

Abstract
Usage of cisplatin, a highly potent chemotherapeutic, is limited by its severe nephrotoxicity. Arachidonic acid (ARA)-derived epoxyeicosatrienoic acids (EETs) and soluble epoxide hydrolase (sEH) inhibitors were shown to ameliorate this dose-limiting side effect, but both approaches have some pharmacological limitations. Analogues of EETs are an alternative avenue with unique benefits, but the current series of analogues face concerns regarding their structure and mimetic functionality. Hence, in this study, regioisomeric mixtures of four new ARA alkyl-ethers were synthesized, characterized, and assessed as EET analogues against the concentration- and time-dependent toxicities of cisplatin in porcine proximal tubular epithelial cells. All four ether groups displayed bioisostere activity, ranging from marginal for methoxy- (1), good for n-propoxy- (4), and excellent for ethoxy- (2) and i-propoxy- (3). Compounds 2 and 3 displayed cytoprotective effects comparable to that of an EET regioisomeric mixture (5) against high, acute cisplatin exposures but were more potent against low to moderate, chronic exposures. Compounds 2 and 3 (and 5) acted through stabilization of the mitochondrial transmembrane potential and attenuation of reactive oxygen species, leading to reduced phosphorylation of mitogen-activated protein kinases p38 and JNK and decreased activation of caspase-9 and −3. This study demonstrates that alkoxy- groups are potent and more metabolically stable bioisostere alternatives to the epoxide within EETs that enable sEH-independent activity. It also illustrates the potential of ether-based mimics of EETs and other epoxy fatty acids as promising nephroprotective agents to tackle the clinically relevant side effect of cisplatin, without compromising its antineoplastic function.
Graphical Abstract

INTRODUCTION
cis-Diamminedichloroplatinum (II), commonly known as cisplatin (CDDP), is a potent inorganic antineoplastic agent that is widely used for treatment of several types of cancers.1 However, it has many side effects, the most severe being dose-limiting nephrotoxicity.1 Due to selective basolateral uptake, cisplatin is particularly damaging to the proximal tubules.2 Mitochondrial disruptions,3 generation of reactive oxygen species (ROS),4 activation of mitogen-activated protein kinases (MAPKs),5,6 and recruitment of caspases7 are major implicated molecular modes of toxicity that lead to apoptosis of epithelial tubular cells. Currently, the lack of safe and efficacious therapeutic agents and impractical treatment alternatives make preserving kidney health during CDDP-based chemotherapy especially challenging. Since there is justified pressure on oncologists to administer higher levels of cisplatin to overcome drug resistance in tumors, therapeutics are urgently needed to mitigate its renal toxicity.
Cytochromes P450 (CYPs)-catalyzed epoxidation of polyunsaturated fatty acids (PUFAs) generates mono-epoxide lipid mediators known as epoxy fatty acids (EpFAs). ω−6 Arachidonic acid (ARA, 20:4)-derived epoxyeicosatrienoic acids (EETs) are the most widely studied group of EpFAs.8–10 EETs exhibit primarily tissue-healing and organ-protective activities but are hydrolyzed to less active vicinal diols by the soluble epoxide hydrolase (sEH). sEH inhibitors (sEHIs) stabilize endogenous levels of EETs, enhancing their bioavailability and facilitating their functions. EETs in vitro and the inhibition or knockout of sEH in vivo have been shown to attenuate cisplatin nephrotoxicity.11–13 Thus, this pathway provides a novel and promising therapeutic target to manage the major side effects of cisplatin.
While administration of EETs and sEHIs are the two most common therapeutic approaches, both have certain disadvantages. sEH-mediated degradation of EETs occurs rapidly and hence EETs are poorly bioavailable in vivo following most routes of administration. The activity of sEHIs is dependent on endogenous levels of EETs and other EpFAs. Therefore, under certain conditions of renal or cardiovascular dysfunction where release of membrane fatty acids or CYP metabolism is impaired, EET biosynthesis could be compromised, limiting efficacy of sEHIs. EET analogues (or mimics) are an alternative approach.14,15 They are designed to be more biologically stable than EETs and act independently of endogenous EET generation. Several potential EET analogues are being investigated and the most promising compounds have demonstrated therapeutic effects in multiple disease models. Some EET analogues have also been shown to ameliorate cisplatin nephrotoxicity in vivo.16,17
However, many current EET analogues utilize classic sEHI pharmacophores (i.e., ureas and amides) as epoxide bioisosteres. For molecules where demonstrable mimetic function, verified in systems such as sEH-deficient mesenteric vessels,18 is absent, there is ensuing debate and uncertainty about their functionality as analogues (or dual-acting agents) as opposed to sEHIs. Furthermore, modified chain lengths and altered unsaturation sites in the fatty acid moieties of some of these analogues have raised questions about off-target effects and a diminished therapeutic index. Since an epoxide is a cyclic ether, linear or branched ethers are plausible alternative bioisosteres. Alkoxy groups would allow direct replacement of the epoxide with more metabolically stable functional groups and permit retention of the core molecular backbone. Incidentally, alkoxy groups were employed as epoxide bioisosteres in compounds mimicking the juvenile hormones (JHs) of insects, and these analogues have been utilized successfully for decades as potent growth regulators for pest control.19,20
Thus, in this study, alkoxy-containing EET analogues were synthesized and characterized. Concentration- and time-dependent activities of bioisosteres were screened, relative to the epoxide within EETs, against cisplatin toxicity in the porcine LLC-PK1 proximal tubular epithelial cell (PTEC) line, the standard in vitro model for PTECs. Furthermore, based on the molecular pathways of cisplatin kidney toxicity, the mechanism of therapeutic action for the most promising analogue series was investigated, namely stabilization of mitochondrial dysfunction, anti-oxidative stress action, reduced relative MAPK phosphorylation, and anti-caspase activation.
EXPERIMENTAL PROCEDURE
Reagents and Apparatus.
cis-diamminedichloroplatinum (II) of 99.99% purity (trace metal basis) was purchased from Fisher Scientific Company LLC (Pittsburg, PA, USA). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and CM-H2DCFDA indicator were purchased from Life Technologies (Carlsbad, CA, USA). JC-10 dye, FCCP (Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone), and MitoSOX Red indicator were purchased from Fisher Scientific. Primary antibodies against p38, phospho-p38, SAPK/JNK, and phospho-SAPK/JNK were purchased from Cell Signaling Technology (Danvers, MA, USA). Cyano(6-methoxy-naphthalen-2-yl)methyl trans-[(3-phenyloxiran-2-yl)methyl] carbonate (CMNPC), WST-8 (CCK-8) cell proliferation assay kit, 14,15-EE-5(Z)-E (14,15-Epoxyeicosa-5(Z)-enoic Acid), and Ac-DEVD-AMC were purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Ac-LEHD-pNa and Erythromycin (EM) were purchased from Millipore Sigma (St. Louis, MO, USA). trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) was generated in-house.21 Arachidonic acid methyl ester was purchased from Nu-Chek Prep, Inc. (Elysian, MN, USA). The reagents required for synthesis are commercially available and were purchased from one of the following vendors: Fisher Scientific, Millipore Sigma, or VWR International (Radnor, PA, USA). All chemicals purchased from commercial sources were used as received without further purification. Analytical thin layer chromatography (TLC) was performed on Merck TLC silica gel 60 F254 plates (Darmstadt, Germany) and spots were revealed via development with a potassium permanganate stain. Flash chromatography was performed on silica gel (230–400 Mesh, Grade 60) from Fisher Scientific. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a 400 MHz Bruker Avance III HD Nanobay NMR spectrometer. Multiplicity is described by the abbreviations, s = singlet, t = triplet, m = multiplet. High resolution electrospray ionization mass spectrometry (HRESIMS) data were recorded on a Thermo Q-Exactive High-field Orbitrap mass spectrometer equipped with an electrospray ionization (ESI) source operating in positive ion mode. High-performance liquid chromatography-mass spectrometry (HPLC-MS) traces and product ion spectra (Figures S1–S5, Supporting Information) were recorded on an Agilent 1200 SL HPLC, coupled to a SCIEX 4000 Q-TRAP tandem mass spectrometer, equipped with an ESI source, and operating in positive ion mode. For HPLC-MS analyses, compounds 1-5 were injected onto a reverse-phase 2.1 × 150 mm 1.8-μm Zorbax Eclipse C18 RRHT column, held at 50 °C. Mobile phase A was water with 0.1% glacial acetic acid (AA) and mobile phase B was acetonitrile with 0.1% glacial AA. A gradient elution was employed, and the chromatographic run was optimized to be 90 min for separation of peaks. Gradient conditions and MS source parameters are described in the Supporting Information (Tables S1, S2).
Synthesis and characterization of mono-ethers (1-4, regioisomeric mixtures) of arachidonic acid methyl ester.
The general synthesis is illustrated with the representative ethoxyeicosatrienoic methyl esters (2). Arachidonic acid methyl ester (1.10 g, 3.45 mmol, 1.0 equiv.) was dissolved in ethanol (5 mL) and quickly added to a stirring solution of Hg(OAc)2 (1.65 g, 5.18 mmol, 1.5 equiv.) in ethanol (12 mL). The mixture was stirred under nitrogen or argon overnight at room temperature. The mixture was then cooled to 0 °C (via ice bath), and a solution of NaBH4 (0.26 g, 6.90 mmol, 2.0 equiv.) in water (10 mL) was slowly added. After stirring for 10 min, 10 drops of acetic acid were added, the stirring was stopped, and the solution was allowed to settle. The supernatant was collected, and the mercury precipitate was washed with ethanol (2 × 10 mL). The combined supernatant fractions were concentrated in vacuo, reconstituted in hexanes (20 mL) and saturated aq. NaHCO3 (15 mL), after which the organic layer was isolated. The aqueous layer was extracted with hexanes (3 × 10 mL). The combined organic layers were washed once with aq. NaHCO3, dried (MgSO4), and concentrated in vacuo. The crude mixture was reconstituted in hexanes and, by monitoring fractions with TLC, purified using flash chromatography (ethyl acetate-hexanes 4:96 → 8:92) to yield the mono-ethyl-ethers of arachidonic acid methyl ester. Compound structures of 1-4 were consistent with the acquired 1H NMR spectra, including ratios of olefinic protons, ether protons, and other indicator peaks (e.g., methyl ester, terminal methyl) on integration. Observed high resolution m/z of adduct ions concurred with calculated values. Relative retention times were also consistent with the polarity of the corresponding alkoxy- substituent.
Compounds 1-4 are complex mixtures expected to contain eight possible regioisomers as the alkoxy- group can be inserted at one of two positions across each of the four ARA olefins. Furthermore, every regioisomer would possess a chiral center, suggesting sixteen potential stereoisomers. Hence, HPLC-MS traces predictably revealed multiple peaks of the parent m/z that eluted as a group within a narrow time window. Similarly, the number of peaks detected by 13C NMR for 1-4 exceeded the number of carbons present within an individual isomer of a group. Slight differences in chemical shifts for an equivalent carbon in different regioisomers would account for these “duplicate” or “triplicate” peaks since the position of the carbon in question varies depending on specific molecule.
Methoxyeicosatrienoic methyl esters (1, regioisomer mixture).
Yield 9% (112 mg); Colorless oil; 1H NMR (400 MHz, CDCl3) δ 5.46–5.36 (6H, m, vinylic), 3.69 (3H, s, CO2Me), 3.37 (3H, s, OMe) 3.24–3.14 (1H, m), 2.85–2.78 (3H, m), 2.37–2.21 (3H, m), 2.17–2.02 (4H, m), 1.75–1.67 (2H, m), 1.58–1.45 (3H, m), 1.42–1.26 (7H, m), 0.91 (3H, t, J = 7.2 Hz, Me); 13C NMR (100 MHz, CDCl3) δ 174.1, 132.1, 130.5, 130.5, 130.4, 130.4, 130.1, 130.0, 129.9, 129.8, 129.7, 129.5, 129.5, 129.2, 129.2, 129.1, 128.9, 128.9, 128.9, 128.6, 128.5, 128.4, 128.2, 128.2, 128.2, 128.1, 127.9, 127.8, 127.8, 127.6, 127.5, 126.4, 126.0, 125.8, 125.7, 125.4, 125.1, 80.9, 80.6, 80.4, 80.3, 80.2, 80.2, 79.9, 56.6, 56.6, 56.5, 56.4, 51.5, 33.6, 33.6, 33.6, 33.5, 33.5, 33.5, 33.4, 33.3, 32.1, 31.9, 31.6, 31.5, 31.1, 31.1, 31.0, 31.0, 29.5, 29.4, 29.4, 29.3, 29.3, 27.4, 27.2, 27.2, 27.2, 26.6, 26.6, 25.8, 25.8, 25.6, 25.6, 25.6, 24.9, 24.8, 24.8, 23.2, 23.2, 23.1, 23.1, 22.7, 22.6, 22.6, 14.1, 14.1; HRESIMS observed m/z 373.2738 [M + Na]+ (calculated m/z for C22H38NaO3+, 373.2719); HPLC-MS observed multiple peaks of m/z 351 [M + H]+ (C22H39O3+), retention times (tR) 39.2 – 41.5 min
Ethoxyeicosatrienoic methyl esters (2, regioisomer mixture).
Yield 22% (280 mg); Colorless oil; 1H NMR (400 MHz, CDCl3) δ 5.43–5.33 (6H, m, vinylic), 3.66 (3H, s, CO2Me), 3.60–3.39 (2H, m, O-CH2) 3.30–3.20 (1H, m), 2.81–2.77 (3H, m), 2.34–2.21 (3H, m), 2.14–2.00 (4H, m), 1.74–1.66 (2H, m), 1.57–1.43 (3H, m), 1.40–1.26 (7H, m), 1.21–1.18 (3H, m, CH3-CH2-O), 0.89 (3H, t, J = 7.0 Hz, Me); 13C NMR (100 MHz, CDCl3) δ 174.0, 131.9, 130.3, 130.2, 130.1, 129.9, 129.6, 129.5, 129.3, 129.2, 129.1, 128.9, 128.9, 128.8, 128.7, 128.6, 128.4, 128.1, 128.0, 127.9, 127.8, 127.8, 127.6, 125.9, 125.3, 78.7, 78.6, 78.5, 78.4, 64.4, 64.3, 64.2, 64.1, 51.4, 34.1, 34.1, 34.0, 34.0, 33.5, 33.4, 33.4, 32.0, 31.9, 31.8, 31.8, 31.7, 31.5, 31.5, 29.4, 29.3, 29.3, 29.2, 27.4, 27.2, 27.2, 27.1, 26.7, 26.6, 26.5, 25.8, 25.7, 25.6, 25.6, 25.6, 25.5, 25.0, 24.8, 24.7, 23.3, 23.3, 23.2, 22.7, 22.6, 22.5, 15.7, 15.6, 14.1, 14.0; HRESIMS observed m/z 387.2900 [M + Na]+ (calculated m/z for C23H40NaO3+, 387.2875); HPLC-MS observed multiple peaks of m/z 365 [M + H]+ (C23H41O3+), tR 51.2 – 54.1 min
i-Propoxyeicosatrienoic methyl esters (3, regioisomer mixture).
Yield 16% (210 mg); Colorless oil; 1H NMR (400 MHz, CDCl3) δ 5.44–5.33 (6H, m, vinylic), 3.67 (3H, s, CO2Me), 3.64–3.59 (1H, m, O-CH), 3.36–3.27 (1H, m), 2.84–2.77 (3H, m), 2.34–2.21 (3H, m), 2.14–2.03 (4H, m), 1.73–1.67 (2H, m), 1.52–1.42 (3H, m), 1.38–1.26 (7H, m), 1.16–1.12 (6H, m, (CH3)2-CH-O), 0.89 (3H, t, J = 7.4 Hz, Me); 13C NMR (100 MHz, CDCl3) δ 174.0, 130.4, 130.2, 130.2, 129.4, 128.9, 128.9, 128.8, 128.8, 128.8, 128.7, 128.5, 128.3, 128.2, 128.1, 128.0, 127.8, 127.8, 127.7, 127.7, 127.6, 127.5, 125.5, 76.7, 76.4, 76.2, 69.7, 69.7, 69.6, 69.6, 69.4, 51.4, 51.4, 34.7, 34.7, 34.6, 33.5, 33.4, 32.7, 32.7, 32.6, 32.1, 31.6, 31.5, 31.5, 29.4, 29.3, 29.3, 27.4, 27.2, 27.2, 26.6, 26.6, 26.5, 25.8, 25.8, 25.7, 25.6, 25.6, 25.5, 25.2, 24.8, 24.7, 23.4, 23.4, 23.3, 23.2, 23.1, 23.1, 23.0, 22.9, 22.9, 22.7, 22.6, 22.6, 22.5, 14.1, 14.0; HRESIMS observed m/z 401.3049 [M + Na]+ (calculated m/z for C24H42NaO3+, 401.3032); HPLC-MS observed multiple peaks of m/z 379 [M + H]+ (C24H43O3+), tR 61.3 – 65.4 min
n-Propoxyeicosatrienoic methyl esters (4, regioisomer mixture).
Yield 2% (10 mg); Colorless oil; 1H NMR (400 MHz, CDCl3) δ 5.46–5.36 (6H, m, vinylic), 3.69 (3H, s, CO2Me), 3.52–3.22 (3H, m, H2C-O-CH), 2.85–2.78 (3H, m), 2.35–2.22 (3H, m), 2.18–2.01 (4H, m), 1.76–1.69 (2H, m), 1.62–1.48 (4H, m), 1.39–1.27 (8H, m), 0.97–0.86 (6H, m, Me); 13C NMR (100 MHz, CDCl3) δ 174.1, 131.8, 130.4, 130.3, 130.3, 130.2, 129.9, 129.2, 129.1, 128.9, 128.8, 128.7, 128.6, 128.4, 128.2, 128.0, 127.9, 127.8, 127.5, 126.3, 125.7, 125.3, 78.9, 78.8, 78.7, 78.6, 70.9, 70.8, 70.6, 51.4, 34.2, 34.1, 34.0, 34.0, 34.0, 33.9, 33.5, 33.5, 33.4, 32.0, 31.9, 31.8, 31.7, 31.7, 31.7, 31.6, 31.5, 29.7, 29.5, 29.4, 29.3, 29.3, 27.4, 27.3, 27.2, 26.8, 26.6, 26.5, 25.8, 25.8, 25.6, 25.5, 25.1, 24.8, 24.7, 23.5, 23.4, 23.3, 23.3, 23.2, 23.2, 22.7, 22.6, 22.5, 14.1, 14.0, 10.8, 10.7; HRESIMS observed m/z 401.3026 [M + Na]+ (calculated m/z for C24H42NaO3+, 401.3032); HPLC-MS observed multiple peaks of m/z 379 [M + H]+ (C24H43O3+), tR 68.3 – 71.7 min
Synthesis of mono-epoxides (5, regioisomeric mixture) of arachidonic acid methyl ester.
Arachidonic acid methyl ester (1.10 g, 3.45 mmol, 1.0 equiv.) was dissolved in dichloromethane (DCM, 2 mL) and quickly added to a vigorously stirring solution of 70% meta-chloroperoxybenzoic acid (171 mg, 0.69 mmol, 0.20 equiv.) in DCM (30 mL). The reaction was stirred for 90 min at room temperature and quenched with saturated aq. Na2CO3. The mixture was extracted with diethyl ether (4 × 10 mL), dried (MgSO4), and concentrated in vacuo. The crude mixture was reconstituted in hexanes and, by monitoring fractions with TLC, purified using flash chromatography (ethyl acetate-hexanes 5:95 → 10:90) to yield the mono-epoxides of arachidonic acid methyl ester.
Epoxyeicosatrienoic methyl esters (5, regioisomer mixture).
Yield 88% (204 mg); Colorless oil; 1H NMR (400 MHz, CDCl3) δ 5.55–5.38 (6H, m, vinylic), 3.70 (3H, s, CO2Me), 2.99–2.95 (2H, m), 2.87–2.82 (3H, m), 2.46–2.22 (5H, m), 2.16–2.05 (3H, m), 1.78–1.69 (3H, m), 1.38–1.28 (6H, m), 0.91 (3H, t, J = 7.0 Hz, Me); 13C NMR (100 MHz, CDCl3) δ 174.1, 132.9, 131.4, 131.0, 130.6, 130.4, 129.2, 129.0, 128.8, 128.5, 127.8, 125.0, 124.6, 124.4, 124.0, 123.6, 57.2, 56.4, 51.5, 33.4, 31.7, 31.5, 29.2, 27.8, 27.4, 27.2, 26.8, 26.6, 26.3, 26.2, 25.8, 25.6, 24.8, 22.6, 22.5, 14.1, 14.0; HRESIMS observed m/z 357.2429 [M + Na]+ (calculated m/z for C21H34NaO3+, 357.2406); HPLC-MS observed multiple peaks of m/z 335 [M + H]+ (C21H35O3+), tR 22.0 – 24.4 min. Individual yields of 14,15-, 11,12-, 8,9-, and 5,6-EET methyl ester were 68 mg, 54 mg, 48 mg, and 34 mg, respectively, as per the corresponding regioisomeric ratio of 2.0:1.6:1.4:1.0 in the mixture.
The methyl esters of the ARA alkyl-ethers and epoxide were employed as the prodrug for therapeutic applications in cells, due to their chemical and pharmacological advantages over the carboxylic acid form.22 Stock solutions were prepared in ethanol, flushed with nitrogen or argon, and stored at −80 °C for long term use.
Soluble Epoxide Hydrolase Inhibition Assay.
Purified, recombinant human sEH (hsEH) was obtained utilizing a baculovirus expression system and affinity chromatography,23 and the inhibition assay was performed as previously described.24 Briefly, a fluorometric assay that measures hydrolysis of the nonfluorescent sEH substrate CMNPC (5 μM) to the fluorescent 6-methoxynaphthaldehyde, in the presence of different concentrations of compounds 1-5, was employed. Fluorescence was measured at λEx/λEm = 330 nm/465 nm using a Molecular Devices M2 microplate reader, and the respective IC50 values (μM) against the hsEH were derived.
Cell Cultures.
Porcine kidney proximal tubular epithelial cells (LLC-PK1) and human liver hepatocellular carcinoma (HepG2) cells were purchased from American Type Culture Collection (Manassas, VA, USA). LLC-PK1 cells were cultured in M199 medium, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS). HepG2 cells were cultured in Dulbecco’s Modified Eagle Medium, supplemented with 10% FBS and 1% PS. Cultures were maintained in a humidified incubator at 37 °C, under an atmosphere of 5% CO2/95% air.
MTT Cell Viability Assay.
LLC-PK1 or HepG2 cells were seeded in sterile, transparent 96-well microplates at a density of 1 × 104 cells/well and incubated overnight. In general, cells were pre-treated for 1 h, in the presence or absence of 14,15-EE-5(Z)-E (3 μM), with the vehicle (EtOH), ARA methyl ester (0.1–10 μM), alkoxy-ARA methyl ester (0.1–10 μM, regioisomeric mixture) with or without EM (20 μM), or EET methyl ester (0.1–10 μM, regioisomeric mixture) with or without t-AUCB (10 μM). This was followed by exposure to cisplatin (5–20 μM) or culture media for 24–120 h. Following treatment regimens, the cell media was replaced with 100 μL/well of fresh media containing the MTT reagent (0.5 mg/mL) and incubated for 4 h in the cell culture incubator. The MTT solution was aspirated, 150 μL/well of 4 mM HCl in isopropanol was added, and plates were shaken for 15 min to dissolve the formazan crystals. Finally, absorbance was read at λAbs = 570 nm using a Molecular Devices M2 microplate reader.
CCK-8 Cell Proliferation Assay.
LLC-PK1 or HepG2 cells were seeded in sterile, transparent 96-well microplates at a density of 1 × 104 cells/well and incubated overnight. Cells were then pre-treated for 1 h with the vehicle (EtOH), alkoxy-ARA methyl ester (1 μM, regioisomeric mixture), or EET methyl ester (1 μM, regioisomeric mixture), followed by exposure to cisplatin (20 μM) for 24 h. The CCK-8 assay was then conducted as per the manufacturer’s instructions. Briefly, 10 μL of the CCK-8 working solution was added to the 100 uL/well cultures, plates were gently shaken for 1 min, and incubated for 2 h in the cell culture incubator. After 1 min of gentle shaking, absorbance was read at λAbs = 450 nm using a Molecular Devices M2 microplate reader.
JC-10 Mitochondrial Transmembrane Potential Assay.
LLC-PK1 cells were seeded in sterile, clear-bottom black 96-well microplates at a density of 1 × 104 cells/well and incubated overnight. Cells were pre-treated for 1 h with the vehicle (EtOH), alkoxy-ARA methyl ester (1 μM, regioisomeric mixture), or EET methyl ester (1 μM, regioisomeric mixture), followed by exposure to cisplatin (20 μM) or culture media for 6, 24, or 48 h. Cells were then washed once with the wash solution (Hanks’ Balanced Salt Solution with Ca2+, Mg2+, 0.1% Pluronic F-68). 10 μM JC-10 dye (in wash solution) was added (under dark) and cells were incubated for 1 h in the cell culture incubator. After cells were washed twice, 100 μL/well of wash solution was added and fluorescence was immediately read at λ1Ex/λ1Em = 492 nm/575 nm (red aggregates) and λ2Ex/λ2Em = 492 nm/525 nm (green monomers) using a Tecan Infinite Pro microplate reader. The ratio of red to green fluorescence was used as a measure of the mitochondrial transmembrane potential. FCCP (10 μM), a potent uncoupler of oxidative phosphorylation, was utilized as a positive control for the loss of mitochondrial membrane potential.
CM-H2DCFDA General Reactive Oxygen Species Quantitation Assay.
LLC-PK1 cells were seeded in sterile, clear-bottom black 96-well microplates at a density of 1 × 104 cells/well and incubated overnight. Cells were pre-conditioned with 5 μM CM-H2DCFDA for 30 min in the cell culture incubator and washed thrice with phosphate buffered saline (PBS). Cells were then pre-treated for 1 h with the vehicle (EtOH), alkoxy-ARA methyl ester (1 μM, regioisomeric mixture), or EET methyl ester (1 μM, regioisomeric mixture), followed by exposure to cisplatin (20 μM) for 24 h. After cells were washed twice with PBS, 100 μL/well of PBS was added and fluorescence was immediately measured at λEx/λEm = 485 nm/535 nm using a Tecan Infinite Pro microplate reader.
MitoSOX Red Mitochondrial Superoxide Detection Assay.
LLC-PK1 cells were seeded in sterile, clear-bottom black 96-well microplates at a density of 1 × 104 cells/well and incubated overnight. Cells were pre-conditioned with 2.5 μM MitoSOX Red for 10 min in the cell culture incubator and washed thrice with PBS. Cells were then pre-treated for 1 h with the vehicle (EtOH), alkoxy-ARA methyl ester (1 μM, regioisomeric mixture), or EET methyl ester (1 μM, regioisomeric mixture), followed by exposure to cisplatin (20 μM) for 24 h. After cells were washed twice with PBS, 100 μL/well of PBS was added and fluorescence was immediately measured at λEx/λEm = 510 nm/580 nm using a Tecan Infinite Pro microplate reader.
Western Blotting for Mitogen-Activated Protein Kinases.
LLC-PK1 cells were seeded in sterile 6-well plates at a density of 3 × 105 cells/well and incubated overnight. Cells were pre-treated for 1 h with the vehicle (EtOH), alkoxy-ARA methyl ester (1 μM, regioisomeric mixture), or EET methyl ester (1 μM, regioisomeric mixture), followed by exposure to cisplatin (20 μM) for 6 or 24 h. Cells were rinsed thrice with ice-cold PBS and harvested with ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer (containing a freshly added protease/phosphatase inhibitor cocktail, 1:100). Following sonication on ice for 30 min and centrifugation at 13000 rpm and 4 °C for 25 min, the supernatants were collected and protein concentrations in lysates were normalized using a Bicinchoninic Acid (BCA) assay and a bovine serum albumin (BSA) standard. After lysate aliquots were denatured and reduced in loading buffer, 15 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked for 1 h with 5% non-fat milk or BSA in tris buffered saline-0.1% tween 20 (TBS-t) at room temperature and incubated overnight with the primary antibodies (1:1000 in TBS-t containing 2% non-fat milk or BSA) at 4 °C. Following quadruplicate washes with TBS-t, blots were incubated with the horseradish peroxidase-linked secondary antibody (1:10000 in TBS-t containing 2% non-fat milk or BSA) for 1 h at room temperature. After another set of quadruplicate washes, membranes were exposed to an enhanced chemiluminescence substrate (under dark) for 2 min and bands were visualized with a charged-coupled device imager.
Caspase −3 and −9 Activity Assays.
LLC-PK1 cells were seeded in sterile 12-well plates at a density of 1 × 105 cells/well and incubated overnight. Cells were pre-treated for 1 h with the vehicle (EtOH), alkoxy-ARA methyl ester (1 μM, regioisomeric mixture), or EET methyl ester (1 μM, regioisomeric mixture), followed by exposure to cisplatin (20 μM) for 24 h. Cells were rinsed twice with ice-cold PBS and harvested with ice-cold RIPA buffer (without protease inhibitors). Following cold centrifugation, the supernatants were collected and kept on ice. 30 uL of supernatants were incubated with 30 μL of caspase activity buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10% glycerol, 1 mM EDTA, 10 mM DTT) containing Ac-DEVD-AMC (20 μM, fluorogenic caspase-3 substrate) or Ac-LEHD-pNa (100 μM, chromogenic caspase-9 substrate) for 90 min at 37 °C. Caspase activity was estimated by reading fluorescence at λEx/λEm = 345 nm/445 nm (Ac-DEVD-AMC) or absorbance at λAbs = 400 nm (Ac-LEHD-pNA) using a Tecan Infinite Pro microplate reader. The activities of caspases were normalized relative to total protein present in lysates (quantified by a BCA assay).
Statistics.
The results are reported as the means ± SEM. Significant differences between groups were determined by One-way analysis of variance, followed by Holm-Sidak multiple comparison testing. A p-value < 0.05 (i.e., significance level α) was considered statistically significant.
RESULTS AND DISCUSSION
Design of Arachidonic acid Alkyl-Ethers.
Four different alkoxy groups (Table 1) were explored as potential bioisosteres for the epoxide within EETs. Due to the presence of olefins in the ARA backbone, an oxymercuration-demercuration synthetic method was employed to transform the alkenes to alkyl-ethers (Scheme 1). Subsequently, the mono-ethers of ARA (regioisomeric mixtures) were isolated to obtain alkoxy-containing mimics of the mono-epoxy EETs. Different alkoxy groups were introduced by varying the alcohol solvent (Scheme 1), which served as the nucleophile reacting with the mercurinium ion to form the corresponding ether. More specifically, methanol, ethanol, isopropanol, and n-propanol were employed to synthesize the methoxy- (1), ethoxy- (2), i-propoxy- (3), and n-propoxy- (4) analogues, respectively.
Table 1.
Structures of Four Alkoxy Bioisosteres of the Epoxide
| Bioisostere | Structure of Functional |
| Group | |
|
| |
| Methoxy 1 |
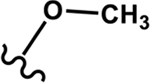
|
| Ethoxy 2 |
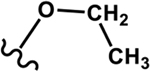
|
|
i-Propoxy 3 |
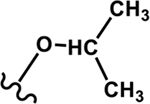
|
|
n-Propoxy 4 |
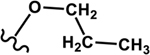
|
|
| |
| Comparison | Structure of Functional |
| Group | Group |
|
| |
| Epoxy 5 |
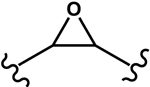
|
Scheme 1.

General synthetic route for generation of mono-alkyl-ethers of arachidonic acid methyl ester (1-4, regioisomeric mixturesa). aDepicted by the representative terminal regioisomer.
Initially, a concentration-response of cisplatin toxicity in LLC-PK1 cells was run (Figure S6, Supporting Information) to assess the optimal screening concentrations of cisplatin. These were determined to be 5, 10, and 20 μM for a low, moderate, and high concentration, respectively.
Ether Bioisostere Screening.
Cell protective activity of the four alkoxy- EET analogues was tested against acute exposure (24 h) to a high concentration (20 μM) of cisplatin and compared to that of an EET control group (5, regioisomeric mixture) to gauge bioisostere functionality relative to the epoxide. Firstly, a concentration-response of therapeutic effect against cisplatin toxicity was conducted (Figure 1). Compound 1 did not show significant protective activity at any of the concentrations tested (Fig. 1A). It was suspected that the generally faster rate of CYP-mediated O-dealkylation for methyl substituted ethers, relative to longer chain alkyl counterparts,25 might be one factor that limits its action. This hypothesis was tested by co-pre-treating 1 with EM, a CYP inhibitor, to restrict potential metabolic inactivation. However, 1 was unable to significantly maintain cell viability even when incubated with the CYP inhibitor (Figure S7, Supporting Information), suggesting the lack of effect might be due to low intrinsic efficacy of the methoxy- group rather than its metabolically labile nature. On the other hand, 1 μM of 2 and 3 significantly shielded cells from cisplatin-induced toxicity (Fig. 1B, 1C, 1F), with an efficacy comparable to that of 1 μM 5 (Fig. 1E, 1F). 1 μM of 4 also displayed good bioactivity (Fig. 1D), but its magnitude of effect was smaller than that of 2, 3, and 5 (Fig. 1F). Since all the analogues, as well as 5, displayed maximal activity at 1 μM, a concentration of 1 μM was considered optimal and utilized for further experiments. To confirm that the observed effects were independent of the assay employed (i.e., MTT), a supporting viability assay (CCK-8) was also conducted (Figure S8, Supporting Information). The magnitude and trends of therapeutic action remained consistent across the two assays, reaffirming inherent compound activity.
Figure 1.

EET analogues 2-4 exhibit cytoprotective activity against acute cisplatin (CDDP) toxicity in LLC-PK1 proximal tubular cells in a concentration-dependent manner. Cells were pre-treated with the vehicle (EtOH), analogues 1-4 (0.1–10 μM), or compound 5 (0.1–10 μM) for 1 h, followed by exposure to CDDP (20 μM) for 24 h. (A) Compound 1 did not demonstrate protective effects at any concentration while (B) compound 2, (C) compound 3, (D) compound 4, and (E) compound 5 all displayed maximal protective activity at a concentration of 1 μM. (F) 1 μM of 2, 3, 4, and 5 significantly shielded LLC-PK1 cells from CDDP toxicity (One way analysis of variance, Holm-Sidak method, ***p < 0.001; **p < 0.01 vs. EtOH + CDDP group), and the magnitudes of action of 2, 3, and 5 were greater than that of 4 (One way analysis of variance, Holm-Sidak method, #p < 0.05 vs. 4 + CDDP group, α = 0.05).
14,15-EE-5(Z)-E, or 14,15-EEZE, is a selective antagonist of the putative EET receptor26 and was employed to study whether antagonism of EET action could pare activity of analogues 1-4. Compounds 2-5 successfully preserved cell viability against cisplatin in the absence of 14,15-EEZE (Figure 2). Presence of 14,15-EEZE significantly curtailed the magnitude of therapeutic action for 2 and 3, while completely abolishing activity of 5 (Figure 2). Hence, generally, alkyl ethers of ARA appear to act at least partially through agonism of the presumed EET receptor, further validating alkoxy- groups as suitable bioisosteric substitutes for the epoxide of EETs.
Figure 2.

EET receptor antagonist 14,15-Epoxyeicosa-5(Z)-enoic Acid (14,15-EEZE) attenuates protective effects of EET analogues 2 and 3 against cisplatin (CDDP) toxicity in LLC-PK1 proximal tubular cells. Cells were pre-treated with vehicle (EtOH), analogues 1-4 (1 μM), or compound 5 (1 μM), in the absence or presence of 14,15-EEZE (3 μM), for 1 h, followed by exposure to CDDP (20 μM) for 24 h. Compounds 2-5 significantly shielded LLC-PK1 cells from CDDP toxicity in the absence of 14,15-EEZE (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP - 14,15-EEZE group, α = 0.05). Only compounds 2-4 significantly shielded LLC-PK1 cells from CDDP toxicity in the presence of 14,15-EEZE (One way analysis of variance, Holm-Sidak method, *p < 0.05; **p < 0.01 vs. EtOH + CDDP + 14,15-EEZE group, α = 0.05). Co-pre-treatment with 14,15-EEZE reduced efficacy of 2 and 3 and eliminated activity of 5 (One way analysis of variance, Holm-Sidak method, ^p < 0.05; ^^p < 0.01 vs. corresponding - 14,15-EEZE group, α = 0.05).
Additionally, a concentration-response of effect was obtained for the parent molecule ARA (Figure S9, Supporting Information), which did not sustain cell viability against cisplatin toxicity at any tested concentration. Furthermore, compounds 1–5 (and parent ARA) were tested in the absence of cisplatin to account for potential isolated effects on cells. None of the compounds stimulated cell growth, nor were they cytotoxic (Figure S10, Supporting Information). Finally, inhibitory activity of analogues 1-4 towards the human sEH was tested (Table S3, Supporting Information). The weak inhibitory potency (IC50 = 6.7–32.1 μM) verified their poor sEH inhibition, even relative to 5 (IC50 = 3.1 μM), suggesting sEH-independent action.
Time-Dependent Therapeutic Action.
Activity of analogues 1-4, relative to 5, against prolonged exposure (48–120 h) to low or moderate concentrations (5 or 10 μM) of cisplatin was also assessed (Figure 3). Interestingly, 1 displayed previously unseen therapeutic activity at 72 h exposure with 5 μM cisplatin (Fig. 3C). This suggests that a degree of bioisosteric activity is associated with the methoxy- group, particularly under conditions of low acute stress. However, unlike other analogues, its activity was not sustainable against 10 μM cisplatin (Fig. 3A, 3B), or for longer durations (96 h) against 5 μM cisplatin (Fig. 3D). Analogues 2 and 3 continued to demonstrate protective effects comparable to that of 5 for earlier time points, namely 48 h and 72 h exposures to 10 μM and 5 μM cisplatin, respectively (Fig. 3A, 3C). Notably, at later durations (72 and 96 h exposures to 10 μM and 5 μM cisplatin, respectively), their activity in fact surpassed that of EETs as the extent of effects exerted by the latter subsided entirely (Fig. 3B) or partially (Fig. 3D). Co-pre-treating 5 with t-AUCB, an sEHI, abolished this drop-off in bioactivity (Fig. 3B, 3D), suggesting sEH-mediated degradation of EETs impedes their function under chronic toxicity conditions. Accordingly, the necessity for dual 5/sEHI treatment to match the activity of 2 and 3 at later toxicity time points lends confidence to the notion that the ethoxy- and i-propoxy- compounds would possess greater bioavailability than their epoxide equivalents and hence perhaps be more suitable for extended medicinal applications. Analogue 4 also demonstrated therapeutic effects against 5 μM cisplatin (Fig. 3C, 3D), but the magnitude of its activity lagged slightly, relative to that of 2 and 3, at the later (96 h) time point (Fig. 3D). However, it was still significantly more active than 5 (without sEHI) at the latter time-concentration combination, suggesting a biochemical stability advantage over EETs akin to 2 and 3. However, 4 did not display significant activity against 10 μM cisplatin at either the 48 or 72 h time points (Fig. 3A, 3B). None of the compounds were able to maintain cell viability after 96 h and 120 h exposures to 10 μM and 5 μM cisplatin, respectively (Figure S11, Supporting Information).
Figure 3.

EET analogues 1-4 attenuate chronic cisplatin (CDDP) toxicity in LLC-PK1 proximal tubular cells in a time-dependent manner. Cells were pre-treated with vehicle (EtOH), analogues 1-4 (1 μM), or compound 5 (1 μM) for 1 h, followed by exposure to low (5 μM) or moderate (10 μM) concentrations of CDDP for 48, 72, or 96 h. (A) Compounds 2, 3, and 5 significantly maintained viability of LLC-PK1 cells exposed to 10 μM CDDP for 48 h (One way analysis of variance, Holm-Sidak method, *p < 0.05; **p < 0.01 vs. EtOH + CDDP group, α = 0.05). (B) Only analogues 2 and 3 significantly maintained viability of LLC-PK1 cells exposed to 10 μM CDDP for 72 h (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP group, α = 0.05). Their activity surpassed that of 5 (One way analysis of variance, Holm-Sidak method, †p < 0.05; ††p < 0.01 vs. 5 + CDDP group, α = 0.05), as the latter failed to display protective effects. Co-pre-treating 5 with an sEHI (t-AUCB) maintained activity of 5 (One way analysis of variance, Holm-Sidak method, ††p < 0.01 vs. 5 + CDDP group, α = 0.05) and significantly shielded cells (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP group, α = 0.05) (C) All four EET analogues 1-4, along with 5, significantly maintained viability of LLC-PK1 cells exposed to 5 μM CDDP for 72 h (One way analysis of variance, Holm-Sidak method, *p < 0.05; **p < 0.01; ***p < 0.001 vs. EtOH + CDDP group, α = 0.05). (D) Compounds 2, 3, 4, and 5 significantly maintained viability of LLC-PK1 cells exposed to 5 μM CDDP for 96 h (One way analysis of variance, Holm-Sidak method, *p < 0.05; ***p < 0.001 vs. EtOH + CDDP group, α = 0.05). Moreover, the activity of 2-4 was significantly greater than that of 5 (One way analysis of variance, Holm-Sidak method, †††p < 0.001 vs. 5 + CDDP group, α = 0.05). Co-pre-treating 5 with the sEHI bridged the gap in activity (One way analysis of variance, Holm-Sidak method, †††p < 0.001 vs. 5 + CDDP group, α = 0.05) and significantly shielded cells (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP group, α = 0.05). Finally, the cell protective effects of 2 and 3 were greater than that of 4 (One way analysis of variance, Holm-Sidak method, #p < 0.05 vs. 4 + CDDP group, α = 0.05).
Taken together with their effects against the acute toxicity of CDDP, all four alkoxy- EET analogues exhibited bioactivity against cisplatin toxicity in LLC-PK1 cells, ranging from excellent for 2 and 3 to good for 4 to minimal for 1. Consequently, the two most promising analogues (i.e., 2 and 3) were selected for further biological mechanism of action studies.
Prevention of Mitochondrial Transmembrane Potential Collapse.
Cisplatin preferentially accumulates in the mitochondria, enhancing susceptibility of cells with higher mitochondrial densities (e.g., PTECs) to its noxious effects.3,27,28 It proceeds to trigger mitochondrial dysfunction by interfering with the electron transport chain, and perturbations typically occur in a time-dependent manner (Figure 4). For instance, a short exposure (6 h) to a high concentration of cisplatin did not significantly impact the mitochondrial transmembrane potential (ΔΨm) of LLC-PK1 cells (Fig. 4A). However, a longer exposure (24 h) resulted in substantial loss of the ΔΨm (Fig. 4B). Pre-treatment with 2 or 3 (or 5) significantly inhibited decline of the electrochemical gradient, returning it closer to basal levels (Fig. 4B), signifying that the alkoxy-based analogues of EETs exert mitochondrial protective effects. Previously, EETs were shown to reduce loss of ΔΨm and thus mitigate stressor-induced mitochondrial damage in cardiac cells29 and hippocampal astrocytes,30 further underlining the involvement of mitochondrion-mediated action for this class of molecules. Moreover, the cell viability assays employed in this study are contingent on activity of mitochondrial reductases and hence the parallel cell-protective effects of these compounds (Fig. 1F) provide yet more evidence of a mitochondria-dependent pathway. An excessively long (48 h) exposure resulted in a nearly complete gradient collapse and pre-treatment with 2 or 3 (or 5) was unable to preserve the ΔΨm (Fig. 4C). This finding aligns closely with the observation that exposure to a high level of cisplatin for 48 h was entirely lethal to LLC-PK1 cells and pre-treatment with the compounds could not sustain viability (Figure S12, Supporting Information). Finally, none of the compounds affected the ΔΨm of cells in the absence of cisplatin (Figure S13, Supporting Information).
Figure 4.

EET analogues 2 and 3 protect against cisplatin (CDDP)-induced mitochondrial transmembrane potential (ΔΨm) decline in LLC-PK1 proximal tubular cells. (A) The ΔΨm was not significantly affected in cells exposed to CDDP (20 μM) for 6 h (B) Pre-treatment with compound 2 (1 μM), 3 (1 μM), or 5 (1 μM) preserved mitochondrial health by significantly mitigating loss of the mitochondrial electrochemical gradient in cells exposed to CDDP (20 μM) for 24 h, relative to vehicle control (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP group, α = 0.05) (C) Pre-treatment with 2 (1 μM), 3 (1 μM), or 5 (1 μM) failed to mitigate the collapse of the ΔΨm in cells exposed to CDDP (20 μM) for 48 h. A depolarized ΔΨm was validated by FCCP (10 μM), a positive control (One way analysis of variance, Holm-Sidak method, &&&p < 0.001 vs. Control group, α = 0.05).
Reduction of General and Mitochondrial Reactive Oxygen Species Generation.
Cisplatin is known to induce oxidative stress,4,31 stimulating the generation of reactive oxygen species (ROS), particularly superoxide anion (O2•−), H2O2, and hydroxyl radical (•OH), through both mitochondrial and non-mitochondrial pathways. Disruptions to the mitochondrial respiratory complexes are a major source of ROS. Non-mitochondrial sources include depletion of glutathione and activation of NADPH and xanthine-xanthine oxidases, which in turn can further exacerbate the mitochondrial dysfunction. Thus, effects of the EET analogues were assessed against both total cellular ROS and mitochondria-specific ROS, expressly O2•− (Figure 5). Predictably, cisplatin increased the general ROS in LLC-PK1 cells (Fig. 5A), as well as the O2•−generated in mitochondria (Fig. 5B). Compound 2 and 3 (as well as 5) lessened both surges, implying oxidative stress-alleviating activity that could be both mitochondria-targeted and whole cell divergent.
Figure 5.

EET analogues 2 and 3 attenuate cisplatin (CDDP)-induced generation of reactive oxygen species (ROS) in LLC-PK1 proximal tubular cells. Pre-treatment with compound 2 (1 μM), 3 (1 μM), or 5 (1 μM) significantly decreased the augmented levels of (A) general cellular ROS and (B) mitochondria-specific superoxide anion in LLC-PK1 cells exposed to CDDP (20 μM) for 24 h, relative to vehicle control (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP group, α = 0.05).
Decreased Ratio of Phosphorylated to Total p38 and JNK.
Involvement of MAPKs, namely p38 and Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK), has been closely linked to the nephrotoxic effects of cisplatin.5,6 MAPKs are key regulators of the mitochondria-mediated intrinsic cellular death pathway.32 They act through recruitment of pro-apoptotic regulators and suppression of anti-apoptotic function, sensitizing the mitochondria towards cell death and activating the downstream caspase-9 pathway. Phosphorylation of MAPKs triggers their activity and cisplatin-generated ROS (in particular •OH) are the primary cellular stressors considered to initiate this MAPK signaling. Accordingly, little to no levels of the phosphorylated MAPK variant were detectable in LLC-PK1 cells after only a brief period (6 h) of cisplatin exposure (Figure S14, Supporting Information), a time point at which mitochondrial integrity was intact (Fig. 4A). However, following an extended exposure (24 h), under conditions of verifiable cisplatin-induced mitochondrial disruption and ROS upregulation (Fig. 4B, 5), markedly augmented levels of the phosphorylated p38 and JNK/SAPK forms (relative to total variants) were observed (Figure 6). The ratios were significantly reduced by pre-treatments with 2, 3, or 5 (Figure 6), denoting the capacity of alkoxy- EET analogues to terminate the MAPK signaling upregulated by cisplatin.
Figure 6.

EET analogues 2 and 3 diminish cisplatin (CDDP)-induced MAPK signaling in LLC-PK1 proximal tubular cells. Pre-treatment with compound 2 (1 μM), 3 (1 μM), or 5 (1 μM) significantly reduced the higher ratio of phosphorylated variant to total variant for MAPKs (A) p38 and (B) JNK/SAPK in LLC-PK1 cells exposed to CDDP (20 μM) for 24 h, relative to vehicle control. Results are representative of three independent experiments (One way analysis of variance, Holm-Sidak method, *p < 0.05; ***p < 0.001 vs. EtOH + CDDP group, α = 0.05).
Inhibition of Caspase-9 and −3 Activation.
Initiation of caspase-9 and ensuing recruitment of caspase-3 is a primary mode of cisplatin-induced cell death.7 Disruptions to mitochondrial health, through direct agitations, oxidative damage, and MAPK-mediated pro-apoptotic signaling, trigger the caspase-9 pathway, leading to activation of the executioner caspase-3. Hence, as expected, activity of both caspase-9 and caspase-3 was greatly elevated with exposure to cisplatin (Figure 7). This activity was ablated by 2 and 3 (as well as 5), suggesting the ability of these compounds to mitigate cisplatin-induced cell death by obstructing the downstream caspase stimulation.
Figure 7.

EET analogues 2 and 3 curb cisplatin (CDDP)-induced caspase activity in LLC-PK1 proximal tubular cells. Pre-treatment with compound 2 (1 μM), 3 (1 μM), or 5 (1 μM) significantly reduced the elevated normalized activity of (A) caspase-9 and (B) caspase-3 in LLC-PK1 cells exposed to CDDP (20 μM) for 24 h, relative to vehicle control (One way analysis of variance, Holm-Sidak method, ***p < 0.001 vs. EtOH + CDDP group, α = 0.05).
Preservation of Cytotoxicity Against Cancer Cells.
While attenuating the off-target renal toxicity of cisplatin is vitally important, ensuring that the pharmacological intervention does not dilute the desired chemotherapeutic action is a key consideration. Hence, the effects of compounds 1-5 on human liver hepatocellular carcinoma cells (HepG2) were assessed (Figure 8). Compounds 1-5 did not shield liver cancer cells from the toxic effects of cisplatin (Fig. 8A), nor did they stimulate cancer cell growth in the absence of cisplatin (Fig. 8B). The findings were reinforced with a secondary assay (CCK-8) to ensure the lack of hindrance was assay-independent (Figure S8, Supporting Information). This implies these compounds do not interfere with the cancer cell-targeted toxicity of cisplatin, potentially expanding its therapeutic index and capacity for use.
Figure 8.

EET analogues 1-4 do not affect the cytotoxicity of cisplatin (CDDP) towards HepG2 liver cancer cells. (A) Pre-treatment with analogues 1-4 (1 μM) or compound 5 (1 μM) did not exert protective effects in HepG2 cells exposed to CDDP (20 μM) for 24 h. (B) Compounds 1-5 (1 μM) had no significant influence on viability of HepG2 cells.
CONCLUSIONS
Here we report new EET mimics developed as potential lead therapeutics against the off-target nephrotoxicity of the common cancer drug cisplatin. They employ alkoxy- substituents as replacement groups for the chemically stable but enzymatically labile epoxide. Since alkoxy groups are so chemically similar to the epoxide, alkyl-ethers of ARA potentially orient at the putative receptor in a manner akin to EETs. This study emphasized assessing the bioisosteric activity of common alkoxy groups in pig derived PTECs, as opposed to investigating pure regioisomers. Thus, regioisomeric mixtures of four alkoxy-ARA analogues of EETs were screened against cisplatin renal toxicity in vitro, relative to an EET regioisomer mixture. All bioisosteres exhibited activity, to varying degrees. The ethoxy- and i-propoxy- compounds were the most efficacious in this model and, importantly, proved more effectual than EETs at mitigating effects of prolonged cisplatin exposures. They protected PTECs from cisplatin toxicity through the same molecular modes as EETs, specifically by blocking mitochondrial dysfunction and oxidative stress and curbing subsequent MAPK signaling and caspase activation, while not influencing cytotoxicity of cisplatin against the target cancer cells. Application of an EET receptor antagonist diminished their efficacy, further authenticating mimetic functionality.
In earlier studies on alkoxy- mimics of insect JHs, large differences in biological potency were observed for the ether groups across different target species. For instance, the methoxy- was highly potent on dipterous insects (flies), relative to the weakly active ethoxy-, i-propoxy-, and n-propoxy-. In contrast, high activity of ethoxy- and n-propoxy- was seen in coleopterous insects (beetles), compared to low potency for methoxy and i-propoxy. Interestingly, the i-propoxy analogue of EETs was of high activity in this study but the homologous mimic of JH was over 100-fold less active than the n-propoxy in beetles. Hence, uniform recognition across systems cannot be assumed, and species differences and the specific bioassay of interest should be taken into account when evaluating bioisosteric potential.
More recently, there is growing interest in ω−3 EpFAs, namely eicosapentaenoic acid (EPA, 20:5)-derived epoxyeicosatetraenoic acids (EEQs) and docosahexaenoic acid (DHA, 22:6)-derived epoxydocosapentaenoic acids (EDPs), and their synthetic analogues.33–35 Thus, as a next step, the alkoxy- analogues of these EpFAs could also be examined, especially since EEQs and EDPs are more potent than EETs in certain disease models.36,37 Moreover, activity of individual regioisomers within the most promising series should eventually be explored.
Finally, while sEH-mediated hydrolysis is the primary mode of EpFA degradation, EpFAs are also metabolized by a variety of pathways including β-oxidation, chain elongation, ω/ω−1 hydroxylation, and secondary metabolism.38 EpFA analogue design generally attempts to block these routes as well since they become major contributors to degradative metabolism once the hydrolytic role of sEH is blocked. Hence, the alkoxy- bioisostere approach might facilitate employment of ideal scaffolds that lead to further improved EpFA analogues. Thus, overall, future work will highlight the importance of identifying the appropriate regio- and, possibly, stereoisomer for medicinal optimization and targeted biology.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by National Institutes of Health grants, National Institute of Environmental Health Sciences (NIEHS) RIVER Award R35 ES030443-01, NIEHS Superfund Award P42 ES004699, and S10 OD025271-01A1. N.S. thanks the NIEHS/UC Davis Superfund Research Program for financial support in the form of a GSR fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information. HPLC traces and product ion spectra for compounds 1-5, HPLC gradient, Q-TRAP MS source parameters, optimization of cisplatin screening concentrations, influence of CYP inhibitor on activity of compound 1, CCK-8 assay-facilitated supporting viability assessments, concentration-response of activity for parent ARA, isolated effects on PTEC viability, IC50 values against human sEH, endpoints of therapeutic action against low-moderate, chronic and high, acute cisplatin exposures, isolated effects on PTEC mitochondrial health, immunoblots for MAPKs at an early exposure time point
REFERENCES
- (1).Manohar S, and Leung N (2018) Cisplatin nephrotoxicity: a review of the literature. J. Nephrol. 31, 15–25. [DOI] [PubMed] [Google Scholar]
- (2).Yao X, Panichpisal K, Kurtzman N, and Nugent K (2007) Cisplatin nephrotoxicity: a review. Am. J. Med. Sci. 334, 115–124. [DOI] [PubMed] [Google Scholar]
- (3).Kruidering M, Van de Water B, de Heer E, Mulder GJ, and Nagelkerke JF (1997) Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 280, 638–649. [PubMed] [Google Scholar]
- (4).Chirino YI, and Pedraza-Chaverri J (2009) Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 61, 223–242. [DOI] [PubMed] [Google Scholar]
- (5).Ramesh G, and Reeves WB (2005) p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am. J. Physiol.: Renal Physiol 289, F166–174. [DOI] [PubMed] [Google Scholar]
- (6).Francescato HD, Costa RS, Júnior FB, and Coimbra TM (2007) Effect of JNK inhibition on cisplatin-induced renal damage. Nephrol Dial Transport. 22, 2138–2148. [DOI] [PubMed] [Google Scholar]
- (7).Kaushal GP, Kaushal V, Hong X, and Shah SV (2001) Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 60, 1726–1736. [DOI] [PubMed] [Google Scholar]
- (8).Elmarakby AA (2012) Reno-protective mechanisms of epoxyeicosatrienoic acids in cardiovascular disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 302, R321–330. [DOI] [PubMed] [Google Scholar]
- (9).Inceoglu B, Bettaieb A, Haj FG, Gomes AV, and Hammock BD (2017) Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 133, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Imig JD (2018) Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol. Ther. 192, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, and Hammock BD (2009) Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol. Toxicol. 25, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu Y, Webb HK, Fukushima H, Micheli J, Markova S, Olson JL, and Kroetz DL (2012) Attenuation of Cisplatin-Induced Renal Injury by Inhibition of Soluble Epoxide Hydrolase Involves Nuclear Factor κB Signaling. J Pharmacol. Exp. Ther. 341, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu Y, Lu X, Nguyen S, Olson JL, Webb HK, and Kroetz DL (2013) Epoxyeicosatrienoic acids prevent cisplatin-induced renal apoptosis through a p38 mitogen-activated protein kinase-regulated mitochondrial pathway. Mol. Pharmacol. 84, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sudhahar V, Shaw S, and Imig JD (2010) Epoxyeicosatrienoic acid analogs and vascular function. Curr. Med. Chem. 17, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Campbell WB, Imig JD, Schmitz JM, and Falck JR (2017) Orally Active Epoxyeicosatrienoic Acid Analogs. J. Cardiovasc. Pharmacol. 70, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Khan MAH, Liu J, Kumar G, Skapek SX, Falck JR, and Imig JD (2013) Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 27, 2946–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Imig JD, Hye Khan MA, Burkhan A, Chen G, Adebesin AM, and Falck JR (2021) Kidney-Targeted Epoxyeicosatrienoic Acid Analog, EET-F01, Reduces Inflammation, Oxidative Stress, and Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 22, 2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Olearczyk JJ, Field MB, Kim I-H, Morisseau C, Hammock BD, and Imig JD (2006) Substituted Adamantyl-Urea Inhibitors of the Soluble Epoxide Hydrolase Dilate Mesenteric Resistance Vessels. J. Pharm. Exp. Ther. 318, 1307. [DOI] [PubMed] [Google Scholar]
- (19).Henrick CA, Staal GB, and Siddall JB (1973) Alkyl 3,7,11-trimethyl-2,4-dodecadienoates, a new class of potent insect growth regulators with juvenile hormone activity. J. Agric. Food Chem. 21, 354–359. [DOI] [PubMed] [Google Scholar]
- (20).Hammock BD, Gill SS, and Casida JE (1974) Synthesis and morphogenetic activity of derivaties and analogs of aryl geranyl ether juvenoids. J. Agric. Food Chem. 22, 379–385. [DOI] [PubMed] [Google Scholar]
- (21).Hwang SH, Tsai H-J, Liu J-Y, Morisseau C, and Hammock BD (2007) Orally Bioavailable Potent Soluble Epoxide Hydrolase Inhibitors. J. Med. Chem. 50, 3825–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Singh N, Barnych B, Wagner KM, Wan D, Morisseau C, and Hammock BD (2021) Adrenic Acid-Derived Epoxy Fatty Acids Are Naturally Occurring Lipids and Their Methyl Ester Prodrug Reduces Endoplasmic Reticulum Stress and Inflammatory Pain. ACS Omega 6, 7165–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Newman JW, Morisseau C, Harris TR, and Hammock BD (2003) The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc. Natl. Acad. Sci. U.S.A. 100, 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Morisseau C, and Hammock BD (2007) Measurement of soluble epoxide hydrolase (sEH) activity. Curr. Protoc. Toxicol. Chapter 4, Unit 4.23. [DOI] [PubMed] [Google Scholar]
- (25).Silverman RB, and Holladay MW (2014) Chapter 8 - Drug Metabolism, In The Organic Chemistry of Drug Design and Drug Action (Third Edition) (Silverman RB, and Holladay MW, Eds.) pp 357–422, Academic Press, Boston. [Google Scholar]
- (26).Batchu SN, Lee SB, Qadhi RS, Chaudhary KR, El-Sikhry H, Kodela R, Falck JR, and Seubert JM (2011) Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br. J. Pharmacol. 162, 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cullen KJ, Yang Z, Schumaker L, and Guo Z (2007) Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J. Bioenerg. Biomembr. 39, 43–50. [DOI] [PubMed] [Google Scholar]
- (28).Qian W, Nishikawa M, Haque AM, Hirose M, Mashimo M, Sato E, and Inoue M (2005) Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am. J. Physiol: Cell Physiol. 289, C1466–1475. [DOI] [PubMed] [Google Scholar]
- (29).Katragadda D, Batchu SN, Cho WJ, Chaudhary KR, Falck JR, and Seubert JM (2009) Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J. Mol. Cell Cardiol. 46, 867–875. [DOI] [PubMed] [Google Scholar]
- (30).Sarkar P, Zaja I, Bienengraeber M, Rarick KR, Terashvili M, Canfield S, Falck JR, and Harder DR (2014) Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am. J. Physiol.: Heart Circ. Physiol. 306, H475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kawai Y, Nakao T, Kunimura N, Kohda Y, and Gemba M (2006) Relationship of intracellular calcium and oxygen radicals to Cisplatin-related renal cell injury. J. Pharmacol. Sci. 100, 65–72. [DOI] [PubMed] [Google Scholar]
- (32).Yue J, and López JM (2020) Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 21, 2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Adebesin AM, Wesser T, Vijaykumar J, Konkel A, Paudyal MP, Lossie J, Zhu C, Westphal C, Puli N, Fischer R, Schunck W-H, and Falck JR (2019) Development of Robust 17(R),18(S)-Epoxyeicosatetraenoic Acid (17,18-EEQ) Analogues as Potential Clinical Antiarrhythmic Agents. J. Med. Chem. 62, 10124–10143. [DOI] [PubMed] [Google Scholar]
- (34).Berlin S, Goette A, Summo L, Lossie J, Gebauer A, Al-Saady N, Calo L, Naccarelli G, Schunck WH, Fischer R, Camm AJ, and Dobrev D (2020) Assessment of OMT-28, a synthetic analog of omega-3 epoxyeicosanoids, in patients with persistent atrial fibrillation: Rationale and design of the PROMISE-AF phase II study. Int. J. Cardiol. Heart Vasc. 29, 100573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Darwesh AM, Bassiouni W, Adebesin AM, Mohammad AS, Falck JR, and Seubert JM (2020) A Synthetic Epoxydocosapentaenoic Acid Analogue Ameliorates Cardiac Ischemia/Reperfusion Injury: The Involvement of the Sirtuin 3-NLRP3 Pathway. Int. J. Mol. Sci. 21, 5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, and Hammock BD (2010) Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J. Lipid Res. 51, 3481–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wagner K, Vito S, Inceoglu B, and Hammock BD (2014) The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. 113–115, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Falck JR, Kodela R, Manne R, Atcha KR, Puli N, Dubasi N, Manthati VL, Capdevila JH, Yi XY, Goldman DH, Morisseau C, Hammock BD, and Campbell WB (2009) 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J. Med. Chem. 52, 5069–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.