

Abstract
Chronic wounds (e.g. diabetic wounds, pressure wounds, vascular ulcers, etc.) do not usually heal in a timely and orderly manner but rather last for years and may lead to irreversible adverse events, resulting in a substantial financial burden for patients and society. Recently, a large amount of evidence has proven that cellular senescence has a crucial influence on chronic nonhealing wounds. As a defensive mechanism, cell senescence is a manner of cell-cycle arrest with increased secretory phenotype to resist death, preventing cells from stress-induced damage in cancer and noncancer diseases. A growing amount of research has advanced the perception of cell senescence in various chronic wounds and focuses on pathological and physiological processes and therapies targeting senescent cells. However, previous reviews have failed to sum up novel understandings of senescence in chronic wounds and emerging strategies targeting senescence. Herein, we discuss the characteristics and mechanisms of cellular senescence and the link between senescence and chronic wounds as well as some novel antisenescence strategies targeting other diseases that may be applied for chronic wounds.
Keywords: Chronic wounds, Senescence, Senolytics, Targeted therapies, Diabetic wound, Pressure wound, Vascular ulcer
Highlights.
The definition and characteristics of senescent cells are summarized in detail.
The current bottleneck problems in the treatment of chronic wounds are introduced.
The link between senescence and chronic wounds provides a prospective strategy to treat chronic wounds.
The latest treatment methods targeting senescence in various fields can also be applied to treating chronic wounds.
Background
Chronic wounds are pathological wounds that do not heal in a timely manner. It is considered a ‘silent epidemic’ that imposes a significant physical, psychological, social and economic burden on individuals and the whole health system [1]. Chronic wounds are usually divided into three main categories: diabetic ulcers, vascular ulcers and pressure ulcers, which can last for months to years and frequently recur [2]. Some severe ulcers can ultimately lead to amputation. The failure of wound healing contributes to pathological and physiological phenomena, but the underlying molecular mechanisms vary and are even unclear in different chronic wounds [3]. Currently, speculation regarding the pathogenesis of chronic wounds is primarily based on four theories: local tissue hypoxia, biofilm formation in the wound, recurring ischaemia–reperfusion injury and regression of stress response in aged patients [4]. Chronic wounds are not present in experimental animals and it has been challenging to establish an actual chronic wound model in animals. Thus, the limited understanding of chronic wounds mostly comes from the observation and biopsy of patients. However, the precise pathogenesis of chronic wounds is complex and involves many different molecular pathways that can only be clearly investigated in patients suffering from this disease. Recent research has demonstrated that senescent cells present in some chronic wounds impede the process of wound healing [5]. Furthermore, it is reported that eliminating senescent cells or preventing cell senescence can reduce the impact of chronic wounds [6,7].
Current research shows that in different situations, the effects of senescence vary or are even opposite. In general, it is beneficial to transiently induce senescence during tissue remodelling, because the body can eliminate damaged cells in this way. On the other hand, the prolonged presence of senescence or the inability to effectively remove senescent cells is pernicious. As a distinct form of cell-cycle arrest, senescence functions in diverse physiological processes and a in range of age-related disease pathologies [8]. To some extent, senescence is also a kind of antitumour mechanism triggered by a series of internal or external factors, such as oncogene activation and stress stimulation [9]. Previously, the effects of senescence have been thought to be mostly limited to cell damage and stress. However, the recent recognition of senescence in embryonic development and normal somatic cells has further developed this view. It has been discovered that senescence occurs in multiple locations during vertebrate embryonic development [10]. Furthermore, senescence also play a role in the physiological processess of adult tissues, in particular normal macrophages and placenta syncytiotrophoblasts [11]. In age-related diseases, senescence is closely related to neurodegenerative diseases (such as Parkinson’s and Alzheimer’s) [12]. Studies have shown that senescent blood vessel changes accompany or even precede the development of Alzheimer’s disease, which increases the possibility that senescence may have a pathogenic effect [13].
As reported previously, the kernel of chronic wounds is chronological inflammation, and senescence, a physiological process typically associated with reduced proliferative capacity, plays an important role in it [14]. In chronic wounds, senescent cells cannot be cleared in time, leading to a continuous increase of inflammatory factors in the wound environment and a decrease in cell proliferation [15]. However, in different chronic wounds, the underlying pathologies of senescence are intricate or even unclear. Increasing evidence has demonstrated that the accumulation of senescent cells in chronic wounds impedes the healing process. In the past, researchers have suggested that prolonged non-healing of wounds and exposure to exudate may affect cell viability and lead to senescence in the wound bed. However, recent studies have shown that senescence may lead to long-term nonhealing of wounds. Mahmoudi et al. found that senescent fibroblasts affect cell reprogramming efficiency and wound healing rates, and their secretion of inflammatory cytokines (including tumour necrosis factor [TNF]) is a critical contributor [16]. In general, the ability of skin wounds to heal decreases with age [17], primarily due to the following reasons. First, senescence leads to an increased degree of platelet aggregation after injury and decreased vascular permeability, resulting in the inability of inflammatory cells (such as lymphocytes and macrophages) to infiltrate in a timely manner [18]. Although a large number of macrophages are recruited during the ageing process, senescent macrophages have reduced phagocytic ability and produce fewer growth factors, which generally impairs the immune response [19]. In addition, senescence decreases the migration and proliferation capacity of keratinocytes and fibroblasts. Senescent fibroblasts also secrete excessive matrix metalloproteinases (MMPs) that cause alterations in the composition of the extracellular matrix (ECM) [20]. Furthermore, the angiogenesis potential of senescent endothelial cells significantly decreases and produces a large number of proinflammatory secretory phenotypes. Therefore, there is a significant reduction in the revascularisation of chronic wounds, on which the wound healing process closely depends [21]. Although the accumulation of senescent cells might cause chronic wounds that are difficult to heal, there is currently no clear relationship between senescence and chronic wounds.
Previous reviews failed to sum up the new mechanisms of senescence in chronic wounds, and emerging strategies targeting senescence must also be updated. In this review, we not only summarize the mechanisms and characteristics of senescence but also explore the link between senescence and chronic wounds. In addition, we underline some novel antisenescence strategies targeting other diseases with potential future applications for chronic wounds.
Review
Definition and characteristics of senescence
Cellular senescence, a seminal discovery in 1961, is a steady cell-cycle arrest that takes place in diploid cells and restrains their ability to proliferate [22]. Hayflick and Moorhead described this phenomenon for the first time. They found that cultured human diploid fibroblasts would stop growing after dividing to the maximum number [23]. This kind of cell-cycle withdrawal is induced to prevent genomic instability and the consequent accumulation of DNA damage, as telomeres continue to shorten with each cell division [24]. However, this view has been challenged by the fact that these features have been found in other cells [25,26]. Thus, cellular senescence has been identified to respond to numerous stressors (e.g. genotoxic stress, metabolic alternation, oxidative stress, mitochondrial dysfunction, oncogene activation, etc.) [8,27,28]. Senescence differs from quiescence and terminal differentiation in that this irreversible cell-cycle arrest has a distinct phenotype [29]. Therefore, it seems that senescence could be an effective antitumour approach due to its antiproliferative potential. Although senescence is permanent, the cell-cycle can restart in the case of certain conditions, especially in tumour cells [30].
Morphological characteristics
Since Hayflick and Moorhead discovered the senescent cell in 1961, scientists have struggled to identify global and straightforward hallmarks that characterize the senescence stage [31]. Following impairments to DNA, cells activate a radical, excessive or irreparable response, the DNA damage response (DDR) [32]. The DDR initiates cellular senescence and cyclin-dependent kinase inhibitors (CDKI) are involved in the process. It also promotes the expression of antiapoptotic genes in senescent cells and alters metabolic rates and endoplasmic reticulum stress. In addition, it can enhance the secretion of inflammatory and related tissue remodelling factors [33–35]. These signalling pathways are responsible for morphological aberrations in senescent cells, such as enlarged and flattened morphology, accumulation of lysosomal and mitochondria and altered cell membrane composition [36].
Senescence-associated beta-galactose
The most common marker of senescence is senescence-associated beta-galactosidase (SA-β-Gal), which can be measured with X-gal (an artificial substrate) at pH 6.0. This marker can recognize senescence in cultured cells and mammalian tissues [37]. Since SA-β-Gal is not specific, this measurement has restrictions. Thus, other markers such as p16 and p53 can be used alone or in combination to identify senescent cells [37].
Cyclin-dependent kinase inhibitors
CDKIs are major regulators of cell-cycle processes. The CDKIs regulating cell cycle arrest in senescent cells are CDKN2B (p15INK4b), CDKN2A (p16INK4a) and CDKN1A (p21CIP). p16INK4A, a selective inhibitor of CDK4 and CDK6, is commonly used as a specific marker for the recognition of senescence [38]. It is highly expressed in response to particular stress stimuli such as tumour, trauma and ageing, but is usually absent in healthy young tissues [39]. The transcriptional activity of p16 has been widely used to prove the presence of senescence in vivo [40]. The gene CDKN2B, encoding p15INK4B, is closely linked to the CDKN2A gene encoding p16INK4A on the chromosome [41]. Therefore, the function and structure of p15 is comparable to that of p16, but it has received less interest in the context of ageing [42]. p21 can inhibit a series of CDKs and, paradoxically, it is necessary for the cell-cycle process [43]. Although p21 is over expressed in response to senescence-inducing stress, it is hard to use as a particular senescence marker as p21 expression is partially influenced by the DDR and modulated mainly through transactivation of p53 [44].
Senescence-associated secretory phenotype
Although senescent cells have a stable cell-arrest state, they have activated metabolism and upregulate a wide range of genes. Senescent cells secrete chemokines, proinflammatory cytokines, growth factors and ECM remodelling factors that have a positive or negative impact on different pathological processes (e.g. wound healing and cancer) [45]. This phenotype, termed the ‘senescence-associated secretory phenotype’ (SASP), is an essential feature in distinguishing senescent cells from nonsenescent, quiescent and terminally differentiated cells [46]. The SASP is mainly a feature of the DDR that induces cell senescence, as SASP will not present in cells with senescence that is naturally induced by p16 and p21 overexpression [47,48]. This phenotype is mediated by a variety of factors, including nuclear factor-κB (NF-κB) [49], CCAAT/enhancer-binding protein β transcription factors [50], p38MAPK [51] and mammalian target of rapamycin (mTOR) signalling [52,53]. The SASP is largely a transcriptional programme regulated by the transcription factor NF-κB [54]. The DDR is the major trigger of NF-κB activation, but the cGAS/STING pathway has also recently been found to be involved [55,56]. In addition, the Jak2/Stat3 pathway regulates a diverse set of cytokines, including granulocyte macrophage-colony stimulating factor (GM-CSF), interleukin-10 (IL-10), IL-13, C-X-C motif ligand 1 (CXCL1)/CXCL2 and macrophage-colony stimulating factor (M-CSF) [57]. While the majority of SASP factors can only affect neighbouring cells and cause them to senesce, several SASP factors (IL-1 and IL-6) possess cell-autonomous functions and intensify their own senescence condition [58]. A number of SASP factors have been reported to play a key role in maintaining cell-cycle arrest in senescent cells, which may enable senescent cells to exhibit tumour suppressive function [50,59]. In addition, SASP factors have been proven to contribute to embryonic development, facilitate tissue remodelling and repair and improve immunosurveillance [45,60]. On the other hand, if the SASP exists persistently and chronically, it can lead to some disease states, such as inflammation and ageing [61]. The SASP, as a characteristic of senescence, has complicated and adverse biological effects, remarkably linked to age and chronic diseases in autocrine and paracrine manners. Clearly, the SASP plays a significant role in the pathophysiological process of senescence. However, it is not specific enough and is heterogeneous as a hallmark of senescence.
Challenges in chronic wound healing
Wound healing is artificially divided into four phases: haemostasis, inflammation, proliferation and remodelling, which proceed in a timely and sequential manner [62]. In most clinical situations, wound healing can be implemented appropriately with thoroughly organized and harmonized cellular processes. Chronic nonhealing wounds cannot progress in an orderly and timely manner. They stall in the inflammatory phase, leading to persistent inflammation, consequently impeding wound healing. The vast majority of chronic wounds include, but are not limited to, pressure ulcers, diabetic ulcers and vascular ulcers (venous and arterial ulcers). Although the aetiology of chronic trauma is variable, some common features are shared at the molecular level, such as secretion of pro-inflammatory cytokines, production of high levels of reactive oxygen species (ROS), chronic infection, accumulation of senescent cells and dysfunctional stem cells [63].
Histological assay of chronic venous ulcers has demonstrated a hyperproliferative epidermis accumulated at the margins of the wound, with an exudate containing necrotic debris covering the base of the ulcer. Where granulation tissue should have been present, the neovascularization did not sprout but was wrapped in fibrin cuffs. The mechanisms responsible for this include a reduced level of growth factors and their receptors, destruction of the ECM, decreased proliferative capacity of resident cells and inadequate recruitment of stem cells [64]. In particular, the abnormal expression of the vascular endothelial growth factor receptor has been shown to be associated with these ulcers [65]. Hence, most chronic wounds have a large infiltration of inflammatory cells (especially neutrophils), but only a small number of myofibroblasts and a lack of neovascularization Hence, most chronic wounds, have few myofibroblasts but heavy inflammatory infiltrates, particularly neutrophils [66].
Tissue injury, microorganisms and uncleared necrotic debris constantly stimulate the influx of immune cells. Although the role of each macrophage population is poorly understood, disruption in the transition from the proinflammatory (M1) to the anti-inflammatory (M2) state has been associated with impaired wound healing in animal models. The failure of macrophages to switch from M1 to M2 type is due to two major elements: (1) macrophage dysfunction in pathological states and (2) high levels of biomolecules in the wound microenvironment, such as advanced glycation end-products, hyperglycaemia, oxidative stress products, inflammatory factors and wound microbes [67]. Thus, the inflammatory cytokine cascade extends and exists continually for a prolonged time, resulting in increased proteases that exist persistently [63].
In chronic wounds, increased levels of tissue-degrading MMPs surpass that of the inhibitors, contributing to degradation of the ECM and reduction in growth factors. Concurrently, the upregulation of cytokines such as TNF-α contributes to a reduction in the secretion of inhibitors for the synthesis of metalloproteinases by fibroblasts, which delays wound healing [68]. The increase in MMPs prevents the wound healing process from entering the proliferative phase and thus more inflammatory cells and cytokines are accumulated, resulting in stasis at the inflammation phase [69].
Furthermore, another challenge of chronic wounds is the accumulation of senescent cells, with damaged proliferative capacities and altered secretory phenotype, which prevents cells from responding to classical wound healing signals [15,70,71]. Senescence in chronic wounds, a cell-cycle arrest form, is due to oxidative stress resulting in the DDR, which impedes normal intracellular molecular pathways [5,72]. Wilkinson et al. discovered that macrophages in diabetic mice undergo senescence and have a reduced anti-inflammatory M2 phenotype. In addition, these macrophages secreted high levels of the C-X-C motif chemokine receptor 2 (CXCR2) ligand, a marker of fibroblast fibrosis induction, and promoted fibroblast senescence [73]. Thus, macrophage senescence not only undermines polarization from a proinflammatory to an anti-inflammatory phenotype that supports the healing process but also, through paracrine effects, affects the biofunction of the wound microenvironment and other cells.
There are several topical dressings used in clinical practice. However, few dressings effectively promote chronic wound healing, and clinicians routinely use approaches grounded in experience. Some products, such as Integra and OxyHeal1000, have been used to treat chronic wounds, but the efficacy is unsatisfactory and prolonged treatment is required. Bioengineered scaffolds containing living cells have shown a promising impact on chronic wounds. In addition, cellular strategies are more effective and safer in treating diabetes, age-related wounds and other nonhealing wounds. Despite using advanced treatment and care, 15–20% of chronic wounds heal improperly using the above treatments. It is vital not only to cure the topical wounds but also to improve the systematic disorder, such as diabetic wounds and peripheral arterial disease. Even more improtant is that treatment should be chosen according to the actual situation to guarantee the effectiveness of the treatment. Many challenges remain in determining the pathological processes and modulating factors involved in chronic wounds, including sophisticated molecular signals, different processes and cell types, as well as the absence of a suitable animal model to study this complex condition [3].
Senescence in chronic wound healing
An understanding of the pathophysiological mechanism of chronic wounds is essential in clinical practice, where an effective cure decides life and death [74]. Most chronic wounds are classified as one of three types: diabetic wounds, pressure ulcers and vascular ulcers. Despite the various aetiologies of these chronic wounds, there are also fairly consistent similarities between them. For example, the vast majority of chronic wounds show increased inflammatory factors, decreased cells proliferation and migration and increased ECM proteases, among other features [63]. These features are induced by senescent cells, but the actual relationship between senescence and chronic wounds remain incompletely understood. Although the link between chronic wounds and senescence is not well established, an increasing number of studies provide a more profound understanding of the role of senescence in chronic wounds [9,61]. It is universally acknowledged that aged skin is more vulnerable to developing chronic wounds than young skin is, possibly because of cellular senescence. The main factors (including SASP, ROS, immune system dysfunction and stem cell dysfunction) by which senescent cells impede chronic wound healing are as follows.
Senescent cells produce a set of SASPs that affects different aspects of wound repair in a direct or indirect manner, including matrix remodelling, angiogenesis and cell growth, by regulating the adjacent microenvironment [46,50,59]. Major SASP components influence wound healing by targeting NF-κB. In general, SASP leads to matrix proteolysis and increasing inflammation, which are the main features of ageing and diabetes [75].
Accumulation of oxidative damage is another contributor to the normal replicative ageing process related to senescence. For instance, human diploid fibroblasts and endothelial cells go through senescence in the presence of elevated levels of ROS. Nevertheless, replicative capability can be promoted by lower oxygen tension in culture conditions [76–78]. In these cases, inflammatory signals set off an immune response, which causes the removal of deficient cells. However, these processes of clearance are damaged in aged tissues, leading to an intensive immune response, sterile inflammation, which leads to many prolonged pathological processes [79].
In normal conditions, macrophages clear the senescent cells dependent on immunosurveillance after inflammation. Conversely, senescent cells cannot be removed in chronic wounds and continue to cause increased secretion of proinflammatory cytokines and reduced cell proliferation [15]. During ageing and injury, there is a transformation from M1 macrophages to M2 macrophages that is associated with reduced immune activity, tumour promotion and damaged chemotaxis or phagocytosis. The impaired capacity of macrophages make them susceptible to migrating to where senescent cells accumulate, which is linked to the decreased chemotaxis of macrophages in chronic wounds [80–82].
Furthermore, stem cell function in chronic wounds is also impaired [83,84]. As previously reported, researchers transplanted senescent cells into the skin, skeletal and muscle tissues of mice with neuron-specific genes for immune deficiency. After 3 weeks, the fibroblasts and myofibres in the dermal tissue expressed different senescent hallmarks in the area where senescent cells were transplanted. In contrast, there were no markers in the area without injected senescent cells [85]. Hence, inhabitant senescent cells can induce stem cell dysfunction during the chronic wound healing process. Stem cells proliferate and function at every stage of wound healing. Therefore, the impairment of stem cells can result in nonhealing wounds. It has been shown that mesenchymal stem cells play a vital role in wound healing. They can be recruited into the circulatory system and grafted into tissue remodelling [86]. In a nutshell, although stem cells are impaired in chronic wounds, their function in wound healing is indispensable, and more attention and efforts are needed to address these challenging observations (Figure 1) [84,87].
Figure 1.

Roles of senescence in chronic wound healing. The main factors by which senescent cells impede the healing of chronic wounds include the SASP, ROS and immune system dysfunction, among others. SASP senescence-associated secretory phenotype, ROS reactive oxygen species. (The figure was generated with Bio Render)
Targeting senescent cells is a promising therapy
In tissue-ageing pathology, the comprehensive biological effects of senescence make therapy that targets senescence a prospective strategy for many age-related diseases. Several promising strategies to either clear the senescent cells or decrease their defective impacts are in development [88]. Recent approaches are mostly committed to developing pharmacologic agents that can mediate the cell death of senescent cells, termed ‘senolytics’ [89]. Studies in this field are mainly based on the potential cellular or molecular mechanisms underlying senescence. These therapies focus on distinct features of senescent cells that differ from other normal cells in the tissue [88].
Caloric restriction
It has been well-recorded that the effects of caloric restriction both prolong life expectancy and postpone the beginning of age-related diseases [90]. Caloric restriction may be a physical method of modulating senescence. It has been demonstrated to minimize the senescence of cardiac, hepatocyte and intestinal crypt cells in vivo [91,92]. From an epigenetic point of view, caloric restriction allows DNA to avoid age-related defects, such as methylation [93]. It also reduces senescence epigenetically by upregulating the sirtuin pathway to some extent, elevating anti-apoptosis and anti-inflammatory mechanisms [94]. In addition, sirtuins may prevent an age-related recession in skin tissue repair because insufficiency of SIRT1 aggravates the healing process in diabetic wounds [95]. Effects following caloric restriction include deceleration of metabolic procedures that contribute to cellular senescence (Figure 2) [86], intensification of antioxidant production, increased autophagy and removal of intracellular components [96]. Although caloric restriction wields many life benefits, it has insufficient feasibility as an effective clinical treatment as it requires high patient compliance. For the above reasons, drugs known as senolytics, that directly target senescence, are more appealing clinical interventions.
Figure 2.
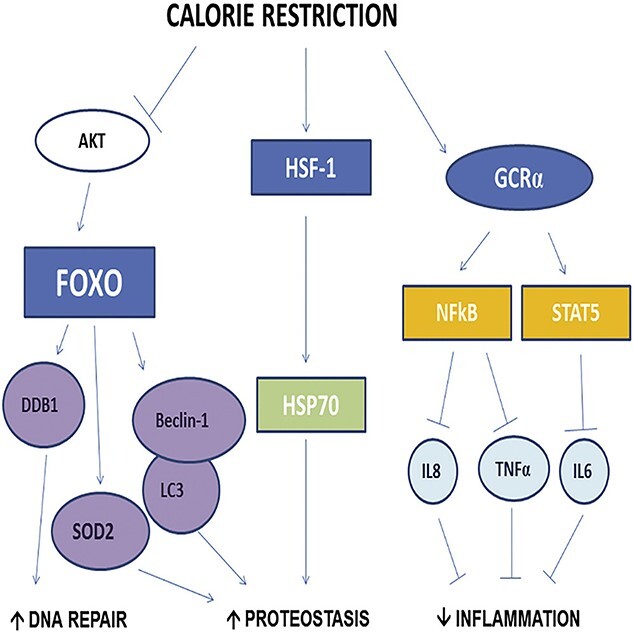
Calorie restriction modulated upstream and downstream regulatory pathways of AKT/FOXO and HSF1/HSP70, and the glucocorticoid receptor pathway in human skeletal muscle. AKT protein kinase B, HSF-1 heat shock factor 1, GCR-α glucocorticoid receptor alpha, FOXO forkhead box O, HSP70 heat shock protein 70, DDB1 damage specific DNA-binding protein 1, SOD2 superoxide dismutase 2, NF-κB nuclear factor-κB, STAT5 signal transducer and activator of transcription 5, LC3 microtubule-associated protein 1 light chain 3, TNF-α tumour necrosis factor, IL-6 interleukin-6, IL-8 interleukin-8. (Reprinted with permission from Ref. [162] © 2016 Cell Press)
Blocking the prosurvival pathway
Senolytics can reverse the immunity of senescent cells to apoptosis [97]. Senescent cells promote prosurvival pathways, especially BCL-2. Currently, several prosurvival pathways have been determined as a direct target to eliminate senescent cells. These prosurvival pathways upregulated by senescence include the p53/p21 axis, the BCL-2 protein family, receptor tyrosine kinases, the phosphatidylinositol 3-kinase (PI3K)/AKT axis, HSP90 and HIF-1α proteins [98–103]. Currently, the majority of senolytics that block the pro-survival pathway are agents that directly target members of the BCL-2 protein family. Researchers have conducted thorough studies of antiapoptotic agents targeting the BCL-2 protein family at the molecular level (Figure 3) [104]. Blocking these proteins may activate programmed cell death in senescent cells [89]. The BCL-2 protein family has been extensively studied for cancer treatment, providing repurposing opportunities for candidate compounds that might be used as senolytics. For instance, ABT-737 is a BH3 domain analogue that blocks members of the antiapoptotic family (including BCL-W, BCL-XL and BCL-2) and induces senescent cells to undergo apoptosis via the BH3 domain [105,106]. ABT-737 was reported to be effective in removing senescent cells both induced by irradiation and transgenosis [102].
Figure 3.
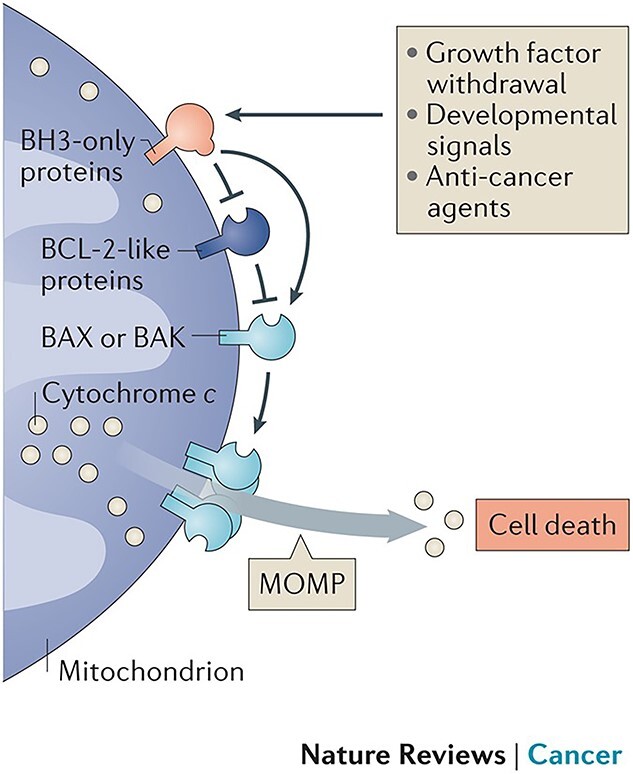
Bcl-2 family members have an antiapoptotic function by blocking Bax/Bak directly in mitochondrial-mediated apoptosis. BCL-2 B-cell lymphoma 2, BH3 Bcl-2 homology 3, BAX BCL-2 associated X, BAK BCL-2 antagonist killer, MOMP mitochondrial outer membrane permeabilization. (Reprinted with permission from Ref. [104] © 2016 Nature Reviews Cancer)
ABT-263, also called navitoclax, is a next-generation analogue of ABT-737 [107]. Other researchers have further established senescent stem cells induced by sublethal irradiation. They have proven that these senescent cells are more sensitive to ABT-263 than nonsenescent cells are [100]. ABT-263 also has the ability to diminish senescent macrophages from atherosclerotic lesions, consequently halting the disease process [108]. It is worth noting that therapy using the inhibitors of BCL-W, BCL-XL and BCL-2will cause haematological toxicity (Figure 4). In contrast to other BCL family proteins, they have the capability of causing an off-target problem in the clinic, such as thrombocytopenia and neutropenia [109,110]. These cytotoxicity issues may hamper the use of BCL-2 family inhibitors in routine clinical practice. Thus, more specific candidates are required and lower-toxicity inhibitors are being tested. Other BCL-XL inhibitors, such as A1331852 and A1155463, are expected to induce less toxicity to nonsenescent cells [111]. A1331852 can reduce liver damage and fibrosis by inducing apoptosis of senescent cells in a transgenic mouse model [112]. However, the above reagent may still cause thrombocytopenia, which is attributed to the presence of platelets that also rely on BCL-XL. Furthermore, in some cases, higher-level expression of BCL-2 and BCL-W might be less powerful for eliminating senescent cells [100,102]. Injection of the BCL-2 targeting compound UBX0101 into the articulations of aged mice can effectively remove senescent cells from the synovium and articular cartilage and alleviate symptoms of osteoarthritis [113]. Taken together, although the side effects might restrain the clinical use to a large extent, targeting the BCL-2 family currently remains an effective molecular strategy to eliminate senescent cells.
Figure 4.
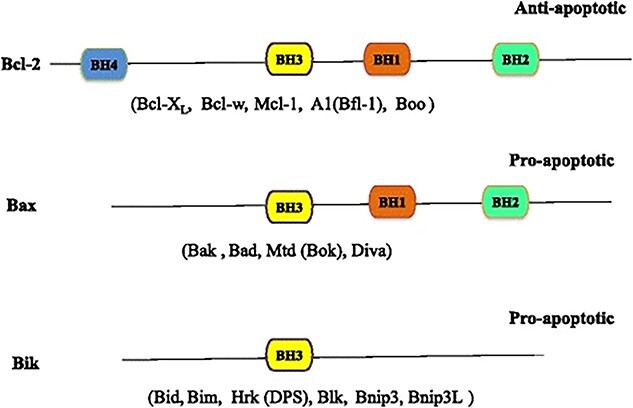
The BCL-2 family proteins can be classified into three categories by BH domains, including the antiapoptotic Bcl-2 and the proapoptotic proteins Bax and Bik, which do not have the BH4 structural domain. Bcl-2 B-cell lymphoma-2, BH BCL-2 homology, Bcl-XL B-cell lymphoma-extra large, Bcl-w BCL-2-like 2, Mcl-1 myeloid cell leukemia sequence 1, Bfl-1 BCL-2 related protein A1, Bax BCL-2-associated X protein, Bak BCL-2 antagonist/killer 1, Bad BCL-2-associated agonist of cell death, Bok Bcl-2-related ovarian killer, Bik BCL2-interacting killer, Bid BH3 interacting domain death agonist, Bim BCL-2-like 11, Hrk BCL-2 family members including harakiri, Blk B-lymphoid tyrosine kinase, Bnip3 BCL-2/adenovirus E1B 19KD interacting protein 3, Bnip3L BNIP3-like (Reprinted with permission from Ref. [109] © 2015 J Hematol Oncol)
Combination use of senolytics
There is an alternative approach to avoiding the toxic effects of BCL-2 protein family inhibitors, i.e. combining low-dose synergistic reagents that target other prosurvival pathways. In fact, it was not a single drug that was initially found to possess antisenescence effects, but a combination of two compounds: dasatinib (a tyrosine kinase inhibitor) and quercetin (a flavonoid p53 activator) [101]. Moreover, it has been suggested that the joint use of senolytics in a low dose might be a more effective and less deleterious substitute [88]. These two agents, dasatinib and quercetin, have been used to treat malignant blood system diseases, such as leukaemia. Combination treatment with dasatinib and quercetin can increase the life span, ameliorate the frail condition and improve vascular function in old mice. Roos et al. demonstrated that dasatinib and quercetin reduced products of senescent cells in the medial layer of the aorta from hypercholesterolaemic and aged mice [114]. This combination therapy has also shown optimal results in an open-label phase 1 pilot study of diabetic kidney disease. The findings showed that dasatinib and quercetin could reduce the expression of p16INK4A and p21CIP1 and decrease the accumulation of senescent cells in adipose tissue. These eliminated cells have high SA-β-Gal activity and limited the replicative potential of adipocyte progenitors. The combination can also reduce the level of macrophages in adipose tissue that are activated by senescent cells [115]. Furthermore, they have a significant effect on idiopathic pulmonary fibrosis induced by bleomycin. Schafer et al. demonstrated that fibrotic lung disease is partly mediated by senescent cells. In the idiopathic pulmonary fibrosis model induced by bleomycin, the combination use of dasatinib and quercetin can selectively kill senescent fibroblasts [116]. However, the cocktail treatment failed to improve lung fibrosis visibly, suggesting that eliminating senescent cells with senolytics may be a more effective intervention for the early disease state. Researchers have also demonstrated that senescent cells have antiresorptive and antianabolic characteristics. For example, the combination of dasatinib and quercetin prevented bone loss associated with age, suggesting that eradication of senescent cells could become a novel treatment for osteoporosis [117]. Nevertheless, combination senolytics also have some limitations that impede the transformation to the clinic. First, both agents aim at a long list of molecular pathways and may modulate diverse pathways in various tissues and organs through combination therapy. Without a thorough understanding of the mechanisms, this strategy could lead to detrimental effects after long-term application. Other agents have recently been suggested to possess senolytic functions, including fisetin and piperlongumine [111,118], the former of which has been tested for its latent senolytic effects and its ability to clear senescent cells and even recover some main functions in aged mice [119].
Immune therapy
Senescent cells are subject to immune surveillance mechanisms attributed to the SASP, which is one of the most notable senescence characteristics. The SASP contains many proinflammatory chemokines, cytokines that can recruit and activate different immune cells such as NK cells, monocytes, macrophages and T cells. In addition, senescent cells can also be recognised by the specific immune cell via the upregulated specific immune ligands on their cell membranes [120]. In a hepatocellular carcinoma mouse model, activation of endogenous p53 can induce cells into a senescence programme instead of apoptosis. Reactivating the p53 pathway also triggers an innate immune response that targets senescent cells, leading to tumour clearance [121]. Senescent cells also provide a functional contribution to noncancer diseases. Krizhanovsky et al. determined that senescent cells derived from hepatic stellate cells were present in fibrotic livers. Senescent stellate cells exhibit phenotypes including cell-cycle arrest, decreased secretion of ECM components, increased secretion of ECM-degrading enzymes and, more importantly, improved immune surveillance. Consequently, natural killer (NK) cells eliminate senescent stellate cells and thereby promote the alleviation of fibrosis [122]. The persistence of senescent cells can likely be attributed to immune system senescence and the accompanying weakened immunological functions in some chronic conditions. Therefore, strengthening the immune system, for instance by enhancing the recognition of senescence, may assist in the clearance of senescent cells. To support this concept, the immune stimulator polyI:C can promote NK cells to remove senescent cells in fibrotic livers [122]. However, such a robust immune stimulant in vivo would overwhelm the immune system impaired by ageing. Thus, more specific immune mediators are needed. Natural killer cell receptor (NKG2D), the immune recognition molecule, is upregulated in senescent cells and is necessary for adequate NK-mediated cytotoxic clearance of senescent fibroblasts [123]. Accordingly, NKG2D expression in senescent cells is a prospective immunotherapy approach.
Researchers have found that premalignant senescent hepatocytes secrete cytokines and chemokines, resulting in senescent cells subject to immune-mediated elimination (senescence surveillance). This kind of immunological process relies on a CD4(+) T-cell-mediated adaptive response. Senescence induced by the expression Nras (G12V) in the liver results in the presence of RAS-specific Th1 lymphocytes; thus, CD4(+) T cells collaborate with mononuclear macrophages to implement the elimination of senescent hepatocytes, demonstrating that senescent surveillance is vital for tumour suppression in vivo [124]. However, other evidence has shown that senescent cells could escape the identification and clearance of NK cells and T cells via a high level of the nonclassical major histocompatibility complex molecule HLA-E. The expression of HLA-E is modulated by the p38 MAP kinase signal pathway in vitro [125]. Approaches applied in cancer territory may also be functional, such as engineering T cells to express specific receptors that recognise targeting proteins on the cell surface [126]. Chimaeric antigen receptor-modified T cells have shown encouraging results as a therapy for chronic lymphocytic leukaemia [127]. Thus, studies to discover and identify new senescent cell surface markers will be crucial for the development of immune-regulated approaches applied to targeting senescence. Very recently, a novel senescent cell surface antigen was found. Frescas et al. hypothesised that senescent fibroblasts express a form of membrane-bound malondialdehyde (MDA) modified vimentin that can be recognised by innate immunity, seemingly as a promising method of senescence elimination [128]. Another study using mass spectrometry analysis revealed that dipeptidyl peptidase-4 (DPP4) is preferentially expressed on the membrane of senescent cells. In addition, DPP4 makes senescent cells more vulnerable to NK cells [129]. Overall, the current studies underline the possibility of using the immune system to mitigate senescence in age-related diseases, however, targeting senescent cells through immunotherapy requires profound research. Therefore, identifying novel and specific senescent cell surface markers is essential to further promote the development of an immune-based therapy.
Targeting SASP
A substitute strategy for directly curbing or eliminating senescence is to alleviate pathological conditions caused by the SASP or specific senescence-associated receptors. Although the eradication of senescent cells seems to be an excellent strategy, it might be inappropriate for some situations in which senescent cells are necessary. Under those conditions, the particular targeting of harmful substances of senescent cells may be an alternative approach. The SASP mainly includes cytokines, growth factors inflammatory factors and matrix remodelling proteases. It is closely involved in the damage of tissue homeostasis and the process of age-related pathologies [61]. Normally, NF-κB regulates the SASP transcriptionally [88] and strongly contributes to tissue damage, driving extensive destruction and strengthening senescence [48,130]. Some signalling pathways join the activation of the C/EBPβ pathway, and the NF-κB pathway modulates the complicated secretion of the SASP [49,50,131,132]. Inhibitors of the SASP block essential transcriptional modulators, impeding the signalling pathway and production of the SASP [133]. Metformin, a widely used antidiabetic drug, can block the NF-κB pathway and prevent the secretion of the SASP. Although these studies demonstrate that SASP inhibitors have a beneficial effect on tissue repair, eradicating the SASP might be detrimental to senescent cell clearance and impair the healing process.
Therefore, targeting specific SASP elements that impede healing may be more effective, such as the membrane-bound antibodies IL-1α and specific inhibitors such as compounds against CXCR2 [134]. Orjalo et al. found that cell surface-bound IL-1α modulates two proinflammatory cytokines, IL-6 and IL-8, enhancing the inhibition of senescent growth [135]. Rapamycin, the mTOR inhibitor, was discovered to expand the life span in mice and improve the replicative life span of human keratinocytes and skin fibroblasts in vitro [136–138]. Recently, researchers found that this mTOR inhibitor can selectively decrease the proinflammatory phenotype of senescent cells by blunting the translation of IL-1α (a kind of membrane-bound cytokine) [53,75]. Furthermore, JAK inhibitor was demonstrated to suppress the development of the SASP in senescent human umbilical vein endothelial cells and human preadipocytes, which contributes to the alleviation of senescence-induced frailty. Thus, targeting the Janus kinase pathway might also be a promising strategy for age-related diseases [139]. Another attractive strategy to eliminate these cytokines and chemokines is using neutralizing antibodies, such as siltuximab and tocilizumab, against IL-6 and its receptor, respectively [140–142]. However, these agents cannot be applied clinically yet, and before these agents are used as a therapeutic senolytic, their specific functions for ageing should be proved in detail.
Targeting senescence-associated pathways
Along with the development of new technologies, such as single-cell RNA sequencing, the physiological and pathological changes associated with senescence could be well studied by identifying distinctive transcriptomic characteristics in tissues. Kimmel et al. used single-cell RNA sequencing to compare cellular transcriptomic and heterogeneity changes between young and old mice. They found transcriptional properties of ageing across different cell types and features of ageing unique to each cell type [143]. Angelidis et al. combined proteomics and single-cell transcriptomics to investigate epigenetic changes and chart lung proteomics between young and old mice and to ascertain the function of these phenotypes [144]. The use of single-cell RNA sequencing can assist the identification of senescence and the targeting of specific senescence-linked biomarkers and receptors in pathology and physiology.
Recently, studies have focused on the p53/p21 axis to develop a promising strategy targeting senescence. p53 is a crucial link in the senescence process and the most vulnerable tumour suppressor gene to mutate [145]. Usually, p53 is stable and accumulated by the DDR [27,146]. The transcriptional activity of p53 mediates a number of pathophysiological processes, such as locating transient apoptosis, cell-cycle arrest and senescence [145]. Interactivity between p53 and transcription factor FOXO4 can release p53 from the nucleus and induce apoptosis. Baar et al. designed a FOXO4 peptide that impedes FOXO4 binding with p53 and selectively causes p53 nuclear rejection and endogenous apoptosis in senescent cells [147]. There are also senolytic target options in this pathway [148]. p21 (also called CDKN1A), a principal transcriptional target of p53, is essential for the survival and maintenance of senescent cells [102]. As a CDK inhibitor, p21 can also maintain the viability of senescent cells induced by DNA damage. In addition, senescent cells suffer from multiple DNA injuries, which activates ataxia telangiectasia mutation and NF-κB kinase. Subsequently, these activations lead to cell death. Subsequently, NF-κB activation can induce the production of TNF-α and c-Jun N-terminal kinase (JNK) activation and modulate the survival pathway of senescent cells in a JNK- and caspase-dependent manner. In addition, the knockout of p21 can clear liver senescent stellate cells in the fibrotic liver and mitigate liver fibrosis and collagen production. Senescent cells secrete CXCR2-related SASP in the wounds of aged and diabetic patients [59]. Wilkinson et al. found that in diabetic mice, blocking the potential senescence receptor, CXCR2, with a selective agonist can accelerate wound healing by inhibiting neutrophils and reducing macrophage senescence [75]. These findings indicate that blocking the senescence-involved pathways can also improve senescent pathology.
Nano-based therapies
Over the past two decades, emerging nanotechnology has been applied to medicine, providing an intelligent way to improve the detrimental aspects of conventional medical therapeutics, such as safety, efficiency and sensitivity [149]. Nano-materials have become popular because of their unique ultra-small structure, providing better bioavailability, stability, targeted function and therapeutic efficiency [150]. Based on the fact that SA-β-Gal activity is significantly enhanced in senescent cells, several nanovectors have been put forward for the targeting of senescent cells in vitro and in vivo [151]. For instance, Agostini et al. took advantage of this feature of senescent cells and created a kind of nanoparticle coated with a SA-β-Gal substrate, galacto-oligosaccharides, wrapped around cytotoxic drugs. In this way, senescent cells could be targeted via the hydrolysis of galacto-oligosaccharides by senescence-associated β-galactosidase, and consequently, cargo would be released only in senescent cells (Fig. 5) [152,153]. Furthermore, Xu et al. reported the first form of enzyme-induced self-assembly nanopartical. This nanoparticle is regulated by β-galactosidase and can selectively form hydrogels and nanofibres in senescent cells [154].
Figure 5.

Galactose-modified nanoparticles (GalNP) are made of a mesoporous silica scaffold encapsulated by galacto-oligosaccharides and can be loaded with cargo to target senescent cells. This nanoformulation can be taken up by endocytosis into cells and then released via exocytosis. In senescent cells, the coated galacto-oligosaccharides can be degraded by galactosidase with increased activity and then consequently release the cargo. (Reprinted with permission from Ref. [153] © 2018 EMBO Molecular Medicine)
There are also other strategies for eliminating senescent cells via conjugating drugs with nanostructures. For example, Thapa et al. used the CD9 receptor overexpressed in senescent cells to functionalize calcium carbonate nanoparticles to target senescent cells and eliminate them with loaded rapamycin [155]. In addition, Nguyen et al. conjugated CD9 monoclonal antibodies on the surface of PEGylated liposomes for rapamycin delivery to target senescent cells [156]. Nagesh et al. formulated a tannic acid-docetaxel self-assembly (DSA) nanoformulation as an efficient delivery system for targeting and diminishing cellular senescence in prostate tumours. They also demonstrated that exposure to DSAs could wield an antisenescence function by selectively deregulating the senescence-associated TGFβR1/FOXO1/p21 pathway in vitro and in vivo [157]. Researchers have also investigated quercetin functionalized magnetite nanoparticles (MNPQ). In that study, quercetin surface-functionalized Fe3O4 nanoparticles were synthesised and wielded an antisenescence function by inducing oxidative stress in senescent cells, which demonstrated that MNPQ could reduce the number of senescent cells and limit the secretion of IL-8 and IFN-β via activating AMP-activated protein kinase [158].
Other strategies
Recently, research on targeted senescence has also focused on cellular metabolic reprogramming and intestinal flora metabolism. Studies have found that regulating the metabolism of intestinal microflora or indirectly regulating intestinal microecology with antiaging drugs can mitigate senescence or eliminate senescent cells. Bárcena et al. investigated the role of deregulation of the gut flora in the aging process. They identified specific alterations in the intestinal flora of a mouse model of progeria and found similar changes in the intestines of human patients with progeria. In addition, they transplanted the faecal microbiota of wild-type mice into the gut of a progeria mouse model and found that the healthy life span of prematurely aged mice was extended, and through intestinal metabolomics analysis, they identified secondary bile acids as a key component in the restoration of healthy gut microbiota in ageing mice [159]. Saccon et al. studied the effects of dasatinib and quercetin, a common combination of senolytics, on gut microbes in ageing mice. They found that dasatinib plus quercetin can reduce cellular senescence and inflammation in the gut while altering the gut microbiota, suggesting that combined senolytics could improve the health of older people by reducing ageing, inflammation and microbial dysbiosis in the gut [160]. Trimethylamine-N-oxide (TMAO) is an intestinal microbial metabolite. Ke et al. investigated the relationship between TMAO and vascular senescence and found that circulating levels of TMAO were elevated in elderly and senescent mouse models, and using both in vivo and in vitro experiments demonstrated that TMAO can promote endothelial and vascular senescence. Furthermore, this phenomenon may be associated with the inhibition of SIRT1 expression and increased oxidative stress, which in turn activates the p53/Rb gene [161].
Conclusions
Chronic wounds are a tremendous problem for clinicians and researchers in wound healing. Tissue replacement, implantation materials, cell therapy and growth factors might be novel strategies for accelerating chronic wound healing. However, these treatments are ineffective for some wounds. In chronic wounds, accumulation of senescent cells creates a prolonged inflammation microenvironment, leading to stem cell dysfunction. Increasing evidence in various fields (e.g. cancer, pulmonary fibrosis, atherosclerosis, etc.) concerning targeted therapies has demonstrated that impeding cells from becoming senescent or eliminating them can alleviate the pathological processes of senescence. Similarly, targeting senescent cells can be a promising strategy for treating chronic wounds. Emerging strategies aimed at senescence and its harmful components mainly focus on controlling the stimulus factors, improving the pathological microenvironment and directly targeting senescence. Experimental data reveal the actual function of novel antisenescence therapies in age-related diseases, demonstrating that therapies targeting senescence for nonhealing chronic wounds may be on the horizon. A number of published investigations have determined a clear relationship between senescence and chronic wounds, especially diabetes wounds. In several cases, wound healing was obviously accelerated by eliminating senescent cells or its by-product, SASP. As a result, strategies targeting senescence might be a prospective and effective way to mitigate nonhealing wounds with accelerated repair.
Authors’ contributions
XW and ML contributed to concept generation, literature searching and manuscript writing. ZZ, JM, YG, LC, YP and SY contributed to literature searching and eligible study screening. LY contributed to manuscript preparation and reviewed the final version.
Abbreviations
CKDI: Cyclin-dependent kinase inhibitors; DDR: DNA damage response; DPP4: Dipeptidyl peptidase-4; DSA: docetaxel self-assembly; ECM: Extracellular matrix; IL-10: Interleukin-10; MMPs: Matrix metalloproteinases; MNPQ: Quercetin functionalised magnetite nanoparticles; mTOR: Mammalian target of rapamycin; NF-κB; Nuclear factor-kB; NKG2D: Natural killer cell receptor; ROS: Reactive oxygen species; SA-β-Gal: Senescence-associated beta-galactosidase; SASP: Senescence-associated secretory phenotype; TMAO: Trimethylamine-N-oxide; TNF: Tumour necrosis factor.
Conflicts of interest
The authors declare that they have no competing interests.
Funding
This work was supported by the Science and Technology Innovation Project of Guangdong Province (No. 2018KJYZ005), the Natural Science Foundation of Guangdong Province (No. 2020A151501107), the Natural Science Foundation of Tibet Autonomous Region (No. XZ2017ZR-ZY021) and the Guangdong Province Key Field R&D Programme Project (No. 2020B1111150001).
References
- 1. Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care. 2015;4:560–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. Dis Model Mech. 2014;7:1205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Z, Shi C. Cellular senescence is a promising target for chronic wounds: a comprehensive review. Burns Dent Traumatol. 2020;8:tkaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mustoe TA, O'Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg. 2006;117:35S–41. [DOI] [PubMed] [Google Scholar]
- 5. Telgenhoff D, Shroot B. Cellular senescence mechanisms in chronic wound healing. Cell Death Differ. 2005;12:695–8. [DOI] [PubMed] [Google Scholar]
- 6. Wang Z, Chen Z, Jiang Z, Luo P, Liu L, Huang Y, et al. Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat Commun. 2019;10:2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bian X, Li B, Yang J, Ma K, Sun M, Zhang C, et al. Regenerative and protective effects of dMSC-sEVs on high-glucose-induced senescent fibroblasts by suppressing RAGE pathway and activating Smad pathway. Stem Cell Res Ther. 2020;11:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813–27. [DOI] [PubMed] [Google Scholar]
- 9. Wilkinson HN, Hardman MJ. Wound senescence: a functional link between diabetes and ageing? Exp Dermatol. 2021;30:68–73. [DOI] [PubMed] [Google Scholar]
- 10. Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–18. [DOI] [PubMed] [Google Scholar]
- 11. Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, et al. A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol. 2010;8:e1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cortes-Canteli M, Iadecola C. Alzheimer's disease and vascular aging: JACC focus seminar. J Am Coll Cardiol. 2020;75:942–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prattichizzo F, De Nigris V, Mancuso E, Spiga R, Giuliani A, Matacchione G, et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 2018;15:170–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schultz GS, Sibbald RG, Falanga V, Ayello EA, Dowsett C, Harding K, et al. Wound bed preparation: a systematic approach to wound management. Wound Repair Regen. 2003;11:S1–28. [DOI] [PubMed] [Google Scholar]
- 16. Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature. 2019;574:553–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010;89:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pils V, Terlecki-Zaniewicz L, Schosserer M, Grillari J, Lammermann I. The role of lipid-based signalling in wound healing and senescence. Mech Ageing Dev. 2021;198:111527. [DOI] [PubMed] [Google Scholar]
- 19. Chelvarajan RL, Liu Y, Popa D, Getchell ML, Getchell TV, Stromberg AJ, et al. Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol. 2006;79:1314–27. [DOI] [PubMed] [Google Scholar]
- 20. Gosain A, DiPietro LA. Aging and wound healing. World J Surg. 2004;28:321–6. [DOI] [PubMed] [Google Scholar]
- 21. Dellago H, Preschitz-Kammerhofer B, Terlecki-Zaniewicz L, Schreiner C, Fortschegger K, Chang MW, et al. High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell. 2013;12:446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. 2019;99:1047–78. [DOI] [PubMed] [Google Scholar]
- 23. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. [DOI] [PubMed] [Google Scholar]
- 24. Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27:2801–9. [DOI] [PubMed] [Google Scholar]
- 25. Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. [DOI] [PubMed] [Google Scholar]
- 28. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol. 2016;18:777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–53. [DOI] [PubMed] [Google Scholar]
- 32. Turenne GA, Paul P, Laflair L, Price BD. Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal serine residues of p53. Oncogene. 2001;20:5100–10. [DOI] [PubMed] [Google Scholar]
- 33. Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–8. [DOI] [PubMed] [Google Scholar]
- 34. Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol. 1999;19:1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. [DOI] [PubMed] [Google Scholar]
- 37. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–806. [DOI] [PubMed] [Google Scholar]
- 38. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. [DOI] [PubMed] [Google Scholar]
- 39. Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–61. [DOI] [PubMed] [Google Scholar]
- 42. Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011;50:5566–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010;22:1003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hernandez-Segura A, Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol. 2017;27:2652–2660 e2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. [DOI] [PubMed] [Google Scholar]
- 51. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17:1205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011;25:1245–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hayakawa T, Iwai M, Aoki S, Takimoto K, Maruyama M, Maruyama W, et al. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One. 2015;10:e0116480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. [DOI] [PubMed] [Google Scholar]
- 60. Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. [DOI] [PubMed] [Google Scholar]
- 61. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6:265–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care (New Rochelle). 2015;4:560–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Demidova-Rice TN, Durham JT, Herman IM. Wound healing angiogenesis: innovations and challenges in acute and chronic wound healing. Adv Wound Care (New Rochelle). 2012;1:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhou K, Ma Y, Brogan MS. Chronic and non-healing wounds: the story of vascular endothelial growth factor. Med Hypotheses. 2015;85:399–404. [DOI] [PubMed] [Google Scholar]
- 66. Herrick SE, Sloan P, McGurk M, Freak L, McCollum CN, Ferguson MW. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am J Pathol. 1992;141:1085–95. [PMC free article] [PubMed] [Google Scholar]
- 67. Li M, Hou Q, Zhong L, Zhao Y, Fu X. Macrophage related chronic inflammation in non-healing wounds. Front Immunol. 2021;12:681710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kathawala MH, Ng WL, Liu D, Naing MW, Yeong WY, Spiller KL, et al. Healing of chronic wounds: an update of recent developments and future possibilities. Tissue Eng Part B Rev. 2019;25:429–44. [DOI] [PubMed] [Google Scholar]
- 69. McCarty SM, Percival SL. Proteases and delayed wound healing. Adv Wound Care (New Rochelle). 2013;2:438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stanley A, Osler T. Senescence and the healing rates of venous ulcers. J Vasc Surg. 2001;33:1206–11. [DOI] [PubMed] [Google Scholar]
- 71. Tsourdi E, Barthel A, Rietzsch H, Reichel A, Bornstein SR. Current aspects in the pathophysiology and treatment of chronic wounds in diabetes mellitus. Biomed Res Int. 2013;2013:385641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bitar MS. The GSK-3beta/Fyn/Nrf2 pathway in fibroblasts and wounds of type 2 diabetes: on the road to an evidence-based therapy of non-healing wounds. Adipocyte. 2012;1:161–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilkinson HN, Clowes C, Banyard KL, Matteuci P, Mace KA, Hardman MJ. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. Journal of Investigative Dermatology. 2019;139:1171. [DOI] [PubMed] [Google Scholar]
- 74. Han G, Ceilley R. Chronic wound healing: a review of current management and treatments. Adv Ther. 2017;34:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wilkinson HN, Clowes C, Banyard KL, Matteuci P, Mace KA, Hardman MJ. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J Invest Dermatol. 2019;139:1171–1181 e1176. [DOI] [PubMed] [Google Scholar]
- 76. Ruan Y, Wu S, Zhang L, Chen G, Lai W. Retarding the senescence of human vascular endothelial cells induced by hydrogen peroxide: effects of 17beta-estradiol (E2) mediated mitochondria protection. Biogerontology. 2014;15:367–75. [DOI] [PubMed] [Google Scholar]
- 77. Duan J, Duan J, Zhang Z, Tong T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int J Biochem Cell Biol. 2005;37:1407–20. [DOI] [PubMed] [Google Scholar]
- 78. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Prattichizzo F, De Nigris V, La Sala L, Procopio AD, Olivieri F, Ceriello A. "Inflammaging" as a Druggable target: a senescence-associated secretory phenotype-Centered view of type 2 diabetes. Oxid Med Cell Longev. 2016;2016:1810327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. 2017;15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain. 2016;139:653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kobbe C. Cellular senescence: a view throughout organismal life. Cell Mol Life Sci. 2018;75:3553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ennis WJ, Sui A, Bartholomew A. Stem cells and healing: impact on inflammation. Adv Wound Care (New Rochelle). 2013;2:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cianfarani F, Toietta G, Di Rocco G, Cesareo E, Zambruno G, Odorisio T. Diabetes impairs adipose tissue-derived stem cell function and efficiency in promoting wound healing. Wound Repair Regen. 2013;21:545–53. [DOI] [PubMed] [Google Scholar]
- 85. Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell. 2019;18:e12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sorg H, Tilkorn DJ, Hager S, Hauser J, Mirastschijski U. Skin wound healing: an update on the current knowledge and concepts. Eur Surg Res. 2017;58:81–94. [DOI] [PubMed] [Google Scholar]
- 87. Rodriguez-Menocal L, Salgado M, Ford D, Van Badiavas E. Stimulation of skin and wound fibroblast migration by mesenchymal stem cells derived from normal donors and chronic wound patients. Stem Cells Transl Med. 2012;1:221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ovadya Y, Krizhanovsky V. Strategies targeting cellular senescence. J Clin Invest. 2018;128:1247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. [DOI] [PubMed] [Google Scholar]
- 90. Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Weindruch R, Walford RL. Dietary restriction in mice beginning at 1 year of age: effect on life-span and spontaneous cancer incidence. Science. 1982;215:1415–8. [DOI] [PubMed] [Google Scholar]
- 92. Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hahn O, Gronke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017;18:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bonda DJ, Lee HG, Camins A, Pallas M, Casadesus G, Smith MA, et al. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 2011;10:275–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Thandavarayan RA, Garikipati VN, Joladarashi D, Suresh Babu S, Jeyabal P, Verma SK, et al. Sirtuin-6 deficiency exacerbates diabetes-induced impairment of wound healing. Exp Dermatol. 2015;24:773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl). 2011;89:667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, et al. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017;36:2280–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. [DOI] [PubMed] [Google Scholar]
- 105. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. [DOI] [PubMed] [Google Scholar]
- 106. Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119:5807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. [DOI] [PubMed] [Google Scholar]
- 108. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol. 2015;8:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. King AC, Peterson TJ, Horvat TZ, Rodriguez M, Tang LA. Venetoclax: a first-in-class oral BCL-2 inhibitor for the management of lymphoid malignancies. Ann Pharmacother. 2017;51:410–6. [DOI] [PubMed] [Google Scholar]
- 111. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017;9:955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Moncsek A, Al-Suraih MS, Trussoni CE, O'Hara SP, Splinter PL, Zuber C, et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(−/−) ) mice. Hepatology. 2018;67:247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23:775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23:1072–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 2016;8:2915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14:617–28. [DOI] [PubMed] [Google Scholar]
- 121. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, et al. NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY). 2016;8:328–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. [DOI] [PubMed] [Google Scholar]
- 125. Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun. 2019;10:2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–5. [DOI] [PubMed] [Google Scholar]
- 127. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Frescas D, Roux CM, Aygun-Sunar S, Gleiberman AS, Krasnov P, Kurnasov OV, et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc Natl Acad Sci U S A. 2017;114:E1668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE, et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017;31:1529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012;24:835–45. [DOI] [PubMed] [Google Scholar]
- 132. Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Moiseeva O, Deschenes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12:489–98. [DOI] [PubMed] [Google Scholar]
- 134. Kobbe C. Targeting senescent cells: approaches, opportunities, challenges. Aging (Albany NY). 2019;11:12844–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Horvath S, Lu AT, Cohen H, Raj K. Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging (Albany NY). 2019;11:3238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sodagam L, Lewinska A, Wnuk M, Rattan SIS. Chronic exposure to rapamycin and episodic serum starvation modulate ageing of human fibroblasts in vitro. Biogerontology. 2017;18:841–54. [DOI] [PubMed] [Google Scholar]
- 139. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 2015;112:E6301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Karkera J, Steiner H, Li W, Skradski V, Moser PL, Riethdorf S, et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. Prostate. 2011;71:1455–65. [DOI] [PubMed] [Google Scholar]
- 141. Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fossa A, et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15:966–74. [DOI] [PubMed] [Google Scholar]
- 142. Emery P, Keystone E, Tony HP, Cantagrel A, Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kimmel JC, Penland L, Rubinstein ND, Hendrickson DG, Kelley DR, Rosenthal AZ. Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res. 2019;29:2088–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. 2019;10:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. [DOI] [PubMed] [Google Scholar]
- 147. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to Chemotoxicity and aging. Cell. 2017;169:132–147 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Papismadov N, Gal H, Krizhanovsky V. The anti-aging promise of p21. Cell Cycle. 2017;16:1997–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sajja HK, East MP, Mao H, Wang YA, Nie S, Yang L. Development of multifunctional nanoparticles for targeted drug delivery and noninvasive imaging of therapeutic effect. Curr Drug Discov Technol. 2009;6:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chenthamara D, Subramaniam S, Ramakrishnan SG, Krishnaswamy S, Essa MM, Lin FH, et al. Therapeutic efficacy of nanoparticles and routes of administration. Biomater Res. 2019;23:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–95. [DOI] [PubMed] [Google Scholar]
- 152. Agostini A, Mondragon L, Bernardos A, Martinez-Manez R, Marcos MD, Sancenon F, et al. Targeted cargo delivery in senescent cells using capped mesoporous silica nanoparticles. Angew Chem Int Ed Engl. 2012;51:10556–60. [DOI] [PubMed] [Google Scholar]
- 153. Munoz-Espin D, Rovira M, Galiana I, Gimenez C, Lozano-Torres B, Paez-Ribes M, et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med. 2018;10:e9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Xu T, Cai Y, Zhong X, Zhang L, Zheng D, Gao Z, et al. Beta-galactosidase instructed supramolecular hydrogelation for selective identification and removal of senescent cells. Chem Commun (Camb). 2019;55:7175–8. [DOI] [PubMed] [Google Scholar]
- 155. Thapa RK, Nguyen HT, Jeong JH, Kim JR, Choi HG, Yong CS, et al. Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci Rep. 2017;7:43299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nguyen HT, Thapa RK, Shin BS, Jeong JH, Kim JR, Yong CS, et al. CD9 monoclonal antibody-conjugated PEGylated liposomes for targeted delivery of rapamycin in the treatment of cellular senescence. Nanotechnology. 2017;28:095101. [DOI] [PubMed] [Google Scholar]
- 157. Nagesh PKB, Chowdhury P, Hatami E, Kumari S, Kashyap VK, Tripathi MK, et al. Cross-linked polyphenol-based drug Nano-self-assemblies engineered to blockade prostate cancer senescence. ACS Appl Mater Interfaces. 2019;11:38537–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lewinska A, Adamczyk-Grochala J, Bloniarz D, Olszowka J, Kulpa-Greszta M, Litwinienko G, et al. AMPK-mediated senolytic and senostatic activity of quercetin surface functionalized Fe3O4 nanoparticles during oxidant-induced senescence in human fibroblasts. Redox Biol. 2020;28:101337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Barcena C, Valdes-Mas R, Mayoral P, Garabaya C, Durand S, Rodriguez F, et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat Med. 2019;25:1234–42. [DOI] [PubMed] [Google Scholar]
- 160. Saccon TD, Nagpal R, Yadav H, Cavalcante MB, Nunes ADC, Schneider A, et al. Senolytic combination of Dasatinib and quercetin alleviates intestinal senescence and inflammation and modulates the gut microbiome in aged mice. J Gerontol A Biol Sci Med Sci. 2021;76:1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ke Y, Li D, Zhao M, Liu C, Liu J, Zeng A, et al. Gut flora-dependent metabolite trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic Biol Med. 2018;116:88–100. [DOI] [PubMed] [Google Scholar]
- 162. Yang L, Licastro D, Cava E, Veronese N, Spelta F, Rizza W, et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 2016;14:422–8. [DOI] [PubMed] [Google Scholar]