

Abstract
In animals, systemic control of metabolism is conducted by metabolic tissues and relies on the regulated circulation of a plethora of molecules, such as hormones and lipoprotein complexes. MicroRNAs (miRNAs) are a family of post-transcriptional gene repressors that are present throughout the animal kingdom and have been widely associated with the regulation of gene expression in various contexts, including virtually all aspects of systemic control of metabolism. Here we focus on glucose and lipid metabolism and review current knowledge of the role of miRNAs in their systemic regulation. We survey miRNA-mediated regulation of healthy metabolism as well as the contribution of miRNAs to metabolic dysfunction in disease, particularly diabetes, obesity and liver disease. Although most miRNAs act on the tissue they are produced in, it is now well established that miRNAs can also circulate in bodily fluids, including their intercellular transport by extracellular vesicles, and we discuss the role of such extracellular miRNAs in systemic metabolic control and as potential biomarkers of metabolic status and metabolic disease.
Animal life appeared approximately 600 million years ago, and several challenges had to be overcome. Among them, there was the challenge of organizing cells into specialized structures of the body. But an equally important challenge was ensuring that all cells coordinate their common metabolism of the nutrients consumed as a whole by the organism. This coordination necessitated a body-scale regulatory system that controlled the fundamental metabolic biochemistry operating in every cell.
Novel classes of genes also appeared with the advent of multicellular life, and one of the most prevalent of these classes is the one that encodes microRNAs (miRNAs). miRNAs are highly conserved in both plants and animals, but they appear to have emerged independently in each kingdom. In animals, miRNAs act as post-transcriptional inhibitors of gene expression. They associate with proteins of the Argonaute (Ago) family to form an RNA-induced silencing complex (RISC or miRISC) that targets specific mRNA transcripts1. Specificity is provided by base pairing between the single-stranded miRNA in the RISC and the mRNA, typically in the 3′ untranslated region (UTR) of the message. The base pairing is rarely 100% between miRNA and mRNA, and there is a strong bias for pairing between seven bases at the 5′ end of the miRNA (called the ‘seed sequence’) and the mRNA2. This physical association causes the mRNA to be translated into protein with lower yield and the mRNA to turn over more rapidly3. The net result is a reduction in protein output from a given gene, albeit this reduction is often modest in magnitude4,5.
Argonaute (Ago) family.
A class of proteins conserved in eukaryotes that associate with small RNAs such as PIWI-interacting RNAs, microRNAs and siRNAs, and together the RNA–induced silencing complex acts upon DNA or RNA targets.
Seed sequence.
A heptamer sequence located at nucleotides 2–7 relative to the 5′ end of a microRNA. The seed sequence is essential for binding of the microRNA in the RNA-induced silencing complex to a target mRNA, with the binding site typically located in the 3′ untranslated region.
miRNAs are expressed by genes that encode a primary RNA transcript (pri-miRNA) containing one or more short stem–loop structures. These structures are cleaved out of the pri-miRNA by the nuclear enzyme Drosha to form pre-miRNAs6–8, and the pre-miRNAs are processed in the cytoplasm by the enzyme Dicer to form 21-nucleotide duplexes9–11. Association with an Ago protein leads to unwinding and retention of one 21-nucleotide strand of the miRNA to form the RISC1. Typically, one particular strand is favoured by the RISC, but some miRNA duplexes donate both strands to form separate and complementary RISCs12. It was initially believed that miRNAs regulated expression of their target mRNAs only in the cells that produced them. However, in 2007 it was found that miRNAs can be secreted from cells into the circulatory system13.
Owing to the progress in small RNA sequencing, there are now hundreds of annotated miRNA genes per species across the animal kingdom14–18. Invertebrate species typically contain 100–200 annotated miRNAs, and the number is higher in vertebrate species. Humans have the highest number of documented miRNA genes, which currently stands at 556 (REF.18). Many miRNAs can be grouped into families on the basis of their mature sequences19. miRNAs in a family can share almost total sequence identity but many share just their seed sequence20. Of the 556 miRNAs confidently annotated in humans16,18, it is estimated that approximately 300 of them fall into 177 distinct miRNA families20. Of these, 27 families are conserved among all bilateral animals, suggesting that they arose in a common ancestor approximately 500 million years ago.
Bilateral animals.
Animals with bilateral symmetry as an embryo. Bilateral symmetry is where the body has a left side and a right side that are mirror images of one another. Animals that do not fall into this category include sponges, ctenophores, placozoans and cnidarians.
Shortly after miRNAs were shown to be widespread throughout the animal kingdom, it was found experimentally that they regulate various aspects of animal metabolism. Here we summarize our current knowledge of miRNA interactions with two fundamental branches of metabolism: glucose and lipid metabolism. We outline the basic regulation of glucose and lipid metabolism, highlighting their connections with miRNAs in both normal physiology and disease, using multiple, well-documented examples (TABLES 1,2). We focus on glucose and lipid metabolism, since our knowledge of how they are regulated by miRNAs is perhaps the deepest of all aspects of metabolism. This Review also focuses more attention on miRNA regulation in the pancreas and liver, being organs that have central roles in lipid and glucose metabolism. Implications of miRNA-mediated regulation in other important tissues, such as fat, muscle and the brain, can be found in other reviews21–23. Finally, we review studies of circulating miRNAs, examining both their normal functions and the emerging use of these as markers for disease.
Table 1 |.
Key mirNas associated with metabolic regulation in mammals
| mirNa | Tissue of origin | Function and key targets |
|---|---|---|
| let-7 | Muscle | Represses IGF1R, INSR and IRS2 in insulin signal transduction |
| miR-7 | β-Cells of pancreas; liver | Represses Pax6, CAPZA1 and SNCA (pancreas). Activates SREBP1 and SREBP2 (liver). Regulates both lipid and glucose metabolism |
| miR-15b | Liver | Represses insulin receptor |
| miR-19a | Liver | Represses PTEN in insulin signal transduction |
| miR-24 | β-Cells of pancreas | Represses repressors of insulin transcription |
| miR-26a | β-Cells of pancreas | Represses repressors of insulin transcription and CACNA1C in the pancreas. Represses PKCθ in liver (when delivered by extracellular vesicles) |
| miR-27a | Muscle; liver | Represses GP, GAA and PGM to inhibit glycogenolysis in muscle. Represses SCD1 and regulates lipid metabolism in hepatocytes |
| miR-29 | β-Cells of pancreas; liver | Represses MCT1 (pancreas). Represses gluconeogenesis (liver) |
| miR-30c | Liver | Represses MTP in LDL biogenesis |
| miR-30d | β-Cells of pancreas | Represses repressors of insulin transcription |
| miR-33 | Liver | Represses ABCA1, ABCB11, ATP8B1, IRS2, GP, PGM, PCK1 and G6PC |
| miR-34a | Liver | Represses HNF4A and PPARα in lipid metabolism |
| miR-99b | Adipose tissue | Represses FGF21 in liver and muscle |
| miR-103 | Adipose tissue | Represses caveolin in insulin signal transduction |
| miR-107 | Adipose tissue | Represses caveolin in insulin signal transduction |
| miR-122 | Liver | Regulates liver-specific gene expression |
| miR-124a | β-Cells of pancreas | Represses NEUROD1 in insulin expression |
| miR-128-1 | Liver; adipose tissue; muscle | Represses PPAR genes, ABCA1 and LDLR in lipid metabolism |
| miR-130ab | β-Cells of pancreas | Represses PDHA1 and GCK, inhibiting glucose-stimulated insulin release |
| miR-143 | Liver | Represses ORP8 in lipid metabolism |
| miR-144 | Liver | Represses ABCA1 in lipid metabolism |
| miR-148 | β-Cells of pancreas; liver | Represses repressors of insulin transcription. Represses ABCA1 and LDLR in lipid metabolism |
| miR-152 | β-Cells of pancreas | Represses PDHA1 and GCK, inhibiting glucose-stimulated insulin release |
| miR-155 | Adipose tissue | Represses PPARγ in liver and muscle in lipid metabolism (when delivered by extracellular vesicles) |
| miR-186 | β-Cells of pancreas | Represses repressors of insulin transcription |
| miR-187 | β-Cells of pancreas | Represses HIPK1 in insulin secretion |
| miR-200 | β-Cells of pancreas | Represses DNAJC3 and XIAP to control cell survival |
| miR-204 | β-Cells of pancreas | Represses GLP1R in insulin expression |
| miR-223 | Liver | Represses SR-BI and cholesterol biosynthesis genes |
| miR-375 | β-Cells of pancreas | Represses myotrophin in insulin secretion |
| miR-378 | Liver | Represses p110α of PI3K in insulin signal transduction |
| miR-451-1 | Liver | Represses gluconeogenesis |
| miR-466b | Liver | Represses PEPCK |
| miR-802 | β-Cells of pancreas; liver | Represses NEUROD1 and FZD5, inhibiting insulin transcription in β-cells. Represses HNF1B in liver, leading to the impairment of insulin signal transduction |
ABCA1, ATP-binding cassette transporter A1; ABCB11, ATP-binding cassette transporter B11; ATP8B1, phospholipid-transporting ATPase 8B1; CACNA1C, voltage-dependent L-type calcium channel subunit-α 1C; DNAJC3, HSP40 family member C3; FZD5, Frizzled 5; GAA, α-glucosidase; GCK, glucokinase; GLP1R, glucagon-like peptide 1 receptor; GP, glycogen phosphorylase; G6PC, glucose 6-phosphatase; HIPK1, homeodomain-interacting protein kinase 1; HNF1B, hepatocyte nuclear factor 1β; HNF4A, hepatocyte nuclear factor 4α; IGF1R, insulin-like growth factor 1 receptor; INSR, insulin receptor; IRS2, insulin receptor substrate 2; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; MCT1, monocarboxylate transporter 1; miRNA, microRNA; MTP, microsomal triglyceride transfer protein; ORP8, oxysterol-binding protein-related protein 8; PCK1, phosphoenolpyruvate carboxykinase 1; PDHA1, pyruvate dehydrogenase E1 subunit-α 1; PEPCK, phosphoenolpyruvate carboxykinase; PGM, phosphoglucomutase; PI3K, phosphoinositide 3-kinase; PKCθ, protein kinase Cθ; PPAR, peroxisome proliferator-activated receptor; PTEN, phosphatase and tensin homologue; SCD1, stearoyl-CoA desaturase 1; SR-BI, scavenger receptor type B1; SREBP, serum response element-binding protein; XIAP, X-linked inhibitor of apoptosis protein.
Table 2 |.
mirNas and their roles in metabolic diseases
| mirNa | related disease or condition | Mechanism |
|---|---|---|
| miR-7 | Type 2 diabetes | Overexpression in β-cells inhibits insulin synthesis and secretion |
| miR-15b | Obesity-induced insulin resistance | Overexpression inhibits insulin receptor |
| miR-19 | Obesity-induced insulin resistance | Overexpressed in liver |
| miR-29 | Obesity-induced insulin resistance | Overexpressed in liver |
| miR-33 | Obesity | Its loss promotes overeating |
| miR-34a | Alcoholic and non-alcoholic fatty liver disease | Overexpression in liver disease is associated with repression of PPARα and impaired fatty acid metabolism in the liver |
| miR-103 | Obesity-induced insulin resistance | Overexpressed in liver |
| miR-107 | Obesity-induced insulin resistance | Overexpressed in liver |
| miR-122 | Alcoholic liver disease | Underexpression derepresses HIF1α, leading to increased lipid synthesis mediated by PPARγ |
| miR-128-1 | Obesity | Overexpression inhibits energy expenditure in multiple tissues |
| miR-130 | Type 2 diabetes | Overexpression in β-cells inhibits GCK and PDHA1, interfering with metabolism of glucose and glucose-stimulated insulin secretion |
| miR-143 | Obesity-induced insulin resistance | Overexpression inhibits ORP8, impeding insulin signalling via the PI3K–AKT pathway |
| miR-152 | Type 2 diabetes | Overexpression in β-cells inhibits GCK and PDHA1, interfering with metabolism of glucose and glucose-stimulated insulin secretion |
| miR-187 | Type 2 diabetes | Overexpression in β-cells inhibits HIPK1 and in effect insulin expression and secretion |
| miR-200 | Type 2 diabetes | Overexpression in β-cells inhibits the negative regulators of apoptosis DNAJC3 and XIAP |
| miR-451-1 | Obesity-induced insulin resistance | Overexpressed in liver |
| miR-802 | Type 2 diabetes | Overexpression inhibits HNF1B in liver (interfering with insulin signal transduction) and NEUROD1 and FZD5 and in β-cells (suppressing insulin transcription and secretion) |
DNAJC3, HSP40 family member C3; FZD5, Frizzled 5; GCK, glucokinase; HIF1α, hypoxia-inducible factor-α; HIPK1, homeodomain-interacting protein kinase 1; HNF1B, hepatocyte nuclear factor 1β; miRNA, microRNA; ORP8, oxysterol-binding protein-related protein 8; PDHA1, pyruvate dehydrogenase E1 subunit-α1; PI3K, phosphoinositide 3-kinase; PPAR, peroxisome proliferator-activated receptor; XIAP, X-linked inhibitor of apoptosis protein.
miRNAs in systemic glucose metabolism
Glucose is the primary circulating carbohydrate, and its uptake and metabolism must be tightly regulated to ensure balanced distribution of energy to all cells. This regulation involves signalling by insulin and glucagon — two hormones with opposing activities on blood glucose levels — which occurs between the pancreas and other organs, including the liver, where glucose can be stored in the form of glycogen. Insulin and glucagon are released from the endocrine portion of the pancreas, which is composed of roughly one million islets of langerhans distributed throughout the exocrine portion. There are very few connections in the literature linking miRNAs and glucagon signalling, either because glucagon signalling is not extensively regulated by miRNAs or because it has been little studied24. Therefore, we primarily focus on insulin signalling and its relationship with miRNAs.
Islets of Langerhans.
Regions of the pancreas that contain endocrine cells responsible for glucose homeostasis. Constituting 1–2% of the pancreas volume, there are approximately one million islets distributed throughout the pancreas in density routes.
miRNAs as attenuators of insulin expression and secretion.
Insulin synthesis and secretion from pancreatic β-cells is controlled by circulating glucose levels (FIG. 1). The flux of glucose from the blood into β-cells is proportional to the extracellular glucose concentration. This flux occurs through the glucose transporter GLUT2, and intracellular glucose is metabolized via the glycolytic pathway and the tricarboxylic acid cycle to generate an increase in the ATP/ADP ratio25. Insulin expression is stimulated by the elevated ATP/ADP ratio acting on the kinase ERK, which leads to the binding of key transcription activators, including MafA, NEUROD1 and Pax6, to the insulin gene promoter26,27. The ratio of intracellular ATP to ADP also controls insulin secretion from β-cells (FIG. 1). When the ratio is high, it triggers the closure of ATP-sensitive potassium channels, causing plasma membrane depolarization and calcium influx. The increase in cytosolic calcium stimulates SNARE-dependent fusion of insulin-containing secretory granules with the plasma membrane and subsequent release of insulin into the circulation28.
Fig. 1 |. Insulin synthesis and secretion in the islet β-cell and its control by mirNas.

The influx of glucose and its effects on synthesis and secretion of insulin in pancreatic β-cells. Numbers 1–9 refer to the different steps of insulin release. Glucose enters the cell via the transporter GLUT2 (1). It is then metabolized via glycolysis in the cytosol and the tricarboxylic acid (TCA) cycle in mitochondria to generate ATP (2). This increases the ratio of ATP to ADP in the cell (3). A K+ ion channel senses the elevated ATP/ADP ratio and closes (4), resulting in plasma membrane depolarization (5). This is sensed by a voltage-gated calcium channel, which opens, leading to Ca2+ influx (6). Elevation of calcium level triggers rearrangement of the actin cytoskeleton and docking of insulin secretory granules to the plasma membrane, leading to their fusion and release. In parallel, ERK senses the high ATP/ADP ratio and activates the transcription factor NEUROD1 to potentiate insulin gene transcription (8). Following the first phase of insulin release is a second, more sustained phase, which involves substantial cytoskeletal rearrangements and long-range transport of insulin vesicles from intracellular stores to the plasma membrane. Binding of glucagon-like peptide 1 (GLP1) to GLP1R augments these processes via the cAMP–protein kinase A signal transduction pathway (9). MicroRNAs (miRNAs) expressed in β-cells that are implicated in regulating insulin are mostly involved in antagonizing the various steps in the synthesis and release of insulin. They attenuate GLP1 signal transduction, inhibit insulin gene transcription, reduce calcium influx to the cytosol and lower secretory granule docking and release. However, a small subset of miRNAs have also been shown to promote insulin synthesis. AC, adenylyl cyclase.
SNARE.
Eukaryotic protein family that mediates the fusion of membrane-bound vesicles with a target membrane. Target membranes can include the plasma membrane (exocytosis) and membrane-bound compartments such as the Golgi apparatus.
During fasting conditions, insulin secretion is repressed to ensure its minimal release. Under these conditions, a very small pool of granules is present at the plasma membrane and primed to release insulin in response to an increase in blood glucose levels. When feeding induces glucose uptake by β-cells, further granules are trafficked through the secretory system and docked to target-SNARE sites at the plasma membrane to enable massive insulin secretion. The actin cytoskeleton acts as both a barrier to granule docking under fasting conditions and a gatekeeper to allow docking upon glucose induction29,30. This involves cytoskeletal reorganization.
miRNAs have been implicated in controlling insulin expression and secretion24. One way miRNAs regulate these processes is by targeting key enzymes involved in β-cell glucose catabolism and ATP generation (FIG. 1).miR-130a, miR-130b and miR-152 decrease the levels of glucokinase (GCK) and pyruvate dehydrogenase E1 subunit-α1 (PDHA1)31. GCK acts at the first step of glycolysis. PDHA1 is a subunit of pyruvate dehydrogenase, which converts pyruvate derived from glycolysis to acetyl-CoA in mitochondria. Overexpression of miR-130a, miR-130b and miR-152 lowers the intracellular ATP/ADP ratio and decreases insulin synthesis and secretion.
miRNAs can also regulate insulin expression more directly. miR-7 belongs to one of the 27 basal miRNA families common to all bilateral animals18, and its mature RNA sequence is perfectly conserved between humans and the fruit fly32. Comparative analysis of miR-7 expression in animals has demonstrated that it is highly expressed in cells secreting insulin-like peptides as well as the β-cells of the vertebrate pancreas33–37. Mouse miR-7 inhibits insulin gene transcription by repressing the expression of Pax6 (REFS36,38). In Drosophila melanogaster, miR-7 inhibits transcription of the insulin-like peptide gene Ilp2 via an undetermined mechanism37.
Insulin-like peptides.
The evolutionarily ancient superfamily of peptides that include insulin, insulin-like growth factors and peptides within the invertebrates that fulfil functions homologous to those of vertebrate insulin and insulin-like growth factors.
Mouse miR-124a and miR-802 are also inhibitors of insulin gene transcription, and one of their key targets is NEUROD1 mRNA39,40. miR-124a expression is upregulated by glucose41,42. By contrast, a few other miRNAs, including miR-30d, miR-24, miR-26, miR-148 and miR-186, serve as activators of insulin expression by inhibiting repressors of insulin transcription38,41. miR-30d is induced by glucose, and when its expression is blocked, glucose is unable to stimulate insulin gene expression, arguing that miR-30d is a key mediator of glucose signalling in pancreatic β-cells41.
Insulin granule fusion and release is also under miRNA control (FIG. 1). miR-26a is expressed in β-cells and inhibits expression of voltage-gated L-type calcium channel subunit-α1C (CACNA1C), which mediates calcium influx and insulin granule fusion43. Similarly, miR-802 counteracts calcium influx by repressing the WNT5A receptor FZD5, which signals through Ca2+/calmodulin-dependent protein kinase II(CaMKII) to promote the activity of the high-voltage calcium channel for calcium influx40.
In addition to regulating insulin expression, miR-7 also inhibits the amount of insulin secreted from β-cells by repressing proteins involved in cytoskeletal rearrangement. This function is ancient, dating back to the common ancestor of invertebrates and vertebrates37,44. Remarkably, in mammals and D. melanogaster, miR-7 represses expression of the same homologous gene, cpa in the fly and Capza1 in the mouse37. These encode proteins that bind to the barbed ends of F-actin and prevent actin filaments from polymerizing and depolymerizing45,46. Hence, they inhibit actin filament growth dynamics and stabilize the cytoskeleton. In D. melanogaster, Cpa is required for release of insulin-like peptides and increase of their levels in the circulation37. Another mouse miRNA, miR-375, has a similar effect on insulin granule fusion and release as miR-7 (REF.47). The primary target of miR-375 encodes myotrophin47 (also called V-1), which physically associates with free CAPZA1 and regulates its activity48,49. Myotrophin appears to be unable to bind to CAPZA1 that is on the barbed end of F-actin49. Thus, miR-7 and miR-375 appear to exert their effects on the same step in the exocytosis pathway. In addition, mouse miR-7 also represses expression of SNCA, which acts as a molecular chaper-one to assemble SNARE complexes44, thereby limiting SNARE assembly and insulin granule fusion. Mouse miR-7 also has additional targets that also regulate the actin cytoskeleton, including PFN2, WIPF2, BASP1 and PHACTR1, but there is no evidence that they mediate an effect of miR-7 on insulin secretion44.
Barbed ends of F-actin.
Actin filaments have polarity, with each filament having a barbed end and a pointed end. Actin monomers are added to filaments predominantly at the barbed end, whereas release of monomers from a filament occurs predominantly at the pointed end. In the cell, filaments are continuously turned over by dynamic monomer–filament exchange.
There are other ways that miRNAs act on glucose-stimulated insulin release. One way is to inhibit the expression of disallowed genes — that is, genes that should not be expressed in β-cells for them to function properly50. One such gene encodes the monocarboxylate transporter MCT1, which transports lactate and pyruvate into mitochondria51. One reason for repression of MCT1 is that β-cells normally do not release insulin during vigorous exercise, which generates substantial levels of circulating lactate and pyruvate50. MCT1 is blocked from being expressed in β-cells by three miR-29 paralogues, which directly repress MCT1 mRNA52. Another way that miRNAs act in regulating insulin secretion is through the signalling mediated by the gut-derived incretin hormone glucagon-like peptide 1 (GLP1) (FIG. 1). GLP1 provokes insulin release from β-cells by stimulating their responsiveness to glucose, leading to the amplification of insulin release53. GLP1 is secreted in response to food intake, and upon GLP1 binding its receptor, GLP1R in β-cells, cAMP levels increase and sensitize the cells to glucose. miR-204 is highly enriched in β-cells, where it represses GLP1R expression54. Accordingly, loss of miR-204 enhances responsiveness to GLP1R agonists, increasing insulin secretion.
Incretin hormone.
Incretins are peptides secreted into the circulatory system by specialized enteroendocrine cells in the gut upon nutrient ingestion and absorption. They target the islets of Langerhans, where they augment the response of β-cells to glucose by secreting insulin.
miRNAs as attenuators of insulin signal reception.
The liver and adipose tissue are primary sites for glucose storage and release in response to circulating insulin. miRNAs have particularly prominent roles in hepatic biology55. One role of hepatic miRNAs is to regulate the reception of insulin signalling and its effects on glucose storage and release (FIG. 2). miR-378 and miR-33 function in the liver as a nutrient-responsive brake for insulin signal transduction56,57. miR-33 targets insulin receptor substrate 2 (IRS2), a key component of the insulin signalling pathway56. miR-378 represses expression of p110α, the catalytic subunit of phosphoinositide 3-kinase (PI3K). As PI3K is a key transducer of insulin signalling, sustained overexpression of miR-378 results in insulin resistance57, whereas mir-378-knockout mice exhibit enhanced insulin sensitivity. Since fasting increases hepatic miR-378 levels and refeeding has the opposite effect, the function of this miRNA is thought to enhance the liver’s response to feeding/fasting cues mediated by insulin.
Fig. 2 |. Insulin signal transduction and metabolic responses in peripheral tissues and their control by mirNas.
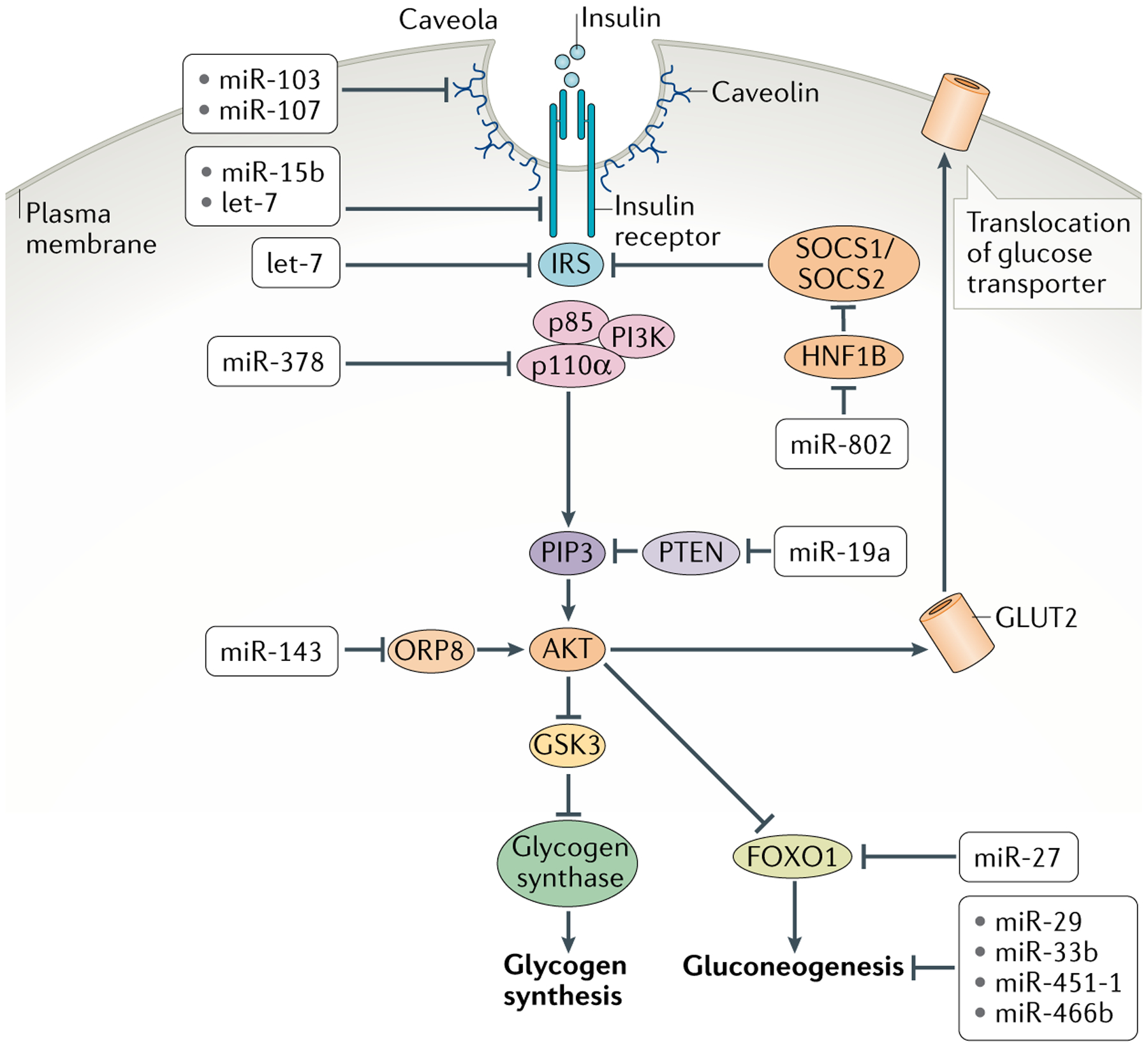
Cells bind circulating insulin via the insulin receptor, which activates insulin receptor substrate (IRS). This in turn recruits association of the regulatory p85 and catalytic p110α subunits of phosphoinositide 3-kinase (PI3K), which becomes activated. Activated PI3K catalyses the addition of phosphate groups to the 3′-OH position in the inositol ring of phosphoinositides to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). Phosphatase and tensin homologue (PTEN) converts PIP3 to hypophosphorylated isoforms. Binding of PIP3 to AKT stimulates its kinase activity, and two of its targets are glycogen synthase kinase 3 (GSK3) and the transcription factor FOXO1, both of which are repressed by phosphorylation. Phosphorylated GSK3 is blocked from inactivating glycogen synthase, resulting in synthesis of glycogen. Phosphorylated FOXO1 is blocked from transcribing genes encoding enzymes in the gluconeogenic pathway, resulting in dampening of gluconeogenesis. AKT additionally promotes glucose uptake by increasing the levels of glucose transporters at the cell membrane. Shown are the various microRNAs (miRNAs) that regulate insulin signal transduction in cells and miRNAs that regulate glycogenesis and gluconeogenesis. HNF1B, hepatocyte nuclear factor 1β; ORP8, oxysterol-binding protein-related protein 8.
Insulin resistance.
A condition where cells of the liver, fat and muscle are not as responsive to insulin doses that elicit normal responses in healthy individuals. Consequently, cells do not absorb glucose from the blood as readily, and the pancreas responds by secreting even more insulin to overcome the weak response to insulin.
miRNAs regulate insulin signalling also in adipose tissue. In adipocytes, miR-103 and miR-107 repress expression of caveolin, resulting in attenuated strength of signals transduced by the insulin receptor58. Finally, skeletal muscle is a major consumer of glucose and relies on insulin signalling for glucose uptake. miRNAs have diverse roles in this tissue as well59. The highly conserved miRNA let-7 is expressed in mouse muscle, where it inhibits multiple components of the insulin signal transduction pathway, including insulin receptor, IRS2 and the receptor for insulin-like growth factor 1 (REF.60). Thus, miRNAs attenuate insulin sensitivity in liver, fat and muscle by weakening the transduction pathway downstream of the insulin signal.
miRNAs in the regulation of glucose metabolism in response to insulin signals.
In addition to regulating systemic glucose metabolism by modulating insulin signal generation and reception, miRNAs also impact the enzymatic responses downstream of insulin signalling in peripheral tissues such as liver and muscle to regulate glucose storage and synthesis. Glucose is stored in liver and muscle as glycogen, and glycogen levels rise in cells stimulated by insulin. This occurs by enhancement of glycogen synthesis (glycogenesis) and reduction of glycogen breakdown (glycogenolysis). miR-19a activates glycogenesis by repressing expression of PTEN in hepatocytes61. PTEN indirectly activates glycogen synthase kinase 3 (GSK3), which is an inhibitor of the enzyme responsible for glycogenesis, glycogen synthase. Thus, by inhibiting PTEN, miR-19a stimulates hepatic glycogenesis. Glycogenolysis is also regulated by miRNAs. Liver cells express miR-33, overexpression of which causes a drop in the levels of glycogen phosphorylase and phosphoglucomutase (PGM), two key glycogenolytic enzymes, leading to glycogen accumulation in the liver62. Glycogen storage is regulated in a similar fashion in the muscle, where expression of miR-27a63 leads to the inhibition of glycogen phosphorylase, PGM and another glycogenolytic enzyme, α-glucosidase (GAA), and in consequence glycogen accumulation.
In concordance with promoting glycogen storage, insulin also instructs cells to stop the biosynthesis of glucose (gluconeogenesis). The main site of gluconeogenesis in the body is the liver, which is also subject to regulation, and specifically, attenuation by miRNAs (FIG. 2). miR-27, miR29 and miR-451-1 decrease hepatic glucose output by directly targeting enzymes in the gluconeogenic pathway or transcription factors that regulate them64–66. Another particularly interesting example of hepatic gluconeogenesis regulation is human miR-33b. miR-33b directly represses expression of two key gluconeogenesis genes encoding phosphoenolpyruvate carboxykinase 1 (PCK1) and glucose 6-phosphatase (G6PC)62. This has the effect of reducing the level of gluconeogenesis. miR-33b is generated from a pri-miRNA that encodes the transcription factor SREBP1, which together with SREBP2 activates gene expression to enhance lipid uptake and fatty acid/sterol synthesis in cells67–69. In addition, SREBP1 expression also promotes hepatic glycogenesis and simultaneously suppresses the expression of gluconeogenic genes in response to postprandial signals from insulin, and its expression is increased in hepatocytes in response to insulin signalling70,71. Since SREBP1 and miR-33b are co-expressed by the same gene, insulin triggers upregulation of both factors in the liver to efficiently suppress gluconeogenesis in glucose-abundant conditions62.
Of interest, miRNA-mediated regulation of gluconeogenesis was linked to the effect of psychological stress on metabolism. Remarkably, the effect was transmitted from male mice who were subjected to stress to their offspring, who were not directly subjected to stress72. These offspring exhibited increased gluconeogenesis and consequent hyperglycaemia, which was linked to elevated levels of phosphoenolpyruvate carboxykinase (PEPCK), a key gluconeogenic enzyme. Mechanistically, it was demonstrated that the sperm of stressed male mice featured hypermethylation at the promoter of the Sfmbt2 gene in their sperm, and this epigenetic mark was maintained in the somatic cells of their offspring72. This methylation inhibited expression of both SFMBT2 and miR-466b, which is encoded in an intron within the Sfmbt2 gene. Accordingly, the livers from offspring of stressed fathers showed lower miR-466b levels, and because miR-466b directly represses PEPCK expression, this partially accounts for the abnormal gluconeogenesis in such mice.
In conclusion, miRNAs generally potentiate the metabolic responses to insulin signalling. This is opposed to their role in regulating insulin signalling, which is mostly centred on the attenuation of insulin signal generation and transduction (TABLE 1). Although this might appear contradictory, cell regulation is often a result of combining inhibition with activation, and the opposing effects of miRNA on insulin action likely reflect the different timescales associated with signal transduction versus enzymatic metabolic responses.
miRNAs in obesity and type 2 diabetes
The widespread availability of high-calorie foods coupled with a sedentary lifestyle is a primary driver that fuels the current epidemic of obesity and type 2 diabetes73. Obesity contributes to contracting type 2 diabetes in that excessive fat accumulation generates insulin resistance by impairing insulin’s ability to regulate liver metabolism74. Various miRNAs have been associated with obesity and type 2 diabetes (TABLE 2).
miRNAs as promoters of obesity.
Several lines of evidence link miRNA to weight control. One of these miRNAs is miR-33, which regulates both hepatic glucose metabolism (see earlier) and systemic lipid metabolism (discussed later). Loss of mir-33 in mice has a profound effect on their feeding behaviour, with abnormally high food intake causing obesity and insulin resistance75.
Another illustrative example is miR-128-1, expression of which, in contrast to miR-33, has been implicated in human obesity. In humans, there are heritable predispositions to becoming obese. One of the contributing genetic loci has been mapped to 2q21.3, which segregates with a lactase gene variant that confers upon adults the ability to digest milk76. This variant was positively selected for in Europeans, and an additional gene element in the locus was co-selected that promotes energy storage77,78. In modern times, this now confers susceptibility to obesity and consequently type 2 diabetes. The mir-128-1 gene is located within the human 2q21.3 locus, and it is also located within syntenic loci in dogs and cattle that promote fat storage in these species79–81. miR-128-1 is broadly expressed in adipose tissue, muscle and liver, where it regulates circulating lipoprotein homeostasis80,82. It also regulates expression of genes encoding PPAR transcription factors as well as other regulators of fatty acid oxidation, mitochondrial energy expenditure and inflammation83. The net effect is that miR-128-1 coordinately restricts gene expression programmes governing energy expenditure across multiple metabolically active organs and tissues. Consequently, loss of miR-128-1 in mice fed a calorie-rich diet resulted in reduced weight gain and less fat accumulation83, which correlated with increased insulin sensitivity. Since humans carrying the 2q21.3 variant show increased chromatin accessibility in the mir-128-1 gene and elevated expression of miR-128-1 (REF.83), it is quite possible that their predisposition to obesity is, at least in part, caused by enhanced expression of miR-128-1, and this miRNA may be a valuable target for obesity management.
Lipoprotein.
Lipoproteins carry cholesterol and triglycerides in the circulation, and they can both deliver and remove lipids from cells to mediate lipid homeostasis. Lipoproteins do not simply associate with lipids into small molecular complexes, but rather are found as lipoprotein particles of various sizes in the blood plasma.
PPAR transcription factors.
A family of nuclear receptor proteins that control the expression of a large number of genes involved in metabolic homeostasis, lipid, glucose and energy metabolism, adipogenesis and inflammation. Endogenous ligands for peroxisome proliferator-activated receptors (PPARs) include free fatty acids, eicosanoids and vitamin B3.
Contribution of miRNAs to insulin resistance.
Other miRNAs propagate obesity-induced insulin resistance. A number of adipose and hepatic miRNAs discussed earlier, including miR-19, miR-29, miR-103, miR-107 and miR-451-1, are abnormally expressed in obese mice58,61,64,65, which correlates with insulin resistance. Other miRNAs are also overexpressed in obese mouse models. miR-15b overexpression directly targets the insulin receptor in obese animal models, and this can be a causal factor in the pathogenesis of hepatic insulin resistance84. Obesity-induced miR-143 overexpression promotes insulin resistance by targeting an oxysterol-binding protein-related-protein, ORP8 (REF.85). This results in inhibition of the PI3K–AKT pathway activation by insulin (FIG. 2). miR-802 is overexpressed in the livers of obese mice and humans, and this promotes insulin resistance and glucose intolerance86. Mechanistically, miR-802 was shown to target expression of the transcription factor HNF1B, leading to the upregulation of two HNF1B-responsive genes, Socs1 and Socs3, which impairs insulin signal transduction by inhibiting phosphorylation of IRS proteins (FIG. 2). For most of the miRNAs discussed above, obesity leads to overexpression of the miRNAs in the liver, which appears to contribute to the pathophysiology of obesity. Genetic ablation of miRISCs from the liver of obese mice alleviated the symptoms of insulin resistance, consistent with liver miRNAs contributing to pathophysiology of obesity-induced glucose intolerance87.
miRNAs as drivers of pancreatic β-cell dysfunction.
When tissues become resistant to insulin, β-cells adapt by secreting larger amounts of insulin to maintain normal glucose levels. If the condition persists, then there is a modest loss of β-cell mass in the pancreas, but the most striking consequence is a reduced capability of β-cells to secrete insulin in response to glucose25,88. In effect, the pancreas can no longer meet the demands set by blood glucose levels, and chronic hyperglycaemia results in type 2 diabetes.
Different stages of the disease are accompanied by dynamic changes in β-cell miRNA expression, and sequencing of small RNAs expressed in the islets of patients with type 2 diabetes identified miRNAs that are misexpressed89. A large number of these miRNAs are transcribed from genes localized to an imprinted region of chromosome 14. This locus becomes hypermethylated with the disease onset, leading to repressed expression of these miRNAs89. Downstream target genes of these miRNAs include IAPP and TP53INP1, genes which have been shown to induce β-cell apoptosis upon overexpression in human islets. Furthermore, expression of miR-187 greatly increases in diabetic islets, and experimental overexpression of miR-187 in rat islets attenuated glucose-stimulated insulin secretion in vitro90. This effect appears to be partially mediated by miR-187 regulation of the kinase HIPK1, which promotes insulin expression and secretion. Patients with diabetes also exhibit elevated expression of miR-130a, miR-130b and miR-152, and consequent reduced expression of PDHA1 and GCK31, which may contribute to the impaired secretion of insulin for reasons previously discussed.
Mouse models of the disease have provided further evidence for the role of miRNA misregulation in defects of glucose-stimulated insulin secretion. miR-7a levels increase in β-cells as they undergo insulin secretory failure44. Since this miRNA normally acts to inhibit insulin synthesis and secretion via several mechanisms as discussed earlier (FIG. 1), its overexpression is expected to contribute to β-cell secretory insufficiency. Another miRNA that shows altered expression associated with obesity is miR-802. The levels of miR-802 increase in the pancreatic islets of obese mice, and this overexpression results in impaired insulin transcription (through downregulation of NEUROD1) and insulin secretion (through downregulation of FZD5)40,91. Expression of the miR-200 family greatly increases in the islets of diabetic mice92. β-Cell death is one feature of type 2 diabetes, and miR-200 overexpression in islets of healthy mice leads to β-cell apoptosis. Mechanistically, miR-200 represses expression of the antiapoptotic and stress resistance gene Dnajc3 and the caspase inhibitor gene Xiap92. Blocking miR-200 expression reduces β-cell death and alleviates the symptoms of type 2 diabetes.
In summary, miRNAs contribute to the onset and progression of type 2 diabetes — they become misexpressed in the course of the disease, leading to misregulation of their targets and various steps in glucose homeostasis. A future challenge is to understand the mechanisms behind misexpression of these miRNAs as a means to potentially counteract these mechanisms for the treatment of type 2 diabetes.
miRNAs and lipid metabolism
The liver has a central role in both the production and the clearance of lipids. It does so in part by regulating the distribution of lipoprotein particles in the body. Low-density lipoprotein (LDL) particles are synthesized in hepatocytes and transport cholesterol, triglycerides and phospholipids to cells of the periphery. The liver is also the main source of high-density lipoprotein (HDL) particles, which are produced as lipid-poor particles (also known as pre-β HDL) and released into the circulation, where they can take up lipids, mostly cholesterol, from the periphery for delivery to the liver to be excreted as bile acids (this process is known as reverse cholesterol transport). The balance of LDL and HDL in circulation is critical for lipid homeostasis. Imbalances that favour the accumulation of cellular cholesterol, such as an excess of LDL or a deficit of HDL, promote atherosclerosis and its associated morbidities: stroke and heart disease. Atherosclerosis is initiated when circulating macrophages take up more lipoprotein-derived cholesterol than they can excrete and consequently convert it to cholesteryl ester. Macrophages bearing lipid droplets of cholesteryl ester are called ‘foam cells’, and they are the hallmark of early atherosclerotic lesions93. HDL can remove cholesterol from macrophages and retard their formation into foam cells94. Our understanding of the genetic regulation of plasma lipoprotein levels has been greatly aided by discoveries of miRNAs that mediate lipid homeostasis.
miRNAs and HDL homeostasis.
miRNAs have been identified as critical regulators of HDL biogenesis (FIG. 3). They do not regulate synthesis of the apolipoproteins constituting HDL. Rather, they modulate the efflux of cholesterol across hepatocyte membranes onto lipid-poor APOA1 to generate hepatic HDLs, which is mediated by the ATP-binding cassette transporters ABCA1 and ABCG1 (REF.95). Many miRNAs have been identified that repress expression of ABCA1 in cell culture, including miR-33, miR-128-1, miR-144 and miR-148a67,80,96–103. These miRNAs have also been functionally tested in mice or monkeys, and their inhibition by antagomir or antisense techniques results in an increase in circulating HDL levels80,99–101,103–105. Therefore, the net effect of these miRNAs is to reduce cholesterol efflux into HDLs.
Fig. 3 |. HDL biogenesis and control by mirNas in hepatocytes.
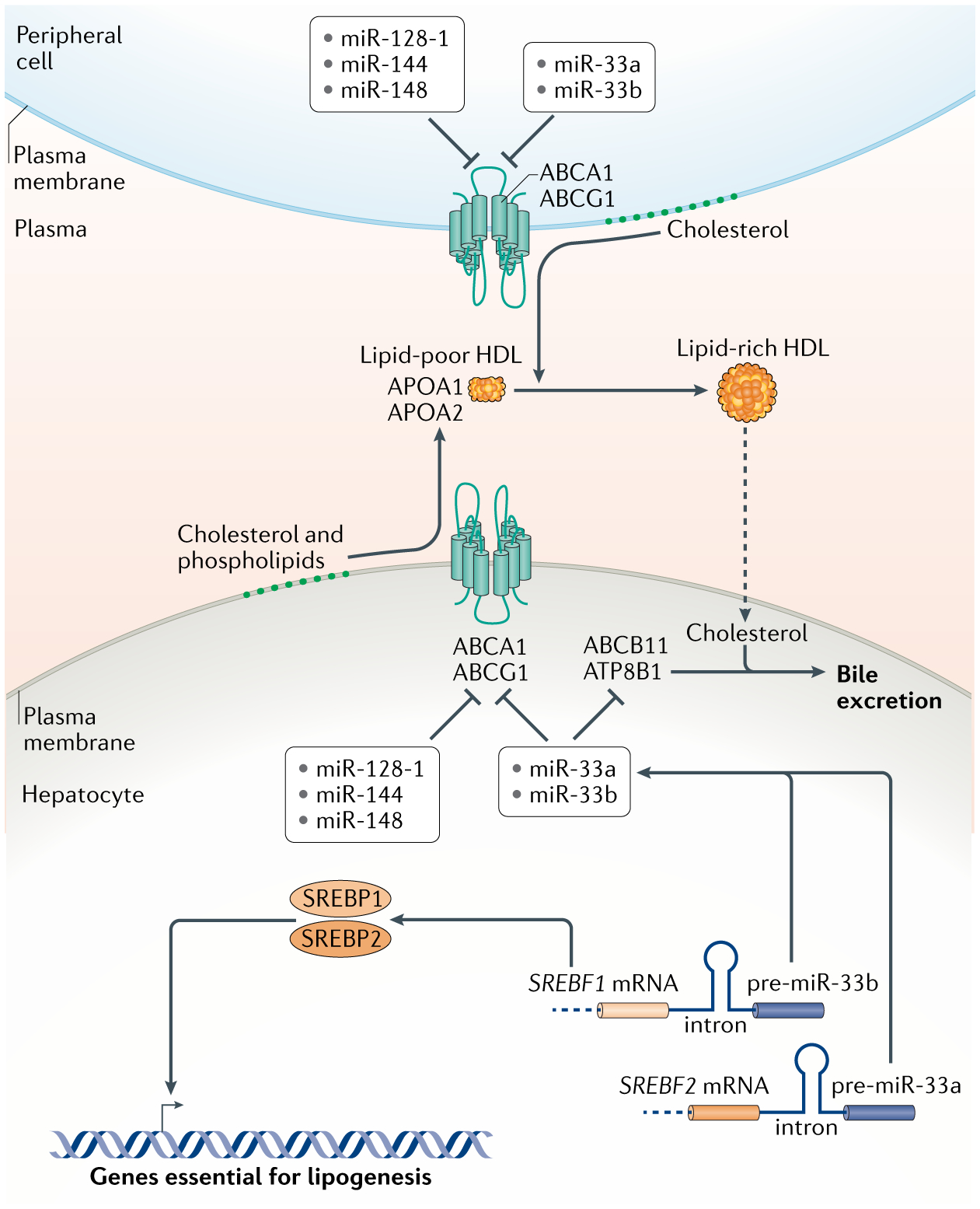
High-density lipoproteins (HDLs) are generated by the association of apolipoproteins APOA1 and APOA2 with lipids. Hepatocytes assemble extracellular lipid-poor HDL particles, which are then enriched with lipids, mostly cholesterol, derived from peripheral cells to make lipid-rich HDL. Loading of lipids into HDLs occurs via the ATP-binding cassetter transporters ABCA1 and ABCG1. The levels of these transporters are inhibited by a number of microRNAs (miRNAs) as shown. Two of these miRNAs, miR-33a and miR-33b, perform this function in concordance with two transcription factors, SREBP1 and SREBP2, which stimulate the synthesis of enzymes in lipogenic pathways. Since the precursors of miR-33a and miR-33b are located in the introns of the genes encoding SREBP2 and SREBP1 respectively, both the proteins and the miRNAs are co-expressed in tandem. miR-33a and miR-33b also inhibit expression of ABCB11 and ATP8B1, which in hepatocytes transport sterols into bile for excretion, allowing clearance of cholesterol derived from lipid-rich HDL returned to the liver after loading in the periphery. Altogether, the coordinated actions of these miRNAs and transcription factors result in accumulation of lipids within hepatocytes.
Apolipoproteins.
Proteins that bind to lipids and form lipoproteins, which circulate in the blood, lymph and cerebrospinal fluid. They not only function to solubilize lipids for transport but also interact with lipoprotein receptors and lipid transport proteins to facilitate lipoprotein uptake and clearance.
Antagomir.
A small synthetic RNA whose purpose is to block the action of a specific microRNA in vivo. An antagomir is fully complementary to a microRNA except for a mismatch or chemical modification at the site of RNA-induced silencing complex cleavage, so as to prevent the antagomir from being cleaved. Antagomirs often have other chemical modifications to inhibit their degradation by ribonucleases.
Of the miRNAs regulating HDL biogenesis and homeostasis, miR-33 has been the most extensively studied. Human miR-33a and miR-33b are intronic miRNAs that are embedded within genes that encode SREBP2 and SREBP1, respectively96–99. miR-33a and miR-33b are co-expressed with the SREBP factors, and increase cellular steady-state lipid levels by repressing ABCA1 (REFS96–99,106) (FIG. 3). They also repress fatty acid oxidation enzymes56,67,107, resulting in lower lipid turnover within cells. Additional miR-33 targets such as ABCB11 and ATP8B1 promote cholesterol excretion from the liver into bile to support cholesterol clearance from the body108,109. An unusual aspect of miR-33 action is that both of its precursor strands are loaded into miRISCs, and even though their sequence composition is complementary, both strands repress the same target genes, including ABCA1 (REF.110). Thus, miR-33 coordinately inhibits the reverse cholesterol transport from the periphery back to the liver and elimination of cholesterol from the body.
miRNAs and LDL homeostasis.
miRNAs regulate production of LDL by modulating hepatic lipid metabolism and lipid loading onto LDL as well as by regulating biogenesis of LDL precursor, called ‘very low density lipoprotein’ (VLDL), in the liver (FIG. 4). miR-30c targets microsomal triglyceride transfer protein (MTP), which is necessary for the lipidation of newly synthesized APOB in the liver to generate VLDL and LDL111. Overexpression of miR-30c in mice reduces the assembly and secretion of these APOB-containing lipoproteins, leading to decreased levels of plasma LDL.
Fig. 4 |. Control of lipid metabolism and LDL biogenesis/turnover by mirNas.
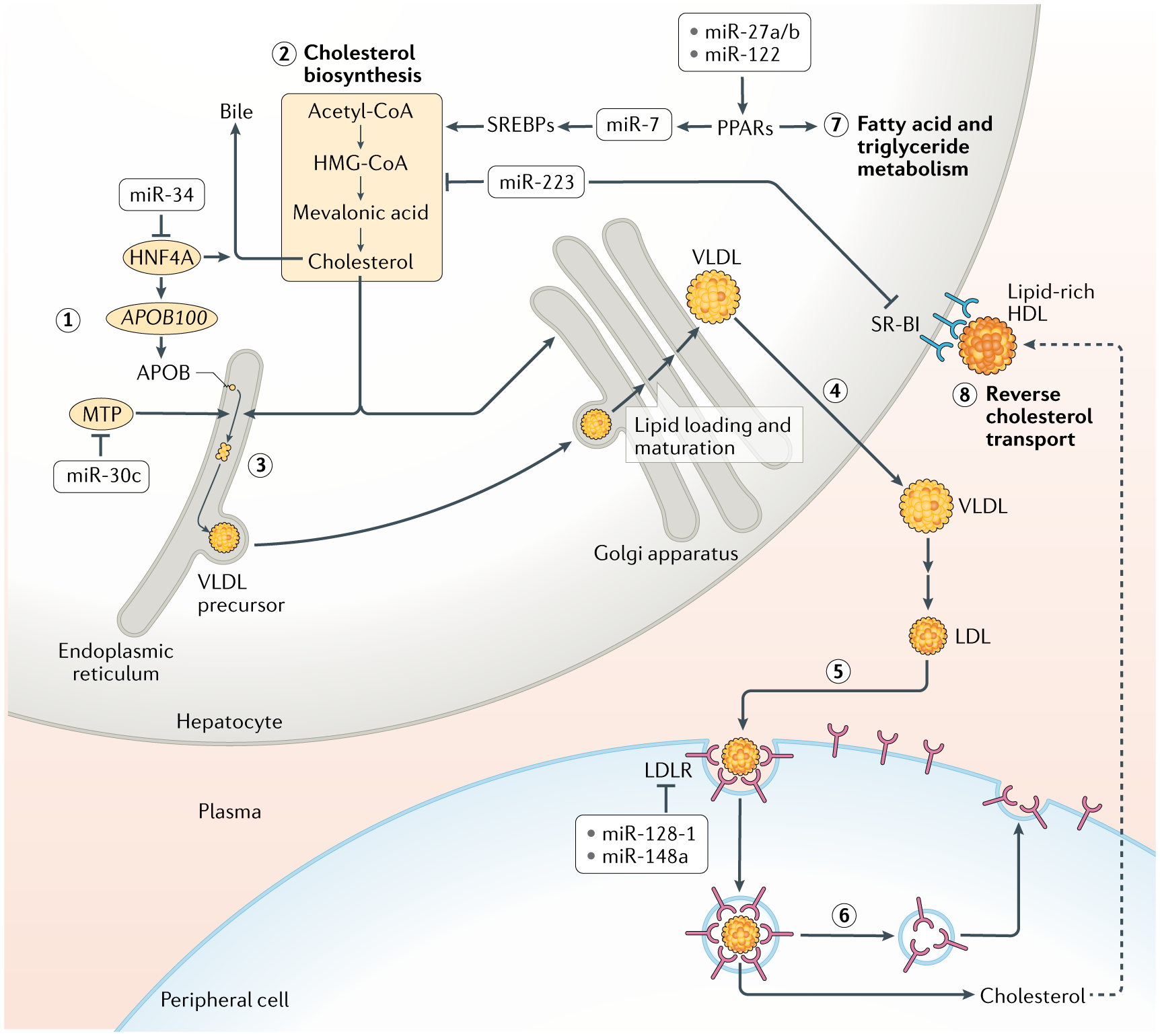
The biogenesis and turnover of low-density lipoproteins (LDL) is shown in steps 1–6. Apolipoprotein B-100 (APOB100) is synthesized and inserted into the endoplasmic reticulum of hepatocytes (1). In parallel, sterol biosynthesis converts the metabolite acetyl-CoA into cholesterol (2). Microsomal triglyceride transfer protein (MTP) transfers lipids to APOB100 to initiate assembly of very-low-density lipoprotein (VLDL) complexes in the endoplasmic reticulum (3). VLDL is shuttled to the Golgi apparatus, where it matures via lipidation, and then is secreted from hepatocytes (4). In the circulatory system, VLDL is oxidized into LDL, and this species reaches cells of the periphery. There, LDL binds to LDL receptor (LDLR) and is endocytosed (5). Sterols and other lipids are transferred out of the endosomes of peripheral cells, where they are utilized (6). The endosomes are recycled to the plasma membrane to redeposit LDLR on the surface. MicroRNAs (miRNAs) regulate virtually all steps of the LDL pathway, as indicated. Moreover, some miRNAs also regulate enzymes that control fatty acid and triglyceride metabolism in the liver, mostly by promoting these processes (7). However, miR-223 represses genes involved in cholesterol biosynthesis. In addition, miR-223 inhibits reverse transport of cholesterol from the peripheral cells back to the liver by repressing hepatocyte expression of the scavenger receptor SR-BI, which binds to cholesterol-loaded HDL and transports cholesterol into the liver (8). The net effect of the action of miR-223 is to reduce hepatic cholesterol levels. HNF4A, hepatocyte nuclear factor 4α; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; PPARs, peroxisome proliferator-activated receptors; SREBPs, sterol regulatory element-binding proteins.
Cells take up lipids from circulating LDL through the receptor LDLR, which is also a target of miRNA regulation. miR-148a is a repressor of LDLR expression, and inhibition of miR-148a in mice increased the clearance of circulating LDL, leading to lower plasma LDL levels80,103. miR-128-1 is also a predicted repressor of LDLR expression, and indeed, inhibition of miR-128-1 resulted in enhanced LDLR expression and promoted LDL clearance from the circulation in mice80,82. It is interesting to note that miR-128-1 and miR-148a also downregulate HDL biosynthesis as discussed above, and so they simultaneously inhibit the opposing branches of circulating lipid transport.
miRNAs and hepatic metabolism of lipids.
The liver is not only the source of lipoprotein biogenesis but also is the major site for lipid metabolism in the body. In the liver, miR-122 is highly abundant112. It regulates various genes involved in hepatic cholesterol and fatty acid synthesis113,114, but is rather more broadly required for the regulation of hepatocyte-specific gene expression than for the targeting of specific metabolic pathways115. Hepatic miR-27b acts as a regulatory hub controlling specifically networks of lipid metabolic genes116. Its paralogue, miR-27a, has also been found to regulate lipid metabolism in the liver117. miR-223 is another regulatory hub that represses genes involved in cholesterol biosynthesis118. Additionally, miR-223 represses hepatocyte expression of the scavenger receptor SR-BI, which binds to cholesterol-loaded HDL and transports cholesterol into the liver119. The net effect of miR-223 action is to reduce hepatic cholesterol levels.
Transcription factor genes are frequently regulated by miRNAs, and this is also observed in relation to lipid metabolism. miR-34a mediates the hepatic response to metabolic stress associated with lipid overload through repression of expression of HNF4A, which is a critical transcription factor in lipid metabolism120. miR-7 activates the expression of SREBP1 and SREBP2 to upregulate genes involved in sterol biosynthesis121. miR-7 is itself activated by PPARα, a transcription factor that regulates fatty acid metabolism. Thus, miR-7 acts to bridge the sterol and fatty acid metabolic pathways in the liver.
In conclusion, miRNAs are involved at many steps in systemic lipid metabolism (TABLE 1).
miRNAs in lipid-associated diseases
Elevated cholesterol levels in blood and peripheral tissues lead to atherosclerosis and its associated morbidities: stroke and heart disease. Abnormal lipid metabolism in the liver leads to liver disease and ultimately liver failure. Some miRNAs involved in lipid metabolism are misregulated in these diseases, and this contributes to disease progression (TABLE 2). A handful of these are being considered as targets for therapeutics (TABLE 3).
Table 3 |.
mirNas as potential biomarkers and therapeutics in metabolic diseases
| mirNa | associated disease or condition | Biomarker or therapeutic |
|---|---|---|
| miR-15a | Insulin resistance | Biomarker |
| miR-20b | Type 2 diabetes | Biomarker |
| miR-23a | Gestational diabetes | Biomarker |
| miR-27a | Non-alcoholic fatty liver disease | miRNA mimic as potential therapeutic |
| miR-30c | Atherosclerosis | miRNA mimic as potential therapeutic |
| miR-33 | Atherosclerosis | Antagomir as potential therapeutic |
| miR-34a | Non-alcoholic fatty liver disease | Biomarker and miRNA mimic as potential therapeutic |
| miR-122 | Non-alcoholic fatty liver disease | Biomarker |
| miR-122 | Alcoholic liver disease | miRNA mimic as potential therapeutic |
| miR-144 | Atherosclerosis | Antagomir as potential therapeutic |
| miR-155 | Type 2 diabetes | Biomarker |
| miR-223 | Gestational diabetes | Biomarker |
| miR-375 | Type 2 diabetes | Biomarker |
miRNA, microRNA.
Roles of miRNAs in deregulated lipoprotein metabolism and atherosclerosis.
Inhibition of miR-33 increases cholesterol transport out of macrophages to the plasma, liver and faeces by more than 80%, suggesting that miR-33 inhibition might prevent foam cell formation and atherosclerosis105. Mouse models for atherosclerosis have been used to examine such a possibility. Mice in which either the Apoe gene or the Ldlr gene is mutated develop atherosclerosis owing to an accumulation of circulatory LDL122,123. When miR-33 was globally mutated or inhibited by antagomirs, there was a regression in the appearance of atherosclerosis in these mice105,124,125. Specific loss of miR-33 in macrophages decreases lipid accumulation and plaque development, arguing for an important role of macrophage miR-33 in the pathogenesis of atherosclerosis125. Derepression of ABCA1 to promote cholesterol efflux from macrophages could be expected as a primary mechanism of atheroprotective effects of miR-33 depletion, but miR-33 inhibition may also have other atheroprotective effects, beyond regulation of HDL levels, such as regulation of macrophage functional polarization126.
Other miRNAs might prove to be suitable therapeutic targets to treat atherosclerosis. miR-144 is a potent inhibitor of cholesterol efflux from a variety of tissues, including macrophages101. Prolonged treatment of Ldlr-mutant male mice with a miR-144 antagomir results in reduced atherosclerosis progression, and even short-term treatment has some atheroprotective benefit127. miR-30c is an inhibitor of LDL biogenesis, and therefore its expression might reduce the development of atherosclerosis111. Indeed, in Apoe-mutant and Ldlr-mutant mouse models, overexpressed miR-30c reduces the development of atherosclerosis111,128.
Implications of miRNAs in liver disease.
Excess accumulation of hepatic triglycerides and fatty acids is characteristic of several liver diseases, including alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD). miR-34a was found to be overexpressed in the course of ALD in mice and humans and contributed to the disease in a mouse model129. miR-34a was also associated with NAFLD, and its inhibition in mice suppressed the disease, which was linked to miR-34a-mediated repression of PPARα and subsequent suppression of fatty acid metabolism in the liver130. Conversely, hepatic expression of miR-122 was downregulated in both mice and human patients with ALD, and restoration of miR-122 expression in hepatocytes had a therapeutic benefit in a mouse model of ALD131. It appears that miR-122 protects the liver from alcohol-induced damage by targeting the transcription factor HIF1α, which regulates the expression of, among other factors, PPARγ — a key regulator of lipid synthesis and an established factor in driving steatosis. Similarly, miR-27a was shown to suppress NAFLD progression in mouse models by downregulating the lipid biosynthetic enzymes fatty acid synthase and stearoyl-CoA desaturase 1 (REFS117,120,130). Thus, miRNAs might be potential targets for treatment of liver diseases associated with lipid accumulation.
Metabolic roles of circulating miRNAs
miRNAs were traditionally thought to act cell autonomously. This belief was overturned when miRNAs were detected in extracellular vesicles secreted from mammalian cells13. Extracellular vesicles larger than 200 nm are called ‘microvesicles’, and are formed by outward budding of the plasma membrane132. Smaller extracellular vesicles are formed either by outward budding of the plasma membrane to secrete ectosomes132 or by fusion of multivesicular bodies (MVBs) with the plasma membrane132. MVBs are formed from late endosomes by inward budding of the endosome membrane to form intralumenal vesicles. When an MVB fuses with the plasma membrane, the internal vesicles are released into the extracellular environment as exosomes. RISCs composed of miRNA and Ago protein are not only free in the cytosol, but a sizeable fraction of RISCs are physically associated with late endosomes133,134. Endosomal association of RISCs and their silencing activity in this cellular compartment is dependent on endosomes maturing into MVBs, upon which the RISCs become localized within the intralumenal vesicles of MVBs133,134. This then provides one route for miRNA secretion, but it is unclear if it is the only route used, and possibly microvesicles and ectosomes also transport miRNAs135. Extracellular vesicles in the blood serum are loaded with miRNAs136,137. This makes circulating miRNAs an exciting emergent field of study for understanding new mechanisms of gene regulation as well as applications for disease diagnosis and treatment.
Circulating miRNAs from adipose tissue and pancreas.
With the identification of circulating miRNAs, an area of focus became understanding the origins, destination and function of these miRNAs. Among metabolic tissues, adipose tissue and pancreatic β-cells have been shown to be important sources of circulating miRNAs.
There is a major role for miRNAs originating from adipose tissue in the regulation of metabolism. Specific knockout of Dicer1 in mouse adipose tissue causes an overall decrease in circulating miRNA levels138,139. Of 653 circulating miRNAs identified, 419 are reduced in their level or eliminated from the circulation in Dicer1-knockout mice. The Dicer1-knockout mice were also shown to develop insulin resistance138, which could be reversed by the transplantation of wild-type fat or injection of extracellular vesicles from wild-type mouse serum into these mice139. One of the circulating miRNAs, miR-99b, was found to be responsible for downregulating expression of the fibroblast growth factor FGF21 in healthy liver and muscle139. These results argue for circulating miRNAs to act as adipokines that regulate glucose homeostasis. Adipose tissue-derived miRNAs may also have a role in metabolic performance in response to aerobic exercise. Mice with adipose tissue-specific loss of Dicer show poor responses to aerobic exercise training, unlike mice with wild-type adipose tissue140. Exercise also increases miRNA levels in adipose tissue, suggesting that these miRNAs might be secreted into extracellular vesicles that act systemically as adipokines to mediate responses to healthy activities such as exercise. In obesity, which is associated with adipose tissue inflammation, adipose tissue macrophages might be an important source of circulating miRNAs. One such miRNA secreted from obese mouse adipose tissue macrophages is miR-155 (REF.141). It inhibits insulin signal transduction by repressing the expression of PPARγ in target tissues such as the liver and muscle.
FGF21.
A member of the fibroblast growth factor family. It is secreted from liver cells into the circulatory system. It binds to a receptor on the surface of cells of the hypothalamus and regulates simple sugar intake and preference for sweet foods.
Adipokines.
Also known as adipocytokines, these molecules are cytokines that are secreted by adipose tissue. Representative adipokines include leptin, Il-6 and TNF.
One prediction is that obesity might cause circulating miRNA imbalance, which could contribute to diabetes progression. Indeed, injection of circulating extracellular vesicles from obese mice into lean mice causes systemic insulin resistance and glucose intolerance in the lean hosts141,142. Even synthetic extracellular vesicles loaded with miRNAs simulating those enriched in obesity have the same effect142. Conversely, injection of extracellular vesicles from lean mice into obese mice alleviates their insulin resistance141. This suggests that extracellular vesicles, loaded with specific miRNAs, could be explored as therapeutics to treat diabetes progression.
Another source of circulating miRNAs is pancreatic β-cells. miRNAs are secreted from β-cells grown in culture, and these can regulate activities of co-cultured β-cells143. If cells are exposed to proinflammatory cytokines, their extracellular vesicle miRNA profile changes, and such extracellular vesicles can induce apoptosis in naive β-cells. miR-26a functions both autonomously in β-cells (to regulate glucose-stimulated insulin secretion as discussed earlier) and non-autonomously in insulin target tissues of the mouse43. Strikingly, miR-26a abundance is greatly reduced in circulating extracellular vesicles from obese mice and humans. When miR-26a was experimentally overexpressed in β-cells of mice, the circulating extracellular vesicles were hyperloaded with miR-26a, which was associated with a marked increased of insulin sensitivity and metabolic homeostasis in liver, muscle and fat43. It was further shown that mouse primary hepatocytes exposed to extracellular vesicles hyperloaded with miR-26a downregulated PKCθ expression and showed increased PI3K–AKT signalling in response to insulin in vitro.
PKCθ.
A member of the protein kinase C (PKC) family, which phosphorylate target proteins at serine and threonine residues. Kinase activation requires a second messenger, and this PKC isoform requires binding to diacylglycerol. It does not require binding to calcium, and thus it is a member of the novel subfamily of PKCs.
In addition to extracellular vesicle-mediated circulation, miRNAs can be loaded into HDL particles and transferred from one region of the body to another144. The mechanisms of this loading are unclear, but it may be occurring extracellularly, as HDL can incorporate miRNAs in vitro, and loading does not require ABCA1 or ABCG1 in vivo144,145. miR-375 is one such HDL-bound miRNA, and its source appears to be islet β-cells145. Under conditions that stimulate insulin secretion such as high postprandial glucose levels, β-cell loading of HDL with miR-375 is inhibited. This implies that miR-375 signals from the islets to other tissues during fasting conditions, but the identity of those target tissues is unknown. Given hepatocyte uptake of HDL-loaded material, the liver seems to be a likely target. Here, delivery of HDL-associated miRNAs was shown to depend on the HDL receptor SR-BI144.
Circulating miRNAs as biomarkers for metabolic status and disease state.
Given the alterations in the levels of circulating miRNAs found in animal models for metabolic disease, one area of interest is to use serum miRNA profiling to diagnose human disease status (TABLE 3). miRNAs whose circulating levels are predictive of obesity, type 2 diabetes, atherosclerosis and other metabolic disorders have been identified to be promising biomarker candidates135,146. In particular, the level of circulating miR-15a increases in patients in a prediabetic state with impaired glucose tolerance147,148. There is also a correlation between miR-15a and the early onset of retinal degeneration, suggesting a potential causative link147,148. The level of another circulating miRNA, miR-20b, is elevated in patients with type 2 diabetes, and it alters insulin signalling and reduces the ability of muscle cells to synthesize glycogen149.
One promising use of circulating miRNAs for diabetes diagnosis is with gestational diabetes mellitus, which is one of the most common complications during pregnancy. Although diagnosis is usually made in the second or third trimester, the onset of gestational diabetes begins in the first trimester150. The levels of blood plasma miRNAs measured in women during the first trimester are strongly correlated with their outcomes related to developing diabetes later in pregnancy151. The levels of two miRNAs in particular, miR-23a and miR-223, are abnormally elevated in women who develop gestational diabetes, and are highly predictive of developing this condition. This example hints at the potential of using circulatory miRNAs as early predictive markers for metabolic dysfunction more broadly.
Conclusions and perspective
In the past 15 years, deep connections have been uncovered between miRNA-mediated gene regulation and systemic regulation of carbohydrate and lipid metabolism (TABLE 1). Moreover, misregulated expression of some of these miRNAs contributes to metabolic disease progression (TABLE 2). Some unexpected lessons have been learned thus far. One is the evolutionary history of miRNAs that have been found to directly regulate systemic glucose and lipid metabolism. Most of these miRNAs arose in the ancestors that were present at the base of the chordate or vertebrate lineage. Some of these miRNAs originated in the ancestor at the base of the vertebrate and invertebrate lineages, which diverged approximately 500 million years ago. In particular, the ancient miRNA miR-7 regulates a gene homologue common to the mouse and the fly, whose protein product physically binds and acts with the target of miR-375. Such conservation of target and biological function of miRNAs is rare in the animal kingdom, which may imply that the common ancestor of vertebrates and invertebrates utilized miRNAs for equivalent metabolic processes, for example to regulate glucose metabolism via insulin-like signalling.
Theoretical and experimental studies have indicated that miRNAs are often used to generate robustness with regard to a given biological process152. In the case of systemic metabolism, feeding is generally a pulse-like event and therefore the system must respond in a similar pulse-like manner to maintain circulating glucose and lipids at fairly uniform levels. Control theory, which is used to model dynamical systems, posits that when a system responds to a transient external stimulus such as feeding, it must rapidly relax to its resting prepulse state to maintain output homeostasis153. Feedback repression is key to this relaxation. It is possible that miRNAs function as such feedback repressors to rapidly relax the pulses of insulin signalling and lipid metabolism to their resting levels. Therefore, it might be that the primary role of metabolism-regulating miRNAs is to control the dynamics of metabolic response and maintain homeostasis.
There is precedent for miRNAs acting on targets in such a manner. miR-7 represses expression of a transcription factor required for photoreceptor differentiation in the developing eye32. This transcription factor is expressed as a pulse within progenitor cells, and miR-7 acts to accelerate the relaxation in its levels to a basal level154. Failure to rapidly reduce the level of the factor results in cells committing errors in lineage restriction. Remarkably, feedback repression by miR-7 is dependent on glucose metabolism in cells. If cells are unable to take up normal amounts of glucose, the decay in levels of the transcription factor is naturally retarded, and miR-7 becomes completely unnecessary for regulating the dynamics of its expression154. It is a general feature of all miRNAs in D. melanogaster that glucose limitation renders them unnecessary for normal development154. It is tempting to speculate that the strength of miRNA-mediated inhibition of insulin signalling might vary not simply according to miRNA concentrations but according to the mutual relationship between miRNAs and the rate of intracellular glucose metabolism. In the future, it will be interesting to investigate how miRNAs affect the dynamics of insulin signalling and responses in vivo. It will also be worthwhile to understand how miRNAs modulate the dynamics of lipid transport during the natural feeding–fasting cycles in vivo.
Finally, in the past few years an exciting role for miRNAs as intercellular signalling molecules and ‘RNA hormones’ has been discovered. They are secreted into the circulation, from where they can be taken up by cells in faraway tissues. Although the fate and roles of such circulatory miRNAs in recipient cells are still poorly understood, there is evidence that they may regulate gene expression and metabolism upon their uptake. These circulatory miRNAs greatly expand the repertoire of signalling and coordination between tissues, and they hold great promise for diagnosis and treatment of metabolic diseases (TABLE 3).
In conclusion, despite extensive studies on miRNAs since their discovery 28 years ago, they continue to surprise us, and we anticipate that uncovering their mechanisms and functions will provide ample research opportunities for many years to come.
Acknowledgements
The authors apologize to those whose work they omitted in this Review due to space limitations. They thank the following funding agencies for support: NIH (T32GM008061 and R35GM118144), NSF (1764421) and the Simons Foundation (597491).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Carthew RW & Sontheimer EJ Origins and mechanisms of miRNAs and siRNAs. Cell 136, 642–655 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filipowicz W, Bhattacharyya SN & Sonenberg N Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet 9, 102–114 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Baek D et al. The impact of microRNAs on protein output. Nature 455, 64–71 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selbach M et al. Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Lee Y et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Denli AM, Tops BB, Plasterk RH, Ketting RF & Hannon GJ Processing of primary microRNAs by the microprocessor complex. Nature 432, 231–235 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Gregory RI et al. The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Grishok A et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106, 23–34 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Hutvagner G et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Ketting RF et al. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 15, 2654–2659 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meijer HA, Smith EM & Bushell M Regulation of miRNA strand selection: follow the leader? Biochem. Soc. Trans 42, 1135–1140 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Valadi H et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9, 654–659 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Chiang HR et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 24, 992–1009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jan CH, Friedman RC, Ruby JG & Bartel DP Formation, regulation and evolution of Caenorhabditis elegans 3’UTRs. Nature 469, 97–101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fromm B et al. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet 49, 213–242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozomara A, Birgaoanu M & Griffiths-Jones S miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fromm B et al. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 48, D1172 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalvari I et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartel DP Metazoan microRNAs. Cell 173, 20–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynn FC Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol. Metab 20, 452–459 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Guller I & Russell AP MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. J. Physiol 588, 4075–4087 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haeusler RA, McGraw TE & Accili D Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol 19, 31–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dalgaard LT & Eliasson L An ‘alpha-beta’ of pancreatic islet microribonucleotides. Int. J. Biochem. Cell Biol 88, 208–219 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Rorsman P & Ashcroft FM Pancreatic beta-cell electrical activity and insulin secretion: of mice and men. Physiol. Rev 98, 117–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melloul D, Marshak S & Cerasi E Regulation of insulin gene transcription. Diabetologia 45, 309–326 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Andrali SS, Sampley ML, Vanderford NL & Ozcan S Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem. J 415, 1–10 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Rorsman P et al. The cell physiology of biphasic insulin secretion. New Physiol. Sci 15, 72–77 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Wang Z & Thurmond DC Mechanisms of biphasic insulingranule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci 122, 893–903 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arous C & Halban PA The skeleton in the closet: actin cytoskeletal remodeling in beta-cell function. Am. J. Physiol. Endocrinol. Metab 309, E611–E620 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Ofori JK et al. Elevated miR-130a/miR130b/miR-152 expression reduces intracellular ATP levels in the pancreatic beta cell. Sci. Rep 7, 44986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Cassidy JJ, Reinke CA, Fischboeck S & Carthew RW A microRNA imparts robustness against environmental fluctuation during development. Cell 137, 273–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wienholds E et al. MicroRNA expression in zebrafish embryonic development. Science 309, 310–311 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Correa-Medina M et al. MicroRNA miR-7 is preferentially expressed in endocrine cells of the developing and adult human pancreas. Gene Expr. Patterns 9, 193–199 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Christodoulou F et al. Ancient animal microRNAs and the evolution of tissue identity. Nature 463, 1084–1088 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kredo-Russo S et al. Pancreas-enriched miRNA refines endocrine cell differentiation. Development 139, 3021–3031 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Agbu P, Cassidy JJ, Braverman J, Jacobson A & Carthew RW MicroRNA miR-7 regulates secretion of insulin-like peptides. Endocrinology 161, bqz040 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that miR-7-mediated regulation of insulin secretion is deeply conserved and suggests that the ancestor of invertebrates and vertebrates used this miRNA to regulate glucose metabolism.
- 38.Melkman-Zehavi T et al. miRNAs control insulin content in pancreatic beta-cells via downregulation of transcriptional repressors. EMBO J. 30, 835–845 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sebastiani G et al. MicroRNA-124a is hyperexpressed in type 2 diabetic human pancreatic islets and negatively regulates insulin secretion. Acta Diabetol. 52, 523–530 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Zhang F et al. Obesity-induced overexpression of miR-802 impairs insulin transcription and secretion. Nat. Commun 11, 1822 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang X, Muniappan L, Tang G & Ozcan S Identification of glucose-regulated miRNAs from pancreatic beta cells reveals a role for miR-30d in insulin transcription. RNA 15, 287–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang L et al. EGF suppresses the expression of miR-124a in pancreatic beta cell lines via ETS2 activation through the MEK and PI3K signaling pathways. Int. J. Biol. Sci 15, 2561–2575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu H et al. Pancreatic beta cell microRNA-26a alleviates type 2 diabetes by improving peripheral insulin sensitivity and preserving beta cell function. PLoS Biol. 18, e3000603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes a miRNA from pancreatic islet cells that not only regulates insulin output autonomously but also circulates in the blood and sensitizes target tissues to respond to insulin, and therefore could be a potential therapeutic.
- 44.Latreille M et al. MicroRNA-7a regulates pancreatic beta cell function. J. Clin. Invest 124, 2722–2735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caldwell JE, Heiss SG, Mermall V & Cooper JA Effects of CapZ, an actin capping protein of muscle, on the polymerization of actin. Biochemistry 28, 8506–8514 (1989). [DOI] [PubMed] [Google Scholar]
- 46.Delalle I, Pfleger CM, Buff E, Lueras P & Hariharan IK Mutations in the Drosophila orthologs of the F-actin capping protein alpha- and beta-subunits cause actin accumulation and subsequent retinal degeneration. Genetics 171, 1757–1765 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poy MN et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432, 226–230 (2004). [DOI] [PubMed] [Google Scholar]; This article describes miR-375 and its role in regulating insulin secretion.
- 48.Taoka M et al. V-1, a protein expressed transiently during murine cerebellar development, regulates actin polymerization via interaction with capping protein. J. Biol. Chem 278, 5864–5870 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Bhattacharya N, Ghosh S, Sept D & Cooper JA Binding of myotrophin/V-1 to actin-capping protein: implications for how capping protein binds to the filament barbed end. J. Biol. Chem 281, 31021–31030 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quintens R, Hendrickx N, Lemaire K & Schuit F Why expression of some genes is disallowed in beta-cells. Biochem. Soc. Trans 36, 300–305 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Zhao C, Wilson MC, Schuit F, Halestrap AP & Rutter GA Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes 50, 361–366 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Pullen TJ, da Silva Xavier G, Kelsey G & Rutter GA miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (MCT1). Mol. Cell Biol 31, 3182–3194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brubaker PL & Drucker DJ Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 145, 2653–2659 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Jo S et al. miR-204 controls glucagon-like peptide 1 receptor expression and agonist function. Diabetes 67, 256–264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szabo G & Bala S MicroRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol 10, 542–552 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davalos A et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl Acad. Sci. USA 108, 9232–9237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu W et al. Hepatic miR-378 targets p110alpha and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat. Commun 5, 5684 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Trajkovski M et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474, 649–653 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Williams AH, Liu N, van Rooij E & Olson EN MicroRNA control of muscle development and disease. Curr. Opin. Cell Biol 21, 461–469 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu H et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 147, 81–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dou L et al. MiR-19a regulates PTEN expression to mediate glycogen synthesis in hepatocytes. Sci. Rep 5, 11602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramirez CM et al. MicroRNA 33 regulates glucose metabolism. Mol. Cell Biol 33, 2891–2902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chemello F et al. Transcriptomic analysis of single isolated myofibers identifies miR-27a-3p and miR-142–3p as regulators of metabolism in skeletal muscle. Cell Rep. 26, 3784–3797 e3788 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Liang J et al. MicroRNA-29a-c decrease fasting blood glucose levels by negatively regulating hepatic gluconeogenesis. J. Hepatol 58, 535–542 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Zhuo S et al. MicroRNA-451 negatively regulates hepatic glucose production and glucose homeostasis by targeting glycerol kinase-mediated gluconeogenesis. Diabetes 65, 3276–3288 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Wang S et al. Micro-RNA-27a/b negatively regulates hepatic gluconeogenesis by targeting FOXO1. Am. J. Physiol. Endocrinol. Metab 317, E911–E924 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Gerin I et al. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J. Biol. Chem 285, 33652–33661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown MS & Goldstein JL The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997). [DOI] [PubMed] [Google Scholar]
- 69.Espenshade PJ & Hughes AL Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet 41, 401–427 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Foretz M, Guichard C, Ferre P & Foufelle F Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc. Natl Acad. Sci. USA 96, 12737–12742 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto T et al. SREBP-1 interacts with hepatocyte nuclear factor-4 alpha and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes. J. Biol. Chem 279, 12027–12035 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Wu L et al. Paternal psychological stress reprograms hepatic gluconeogenesis in offspring. Cell Metab. 23, 735–743 (2016). [DOI] [PubMed] [Google Scholar]; This article shows that transgenerational epigenetic transmission of a glucose metabolic state is mediated by a miRNA.
- 73.Bluher M Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol 15, 288–298 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Kahn BB & Flier JS Obesity and insulin resistance. J. Clin. Invest 106, 473–481 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Price NL et al. Genetic ablation of miR-33 increases food intake, enhances adipose tissue expansion, and promotes obesity and insulin resistance. Cell Rep. 22, 2133–2145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swallow DM Genetics of lactase persistence and lactose intolerance. Annu. Rev. Genet 37, 197–219 (2003). [DOI] [PubMed] [Google Scholar]
- 77.Kettunen J et al. European lactase persistence genotype shows evidence of association with increase in body mass index. Hum. Mol. Genet 19, 1129–1136 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Field Y et al. Detection of human adaptation during the past 2000 years. Science 354, 760–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bovine HapMap C et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wagschal A et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med 21, 1290–1297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Plassais J et al. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun 10, 1489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang WX, Wilfred BR, Hu Y, Stromberg AJ & Nelson PT Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA 16, 394–404 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L et al. A microRNA linking human positive selection and metabolic disorders. Cell 183, 684–701 e614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows how a miRNA gene variant co-selected in humans confers energy efficiency that predisposes humans and mice to obesity.
- 84.Yang WM, Jeong HJ, Park SW & Lee W Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol. Nutr. Food Res 59, 2303–2314 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Jordan SD et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol 13, 434–446 (2011). [DOI] [PubMed] [Google Scholar]
- 86.Kornfeld JW et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of HNF1B. Nature 494, 111–115 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Zhang C et al. Hepatic Ago2-mediated RNA silencing controls energy metabolism linked to AMPK activation and obesity-associated pathophysiology. Nat. Commun 9, 3658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Butler AE et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 (2003). [DOI] [PubMed] [Google Scholar]
- 89.Kameswaran V et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 19, 135–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Locke JM, da Silva Xavier G, Dawe HR, Rutter GA & Harries LW Increased expression of miR-187 in human islets from individuals with type 2 diabetes is associated with reduced glucose-stimulated insulin secretion. Diabetologia 57, 122–128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin T The WNT signalling pathway and diabetes mellitus. Diabetologia 51, 1771–1780 (2008). [DOI] [PubMed] [Google Scholar]
- 92.Belgardt BF et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med 21, 619–627 (2015). [DOI] [PubMed] [Google Scholar]
- 93.Moore KJ, Sheedy FJ & Fisher EA Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol 13, 709–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chinetti G et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med 7, 53–58 (2001). [DOI] [PubMed] [Google Scholar]
- 95.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T & Wang N HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 7, 365–375 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Horie T et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (SREBP2) regulates HDL in vivo. Proc. Natl Acad. Sci. USA 107, 17321–17326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marquart TJ, Allen RM, Ory DS & Baldan A miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl Acad. Sci. USA 107, 12228–12232 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Najafi-Shoushtari SH et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328, 1566–1569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rayner KJ et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328, 1570–1573 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adlakha YK et al. Pro-apoptotic miRNA-128–2 modulates ABCA1, ABCG1 and RXRalpha expression and cholesterol homeostasis. Cell Death Dis. 4, e780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ramirez CM et al. Control of cholesterol metabolism and plasma high-density lipoprotein levels by microRNA-144. Circ. Res 112, 1592–1601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Aguiar Vallim TQ et al. MicroRNA-144 regulates hepatic ATP binding cassette transporter A1 and plasma high-density lipoprotein after activation of the nuclear receptor farnesoid X receptor. Circ. Res 112, 1602–1612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goedeke L et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med 21, 1280–1289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rayner KJ et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478, 404–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rayner KJ et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Invest 121, 2921–2931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Price NL et al. Specific disruption of ABCA1 targeting largely mimics the effects of miR-33 knockout on macrophage cholesterol efflux and atherosclerotic plaque development. Circ. Res 124, 874–880 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rottiers V et al. MicroRNAs in metabolism and metabolic diseases. Cold Spring Harb. Symp. Quant. Biol 76, 225–233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allen RM et al. miR-33 controls the expression of biliary transporters, and mediates statin-and diet-induced hepatotoxicity. EMBO Mol. Med 4, 882–895 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li T, Francl JM, Boehme S & Chiang JY Regulation of cholesterol and bile acid homeostasis by the cholesterol 7alpha-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology 58, 1111–1121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goedeke L et al. A regulatory role for microRNA 33* in controlling lipid metabolism gene expression. Mol. Cell Biol 33, 2339–2352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Soh J, Iqbal J, Queiroz J, Fernandez-Hernando C & Hussain MM MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat. Med 19, 892–900 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chang J et al. miR-122, a mammalian liver-specific microRNA, is processed from HCR mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 1, 106–113 (2004). [DOI] [PubMed] [Google Scholar]
- 113.Krutzfeldt J et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438, 685–689 (2005). [DOI] [PubMed] [Google Scholar]
- 114.Esau C et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 3, 87–98 (2006). [DOI] [PubMed] [Google Scholar]
- 115.Elmen J et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 36, 1153–1162 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vickers KC et al. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology 57, 533–542 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang M, Sun W, Zhou M & Tang Y MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci. Rep 7, 14493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vickers KC et al. MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl Acad. Sci. USA 111, 14518–14523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang L et al. MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol. Cell Biol 33, 1956–1964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu Y et al. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat. Commun 6, 7466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Singaravelu R et al. MicroRNA-7 mediates cross-talk between metabolic signaling pathways in the liver. Sci. Rep 8, 361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Breslow JL Mouse models of atherosclerosis. Science 272, 685–688 (1996). [DOI] [PubMed] [Google Scholar]
- 123.Masucci-Magoulas L et al. A mouse model with features of familial combined hyperlipidemia. Science 275, 391–394 (1997). [DOI] [PubMed] [Google Scholar]
- 124.Horie T et al. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/−mice. J. Am. Heart Assoc 1, e003376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Price NL et al. Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep. 21, 1317–1330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article points out the potential use of anti-miR-33 for reducing the progression of atherosclerosis.
- 126.Ouimet M et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Invest 125, 4334–4348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cheng J et al. MicroRNA-144 silencing protects against atherosclerosis in male, but not female mice. Arterioscler. Thromb. Vasc. Biol 40, 412–425 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Irani S, Iqbal J, Antoni WJ, Ijaz L & Hussain MM microRNA-30c reduces plasma cholesterol in homozygous familial hypercholesterolemic and type 2 diabetic mouse models. J. Lipid Res 59, 144–154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wan Y et al. Regulation of cellular senescence by miR-34a in alcoholic liver injury. Am. J. Pathol 187, 2788–2798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ding J et al. Effect of miR-34a in regulating steatosis by targeting PPARalpha expression in nonalcoholic fatty liver disease. Sci. Rep 5, 13729 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Satishchandran A et al. MicroRNA 122, regulated by GRLH2, protects livers of mice and patients from ethanol-induced liver disease. Gastroenterology 154, 238–252 e237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that the abundant miR-122 is downregulated by alcohol-induced liver disease, and that its artificial expression can protect the liver from disease progression.
- 132.van Niel G, D’Angelo G & Raposo G Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol 19, 213–228 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Gibbings DJ, Ciaudo C, Erhardt M & Voinnet O Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol 11, 1143–1149 (2009). [DOI] [PubMed] [Google Scholar]
- 134.Lee YS et al. Silencing by small RNAs is linked to endosomal trafficking. Nat. Cell Biol 11, 1150–1156 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mori MA, Ludwig RG, Garcia-Martin R, Brandao BB & Kahn CR Extracellular miRNAs: from biomarkers to mediators of physiology and disease. Cell Metab. 30, 656–673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mitchell PS et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl Acad. Sci. USA 105, 10513–10518 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Skog J et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10, 1470–1476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mori MA et al. Altered miRNA processing disrupts brown/white adipocyte determination and associates with lipodystrophy. J. Clin. Invest 124, 3339–3351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Thomou T et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that adipose tissue secretes extracellular vesicles loaded with miRNAs into the circulation, and this can promote insulin sensitivity in target tissues.
- 140.Brandao BB et al. Dynamic changes in DICER levels in adipose tissue control metabolic adaptations to exercise. Proc. Natl Acad. Sci. USA 117, 23932–23941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ying W et al. Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 171, 372–384 e312 (2017). [DOI] [PubMed] [Google Scholar]; This article shows that a major source of circulating miRNAs is adipose tissue, and that circulating miRNAs from obese mice injected into lean mice cause insulin resistance, and vice versa.
- 142.Castano C, Kalko S, Novials A & Parrizas M Obesity-associated exosomal miRNAs modulate glucose and lipid metabolism in mice. Proc. Natl Acad. Sci. USA 115, 12158–12163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that circulating miRNAs from obese mice injected into lean mice cause insulin resistance, and that synthetic extracellular vesicles loaded with miRNAs misexpressed in obesity can mimic the effect.
- 143.Guay C, Menoud V, Rome S & Regazzi R Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun. Signal 13, 17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD & Remaley AT MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol 13, 423–433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sedgeman LR et al. Beta cell secretion of miR-375 to HDL is inversely associated with insulin secretion. Sci. Rep 9, 3803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oses M, Margareto Sanchez J, Portillo MP, Aguilera CM & Labayen I Circulating miRNAs as biomarkers of obesity and obesity-associated comorbidities in children and adolescents: a systematic review. Nutrients 11, 2890 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kamalden TA et al. Exosomal microRNA-15a transfer from the pancreas augments diabetic complications by inducing oxidative stress. Antioxid. Redox Signal 27, 913–930 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sangalli E et al. Circulating microRNA-15a associates with retinal damage in patients with early stage type 2 diabetes. Front. Endocrinol 11, 254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Katayama M et al. Circulating exosomal miR-20b-5p is elevated in type 2 diabetes and could impair insulin action in human skeletal muscle. Diabetes 68, 515–526 (2019). [DOI] [PubMed] [Google Scholar]
- 150.Buchanan TA & Xiang AH Gestational diabetes mellitus. J. Clin. Invest 115, 485–491 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yoffe L et al. Early diagnosis of gestational diabetes mellitus using circulating microRNAs. Eur. J. Endocrinol 181, 565–577 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Ebert MS & Sharp PA Roles for microRNAs in conferring robustness to biological processes. Cell 149, 515–524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Doyle JC, Francis BA & Tannenbaum AR Feedback Control Theory (Courier Corporation, 2013). [Google Scholar]
- 154.Cassidy JJ et al. Repressive gene regulation synchronizes development with cellular metabolism. Cell 178, 980–992 e917 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]