

ABSTRACT
Enterotoxigenic Escherichia coli (ETEC) isolates are genetically diverse pathological variants of E. coli defined by the production of heat-labile (LT) and/or heat-stable (ST) toxins. ETEC strains are estimated to cause hundreds of millions of cases of diarrheal illness annually. However, it is not clear that all strains are equally equipped to cause disease, and asymptomatic colonization with ETEC is common in low- to middle-income regions lacking basic sanitation and clean water where ETEC are ubiquitous. Recent molecular epidemiology studies have revealed a significant association between strains that produce EatA, a secreted autotransporter protein, and the development of symptomatic infection. Here, we demonstrate that LT stimulates production of MUC2 mucin by goblet cells in human small intestine, enhancing the protective barrier between pathogens and enterocytes. In contrast, using explants of human small intestine as well as small intestinal enteroids, we show that EatA counters this host defense by engaging and degrading the MUC2 mucin barrier to promote bacterial access to target enterocytes and ultimately toxin delivery, suggesting that EatA plays a crucial role in the molecular pathogenesis of ETEC. These findings may inform novel approaches to prevention of acute diarrheal illness as well as the sequelae associated with ETEC and other pathogens that rely on EatA and similar proteases for efficient interaction with their human hosts.
KEYWORDS: Escherichia, diarrhea, enterotoxins, mucinase, proteases
INTRODUCTION
Enterotoxigenic Escherichia coli (ETEC) strains are ubiquitous pathogens in low- to middle-income regions, where they are a major cause of morbidity and mortality due to diarrheal illness, particularly among young children (1). In the classical paradigm of ETEC virulence, the organisms adhere to epithelial cells in the small intestine via plasmid-encoded colonization factors, where they deliver heat-labile (LT) and/or heat-stable (ST) enterotoxins that promote the net efflux of salt and water into the intestinal lumen with ensuing watery diarrhea. Nevertheless, some features of ETEC illness suggest that this classical paradigm of molecular pathogenesis is far from complete (2). Notably, ETEC strains cause illness that may range from mild disease to severe diarrhea accompanied by rapid dehydration clinically indistinguishable from cholera (3–6). In addition, ETEC and other bacterial enteric pathogens, including Shigella, have repeatedly been associated with poorly understood nondiarrheal sequelae (7), including environmental enteropathy (8), growth stunting (9–12), malnutrition (13), and cognitive impairment (14, 15).
In addition to the canonical virulence factors, several more recently discovered plasmid-encoded proteins appear to be conserved within the ETEC pathovar (16, 17). Among these is the EatA autotransporter protein (18), a member of the serine protease autotransporters of the Enterobacteriaceae (SPATE) family (19). EatA, which shares approximately 74% identity with the SepA autotransporter from Shigella flexneri (20), has recently been shown to degrade MUC2 (21), the major gel-forming mucin secreted by goblet cells in both the small and large intestine (22). The 110-kDa secreted passenger domain of the EatA autotransporter contains the functional serine protease activity centered on a catalytic triad formed by residues H134, D162, and S267 (18).
MUC2 is a large (∼5,200 amino acids), heavily glycosylated protein with more than 80% of its mass comprised of glycans. The central portion of the protein is organized into PTS domains that are comprised largely of repeated proline (P), threonine (T), and serine (S) residues, where T and S hydroxyl groups serve as sites for O-glycosylation. These densely glycan-rich PTS core regions of MUC2 are protected from proteolytic degradation. The MUC2 apoprotein undergoes end-to-end dimerization at the C-terminal end of the molecule in the endoplasmic reticulum (23), followed by O-glycosylation in the Golgi apparatus and further multimerization via von Willebrand D domains (VWD) in the N-terminal region of the glycoprotein (24, 25). Ultimately, the MUC2 secreted by goblet cells expands to form large layered polymeric net-like structures that serve as a primary mucosal defense against gastrointestinal pathogens and resident microbiota (26).
Interestingly, recent studies of ETEC isolated from a cohort of young Bangladeshi children monitored from birth to 2 years of age (12) demonstrated that the presence of the eatA locus was strongly associated with symptomatic diarrheal disease (27). The studies reported here demonstrate that EatA degradation of mucin plays an essential role in promoting effective pathogen-host interactions central to the molecular pathogenesis of ETEC.
RESULTS
ETEC heat-labile toxin stimulates production of a defensive mucin barrier.
Although the mucin layer in the small intestine is more penetrable than the colon (28), it is possible that it is sufficient to preclude the direct engagement of intestinal epithelial cells with ETEC required for effective toxin delivery (29). In addition, studies of rat intestine (30, 31) and cell lines derived from a colonic cancer (32) suggested that cholera toxin, a homologue of LT, can stimulate goblet cell secretion of mucin. Accordingly, LT treatment of enteroids derived from human small intestine resulted in significantly increased transcription of the MUC2 gene (Fig. 1a), while treatment of spheroids of small intestinal cells, in which the apical surface of cells is oriented facing an internal luminal cavity, resulted in the release of substantial amounts of MUC2 mucin into the lumen compared to untreated controls (Fig. 1b). Similarly, treatment of polarized small intestinal enteroid monolayers with LT and cholera toxin, but not a mutant E112K enzymatically inactive version of LT (mLT), resulted in substantial increases in the secretion of MUC2 mucin (Fig. 1c) and a corresponding increase in integrity (Fig. 1d). Enteroid monolayers treated with either forskolin or LT, but not untreated monolayers, excluded the migration of fluorescent beads to the epithelial surface, suggesting that the induced mucin presents a significant barrier (Fig. 1e and f). Altogether, these data suggest that both LT and cholera toxin provoke intestinal epithelia to reinforce the protective mucin barrier in the small intestine.
FIG 1.

LT stimulates production and secretion of MUC2 mucin barrier by goblet cells in small intestinal epithelia. (a) Transcriptional response (RT-PCR, MUC2) of small intestinal enteroids following treatment with heat-labile toxin (LT; 100 ng/ml overnight) compared to untreated (ø) control cells. Shown are summaries of replicate experiments, each with 3 technical replicates (**, P = 0.02, Mann-Whitney, two-tailed, nonparametric analysis). (b) Small intestinal enteroids (spheroids) in which the apical surface of the enterocytes is oriented to the inside of the sphere. Top panels represent untreated spheroids (ø) where MUC2 (green) is largely contained within goblet cells. Nuclei are pseudocolored white (DAPI) and cell membranes are blue (CellMask). The hashed line depicts the region enlarged below. Bottom, LT treatment of small intestinal spheroids results in MUC2 secretion into the lumen. (c) MUC2 mucin production by polarized differentiated small intestinal enteroids. α-MUC2 immunoblot shows MUC2 present in apical supernatants of (duplicate wells) untreated monolayers (ø) and following treatment with mutant LT (mLT), native LT, or cholera toxin (CT). (d) LT treatment increases the integrity of the mucin barrier overlying enterocytes. Data represent time required for metal beads to traverse mucin in supernatants obtained from LT-treated or control cells (ø). Shown are results of replicate experiments, each with 4 technical replicates (***, P < 0.01 by Mann-Whitney, two-tailed nonparametric testing). (e) Graph depicts gap distances between 1-μm fluorescent beads and small intestinal enteroid monolayer surfaces comparing LT-treated cells to those treated with forskolin-treated (FSK) or untreated (ø) monolayers. Data from duplicate experiments (n = 3 technical replicates) are differentially colored. Each symbol represents the distance (z) between the bead front and the monolayer surface at selected points along the x-y axis. Dashed lines represent median values. ****, P < 0.0001 by Kruskal-Wallis testing. (f) Volume projections of representative confocal z-stack images of human small intestinal monolayers left untreated (ø) or treated with LT or forskolin (FSK). Cells were stained with CellMask (red) and treated overnight, followed by addition of fluorescent beads (1 μm). Confocal acquisition was ∼45 min after addition of beads.
EatA promotes bacterial migration through the secreted mucin network.
To explore the hypothesis that the secreted EatA protease facilitates pathogen access to host enterocytes by degrading the protective mucin barrier, we first examined the impact of EatA on the viscosity of the mucin matrix. Treatment of purified intestinal mucin with wild-type (wt) recombinant passenger domain (rEatAp; Fig. 2a) resulted in significant reduction in viscosity (Fig. 2b) relative to either untreated mucin or mucin treated with proteolytically inactive passenger domain (H134R). Likewise, polyclonal IgG against EatAp prevented reductions in integrity (Fig. 2c). Next, we found that wild-type ETEC migrated significantly faster through purified mucin than the eatA mutant (Fig. 2d), suggesting that mucin serves as a substantial barrier to ETEC migration. Indeed, in ex vivo explants of human small intestine (Fig. 2e; see also Movie S1 in the supplemental material), we found that wild-type ETEC efficiently penetrated the mucin layer to engage the epithelium, while the eatA mutant was excluded from contact with the epithelial surface. In addition, we used small intestinal enteroids in which the O-linked MUC2 sugars were metabolically labeled with azide-modified galactosamine (GalNAz) followed by click conjugation with the alkyne flurophore (DBCO-Cy3) to highlight the secreted mucin (red). Again, we found that the eatA mutant was trapped by small intestinal mucin (Fig. 2f, Movie S2).
FIG 2.
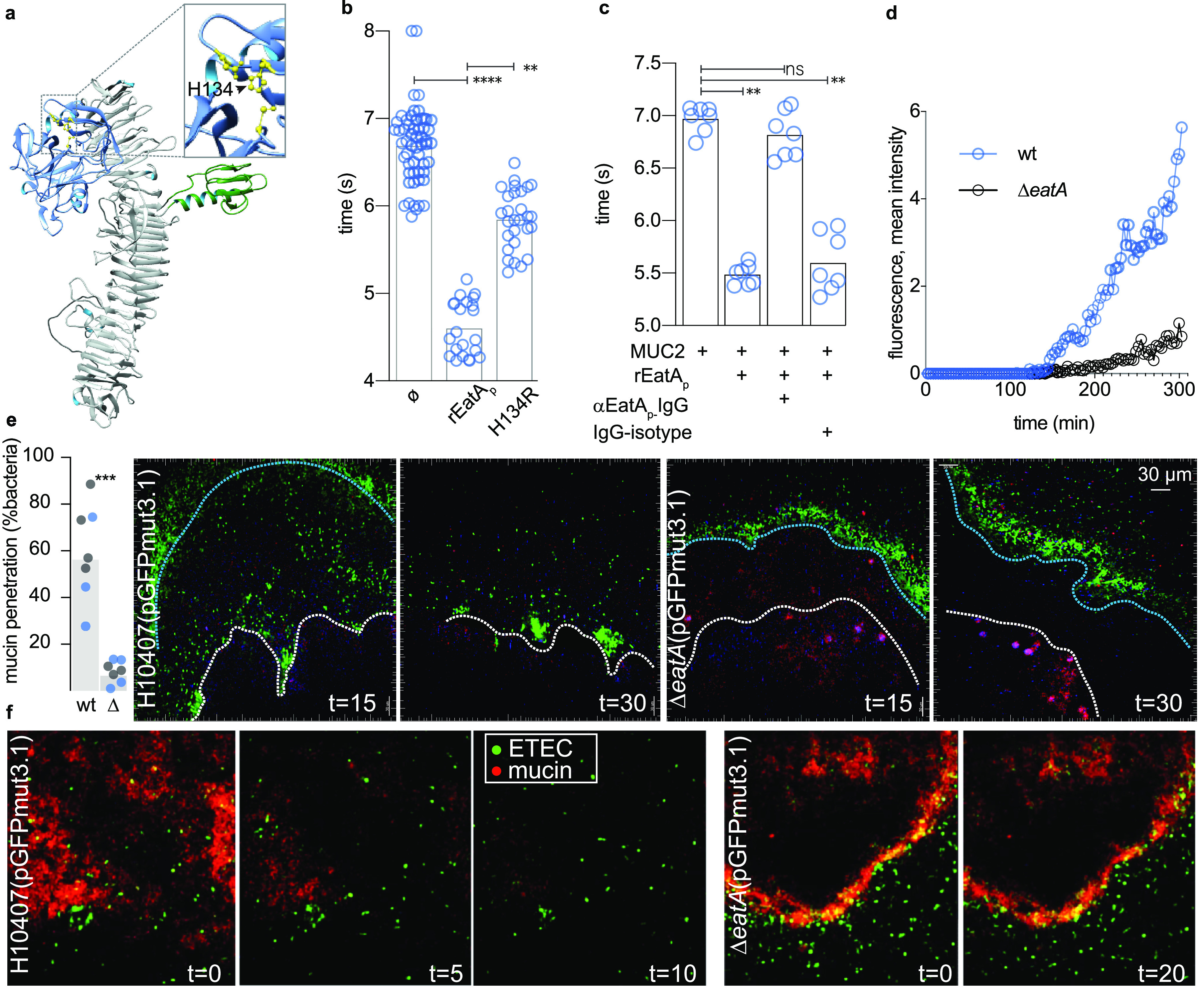
EatA reduces mucin viscosity and promotes bacterial migration through intestinal mucin. (a) Putative EatA passenger domain structure, protease subdomain (blue), showing active site with the H134, D162, S267 catalytic triad highlighted in yellow (inset) and the domain of unknown function (DUF) in green. (b) Wild-type recombinant EatA passenger domain (rEatAp), but not the passenger bearing a mutation in the serine protease catalytic triad (H134R), reduces the integrity of purified MUC2 mucin. Symbols represent technical replicates combined from a total of n = 5 experimental replicates. (c) Antibodies against rEatAp inhibit EatA effects on mucin integrity. (Symbols indicate technical replicates from n = 2 experiments.) (d) EatA is required for efficient ETEC migration through purified MUC2 mucin. Each symbol represents average accumulated fluorescence intensity of gfpmut3.1-expressing wild-type and eatA mutant bacteria over time following introduction into μ-channel slides (ibidi μ-slide vi 0.4). Data represent progression of fluorescently labeled bacteria along their respective channels in images acquired ∼14 mm from the point of entry. (e) EatA is required for penetration of the mucin layer in human intestinal explants in vitro. The graph at the left depicts the percentage of wt and eatA mutant (Δ) bacteria expressing pGFPmut3.1 that have penetrated the mucin layer at 15 min after infection (3 technical replicates in each of two independent experiments from separate tissue donors are shown by color). P = 0.0006 by analysis of variance (ANOVA). Shown on the right are representative images acquired by two-photon microscopy of bacteria (green) and penetration into the mucin layer over time. The edge of the intestinal mucin is depicted by the blue line and the surface of the intestine by the white line. Intestinal cells autofluoresce purple. (f) Two-photon microscopy images of small intestinal enteroids metabolically labeled with GalNAz to highlight secreted mucin (red).
EatA is required for efficient pathogen-host interaction and toxin delivery.
Efficient delivery of ETEC enterotoxin requires intimate interaction of the bacteria with the surface of target intestinal epithelial cells (IECs) (29). We found that in the absence of eatA, ETEC bacteria were incapable of effectively migrating through MUC2 purified from small intestinal enteroids and were as impaired as an immotile (fliC) mutant, while pretreatment of MUC2 with rEatAp restored migration (Fig. 3a). Similarly, adherence of the eatA mutant ETEC to target small intestinal enteroid epithelial cells (Fig. 3b, Fig. S1) was significantly impaired relative to wild-type ETEC, as was delivery of heat-labile toxin (Fig. 3c). Collectively, these data suggest that eatA plays an essential role in promoting effective ETEC pathogen-host interactions.
FIG 3.

Both EatA and motility are required to penetrate MUC2 and engage small intestinal epithelia. (a) Shown are data from Transwell mucin migration assays in which wild-type H10407 (wt) or the eatA or flagellin (fliC) mutants (∼105 CFU) were added to the upper chamber containing MUC2 mucin harvested from small intestinal enteroids. Data are expressed as the percentage of inoculum recovered from the lower chamber after 2 h. rEatAp represents complementation with exogenous EatA passenger domain (50 μg/ml) added at the time of infection. Bar graphs at the right demonstrate mean recovery ± standard errors of the means in the absence of MUC2. (b) EatA facilitates bacterial access to epithelial cells. Shown are wt or mutant bacteria adherent to small intestinal enteroid monolayer epithelial cells. The eatA mutant was complemented with either a recombinant eatA expression plasmid (pTW5016) or the vector control (pWSK29). Shown are the results of three replicate experiments (separated by color) with each symbol (n = 40) representing a technical replicate. (c) EatA is required for efficient ETEC delivery of heat-labile toxin to enteroid monolayers. (Shown are cAMP levels in target epithelial cells infected with wild-type or mutant bacteria 2 h after infection.) P values in each panel reflect comparisons by ANOVA with nonparametric Kruskal-Wallis testing (*, P = 0.03; **, P = 0.002; ***, P = 0.0002; ****, P < 0.0001).
Identification of a subdomain required for binding and degradation of MUC2 mucin.
MUC2 mucin produced by goblet cells in the small intestine is heavily glycosylated. Interestingly, recent structural analysis of SepA suggested that an 80-amino-acid subdomain within the passenger domain could represent a carbohydrate binding module (CBM) (33). To investigate the importance of the corresponding region in EatA (Fig. 4a) to virulence, we first replaced the passenger subdomain of unknown function encompassed by amino acids E541 to S616 with a stretch of 8 histidines (DUF::H8). The resulting protein was secreted as efficiently as the parent molecule (Fig. 4b) and retained the ability to degrade the AAPL synthetic peptide (Fig. 4c). While the passenger domain containing a single point mutation in the catalytic triad (H134R) colocalized with MUC2 in sections of human intestine (Fig. 4d), we observed appreciably less interaction between MUC2 and the DUF::H8 mutant in sections of human ileum (Fig. 4e, Fig. S2) and in the lumen of small intestinal spheroids (Fig. 4f). Similarly, the DUF::H8 passenger protein exhibited only weak interaction with purified MUC2 (Fig. 4g), and the mutant protein was incapable of efficiently degrading MUC2 (Fig. 4h, Fig. S3A). Importantly, antibodies specific to the E541-S616 peptide inhibited the activity of the parent molecule (Fig. S3B to D). Complementation of the eatA mutant in trans with a plasmid expressing the DUF::H8 protein also failed to restore ETEC interaction with target epithelial cells (Fig. 4i), further suggesting that this region is essential to EatA function. Although interactions of EatA with heavily glycosylated MUC2 molecules may involve carbohydrate moieties, we did not observe significant binding to glycan microarrays (Data Set S2) with either the full-length recombinant EatA passenger domain or the mutant lacking the putative carbohydrate binding module encompassed by the E541-S616 subdomain. Likewise, nanoparticles in which Spy-tagged EatA (K535-S616) was conjugated to the surface of SpyCatcher-mi3 (34, 35) were not sufficient to direct MUC2 binding (Fig. S4). Collectively, these studies suggest that while this region of the passenger domain is critical for mucin interactions, it likely acts in concert with the proteolytic subdomain to engage and degrade MUC2.
FIG 4.
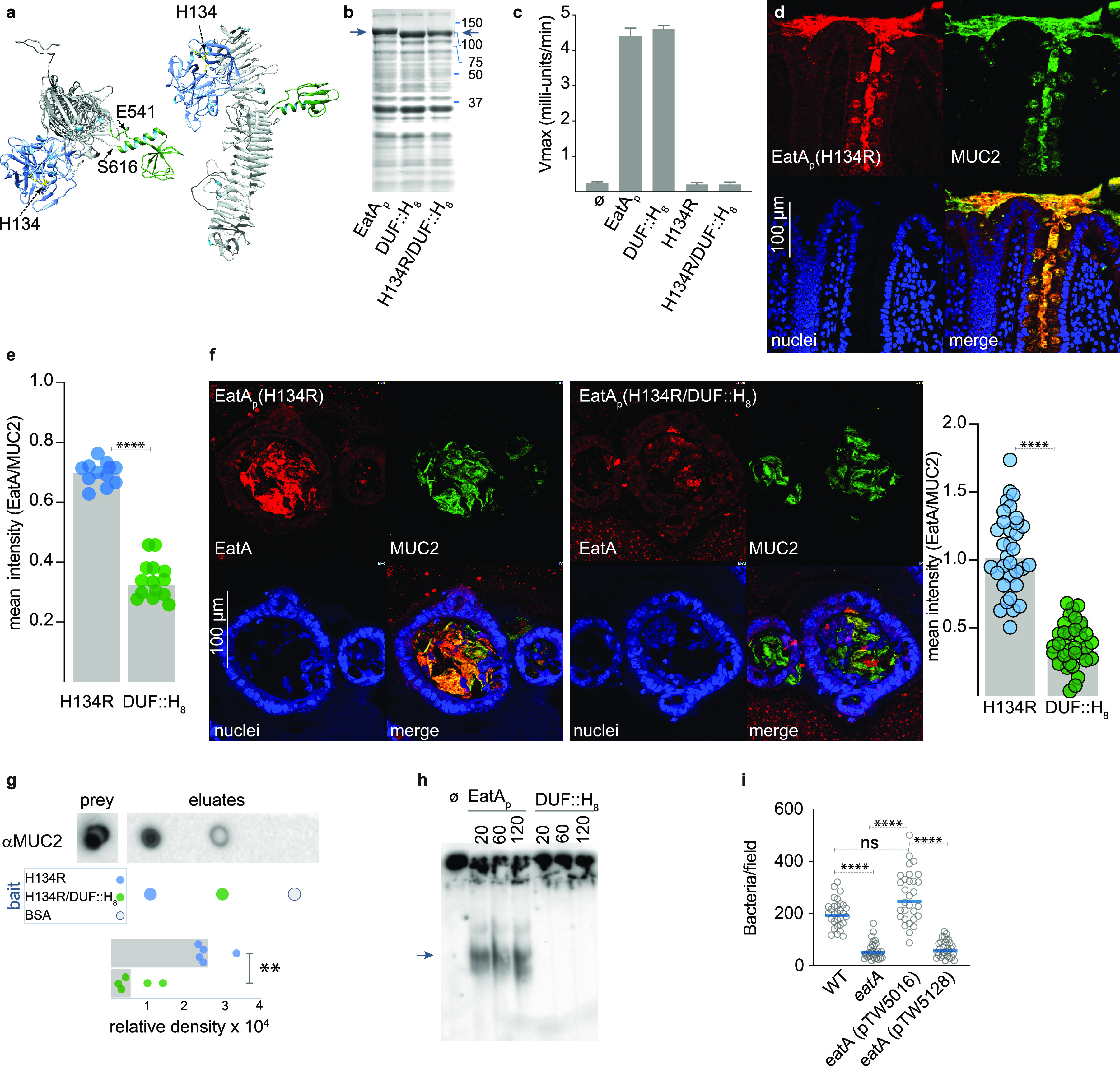
(a) Model of the EatA passenger domain showing the relative location of the protease (blue) and putative CBM (green). (b) Coomassie-stained SDS-PAGE of TCA-precipitated culture supernatants showing migration of the recombinant passenger domain (arrows) from E. coli Ig10B expressing EatA, EatA bearing a polyhistidine replacement of the domain of unknown function (DUF::H8), and the H134R/DUF::H8 double mutant passenger protein. (c) The domain of unknown function is not required for proteolytic activity. Shown are the results of AAPL oligopeptide cleavage experiments for using the wild-type EatA passenger protein (EatAp) and mutant passenger proteins. Results represent means and standard deviations from 3 separate experiments. (d) EatA passenger domain colocalizes with MUC2 mucin. Panels show representative confocal images of proteolytically inactive EatA (H134R) passenger domain interacting with MUC2 secreted into the lumen of human intestinal explants and retained in goblet cells. (e) Optimal binding to small intestinal MUC2 requires the subdomain from E541-S616. The graph depicts colocalized binding of H134R or H134R/DUF::H8 protein to MUC2 in sections of small intestine shown in Fig. S2. (f) Binding to secreted MUC2 requires the E541-S616 region. Shown are maximal intensity projections of confocal microscopy z-stack images of sections of small intestinal spheroids stimulated with heat-labile toxin to stimulate release of MUC2 into the lumen. Replicate daughter sections were incubated with the H134R passenger protein (left) or the DUF::H8 mutant protein (right). The graph depicts binding of EatA H134R and DUF::H8 passenger domains relative to target MUC2 (n = 33 regions from 30 spheroids; ****, P < 0.0001, two-tailed Mann-Whitney). (g) MUC2 pulldown using EatA(H134R) passenger domain or the H134R/DUF::H8 passenger domain as bait. The graph represents a summary of three replicate experiments (**, P = 0.008) in two-tailed Mann-Whitney nonparametric testing. (h) MUC2 degradation requires the domain of unknown function. (i) Optimal ETEC engagement of small intestinal enteroids requires the E541-Q617 region of the passenger domain. Shown are the combined results of three experimental replicates, each with 10 technical replicates/experiment (n = 30/group). ****, P < 0.0001 by ANOVA (Kruskal-Wallis).
DISCUSSION
The studies presented here potentially provide a biological basis for the recent finding that the presence of eatA is strongly associated with development of symptomatic disease in young children infected with ETEC (27) and suggest that mucin degradation plays a critical role for these pathogens. It is clear from earlier studies that enterotoxin delivery by these bacteria requires direct enterocyte engagement (29), and the data presented here indicate that in the absence of an effective mucin-degrading enzyme, even the relatively loose layer of mucin overlying the small intestinal epithelia appears to be sufficient to mitigate interaction of ETEC with the host.
This barrier is further reinforced by the host response to LT. Using enteroids propagated from human small intestine, we show that LT and the closely related cholera toxin are potent secretagogues for MUC2 mucin. Therefore, while the layer of mucin overlying small intestinal epithelia is thought to be more penetrable relative to that in the colon, it does not appear to be static, and EatA may have evolved to counter what would otherwise be an effective host defense strategy.
These data may also add another layer of complexity to the interpretation of epidemiologic studies that to date have defined the ETEC pathovar simply by the presence of heat-labile and heat-stable toxins. The large proportion of asymptomatic ETEC colonization in these studies potentially confounds attempts to accurately assess the burden of diarrheal illness attributable to ETEC (36). The present studies provide further data in support of the concept that ETEC are not equally equipped to cause disease. Notably, eatA originally discovered on a virulence plasmid of H10407 (18), a strain isolated from a patient with cholera-like illness in Bangladesh, has been identified subsequently in other strains with clear metadata linking them to severe cholera-like illness (5, 6). H10407 has been the subject of multiple controlled human infection studies, largely because relative to other strains it reliably causes significant diarrheal illness (37–39).
The presence of SepA, a homologue that shares more than 70% identity with EatA, has been strongly linked to virulence of other pathogens, including enteroaggregative E. coli (EAEC) (40, 41). Indeed, in the interrogation of more than 90 genomes from EAEC strains collected in the Global Enteric Multicenter Study (GEMS) (1) for potential virulence genes, sepA was recently shown to have the strongest association with the development of moderate-to-severe diarrheal illness (41). The role of SepA in the virulence of Shigella species, including Shigella flexneri 5a, where SepA was originally discovered (20) on the large virulence plasmid, is currently unknown. However, given that some Shigella flexneri 2a as well as other Shigella species, including Sonnei species, make EatA (42), these pathogens might also utilize these proteases to degrade the thicker layer of mucin in the colon.
Collectively, the information emerging from molecular epidemiology studies (27), as well as the data presented here, suggest that EatA plays a crucial role in the molecular pathogenesis of ETEC. These findings may inform novel approaches to prevention of acute diarrheal illness as well as the sequelae associated with ETEC and other pathogens that rely on EatA and similar proteases for efficient interaction with human hosts.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains used in this study are presented in Table S1. Bacterial cultures were routinely incubated with aeration at 37°C in Luria-Bertani (LB) broth (10 g tryptone, 5 g yeast extract, and 10 g NaCl per liter) or on LB agar plates unless indicated otherwise. Antibiotics were added as appropriate. For infection assays, overnight-grown liquid cultures diluted to 1:100 in LB broth were incubated for ∼2 h to mid-log phase.
Propagation of enteroids and epithelial monolayer culture.
Detailed procedures for growth and maintenance of these cells have been described previously (43, 44). Briefly, cryopreserved cells were resuspended in Matrigel (BD Biosciences), and 15 μl of the suspension was seeded into each well of a 48-well tissue culture plate. Cells were maintained in 50% conditioned medium (CM) consisting of a 1:1 mixture of L-WRN conditioned medium and primary tissue culture medium, advanced Dulbecco’s modified Eagle’s medium (DMEM)-F12 containing 20% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin, supplemented with 10 μM of Y-27632 (ROCK inhibitor; Tocris Bioscience) and 10 μM SB431542 (TGFBR1 inhibitor; Tocris Bioscience). To obtain a polarized epithelial monolayer, cells were pooled from 10 wells, washed in phosphate-buffered saline (PBS), and trypsinized before seeding onto a 75-mm, 0.4-μm-pore polycarbonate membrane (Transwell, Corning) coated with type IV collagen (34 μg/ml) in sterile cell culture water. CM was added to upper and lower wells and maintained at 37°C, 5% CO2 for 5 days. Fresh CM was added every 2 days. Following confluence, medium was replaced with cell differentiation medium (DM) containing 5% CM in DMEM-F12 medium supplemented with 20% FBS, 2 mM l-glutamine, and 10 μM Y-27632. Fresh DM was added daily for 2 days for monolayer differentiation prior to treatment with toxin.
Spheroid propagation and processing.
For spheroid cultures, cells suspended in Matrigel were seeded as 15-μl droplets into individual wells of a 48-well tissue culture plate and grown for 3 days in 50% CM. Spheroids were then diluted in 150 μl Matrigel (1:10 dilution from initial volume), reseeded as 50-μl droplets in a 24-well tissue culture plate, and grown for 5 days in 50% CM. Finally, CM was replaced with cell differentiation medium containing 5% CM and grown for 3 days. Mucin production was stimulated by the addition of E. coli LT (100 ng/ml). Resulting spheroids were fixed with 4% paraformaldehyde (PFA) for 30 min at 37°C and 30 min at room temperature, washed 3× with PBS, and embedded in 5% Luria agar prior to being processed as paraffin blocks for sectioning.
Confocal microscopy.
For EatA binding, spheroid sections were deparaffinized, hydrated, unmasked, and incubated with 10 μg/ml of EatAH134R or EatAH134R-DUF-his at 4°C overnight. Following incubation, unbound EatA was washed off by rinsing the slides 3× with PBS. Muc2 mucin and EatA signals were detected with anti-Muc2 rabbit polyclonal (1:200; sc-15334; Santa Cruz) and anti-EatA mouse polyclonal (1:100; in-house) primary antibodies, followed by fluorescence-tagged goat anti-rabbit IgG Alexa Fluor 488 (1:200; Invitrogen) and fluorescence tagged goat anti-mouse IgG Alexa Fluor 594 (1:200; Invitrogen) secondary antibodies. Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000) and mounted using prolong gold antifade reagent (Invitrogen). Images were captured and analyzed on a Nikon C2 confocal microscope equipped with NIS-Elements AR 5.11.01 software (Nikon).
Human intestinal explants and two-photon imaging.
Freshly isolated sections of human small and large intestine were obtained at ileocolonic resection surgery from the Digestive Diseases Research Core Center at Washington University (DDRCC). The muscle layer was carefully removed, and the tissues were supported on a 70-μm cell strainer mesh placed in a custom imaging chamber. The tissue was perfused from the serosal side with oxygenated DMEM throughout imaging. Wild-type (jf2450) and eatA mutant (jf4738) green fluorescent protein (GFP)-expressing bacteria were grown in Luria broth containing ampicillin (100 μg/ml) overnight at 37°C, 200 rpm, and then diluted 1:100 and grown to an optical density at 600 nm (OD600) of ∼0.125 immediately prior to the experiment. Four hundred microliters of undiluted culture was then mixed with 50 μl of a 1:100 dilution of 1.75-μm fluorescent microspheres (19392; Polysciences); 450 μl of the bead/culture suspension was added to the epithelial surface of individual tissue sections. Images were acquired using a custom-built two-photon microscope in the Washington University In Vivo Imaging Core (IVIC) equipped with an Olympus XLUMPLFLN 20×, 1.0-numeric-aperture water-immersion objective. Fluorescence was excited using a Ti-Sapp laser (Chameleon Vision II; Choherent) tuned to 890 nm and emission collected using 495/540/570-nm dichroic filters (Semrock) and 3 head-on bi- and multialkali PMTs (Hamamatsu). Slidebook digital microscopy software (Intelligent Imaging Innovations) was used for hardware control and acquisition of two-dimensional time-lapse recordings and z-stack images. Imaris 9.5 (Oxford Instruments) was used for data rendering and analysis.
Metabolic labeling of enteroids.
Enteroid monolayers were metabolically labeled overnight with medium containing 50 μM GalNaz (N-azidoacetylgalactosamine, tetra-acylated; CLK-1086; Jena Biosciences). After washing with fresh media, resulting azide-functionalized glycoconjugates were reacted with DBCO-Cy3 (Kerafast), and nuclei were stained with Hoechst 3342 (B2261; Sigma).
Mucin purification.
Human MUC2 mucin was recovered from culture supernatants of LS174T cells (ATCC CL-188) by ultrafiltration as previously described (21).
Mucin migration assays.
To evaluate bacterial migration through a mucin network, mucin was first harvested from human small intestinal enteroid monolayers. Approximately 48 h after differentiation, medium was aspirated and mucin was carefully retrieved using a 1.8-cm-wide cell scraper (Corning), leaving the cell monolayer undisturbed; 200 μl of the harvested mucin was dispensed onto 3-μm polycarbonate Transwell membranes in a 24-well plate. Five hundred microliters of base medium (Advanced DMEM-F12 containing 20% FBS and 2 mM l-glutamine) was added to the bottom wells. Fifty microliters of bacterial suspension containing 105 CFU was dispensed into the wells containing mucin, and plates were incubated at 37°C, 5% CO2, for 2 h. Medium was then collected from the bottom well, and serial dilutions were plated on Luria agar with appropriate antibiotics. Purified EatA protein was added, where indicated, at a final concentration of 50 μg/ml.
To evaluate the impact of EatA on migration through mucin to the epithelial surface, the upper chamber of monolayer cultures of small intestinal enteroids was inoculated by with ∼105 CFU of wild-type or mutant bacteria. Plates were then incubated at 37°C in 5% CO2. After 2 h, medium was gently aspirated and washed 3 times with prewarmed medium to remove unbound bacteria, and cells were then fixed with 4% paraformaldehyde for 30 min at room temperature. The fixed samples were then blocked with PBS containing 1% bovine serum albumin (BSA) for 30 min and immunostained with rabbit anti-O78 antibody followed by cross-absorbed goat anti-rabbit Alexa Fluor 594-conjugated IgG (H+L) (A-11072; ThermoFisher). Membranes with fluorescently labeled samples were excised and mounted onto glass slides with ProLong Gold (P36930; ThermoFisher Scientific) and imaged with a Zeiss Axio Imager M2 plus wide-field fluorescence microscope, capturing 10 random fields per membrane to enumerate bacteria/field.
To evaluate the impact of EatA on kinetics of ETEC migration through mucin, equal numbers of GFP-expressing eatA mutant (jf4737) or wild-type H10407 (jf2451) were loaded into adjacent wells of μ-channel slides (ibidi, v. 0.4) containing MUC2+ concentrated supernatants from LS174T cells. Slides were maintained at 37°C, 5% CO2, and 80% humidity in a stage-top environmental chamber (ibidi). Confocal images were collected at 3-min intervals at distances of 1.8, 4.5, 8.8, and 13.7 mm from the inoculation well, and mean fluorescence intensity was recorded over time to assess migration of GFP-expressing bacteria.
Mucin integrity assays.
To examine the impact of EatA and EatA mutants on mucin integrity, we used a microscale rolling-ball viscometer apparatus as described by Tang (45). Briefly, purified mucin treated with protease or controls was loaded into a 100-mm glass capillary tube (Smurray G119/02) sealed at one end with modeling clay (Plastalina; Van Aken); 0.5-mm steel balls were introduced into the open end of the tube and maintained in place with a magnet. Tubes were maintained on a 20° incline, and the time required for the ball to traverse a distance of 80 mm following release of the magnet was recorded, averaging a minimum of 4 technical replicates/experiment.
Immunoblotting.
MUC2 immunoblotting was performed as described previously (21). Briefly, after EatA treatment, samples were separated by discontinuous 4% Tris-glycine SDS-PAGE, the gel was reduced by agitation for 10 min in a 10 mM DTT solution in Tris-glycine-isopropanol blotting buffer to improve transfer of the high-molecular-mass MUC2, and then samples were blotted onto nitrocellulose, blocked with 5% (wt/vol) milk in PBST, and probed with anti-MUC2 rabbit polyclonal (H-300; sc-15334; Santa Cruz) (1:2,000), followed by goat anti-rabbit horseradish peroxidase (HRP)-conjugated sc-2030 (Santa Cruz) (1:5,000). Bound antibody was visualized by chemiluminescence using Clarity ECL Western blot substrate (Bio-Rad).
MUC2 dot blotting.
MUC2 dot blots were performed with supernatants from the apical compartment of small intestine enteroid monolayers grown as described above. Briefly, cells were grown in Transwells to confluence and differentiated for 2 days, and then duplicate wells were treated overnight with 100 ng/ml of native LT, mutant LT (mLT; E112K), or cholera toxin (CT). Supernatants were collected and spun down at 10,000 × g for 1 min to remove cellular debris. Serial dilutions (1:2 to 1:64) of the corresponding supernatants were performed in PBS. Serial dilutions (1:2 to 1:128) of concentrated LS174T supernatant containing MUC2 (described above) were used as positive controls. Two microliters of each dilution was spotted onto a nitrocellulose membrane, allowed to dry, blocked with 5% (wt/vol) milk in PBST, and probed with anti-MUC2 (clone F2; sc-515032; Santa Cruz) (1:500) followed by horse anti-mouse HRP conjugate (7076; Cell Signaling) (1:1,000). Bound antibody was visualized by chemiluminescence using Clarity ECL Western blot substrate (Bio-Rad).
Glycan array screening.
Glycan arrays were used as previously described (44) to examine the potential interaction of the EatA passenger domain with carbohydrates. Briefly, these glycan arrays that contained 737 separate features (including control spots) were fabricated, validated, and analyzed as reported earlier (46–49). After blocking overnight at 4°C, using 3% (wt/vol) BSA in PBS (200 μl/well), slides were washed 6 times with PBST (PBS with 0.05% Tween 20, 200 μl/well). Biotinylated rEatA-6×His, or biotinylated rEatAp-DUF::His8, was diluted to final concentrations of 5 and 50 μg/ml in lectin buffer containing 20 mM Tris, 150 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, pH 7.4, with 3% BSA and 1% human serum albumin (Millipore Sigma). In one pair of assays, 100 μl of each sample was incubated on the array for 2 h (37°C, 100 rpm). The arrays were washed and incubated (1 h at 37°C; 100 rpm) with Cy3-labeled streptavidin (S6402; Millipore Sigma) at a final concentration of 2 μg/ml in lectin buffer (1 h at 37°C; 100 rpm). In a second pair of experiments, each sample was precomplexed with streptavidin (10:1 protein-streptavidin), followed by incubation on the array for 2 h (37°C, 100 rpm). Slides were then washed 7 times in PBST (200 μl/well), immersed in wash buffer (5 min at room temperature), and centrifuged (5 min at 200 × g). Slide scanning was performed on an InnoScan 1100AL fluorescence scanner (Innopsys; Chicago, IL) at 5-μm resolution (excitation at 532 nm and emission at 575 nm) and data analyzed using GenePix Pro 7.0 (46). A complete list of array components is available in Data Set S1 in the supplemental material.
Molecular cloning.
To produced properly folded polyhistidine-tagged EatA passenger domains, the eatA gene was first amplified in two fragments using primer pairs jf110414.1/jf02215.5 and jf022515.6/jf022515.7 (Table S3), yielding amplicons NcoI-eatA5-2588His-XhoI and XhoI-eatA259-4095-HindIII, respectively. These fragments were sequentially introduced into the corresponding restriction sites on pBAD/myc-HisB (Table S2) to produce pTV001, which encodes EatA bearing a polyhistidine tag at a permissive site between amino acids 86 and 87. Similarly, a plasmid was constructed to replace the region encoding the domain of unknown function corresponding to E541-S616 of EatA with an 8× polyhistidine tag. Briefly, primer sets jf110414.1/jf061615.3 and jf061615.2/jf022515.7 were used to generate amplicons NcoI-eatA5-16209His-XhoI and XhoI-eatA1849-4095-HindIII, respectively. These fragments were then sequentially cloned into the corresponding sites on pBAD/myc-HisB, yielding pTW5126.
To generate the eatA complementation plasmid, pTW5016, full-length eatA DNA sequence was amplified from H10407 with primer set jf081718.1 and jf081718.2. The PCR product was ligated to a low-copy-number plasmid vector pWSK29 (BamHI/SalI) by Gibson assembly (New England Biolabs). Similarly, pTW5128 was constructed by amplifying the mutated eatA sequence from pTW5126 with primers jf071519.1 and jf071519.2. The amplicon was then ligated into PstI-digested pTW5016 by Gibson assembly.
The region corresponding to K535-P618 of EatA was amplified using primers jf070220.1/0.2 and cloned by Gibson assembly to replace the mClover3 sequence of pSpyTag003-mClover3, which was amplified using primers jf070220.3/0.4. The DNA sequence of the resulting plasmid pJMF0820 confirmed placement of the region encoding the EatA subdomain in-frame with 5′ SpyTag and 3′ 6×His tags.
Plasmids pTV001 and pTW5126 are deposited with Addgene with accession numbers 130265 and 129732, respectively.
Recombinant protein expression and antibody purification.
To prepare untagged recombinant native (rEatAp) and inactivated (rEatAp-H134R) proteins, passenger domains proteins were expressed from strains jf1960 and jf1975 (Table S1), and concentrated culture supernatants were then purified by anion exchange followed by size exclusion chromatography, as previously described (21). The mutant passenger domain in which the domain of unknown function was replaced by a polyhistidine tag (rEatAp-DUF::His8) was expressed from jf5132. Concentrated culture supernatant was then exchanged into PBS and the secreted recombinant protein purified using immobilized metal affinity chromatography, as described previously (50).
Production of SpyCatcher-EatAK535-P618 nanoparticles.
BL21-CodonPlus (DE3)-RIPL transformed with pJMF0820 was grown overnight in Luria-Bertani (LB) medium containing 50 μg/ml kanamycin at 37°C. The following morning, saturated cultures were diluted 1:100 in 1 liter of fresh LB medium supplemented with kanamycin and 0.8% glucose (wt/vol) and grown at 37°C, 200 rpm, to an A600 of ∼0.5. Cultures were then induced with isopropyl β-d-1-thiogalactopyranoside (IPTG; 0.5 mM) and incubated at 30°C for an additional 4.5 h. Following centrifugation, bacterial pellets were frozen at −80°C. Each of 4 pellets from 250 ml of culture was resuspended in 30 ml of immobilized metal affinity chromatography (IMAC) lysis buffer containing 75 mM sodium phosphate, pH 7.5, 500 mM NaCl, 1 mM imidazole, cOmplete mini EDTA-free protease cocktail (Roche), 1 mg lysozyme, 1 mg DNase, and 0.5% Triton X-100. Following sonication and centrifugation at 20,000 × g for 20 min, polyhistidine-tagged recombinant proteins were purified from the clarified lysates by IMAC as previously described (35), yielding SpyTagged-EatAK535-P618.
His6-SpyCatcher-mi3 was produced as previously described (34). Briefly, after transformation of BL21-CodonPlus (DE30)-RIPL cells to kanamycin resistance, transformants were grown overnight in Luria-Bertani broth containing kanamycin (50 μg/ml), chloramphenicol (15 μg/ml), and streptomycin (10 μg/ml), diluted 1:100 into 4× 500-ml flasks with fresh medium, allowed to grow at 37°C to an OD600 of ∼0.7, and then induced with 0.5 mM IPTG overnight at room temperature at 250 rpm. Bacteria were harvested by centrifugation and lysed in 30 ml of IMAC lysis buffer. HisSpyCatcher-mi3 was then purified using affinity precipitation by the addition of nickel chloride to clarified lysates at a final concentration of 200 μM and incubation at room temperature for 10 min. Precipitated protein was collected by centrifugation at 11,000 × g for 10 min and the resulting pure protein pellet redissolved in 10 ml of 50 mM Tris, pH 9, 10 mM EDTA. Polyclonal IgG from the sera of mice previously immunized with rEatAp (21), or normal mouse controls, was purified by protein G affinity chromatography.
Antibody preparation.
Antibodies were purified from the sera of 20 mice that were vaccinated intranasally three times 2 weeks apart with 20 μg of recombinant EatA passenger domain (rEatAp) mixed with 1 μg of heat-labile toxin, as described previously (21). Two weeks after the final dose, mice were sacrificed and serum collected. The sera were screened for αEatAp IgG antibodies by enzyme-linked immunosorbent assay (ELISA), and 10 sera with the highest αEatAp titers were pooled. To isolate antibodies specific to the EatA domain of unknown function from K535-P618, an affinity column was prepared by coupling 3.6 mg of the SpyTag-EatAK535-P618-His protein to AminoLink plus resin (Thermo) in 100 mM sodium citrate, 50 mM sodium carbonate, pH 10, at room temperature in a 10-ml glass column for 4 h. The resin was then washed with 5 ml of PBS, and 5 ml 50 mM sodium cyanoborohydride in PBS was added and allowed to react overnight at 4°C. The resin was then washed with 4 ml of quenching buffer (1 M Tris, pH 7.4); 2 ml of quenching buffer containing 50 mM sodium cyanoborohydride was added and the suspension mixed at room temperature for 30 min. The resin was then washed with 10 ml PBS prior to antibody capture. Pooled αEatAp sera were diluted 1:1 with PBS and mixed with the resin for 1 h at room temperature. The resin was washed three times with 5 ml of PBS, and bound antibodies were then eluted in 10 ml 100 mM glycine, pH 2.7; 1-ml fractions of eluate were collected in tubes containing 50 μl of neutralization buffer (1 M Tris, pH 9). Fractions containing αDUF antibodies and examined by ELISA were then pooled and dialyzed against PBS.
Toxin delivery assays.
Enteroid-derived monolayer of polarized cells was obtained in a 24-well cell culture plate using the method described above. Cells cultures were treated with phosphodiesterase (PDE) inhibitors vardenafil (Y0001647; Millipore Sigma), cilostazol (PHR1503; Millipore Sigma), and rolipram (R6520; Millipore Sigma) 1 h prior to infections at a final concentration of 25 μM. The cells were then inoculated by dispensing 100 μl containing ∼105 CFU to individual wells. The samples were incubated at 37°C in a 5% CO2 incubator for 2 h, after which the medium was replaced with fresh, prewarmed medium containing PDE inhibitors and incubated for 1 h. Cellular cyclic AMP (cAMP; Arbor Assays) levels were used as a readout for the efficiency of LT delivery as previously described (51).
Oligopeptide cleavage.
p-Nitroanilide substrate cleavage was carried out as previously described (18). Briefly, 1 mM N-succinyl-Ala-Ala-Pro-Leu-p-nitroanilide (S8511; Sigma) dissolved in 100 mM morpholinepropanesulfonic acid, pH 7.3, 200 mM NaCl was digested at 37°C with 15 μg of enzyme in a final volume of 300 μl. The reaction was monitored at 405 nm and initial rates expressed as measured optical density (mOD)/min.
Molecular modeling.
The structure of the EatA passenger domain (GenBank accession no. AAO17297.1, amino acids 57 to 1062) was aligned with the corresponding region of SepA (33) using the Multialign Viewer extension (52) within UCSF Chimera (53) v1.13.1, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. Theoretical homology models of the EatA passenger were generated against the SepA template Protein Data Bank entry 5J44 using the Chimera interface to Modeller (54).
Ethics statement.
Studies included here involving human tissues were approved by the Institutional Review Board at Washington University in Saint Louis School of Medicine. Deidentified human small intestinal enteroids and fresh explants of human intestine were obtained from the DDRCC under approved protocols 201406083 and 201804112, respectively. Mouse studies were conducted under protocol 20-0438, approved by the Institutional Animal Care and Use Committee at Washington University in Saint Louis School of Medicine.
ACKNOWLEDGMENTS
J.M.F. was supported by funding from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) R01 AI126887, R01 AI089894, and U01 AI095473 and the Department of Veterans Affairs (5I01BX001469-05). Research conducted by A.S. was also supported by National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number T32AI007172. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Veterans Affairs. B.A. and M.P. were supported in part by the Institute for Public Health Summer Scholars Program at Washington University, in Saint Louis. M.J.M. was supported by funding from NIH/NIAID R01 AI077600. Intestinal biopsy specimens were obtained through the Biobank Core of the Digestive Disease Research Core Center (DDRCC) supported by NIH Washington University DDRCC grant no. NIDDK P30 DK052574, and two-photon imaging experiments were conducted at Washington University School of Medicine, In Vivo Imaging Core (IVIC). S.V.D.P. and G.C.H. were supported by the Swedish Research Council (Vetenskapsrådet 2017-00958, 2020-02536).
Footnotes
Supplemental material is available online only.
Contributor Information
James M. Fleckenstein, Email: jflecken@wustl.edu.
Manuela Raffatellu, University of California San Diego School of Medicine.
REFERENCES
- 1.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Fleckenstein JM, Kuhlmann FM. 2019. Enterotoxigenic Escherichia coli infections. Curr Infect Dis Rep 21:9. 10.1007/s11908-019-0665-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sack RB. 2011. The discovery of cholera-like enterotoxins produced by Escherichia coli causing secretory diarrhoea in humans. Indian J Med Res 133:171–180. [PMC free article] [PubMed] [Google Scholar]
- 4.Sack RB, Gorbach SL, Banwell JG, Jacobs B, Chatterjee BD, Mitra RC. 1971. Enterotoxigenic Escherichia coli isolated from patients with severe cholera-like disease. J Infect Dis 123:378–385. 10.1093/infdis/123.4.378. [DOI] [PubMed] [Google Scholar]
- 5.Vicente AC, Teixeira LF, Iniguez-Rojas L, Luna MG, Silva L, Andrade JR, Guth BE. 2005. Outbreaks of cholera-like diarrhoea caused by enterotoxigenic Escherichia coli in the Brazilian Amazon Rainforest. Trans R Soc Trop Med Hyg 99:669–674. 10.1016/j.trstmh.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Finkelstein RA, Vasil ML, Jones JR, Anderson RA, Barnard T. 1976. Clinical cholera caused by enterotoxigenic Escherichia coli. J Clin Microbiol 3:382–384. 10.1128/jcm.3.3.382-384.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khalil I, Troeger CE, Blacker BF, Reiner RC, Jr.. 2019. Capturing the true burden of Shigella and ETEC: the way forward. Vaccine 37:4784–4786. 10.1016/j.vaccine.2019.01.031. [DOI] [PubMed] [Google Scholar]
- 8.George CM, Burrowes V, Perin J, Oldja L, Biswas S, Sack D, Ahmed S, Haque R, Bhuiyan NA, Parvin T, Bhuyian SI, Akter M, Li S, Natarajan G, Shahnaij M, Faruque AG, Stine OC. 2018. Enteric infections in young children are associated with environmental enteropathy and impaired growth. Trop Med Int Health 23:26–33. 10.1111/tmi.13002. [DOI] [PubMed] [Google Scholar]
- 9.Black RE, Brown KH, Becker S. 1984. Effects of diarrhea associated with specific enteropathogens on the growth of children in rural Bangladesh. Pediatrics 73:799–805. [PubMed] [Google Scholar]
- 10.Lee G, Paredes Olortegui M, Penataro Yori P, Black RE, Caulfield L, Banda Chavez C, Hall E, Pan WK, Meza R, Kosek M. 2014. Effects of Shigella-, Campylobacter- and ETEC-associated diarrhea on childhood growth. Pediatr Infect Dis J 33:1004–1009. 10.1097/INF.0000000000000351. [DOI] [PubMed] [Google Scholar]
- 11.Anderson JDT, Bagamian KH, Muhib F, Amaya MP, Laytner LA, Wierzba T, Rheingans R. 2019. Burden of enterotoxigenic Escherichia coli and shigella non-fatal diarrhoeal infections in 79 low-income and lower middle-income countries: a modelling analysis. Lancet Glob Health 7:e321–e330. 10.1016/S2214-109X(18)30483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qadri F, Saha A, Ahmed T, Al Tarique A, Begum YA, Svennerholm AM. 2007. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect Immun 75:3961–3968. 10.1128/IAI.00459-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platts-Mills JA, Taniuchi M, Uddin MJ, Sobuz SU, Mahfuz M, Gaffar SA, Mondal D, Hossain MI, Islam MM, Ahmed AS, Petri WA, Haque R, Houpt ER, Ahmed T. 2017. Association between enteropathogens and malnutrition in children aged 6–23 mo in Bangladesh: a case-control study. Am J Clin Nutr 105:1132–1138. 10.3945/ajcn.116.138800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MAL-ED Network Investigators. 2018. Early childhood cognitive development is affected by interactions among illness, diet, enteropathogens and the home environment: findings from the MAL-ED birth cohort study. BMJ Glob Health 3:e000752. 10.1136/bmjgh-2018-000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guerrant RL, DeBoer MD, Moore SR, Scharf RJ, Lima AA. 2013. The impoverished gut–a triple burden of diarrhoea, stunting and chronic disease. Nat Rev Gastroenterol Hepatol 10:220–229. 10.1038/nrgastro.2012.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Q, Qadri F, Kansal R, Rasko DA, Sheikh A, Fleckenstein JM. 2015. Conservation and immunogenicity of novel antigens in diverse isolates of enterotoxigenic Escherichia coli. PLoS Negl Trop Dis 9:e0003446. 10.1371/journal.pntd.0003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhlmann FM, Martin J, Hazen TH, Vickers TJ, Pashos M, Okhuysen PC, Gomez-Duarte OG, Cebelinski E, Boxrud D, Del Canto F, Vidal R, Qadri F, Mitreva M, Rasko DA, Fleckenstein JM. 2019. Conservation and global distribution of non-canonical antigens in Enterotoxigenic Escherichia coli. PLoS Negl Trop Dis 13:e0007825. 10.1371/journal.pntd.0007825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel SK, Dotson J, Allen KP, Fleckenstein JM. 2004. Identification and molecular characterization of EatA, an autotransporter protein of enterotoxigenic Escherichia coli. Infect Immun 72:1786–1794. 10.1128/IAI.72.3.1786-1794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruiz-Perez F, Wahid R, Faherty CS, Kolappaswamy K, Rodriguez L, Santiago A, Murphy E, Cross A, Sztein MB, Nataro JP. 2011. Serine protease autotransporters from Shigella flexneri and pathogenic Escherichia coli target a broad range of leukocyte glycoproteins. Proc Natl Acad Sci USA 108:12881–12886. 10.1073/pnas.1101006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjelloun-Touimi Z, Sansonetti PJ, Parsot C. 1995. SepA, the major extracellular protein of Shigella flexneri: autonomous secretion and involvement in tissue invasion. Mol Microbiol 17:123–135. 10.1111/j.1365-2958.1995.mmi_17010123.x. [DOI] [PubMed] [Google Scholar]
- 21.Kumar P, Luo Q, Vickers TJ, Sheikh A, Lewis WG, Fleckenstein JM. 2014. EatA, an immunogenic protective antigen of enterotoxigenic Escherichia coli, degrades intestinal mucin. Infect Immun 82:500–508. 10.1128/IAI.01078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson ME, Sjovall H, Hansson GC. 2013. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol 10:352–361. 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asker N, Axelsson MA, Olofsson SO, Hansson GC. 1998. Dimerization of the human MUC2 mucin in the endoplasmic reticulum is followed by a N-glycosylation-dependent transfer of the mono- and dimers to the Golgi apparatus. J Biol Chem 273:18857–18863. 10.1074/jbc.273.30.18857. [DOI] [PubMed] [Google Scholar]
- 24.Godl K, Johansson ME, Lidell ME, Morgelin M, Karlsson H, Olson FJ, Gum JR, Jr, Kim YS, Hansson GC. 2002. The N terminus of the MUC2 mucin forms trimers that are held together within a trypsin-resistant core fragment. J Biol Chem 277:47248–47256. 10.1074/jbc.M208483200. [DOI] [PubMed] [Google Scholar]
- 25.Javitt G, Khmelnitsky L, Albert L, Bigman LS, Elad N, Morgenstern D, Ilani T, Levy Y, Diskin R, Fass D. 2020. Assembly mechanism of mucin and von Willebrand factor polymers. Cell 183:717–729. 10.1016/j.cell.2020.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. 2008. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105:15064–15069. 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhlmann FM, Laine RO, Afrin S, Nakajima R, Akhtar M, Vickers T, Parker K, Nizam NN, Grigura V, Goss CW, Felgner PL, Rasko DA, Qadri F, Fleckenstein JM. 2021. Contribution of noncanonical antigens to virulence and adaptive immunity in human infection with enterotoxigenic E. coli. Infect Immun 10.1128/IAI.00041-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ermund A, Schutte A, Johansson ME, Gustafsson JK, Hansson GC. 2013. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer's patches. Am J Physiol Gastrointest Liver Physiol 305:G341–G347. 10.1152/ajpgi.00046.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorsey FC, Fischer JF, Fleckenstein JM. 2006. Directed delivery of heat-labile enterotoxin by enterotoxigenic Escherichia coli. Cell Microbiol 8:1516–1527. 10.1111/j.1462-5822.2006.00736.x. [DOI] [PubMed] [Google Scholar]
- 30.Forstner JF, Roomi NW, Fahim RE, Forstner GG. 1981. Cholera toxin stimulates secretion of immunoreactive intestinal mucin. Am J Physiol 240:G10–G16. 10.1152/ajpgi.1981.240.1.G10. [DOI] [PubMed] [Google Scholar]
- 31.Roomi N, Laburthe M, Fleming N, Crowther R, Forstner J. 1984. Cholera-induced mucin secretion from rat intestine: lack of effect of cAMP, cycloheximide, VIP, and colchicine. Am J Physiol 247:G140–G148. 10.1152/ajpgi.1984.247.2.G140. [DOI] [PubMed] [Google Scholar]
- 32.Epple HJ, Kreusel KM, Hanski C, Schulzke JD, Riecken EO, Fromm M. 1997. Differential stimulation of intestinal mucin secretion by cholera toxin and carbachol. Pflugers Arch 433:638–647. 10.1007/s004240050325. [DOI] [PubMed] [Google Scholar]
- 33.Maldonado-Contreras A, Birtley JR, Boll E, Zhao Y, Mumy KL, Toscano J, Ayehunie S, Reinecker HC, Stern LJ, McCormick BA. 2017. Shigella depends on SepA to destabilize the intestinal epithelial integrity via cofilin activation. Gut Microbes 8:544–560. 10.1080/19490976.2017.1339006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruun TUJ, Andersson AC, Draper SJ, Howarth M. 2018. Engineering a rugged nanoscaffold to enhance plug-and-display vaccination. ACS Nano 12:8855–8866. 10.1021/acsnano.8b02805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeble AH, Turkki P, Stokes S, Khairil Anuar INA, Rahikainen R, Hytonen VP, Howarth M. 2019. Approaching infinite affinity through engineering of peptide-protein interaction. Proc Natl Acad Sci USA 116:26523–26533. 10.1073/pnas.1909653116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanata CF, Black RE. 2018. Estimating the true burden of an enteric pathogen: enterotoxigenic Escherichia coli and Shigella spp. Lancet Infect Dis 18:1165–1166. 10.1016/S1473-3099(18)30546-2. [DOI] [PubMed] [Google Scholar]
- 37.Porter CK, Riddle MS, Alcala AN, Sack DA, Harro C, Chakraborty S, Gutierrez RL, Savarino SJ, Darsley M, McKenzie R, DeNearing B, Steinsland H, Tribble DR, Bourgeois AL. 2016. An evidenced-based scale of disease severity following human challenge with enteroxigenic Escherichia coli. PLoS One 11:e0149358. 10.1371/journal.pone.0149358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harro C, Chakraborty S, Feller A, DeNearing B, Cage A, Ram M, Lundgren A, Svennerholm AM, Bourgeois AL, Walker RI, Sack DA. 2011. Refinement of a human challenge model for evaluation of enterotoxigenic Escherichia coli vaccines. Clin Vaccine Immunol 18:1719–1727. 10.1128/CVI.05194-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coster TS, Wolf MK, Hall ER, Cassels FJ, Taylor DN, Liu CT, Trespalacios FC, DeLorimier A, Angleberger DR, McQueen CE. 2007. Immune response, ciprofloxacin activity, and gender differences after human experimental challenge by two strains of enterotoxigenic Escherichia coli. Infect Immun 75:252–259. 10.1128/IAI.01131-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boisen N, Scheutz F, Rasko DA, Redman JC, Persson S, Simon J, Kotloff KL, Levine MM, Sow S, Tamboura B, Toure A, Malle D, Panchalingam S, Krogfelt KA, Nataro JP. 2012. Genomic characterization of enteroaggregative Escherichia coli from children in Mali. J Infect Dis 205:431–444. 10.1093/infdis/jir757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boisen N, Osterlund MT, Joensen KG, Santiago AE, Mandomando I, Cravioto A, Chattaway MA, Gonyar LA, Overballe-Petersen S, Stine OC, Rasko DA, Scheutz F, Nataro JP. 2020. Redefining enteroaggregative Escherichia coli (EAEC): genomic characterization of epidemiological EAEC strains. PLoS Negl Trop Dis 14:e0008613. 10.1371/journal.pntd.0008613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson M, Sansonetti PJ, Marteyn BS. 2016. Shigella diversity and changing landscape: insights for the twenty-first century. Front Cell Infect Microbiol 6:45. 10.3389/fcimb.2016.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, Ciorba MA, Stappenbeck TS. 2015. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64:911–920. 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar P, Kuhlmann FM, Chakraborty S, Bourgeois AL, Foulke-Abel J, Tumala B, Vickers TJ, Sack DA, DeNearing B, Harro CD, Wright WS, Gildersleeve JC, Ciorba MA, Santhanam S, Porter CK, Gutierrez RL, Prouty MG, Riddle MS, Polino A, Sheikh A, Donowitz M, Fleckenstein JM. 2018. Enterotoxigenic Escherichia coli blood group A interactions intensify diarrheal severity. J Clin Invest 128:3298–3311. 10.1172/JCI97659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang JX. 2016. Measurements of fluid viscosity using a miniature ball drop device. Rev Sci Instrum 87:e054301. 10.1063/1.4948314. [DOI] [PubMed] [Google Scholar]
- 46.Campbell CT, Zhang Y, Gildersleeve JC. 2010. Construction and use of glycan microarrays. Curr Protoc Chem Biol 2:37–53. 10.1002/9780470559277.ch090228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oyelaran O, McShane LM, Dodd L, Gildersleeve JC. 2009. Profiling human serum antibodies with a carbohydrate antigen microarray. J Proteome Res 8:4301–4310. 10.1021/pr900515y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manimala JC, Roach TA, Li Z, Gildersleeve JC. 2007. High-throughput carbohydrate microarray profiling of 27 antibodies demonstrates widespread specificity problems. Glycobiology 17:17C–23C. 10.1093/glycob/cwm047. [DOI] [PubMed] [Google Scholar]
- 49.Manimala JC, Roach TA, Li Z, Gildersleeve JC. 2006. High-throughput carbohydrate microarray analysis of 24 lectins. Angew Chem Int Ed Engl 45:3607–3610. 10.1002/anie.200600591. [DOI] [PubMed] [Google Scholar]
- 50.Fleckenstein JM, Roy K. 2009. Purification of recombinant high molecular weight two-partner secretion proteins from Escherichia coli. Nat Protoc 4:1083–1092. 10.1038/nprot.2009.87. [DOI] [PubMed] [Google Scholar]
- 51.Sheikh A, Tumala B, Vickers TJ, Alvarado D, Ciorba MA, Bhuiyan TR, Qadri F, Singer BB, Fleckenstein JM. 2020. CEACAMs serve as toxin-stimulated receptors for enterotoxigenic Escherichia coli. Proc Natl Acad Sci USA 117:29055–29062. 10.1073/pnas.2012480117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. 2006. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 7:339. 10.1186/1471-2105-7-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 54.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download iai.00572-21-s0001.pdf, PDF file, 7.7 MB (7.7MB, pdf)
Supplemental material. Download iai.00572-21-s0002.xlsx, XLSX file, 0.07 MB (69.9KB, xlsx)
Supplemental material. Download iai.00572-21-s0003.xlsx, XLSX file, 0.09 MB (91.2KB, xlsx)
Supplemental material. Download iai.00572-21-s0004.mov, MOV file, 9.0 MB (9MB, mov)
Supplemental material. Download iai.00572-21-s0005.mov, MOV file, 6.4 MB (6.4MB, mov)