhoPQ two-component system in tolerance to the β-lactam antibiotic meropenem in Enterobacterales. We found that PhoPQ is induced by meropenem treatment and promotes an increase in 4-amino-4-deoxy-L-aminoarabinose [L-Ara4N] modification of lipid A, the membrane anchor of LPS. L-Ara4N modifications likely enhance structural integrity, and consequently tolerance to meropenem, in several Enterobacterales species. Importantly, mutational inactivation of the negative PhoPQ regulator mgrB (commonly selected for during clinical therapy with the last-resort antibiotic colistin, an antimicrobial peptide [AMP]) results in dramatically enhanced tolerance, suggesting that AMPs can collaterally select for meropenem tolerance via stable overactivation of PhoPQ. Lastly, we identify histidine kinase inhibitors (including an FDA-approved drug) that inhibit PhoPQ-dependent LPS modifications and consequently potentiate meropenem to enhance lysis of tolerant cells. In summary, our results suggest that PhoPQ-mediated LPS modifications play a significant role in stabilizing the OM, promoting survival when the primary integrity maintenance structure, the cell wall, is removed.
Author summary
Treating an infection with an antibiotic often fails, resulting in a tremendous public health burden. One understudied likely reason for treatment failure is the development of “antibiotic tolerance”, the ability of bacteria to survive normally lethal exposure to an antibiotic. Here, we describe a molecular mechanism promoting tolerance. A bacterial stress sensor (PhoPQ) is activated in response to antibiotic (meropenem) treatment and consequently strengthens a bacterial protective “shell” to enhance survival. We also identify inhibitors of this mechanism, opening the door to developing compounds that help antibiotics work better against tolerant bacteria.
Introduction
The rapid rise of antibiotic treatment failure threatens our ability to prevent and control bacterial infections. Antibiotic resistance, the continued proliferation of bacteria in the presence of the antibiotic, can often explain failure of clinical therapy. However, the response to an antibiotic is oftentimes more nuanced than a simple dichotomy of resistance vs. susceptibility. Bacteria can survive treatment in a non- or slowly-proliferating state, readily reverting to healthy growth after removal of the antibiotic (such as the end of a treatment course), and this is typically referred to as “antibiotic tolerance” [1–3]. Importantly, tolerance to antibotics has been shown to enhance the evolution of outright resistance mechanisms [4–6], and can thus serve as both a direct and indirect contributor to treatment failure.
β-lactams are the most widely-prescribed antibiotic class used to treat bacterial infections. The β-lactam ring inhibits the activity of the penicillin-binding proteins (PBPs) through covalent modification of a catalytic residue. PBPs are enzymes that synthesize the cell wall, an essential structure composed mainly of the polysaccharide peptidoglycan (PG). In many well-studied model organisms, PBP inhibition induces cell wall degradation and often subsequent lysis through the action of cell wall degrading enzymes (collectively referred to as “autolysins”) in a poorly-understood manner [2]. While lysis is the canonical response of model organisms like Escherichia coli K12, many formally β-lactam susceptible clinical isolates of Gram-negative pathogens (including prominent clinical isolates belonging to the Enterobacterales, like Klebsiella spp. and Enterobacter spp.) exhibit a unique type of β-lactam tolerance [7–9]. Like E. coli, these cells digest their PG upon exposure to β-lactams. However, instead of lysing, these pathogens survive antibiotic-induced cell wall degradation by forming viable, non-dividing, cell wall-deficient spheroplasts, which presumably rely on the outer membrane to counter their internal turgor. Interestingly, spheroplasts do not absolutely require osmotic stabilization and form in diverse types of growth media, including human serum [9]. This cell wall-deficient phenotype is reminiscent of so-called L-forms [10–12], with the notable distinction that spheroplasts do not divide in the presence of the antibiotic.
Remarkably, spheroplasts formed in response to the carbapenem antibiotic meropenem readily resume growth and revert to wild-type rod shape when the β-lactam is removed from the growth medium [7,9]. Little is known about the molecular mechanisms that facilitate spheroplast formation and survival. In Vibrio cholerae, the two-component system (TCS) VxrAB is essential for spheroplast recovery by upregulating cell wall synthesis and downregulating iron uptake into the cells, mitigating toxic free iron accumulation induced by β-lactam treatment and allowing the cell to avoid damage by oxidative stress [13,14]. Many questions remain, however, as to how the cell envelope maintains its integrity without a cell wall, the essential structure canonically thought to protect the cell against immense turgor pressure.
In this study, we used Enterobacter cloacae as a model Gram-negative pathogen to investigate genetic factors that contribute to bacterial tolerance to meropenem, which is used as a last-resort β-lactam to treat multidrug-resistant bacterial infections [15–17]. We first show that tolerance is dependent on outer membrane modifications (specifically 4-amino-4-deoxy-L-aminoarabinose [L-Ara4N]) induced by the PhoPQ TCS, an important cell envelope stress sensor that has previously been shown to respond to magnesium limitation, cationic antimicrobial peptide exposure, osmotic challenge, and pH changes [18]. Both PhoPQ regulon transcription and modification of the lipopolysaccharide lipid A domain are induced by meropenem treatment, suggesting a specific response to perturbations of PG synthesis in E. cloacae. These findings represent a novel mechanism of β-lactam tolerance in clinically relevant Enterobacterales, as well as an expanded role for the PhoPQ TCS.
Results
The PhoPQ TCS system regulates carbapenem tolerance
We previously showed that many Gram-negative pathogens are highly tolerant to meropenem. Upon treatment, tolerant cells do not appreciably lyse. Instead, they form viable, enlarged, non-replicating spheroplasts that are devoid of detectable cell wall material [9]. Meropenem-induced spheroplast formation is quantifiable as an OD600 increase (Fig 1A) with only a moderate concomitant decrease in survival, as measured by colony-forming units (CFU) (Fig 1B). In contrast, non-tolerant bacteria like many E. coli isolates rapidly lyse in the presence of meropenem, indicated by a decrease in both OD600 and survival (Fig 1A and 1B). Since spheroplast integrity is presumably maintained by the outer membrane, rather than the cell wall, we hypothesized that the strength of the outer membrane might correlate with tolerance. To test this, we repeated the killing experiments in the presence of the known outer membrane fortifying agents Mg2+ and Ca2+, which link adjacent lipopolysaccharide molecules by forming ionic bridges between phosphate groups on the lipid A domain [19,20]. Addition of either divalent cation (Mg2+ or Ca2+) supported spheroplast formation with a concomitant reduction in lysis during meropenem treatment in a concentration-dependent manner (S1A and S1B Fig), particularly at very high concentrations. Furthermore, combinatorial addition of excess Ca2+ and Mg2+ completely prevented lysis (S1A Fig). Thus, divalent cations prevent lysis during meropenem treatment, potentially through increasing the mechanical load-bearing capacity of the outer membrane through lipopolysaccharide crosslinking to protect the spheroplast structure.
Fig 1. Enterobacter cloacae ATCC 13047 is highly meropenem tolerant.
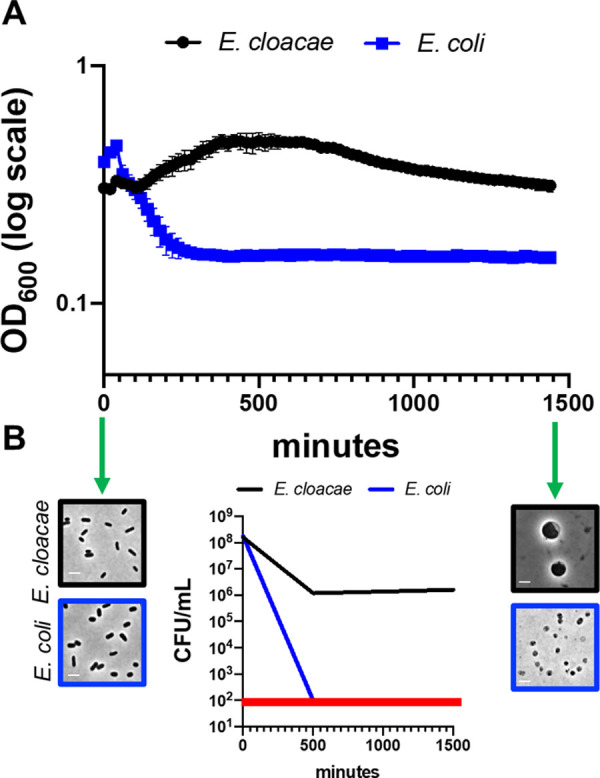
(A) Representative experiment demonstrating changes in OD600 measurements following meropenem treatment in E. cloacae relative to E. coli. Error bars represent the average of 3 technical replicates +/- standard deviation. (B) Survival was calculated as CFU/mL from the experiment depicted in (A). The red line denotes the limit of detection. Phase images from the same experiment show cells before and after meropenem exposure to illustrate spheroplast formation in E. cloacae and lysis in E. coli (only cell debris is visible in phase image). Scale bars, 2 μm.
When divalent cations are limiting, many Gram-negative pathogens induce outer membrane modifications (hyperacylation and/or increasing the positive charge of lipid A) to functionally substitute for divalent ionic bridges between lipopolysaccharide molecules [21]. In many Enterobacterales, these modifications are controlled by the well-studied PhoPQ two-component system (TCS) [18]. As shown in E. cloacae [22] and other Enterobacterales [23,24], the PhoPQ TCS directly regulates expression of the arn operon, which encodes enzymes that synthesize and transfer positively charged L-Ara4N moieties to lipid A, and partially regulates expression of pagP (coding for an outer membrane acyltransferase) [25]. In our strain of E. cloacae, the PhoPQ system is required to sustain “heteroresistance”, i.e. background resistance of a small subpopulation of cells against therapeutic antimicrobial peptides like colistin [22,26,27]. To test whether the PhoPQ TCS contributes to meropenem tolerance, we measured spheroplast formation and stability over 24 hours in ΔphoPQ. Strikingly, OD600 declined sharply in ΔphoPQ relative to wild type, which could be fully complemented by ectopic expression of phoPQ (Fig 2A). The decline in spheroplast formation correlated with a robust 10-fold decrease in ΔphoPQ viability (measured by CFU/mL) relative to wild type (Fig 2B). Step-wise titration of Ca2+ and/or Mg2+ markedly enhanced ΔphoPQ tolerance (S1C and S1D Fig), suggesting decreased tolerance (spheroplast formation) in ΔphoPQ is due to its inability to stabilize adjacent lipopolysaccharide molecules in the outer membrane [28]. To corroborate the involvement of PhoPQ, we sought to either enhance or reduce its activity and measured the effect of such perturbations on tolerance. PhoQ is antagonized by the small periplasmic MgrB protein; mgrB overexpression is thus expected to result in suppression of PhoPQ induction [29]. Indeed, mgrB overexpression from a plasmid reduced spheroplast formation (proxied by OD600 measurements) (Fig 2C) in the wild type, closely resembling the ΔphoPQ phenotype (Fig 2A). We next tested a strain deleted in mgrB. Modified lipid A structures in ΔmgrB were confirmed using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (S3A and S3B Fig), and we also phenotypically validated this mutant. Since the PhoPQ system mediates colistin resistance and heteroresistance, the ΔmgrB mutant (where PhoPQ regulon baseline levels are elevated) should be more resistant against colistin. As expected, colistin MIC was higher in the mutant (≥128 μg/mL) than in the wild type (16 μg/mL). Exposing ΔmgrB to meropenem resulted in an increase in OD600 (S2A Fig) that coincided with an approximately 1000-fold increased survival to meropenem relative to wild type (Fig 2B). Lastly, two fully colistin-susceptible (non-heteroresistant) clinical isolates of E. cloacae from our collection exhibited a low-tolerance phenotype compared to ATCC 13047 (S2C Fig), consistent with the idea that the same OM modifications that cause colistin resistance also contribute to meropenem tolerance.
Fig 2. The PhoPQ system promotes meropenem tolerance in E. cloacae.

(A) Spheroplast formation in response to meropenem treatment. Overnight cultures were diluted 10-fold into fresh LB medium containing 10 μg/mL meropenem and OD600 was measured. (B) Fraction of population surviving after 24 hours of meropenem exposure from experiments as described in (A). (C) MgrB overexpression reduces tolerance. Cells were treated as described in (A), but with the addition of 0.2% arabinose (inducer). pHERD; empty vector. (D) The arn operon is required for meropenem tolerance. Experiments were conducted as described in (A). Data in each graph represent the average of 3 replicates +/- standard deviation; additional biological replicates are shown in S2B Fig. Statistical significance for survival fraction determined by one-way ANOVA of log transformed data, followed by Tukey’s correction for multiple comparisons (ns, not significant; ****, p ≤ 0.0001).
To dissect individual contributions of PhoPQ-regulated genes to tolerance, we created mutants in the pagP and arn loci. Specifically, we deleted arnT, which encodes the L-ara4N transferase that is necessary for aminoarabinose addition to lipid A [30]. While ΔpagP displayed spheroplast formation levels similar to wild type (Fig 2D), ΔarnT exhibited a drop in OD600 reminiscent of ΔphoPQ (Figs 2D and S2B) and a concomitant 100-fold decrease in CFU/mL relative to wild type (Fig 2B). Notably, the survival defect, as measured by CFU/mL, was consistently more pronounced in ΔarnT vs. ΔphoPQ (Fig 2B), suggesting either that residual L-Ara4N modification is retained in the absence of PhoPQ through basal expression of arnT, or that phoPQ induction in the absence of arnT is detrimental for an unknown reason.
Meropenem exposure induces the PhoPQ regulon
Expression of the arn operon in E. cloacae is directly regulated by phosphorylated PhoP [22]. Because we observed that PhoPQ was necessary for arn-mediated tolerance, we asked whether meropenem induced arn transcription in a PhoPQ-dependent manner. To test this, E. cloacae cells were exposed to meropenem for 30 minutes, after which arnB (the first gene in the arn operon and thus the most direct readout of promoter activity [22,31]), pagP, and phoP transcript levels were quantified (Fig 3A). Relative expression was calculated using 16s rRNA as an internal control. phoP, which is autoregulated [32], showed 2.5-fold higher expression in meropenem-treated cells relative to untreated. Additionally, pagP expression was 5-fold higher, and arnB expression was 6-fold higher after meropenem treatment. These results support a model where PhoPQ signaling, as well as transcription of its regulon, is induced in response to meropenem treatment.
Fig 3. Expression of arnB in response to meropenem treatment is dependent on PhoPQ.
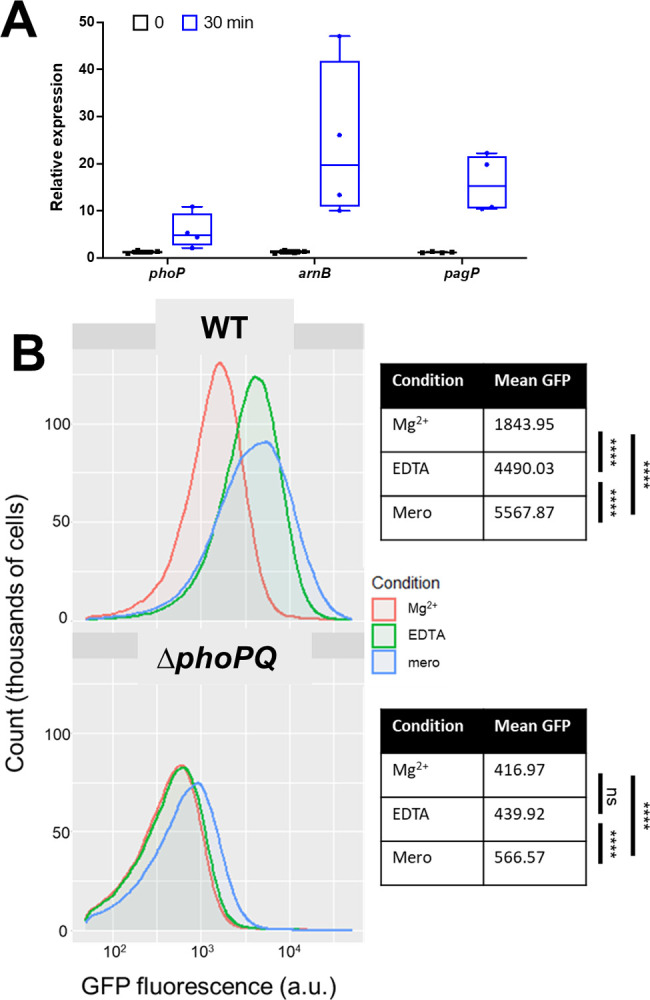
(A) Relative expression reverse-transcription quantitative PCR (qRT-PCR) of PhoPQ regulon transcripts after meropenem exposure. Each experiment was independently replicated three times (individual data points of the three experiments are reported here). (B) Strains carrying transcriptional ParnB:msGFP fusions were exposed to indicated conditions and analyzed on a C6 Accuri flow cytometer. Statistical difference between populations was determined with a one-way ANOVA followed by Tukey’s correction for multiple comparisons (ns, not significant; ****, p ≤ 0.0001).
To corroborate the qRT-PCR findings, we also constructed a fluorescent transcriptional reporter, fusing the arnB promoter with msfGFP, followed by fluorescence measurements upon exposure to meropenem (Fig 3B). As a control, we first exposed cells to EDTA, which chelates divalent cations to destabilize the outer membrane and consequently activates PhoPQ [33]. As expected, ParnB:msfGFP was induced by EDTA treatment in a phoPQ-dependent way (Fig 3B). Interestingly, meropenem treatment also robustly activated the ParnB:msfGFP reporter, where a significant 3-fold fluorescence increase was measured, comparable with the effect of EDTA treatment. In contrast, meropenem only slightly (but reproducibly) increased arnB expression in the ΔphoPQ mutant under the same conditions. Thus, arn transcription is induced by meropenem in a primarily PhoPQ-dependent manner.
Meropenem treatment promotes formation of L-Ara4N-modified lipid A species in a PhoPQ-dependent manner
The lipopolysaccharide lipid A domain is modified with L-Ara4N in a PhoPQ-dependent manner when Mg2+ is limiting [22]. To determine if the E. cloacae lipid A structure is modified with L-Ara4N in response to meropenem treatment, we isolated lipid A from treated and untreated cultures, which we then analyzed using MS and thin-layer chromatography (TLC). A distinct shift in lipid A structures was evident following 3 hours of meropenem treatment, where an increase in L-Ara4N modified vs. unmodified forms was observed (Fig 4A and 4B). Notably, the lipid A species that dominated before treatment (hexa-acylated, bis-phosphorylated, m/z = 1825.25) decreased in abundance in favor of Arn- and PagP-modified lipid A. Doubly Arn/PagP-modified lipid A (m/z = 2114.14) was also produced following treatment. Notably, while L-Ara4N modification of lipid A was PhoPQ-dependent, PagP-dependent lipid A acylation was not (see below for discussion). Quantitative TLC supported the MS results and revealed a 12.21 (+/- 1.13)-fold increase in single-modified L-Ara4-N lipid A in a PhoPQ-dependent manner (Fig 4C).
Fig 4. Analysis of E. cloacae lipid A after meropenem treatment.

(A) MALDI-TOF MS analysis of lipid A extracted from wild-type or ΔphoPQ E. cloacae strains. L-Ara4N modifications are illustrated in red, while C16:0 additions are green. Numbered labels that are both red and green contain both modifications. Each experiment was independently replicated three times, and one representative data set was reported. (B) Predicted lipid A chemical structures in wild type and ΔphoPQ E. cloacae. (C) 32P-radiolabelled lipid A was extracted from treated or untreated wild-type or ΔphoPQ E. cloacae. Lipids were separated based on hydrophobicity using thin-layer chromatography. Red circle denotes L-Ara4N modification, while the green line indicates C16:0 addition.
E. cloacae encodes the principal putative PagP acyltransferase Ecl_03072 (henceforth “PagP”). Lipid A extracted from ΔpagP lacked the acyl chain induced upon meropenem treatment (S4A Fig), suggesting that PagP removes palmitate (C16:0) from surface-exposed glycerophospholipids and transfers it to lipid A, as previously shown [34,35]. We also confirmed that meropenem-induced L-Ara4N modification was dependent on the arn operon (S4B Fig). Furthermore, MS analysis of ΔphoPQ ΔpagP (S4C Fig) and ΔarnT ΔpagP (S4D Fig) lipid A revealed that the mutants produced lipid A structures lacking all modifications following meropenem treatment, confirming that PagP and the arn operon products coordinate E. cloacae lipid A modifications in response to meropenem treatment. Interestingly, meropenem-induced hyperacylation was absent in ΔmgrB cells (S3A Fig). One potential explanation is that meropenem-induced hyperacylation is a result of outer membrane glycerophospholipid accumulation in the outer leaflet of spheroplasts, a condition known to activate PagP enzymatic activity post-translationally [33,36]. This model is also consistent with our observation that while PagP is partially under genetic control of PhoPQ [22], meropenem-induced hyperacylation is independent of PhoPQ (Fig 4A and 4C). The absence of PagP-dependent modification in ΔmgrB might indicate increased outer membrane strength (and concomitant reduction in glycerophospholipid accumulation in the outer leaflet of the outer membrane) in this background. Further analysis of spheroplast membrane composistion is necessary to support this model.
Based on the structural studies and our genetic evidence, we suggest PhoPQ-dependent tolerance is primarily mediated via L-Ara4N addition to lipid A. Presumably, L-Ara4N lipid A modification increases the structural integrity of the outer membrane through stabilization of lateral lipopolysaccharide interactions [28], which protects spheroplasts from high levels of internal turgor.
Colistin exposure primes E. cloacae for meropenem tolerance
Cationic antimicrobial peptides (CAMPs) are known inducers of the PhoPQ TCS [37]. Since our data above suggest that PhoPQ induction promotes tolerance, we hypothesized that pre-exposure to the CAMP colistin “primes” E. cloacae for tolerance to meropenem, potentially by inducing PhoPQ or by selecting for cells that have a higher baseline level of PhoPQ induction. To test this, we measured the extent to which E. cloacae was killed by meropenem with and without prior growth in medium containing supra-MIC colistin; this is expected to enrich specifically for the heteroresistant (more heavily OM modified) subpopulation in E. cloacae ATCC 13047 [22]. Interestingly, after pre-exposure to colistin, the fraction of cells surviving meropenem treatment was reproducibly (and statistically significantly) 3.5-fold greater (Fig 5). This suggests that CAMPs have the potential to induce tolerance to β-lactam antibiotics, but that the temporal conditions of treatment may determine the extent to which this effect is significant.
Fig 5. Colistin primes E. cloacae for meropenem tolerance.
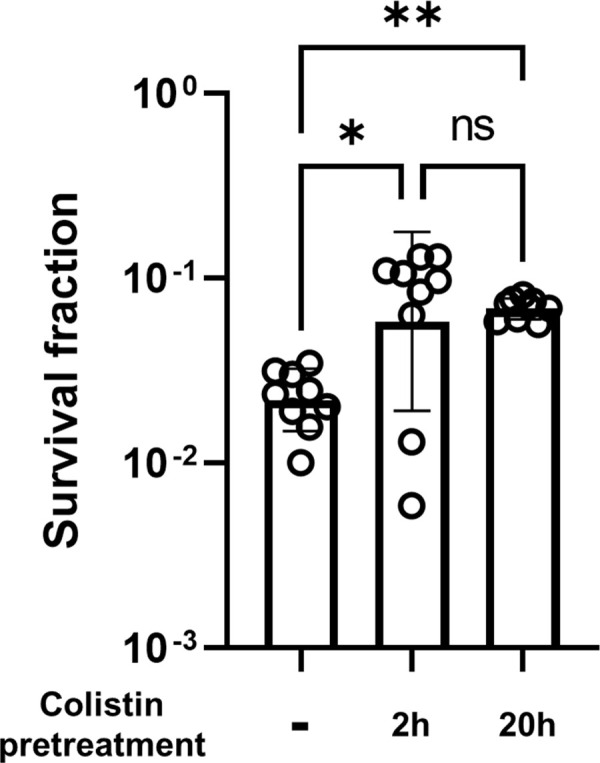
Overnight cultures were grown in LB +/- 20 μg/mL colistin, then diluted 10-fold into LB +/- 20 μg/mL colistin. Colistin pretreatment value represents total time exposed to colistin before exposure to 10 μg/mL meropenem. Cultures were then treated like other meropenem killing experiments (see Materials and Methods). Survival fraction was calculated by dividing CFU/mL at 3h meropenem exposure by CFU/mL prior to meropenem exposure. Each bar represents the mean of 9 biological replicates, error bars represent standard deviation. Statistical significance determined by one-way ANOVA of log transformed data, followed by Tukey’s correction for multiple comparisons (ns, not significant; *, p ≤ 0.05; **, p ≤ 0.01).
Outer membrane modifications are conserved β-lactam tolerance determinants in other Enterobacterales
We next sought to establish whether outer membrane modifications might promote tolerance in other Enterobacterales. We first turned to Klebsiella pneumoniae and used a hypertolerant clinical isolate (Kp 1084) and its ΔphoPQ derivative for killing experiments. We observed a striking, 10- (6 hours) to 105-fold (24 hours) decrease in viability in the presence of meropenem, which could be fully complemented by expressing PhoP in trans (Fig 6A). We also analyzed a well-characterized E. coli K12 variant, WD101, engineered to constitutively upregulate the PmrAB two-component system [38] (which induces L-Ara4N modification of lipid A in E. coli) to test the hypothesis that outer membrane modifications increase tolerance in E. coli (Fig 6B). WD101 exhibited a dramatic, 10,000-fold increase in survival after 24 hours of meropenem exposure compared to the wild type parental strain, further supporting a role for outer membrane modifications in meropenem tolerance beyond E. cloacae ATCC13047.
Fig 6. A conserved mechanism for meropenem tolerance in Enterobacterales.
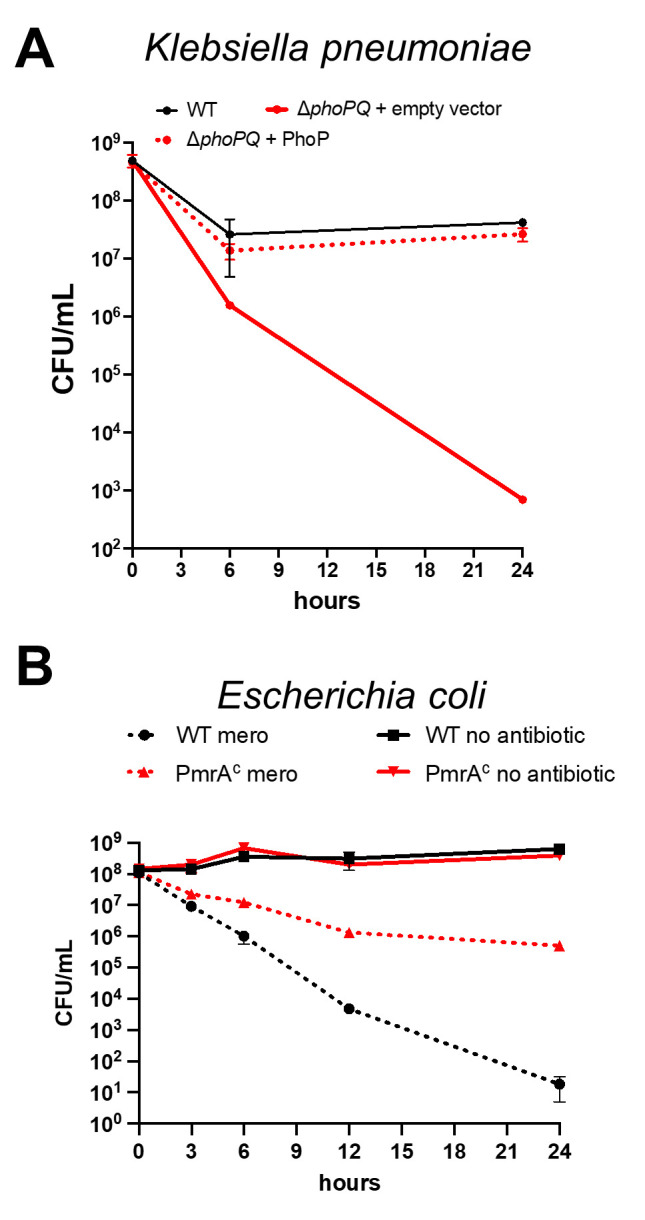
(A) Klebsiella pneumoniae 1084 carrying empty vector, and its ΔphoPQ derivative carrying either empty vector or pBAD33phoP (“ΔphoPQ + PhoP”) was diluted 10-fold into BHI medium containing 10 μg/mL meropenem (100 x MIC) and 0.2% arabinose for induction. Data represent averages of 3 replicates +/- standard deviation (B) E. coli strain W3110 (WT) or strain WD101, which has a constitutively active chromosomal copy of pmrA (pmrAC), were cultured in N-minimal medium and treated with or without meropenem. Cultures were incubated statically at 37°C. CFU were enumerated at indicated time points. Error bars indicate standard deviation. Each experiment was independently replicated three times in triplicate, and one representative data set was reported.
A small molecule histidine kinase inhibitor synergizes with meropenem and colistin in vitro
Tolerance is likely an under appreciated contributor to antibiotic treatment failure. Antibiotic adjuvants that promote killing of tolerant cells thus have the potential to find a prominent place in our antibiotic armamentarium. Since histidine kinases like PhoQ are in principle targetable by small molecules, we tested whether his-kinase inhibitors synergized with meropenem. To this end, we turned to a previously developed suite of small molecules with potent histidine kinase inhibitory activity [39] and tested them in combination with meropenem. The anti-Amyotrophic Lateral Sclerosis (ALS) drug Riluzole, as well as its derivative Rilu-2, exhibited potent, concentration-dependent synergy in combination with meropenem to rapidly lyse tolerant E. cloacae cells in vitro (Figs 7A and S5A). We also verified that Rilu-2 inhibited the formation of the L-ara4N lipid A structure using MALDI-TOF MS (Fig 7B). The peak corresponding to this modification (m/z 1876.29) was completely absent in Rilu-2-treated and ΔphoPQ cells. Since the PhoPQ system is primarily recognized for its contribution to CAMP resistance in many Enterobacterales, we next tested the Rilu compounds’ ability to synergize with colistin. As expected, Rilu-2 and Riluzole indeed potentiated colistin-mediated killing (S5B Fig), lending additional support to a PhoQ-inhibitory role of these compounds and also confirming previous results in Salmonella [40]. Importantly, Riluzole is an FDA-approved treatment for ALS and could thus readily serve as an adjuvant against both meropenem-tolerant and colistin-resistant Enterobacterales.
Fig 7. Rilu compounds synergize with meropenem to expedite E. cloacae killing.

(A) Rilu compounds potentiate meropenem-induced lysis against E. cloacae. Overnight cultures were diluted 10-fold into fresh growth medium containing meropenem (10 μg/mL) and increasing concentrations of Riluzole or its derivative Rilu-2. Data represent the average of 3 technical replicates +/- standard deviation. (B) MALDI-MS analysis of lipid A isolated from untreated wild-type E. cloacae, cells treated with RILU-2 or ΔphoPQ. m/z corresponding with L-Ara4N modifications are illustrated in red. Relevant lipid A chemical structures are shown. Each experiment was independently replicated three times, and one representative data set is reported.
Discussion
While much work has been done in Enterobacterales to elucidate mechanisms of antibiotic resistance and persistence, the genetic and molecular determinants of tolerance, and especially spheroplast formation, have remained poorly understood. In contrast to resistance (continued growth) and persistence (dormancy), carbapenem-tolerant populations are initially susceptible to treatment (i.e., lose their cell wall). This phenotype is reminiscent of so-called “L-forms” [10], with the notable difference that spheroplasts do not replicate in this state, while L-forms do. This is likely a consequence of L-forms being able to “escape” the outer membrane to then proliferate through stochastic membrane blebs [41,42].
The remarkable ability of spheroplasts to survive without their PG layer lends support to the recent realization that the outer membrane has load-bearing capabilities [43] and prompted us to interrogate the molecular mechanism of outer membrane stabilization during antibiotic exposure. Our data suggest that L-Ara4N addition to lipid A is a key factor in spheroplast integrity. We propose that lipid A molecules with a positive charge increase outer membrane stability due to the enhancement of electrostatic interactions between adjacent lipopolysaccharide molecules on the surface-exposed face of the outer membrane [28]. Of note, the L-Ara4N modification has been previously implicated in resistance to CAMPs, such as colistin [22]. In this context, it is worrisome that colistin therapy can select for mgrB mutations [44–46], which we demonstrate here also confers high meropenem tolerance in addition to colistin resistance. Thus, treatment with antimicrobial peptide analogs (and we speculate that this may potentially apply to innate AMPs as well) can select for bacteria that are stably tolerant to subsequent therapy with a β-lactam. Our data also suggest that colistin (and, by extension, potentially AMPs of the innate immune system) can induce transient β-lactam tolerance, e.g., through induction of the PhoPQ system and perhaps other protective stress responses.
However, the relationship between PhoPQ induction and tolerance is not absolute; a significant proportion of both colistin-pretreated and mgrB-deleted cells still lyse in the presence of meropenem. It is thus likely that colistin heteroresistant subpopulations are not the same as carbapenem tolerant cells. In our pre-treatment experiments with colistin, the PhoPQ system should be induced in all cells to similar degrees, yet only a fraction (0.1%) of these cells was also meropenem tolerant. We speculate that β-lactam tolerance and colistin resistance, while relying on the same basic outer membrane modification, may each require a specific fraction of lipopolysaccharide molecules to be modified. Thus, within a sample, there may be a limited subset of cells exhibiting the correct amount of modification to enable both colistin resistance and meropenem tolerance (or even just optimal meropenem tolerance). This would explain why we observe only partial overlap between these two phenomena.
In summary, this work demonstrates a novel genetic determinant of carbapenem tolerance in clinically relevant Enterobacterales. Despite being a well-known regulator of polymyxin resistance, the PhoPQ two-component system was not previously known to respond or mediate tolerance to carbapenem treatment. As tolerance (and spheroplast formation in particular) is a possible culprit for antibiotic treatment failure [2,3,47], our results suggest a potential for combination therapies with histidine kinase inhibitors to increase the efficacy of carbapenems.
Materials and methods
Bacterial strains and growth
All strains/plasmids and primers used in this study are listed in S1 and S2 Tables. All strains were initially grown from freezer stocks on solid agar at 37°C. Isolated colonies were used to inoculate Luria-Bertani (LB), Brain heart infusion (BHI) or N-minimal medium (0.1M Bis-Tris, pH 7.5, 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 M K2SO4, 1 mM KH2PO4, 0.10% casamino acids 0.2% glucose, 0.0002% thiamine, 15 μM FeSO4, 10 mM MgSO4) at 37°C. Where required, kanamycin was used at 50 μg/mL, meropenem was used at 10 μg/mL (300 x MIC; MICATCC13047 = 0.03 μg/mL) and colistin was used at 20 μg/mL (1.25 x MIC; MICATCC13047 = 16 μg/mL), unless noted otherwise. K. pneumoniae 1084 is an isolate (AR0080) from the CDC AR isolate bank (https://wwwn.cdc.gov/arisolatebank/Search), cured of its imipenemase via spontaneous loss.
Meropenem killing experiments
Unless noted otherwise, killing experiments were conducted in 100-well honeycomb plates in a Bioscreen C growth curve analyzer (Growth Curves USA, Piscataway NJ). Overnight cultures were diluted 10-fold into fresh LB medium containing meropenem (10 μg/mL, 300x MIC) and transferred to honeycomb plates (200 μL volume/culture). OD600 was measured by plate reader; at indicated timepoints, the experiment was paused and an aliquot was removed for CFU/mL determination or microscopy. Rilu compounds were dissolved in DMSO as 50 mM (Rilu-2) or 500 mM (Riluzole) stocks and added directly to the LB medium containing meropenem at the indicated concentrations.
For K. pneumoniae killing experiments, we employed our standard tolerance assay as described previously [9], but with 0.2% arabinose present for induction of plasmid-borne PhoP.
Colistin MIC experiment
Cultures were grown overnight at 37°C shaking, then diluted 1000-fold into fresh LB. Subcultures were grown for 1 hour at 37°C shaking before being diluted 1000-fold again into fresh LB to create a “seed culture”. 100 μL of seed culture was subsequently diluted 2-fold into a 96-well plate containing colistin concentrations ranging 0.25–128 μg/mL. Reported values are medians of 4 technical replicates.
qRT-PCR
Strains were grown in 5 mL LB overnight at 37°C. 500 uL of overnight cultures was added to 4.5 mL pre-warmed BHI broth, either water or 10 μg/mL meropenem was added and cultures were incubated statically at 37°C for 0 and 30 mins. Following incubation, cells were harvested via centrifugation and resuspended in 500 μl RNA later and stored in -80°C prior to RNA extraction. Relative-abundance quantitative PCR (qPCR) was performed as previously described [48,49]. In brief, the Sybr Fast One-Step qRT-PCR kit (Kapa Biosystems) was used with 16S rDNA as the internal reference. The PCR was performed using the Bio-Rad CFX Connect Real-Time PCR System. Relative expression levels were calculated using the ΔΔCt method [50], with normalization of gene targets to 16S rDNA signals.
Flow cytometry GFP measurements
Cultures of strains harboring transcriptional ParnB:msfGFP fusions were grown overnight in LB supplemented with 10 mM MgSO4. Overnight cultures were then washed 2x in fresh LB before 10-fold dilution into fresh LB medium containing MgSO4 (10 mM), Ethylenediaminetetraacetic acid (1 mM), or meropenem (10 μg/mL). Cultures were incubated statically for 3 hours at 37°C. Then, 500 μL of culture was harvested and run through a C6 Accuri flow cytometer (BD Biosciences) until 100,000 events (cells) had been analyzed. Mean green fluorescence as measured by the FL1-A channel was used as a readout for GFP.
Mutant construction
E. cloacae subsp. cloacae 13047 mutant strains (phoPQ, arn and arnT) were constructed as previously described using recombineering with the plasmid pKOBEG [22,51]. Briefly, linear PCR products were amplified from pKD3 and transformed into E. cloacae ATCC 13047/pKOBEG strain by electroporation and plated on chloramphenicol selective media. Selected clones were transformed with pCP20 to cure the antibiotic resistance cassette. All mutants were verified by PCR.
pagP was deleted using the Wanner method as described previously [22]. Briefly, the chl resistance cassette was amplified from pKD3 using primers TDP1532/TDP1533, which contain 75 bp flanking homology overhangs. The resulting PCR product was electroporated into E. cloacae ATCC13047 expressing lambda red recombinase from pACBSR-hyg [52] (a hygromycin-resistant derivative of pKD46 [53]). Mutants were selected on chloramphenicol (100 μg/mL) and verified by PCR.
Other mutants were constructed using either lambda red recombinase [53] or the suicide vector pTox [54]. The mgrB gene was deleted using the suicide plasmid pTox5 as described in [54]. ~700 bp upstream and downstream flanking homology regions were amplified from ATCC13047 using primers TDP1767/68 and TDP1769/70, and cloned into pTox5 (digested with EcoRV) using isothermal assembly [55]. Successful pTox5ΔmgrB were conjugated into ATCC13047 using the E. coli donor strain MFD lambda pir; successful recombinants were selected on plates containing 100 μg/mL chloramphenicol. Upon single colony purification, colonies were directly streaked out on an M9 minimal medium plate containing 0.2% casamino acids and 1% rhamnose, followed by incubation at 30°C for 24–36 hours. Mutants were tested using primers TDP1771/72.
The K. pneumoniae ΔphoPQ mutant was constructed using pTox5. Upstream and downstream homology regions were amplified using primers TC120/TC344 and TC345/TC127. Allelic exchange was conducted as described above. Mutants were tested using flanking primers TC166/167. For the complementation construct, the PhoP open reading frame was synthesized as a gene block (gBlock TC391) with a 3x Flag tag (Twist Biosciences, South San Francisco, USA), and cloned into pBAD33 using isothermal assembly.
Lipid A isolation and mass spectrometry
Isolation of lipid A for analysis was performed as previously described [56] with slight modifications. To analyze lipid A after meropenem treatment, overnight cultures grown in BHI broth were diluted 1:10 in pre-warmed media with or without meropenem statically for 3 h. To assess Rilu-dependent modification of lipid A, 12.5 mL of E. cloacae was grown to OD600 1.0. Rilu-2 was used at a final concentration of 200 μM. Bacteria were harvested and lipid A extraction was carried out by mild-acid hydrolysis as previously described [57]. For mass spectrometry (MS), data were collected on a MALDI-TOF (Axima Confidence, Shimadzu) mass spectrometer in the negative mode, as previously done [22].
For quantification of lipid A, cultures were grown with 2.5 μCi/mL of 32P ortho-phosphoric acid (32P) (Perkin Elmer) and lipid A was extracted. Thin layer chromotagraphy was done in a pyridine, chloroform, 88% formic acid, aqueous (50:50:16:5 v/v) tank for 3 hours. Plates were exposed to a phosphor screen, imaged, and densitometry was used to calculate the percentage of each lipid species. Reported densitometry was calculated using 2 replicates +/- standard deviation. For lipid A structural comaprisons, purified 32P-lipid A from E. coli W3110, WD101 [58] were used in each experiment. These strucutres were comparable to previously published E. cloacae structures [26] and validated by mass spectrometry.
Supporting information
Wild type (WT) (A-B) or its ΔphoPQ derivative (C-D) were treated as described in Fig 1A with addition of the indicated concentrations of (A,C) MgSO4 (Mg2+) or (B,D) CaCl2 (Ca2+). Data represent the average of 3 replicates +/- standard deviation.
(TIF)
(A) An mgrB mutation promotes a moderate increase in mass increase during meropenem exposure. (B) Experiments were conducted as described in Fig 1A legend; each graph represents experiments conducted on a different day. In addition, data in each graph represent the average of 3 biological replicates +/- standard deviation. (C) Fraction of colistin-susceptible populations surviving after 24 hours of 10 μg/mL meropenem exposure. Statistical significance determined by one-way ANOVA of log transformed data, followed by Tukey’s correction for multiple comparisons (ns, not significant; ***, p ≤ 0.001).
(TIF)
(A) MALDI-MS analysis of lipid A isolated from E. cloacae ΔmgrB. m/z corresponding with L-Ara4N modifications are illustrated in red. Each experiment was independently replicated three times, and one representative data set was reported. (B) Relevant lipid A chemical structures are shown.
(TIF)
(A) MALDI-MS analysis of lipid A isolated from ΔpagP, (B) Δarn (full operon deletion), (C) ΔphoPQ ΔpagP and (D) ΔarnT ΔpagP. m/z corresponding with L-Ara4N modifications are illustrated in red, while structures with altered acyl chain patterns are illustrated in green. Each experiment was independently replicated three times, and one representative data set was reported.
(TIF)
(A) Rilu compounds do not cause lysis, but (B) potentiate colistin mediate killing. Experiments were conducted as described in Fig 1A legend. Data represent the average of 3 replicates +/- standard deviation.
(TIF)
(DOCX)
(DOCX)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Research on tolerance in the Dörr lab is supported by National Institutes of Health (NIH/NIAID) grant R01AI143704 to TD. Research in the Boll lab is supported by National Institutes of Health (NIH/NIGMS) grant GM143053 to JMB. Histidine kinase inhibitor research in the Carlson lab supported by National Institutes of Health (NIH/NIGMS) grant GM134538-01A1 and the UMN Office of Academic Clinical Affairs to EEC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Brauner A, Fridman O, Gefen O, Balaban NQ. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol. 2016;14(5):320–30. Epub 2016/04/16. doi: 10.1038/nrmicro.2016.34 . [DOI] [PubMed] [Google Scholar]
- 2.Dorr T. Understanding tolerance to cell wall-active antibiotics. Ann N Y Acad Sci. 2021;1496(1):35–58. Epub 2020/12/05. doi: 10.1111/nyas.14541 ; PubMed Central PMCID: PMC8359209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westblade LF, Errington J, Dorr T. Antibiotic tolerance. PLoS Pathog. 2020;16(10):e1008892. Epub 2020/10/16. doi: 10.1371/journal.ppat.1008892 ; PubMed Central PMCID: PMC7561168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin-Reisman I, Brauner A, Ronin I, Balaban NQ. Epistasis between antibiotic tolerance, persistence, and resistance mutations. Proc Natl Acad Sci U S A. 2019;116(29):14734–9. Epub 2019/07/03. doi: 10.1073/pnas.1906169116 ; PubMed Central PMCID: PMC6642377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. Antibiotic tolerance facilitates the evolution of resistance. Science. 2017;355(6327):826–30. Epub 2017/02/12. doi: 10.1126/science.aaj2191 . [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Gefen O, Ronin I, Bar-Meir M, Balaban NQ. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science. 2020;367(6474):200–4. Epub 2020/01/11. doi: 10.1126/science.aay3041 . [DOI] [PubMed] [Google Scholar]
- 7.Dorr T, Davis BM, Waldor MK. Endopeptidase-mediated beta lactam tolerance. PLoS Pathog. 2015;11(4):e1004850. Epub 2015/04/18. doi: 10.1371/journal.ppat.1004850 ; PubMed Central PMCID: PMC4401780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monahan LG, Turnbull L, Osvath SR, Birch D, Charles IG, Whitchurch CB. Rapid conversion of Pseudomonas aeruginosa to a spherical cell morphotype facilitates tolerance to carbapenems and penicillins but increases susceptibility to antimicrobial peptides. Antimicrob Agents Chemother. 2014;58(4):1956–62. Epub 2014/01/15. doi: 10.1128/AAC.01901-13 ; PubMed Central PMCID: PMC4023726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cross T, Ransegnola B, Shin JH, Weaver A, Fauntleroy K, VanNieuwenhze MS, et al. Spheroplast-Mediated Carbapenem Tolerance in Gram-Negative Pathogens. Antimicrob Agents Chemother. 2019;63(9). Epub 2019/07/10. doi: 10.1128/AAC.00756-19 ; PubMed Central PMCID: PMC6709500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Errington J, Mickiewicz K, Kawai Y, Wu LJ. L-form bacteria, chronic diseases and the origins of life. Philos Trans R Soc Lond B Biol Sci. 2016;371(1707). Epub 2016/09/28. doi: 10.1098/rstb.2015.0494 ; PubMed Central PMCID: PMC5052740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercier R, Kawai Y, Errington J. General principles for the formation and proliferation of a wall-free (L-form) state in bacteria. Elife. 2014;3. Epub 2014/10/31. doi: 10.7554/eLife.04629 ; PubMed Central PMCID: PMC4244569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Errington J. Cell wall-deficient, L-form bacteria in the 21st century: a personal perspective. Biochem Soc Trans. 2017;45(2):287–95. Epub 2017/04/15. doi: 10.1042/BST20160435 ; PubMed Central PMCID: PMC5390494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin JH, Choe D, Ransegnola B, Hong HR, Onyekwere I, Cross T, et al. A multifaceted cellular damage repair and prevention pathway promotes high-level tolerance to beta-lactam antibiotics. EMBO Rep. 2021;22(2):e51790. Epub 2021/01/20. doi: 10.15252/embr.202051790 ; PubMed Central PMCID: PMC7857431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorr T, Alvarez L, Delgado F, Davis BM, Cava F, Waldor MK. A cell wall damage response mediated by a sensor kinase/response regulator pair enables beta-lactam tolerance. Proc Natl Acad Sci U S A. 2016;113(2):404–9. Epub 2015/12/30. doi: 10.1073/pnas.1520333113 ; PubMed Central PMCID: PMC4720315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paterson DL. Recommendation for treatment of severe infections caused by Enterobacteriaceae producing extended-spectrum beta-lactamases (ESBLs). Clin Microbiol Infect. 2000;6(9):460–3. Epub 2001/02/13. doi: 10.1046/j.1469-0691.2000.00107.x . [DOI] [PubMed] [Google Scholar]
- 16.Torres JA, Villegas MV, Quinn JP. Current concepts in antibiotic-resistant Gram-negative bacteria. Expert Review of Anti-infective Therapy. 2007;5(5):833–43. doi: 10.1586/14787210.5.5.833 [DOI] [PubMed] [Google Scholar]
- 17.Paterson DL, Bonomo RA. Extended-spectrum beta-lactamases: a clinical update. Clin Microbiol Rev. 2005;18(4):657–86. Epub 2005/10/15. doi: 10.1128/CMR.18.4.657-686.2005 ; PubMed Central PMCID: PMC1265908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groisman EA, Duprey A, Choi J. How the PhoP/PhoQ System Controls Virulence and Mg(2+) Homeostasis: Lessons in Signal Transduction, Pathogenesis, Physiology, and Evolution. Microbiol Mol Biol Rev. 2021;85(3):e0017620. Epub 2021/07/01. doi: 10.1128/MMBR.00176-20 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder S, Kim D, McIntosh TJ. Lipopolysaccharide Bilayer Structure: Effect of Chemotype, Core Mutations, Divalent Cations, and Temperature. Biochemistry. 1999;38(33):10758–67. doi: 10.1021/bi990867d [DOI] [PubMed] [Google Scholar]
- 20.Garidel P, Rappolt M, Schromm AB, Howe J, Lohner K, Andra J, et al. Divalent cations affect chain mobility and aggregate structure of lipopolysaccharide from Salmonella minnesota reflected in a decrease of its biological activity. Biochim Biophys Acta. 2005;1715(2):122–31. Epub 2005/09/03. doi: 10.1016/j.bbamem.2005.07.013 . [DOI] [PubMed] [Google Scholar]
- 21.Henderson JC, Zimmerman SM, Crofts AA, Boll JM, Kuhns LG, Herrera CM, et al. The Power of Asymmetry: Architecture and Assembly of the Gram-Negative Outer Membrane Lipid Bilayer. Annual Review of Microbiology. 2016;70(1):255–78. doi: 10.1146/annurev-micro-102215-095308 [DOI] [PubMed] [Google Scholar]
- 22.Kang KN, Klein DR, Kazi MI, Guerin F, Cattoir V, Brodbelt JS, et al. Colistin heteroresistance in Enterobacter cloacae is regulated by PhoPQ-dependent 4-amino-4-deoxy-l-arabinose addition to lipid A. Mol Microbiol. 2019;111(6):1604–16. Epub 2019/03/16. doi: 10.1111/mmi.14240 ; PubMed Central PMCID: PMC6561824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitrophanov AY, Jewett MW, Hadley TJ, Groisman EA. Evolution and Dynamics of Regulatory Architectures Controlling Polymyxin B Resistance in Enteric Bacteria. PLOS Genetics. 2008;4(10):e1000233. doi: 10.1371/journal.pgen.1000233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin QY, Tsai YL, Liu MC, Lin WC, Hsueh PR, Liaw SJ. Serratia marcescens arn, a PhoP-regulated locus necessary for polymyxin B resistance. Antimicrob Agents Chemother. 2014;58(9):5181–90. Epub 2014/06/25. doi: 10.1128/AAC.00013-14 ; PubMed Central PMCID: PMC4135846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bishop RE, Kim SH, El Zoeiby A. Role of lipid A palmitoylation in bacterial pathogenesis. J Endotoxin Res. 2005;11(3):174–80. Epub 2005/06/14. doi: 10.1179/096805105X35242 . [DOI] [PubMed] [Google Scholar]
- 26.Band VI, Crispell EK, Napier BA, Herrera CM, Tharp GK, Vavikolanu K, et al. Antibiotic failure mediated by a resistant subpopulation in Enterobacter cloacae. Nat Microbiol. 2016;1(6):16053. Epub 2016/08/31. doi: 10.1038/nmicrobiol.2016.53 ; PubMed Central PMCID: PMC5154748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Band VI, Weiss DS. Heteroresistance: A cause of unexplained antibiotic treatment failure? PLoS Pathog. 2019;15(6):e1007726. Epub 2019/06/07. doi: 10.1371/journal.ppat.1007726 ; PubMed Central PMCID: PMC6553791 applications related to heteroresistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice A, Wereszczynski J. Atomistic Scale Effects of Lipopolysaccharide Modifications on Bacterial Outer Membrane Defenses. Biophys J. 2018;114(6):1389–99. Epub 2018/03/29. doi: 10.1016/j.bpj.2018.02.006 ; PubMed Central PMCID: PMC5883967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lippa AM, Goulian M. Feedback Inhibition in the PhoQ/PhoP Signaling System by a Membrane Peptide. PLOS Genetics. 2009;5(12):e1000788. doi: 10.1371/journal.pgen.1000788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem. 2001;276(46):43122–31. Epub 2001/09/06. doi: 10.1074/jbc.M106961200 . [DOI] [PubMed] [Google Scholar]
- 31.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, et al. PmrA–PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Molecular Microbiology. 1998;27(6):1171–82. doi: 10.1046/j.1365-2958.1998.00757.x [DOI] [PubMed] [Google Scholar]
- 32.Soncini FC, Véscovi EG, Groisman EA. Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. Journal of Bacteriology. 1995;177(15):4364–71. doi: 10.1128/jb.177.15.4364-4371.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia W, El Zoeiby A, Petruzziello TN, Jayabalasingham B, Seyedirashti S, Bishop RE. Lipid trafficking controls endotoxin acylation in outer membranes of Escherichia coli. J Biol Chem. 2004;279(43):44966–75. Epub 2004/08/21. doi: 10.1074/jbc.M404963200 . [DOI] [PubMed] [Google Scholar]
- 34.Hwang PM, Choy W-Y, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, et al. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proceedings of the National Academy of Sciences. 2002;99(21):13560–5. doi: 10.1073/pnas.212344499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. Transfer of palmitate from phospholipids to lipid A in outer membranes of Gram-negative bacteria. The EMBO Journal. 2000;19(19):5071–80. doi: 10.1093/emboj/19.19.5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malinverni JC, Silhavy TJ. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc Natl Acad Sci U S A. 2009;106(19):8009–14. Epub 2009/04/23. doi: 10.1073/pnas.0903229106 ; PubMed Central PMCID: PMC2683108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, et al. Recognition of Antimicrobial Peptides by a Bacterial Sensor Kinase. Cell. 2005;122(3):461–72. doi: 10.1016/j.cell.2005.05.030 [DOI] [PubMed] [Google Scholar]
- 38.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CR. Accumulation of a polyisoprene-linked amino sugar in polymyxin-resistant Salmonella typhimurium and Escherichia coli: structural characterization and transfer to lipid A in the periplasm. J Biol Chem. 2001;276(46):43132–44. Epub 2001/09/06. doi: 10.1074/jbc.M106962200 . [DOI] [PubMed] [Google Scholar]
- 39.Wilke KE, Francis S, Carlson EE. Inactivation of Multiple Bacterial Histidine Kinases by Targeting the ATP-Binding Domain. ACS Chemical Biology. 2015;10(1):328–35. doi: 10.1021/cb5008019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thielen MK, Vaneerd CK, Goswami M, Carlson EE, May JF. 2-Aminobenzothiazoles Inhibit Virulence Gene Expression and Block Polymyxin Resistance in Salmonella enterica. Chembiochem. 2020;21(24):3500–3. Epub 2020/08/05. doi: 10.1002/cbic.202000422 ; PubMed Central PMCID: PMC7765688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mercier R, Kawai Y, Errington J. Excess Membrane Synthesis Drives a Primitive Mode of Cell Proliferation. Cell. 2013;152(5):997–1007. doi: 10.1016/j.cell.2013.01.043 [DOI] [PubMed] [Google Scholar]
- 42.Billings G, Ouzounov N, Ursell T, Desmarais SM, Shaevitz J, Gitai Z, et al. De novo morphogenesis in L-forms via geometric control of cell growth. Molecular Microbiology. 2014;93(5):883–96. doi: 10.1111/mmi.12703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rojas ER, Billings G, Odermatt PD, Auer GK, Zhu L, Miguel A, et al. The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature. 2018;559(7715):617–21. Epub 2018/07/20. doi: 10.1038/s41586-018-0344-3 ; PubMed Central PMCID: PMC6089221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poirel L, Jayol A, Bontron S, Villegas M-V, Ozdamar M, Türkoglu S, et al. The mgrB gene as a key target for acquired resistance to colistin in Klebsiella pneumoniae. Journal of Antimicrobial Chemotherapy. 2014;70(1):75–80. doi: 10.1093/jac/dku323 [DOI] [PubMed] [Google Scholar]
- 45.Cannatelli A, D’Andrea MM, Giani T, Pilato VD, Arena F, Ambretti S, et al. In Vivo Emergence of Colistin Resistance in Klebsiella pneumoniae Producing KPC-Type Carbapenemases Mediated by Insertional Inactivation of the PhoQ/PhoP mgrB Regulator. Antimicrobial Agents and Chemotherapy. 2013;57(11):5521–6. doi: 10.1128/AAC.01480-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez-Camacho E, Gomez-Gil R, Tobes R, Manrique M, Lorenzo M, Galvan B, et al. Genomic analysis of the emergence and evolution of multidrug resistance during a Klebsiella pneumoniae outbreak including carbapenem and colistin resistance. J Antimicrob Chemother. 2014;69(3):632–6. Epub 2013/10/25. doi: 10.1093/jac/dkt419 . [DOI] [PubMed] [Google Scholar]
- 47.Roberts D, Higgs E, Rutman A, Cole P. Isolation of spheroplastic forms of Haemophilus influenzae from sputum in conventionally treated chronic bronchial sepsis using selective medium supplemented with N-acetyl-D-glucosamine: possible reservoir for re-emergence of infection. Br Med J (Clin Res Ed). 1984;289(6456):1409–12. Epub 1984/11/24. doi: 10.1136/bmj.289.6456.1409 PubMed Central PMCID: PMC1443692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies BW, Bogard RW, Mekalanos JJ. Mapping the regulon of Vibrio cholerae ferric uptake regulator expands its known network of gene regulation. Proc Natl Acad Sci U S A. 2011;108(30):12467–72. Epub 2011/07/14. doi: 10.1073/pnas.1107894108 ; PubMed Central PMCID: PMC3145737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boll JM, Crofts AA, Peters K, Cattoir V, Vollmer W, Davies BW, et al. A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proceedings of the National Academy of Sciences. 2016;113(41):E6228–E37. doi: 10.1073/pnas.1611594113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 51.Guérin F, Isnard C, Cattoir V, Giard JC. Complex Regulation Pathways of AmpC-Mediated β-Lactam Resistance in Enterobacter cloacae Complex. Antimicrobial Agents and Chemotherapy. 2015;59(12):7753–61. doi: 10.1128/AAC.01729-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang TW, Lam I, Chang HY, Tsai SF, Palsson BO, Charusanti P. Capsule deletion via a lambda-Red knockout system perturbs biofilm formation and fimbriae expression in Klebsiella pneumoniae MGH 78578. BMC Res Notes. 2014;7:13. Epub 2014/01/09. doi: 10.1186/1756-0500-7-13 ; PubMed Central PMCID: PMC3892127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–5. Epub 2000/06/01. doi: 10.1073/pnas.120163297 ; PubMed Central PMCID: PMC18686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lazarus JE, Warr AR, Kuehl CJ, Giorgio RT, Davis BM, Waldor MK. A New Suite of Allelic-Exchange Vectors for the Scarless Modification of Proteobacterial Genomes. Appl Environ Microbiol. 2019;85(16). Epub 2019/06/16. doi: 10.1128/AEM.00990-19 ; PubMed Central PMCID: PMC6677854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 2009;6(5):343–5. doi: 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 56.Kang KN, Kazi MI, Biboy J, Gray J, Bovermann H, Ausman J, et al. Septal Class A Penicillin-Binding Protein Activity and ld-Transpeptidases Mediate Selection of Colistin-Resistant Lipooligosaccharide-Deficient Acinetobacter baumannii. mBio. 2021;12(1). Epub 2021/01/07. doi: 10.1128/mBio.02185-20 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Z, Lin S, Cotter RJ, Raetz CR. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-L-arabinose, phosphoethanolamine and palmitate. J Biol Chem. 1999;274(26):18503–14. Epub 1999/06/22. doi: 10.1074/jbc.274.26.18503 . [DOI] [PubMed] [Google Scholar]
- 58.Needham BD, Trent MS. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol. 2013;11(7):467–81. Epub 2013/06/12. doi: 10.1038/nrmicro3047 ; PubMed Central PMCID: PMC6913092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Wild type (WT) (A-B) or its ΔphoPQ derivative (C-D) were treated as described in Fig 1A with addition of the indicated concentrations of (A,C) MgSO4 (Mg2+) or (B,D) CaCl2 (Ca2+). Data represent the average of 3 replicates +/- standard deviation.
(TIF)
(A) An mgrB mutation promotes a moderate increase in mass increase during meropenem exposure. (B) Experiments were conducted as described in Fig 1A legend; each graph represents experiments conducted on a different day. In addition, data in each graph represent the average of 3 biological replicates +/- standard deviation. (C) Fraction of colistin-susceptible populations surviving after 24 hours of 10 μg/mL meropenem exposure. Statistical significance determined by one-way ANOVA of log transformed data, followed by Tukey’s correction for multiple comparisons (ns, not significant; ***, p ≤ 0.001).
(TIF)
(A) MALDI-MS analysis of lipid A isolated from E. cloacae ΔmgrB. m/z corresponding with L-Ara4N modifications are illustrated in red. Each experiment was independently replicated three times, and one representative data set was reported. (B) Relevant lipid A chemical structures are shown.
(TIF)
(A) MALDI-MS analysis of lipid A isolated from ΔpagP, (B) Δarn (full operon deletion), (C) ΔphoPQ ΔpagP and (D) ΔarnT ΔpagP. m/z corresponding with L-Ara4N modifications are illustrated in red, while structures with altered acyl chain patterns are illustrated in green. Each experiment was independently replicated three times, and one representative data set was reported.
(TIF)
(A) Rilu compounds do not cause lysis, but (B) potentiate colistin mediate killing. Experiments were conducted as described in Fig 1A legend. Data represent the average of 3 replicates +/- standard deviation.
(TIF)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.