

Abstract
Background:
Nuclear protein transport is essential in guiding the traffic of important proteins and RNAs between the nucleus and cytoplasm. Export of proteins from the nucleus is mostly regulated by Exportin 1 (XPO1). In cancer, XPO1 is almost universally hyperactive and can promote the export of important tumor suppressors to the cytoplasm. Currently, there are no studies evaluating XPO1 amplifications and mutations in NSCLC and the impact on outcomes.
Methods:
Tumor samples were analyzed using next-generation sequencing (NGS) (NextSeq, 592 Genes), immunohistochemistry (IHC), and whole transcriptome sequencing (WTS, NovaSeq) (Caris Life Sciences, Phoenix, AZ). Survival was extracted from insurance claims data and calculated from time of tissue collection to last contact using Kaplan-Meier estimate.
Results:
Among 18,218 NSCLC tumors sequenced, 26 harbored XPO1 mutations and 24 had amplifications. XPO1 mutant tumors were more likely to have high TMB (79% vs. 52%, p = 0.007) and less likely to have high PD-L1 (32% vs. 68%, p = 0.03). KRAS co-mutations were seen in 19% (n = 5) and EGFR co-mutations were rare (n = 2). Among the 17,449 NSCLC tumors with clinical data, there were 24 XPO1 mutant. Comparison of survival between XPO1 mutant and WT showed a negative association with a hazard ratio (HR) of 1.932 (95% CI: 1.144- 3.264 p = 0.012). XPO1 amplification was not associated with survival.
Conclusions:
XPO1 pathogenic mutations were associated with a poor survival in NSCLC. Although XPO1 mutations are rare in NSCLC, further studies to assess its associations with treatment responses are warranted.
Keywords: NSCLC, Nuclear Protein Transport, XPO1, NGS, Tumor Mutational Burden
Background:
Nuclear protein transport is an essential process guiding the organized trafficking of important proteins and some RNA between the nucleus and cytoplasm of the cell. Such transport mediated spatial distribution is essential for proper protein function.1 Any entity larger than 40 kDa moves between the nucleus and cytoplasm with the help of specialized transporters belonging to the karyopherin family.2 The import of proteins from the cytoplasm to the nucleus is governed by importins that recognize the nuclear localization signal in the cargo.3 On the other hand, the export of proteins that contain a nuclear export signal (NES) from the nucleus is almost exclusively regulated by exportin 1 (XPO1), also known as chromosome region maintenance 1 (CRM1). XPO1 recognizes a NES in the cargo protein, and this process is facilitated by guanine exchange factors.4 Hundreds of cellular proteins have been identified to carry XPO1 recognizable NES5 6 while most of the NES are yet to be identified in over a thousand potential XPO1 cargos.7
XPO1 functions as the sole exporter of several tumor suppressor proteins and cell cycle regulators. Studies have shown that in cancer, hyperactive XPO1 causes unusual export of important tumor suppressors including p53, RB1, p21Cip1 and p27KIP1 to the cytoplasm leading to their functional inactivation.8 9 For example, the mislocalization of p27KIP1 also known as cyclin- dependent kinase inhibitor 1B has been associated with tumor promotion in MCF7 breast cancer cells via RhoA suppression and AKT signaling activation.10 Overexpression of XPO1 can be observed in patients with cancers and can be associated with disease progression, therapy resistance, and shorter overall survival (OS) or progression free survival (PFS).11
Earlier studies have shown that mutation in XPO1 (XPO1E571) appears to be a recurrent phenomenon in hematological malignancies.12 Missense substitution mutations have crucial roles in several steps of oncogenesis in various cancer types.13 14 These mutations can cause an alteration in XPO1 in the hydrophobic NES-binding groove, leading to preferential recognition of NESs that contain more negatively charged C-terminal ends as well as mitotic defects.13 15 16 The presence of XPO1E571K mutation in cell- free DNA appears to be associated with a shorter PFS in patients with classical Hodgkin lymphoma.17 Overexpression of XPO1 is also common in hematologic malignancies. The XPO1E571 mutation can act as “oncogenic driver”, possibly through the modulation of NFAT and NFAT-related signaling pathways in chronic lymphocytic leukemia.18 19
XPO1 is often overexpressed in multiple myeloma and correlates with increased bone disease and shorter survival.20 XPO1 is also overexpressed in diffuse large B cell lymphoma (DLBCL) and is known to correlate with worse prognosis.21
XPO1 is aberrantly amplified in NSCLC and such amplification has been linked to poor overall survival (OS).22 While the aberrant amplification of XPO1 is well recognized, the underlying mechanism for such amplification is not known.2 23 XPO1 amplification has been associated with drug resistance resulting from export of drug targets such as topoisomerase II24 and galectin-3. 25
Data on the impact of XPO1 mutations and amplifications on NSCLC clinical outcomes and therapy resistance is limited. In this study, we retrospectively analyzed de-identified pathological and molecular information from 18,218 NSCLC samples that underwent NGS with Caris Life Sciences to describe the prevalence of XPO1 mutations and amplifications in NSCLC. Tumor mutational burden (TMB) as well as PD-L1 expression were also assessed.
Methods:
Tissue samples:
Consecutive NSCLC tumors were analyzed by Caris Life Sciences (Phoenix, AZ) as part of routine comprehensive molecular profiling. Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques.
Next generation sequencing (NGS):
NGS was performed on genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples using the NextSeq platform (Illumina, Inc., San Diego, CA). A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with > 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of > 500X and an analytic sensitivity of 5%. The full list of genes assayed can be found elsewhere.26 The copy number alteration (CNA) of each exon was determined by calculating the average depth of the sample along with the sequencing depth of each exon and comparing this calculated result to a pre-calibrated value and copy number of 6.0 or higher was considered amplification. TMB was measured (592 genes and 1.4 megabases [MB] sequenced per tumor) by counting all non-synonymous missense mutations found per tumor that had not been previously described as germline alterations. The threshold to define TMB-high was greater than or equal to 10 mutations/MB.
Immunohistochemistry (IHC):
IHC was performed on full FFPE sections of glass slides. Slides were stained using automated staining techniques, per the manufacturer’s instructions, and were optimized and validated per CLIA/CAO and ISO requirements. For PD-L1 IHC, the primary PD-L1 antibody clone was 22c3 (Dako). Tumor Proportion Score (TPS) was measured, which is the percentage of viable tumor cells showing partial or complete membrane staining at any intensity. The tumor was considered positive if TPS ≥ 1% (high PD-L1 expression if TPS ≥ 50%).
Gene fusion detection:
Gene fusions were detected by RNA sequencing using either the ArcherDx fusion assay (Archer FusionPlex Solid Tumor panel) or whole transcriptome sequencing assay (Illumina NovaSeq platform (Illumina, Inc., San Diego, CA). Variants of genes were pre-determined for their cancer-related and clinical significance interpreted by board-certified molecular geneticists and categorized as pathogenic (P), presumed pathogenic (PP), variant of unknown significance (VUS), presumed benign (PB), or benign (B), according to ACMG (American College of Medical Genetics and Genomics) standards.27
Survival analysis:
Survival analysis was performed using real-world evidence from insurance claims data and calculated from time of tissue collection to last contact using Kaplan-Meier survival analysis. Statistical significance was determined using chi-square and Wilcoxon rank sum test and adjusted for multiple comparisons.
Ethics statement:
This study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont report, and U.S. Common rule. In keeping with 45 CFR 46.101(b) (4), this study was performed utilizing retrospective, deidentified clinical data. Therefore, this study was considered IRB exempt and no patient consent was necessary from the subjects.
Results:
XPO1 genomic landscape in NSCLC:
Among 18,218 NSCLC tumors sequenced, 26 (0.14%) harbored XPO1 mutations. The most common protein changes were at E571K (n =4) and Q626X (n =3) followed by one each of 19 different mutations as shown in Figure 1A. Table 1 shows the baseline demographics of patients based on XPO1 status of mutated, amplified and wild type. There was no significant gender or age prevalence between different XPO1 statuses.
Figure 1A.

Pie chart of XPO1 pathogenic mutational distribution. Figure 1B. Oncoprint for the top concurrent mutations in XPO1 mutant tumors.
Table 1.
Baseline demographics based on XPO1 status.
| XPO1 Status | Male (N) | Avg age Male | Female (N) | Avg age Female | Total |
|---|---|---|---|---|---|
| Mutated | 12 | 69.3 | 14 | 70.9 | 26 |
| Amplified | 11 | 69.9 | 13 | 74.5 | 24 |
| Wild Type | 9109 | 68.3 | 9059 | 67.7 | 18168 |
| Total | 9132 | 68.3 | 9086 | 67.7 | 18218 |
In the XPO1 mutant cohort, 79.2% (n=19) were considered TMB-high with a cutoff of ≥10 mutations/megabase and 31.6% (N=6) were PD-L1 positive (22c3 antibody with TPS cutoff ≥1). When comparing XPO1 mutant tumors to wild type, XPO1 mutant tumors showed a significantly higher median TMB (17.5 mt/Mb vs 10.0 mt/Mb) than wild type tumors. In the analyzed cohort, 24 tumors had XPO1 amplifications. XPO1 amplifications were mutually exclusive of XPO1 mutations.
In the XPO1 amplified cohort, 52.2% were TMB-high with a cutoff of ≥10 and 68.2% were PD-L1 positive (22c3 antibody with TPS cutoff ≥1). No significant difference was observed in TMB values for XPO1 amplified vs non-amplified tumors. XPO1 mutant tumors were more likely to be TMB high (79.2% vs. 51.8%, p = 0.007) and less likely to have high PD-L1 (31.6% vs. 56.3%, p = 0.03) when compared to XPO1 wild type tumors, whereas such correlations were not observed between XPO1 amplified versus XPO1 wild type tumors (Table 2).
Table 2.
Comparative analysis for IO-markers based on XPO1 status.
| MT vs WT | Amp vs WT | MT vs Amp | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Test | Positive | Negative | Total | % XPO1 WT | Positive | Negative | Total | % XPO1 MT | Positive | Negative | Total | % XPO Amplified | p-value | q-value | p-value | q-value | p-value | q-value |
| TMB >=10 | 9002 | 8361 | 17363 | 51.8% | 19 | 5 | 24 | 79.2% | 12 | 11 | 23 | 52.2% | 0.01 | 0.72 | 0.97 | 1 | 0.05 | 0.69 |
| MSI-H/dMMR | 122 | 17764 | 17886 | 0.7% | 0 | 24 | 24 | 0.0% | 0 | 24 | 24 | 0.0% | 1 | 1 | 1 | 1 | 1 | 1 |
| IHC-PD-L1 | ||||||||||||||||||
| (22c3) | 9386 | 7285 | 16671 | 56.3% | 6 | 13 | 19 | 31.6% | 15 | 7 | 22 | 68.2% | 0.03 | 0.77 | 0.26 | 1 | 0.02 | 0.43 |
Co-mutation analysis:
Among XPO1 mutated tumors, TP53 was the most common concurrent mutations with 80% (n =20 out of n= 25 tested for TP53). STK11 was the second most common co-alteration seen at 26.9% (n =7 out of n =26 tested for STK11). Also demonstrated were KRAS mutations in 19% (n = 5). EGFR mutations were rare (n = 2), and no targetable fusions were seen (Figure 1B). Among XPO1 amplified tumors, most tumors were positive for TP53 at 95.7% (n =22 out of n =23 tested for TP53). The second most common co-alterations were CDKN2A and NFE2L2, both seen in 16.7% (n =4 out of n =24 tested). Comparative analysis on common co-occurring alterations based on XPO1 status is shown in Figure 2 and Supplementary Table 1.
Figure 2.

Comparative analysis on common co-occurring alterations based on XPO1 status.
Real-world analysis:
Among the 17,449 NSCLC tumors with clinical data, there were 24 XPO1 mutant tumors with no histology imbalance observed in mutant vs. wild type (WT). Baseline characteristics of histology, sex and age for both XPO1 mutant as well as wild type are shown in Figure 3 and Supplementary Table 2. Comparison of OS in the NSCLC group between XPO1 mutant and WT showed a negative association with a hazard ratio (HR) of 1.932 (95% CI: 1.144- 3.264 p = 0.012) as shown in Figure 3. XPO1 amplification was not associated with survival.
Figure 3.
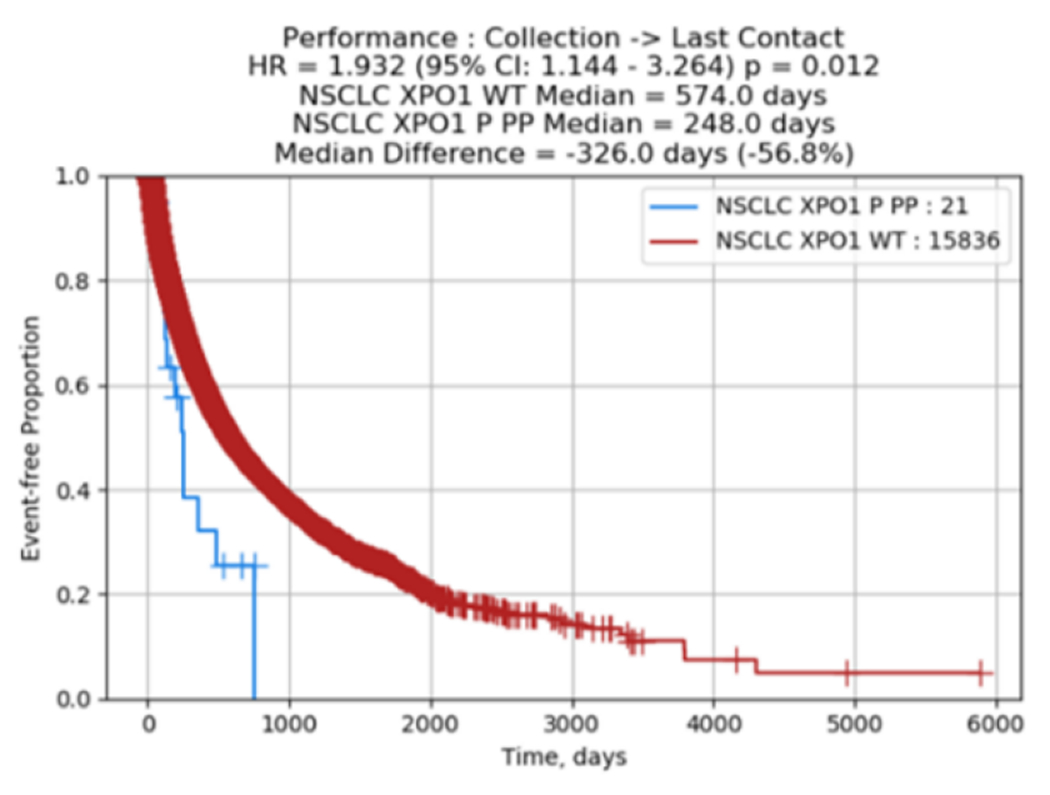
Clinical outcome data on XPO1 in NSCLC.
Comparing the OS within the subgroup with confirmed adenocarcinoma histology (9,973 XPO1 WT and 14 XPO1 mutant) showed a similar negative correlation in survival with a HR of 2.156 (95% CI: 1.027- 4.525 P = 0.037) as shown in Supplementary Figure 1.
As harboring a driver alteration could improve the prognosis, we did an additional analysis evaluating the outcomes of XPO1 WT excluding driver alterations (ALK fusions, RET fusions, ROS1 fusions, BRAF mutations, NTRK 1/2/3 fusions, MET amplifications, NRG1 fusions, HER2 mutations and EGFR mutations). Comparison of OS in the NSCLC group between XPO1 mutant and WT excluding driver alterations continued to show a negative association with a hazard ratio (HR) of 1.837 (95% CI: 1.087-3.104, p = 0.021) as shown in the Supplementary Figure 2. Similar negative correlation in survival was seen with a HR of 1.997 when the analysis was performed in those with adenocarcinoma histology (Supplementary Figure 3).
Discussion:
This is the first study identifying the mutation spectrum in XPO1 in NSCLC. In our study, we did not observe any overlap of XPO1 mutations and amplification. Both mutations and amplification were rare occurrence, and there were no significant gender or age prevalence between different XPO1 status (mutated, amplified or wild type).
In our study, a negative association in OS was found with those with XPO1 mutant tumors when compared to WT in both the NSCLC cohort as well as the adenocarcinoma cohort. As previously shown in a cohort of 92 lung adenocarcinoma patients, cytoplasmic immunoreactivity of p63 protein, a member of the p53 tumor suppressor family, was associated with shorter OS.28 It is speculated that mutations in XPO1 may lead to dysregulated export of these important tumor suppressor proteins into the cytoplasm, resulting in tumorigenesis and disease progression. Tumors with XPO1E571 mutation have been shown to display genetic and epigenetic modulations in cancer cells,18 which may result in resistance to chemotherapeutic drugs.
After the early failures with irreversible XPO1 inhibitors such as leptomycin B, consensus-induced fit docking (cIFD) approaches have since led to the development a novel class of reversible XPO1 inhibitors known as selective inhibitor of nuclear export (SINE).29 30 SINE compound such as selinexor (XPOVIO) and related agents verdinexor can efficiently block XPO1 mediated nuclear export thereby re-aligning the tumor suppressor in the correct compartment leading to restoration of their function. Selinexor has been evaluated in several phase I, II, III studies and received FDA approval for penta-refractory multiple myeloma31 and diffuse large B cell lymphoma. 11 XPO1 blockade in multiple myeloma and DLBCL is thought to re-establish the effects of multiple tumor suppressor proteins by forcing their nuclear retention, and potentially reversing chemotherapy resistance.32 It has been demonstrated that SINE compounds exert its inhibitory activity to the cells irrespective of their mutational status,13 which is also evident from SINE-bound XPO1 crystal structures.18
Earlier studies have shown that SINE can block lung tumor growth in NSCLC cell lines and xenografts, both alone and with cisplatin33 or targeted agents such as Bcl-xL inhibitor A-133185234 and PARP inhibitor BMN673.35 One efficient approach when considering combination therapy would be to target genomic co-alterations. In our study, although TP53 was the most common concurrent genomic mutation found with XPO1 mutated patients, we also found KRAS mutations (19%) and EGFR mutations (8%). Interestingly, in a multigenomic screen of 4,725 biological processes in 106 human NSCLC cell lines, the nuclear transport machinery was found as the sole process-level discriminator of statistical significance exhibiting synthetic-lethal interactions in KRAS-driven cancers.36 Indeed, there is an ongoing phase I/II study to evaluate the efficacy of selinexor in combination with docetaxel for patients with previously treated KRAS mutant NSCLC (NCT03095612). XPO1 is also thought to be involved in EGFR TKI resistance. The involvement of XPO1 in gefitinib resistance has been speculated to be due to the presence of four NES signals in the DEAD box DNA helicase DDX17 protein.37 Importantly, SINE have shown efficacy against EGFR inhibitor resistant NSCLC cell lines.38 These proof-of-concept studies indicate that XPO1 alone and in combination could be a valid therapeutic target for NSCLC.
Although our study demonstrated that XPO1 mutant NSCLC tumors were more likely to be TMB high (79.2% vs. 51.8%, p = 0.007) when compared to XPO1 wild type tumors, the efficacy of immunotherapy is unclear in this setting, especially as XPO1 mutant tumors were less likely to have high PD-L1 (31.6% vs. 56.3%, p = 0.03) when compared to XPO1 wild type tumors.
The prevalence of XPO1 mutations were relatively low in this large cohort. This study utilized real-world evidence data that lack some elements of clinical history and outcomes were inferred based on time from tissue collection to date of last contact. NGS was performed at varying time points during the course of the disease and treatments. Therefore, we were unable to distinguish if XPO1 alterations were baseline characteristics prior to any treatment or actually reflects acquired resistance. Additionally, these studies could have certainly benefitted by IHC validation of XPO1 over-expression in NSCLC tissue. Nevertheless, an independent group has shown higher expression XPO1 in 10 lung tumor tissues from smokers and 10 matched tumor-adjacent histologically normal lung tissues and in TMA on a testing set of 59 lung tumor tissues and their matched adjacent histologically normal tissues39.
Despite these limitations, we were able to determine the characteristics of XPO1 alterations in NSCLC. Further studies to investigate the underlying biological and molecular mechanism to account for the association of XPO1 mutations with negative clinical survival outcomes would be warranted. Finally, more detailed examination of the clinical effects of other co-existing mutations along with XPO1 alterations is eagerly awaited.
Conclusions:
Presence of XPO1 pathogenic mutations was associated with a poor OS in both the entire NSCLC cohort and the adenocarcinoma subgroup. Further studies of this negative association at the molecular level along with effect of other co-existing mutations may lead to development of novel treatment strategies.
Supplementary Material
Supplementary Table 1. Comparative analysis on common co-occurring alterations based on XPO1 status.
Supplementary Table 2. Clinical outcome data on XPO1 in NSCLC.
In cancer, hyperactive XPO1 promotes the export of important tumor suppressors.
Among 18,218 NSCLC tumors, 26 harbored XPO1 mutations and 24 had amplifications.
XPO1 mutant tumors were more likely to have high TMB.
KRAS co-mutations were seen in 19%.
XPO1 pathogenic mutations were associated with a poor survival in NSCLC.
Acknowledgements:
Work in the lab of ASA is supported by NIH R37-CA215427.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Mor A, White MA, Fontoura BMA. Nuclear trafficking in health and disease. Curr Opin Cell Biol. 2014; 28:28–35. doi: 10.1016/j.ceb.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muqbil I, Azmi AS, Mohammad RM. Nuclear Export Inhibition for Pancreatic Cancer Therapy. Cancers (Basel). 2018; 10(5):138. doi: 10.3390/cancers10050138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soniat M, Chook YM. Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem J. 2015; 468(3):353–62. doi: 10.1042/BJ20150368. [DOI] [PubMed] [Google Scholar]
- 4.Azmi AS. The evolving role of nuclear transporters in cancer. Semin Cancer Biol. 2014; 27:102. doi: 10.1016/j.semcancer.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Uddin MH, Zonder JA, Azmi AS. Exportin 1 inhibition as antiviral therapy. Drug Discov Today. 2020; 25(10):1775–1781. doi: 10.1016/j.drudis.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu S, Huang H, Horton P, Juan H. ValidNESs: a database of validated leucine-rich nuclear export signals. Nucleic Acids Res. 2013; 41(Database issue): D338–43. doi: 10.1093/nar/gks936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sendino M, Omaetxebarria MJ, Prieto G, Rodriguez JA, Using a simple cellular assay to map NES motifs in cancer-related proteins, gain insight into CRM1-mediated NES export, and search for NES-harboring micropeptides. Int J Mol Sci. 2020; 21(17):6341. doi: 10.3390/ijms21176341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azmi AS, Mohammad RM. Targeting Cancer at the Nuclear Pore. J Clin Oncol. 2016; 34(34):4180–4182. doi: 10.1200/JCO.2016.67.5637. [DOI] [PubMed] [Google Scholar]
- 9.Ishizawa J, Kojima K, Flail N Jr, Tabe Y, Andreeff M. Expression, function, and targeting of the nuclear exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol Ther. 2015; 153:25–35. doi: 10.1016/j.pharmthera.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu FY, Wang SE, Sanders ME, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006; 66(4):2162–72. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- 11.Azmi AS, Uddin MH, Mohammad RM. The nuclear export protein XPO1- from biology to targeted therapy. Nat Rev Clin Oncol. 2021; 18(3):152–169. doi: 10.1038/s41571-020-00442-4. [DOI] [PubMed] [Google Scholar]
- 12.Taylor J, Sendino M, Gorelick AN, et al. Altered Nuclear Export Signal Recognition as a Driver of Oncogenesis. Cancer Discov. 2019; 9(10):1452–1467. doi: 10.1158/2159-8290.CD-19-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jardin F, Pujals A, Pelletier L, et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B- cell lymphoma. Am. J. Hematol 2016; 91: 923–930. doi: 10.1002/ajh.24451. [DOI] [PubMed] [Google Scholar]
- 14.Maracaja DLV, Puthenpura V, Pels SG, et al. EBV- positive primary large B- cell lymphoma: the role of immunohistochemistry and XPO1 in the diagnosis of mediastinal lymphomas. Appl. Immunohistochem. Mol. Morphol 2020; 28(10):725–730. 10.1097/PAI.0000000000000820. [DOI] [PubMed] [Google Scholar]
- 15.García- Santisteban I, Arregi I, Alonso-Mariňo M, et al. A cellular reporter to evaluate CRM1 nuclear export activity: functional analysis of the cancer- related mutant E571K. Cell Mol. Life Sci 2016; 73(24): 4685–4699. doi: 10.1007/s00018-016-2292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumhardt JM, Walker JS, Lee Y, et al. Recognition of nuclear export signals by CRM1 carrying the oncogenic E571K mutation. Mol. Biol. Cell 2020; 31(17):1871–1891. doi: 10.1091/mbc.E20-04-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camus V, Miloudi H, Taly A, Sola B, Jardin F. XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol. Oncol 2017; 10(1):47. doi: 10.1186/s13045-017-0412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker JS, Hing ZA, Harrington B. Recurrent XPO1 mutations alter pathogenesis of chronic lymphocytic leukemia. J Hematol Oncol. 2021; 14(1):17. doi: 10.1186/s13045-021-01032-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conforti F, Wang Y, Rodriguez JA, Alberobello AT, Zhang Y, Giaccone G. Molecular Pathways: Anticancer Activity by Inhibition of Nucleocytoplasmic Shuttling. Clin Cancer Res. 2015; 21(20):4508–13. doi: 10.1158/1078-0432. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt J, Braggio E, Kortuem KM, et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013;27:2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo B, Huang L, Gu Y, et al. Expression of exportin-1 in diffuse large B-cell lymphoma: immunohistochemistry and TCGA analyses. Int J Clin Exp Pathol 2018; 11: 5547–60 [PMC free article] [PubMed] [Google Scholar]
- 22.The Human Protein Atlas. https://www.proteinatlas.org/ last accessed on 5/10/2021.
- 23.Waldmann I, Spillner C, Kehlenback RH. The nucleoporin-like protein NLP1 (hCG1) promotes CRM1-dependent nuclear protein export. J Cell Sci. 2012;125(Pt 1):144–54. doi: 10.1242/jcs.090316. [DOI] [PubMed] [Google Scholar]
- 24.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takenaka Y, Fukumori T, Yoshii T, et al. Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol. 2004;24:4395–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.I profile for U.S. (excluding New York). 2019. Available from: http://www.carismolecularintelligence.com/solid_tumors
- 27.Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017; 19(10): 1105–1117. doi: 10.1038/gim.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narahashi T, Niki T, Wang T, et al. Cytoplasmic localization of p63 is associated with poor patient survival in lung adenocarcinoma. Histopathology. 2006;49(4):349–57. doi: 10.1111/j.1365-2559.2006.02507.x. [DOI] [PubMed] [Google Scholar]
- 29.Kalid O, Warshaviak DT, Shechter S, Sherman W, Shacham S. Consensus Induced Fit Docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J Comput Aided Mol Des. 2012; 26(11):1217–28. doi: 10.1007/s10822-012-9611-9. [DOI] [PubMed] [Google Scholar]
- 30.Parikh K, Cang S, Sekhri A, Liu D. Selective inhibitors of nuclear export (SINE)--a novel class of anti-cancer agents. J Hematol Oncol. 2014; 7:78. doi: 10.1186/s13045-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chari A, Vogl DT, Gavriatopoulou M, et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N Engl J Med. 2019; 381(8): 727–738. doi: 10.1056/NEJMoa1903455. [DOI] [PubMed] [Google Scholar]
- 32.Laín S, Xirodimas D, Lane DP. Accumulating active p53 in the nucleus by inhibition of nuclear export: a novel strategy to promote the p53 tumor suppressor function. Exp Cell Res 1999; 253: 315–24. [DOI] [PubMed] [Google Scholar]
- 33.Sun H, Hattori N, Chien W, et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer. 2014; 111(2):281—291. doi: 10.1038/bjc.2014.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Z, Liu J, Yang C, Zhao M, Xiong Z. XPO1 inhibitor KPT-330 synergizes with Bcl-xL inhibitor to induce cancer cell apoptosis by perturbing rRNA processing and Mcl-1 protein synthesis. Cell Death Dis. 2019;10(6): 395. doi: 10.1038/s41419-019-1627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Sun T, Meng Z, et al. XPO1 inhibition synergizes with PARP1 inhibition in small cell lung cancer by targeting nuclear transport of FOXO3a. Cancer Lett. 2021; 503:197–212. doi: 10.1016/j.canlet.2021.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, McMillan E, Kim HS, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature. 2016;538:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li K, Mo C, Gong D, et al. DDX17 nucleocytoplasmic shuttling promotes acquired gefitinib resistance in non-small cell lung cancer cells via activation of β-catenin. Cancer Lett. 2017; 400:194–202. doi: 10.1016/j.canlet.2017.02.029. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Han X, Wang J, Yao J, Shi Y. Antitumor effects of a novel chromosome region maintenance 1 (CRM1) inhibitor on non-small cell lung cancer cells in vitro and in mouse tumor xenografts. PLoS One. 2014;9:e89848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao W, Lu C, Chen L, Keohavong P. Overexpression of CRM1: A characteristic feature in a transformed phenotype of lung carcinogenesis and a molecular target for lung cancer adjuvant therapy. J Thorac Oncol. 2015;10(5):815–825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Comparative analysis on common co-occurring alterations based on XPO1 status.
Supplementary Table 2. Clinical outcome data on XPO1 in NSCLC.