

Abstract
Overexpression of heat shock protein 90 (Hsp90) is common in various types of cancer. In cutaneous melanoma, a cancer with one of the high levels of Hsp90 overexpression, such expression was correlated with a panel of protein kinases, thus offering an opportunity to identify Hsp90-based multi-kinase inhibitors for novel cancer therapies. Towards this goal, we utilized a 2,4-dihydroxy-5-isopropylbenzate-based Hsp90 inhibitor scaffold and thieno[2,3-d]pyrimidine-based kinase inhibitor scaffold to develop a Hsp90-inhibiting compound library. Our inhibitory compound named 8m inhibited Hsp90 and PI3Kα with an IC50 value of 38.6 nM and 48.4 nM, respectively; it displayed improved cellular activity which could effectively induce cell cycle arrest and apoptosis in melanoma cells and lead to the inhibition of cell proliferation, colony formation, migration and invasion. Our results demonstrated 8m to be a promising lead compound for further therapeutic potential assessment of Hsp90/PI3Kα dual inhibitors in melanoma targeted therapy.
Keywords: Apoptosis, melanoma, hsp90, PI3Kα, kinase inhibitor
1. Introduction
Melanoma is a type of skin cancer with one of the highest morality rates. This disease accounts for over half of all cutaneous cancer-related deaths but is only one-tenth of newly diagnosed skin cancers1,2. The incidence rate of melanoma has risen continually in the past twenty years. Although early-stage melanoma patients usually can be cured by dermatologic surgery, their prognosis varies by tumor stages and mutation status3. In general, the stages of melanoma are characterized using the TNM system from the American Joint Committee on Cancer (AJCC). Here, we referred to The Cancer Genome Atlas (TCGA) network to help describe the genomic mutation landscape of cutaneous melanoma using biopsies from 331 patients4–7.
Most melanoma can be classified into four driver mutations using biostatistical analysis: B-Raf mutated, Ras mutated, NF1 mutated and Triple-WT (wild-type)8. B-Raf, Ras or NF1 mutated patients often also have activation of MAPK pathway, and Triple-WT patients usually have activated receptor tyrosine kinase (RTK) and serine/threonine kinase (STK), such as c-kit, CDK family, PI3K family and Akt family, and so on5. Some targeted therapy approaches, e.g. vemuragenib (B-Raf inhibitor), cobimetinib (MEK inhibitor) and Yervoy (CTLA-4 mono-antibody) have been approved for melanoma therapy9–12. However, there are still many melanoma patients who do not response to these therapies.
Hsp90 is an important molecular chaperone protein that directly binds to transition state conformations during the late stages of protein assembly. It usually interacts with various client proteins to construct multi-chaperone protein complexes and regulates protein-protein interaction networks to carry out cellular functions of oncogenic signaling transduction factors. Hsp90 is often overexpressed in tumor tissues because it maintains the correct folding of various client oncoproteins.
Hsp90 inhibitors are structurally diversified by their scaffold, e.g. benzoquinone from geldanamycin, resorcinol derivatives, purine-based Hsp90 inhibitors and 2-aminobenzamide derivatives. These four major classes of Hsp90 inhibitors bind to the N-terminal Hsp90 domain13–15. Marcu et al. reported coumarin-based novobiocin binding at the second ATP site of Hsp90 chaperone C-terminal domain16–19. There are more than twenty Hsp90 N-terminal domain inhibitors at different stages of clinical trials, however, no Hsp90 inhibitor has ever been approved by FDA. The potential toxicities and knowledge gaps in how to classify patients who can benefit from Hsp90 inhibitory therapy have limited the clinical applications of Hsp90 inhibitors.
There have been some advances achieved by combining Hsp90 inhibitors and other targeted therapeutics, e.g. HER2 monoantibody trastuzumab, ALK inhibitor crizotinib, and so on20–29. It is therefore reasonable to discover novel inhibitors targeting Hsp90 and the chaperone protein simultaneously, we shall focus on the discovery multitarget small molecular inhibitors of Hsp90 and its oncogenic client kinase proteins.
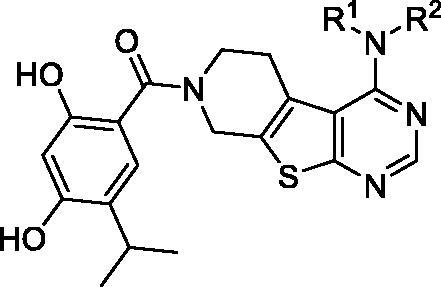
In the current study, we report the design, synthesis and molecular mechanism studies of Hsp90/protein kinases multi-inhibitors. Considering the structural scaffolds of previously reported Hsp90 inhibitors and kinase inhibitors in clinical study or application studies, we employed the 2,4-dihydroxy-5-isopropylbenzate fragment for Hsp90 inhibition and the thieno[2,3-d]pyrimidine scaffold for kinase inhibition, respectively. The association between 4-substituted groups on the thieno[2,3-d]pyrimidine scaffold and in vitro kinase inhibition, cell proliferation, apoptosis, migration and invasion was measured as well as evaluation of an in vivo xenograft model and the molecular mechanisms of these novel Hsp90/PI3K dual inhibitors as determined by protein immunoblotting and immunoprecipitation assays.
2. Materials and methods
2.1. Cell lines and reagents
B16, HepG2, A549, SW620, MCF-7 and Hela cells were obtained from ATCC and maintained by State Key Laboratory of Biotherapy, Sichuan University in DMEM medium supplemented with 10% fetal bovine serum (FBS, GIBCO, Australia). MTT was purchased from Sigma-Aldrich. Antibodies against hsp90α, stat3 were purchased from Genetex. Antibodies against Bcl-2, Bim, caspase-3, caspase-8, caspase-9, FasL, p27, c-myc, N-cadherin, EGFR, pEGFR(T1068), pEGFR(T1173), C-Raf, pC-Raf, ERK1/2, pERK1/2, Akt, pAkt(S308), pAkt(S473), P90RSK, pP90RSK, Met, pMet, PI3K, B-Raf, pB-Raf, hif1-α were purchased from Cell Signaling Technology. Antibodies against poly polymerase (PARP), FasL, JNK, pJNK, Met, pMet were purchased from Abcam. Antibodies against p21, β-actin, cytochrome c, Bad, Bax, E-cadherin, vimentin, MMP2, MMP9, ZEB1, β-catenin, hsp90β, hsp70, p53, mdm2, cdk2, cdk4, cdk6, cdc37, MNK1, ERK5, GAPDH and Secondary antibodies (HRP-conjugated sheep anti-rabbit antibodies or HRP-conjugated sheep anti-mouse antibodies) for western blot were obtained from Proteintech. Protein A/G Mix Magnetic Beads for Immunoprecipitation were purchased from Millipore. The detailed synthesis, characterization and in vitro biological procedures of compounds 8a–n were described in Supplementary materials.
2.2. Molecular docking
The molecular docking was performed by the CDOCKER modules embedded in Accelrys Discovery Studio 3.5 Software, with CHARMm27 force field and MMFF94 charge. The protein structures of the Hsp90 and PI3Kα were retrieved from PDB database. The residues within 1.0 nm around the co-crystallized ligands at the ATP-binding sites were considered as interacted residues for docking sphere definition. The other parameters were set to default values.
2.3. The synthesis and characterization of key intermediates and compounds 8a-n
6-(tert-butyl) 3-ethyl 2-amino-4,7-dihydrothieno[2,3-c]pyridine-3,6(5H)-dicarboxylate (1)
To a mixture of N-Boc-4-piperidone (10.00 g, 50 mmol), ethyl cyanoacetate (5.66 g, 50.0 mmol), and sulfur (1.60 g, 50.0 mmol) in absolute ethanol (100 ml), triethylamine (10 ml) was added and refluxed for 12 h; the reaction mixture was concentrated and the residue was partitioned between water and ethyl acetate. The organic layer was separated, and concentrated, and the crude product was recrystallized with ethanol (150 ml), to give 1. Yield 86%, white powder. 1H NMR (400 MHz, Chloroform-d) δ 6.02 (s, 2H), 4.35 (s, 2H), 4.26 (q, J = 7.0 Hz, 2H), 3.61 (t, J = 5.7 Hz, 2H), 2.80 (t, J = 5.7 Hz, 2H), 1.48 (s, 9H), 1.34 (t, J = 7.0 Hz, 3H).
tert-butyl 4-oxo-3,5,6,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(4H)-carboxylate (2)
A mixture of (1) (3.26 g, 10.0 mmol) and formamidine acetate (1.56 g, 15.0 mmol) was stirred in DMF for 12 h at 100 °C. The reaction mixture was cooled, water was added, and the precipitate formed was collected by filtration, and washed thoroughly with water to give 2, Yield, 90%. 1H NMR (400 MHz, Chloroform-d) δ 11.99 (s, 1H), 7.99 (s, 1H), 4.66 (s, 2H), 3.74 (t, J = 5.8 Hz, 2H), 3.13 (t, J = 5.8 Hz, 2H), 1.50 (s, 9H).
tert-butyl 4-chloro-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (3)
A mixture of (2) (16.32 g, 50.0 mmol), SOCl2 (100 ml) and DMF(2 ml) was refluxed for 4 h. The reaction mixture was cooled in an ice bath and then carefully neutralized by the addition of aqueous sodium bicarbonate solution. The resulting mixture was extracted with ethyl acetate, the organic layer was separated, dried overMgSO4, and concentrated, and the crude product was purified by silica gel column chromatography using a mixture of petroleum ether: dichloromethane (5:1), to give 3, Yield, 85%, white powder. 1H NMR (400 MHz, Chloroform-d) δ 8.77 (s, 1H), 4.74 (s, 2H), 3.80 (t, J = 5.8 Hz, 2H), 3.21 (t, J = 5.8 Hz, 2H), 1.51 (s, 9H).
tert-butyl 4-((3-ethylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4a)
To a suspension of 60% NaH(120 mg, 3 mmol) in anhydrous 1,4-dioxane(10 ml), was added, a solution of 2-ethylaniline (242 mg, 2 mmol) in anhydrous 1,4-dioxane(5 ml), the reaction mixture was stirred for 20 min at room temperature and then compound (3) (325 mg, 1 mmol) in 1,4-dioxane (5 ml) was added to the above reaction mixture, the mixture was stirred for another 2 h at 90 °C, at the end of the reaction, the excessive NaH was quenched by H2O, the mixture was concentrated and the residue was partitioned between water and ethyl acetate. The organic layer was separated, and concentrated, and then the crude product was purified by silica gel column chromatography using a mixture solvent of petroleum ether: ethylacetate (5:1), to give 4a. Yield 55%, light yellow powder, m.p. 159.0–160.4 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.45 (s, 1H), 7.34 – 7.27 (m, 2H), 7.21 (td, J = 7.4, 1.4 Hz, 1H), 4.72 (t, J = 2.0 Hz, 2H), 3.86 (t, J = 5.7 Hz, 2H), 3.13 (t, J = 5.7 Hz, 2H), 2.66 (q, J = 7.6 Hz, 2H), 1.51 (s, 9H), 1.26 (t, J = 7.6 Hz, 3H).
tert-butyl 4-((4-bromophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4b)
Compound (4b) was synthesized from 4-bromoaniline and compound (3), in a manner similar to (4a). Yield 66%, Light yellow powder, m. p. 178.9–181.7 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.52 (s, 1H), 7.58 – 7.46 (m, 4H), 6.94 (s, 1H), 4.71 (s, 2H), 3.85 (t, J = 5.7 Hz, 2H), 3.14 (d, J = 5.7 Hz, 2H), 1.51 (s, 8H).
tert-butyl 4-((3-chlorophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4c)
Compound (4c) was synthesized from 3-chloroaniline and compound (3), in a manner similar to (4a). Yield 65%, Light yellow powder, m. p. 180.4–181.5 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.55 (s, 1H), 7.82 (s, 1H), 7.48 (s, 1H), 7.29 (t, J = 8.1 Hz, 1H), 7.11 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 6.98 (s, 1H), 4.72 (s, 2H), 3.86 (t, J = 5.7 Hz, 2H), 3.14 (t, J = 5.7 Hz, 2H), 1.51 (s, 9H).
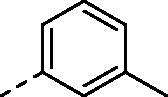
tert-butyl 4-(m-tolylamino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4d)
Compound (4d) was synthesized from m-toluidine and compound (3), in a manner similar to (4a). Yield 59%, Light yellow powder, m. p. 150.7–152.4 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.51 (s, 1H), 7.45 (dd, J = 8.0, 2.2 Hz, 1H), 7.41 (d, J = 1.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 6.97 (ddt, J = 7.6, 1.7, 0.9 Hz, 1H), 6.94 (s, 1H), 4.70 (s, 2H), 3.85 (t, J = 5.7 Hz, 2H), 3.13 (t, J = 5.7 Hz, 2H), 2.39 (s, 3H), 1.51 (s, 9H).
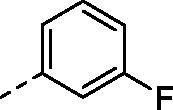
tert-butyl 4-((3-fluorophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4e)
Compound (4e) was synthesized from 3-fluoroaniline and compound (3), in a manner similar to (4a). Yield: 69%, Light yellow powder, m. p. 164.4–165.0 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.55 (s, 1H), 7.73 (d, J = 11.0 Hz, 1H), 7.32 (td, J = 8.1, 6.3 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 7.03 (s, 1H), 6.83 (tdd, J = 8.2, 2.5, 1.0 Hz, 1H), 4.72 (s, 2H), 3.86 (t, J = 5.7 Hz, 2H), 3.14 (t, J = 5.7 Hz, 2H), 1.51 (s, 9H).
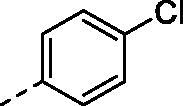
tert-butyl 4-((4-chlorophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4f)
Compound (4f) was synthesized from 4-chloroaniline and compound (3), in a manner similar to (4a). Yield: 68%, Light yellow powder, m. p. 183.7–185.6 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.52 (s, 1H), 7.64 – 7.57 (m, 2H), 7.40 – 7.32 (m, 2H), 6.95 (s, 1H), 4.71 (s, 2H), 3.86 (t, J = 5.7 Hz, 2H), 3.15 (t, J = 5.7 Hz, 2H), 1.51 (s, 9H).
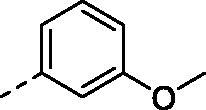
tert-butyl 4-((3-methoxyphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4g)
Compound (4g) was synthesized from 3-methoxyaniline and compound (3), in a manner similar to (4a). Yield: 58%, Light yellow powder, m. p. 136.3–139.6 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.52 (s, 1H), 7.41 (t, J = 2.3 Hz, 1H), 7.29 (t, J = 8.1 Hz, 1H), 7.10 (ddd, J = 8.0, 2.1, 0.9 Hz, 1H), 6.97 (s, 1H), 6.70 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 4.71 (s, 2H), 3.89 – 3.81 (m, 5H), 3.14 (d, J = 5.7 Hz, 2H), 1.51 (s, 9H).
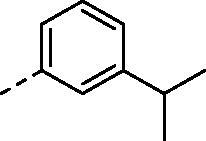
tert-butyl 4-((3-isopropylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4h)
Compound (4h) was synthesized from 3-isopropylaniline and compound (3), in a manner similar to (4a). Yield: 56%, White powder, m. p. 115.7–117.2 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.51 (s, 1H), 7.53 (dt, J = 8.1, 1.6 Hz, 1H), 7.38 (t, J = 1.9 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.04 (dt, J = 7.7, 1.3 Hz, 1H), 6.96 (s, 1H), 4.71 (s, 2H), 3.86 (t, J = 5.7 Hz, 2H), 3.16 (d, J = 5.6 Hz, 2H), 2.94 (hept, J = 6.9 Hz, 1H), 1.51 (s, 9H), 1.28 (d, J = 7.0 Hz, 6H).
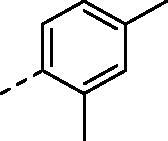
tert-butyl 4-((2,4-dimethylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4i)
Compound (4i) was synthesized from 2,4-dimethylaniline and compound (3), in a manner similar to (4a). Yield: 61%, Yellow powder, m. p. 159.6–161.3 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.44 (s, 1H), 7.58 (s, 1H), 7.16 – 7.02 (m, 2H), 6.70 (s, 1H), 4.71 (s, 2H), 3.85 (t, J = 5.7 Hz, 2H), 3.13 (t, J = 5.7 Hz, 2H), 2.34 (s, 3H), 2.27 (s, 3H), 1.51 (s, 9H).
tert-butyl 4-(piperidin-1-yl)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4k)
Compound 3 (325 mg, 1.0 mmol) was dissolved in piperidine (15 ml), the mixture was stirred at 100° C for 1 h. after the reaction solution was cooled, ethyl acetate (100 ml) were added, and then the organic layer was washed with water, and then dried over MeSO4, filtered and evaporated and then the crude product was purified by silica gel column chromatography using a mixture of petroleum ether: ethyl acetate (3:1), to give 4k, yield: 91%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.52 (s, 1H), 4.71 (s, 2H), 3.67 (s, 2H), 3.41 – 3.31 (m, 4H), 3.05 (s, 2H), 1.78 – 1.65 (m, 6H), 1.52 (s, 9H).
tert-butyl 4-(pyrrolidin-1-yl)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4l)
Compound (4l) was synthesized from pyrrolidine and compound (3), in a manner similar to (4k). Yield: 93%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 4.68 (s, 2H), 3.69 (t, J = 6.6 Hz, 4H), 3.62 (s, 2H), 2.99 (s, 2H), 1.99 – 1.87 (m, 4H), 1.52 (s, 9H).
tert-butyl 4-morpholino-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4m)
Compound (4m) was synthesized from morpholine and compound (3), in a manner similar to (4k). Yield: 95%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 4.72 (s, 2H), 3.93 – 3.83 (m, 4H), 3.69 (s, 2H), 3.50 – 3.41 (m, 4H), 3.04 (s, 2H), 1.52 (s, 9H).
tert-butyl 4-(dimethylamino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (4n)
Compound (4n) was synthesized from 30% dimethylamine in methanol and compound (3), in a manner similar to (4k). Yield: 89%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H), 4.71 (s, 2H), 3.65 (t, J = 4.7 Hz, 2H), 3.06 (s, 6H), 3.04 (s, 2H), 1.52 (s, 9H).
Methyl 2,4-dihydroxy-5-isopropylbenzoate (5)
A mixture of methyl 2,4-dihydroxybenzoate (1.68 g, 10.0 mmol), 2-bromopropane (1.87 ml, 20.0 mmol), and aluminum chloride (2.65 g, 20.0 mmol) in CH2Cl2 was stirred at 50° C for 24 h under argon. 2-bromopropane (1.87 ml, 20.0 mmol) was added to the reaction mixture two times every 8 h. The mixture was neutralized with 15% NaOH to pH 5, concentrated under reduced pressure, and then extracted with ethyl acetate. The organic layer was washed with saturated NaHCO3 solution, dried over MeSO4, concentrated under reduced pressure, and purified by column to afford 5, yield 45%, colorless oil. 1H NMR (400 MHz, Chloroform-d) δ10.8 (s, 1H), 7.64 (s, 1H), 6.34 (s, 1H), 5.50 (s, 1H), 3.90 (s, 3H), 3.15–3.08 (m, 1H), 1.25 (d, J = 10.8 Hz, 6H).
2,4-bis(benzyloxy)-5-isopropylbenzoic acid (6)
To a solution of methyl 2,4-dihydroxy-5-isopropylbenzoate (2.10 g, 10.0 mmol) and potassium carbonate (4.14 g, 30.0 mmol) in DMF (50 ml) was added benzyl chloride (2.53 ml, 22.0 mmol). The suspension was heated to 80 °C for 16 h under a nitrogen atmosphere. Ethyl acetate (100 ml) and water (100 ml) were added, and then the organnic layer was washed with water, and then dried over MeSO4, filtered and evaporated and then the crude product was purified by silicagel column chromatography using a mixture of petroleum ether:ethyl acetate (8:1), to give methyl 2,4-bis (benzyloxy)-5-isopropylbenzoate; and then a solution of THF: Methanol: Water 100 ml, (3:1:1) mixed by methyl 2,4-bis (benzyloxy)-5-isopropylbenzoate (3.90 g, 10.0 mmol) and LiOH·H2O (1.26 g, 30.0 mmol) were stirred for 12 h. Ethyl acetate (100 ml) and water (100 ml) were added, then the organic layer was washed with water, dried over MeSO4, filtered and evaporated, the crude product was purified by silica gel column chromatography using a mixture of petroleum ether: ethyl acetate (2:1), to give 6, Yield 81%, white powder. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (brs, lH), 8.04 (s, lH), 7. 46–7.34 (m,10H), 6.57 (s,1H), 5.20 (s,2H), 5.16–5.13 (m, 2H), 5.11 (s, 2H), 2.06 (s, 3H).
N-(3-ethylphenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7a)
Compound 4a (410 mg, 1.0 mmol) was dissolved in ethylacetate (10 ml) and then 9 N HCl (5 ml) was added, the mixture was stirred at room temperature for 30 min. The reaction solution was neutralized with sodium hydrogen carbonate, then alkalized by NaOH to pH = 9. Fifty millilitres of ethylacetate and 30 ml H2O was added, after stirred adequately, the organic layer was separated, and concentrated, and then the crude product was purified by silica gel column chromatography using a mixture solvent of dichloromethane: methanol (20–50:1), to give 7a, yield 90%, White powder, m. p. 178.8–180.9 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.44 (s, 1H), 7.83 (dd, J = 8.4, 1.4 Hz, 1H), 7.22 – 7.18 (m, 2H), 7.20 (td, J = 7.3, 1.4 Hz, 1H), 6.86 (s, 1H), 4.13 (t, J = 2.0 Hz, 2H), 3.31 (t, J = 5.7 Hz, 2H), 3.08 (tt, J = 5.8, 2.0 Hz, 2H), 2.67 (q, J = 7.6 Hz, 2H), 1.25 (d, J = 7.6 Hz, 3H).
N-(4-bromophenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7b)
Compound (7b) was synthesized from compound (4b), in a manner similar to (7a). Yield: 92%, Light yellow powder, m. p. 178.9–181.7 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.52 (s, 1H), 7.58 – 7.46 (m, 4H), 6.94 (s, 1H), 4.71 (s, 2H), 3.85 (t, J = 5.7 Hz, 2H), 3.14 (d, J = 5.7 Hz, 2H), 1.51 (s, 8H).
N-(3-chlorophenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7c)
Compound (7c) was synthesized from compound (4c), in a manner similar to (7a). Yield: 90%, White powder, m. p. 157.6 – 159.6 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.54 (s, 1H), 7.83 (t, J = 2.1 Hz, 1H), 7.50 (ddd, J = 8.2, 2.2, 1.0 Hz, 1H), 7.30 (t, J = 8.1 Hz, 1H), 7.10 (ddd, J = 8.0, 2.0, 0.9 Hz, 1H), 7.04 (s, 1H), 4.13 (t, J = 2.0 Hz, 2H), 3.31 (t, J = 5.7 Hz, 2H), 3.07 (tt, J = 5.7, 2.0 Hz, 2H).
N-(m-tolyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7d)
Compound (7d) was synthesized from compound (4d), in a manner similar to (7a). Yield: 89%, White powder, m. p. 174.9–178.0 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.51 (s, 1H), 7.47 (dd, J = 8.0, 2.2 Hz, 1H), 7.43 (d, J = 1.8 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.00 (s, 1H), 6.97 (ddt, J = 7.6, 1.7, 0.8 Hz, 1H), 4.12 (t, J = 2.0 Hz, 2H), 3.31 (t, J = 5.7 Hz, 2H), 3.08 (tt, J = 5.7, 2.0 Hz, 2H), 2.39 (s, 3H)
N-(3-fluorophenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7e)
Compound (7e) was synthesized from compound (4e), in a manner similar to (7a). Yield: 93%, White powder, m. p. 146.6–150.9 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.54 (s, 1H), 7.74 (dt, J = 11.1, 2.3 Hz, 1H), 7.35 – 7.27 (m, 1H), 7.25 (s, 1H), 7.09 (s, 1H), 6.82 (tdd, J = 8.2, 2.5, 1.1 Hz, 1H), 4.12 (t, J = 2.2 Hz, 2H), 3.31 (td, J = 5.8, 3.0 Hz, 2H), 3.07 (tt, J = 5.8, 1.9 Hz, 2H).
N-(4-chlorophenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7f)
Compound (7f) was synthesized from compound (4f), in a manner similar to (7a). Yield: 92%, Light yellow powder, m. p. 171.3–176.0 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.50 (s, 1H), 7.69 – 7.51 (m, 2H), 7.40 – 7.28 (m, 2H), 7.00 (s, 1H), 4.11 (t, J = 2.0 Hz, 2H), 3.30 (t, J = 5.7 Hz, 2H), 3.06 (tt, J = 5.9, 2.0 Hz, 2H).
N-(3-methoxyphenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7g)
Compound (7g) was synthesized from compound (4g), in a manner similar to (7a). Yield: 90%, White powder, m. p. 179.2–180.5 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.52 (s, 1H), 7.44 (t, J = 2.3 Hz, 1H), 7.29 (d, J = 8.1 Hz, 1H), 7.11 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H), 7.04 (s, 1H), 6.69 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 4.11 (t, J = 2.0 Hz, 2H), 3.85 (s, 3H), 3.30 (t, J = 5.7 Hz, 2H), 3.07 (tt, J = 5.7, 2.0 Hz, 2H).
N-(3-isopropylphenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7h)
Compound (7h) was synthesized from compound (4h), in a manner similar to (7a). Yield: 90%, Light yellow powder, m. p. 144.7–151.6 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.50 (s, 1H), 7.53 (ddd, J = 8.1, 2.3, 1.1 Hz, 1H), 7.40 (t, J = 2.0 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.03 (dt, J = 8.0, 1.5 Hz, 2H), 4.12 (s, 2H), 3.31 (t, J = 5.7 Hz, 2H), 3.09 (tt, J = 5.7, 2.0, 2H), 2.94 (hept, J = 7.0 Hz, 1H), 1.29 (d, J = 7.0 Hz, 6H).
N-(2,4-dimethylphenyl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7i)
Compound (7i) was synthesized from compound (4i), in a manner similar to (7a). Yield: 89%, Light yellow powder, m. p. 164.6–169.0 °C, 1H NMR (400 MHz, Chloroform-d) δ 8.43 (s, 1H), 7.67 – 7.60 (m, 1H), 7.09 – 7.07 (m, 2H), 6.76 (s, 1H), 4.12 (t, J = 2.0 Hz, 2H), 3.30 (t, J = 5.7 Hz, 2H), 3.07 (tt, J = 5.7, 3.6, 2.0 Hz, 2H), 2.33 (s, 3H), 2.26 (s, 3H).
4-(piperidin-1-yl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine (7k)
Compound (7k) was synthesized from compound (4k), in a manner similar to (7a). Yield: 87%,White powder, 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 4.72 (s, 2H), 3.66 (s, 2H), 3.40 – 3.31 (m, 4H), 3.03 (s, 2H), 1.77 – 1.64 (m, 6H).
4-(pyrrolidin-1-yl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine (7l)
Compound (7l) was synthesized from compound (4l), in a manner similar to (7a). Yield: 88%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 4.65 (s, 2H), 3.70 (t, J = 6.6 Hz, 4H), 3.62 (s, 2H), 2.97 (s, 2H), 1.97 – 1.84 (m, 4H).
4-(pyrrolidin-1-yl)-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine (7m)
Compound (7m) was synthesized from compound (4m), in a manner similar to (7a). Yield: 89%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 4.71 (s, 2H), 3.90 – 3.81 (m, 4H), 3.67 (s, 2H), 3.51 – 3.40 (m, 4H), 3.02 (s, 2H).
N,N-dimethyl-5,6,7,8-tetrahydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-4-amine (7n)
Compound (7n) was synthesized from compound (4n), in a manner similar to (7a). Yield: 85%, White powder, 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 4.73 (s, 2H), 3.66 (t, J = 4.7 Hz, 2H), 3.04 (s, 6H), 3.02 (s, 2H).
(2,4-dihydroxy-5-isopropylphenyl)(4-((3-ethylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8a)
Compound (4a) (205 mg, 0.5 mmol) and Compound (6) (188 mg, 0.5 mmol) were dissolved in CH2Cl2 (15 ml) and then EDCI (144 mg, 0.5 mmol) and HOBt (101 mg, 0.5 mmol) was added, the mixture was stirred overnight. CH2Cl2 (50 ml) and H2O (50 ml) was added, the organic layer was separated and concentrated, the crude product was purified by silica gel column chromatography using dichloromethane: acetone (100:1), yield: 73%; The obtained compound (334 mg, 0.5 mmol) was dissolved in THF (10 ml) and then 10% Pd/C (35 mg, 10%mol) was added, the reaction mixture was catalytic hydrogenated for 24 h at room temperature in hydrogen atmosphere. The solvent was concentrated, the residue was purified by silica gel column chromatography using a mixture of dichloromethane: acetone (30:1), to give 8a, yield: 66%, White powder, m. p. 227.2–230.1 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 2H), 8.24 (s, 1H), 8.05 (s, 1H), 7.45 – 7.37 (m, 1H), 7.34 – 7.28 (m, 1H), 7.27 – 7.21 (m, 2H), 6.92 (s, 1H), 6.41 (s, 1H), 4.78 (s, 2H), 3.79 (s, 2H), 3.23 (t, J = 5.8 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 2.56 (q, J = 7.6 Hz, 2H), 1.12 – 1.09 (m, 9H). 13 C NMR (100 MHz, DMSO-d6) δ 168.92, 166.54, 157.13, 156.70, 153.45, 153.30, 140.39, 137.20, 129.40, 128.85, 128.10, 126.72, 126.56, 126.51, 126.33, 126.04, 115.77, 113.88, 102.74, 26.84, 26.34, 24.51, 23.11(2C), 14.44. HRMS (ESI) calculated for C27H28N4O3S [M + H]+ 489.1955, found 489.1953.
(4-((4-bromophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)(2,4-dihydroxy-5-isopropylphenyl)methanone (8b)
Compound (4b) (270 mg, 0.5 mmol) and Compound (6) (188 mg, 0.5 mmol) were dissolved in CH2Cl2 (15 ml) and then EDCI (144 mg,0.5 mmol) and HOBt (101 mg, 0.5 mmol) was added, the mixture was stirred overnight. CH2Cl2 (50 ml) and H2O (50 ml) was added, the organic layer was separated and concentrated, the crude product was purified by silica gel column chromatography using dichloromethane: acetone (100:1), yield: 73%; The obtained compound (334 mg, 0.5 mmol) was dissolved in CH2Cl2 (10 ml) and then (CH3)3SiI (1.00g, 0.68 ml, 0.5 mmol) was added, the reaction mixture was stirred for 24 h at room temperature. Excessive (CH3)3SiI was quenched by MeOH, the mixture was concentrated, and the residue was purified by silica gel column chromatography using a mixture of dichloromethane: acetone (30:1), to give 8b, yield: 33%, Brown powder, m. p. 248.8–250.2 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 8.44 (s, 1H), 8.35 (s, 1H), 7.62 (d, J = 8.6 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H), 6.91 (s, 1H), 6.41 (s, 1H), 4.79 (s, 2H), 3.77 (s, 2H), 3.26 (t, J = 5.7 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.87, 166.63, 157.10, 155.12, 153.30, 152.62, 138.92, 131.67(2C), 130.55, 126.58, 126.37, 126.05, 124.60(2C), 116.91, 115.65, 113.93, 102.68, 26.56, 26.35, 23.10(2C). HRMS (ESI) calculated for C25H23BrN4O3S [M + H]+ 539.0747, found 539.0742
(4-((3-chlorophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)(2,4-dihydroxy-5-isopropylphenyl)methanone (8c)
Compound (8c) was synthesized from compound (4c), in a manner similar to (8b). Yield: 35%, White powder, m. p. 189.9–190.6 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 8.48 (s, 1H), 8.37 (s, 1H), 7.81 (t, J = 2.0 Hz, 1H), 7.62 (dd, J = 7.5, 1.8 Hz, 1H), 7.37 (t, J = 8.1 Hz, 1H), 7.13 (dd, J = 7.9, 1.9 Hz, 1H), 6.92 (s, 1H), 6.41 (s, 1H), 4.80 (s, 2H), 3.78 (s, 2H), 3.25 (t, J = 5.7 Hz, 2H), 3.08 (hept, J = 7.0 Hz, 1H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.88, 167.14, 157.14, 155.01, 153.35, 152.84, 141.22, 133.19, 130.67, 130.45, 126.61, 126.30, 126.07, 123.27, 121.62, 120.67, 117.01, 113.94, 102.71, 26.55, 26.38(2C), 23.10. HRMS (ESI) calculated for C25H23ClN4O3S [M + H]+ 495.1252, found 495.1253.
(2,4-dihydroxy-5-isopropylphenyl)(4-(m-tolylamino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8d)
Compound (8d) was synthesized from compound (4d), in a manner similar to (8a). Yield: 65%, White powder, m. p. 157.6–160.2 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 8.41 (s, 1H), 8.15 (s, 1H), 7.48 (dd, J = 7.9, 2.1 Hz, 1H), 7.43 (t, J = 1.9 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 6.92 – 6.90 (m, 2H), 6.41 (s, 1H), 4.79 (s, 2H), 3.77 (s, 2H), 3.26 (t, J = 5.7 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 2.31 (s, 3H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.87, 166.80, 157.12, 155.49, 153.35, 153.02, 139.50, 138.10, 130.06, 128.75, 126.60, 126.36, 126.06, 124.61, 123.04, 119.73, 116.67, 113.96, 102.70, 26.62, 26.38, 23.10(2C), 21.59. HRMS (ESI) calculated for C26H26N4O3S [M + H]+ 475.1798, found 475.1796.
(2,4-dihydroxy-5-isopropylphenyl)(4-((3-fluorophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8e)
Compound (8e) was synthesized from compound (4e), in a manner similar to (8b). Yield: 34%, White powder, m. p.218.1–219.2 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 8.50 (s, 1H), 8.44 (s, 1H), 7.63 (dt, J = 11.7, 2.2 Hz, 1H), 7.45 (dd, J = 8.1, 1.9 Hz, 1H), 7.38 (td, J = 8.1, 6.6 Hz, 1H), 6.94 – 6.89 (m, 2H), 6.41 (s, 1H), 4.80 (s, 2H), 3.78 (s, 2H), 3.27 (t, J = 5.7 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.91, 165.42, 162.47 (d, J = 240 Hz, 1C), 157.14, 155.04, 153.32, 151.96, 141.00 (d, J = 11 Hz, 1C), 131.06, 130.50(d, J = 9 Hz, 1C), 126.54 (d, J = 13 Hz, 1C), 126.08, 118.51, 118.50, 117.05, 113.88, 110.65 (d, J = 21 Hz, 1C), 109.48 (d, J = 25 Hz, 1C), 102.69, 26.55, 26.37, 23.11(2C). HRMS (ESI) calculated for C25H23FN4O3S [M + H]+ 479.1548, found 479.1550.
(4-((4-chlorophenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)(2,4-dihydroxy-5-isopropylphenyl)methanone (8f)
Compound (8f) was synthesized from compound (4f), in a manner similar to (8b). Yield: 36%, White powder, m. p. 178.0–182.0 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 2H), 8.43 (s, 1H), 8.34 (s, 1H), 7.75 – 7.62 (m, 2H), 7.46 – 7.32 (m, 2H), 6.92 (s, 1H), 6.44 (s, 1H), 4.80 (s, 2H), 3.78 (s, 2H), 3.27 (t, J = 5.8 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.91, 167.02, 157.18, 155.20, 153.40, 152.86, 138.63, 130.43, 128.72(2C), 127.47, 126.58, 126.34, 126.03, 124.11(2C), 116.87, 113.91, 102.74, 26.59, 26.37, 23.11(2C). HRMS (ESI) calculated for C25H23ClN4O3S [M + H]+ 495.1252, found 495.1249.
(2,4-dihydroxy-5-isopropylphenyl)(4-((3-methoxyphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8g)
Compound (8g) was synthesized from compound (4g), in a manner similar to (8a). Yield: 67%, White powder, m. p. 220.4–221.2 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 8.43 (s, 1H), 8.20 (s, 1H), 7.34 – 7.28 (m, 1H), 7.25 (d, J = 4.9 Hz, 2H), 6.92 (s, 1H), 6.67 (td, J = 4.7, 2.6 Hz, 1H), 6.41 (s, 1H), 4.79 (s, 2H), 3.86 – 3.66 (m, 5H), 3.26 (t, J = 5.7 Hz, 2H), 3.08 (hept, J = 6.8 Hz, 1H), 1.12 (d, J = 6.8 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.88, 166.89, 159.91, 157.14, 155.36, 153.37, 152.94, 140.77, 130.24, 129.61, 126.60, 126.33, 126.07, 116.82, 114.73, 113.94, 109.16, 108.25, 102.71, 55.55, 26.58, 26.38, 23.10(2C). HRMS (ESI) calculated for C26H26N4O4S [M + H]+ 491.1748, found 491.1744.
(2,4-dihydroxy-5-isopropylphenyl)(4-((3-isopropylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8h)
Compound (8h) was synthesized from compound (4h), in a manner similar to (8a). Yield: 60%, White powder, m. p. 209.5–212.4 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 8.40 (s, 1H), 8.18 (s, 1H), 7.53 (d, J = 9.0 Hz, 1H), 7.44 (t, J = 2.0 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 6.98 (dt, J = 7.7, 1.3 Hz, 1H), 6.92 (s, 1H), 6.41 (s, 1H), 4.79 (s, 2H), 3.78 (s, 2H), 3.24 (t, J = 6.5 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 2.87 (hept, J = 6.9 Hz, 1H), 1.22 (d, J = 6.9 Hz, 6H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.86, 166.79, 157.12, 155.54, 153.36, 153.01, 149.25, 139.50, 130.01, 128.73, 126.59, 126.39, 126.05, 121.97, 120.59, 120.40, 116.63, 113.95, 102.71, 33.94, 26.64, 26.37, 24.35(2C), 23.10(2C). HRMS (ESI) calculated for C28H30N4O3S [M + H]+ 503.2111, found 503.2112.
(2,4-dihydroxy-5-isopropylphenyl)(4-((2,4-dimethylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8i)
Compound (8i) was synthesized from compound (4i), in a manner similar to (8a). yield: 63%, White powder, m. p. 254.3–255.4 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 9.57 (s, 1H), 8.24 (s, 1H), 7.98 (s, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.09 (s, 1H), 7.03 (d, J = 7.4 Hz, 1H), 6.92 (s, 1H), 6.40 (s, 1H), 4.77 (s, 2H), 3.79 (s, 2H), 3.23 (t, J = 5.7 Hz, 2H), 3.08 (hept, J = 6.9 Hz, 1H), 2.29 (s, 3H), 2.13 (s, 3H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.89, 166.45, 157.10, 156.43, 153.42, 153.30, 135.31, 135.22, 134.22, 131.29, 129.28, 127.12, 127.07, 126.54, 126.39, 126.04, 115.82, 113.91, 102.71, 26.85, 26.35, 23.10(2C), 21.04, 18.45. HRMS (ESI) calculated for C27H28N4O3S [M + H]+ 489.1955, found 489.1953.
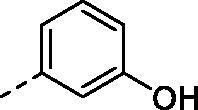
(2,4-dihydroxy-5-isopropylphenyl)(4-((3-hydroxyphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8j)
Compound (8j) was synthesized from compound (4g), in a manner similar to (8b) 48 h. yield: 25%, Yellow powder, m. p. 146.4–148.2 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 2H), 9.37 (s, 1H), 8.42 (s, 1H), 8.12 (s, 1H), 7.18 (s, 1H), 7.11 (t, J = 8.0 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.92 (s, 1H), 6.50 (d, J = 7.9 Hz, 1H), 6.41 (s, 1H), 4.78 (s, 2H), 3.76 (s, 2H), 3.24 (s, 2H), 3.08 (p, J = 7.0 Hz, 1H), 1.12 (d, J = 7.0 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ 168.85, 166.81, 157.94, 157.11, 155.44, 153.35, 152.98, 140.62, 130.10, 129.52, 126.58, 126.41, 126.05, 116.79, 113.95, 113.16, 111.01, 109.48, 102.70, 26.57, 26.36, 23.11(2C). HRMS (ESI) calculated for C25H24N4O4S [M + H]+ 477.1591, found 477.1591.
(2,4-dihydroxy-5-isopropylphenyl)(4-(piperidin-1-yl)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8k)
Compound (8k) was synthesized from compound (4k), in a manner similar to (8a). yield: 67%, White powder, m. p. 196.1–198.5 °C, 1H NMR (400 MHz, DMSO) δ 9.60 (s, 1H), 9.58 (s, 1H), 8.47 (s, 1H), 6.96 (s, 1H), 6.42 (s, 1H), 4.81 (s, 2H), 3.64 (s, 2H), 3.33 (s, 4H), 3.11 – 3.03 (m, 3H), 1.64 (s, 6H), 1.12 (d, J = 6.7 Hz, 6H). 13 C NMR (101 MHz, DMSO) δ 169.12, 168.16, 162.38, 157.04, 153.45, 152.21, 130.84, 127.14, 126.43, 126.03, 119.85, 114.07, 102.74, 51.41 (s, 2C), 33.12, 29.47, 27.60, 26.29, 25.65 (s, 2C), 24.26 (s), 23.10 (s, 2C). HRMS (ESI) calculated for C25H24N4O4S [M + H] + 452.1882, found 452.1879.
(2,4-dihydroxy-5-isopropylphenyl)(4-(pyrrolidin-1-yl)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8l)
Compound (8l) was synthesized from compound (4l), in a manner similar to (8a). yield: 69%, White powder, m. p. 185.6–188.9 °C, 1H NMR (400 MHz, DMSO) δ 9.60 (s, 1H), 9.56 (s, 1H), 8.27 (s, 1H), 6.95 (s, 1H), 6.41 (s, 1H), 4.77 (s, 2H), 3.63 (s, 6H), 3.13 – 3.04 (m, 1H), 3.00 (s, 2H), 1.85 (s, 4H), 1.12 (d, J = 6.7 Hz, 6H). 13 C NMR (101 MHz, DMSO) δ 168.96, 167.85, 158.16, 157.02, 153.32, 151.68, 128.42, 127.17, 126.56, 126.03, 116.45, 114.18, 102.69, 50.91 (s, 2C), 31.75, 29.90, 29.48, 26.32, 25.56 (s, 2C), 23.09 (s, 2C). HRMS (ESI) calculated for C25H24N4O4S [M + H]+ 438.1726, found 438.1730.
(2,4-dihydroxy-5-isopropylphenyl)(4-morpholino-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8m)
Compound (8m) was synthesized from compound (4m), in a manner similar to (8a). yield: 65%, White powder, m. p. 183.4–185.1 °C, 1H NMR (400 MHz, DMSO) δ 9.60 (s, 1H), 9.58 (s, 1H), 8.53 (s, 1H), 6.97 (s, 1H), 6.42 (s, 1H), 4.82 (s, 2H), 3.75 (t, J = 3.7 Hz, 4H), 3.66 (s, 2H), 3.38 (t, J = 3.7 Hz, 4H), 3.16 – 3.02 (m, 3H), 1.13 (d, J = 6.9 Hz, 6H). 13 C NMR (101 MHz, DMSO) δ 169.10, 168.40, 161.91, 157.07, 153.49, 152.22, 131.54, 126.87, 126.47, 126.04, 119.99, 114.02, 102.74, 66.26 (s, 2C), 50.90 (s, 2C), 31.13, 29.48, 27.50, 26.30, 23.09 (s, 2C). HRMS (ESI) calculated for C25H24N4O4S [M + H]+ 454.1675, found 454.1677.
(2,4-dihydroxy-5-isopropylphenyl)(4-(dimethylamino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidin-7(6H)-yl)methanone (8n)
Compound (8n) was synthesized from compound (4n), in a manner similar to (8a). Yield: 66%, White powder, m. p. 189.1–192.2 °C, 1H NMR (400 MHz, DMSO) δ 9.59 (s, 1H), 9.56 (s, 1H), 8.40 (s, 1H), 6.95 (s, 1H), 6.40 (s, 1H), 4.80 (s, 2H), 3.60 (s, 2H), 3.14 – 2.99 (m, 9H), 1.12 (d, J = 6.9 Hz, 6H). 13 C NMR (101 MHz, DMSO) δ 169.05, 168.07, 161.89, 157.01, 153.34, 151.85, 130.02, 127.21, 126.52, 126.03, 118.42, 114.20, 102.70, 41.95 (s, 2C), 31.15, 29.48, 28.43, 26.31, 23.10 (s, 2C). HRMS (ESI) calculated for C25H24N4O4S [M + H]+ 412.1569, found 412.1566.
2.4. Cell proliferation assay
Cells were grown as monolayers at 37 °C in a humidified atmosphere of 5% CO2. To remove adherent cells from the flask for subculturing and counting, cells were washed with PBS without calcium or magnesium, incubated in a small volume of 0.25% trypsin–EDTA solution for 5–10 min, resuspended with culture medium, and centrifuged. All experiments were carried out using cells in the exponential growth phase. Growth inhibition was assessed using an MTT assay as described previously. Cells were seeded (3–6 × 103 cells/well) in 96-well plates with 200 μL complete culture medium for 24 h, followed by the treatments of either DMSO (control) or various concentrations of 8m for 24 or 48 h. After each time point, 200 μL of the culture medium containing 0.5 mg/mL MTT was added to each well and incubated for 3 h at 37 °C. After removal of the supernatant, DMSO was added to the wells and the absorbance was read at 570 nm. All experiments were performed three times in triplicate.
2.5. Cell migration and invasion assays
Cell migration assay was performed using the agarose wound-healing method. In brief, B16 cells were seeded in 6-well plate with DMEM for 1 × 105 cells per well and was further grown overnight. Three lines were cut in each well using a 200 µl pipette tip. Cells were then washed with PBS and serum-free DMEM including various concentrations of drugs or DMSO were added. Evaluation of the cell migration was determined under a light microscope between 0 h and 24 h of treatment.
For the invasion assay, we used Transwell chambers (8-μm pore size, Corning). Specifically, 1 × 105 of B16 cells were suspended in serum-free medium, and then loaded into the Matrigel precoated top-chambers (BD Bioscience, San Jose, CA, USA) and incubated with DMEM culture in the bottom chamber. Crystal violet staining was used to measure the invasiveness of B16 cells. Cell counts were performed in triplicate under a microscope.
2.6. Western blotting analysis
The total protein was extracted from B16 cells, separated on SDS PAGE gel (7.5–15%, according to the molecular weight of protein) and then transferred to PVDF membranes (Millipore, MA, USA). The transferred PVDF membranes were blocked by 5% BSA and then incubated with primary antibodies under ice bath overnight, then the secondary antibodies were added for one hour incubation at room temperature. Specific protein bands were visualized using ECL Western Blotting Substrate (Millipore, MA, USA) and imaged by Chemi Scope 3300 Mini (Clinx Science Instruments, Shanghai, China).
2.7. Subcutaneous tumor xenograft model
Female C57BL/6J mice with 8–12 weeks old were purchased from Beijing Vitalriver Co. Ltd. (Beijing, China). All animals were housed in the SPF animal facility at State Key Laboratory of Biotherapy in individually ventilated cages. The experimental protocols were in compliance with the guidelines of the Animal Ethics Committee (Sichuan University) and were approved by the Animal Ethics Committee of West China Hospital of Sichuan University. Tumor cells lines B16 were injected subcutaneously in the dorsal flank of C57BL/6J mice as single cell suspensions (5 × 105 in 100 μL) in PBS. The tumor sizes were measured every two days from the day after injection of cancer cells. Calculation of tumor volume was according to Vol = a×b2×0.5, where a is the large diameter tumor tissue and b is the small diameter tumor tissue. The tumor tissues were stripped, fixed in formalin, then embedded in paraffin, and sectioned. Tumor tissue sections were stained with TUNEL, EGFR, Akt, pAkt473 and Ki67 for further histologic examination.
3. Results and discussion
3.1. Hsp90 and protein kinases are overexpressed in melanoma
We first observed an Hsp90 mRNA expression pattern in 33 cancer subtypes using the TCGA database30. Hsp90 was significantly overexpressed in ten cancers (fold change > 2.0, p < .01), including cutaneous melanoma (Figure 1(A)). In total, 461 HCC cutaneous melanoma tissues and 558 adjacent or normal skin tissues from the TCGA and GTEx databases were included in this study4. Hsp90 expression levels were significantly upregulated in melanoma tissues compared with normal skin tissues (p < .001, Figure 1(B)). Somatic mutation and expression analysis of Hsp90 in 470 cutaneous melanoma patients suggested that Hsp90 expression dysregulation occurred in about 25% of melanoma patients, only four patients had Hsp90 copy number alterations (one for amplification and three for deep deletion, Figure 1(C))31.
Figure 1.

The mRNA expression pattern of Hsp90 in TCGA (The Cancer Genome Atlas) database, its genetic alteration from cBioPortal web server and protein expression changes from proteinatlas database. (A) The logarithmic data of the Hsp90 mRNA expression TPM (transcripts per million reads) values presented by GEPIA (Gene Expression Profiling Interactive Analysis) database; (B) The mRNA levels in melanoma or normal skin tissues in TCGA and GTEx database; (C) The genetic alterations of Hsp90 were analyzed in 470 melanoma samples based on cBioPortal web server, Hsp90 DNA copy number amplified in one patient and deep deleted in three patients, and Hsp90 mRNA upregulated in 38 cases but downregulated in 74 cases; (D) The Hsp90 mRNA levels in melanoma patients with different stages; (E) The Kaplan–Meier plot of overall survival in melanoma patients with different Hsp90 mRNA levels; (F) The representative Hsp90 protein levels between melanoma and normal skin tissues detected by IHC.
Although there was no difference in expression between T1 to T4 stage melanoma tissues, both had higher expression compared to T0 melanoma tissues (Figure 1(D)). Moreover, there were no significant differences observed in total survival ratios between Hsp90 groups with high expression vs low expression (Figure 1(E)). In order to further validate the protein expression levels of Hsp90 (Figure 1(F)) and protein kinases (Figure 2(A)), expression of Hsp90 and several protein kinases were determined by IHC immunohistochemistry (IHC) of melanoma tissue microarray from the Protein Atlas database32. Relative expression levels of Hsp90 and protein kinases (such as c-Kit, VEGFR2, EGFR and PI3Kα) in both melanoma and normal skin tissues are illustrated in Figure 2(C,D). Both Hsp90 and these protein kinases were overexpressed in melanoma tissues compared to normal skin tissues.
Figure 2.

Identification of overexpressed protein kinases and their relationship to Hsp90 in melanoma tissues. (A) The representative IHC images of VEGFR2, c-Kit, EGFR and PI3Kα in melanoma and normal skin tissues; (B) Kaplan–Meier analysis of overall survival in melanoma patients, the median expression levels of Hsp90 and protein kinases were used as cutoff; (C) The correlation of Hsp90 and PI3Kα mRNA expressions in melanoma patients.
Considering that the oncogenic function of Hsp90 is often related to its chaperone proteins, survival analysis was further performed in Hsp90 combining with several protein kinases (Figure 2(B)). The total survival ratio of those with high levels of Hsp90-kinase expression were significantly worse than those with lower expression.33,34 Correlated expression profile of Hsp90 and protein kinases were also analyzed, PI3Kα was utilized as an example kinase to calculate the Pearson correlation coefficient with Hsp90. A high coefficient of 0.51 was determined to be suggestive of a strong relationship between PI3Kα and Hsp90 (Figure 2(C)).
3.2. Design and synthesis of novel Hsp90/protein kinases inhibitors
Some representative chemical structures of Hsp90 inhibitors, PI3Kα inhibitors and multi-kinase inhibitors are shown in Figure 3(A). The 2,4-dihydroxy-5-isopropylbenzate fragment was observed to be common in a panel of synthetic Hsp90 inhibitors. A 4-substituted pyrimidine fragment is a known scaffold for multi-kinase inhibitors. A2 or 4-alkylation substituted pyrimidine was common in PI3K inhibitors, and the 4-aromatic substituted pyrimidine was suited for tyrosine protein kinases, such as EGFR, VEGFR, c-Kit, etc. Therefore, the role of the 2,4-dihydroxy-5-isopropylbenzate fragment was to maintain binding specificity to Hsp90, and the role of the 4-aromatic substituted pyrimidine fragment was to achieve the potent binding to protein kinases, respectively (Figure 3(B)). The piperido[4’,3’:4,5]thieno[2,3-d]pyrimidine scaffold utilized conjugated Hsp90 and protein kinase binding fragments. The Hsp90 binding fragment could easily form an amide bind to the nitrogen atom of piperidine ring, and the 4-substituted thieno[2,3-d]pyrimidine was a known scaffold to protein kinase inhibitors35–38.
Figure 3.

(A) Representative structures of Hsp90 inhibitors, PI3K inhibitors and multikinase inhibtors; (B) Depiction of the design method of Hsp90/protein kinase dual inhibitors.
The synthetic process for novel Hsp90/protein kinase inhibitors is depicted in Scheme 1. In brief, 6-(tert-butyl) 3-ethyl 2-amino-4,7-dihydrothieno[2,3-c] pyridine-3,6(5H)-dicarboxylate (1) was synthesized from sulfur (3.20 g, 100.0 mmol) and ethyl cyanacetate (11.32 g, 100.0 mmol), into N-Boc-piperidin-4-one (19.90 g, 100.0 mmol) and refluxed for 12 h, then form amidine acetate (6.24 g, 60.0 mmol) and compound 1 (13.00 g, 40.0 mmol) was stirred in DMF for 12 h at 100 °C to give the pyrimidine-4-one ring 2. The pyrimidine-4-one (15.35 g, 50.0 mmol) was transformed to 4-Cl-pyrimidine 3 by SO2Cl2 (100 ml) and DMF (2 ml), and then the 2-ethylaniline (242.39 mg, 2.0 mmol), compound 3 (327.82 mg, 1.0 mmol) and NaH (72 mg, 3 mmol) were stirred in anhydrous 1,4-dioxane(20 ml) for 2 h under 90 °C to achieve tert-butyl 4-((2-ethylphenyl)amino)-5,8-dihydropyrido[4',3':4,5]thieno[2,3-d]pyrimidine-7(6H)-carboxylate (intermediates 4a). For synthesis of 2,4-dihydroxy-5-isopropylbenzoic acid fragment, we used methyl 2,4-dihydroxybenzoate (1.68 g, 10 mmol) as start compound, after the protection of hydroxyl group by benzyl chloride (2.53 ml, 22 mmol), and potassium carbonate (3.20 g, 22 mmol), the 5-isopropyl group was introduced by isopropyl bromide and DIBAL-H to form intermediates 6. The N-Boc group of intermediates 4a was removed by TFA in dichloromethane, and then conjugated to intermediates 6 by the catalyzed by EDC (1.5M) and HOBt (1.5M) in THF under room temperature. After removal of benzyl protection groups of 2,4-dihydroxybenzoate by hydrogen atmosphere and 10% Pd-C or by Me3SiI (8M) in dichloromethane (especially for the structures contained halogen) at room temperature for 48 h., ultimately the targeted compounds 8a–n were successfully synthesized, and characterized by 1H NMR, 13 C NMR and mass spectrum to validate their chemical structures.
Scheme 1.

Synthesis of Compounds 8a–8n.
Reagents and conditions: (a) NCCH2CO2Et, S8, Et3N, EtOH, reflux, 12h; (b) formamidine acetate, DMF, 100 °C,16h; (c) SO2Cl2, reflux, 4h; (d) Aromatic amine, NaH, 1,4-dioxane, 90 °C, 2 h or aliphatic amine, self-solvent, 100 °C, 1 h; (e) 9 N HCl, ethyl acetate, 25 °C, 30 min; (f) aluminum chloride, CH2Cl2, 50 °C, 24 h; (g) benzyl chloride, K2CO3, DMF; LiOH·H2O, 25 °C, 12h; (h) EDCI, HOBt, DCM, overnight and then trimethyliodosilane (TMIS), 25 °C, 24 h or H2, 10% Pd/C, 25 °C, 24 h.
3.3. Structure-activity relationship (SAR) analysis and binding modes of novel Hsp90/PI3Kα dual inhibitors
Protein kinases and the cytotoxicity of B16 melanoma inhibitory potencies of compounds 8a–8n are shown in Table 1. These data include the kinase activity of Hsp90 and several kinases including EGFR, VEGFR2 and PI3Kα, as well as the cellular viability of the B16 melanoma cell line. The half of the maximal inhibitory concentrations (IC50) values for these compounds on Hsp90 suggested that the 4-alkylation substitution was a more favorable mechanism than of 4-aromatic substitution. For compounds 8a–8j, different substituted groups on the 4-phenyl ring resulted different kinase binding potencies and selectivity. Specifically, the 2,4-dimethylphenyl or 3-hydroxylphenyl group at 4-position of thieno[2,3-d]pyrimidine scaffold displayed moderate VEGFR2 inhibitory activities, but did not contribute to B16 cytotoxicity. In general, the Hsp90/PI3Kα dual inhibitor based on 4-alkylation thieno[2,3-d]pyrimidine scaffold demonstrated higher inhibition in Hsp90 and B16 cell proliferation assays than that of Hsp90/RTKs inhibitors. Finally, compound 8m displayed the strongest inhibitory capacity of Hsp90 and PI3Kα with IC50 values of 38.6 and 48.4 nM, respectively.
Table 1.
Kinase inhibition activities and cytotoxicity of compounds 8a–8m.
| |||||||
|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | Hsp90α IC50(nM)a,b | EGFR IC50(nM)a,b | VEGFR2 IC50(nM)a,b | PI3Kα IC50(nM)a,b | B16 GI ratio (%)b |
| 8a |
|
H | 375.3 | 60.7 | >10000 | N.D. | 26.8 |
| 8b |
|
H | 289.6 | 49.9 | >10000 | N.D. | 33.6 |
| 8c |
|
H | 196.3 | 37.1 | >10000 | N.D. | 51.2 |
| 8d |
|
H | 318.2 | 51.8 | >10000 | N.D. | 41.7 |
| 8e |
|
H | 229.0 | 40.2 | >10000 | N.D. | 38.9 |
| 8f |
|
H | 318.5 | 76.6 | >10000 | N.D. | 29.5 |
| 8g |
|
H | 210.4 | 71.0 | >10000 | N.D. | 37.5 |
| 8h |
|
H | 468.2 | 380.1 | >10000 | N.D. | 39.1 |
| 8i |
|
H | 1637.8 | 578.4 | 169.5 | N.D. | 24.8 |
| 8j |
|
H | 207.4 | 146.5 | 715.9 | N.D. | 36.0 |
| 8k |

|
51.9 | N.D. | N.D. | 132.6 | 65.2 | |
| 8l |

|
65.0 | N.D. | N.D. | 168.9 | 60.4 | |
| 8m |

|
38.6 | N.D. | N.D. | 48.4 | 72.5 | |
| 8n | Me | Me | 79.5 | N.D. | N.D. | 567.5 | 46.7 |
IC50 values are determined from KinaseProfiler of Eurofins for Hsp90 and HTRF based method for PI3Kα. The data represent the mean values of three independent experiments.
Each compound is tested in triplicate; the data are presented as the mean values.
The possible binding conformations of 8m to the binding sites of Hsp90 and PI3Kα are depicted in Figure 4. From Figure 4(A,B), it can be observed that 2,4-dihydroxybenzoate could form a stable hydrogen bond with Asp93 of Hsp90, and the thieno[2,3-d]pyrimidine scaffold formed a pi-cation interaction to Lys58. Moreover, 8m formed three stable hydrogen bonds to PI3Kα, at Lys802, Ala805 and Val882. Additionally, 2,4-dihydroxyl-5-isopropyl benzoate formed a pi-cation interaction with Lys890. It was notable that compound 8m also formed stable hydrogen bonds and pi-cation interactions to both Hsp90 and PI3Kα, which might be the source of the better inhibition activity on Hsp90 and PI3Kα than the other inhibitors.
Figure 4.

The binding modes of compound 8m to kinase domains of Hsp90 and PI3Kα, respectively. (A) the surface of binding pocket contoured by the atomic charge of Hsp90; (B) The 2D diagrams of interaction residues between compound 8m and Hsp90; (C) the surface of binding pocket contoured by the atomic charge of PI3Kα; D. The 2D diagrams of interaction residues between compound 8m and PI3Kα.
3.4. Novel Hsp90/PI3Kα dual inhibitors 8m suppressed cell B16 melanoma cell proliferation via inducing apoptosis
The cytotoxic activity of 8m was initially evaluated in several cancer cells and was most potent in melanoma cells (Figure 5(A)). Then we treated a B16 melanoma cell line with 8m for a 24 or 48-h incubation. Our results suggested that IC50 were around 0.56 and 0.47 μg/mL, respectively (Figure 5(B)). Furthermore, results of clone formation assay in 8m-incubated B16 cells indicated a dose-dependent decline manner compared to control group in both the size and number of the colonies (Figure 5(C)).
Figure 5.

The Hsp90/PI3Kα dual inhibitor 8m inhibits the proliferation and induces apoptosis of cancer cell lines (A and B); compound 8m potently inhibited colony formation (B); induced apoptotic cell death (D and E) and the changes on expression levels of apoptosis related proteins (F).
In order to assess the possibility of subroutine cell death underlying 8m treatment, flow cytometry with annexinV/PI dual staining was used to assess the activation of apoptosis39–45. Hoechst 33258 staining also clearly showed that nuclear morphological changes in apoptotic cells, including chromatin condensation and apoptotic bodies, mediated apoptosis (Figure 5(D)). There was a significant increase in the apoptotic positive cells under 24 h compound 8m incubation (Figure 5(E)). As western blot analysis results shown, apoptosis was initiated through the intrinsic pathway and the extrinsic pathway by the treatment of 8m in B1 cells with the cleavage of Caspase-3, Caspase-8, Caspase-9 and PARP (Figure 5(F)).
Compound 8m also induced cell cycle arrest in B16 cells. Cell counts in G2 phase were significantly increased after 8m treatment 8m for 24 h, and cell counts in G1 phase demonstrated a corresponding descent (Figure 6(A)). These results were confirmed by overexpression of p53, p21, p27 proteins and decrease of mdm2, c-myc, CDK2 and CDK6 proteins compared to NS group (Figure 6(D)).
Figure 6.

Compound 8m-induced changes of cell cycle arrest, cell invasion and migration and changes on related proteins. (A) The cell cycle analysis under treatment of various doses of 8m; (B) Changes on cell invasion under treatment of various doses of 8m by transwell assay; (C) Changes on cell migration under treatment of various doses of 8m by wound healing assay; (D) The changes on expression levels of cell cycle related proteins; (E) The changes on expression levels of EMT related proteins.
3.5. 8m inhibited cell migration and invasion in B16 cells
Cellular migration and invasion inhibitory potencies of 8m were determined by Transwell and agarose wound healing assays using B16 cells. For cell invasion assays assays based on Matrigel, Transwells were used to observe a remarkable decrease in B16 cells after incubation with 8m in a dose-dependent manner (Figure 6(B)). The wound areas wounds in 6-well culture plates had minimal changes after applying 8m in a dose-dependent manner after 24 h of exposure. Contrastingly, control plates were significantly healed (Figure 6(C)). Furthermore, the expression levels of tumor migration- and invasion-associated proteins at different concentrations of 8m (20, 40 μg/mL) were examined by western blot. We observed that 8m remarkably down-regulated MMPs, vimentin, N-cadherin, β-catenin, ZEB1 and up-regulated E-cadherin expressions through phenotypic analysis in B16 cells (Figure 6(E)). Taken together, the above results indicated that 8m markedly inhibited the migration and invasion of B16 cells.
3.6. 8m function via PI3K-Akt pathway and signaling pathways associated to Hsp90 client proteins
In order to further validate the dual inhibitory effects on Hsp90/PI3Kα and molecular mechanisms of compound 8 m, the expression levels of total and phosphorylated PI3Kα and Hsp90 family proteins were determined by western blotting of B16 melanoma cell extracts (Figure 7(A)). As expected, 8m indeed inhibited the autophosphorylation of PI3Kα in B16 cells without influencing levels of total PI3Kα. However, the expression levels of Hsp90α and Hsp70 declined with 8m incubation, which partially contributed to 8m inhibition of the interaction between Hsp90 complexed to its client proteins46–48. Western blot analysis of the Hsp90 client proteins demonstrated that the expression levels of many canonical clients, such as cdc37, CDK4, EGFR, bRaf and cRaf were remarkably suppressed by 8m treatment without significantly altering their phosphorylation ratio (Figure 7(B)).
Figure 7.

The molecular mechanisms of compound 8m to Hsp90 and PI3Kα kinase and downstream signaling pathways. (A) The changes on expression or phosphorylation levels of PI3Kα or Hsp90 protein family; (B) The changes on expression or phosphorylation levels of Hsp90 client proteins; (C) The changes on expression or phosphorylation levels of proteins in downstream MAPK signaling pathway; (D) The changes on expression or phosphorylation levels of proteins in downstream PI3K-Akt signaling pathway.
MAPK and PI3K-Akt signaling pathways were important signaling pathways associated with Hsp90 client proteins, which stimulated cell proliferation, oncogene transcription, cell migration and invasion. Western blot analysis showed that 8m inhibited the expression of c-Myc, ERK1/2 and JNK, and suppressed the activation of ERK-MNK/p90RSK signaling axis (Figure 7(C)). In addition, 8m downregulated p-Akt (Ser473), p-Akt (Thr308) and total Akt expression as the results of PI3Kα inhibition, which caused significant expression changes of downstream proteins, such as Bim and MDM2-p53 complex. Taken together, these results indicated that 8m could potently regulate PI3K-Akt signaling as well as Hsp90 client proteins and their downstream effectors so as to inhibit overall melanoma progression.
3.7. 8m suppressed tumor growth by targeting Hsp90 and PI3Kα in vivo
In order to evaluate the antitumor activity of 8m in vivo, a B16 subcutaneous xenograft model was used. We performed this in vivo study using three agents: PI3K inhibitor PI-103, Hsp90 inhibitor Ganetespib and 8m. It was clear that 8m possessed superior tumor growth inhibition as 73%, 57% and 29% tumor growth inhibition ratio were observed, respectively (Figure 8(A)). Moreover, there were remarkable declines in weight decline in all groups (p < .01) (Figure 8(B)).
Figure 8.

8m has an antitumor effect in mouse xenograft models via inducing apoptosis. (A) Mice bearing B16 (n = 8 per group) melanoma xenograft were treated with NS control, PI-103, Ganetespib or 8m once a day. Tumor volume data were presented as mean ± SD. (B) Tumor weight change of mice in different group: (∗) p < 0.05; (∗∗) p < 0.01. (C) Immunohistochemical analysis of EGFR, Akt, p-Akt473, proliferative marker Ki-67 and apoptosis marker TUNEL. Representative images of positive staining are shown.
To further validate whether 8m-mediated tumor growth inhibition was associated with suppressed cell proliferation and Hsp90/PI3Kα dual inhibition, tumor tissues from all groups were processed for the immunofluorescence and IHC analysis using TUNEL and Ki-67, EGFR, Akt and pAkt473, respectively. In all therapeutic groups, we found that 8m achieved had the highest number of apoptotic positive cells, most significant suppression of Ki-67 (p < .01) positive cells, degradation of Hsp90 client protein EGFR, as well as a decline in both expression and phosphorylation levels of PI3Kα downstream Akt protein (p < .01) (Figure 8(C)). In brief, the in vivo results were almost consistent with in vitro antitumor assays and signaling pathway analysis.
4. Conclusion
In summary, we reported on the design, synthesis and biological evaluation of a series of 2,4-dihydroxy-5-isopropylphenyl-thieno[2,3-d]pyrimidin derivatives as novel Hsp90/PI3Kα dual inhibitors in B16 melanoma cells. The most potent compound, 8m, inhibited Hsp90 and PI3Kα with IC50 values at nanomolar levels, suppressing melanoma cell migration, invasiveness and proliferation, induced apoptosis and cell cycle arrest. In addition, the western blotting analysis validated the kinase inhibitory activities of 8m on B16 cells. These results suggested that Hsp90/PI3Kα dual inhibition might be an attractive approach to melanoma targeted therapy.
Detailed binding modes of 8m to Hsp90 and PI3Kα were probed using a molecular docking method. WB analysis results of the Hsp90 and PI3Kα downstream signaling pathways as indicated that both MAPK pathway regulated by Hsp90 client proteins and PI3K-Akt pathway are involved in the novel Hsp90/PI3Kα dual inhibitor 8m for melanoma cell apoptotic death. These results also were validated in subcutaneous B16 xenograft murine model. Collectively, our results provide valuable insights about our library of novel Hsp90/PI3Kα inhibitors which allows us to explore a further integrative translational medicine paradigm. This future direction includes transcriptomic analysis, chemical synthesis and molecular mechanisms to study synthesized Hsp90/PI3Kα dual inhibitors as an attractive lead compound for melanoma targeted therapy.
Supplementary Material
Acknowledgements
The characterization and NMR spectrum of synthesized compounds are supplied in Supplementary material. This material is available free of charge via the Internet.
Funding Statement
We are grateful for financial support from the National Natural Science Foundation of China (21772131 and 81872535), the Fundamental Research Funds of Science & Technology Department of Sichuan Province (2019YFSY0004 and 2017JY0123), China Postdoctoral Science Foundation (No. 2016M602696 and 2016M592694) and the Fundamental Research Funds for the Central Universities and Distinguished Young Scholars of Sichuan University (2015SCU04A13).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin 2016;66:115–32. [DOI] [PubMed] [Google Scholar]
- 4.Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Network . Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guan J, Gupta R, Filipp FV. Cancer systems biology of TCGA SKCM: efficient detection of genomic drivers in melanoma. Sci Rep 2015;5:7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018;173:400–16. e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman RJ, Heilman ER. The pathology of malignant melanoma. Dermatol Clin 2002;20:659–76. [DOI] [PubMed] [Google Scholar]
- 9.Bellet V, Lichon L, Arama DP, et al. Imidazopyridine-fused [1,3]-diazepinones part 2: structure-activity relationships and antiproliferative activity against melanoma cells. Eur J Med Chem 2017;125:1225–34. [DOI] [PubMed] [Google Scholar]
- 10.Borges LJH, Bull ES, Fernandes C, et al. In vitro and in vivo studies of the antineoplastic activity of copper (II) compounds against human leukemia THP-1 and murine melanoma B16-F10 cell lines. Eur J Med Chem 2016;123:128–40. [DOI] [PubMed] [Google Scholar]
- 11.Kalhor-Monfared S, Beauvineau C, Scherman D, Girard C. Synthesis and cytotoxicity evaluation of aryl triazolic derivatives and their hydroxymethine homologues against B16 melanoma cell line. Eur J Med Chem 2016;122:436–41. [DOI] [PubMed] [Google Scholar]
- 12.Karelia DN, Sk UH, Singh P, et al. Design, synthesis, and identification of a novel napthalamide-isoselenocyanate compound NISC-6 as a dual topoisomerase-II alpha and Akt pathway inhibitor, and evaluation of its anti-melanoma activity. Eur J Med Chem 2017;135:282–95. [DOI] [PubMed] [Google Scholar]
- 13.Koca I, Ozgur A, Er M, et al. Design and synthesis of pyrimidinyl acyl thioureas as novel Hsp90 inhibitors in invasive ductal breast cancer and its bone metastasis. Eur J Med Chem 2016;122:280–90. [DOI] [PubMed] [Google Scholar]
- 14.Liang C, Hao H, Wu X, et al. Design and synthesis of N-(5-chloro-2,4-dihydroxybenzoyl)-(R)-1,2,3,4-tetrahydroisoquinoline-3-carboxamides as novel Hsp90 inhibitors. Eur J Med Chem 2016;121:272–82. [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Wang X, Liu H, et al. Design, synthesis and pharmacological evaluation of 4,5-diarylisoxazols bearing amino acid residues within the 3-amido motif as potent heat shock protein 90 (Hsp90) inhibitors. Eur J Med Chem 2017;125:315–26. [DOI] [PubMed] [Google Scholar]
- 16.Jiang F, Guo AP, Xu JC, et al. Identification and optimization of novel 6-acylamino-2-aminoquinolines as potent Hsp90 C-terminal inhibitors. Eur J Med Chem 2017;141:1–14. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Li L, Zhou ZH, et al. Structure-based virtual screening and optimization of modulators targeting Hsp90-Cdc37 interaction. Eur J Med Chem 2017;136:63–73. [DOI] [PubMed] [Google Scholar]
- 18.Marcu MG, Chadli A, Bouhouche I, et al. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem 2000;275:37181–6. [DOI] [PubMed] [Google Scholar]
- 19.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Institute 2000;92:242–8. [DOI] [PubMed] [Google Scholar]
- 20.Cavenagh J, Oakervee H, Baetiong-Caguioa P, et al. A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br J Cancer 2017;117:1295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canella A, Welker AM, Yoo JY, et al. Efficacy of onalespib, a long-acting second-generation HSP90 inhibitor, as a single agent and in combination with temozolomide against malignant gliomas. Clin Cancer Res 2017;23:6215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner AJ, Agulnik M, Heinrich MC, et al. Dose-escalation study of a second-generation non-ansamycin HSP90 inhibitor, onalespib (AT13387), in combination with imatinib in patients with metastatic gastrointestinal stromal tumour. Eur J Cancer 2016;61:94–101. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki R, Kikuchi S, Harada T, et al. Combination of a selective HSP90 alpha/beta inhibitor and a RAS-RAF-MEK-ERK signaling pathway inhibitor triggers synergistic cytotoxicity in multiple myeloma cells. PLoS One 2015;10;e0143847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang W, Wu QD, Zhang M, et al. Novel Hsp90 inhibitor FW-04-806 displays potent antitumor effects in HER2-positive breast cancer cells as a single agent or in combination with lapatinib. Cancer Lett 2015;356:862–71. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann S, Popov N, Zhu J, et al. Strong radiosensitizing effects by combination of Hsp90 inhibitor NVP-AUY922 and dual PI3 K/mTOR inhibitor PI-103 on PTEN knock-down tumor cell lines. Strahlenther Onkol 2015;191:S46. [Google Scholar]
- 26.Rao A, Lowe DB, Storkus WJ. Shock block for improved immunotherapy. Oncoimmun 2012;1:1427–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MH, Chiang KC, Cheng CT, et al. Antitumor activity of the combination of an HSP90 inhibitor and a PI3K/mTOR dual inhibitor against cholangiocarcinoma. Oncotarget 2014;5:2372–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao A , Taylor JL, Chi-Sabins N, Kawabe M, Gooding WE, Storkus WJ. Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve recruitment of therapeutic T cells. Cancer Res 2012;72:3196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen C, Zingales S, Wang T, et al. Synthesis and in vitro evaluation of 4-substituted furano 3,2-c tetrahydroquinolines as potential anti-cancer agents. J Enzyme Inhib Med Chem 2016;31:853–8. [DOI] [PubMed] [Google Scholar]
- 30.Liu JF, Lichtenberg T, Hoadley KA, et al. Network, an integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018;173:400–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao JJ, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhlen M, Fagerberg L, Hallstrom BM, et al. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 33.Al-Sha'er MA, Mansi I, Khanfar M, Abudayyh A. Discovery of new heat shock protein 90 inhibitors using virtual co-crystallized pharmacophore generation. J Enzyme Inhib Med Chem 2016;31:64–77. [DOI] [PubMed] [Google Scholar]
- 34.Morrison R, Al-Rawi JMA. Synthesis, structure elucidation, DNA-PK, PI3K, anti-platelet and anti-bacteria activity of linear 5, 6, and 10-substituted-2-morpholino-chromen-oxazine-dione and angular 3, 4, 6-substituted-8-morpholino-chromen-oxazine-2,10-dione. J Enzyme Inhib Med Chem 2016;31:86–95. [DOI] [PubMed] [Google Scholar]
- 35.Ouyang L, Zhang L, Liu J, et al. Discovery of a small-molecule bromodomain-containing protein 4 (BRD4) inhibitor that induces AMP-activated protein kinase-modulated autophagy-associated cell death in breast cancer. J Med Chem 2017;60:9990–10012. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y, Zheng YX, Jiang QL, et al. Integrated bioinformatics, computational and experimental methods to discover novel Raf/extracellular-signal regulated kinase (ERK) dual inhibitors against breast cancer cells. Eur J Med Chem 2017;127:997–1011. [DOI] [PubMed] [Google Scholar]
- 37.He G, Wu FB, Huang W, et al. One-Pot asymmetric synthesis of substituted tetrahydrofurans via a multicatalytic Benzoin/Michael/Acetalization cascade. Adv Synth Catal 2014;356:2311–9. [Google Scholar]
- 38.Zhou R, Huang W, Ren W, et al. Synthesis of cis or trans 4-heteroaromatic substituted furano and pyrano[3,2-C]tetrahydroquinolines by one-pot imino diels-alder reactions. Heterocycles 2013;87:2495–500. [Google Scholar]
- 39.Zhao Q, Peng C, Huang H, et al. Asymmetric synthesis of tetrahydroisoquinoline-fused spirooxindoles as Ras- GTP inhibitors that inhibit colon adenocarcinoma cell proliferation and invasion. Chem Commun 2018;54:8359–62. [DOI] [PubMed] [Google Scholar]
- 40.Zhang YH, Wang CT, Huang W, et al. Application of organocatalysis in bioorganometallic chemistry: asymmetric synthesis of multifunctionalized spirocyclic pyrazolone-ferrocene hybrids as novel RalA inhibitors. Org Chem Front 2018;5:2229–33. [Google Scholar]
- 41.Yang MC, Peng C, Huang H, et al. Organocatalytic asymmetric synthesis of spiro-oxindole piperidine derivatives that reduce cancer cell proliferation by inhibiting MDM2-p53 interaction. Org Lett 2017;19:6752–5. [DOI] [PubMed] [Google Scholar]
- 42.Ke BW, Tian M, Li JJ, et al. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Med Res Rev 2016;36:983–1035. [DOI] [PubMed] [Google Scholar]
- 43.Zhou R, Wu QJ, Guo MR, et al. Organocatalytic cascade reaction for the asymmetric synthesis of novel chroman-fused spirooxindoles that potently inhibit cancer cell proliferation. Chem Commun 2015;51:13113–6. [DOI] [PubMed] [Google Scholar]
- 44.Leng HJ, Peng F, Zingales S, et al. Core-scaffold-inspired asymmetric synthesis of polysubstituted chiral hexahydropyridazines that potently inhibit breast cancer cell proliferation by inducing apoptosis. Chem-Eur J 2015;21:18100–8. [DOI] [PubMed] [Google Scholar]
- 45.Wu QJ, Li GY, Deng SY, et al. Enhanced antitumor activity and mechanism of biodegradable polymeric micelles-encapsulated chetomin in both transgenic zebrafish and mouse models. Nanoscale 2014;6:11940–52. [DOI] [PubMed] [Google Scholar]
- 46.Mellatyar H, Talaei S, Pilehvar-Soltanahmadi Y, et al. Targeted cancer therapy through 17-DMAG as an Hsp90 inhibitor: overview and current state of the art. Biomed Pharmacother 2018;102:608–17. [DOI] [PubMed] [Google Scholar]
- 47.Gupta SD, Snigdha D, Mazaira GI, et al. Molecular docking study, synthesis and biological evaluation of Schiff bases as Hsp90 inhibitors. Biomed Pharmacother 2014;68:369–76. [DOI] [PubMed] [Google Scholar]
- 48.Huang YH, Lei J, Yi GH, et al. Coroglaucigenin induces senescence and autophagy in colorectal cancer cells. Cell Proliferation 2018;51:e12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.