

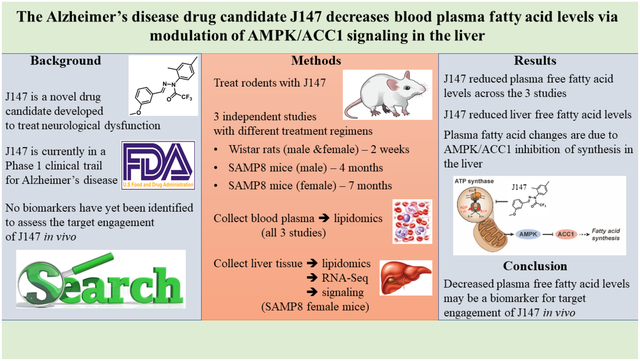
Abstract
J147 is a novel drug candidate developed to treat neurological dysfunction. Numerous studies have demonstrated the beneficial effects of J147 in cellular and animal models of disease which has led to the transitioning of the compound into human clinical trials. However, no biomarkers for its target engagement have been identified. Here, we determined if specific metabolites in the plasma could be indicative of J147’s activity in vivo. Plasma lipidomics data from three independent rodent studies were assessed along with liver lipidomics data from one of the studies. J147 consistently reduced plasma free fatty acid (FFA) levels across the independent studies. Decreased FFA levels were also found in the livers of J147-treated mice that correlated well with those in the plasma. These changes in the liver were associated with activation of the AMP-activated protein kinase/acetyl-CoA carboxylase 1 signaling pathway. A reduction in FFA levels by J147 was confirmed in HepG2 cells, where activation of the AMPK/ACC1 pathway was seen along with increases in acetyl-CoA and ATP levels which correlated with enhanced cellular bioenergetics. Our data show that J147 targets liver cells to activate the AMPK/ACC1 signaling pathway and preserve energy at the expense of inhibiting FFA synthesis.
Keywords: dementia, aging, blood biomarkers, liver, lipidomics, drug discovery
Graphical abstract
1. Introduction
J147 is a novel drug candidate for Alzheimer’s disease (AD) that was purposely developed to target aging processes in the brain. This was accomplished by using a drug discovery paradigm based on phenotypic screening assays for aspects of old age-associated neurodegeneration and brain pathology, including energy failure and mitochondrial dysfunction1,2. It has been shown that J147 is very protective in transgenic AD animal models3–5, and that it slows down the aging process in the brains of rapidly aging senescence-accelerated prone 8 (SAMP8) mice when administered both early6 and late in life7. The SAMP8 mice are a model of accelerated aging that develop a progressive, age-associated decline in brain function as well as a number of brain pathologies similar to human AD patients8–10.
The molecular target of J147 was identified as the alpha subunit of ATP synthase through which it engages a neuroprotective response involving the activation of AMP-activated protein kinase (AMPK)11. ATP synthase is also an anti-aging target in C. elegans12. It was later demonstrated that J147 reduces physiological aging in the brain by maintaining mitochondrial homeostasis via the AMPK/acetyl-CoA carboxylase 1 (ACC1) signaling pathway7. As an outcome of these studies, J147 entered a Phase I clinical trial for AD, which is expected to be completed in 2022 (NCT03838185).
One of the main challenges in clinical testing is to ensure that the dose of the drug candidate chosen to be administered to humans generates sufficient levels in the organism to interact with the therapeutic target, what is referred to as target engagement. Along with pharmacokinetics and safety, finding biomarkers for target engagement is thus a critical technical determinant of drug discovery effectiveness and clinical success13,14. Not only does this parameter confirm the action of the drug candidate in vivo, but it also provides information on the validity of a given target for a specific disease14.
There are several methods that can be used to measure target engagement in humans, with some of the more complex ones involving radiotracers. One of the simplest and least invasive approach is to determine substrate or product changes in the blood13. Although J147 activates the AMPK/ACC1 pathway by targeting the ATP synthase7, no biomarkers for its target engagement have been described. It was unknown whether potential biomarkers for ATP synthase modulation could be found in the blood. Given that J147 is already in human clinical trials, it is critical to accelerate the discovery of markers that provide evidence for the activity of J147 in vivo.
We have previously reported on the use of large-scale metabolomics to assess the contribution of aging to dementia using J147 in a chemical biology approach6,7. As part of these two studies, a large number of metabolites spanning diverse biochemical pathways were analyzed from the plasma of male and female SAMP8 mice at different ages following treatment with J147. One of the most significant differences that was found to be consistent between the studies was an increase in several species of free fatty acids (FFAs) with age that was strongly inhibited by J147. We later showed that CAD031, a chemical derivative of J147, was capable of reducing the levels of several FFAs in the plasma of wildtype C57BL/6 mice15. Given that J147 inhibits ACC17, the enzyme responsible for converting acetyl-CoA into malonyl-CoA (which is then used to synthesize FFAs), we asked if the effects of J147 on plasma FFAs could be explained by the regulation of the AMPK/ACC1 axis in liver. The implications of our findings to the discovery of biomarkers for target engagement of J147 are discussed.
2. Methods
2.1. Materials
All reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated.
2.2. Rodents
All experiments were performed in accordance with the US Public Health Service Guide for Care and Use of Laboratory Animals and protocols were approved by the IACUC at the Salk Institute. Rodents were randomly assigned to experimental groups and the number per group was determined based on previous experiments6,16 and was sufficient to attain statical power. The SAMP8 line was acquired from Harlan Laboratories (U.K.), and the experimental approaches carried out to generate the tissues in this study were detailed previously7. Adult male and female Wistar rats (Rattus norvegicus) were obtained from Charles River.
2.3. Tissue preparation
Mice were euthanized by deep anesthesia with ketamine/xylazine (i.p., 100 mg/kg and 10 mg/kg) and their blood was collected by cardiac puncture. After perfusing with PBS, their brains and livers were removed. Tissue was prepared for Western blotting, RNA extraction, and metabolomic analysis as previously described6,7.
2.4. Lipidomic analysis
Metabolite measurement and analysis were conducted at Metabolon as previously described6,17. For statistical analyses and data display, any missing values were assumed to be below the limits of detection and imputed with the compound minimum (minimum value imputation).
2.5. Western blotting
Western blots were performed as described previously6. The primary antibodies used were phospho-ACC1 (280 kDa, Cat#3661, RRID: AB_330337), total ACC1 (265 kDa, Cat#4190, RRID: AB_10547752), phospho-AMPK (62 kDa, Cat#2535, RRID: AB_331250), and total AMPK (62 kDa, Cat#2793, RRID: AB_915794) from Cell Signaling Technology. Horseradish peroxidase-conjugated secondary antibodies goat anti-rabbit (Cat#172–1019, RRID: AB_11125143) or goat anti-mouse (Cat#170–6516, RRID: AB_11125547), from BioRad were used for detection of antibody binding.
2.6. Whole transcriptome analysis
RNA was isolated from the livers of SAMP8 mice using the RNeasy Plus Universal mini kit (Qiagen). RNA-Seq libraries were prepared using the Illumina TruSeq Stranded mRNA Sample Prep Kit according to the manufacturer’s instructions. Briefly, poly-A RNA was selected using poly dT-beads. mRNA was then fragmented and reverse transcribed. cDNA was end-repaired, adenylated and ligated with Illumina adapters with indexes. Adapter-ligated cDNA was then amplified. Libraries were pooled and sequenced single-end 50 base-pair (bp) on the Illumina HiSeq 2500 platform.
Sequencing reads were mapped to the mm10 mouse genome using the spliced aligner STAR (2.5.3a) with default parameters18. Only uniquely aligned reads were considered for downstream analysis. Expression values were quantified using Homer (4.9.1,19 by counting reads mapped across all gene exons of RefSeq genes and mitochondrial encoded genes of Ensembl reference. The differential expression (DE) analysis was performed by DESeq2 (v1.18.1,20). Briefly, library size factor was estimated by the default “estimateSizeFactors” function. The dispersion was estimated by the default “estimateDispersion” function. Then the default Wald test was used to identify DE genes at cutoffs of a false discovery rate (FDR) < 0.05 and an absolute log2 fold-change > 0.5.
2.7. Bioinformatics analysis
Pathway analysis was performed using the DAVID database (21 &22) using default parameters and total protein-coding genes as background. Significantly enriched terms were identified with FDR < 0.05.
Principle Component Analysis (PCA) was performed with R function “prcomp” on the z-scaled Metabolon normalized imputed values for the metabolome, and log-transformed counts per million (CPM) values for the transcriptome. Values were centered at zero for PCA and ellipses were drawn to indicate the 95% confidence interval assuming a student distribution of the top 2 PCs. The top 500 most variable genes and total metabolites were used for PCA.
PCA plots were generated using R [R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/] and R package “ggplot2” (https://cran.r-project.org/web/packages/ggplot2/index.html). Heatmaps were generated using R package “gplots” (https://cran.r-project.org/web/packages/gplots/index.html) on the row-wise z-scaled values used of Metabolon normalized imputed values for metabolites, and log-transformed fragment per kilobase per million mapped reads (FPKM) for transcriptome. The clustering dendrogram of the heatmap was computed using “ward.D2” algorithm. Venn diagrams were generated using R package “VennDiagram” (https://cran.r-project.org/web/packages/VennDiagram/index.html).
2.8. HepG2 cell culture
HepG2 cells23 were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM, 11995–065) containing 25 mM glucose, 4 mM L-glutamine, 10% fetal bovine serum, 1% nonessential amino acids, 100U/mL penicillin, and 100 μg/mL streptomycin (all reagents; Gibco). Cells were seeded at a density of 30,000/cm2 and cultured for 4 days prior to treatment. J147 dissolved in dimethyl sulfoxide (DMSO; Avantor) was tested at 1 μM. For signaling experiments (Figure 4E), media was changed to serum-free conditions 24 hours prior to treatment. For Western blotting, cells were washed once in phosphate buffered saline (PBS −/−), scraped directly into sample buffer, and boiled immediately to preserve protein phosphorylation status.
2.9. ATP assay kit
ATP levels were measured by bioluminescence according to the manufacturer’s instructions (Molecular Probes, A22066).
2.10. FFAs assay kit
Total free fatty acids were quantified using the EnzyChrom™ Free Fatty Acid Assay Kit according to the manufacturer’s instructions (BioAssay Systems, EFFA-100).
2.11. Seahorse XF analysis
The assay procedure was previously described with some adaptations24. Cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were assayed with a XF Cell Mito Stress Test (Cat# 103015) or a XF Glycolysis Stress Test (Cat# 103020) (Agilent Technologies) using a Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA), according to the manufacturer’s guidelines. Analyses were conducted using Wave software and XF Report Generators (Agilent Technologies). The sensor cartridge for the XFe analyzer was hydrated at 37 °C one day before the experiment.
HepG2 cells at 5,000 cells/well were seeded onto the Seahorse XFe96 plates under normal culture conditions. The next day, the culture medium in the plates was replaced with complete Seahorse XF DMEM assay medium, and the cells were pretreated with J147 at 1 μM or vehicle (for 4 hr at 37 °C) prior to the assays according to the manufacturer’s instructions. For the Cell Mito Stress Test, the complete Seahorse XF base DMEM medium was supplemented with 10 mM glucose, 1 mM pyruvate, and 2 mM L-glutamine, at pH 7.4; chemicals were used at the following concentrations: 1.5 μM oligomycin, 2 μM FCCP, and 0.5 μM of a mixture of rotenone and antimycin A. For the Glycolysis Stress Test, the complete Seahorse XF base DMEM medium was supplemented with 2 mM L-glutamine, at pH 7.4; chemicals were used at the following concentrations: 10 mM glucose, 1 μM oligomycin, and 50 mM 2-deoxy-glucose (2-DG). OCR and ECAR data were normalized to total protein/well. Each condition was analyzed in 20–30 wells.
2.12. Quantitation of acetyl-coenzyme A
Samples (tissue or cell pellets) were lysed by sonication in 5% perchloric acid (10 mg of tissue per 100 μl). Immediately after sonication, an equal volume of 0.3 M sodium bicarbonate was added. After centrifugation at 15,000 × g, the supernatant was desalted employing a Bond-Elut column (50 mg C-18 silica, Agilent Technologies). Salts were removed by washing with 25 mM sodium bicarbonate. The analyte fraction was eluted with 25 mM sodium bicarbonate/30% acetonitrile. The eluate (150 μl) was concentrated in vacuo using a Speed-Vac Concentrator to 75 μl. Ten microliters were injected onto a Vydac C-18 reversed phase HPLC Column and eluted with a gradient from 0 to 30 % acetonitrile in 15 mM ammonium formate. The eluant was electrosprayed into a triple quadrupole mass spectrometer (Thermo Quantiva) and analyzed by multiple-reaction monitoring.
2.13. Data and statistical analysis
Metabolomic data are presented as scatter plots of the log fold change for the respective groups and the results were compared using the Wilcoxon matched-pairs signed rank test. Gene expression quantitation is presented as scatter plots of DESeq2 (v1.18.1,20) normalized counts and statistics were based on DESeq2 pair-wise comparisons using the default normalization (rlog), Wald test, with adjusted p-values shown. For assessment of protein, ATP, and acetyl-CoA levels in liver and HepG2 cells, as well as free fatty acid levels in HepG2 cells, data are presented as group mean ± standard error of the mean (SEM). Statistical analysis of the groups was performed using one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. Cellular bioenergetic data are presented as group mean ± SEM. Statistical analysis of the groups was performed using an unpaired two-tailed t-test. Where appropriate, experiments were performed at least three independent times. GraphPad Prism 8 was used and exact p-values are indicated. The threshold for statical significance was set at p < 0.05, and the p-values that reached the threshold are highlighted in red. We prospectively selected Dixon’s Q test (one-sided) as the criteria used for excluding any data from analysis.
2.14. Role of the funding source
This study’s sponsors had no involvement in the collection, analysis, or interpretation of the data; writing of the report; or the decision to submit the paper for publication.
3. Results
3.1. J147 reduces plasma FFA levels
In order to investigate potential biomarkers for evaluating target engagement by J147, we analyzed data from two large-scale metabolomic studies that were performed previously on plasma of SAMP8 mice treated with J1476,7. The first study was carried out with male SAMP8 mice that were fed with J147 or control diets from 3 to 10 months of age6. In the second study, we used 9-month-old female SAMP8 mice to address the effects of J147 at an advanced stage of the SAMP8 phenotype, using treatment for 4 months and sacrifice at 13 months of age7. In both studies, an integrative multi-omics approach was used to investigate changes in RNA, metabolites, and proteins in the brain with aging. It was found that J147 prevented or reduced many physiological aspects of aging in the brain along with maintaining cognitive function. Global metabolic profiling was carried out in parallel with plasma to assess the possible therapeutic effects of J147 on whole body health. Consistently, J147 significantly decreased the levels of multiple FFA species (long chain, medium chain, and polyunsaturated) in the plasma of both male and female SAMP8 mice that were elevated with aging (Figure 1A and B,6,7). Dots represent the log fold change of individual FFAs.
Figure 1: J147 regulates the levels of free fatty acids (FFAs) in the plasma and liver of SAMP8 mice.
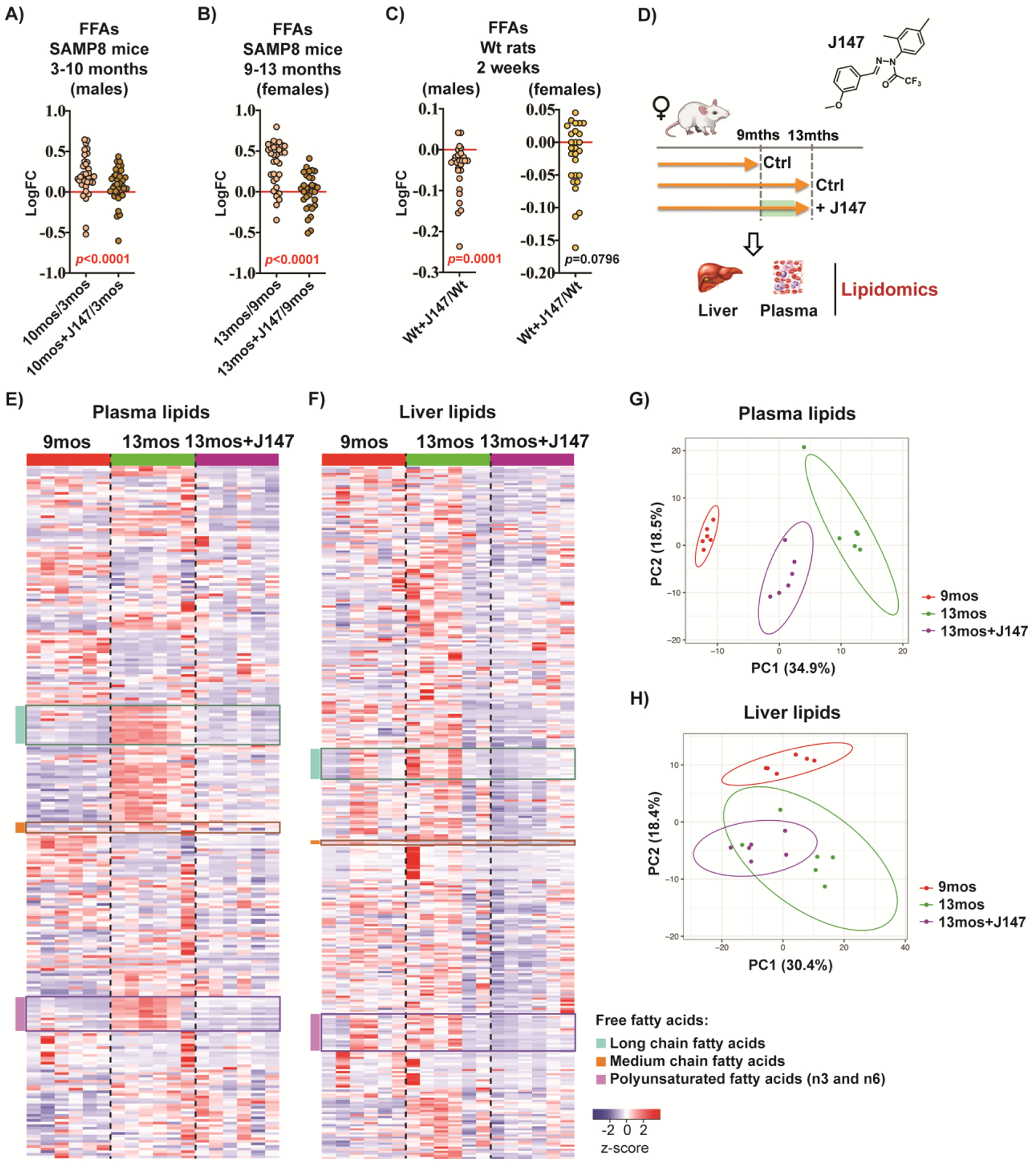
(a) FFA levels in plasma of male SAMP8 mice. Data are presented as the log fold change of 10-month-old to 3-month-old mice or 10-month-old J147-treated mice to 3-month-old untreated mice (n = 5/group). (b) FFA levels in plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). (c) FFA levels in plasma of male and female Wistar rats. Data are presented as the log fold change of J147-treated rats to untreated rats. All rats were the same age at the time of the study (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. (a-c) Dots represent the log fold change of individual FFAs. (d) Schematic diagram of the female SAMP8 mouse study that was used to compare the lipidomic profiles of plasma to liver. (e) Heatmap of 266 plasma lipids from the female SAMP8 mouse study (n = 6/group). (f) Heatmap of 339 liver lipids from the female SAMP8 mouse study (n = 6/group). z-score = row-wise z-scaled Metabolon normalized imputed spectrum values. FFAs highlighted in three color blocks. (g) Principal component analysis of all plasma lipids (n = 6/group). (h) Principal component analysis of all liver lipids (n = 6/group). Ellipses show the 95% confidence interval assuming a t-distribution of the top 2 principal components.
We then measured the levels of a similar subset of FFAs in the plasma of adult male and female Wistar rats fed with J147 for 2 weeks. Consistent with the mouse studies, we found that J147 induced a significant decrease in the levels of most FFAs analyzed in the plasma of the males. The female plasma also displayed decreased levels of FFAs but did not reach significance (Figure 1C, Table S1).
Because the liver is a key regulator of systemic lipid metabolism25, we next compared the lipidomic profile of the plasma from the SAMP8 female study to that of the liver from the same mice (Figure 1D) (no liver was available from the other studies). Several major lipid classes were assessed including short, medium, long chain, and polyunsaturated fatty acids; branched, dicarboxylate, amino, keto, monohydroxy, and dihydroxy fatty acids; ketone bodies; eicosanoids; endocannabinoids; phospholipids; glycerides; sphingolipids; and sterols. It was found that SAMP8 mice developed an overall increase in lipid metabolites in the liver and the plasma between 9 and 13 months of age (Figure 1E and F). Importantly, J147 prevented these increases in both tissues. Principal component analysis (PCA) of all lipids confirmed the overall changes in lipids with aging as well as the preventive effects of J147 in both the plasma and liver (Figure 1G and H). These changes were particularly noticeable in the different classes of FFAs (boxes in Figure 1E and F).
3.2. Changes in plasma FFA levels correlate with changes in liver FFA levels
The two largest classes of FFAs measured were the long chain and polyunsaturated FFAs. Figure 2A shows that metabolites in these two classes significantly increased with aging in both the plasma and the liver and that these increases were significantly prevented by J147. Importantly, in the liver, not only did J147 prevent the increase in FFAs but it actually lowered the levels below that of the 9-month-old baseline control. This observation indicates that J147 may have a drug-specific effect on hepatic FA metabolism. A careful analysis showed that the effects of J147 on both the plasma and the liver extended to other lipid classes such as phospholipids (Figure S1) and secondary bile acid metabolites (Figure S3, Table S2,7) although these results were not as clear-cut as with the FFAs.
Figure 2: Changes in plasma FFA levels correlate with changes in liver FFA levels.
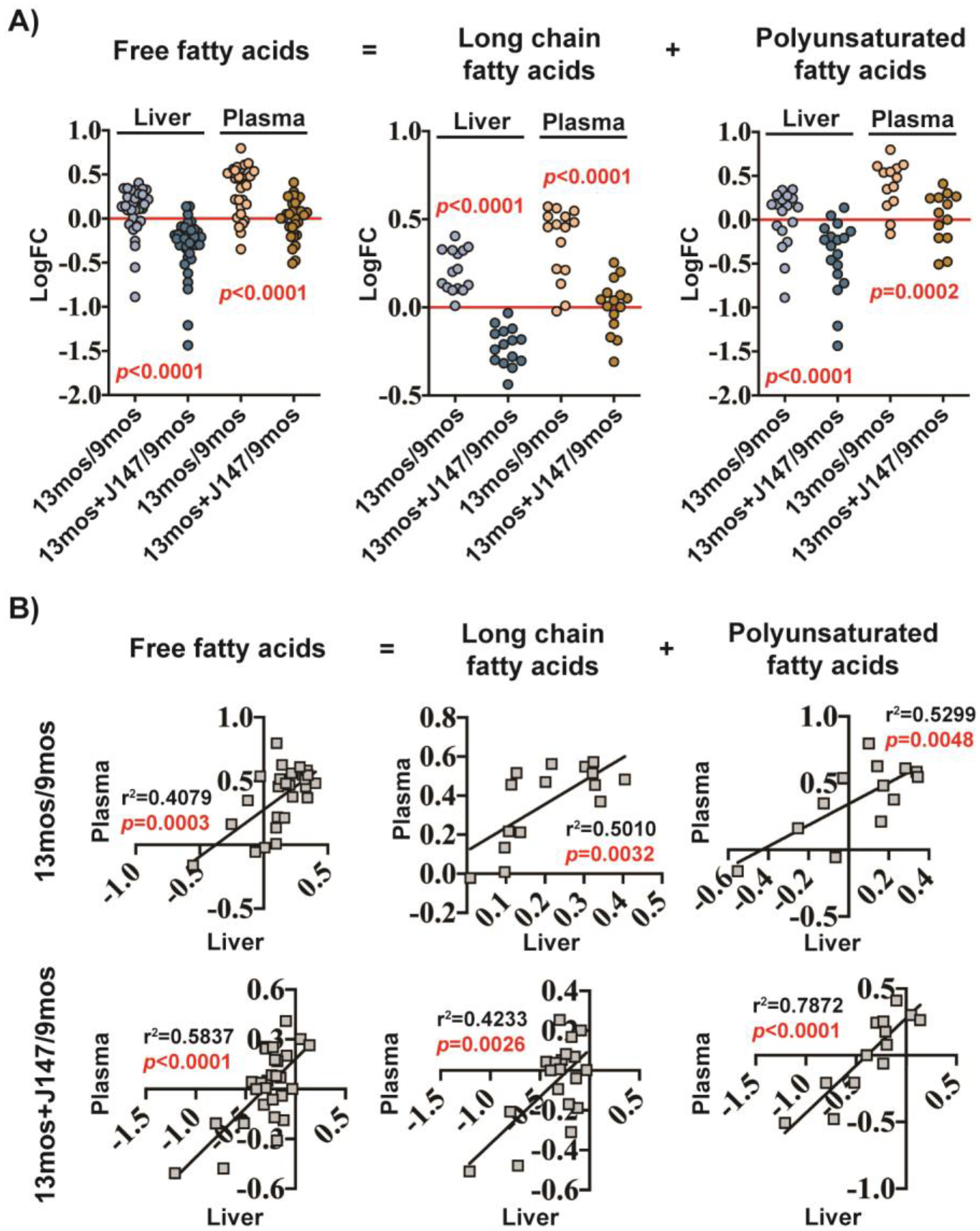
(a) FFA levels in the liver and plasma of female SAMP8 mice, plotted as total free fatty acids, long chain fatty acids, or polyunsaturated fatty acids. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual FFAs. (b) Correlation plots of FFA levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Given that the liver is a regulator of FA synthesis25, we asked if the changes in FFA levels observed in the plasma could be related to alterations in lipid biosynthetic pathways in the liver. To begin to address this question, we correlated the fold changes in individual FFAs observed in the plasma between 9 and 13 months of age in the SAMP8 mice with those in the liver over the same time frame. A significant correlation was detected (Figure 2B top row), indicating that the increases in plasma FFAs with aging might be explained by increases in the liver. The prevention of these increases by J147 also correlated very well (Figure 2B bottom row), suggesting that J147 is exerting its inhibitory effects on plasma FFAs via the liver.
Although not significantly altered with aging or J147, some other lipid classes also showed a good correlation between the liver and the plasma (Figures S1, S2 and S3, Table S2,7), consistent with the key role of the liver in regulating systemic lipid metabolism.
Altogether, these data indicate that the reduction in plasma FFAs by J147 is associated with a specific effect on hepatic lipid biosynthesis by the drug candidate.
3.3. J147 activates the AMPK/ACC1 pathway in the liver
In order to investigate the molecular signaling pathways underlying the effects of J147 on the liver, we conducted an RNA-Seq analysis of liver tissue from the same study with 9- and 13-month-old SAMP8 mice. PCA of gene expression showed that although there was not a clear distinction between the 9- and 13-month-old groups, J147 treatment elicited a very strong and specific effect (Figure 3A).
Figure 3: Transcriptomic analysis of liver tissue from SAMP8 mice.
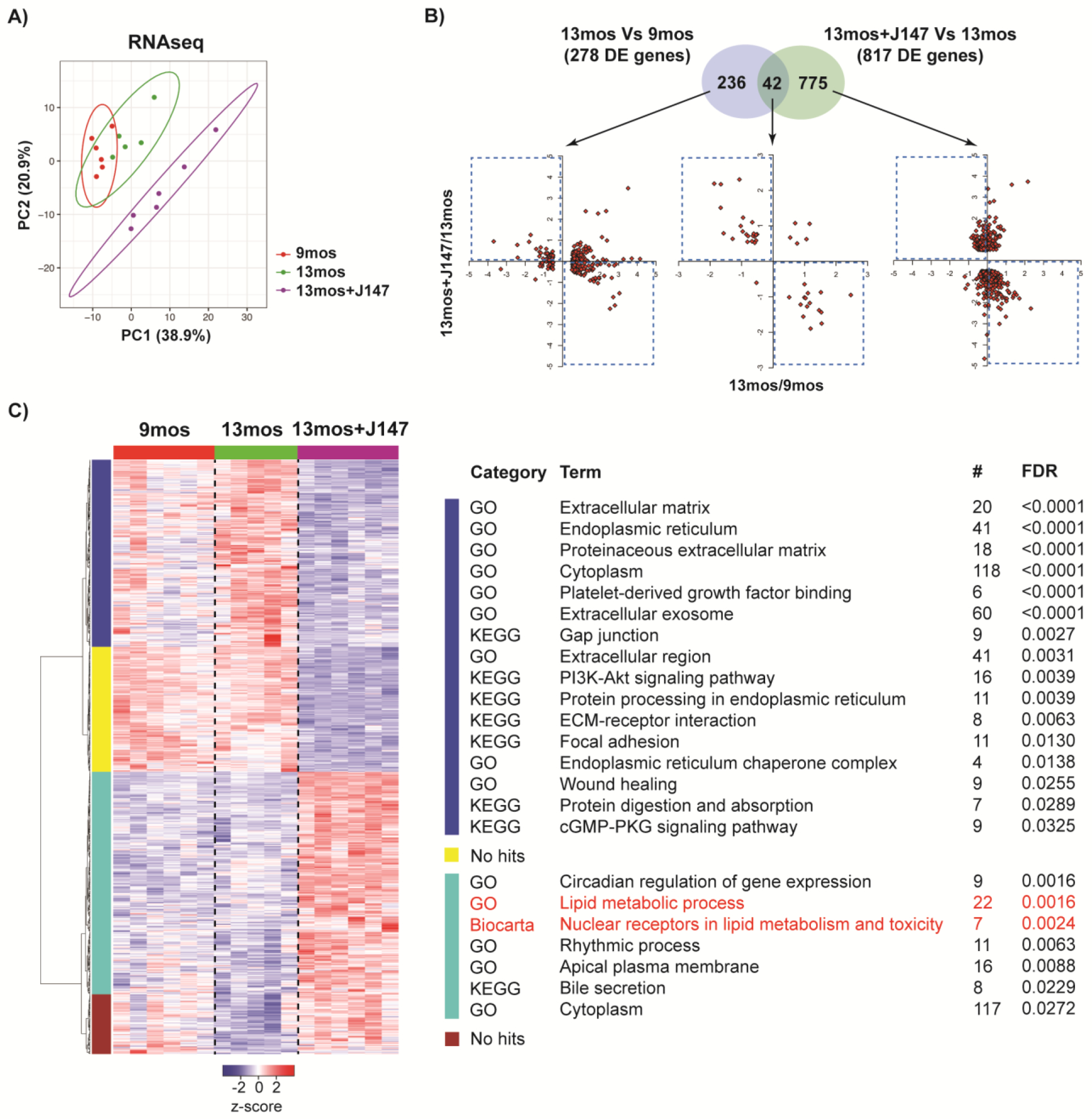
(a) Principal component analysis of the top 500 most variable liver genes (n = 5–6/group). Ellipses show the 95% confidence interval of a t-distribution of the top 2 principal components. (b) Venn diagram illustrating shared and uniquely affected genes between the 13-month-old vs 9-month-old mice and the J147-treated 13-month-old vs untreated 13-month-old mice (n = 5–6/group). Correlation of gene expression for each group of genes is presented on the bottom of the panel (units are −log2(fold change)). (c) (left panel) A clustered heatmap representation of the 817 differentially expressed genes found when comparing the J147-treated 13-month-old mice to untreated 13-month-old mice (n = 5–6/group). z-score = row-wise z-scaled log2 transformed RNA-Seq fragment per kilobase per million mapped reads (FPKM) values. (right panel) Pathway analysis of each major cluster using GO, KEGG, or Biocarta terms.
While only 278 genes were differentially expressed (DE) between the 9- and 13-month-old SAMP8 liver, a total of 817 DE genes were found when comparing the untreated 13-month-old group to the J147-treated 13-month-old group (Figure 3B). Only 42 DE genes overlapped between these two comparisons. Most of these genes (36) were associated with reversal changes by J147 (Figure 3B, middle graph, reversal quadrants). Reversal changes were observed with genes involved in collagen fibrillogenesis and binding, cytoskeletal and extracellular matrix (ECM) organization and maintenance, catalyzed reductions and cellular detoxification, and cellular signaling (Table S3).
A clustered heatmap representation of all of the 817 genes that were differentially expressed between the untreated and J147-treated 13-month-old mice is shown in Figure 3C. Pathway analysis of each major cluster identified the largest group (turquoise blue) as being associated with lipid metabolism (highlighted in red). Interestingly, the expression of these genes was unaltered with aging but strongly increased with J147 treatment.
The second largest cluster (dark blue) is comprised of genes involved in the ECM, endoplasmic reticulum, exosome, focal adhesion, and wound healing. Increased expression of these genes was observed with aging and J147 treatment lowered their expression below that of the 9-month-old baseline control group. Aging is a risk factor for the development of chronic liver disease which is characterized by parenchymal injury, sustained activation of the inflammatory and wound healing responses, and activation of liver fibrogenesis. Liver fibrogenesis is responsible for the excess accumulation of ECM components26. Thus, we focused on the gene transcripts in this cluster associated with the ECM and wound healing to better understand how J147 treatment affects the aging SAMP8 liver, which has been described as a model of liver disease27,28. There were overlaps between these terms, resulting in a total of 30 genes (Figure 4). Transcripts that code for the most abundant ECM proteins, collagen and elastin, showed decreased expression with J147 treatment. This was observed across 6 different collagen gene products. We also observed decreased expression of tubulin beta-B4 chain, the endoplasmic reticulum stress-related messenger Hspa5, and growth factors Tgfb3, Ltbp2, Npnt, and Pdgfrb.
Figure 4: J147 preserves the expression of genes related to the extracellular matrix and wound healing in old SAMP8 liver.
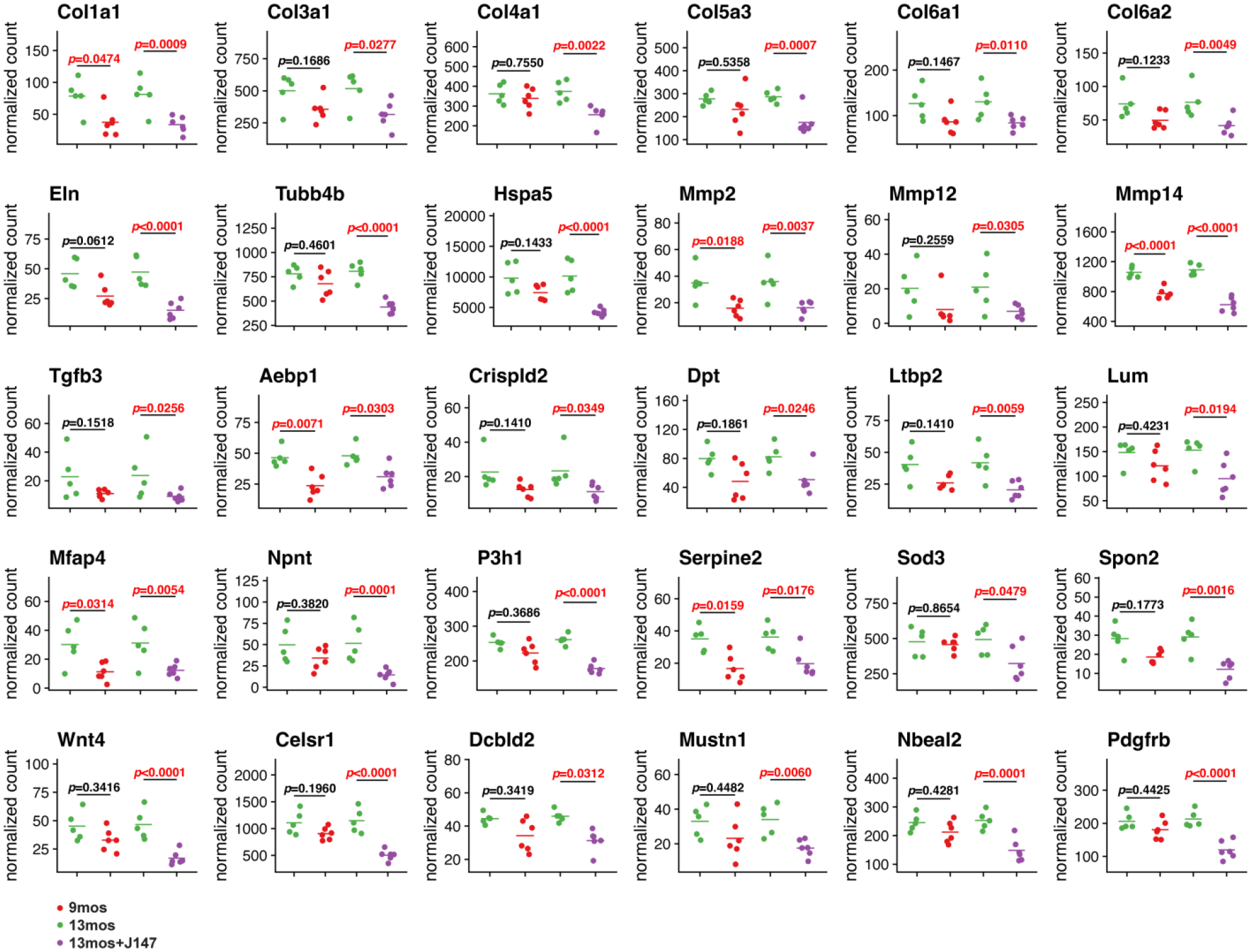
Scatter plots of gene expression in 9-month-old (red), 13-month-old (green), and J147-treated 13-month-old (purple) mouse livers. DESeq220 median of ratios normalization was used to plot expression values while significance was determined using the DESeq2 Wald test assuming a pair-wise design with FDR adjusted p-values shown (n= 5–6/group).
To gain a better understanding of the cellular response to J147’s modulatory effects on ATP synthase and its downstream target AMPK, we assessed potential signaling cascades. We previously found that J147 activated AMPK in a variety of cell culture models and in vivo in the brain (Figure 5A)7,11. AMPK activation was associated with the inhibitory phosphorylation of its downstream target ACC1. Therefore, we tested whether J147 modulated AMPK/ACC1 signaling in the livers of the SAMP8 mice. Consistent with our previous findings, J147 also activated AMPK and inhibited ACC1 in the liver as indicated by an increase in their phosphorylation (Figures 5B and S4). Activation of this pathway was associated with increased levels of ATP (Figure 5C), similar to what we reported in nerve cells11. However, in contrast to what we reported in the brain7, mass spectrometric analysis of the liver showed a 6-fold increase in acetyl-CoA levels in old mice which was prevented by J147 treatment (Figure 5D).
Figure 5: J147 activates the AMPK/ACC1 pathway in the liver.
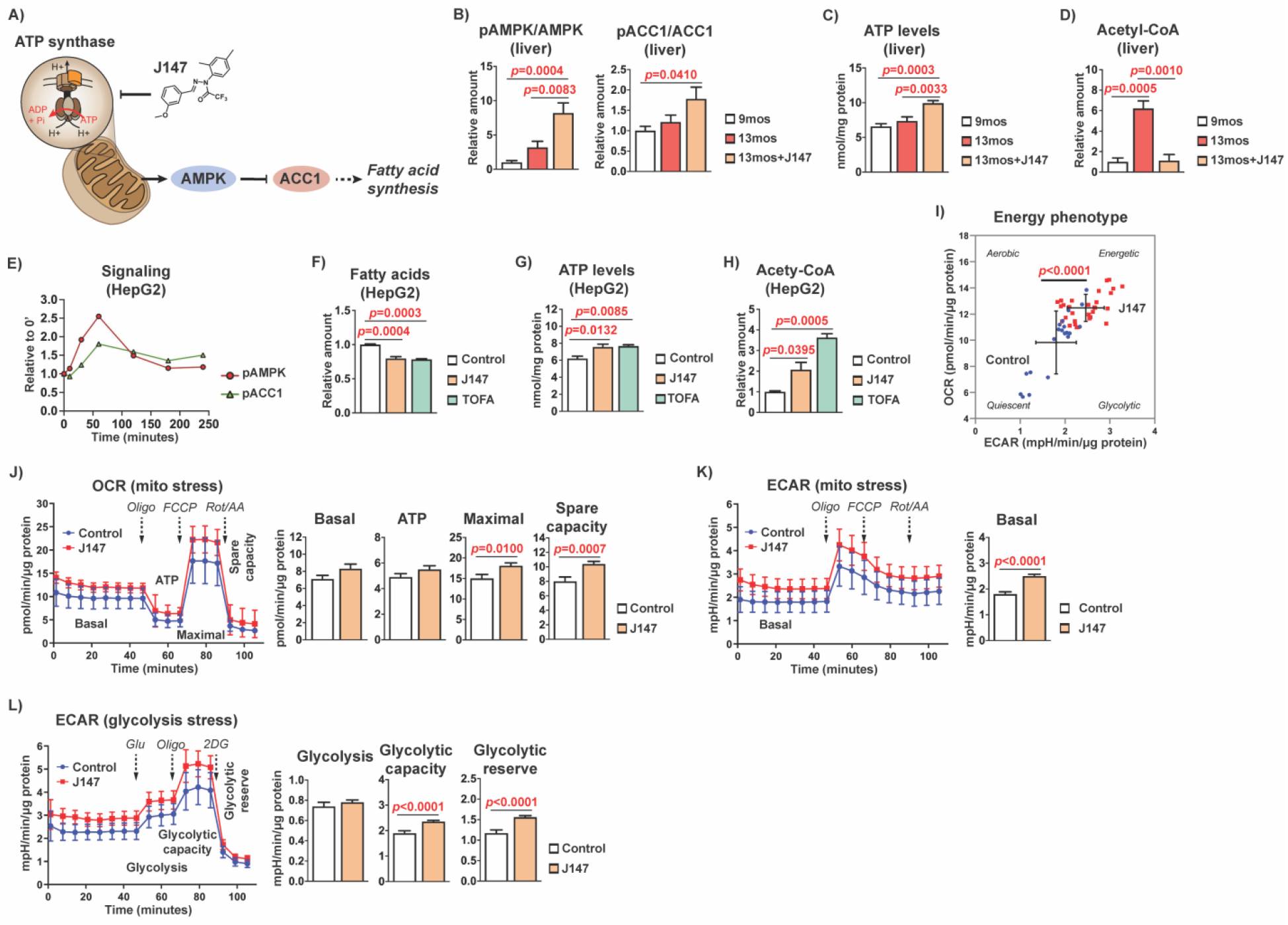
(a) Schematic diagram showing how J147’s inhibitory activity on the mitochondrial ATP synthase affects downstream signaling pathways. (b) Bar graphs of liver protein levels in 9-month-old, 13-month-old, and J147-treated 13-month-old SAMP8 mice (n = 6/group). (c) Bar graph of liver ATP levels in 9-month-old, 13-month-old, and J147-treated 13-month-old SAMP8 mice (n = 6/group). (d) Bar graph of liver acetyl-CoA levels in 9-month-old, 13-month-old, and J147-treated 13-month-old SAMP8 mice (n = 3–4/group). Data are presented as mean ± SEM. Results were compared by one-way ANOVA, followed by Tukey’s multiple comparison test. (e) Time course of activation of the AMPK/ACC1 signaling pathway by 1 μM J147 showing increasing phosphorylation of AMPK (α-Thr172) and ACC1 (Ser79) in HepG2 cells (n = 3/group). (f) Bar graph of fatty acid levels in HepG2 cells following a 24 hr treatment with 1 μM J147 or 10 μg/mL TOFA (5-(tetradecyloxy)-2-furoic acid) (n = 3/group). (g) Bar graph of ATP levels in HepG2 cells following a 24 hr treatment with 1 μM J147 or 10 μg/mL TOFA (n = 3/group). (h) Bar graph of acetyl-CoA levels in HepG2 cells following a 24 hr treatment with 1 μM J147 or 10 μg/mL TOFA (n = 3/group). Data are presented as mean ± SEM. Results were compared by one-way ANOVA, followed by Tukey’s multiple comparison test. (i) Cellular energy phenotype determined by plotting the basal levels of OCR versus ECAR. Aerobic: cells rely predominantly on mitochondrial respiration. Glycolytic: cells utilize predominantly glycolysis. Energetic: cells utilize both metabolic pathways. Quiescent: cells are not very energetic via either metabolic pathway. (j) (left panel) Mitochondrial oxygen consumption rates (OCR) in untreated and J147-treated (4 hr) HepG2 cells. (right panel) Bar graphs for corresponding basal respiration, ATP production, maximal respiration, and spare capacity. (k) (left panel) Mitochondrial extracellular acidification rate (ECAR) in untreated and J147-treated (4 hr) HepG2 cells. (right panel) Bar graph for corresponding basal ECAR. (l) (left panel) Glycolytic extracellular acidification rate (ECAR) in untreated and J147-treated (4 hr) HepG2 cells. (right panel) Bar graphs for corresponding glycolysis levels, glycolytic capacity, and glycolytic reserve. Data are presented as mean ± SEM. Results were compared by an unpaired two-tailed t-test (n = 20–30 wells/group).
To provide further support that our observations were specific to the action of J147 in the liver, we tested J147 in HepG2 cells, a human hepatic cancer cell line frequently used for studies on hepatic lipid metabolism29. J147 also activated the AMPK/ACC1 pathway (Figures 5E and S5), decreased the levels of FFAs (Figure 5F), elevated ATP levels (Figure 5G), and elevated acetyl-CoA levels (Figure 5H) in these cells. Importantly, 5-(tetradecyloxy)-2-furoic acid (TOFA), a competitive inhibitor of ACC1, gave very similar results (Figure 5F–H).
Since J147 increased both ATP and acetyl-CoA levels in the HepG2 cells, we hypothesized that the compound might be modulating cellular metabolism. Therefore, to further elucidate the effects of J147 on the overall bioenergetics of HepG2 cells, we performed Seahorse experiments by measuring the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). The ECAR serves as an indirect measure of extracellular lactate produced by glycolysis. We found that J147 increased both the OCR and ECAR of cells, indicative of an enhancement of cellular bioenergetics (Figure 5I) and consistent with its ability to increase ATP levels (Figure 5G). Using the Mito Stress Test to measure additional parameters of mitochondrial function, we observed that J147 significantly increased the maximal and spare respiratory capacities (Figure 5J) as well as basal ECAR (Figure 5K). The glycolytic function of cells under the same conditions was assessed using the Glycolysis Stress Test, which showed that J147 also increased the glycolytic capacity and glycolytic reserve of cells (Figure 5L).
In summary, our data show that J147 has a specific physiological effect in liver that is associated with the activation of AMPK/ACC1 signaling, preservation of energy, and inhibition of FFA synthesis.
4. Discussion
Biomarkers for target engagement are pivotal for verifying that any drug candidate being tested in humans is acting on its molecular target and having the physiological effects for which it was developed. Lack of this information may not only hinder the interpretation of the outcome of a clinical trial but may also result in substantial financial loss. J147 is now in clinical trials for AD. Although the molecular target of J147 has been identified as the mitochondrial ATP synthase with the resulting activation of the AMPK/ACC1 pathway7,11, no biomarkers for the activity of J147 in vivo have been identified yet. Here, we carried out an investigation into the value of plasma FFAs as a biomarker for target engagement of J147. It was found that long-term treatment with J147 decreased the levels of FFAs in the plasma of SAMP8 mice, likely as a consequence of its inhibitory effect on FA synthesis in the liver via regulation of the AMPK/ACC1 pathway. Moreover, even a two-week treatment in rats resulted in a decrease in plasma FFA levels which was significant in males. This response time further supports the utility of using plasma FFA levels as a biomarker of the activity of J147 since early-stage clinical trials are usually for a short time frame.
Ideally, a biomarker for target engagement should be generated from the tissue that is being targeted14, which often is the main tissue affected in a given disease. In the case of AD, therapeutics are most commonly designed to target pathological features of the disease in the brain. However, J147 was developed based on a phenotypic screening approach using nerve cell lines exposed to toxicities associated with aging and AD1. Its target was only identified several years later through a chemical biology approach, once J147 had already been tested in multiple animal models of aging and dementia11,30. The ATP synthase is not a target exclusive to the brain, but rather an ATP generating complex essential to most cells in an organism. Perhaps because of this, the protective effects of J147 are likely to extend beyond the brain to include other tissues and organs31. In fact, there is a growing awareness of the idea that therapies to treat AD and related disorders may be more beneficial if they provide additional protective effects in peripheral systems, particularly when considering the etiology of AD in the context of aging. This approach to AD drug discovery brings new challenges when considering the discovery of biomarkers for target engagement, as it becomes more difficult to define the tissue from which the biomarker should derive.
We show that decreases in the levels of plasma FFAs could be used as a biomarker for target engagement of J147 in the liver. However, we have not shown that the effects on liver FA metabolism by J147 reflect its effects in the brain. Future studies should be designed to answer this question, particularly if there are FA metabolites that are predominantly generated in the brain that could be used as more specific biomarkers for target engagement of J147 in the brain. However, it is important to note that in both of our SAMP8 studies, the decreases in plasma FFAs were associated with protective effects by J147 in the brain, as measured by a number of physiological parameters including cognition, transcriptomics, metabolomics, and markers of inflammation as well as AD6,7.
Unlike what was found in the brains of SAMP8 mice treated with J147, the effects of the drug candidate in the liver tissue were strongly indicative of a specific effect unrelated to aging, as most of the DE gene expression changes induced by J147 were not correlated with aging. This is consistent with the fact that the liver is a major regulator of lipid metabolism in the body and that J147 targets the AMPK/ACC1 pathway, a key pathway regulating FFA synthesis. It is also possible that some of the decreases in FFAs brought about by J147 treatment are associated with an increase in hepatic FA beta-oxidation. We have not addressed this. It is also not clear why the expression of the DE genes associated with lipid metabolism in the liver was upregulated by J147. Perhaps this upregulation is a response to counterbalance the inhibitory effect of J147 on the levels of FFAs.
The prevention of changes in the expression of DE genes with aging by J147 in the liver was very modest. Only 36 DE genes out of the 278 modified with aging in SAMP8 mice livers were prevented with statistical significance by the drug candidate (Table S3), but many of these genes are involved in pathways associated with liver disease. Furthermore, by focusing on the genes in the dark blue cluster of Figure 3C that were associated with the ECM and wound healing, J147 treatment was seen to strikingly decrease the expression of additional genes implicated in liver disease (Figure 4)26,32. Nonalcoholic fatty liver disease (NAFLD) is a leading form of chronic liver disease with hepatic steatosis not associated with alcohol intake that can lead to secondary complications such as obesity, type 2 diabetes, hepatocarcinoma, and dyslipidemia33,34. There is an increased risk for developing NAFLD with aging34 and SAMP8 mice have been reported to develop fatty liver along with alterations in glucose metabolism and insulin signaling35. Our observation that J147 altered the expression of genes implicated in liver disease justifies further investigation of its potentially beneficial effects in other models of fatty liver diseases.
Another interesting result from our study is the effect of J147 on cellular bioenergetics, namely the increases in respiratory and glycolytic parameters as well as ATP levels, in the HepG2 cells. AMPK’s role in regulating cellular energy balance has been well studied and its activation leads to the promotion of catabolic pathways and inhibition of anabolic pathways, resulting in increased ATP generation36,37. We have previously reported that J147 is able to increase ATP levels in nerve cells by modulating the mitochondrial ATP synthase, leading to a sustained calcium/calmodulin-dependent protein kinase kinase β (CAMKK2)-dependent activation of AMPK11. This response could be coordinated to promote the activation of glycolysis and FA oxidation in addition to decreasing FA synthesis. The latter could lead to an increase in the availability of acetyl-CoA in the mitochondria for the tricarboxylic acid (TCA) cycle and energy production. We have recently shown that J147 increased the levels of acetyl-CoA in nerve cells along with the activation of the AMPK/ACC1 pathway, which was associated with maintenance of mitochondrial function during age-related stresses7. In the present study, we demonstrated that J147 also increased the levels of acetyl-CoA in the human hepatic HepG2 cell line (Figure 5H). While this effect of J147 is consistently shown with short-term treatment across diverse cell lines, the outcome of long-term treatment in disease models appears more complex. Surprisingly, the livers of old SAMP8 mice showed a 6-fold increase in acetyl-CoA levels compared to the young mice but this increase was completely prevented by J147 treatment (Figure 5D), consistent with the ability of J147 to reverse age-related changes in various tissues31. In the SAMP8 brain, we previously reported that acetyl-CoA levels decreased with aging and this was prevented by J147 treatment6,7.
In summary, our data bring important considerations into the discovery of biomarkers for target engagement of the AD drug candidate J147. Although we did not fully validate plasma FFAs as a biomarker for the action of J147 in the brain, we did show that the changes in plasma FFAs with J147 may reflect the physiological effects of J147 in the liver. In the mouse models, these changes were strongly associated with improvements in cognition and multiple neurological markers by J147. Therefore, it will be important to assess how J147 modulates the levels of plasma FFAs in human clinical trials in order to establish an association with its effects on the primary endpoints of the trials. To the best of our knowledge, this is the first study to determine a blood-based biomarker for ATP synthase modulation. Finally, our results further support the testing of J147 in age-related diseases other than AD, such as NAFLD and other hepatic disorders.
Supplementary Material
Figure S1: Changes in plasma and liver lipid metabolites 1. (a) Metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual metabolites. (b) Correlation plots of metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Figure S2: Changes in plasma and liver lipid metabolites 2. (a) Metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual metabolites. (b) Correlation plots of metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Figure S3: Changes in plasma and liver lipid metabolites 3. (a) Metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual metabolites. (b) Correlation plots of metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Figure S4: J147 treatment activates the AMPK/ACC1 pathway in SAMP8 liver. Raw Western blot images of liver from 9-month-old, 13-month-old, and J147-treated 13-month-old SAMP8 mice (n = 6/group).
Figure S5: J147 treatment activates the AMPK/ACC1 pathway in HepG2 cells. Raw Western blot images of a time course of 1 μM J147 activation of the AMPK/ACC1 signaling pathway. Increasing phosphorylation of AMPK (α-Thr172) and ACC1 (Ser79) in HepG2 cells (n = 3/group).
Table S1: List of plasma FFAs in female and male Wistar rats treated with J147.
Table S2: Lipidomics with the liver of 9-month-old, 13-month-old, and J147-treated 13-month-old female SAMP8 mice.
Table S3: List of reversed age-related gene expression changes in the liver by J147 treatment.
Highlights.
J147 decreases free fatty acid levels in both the plasma and liver of rodents
Plasma fatty acid changes are due to AMPK/ACC1 inhibition of synthesis in the liver
AMPK/ACC1 inhibition of fatty acid synthesis in the liver preserves energy levels
Decreased plasma fatty acid levels may be a biomarker for target engagement of J147
Acknowledgements
We thank Joseph Chambers for help with the breeding and husbandry of the mice and Max Shokhirev for help with the manuscript review and editing.
Funding
This work was supported by grants from NIH (RO1 AG069206, RF1 AG054714) to P.M., the Shiley-Marcos Alzheimer’s Disease Research Center at University of California San Diego to A.C., the Paul F. Glenn Center for Biology of Aging Research at the Salk Institute Fellowship to Z.L., a Bundy Foundation Fellowship to D.K., and NIH-NCI CCSG: P30 014195, and the Helmsley Trust to L.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The Salk Institute holds the patent for J147.
Data sharing statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus38 and are accessible through GEO Series accession number GSE166317. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166317)
CRediT authorship contribution statement
Conceptualization, D.K., A.C., P.M. Methodology, D.K., L.H., A.C., Z.L., W.F., P.M. Investigation, D.K., L.H., A.C., Z.L., W.F. Formal data analysis, D.K., L.H., A.C., Z.L. Project administration, D.K., A.C., P.M. Funding acquisition, P.M. Writing – Original draft, D.K., A.C. Writing – Review and editing, D.K., L.H., A.C., Z.L., W.F., P.M.
References
- 1.Prior M, Chiruta C, Currais A, et al. Back to the future with phenotypic screening. ACS Chem Neurosci 2014; 5(7): 503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schubert D, Currais A, Goldberg J, Finley K, Petrascheck M, Maher P. Geroneuroprotectors: Effective Geroprotectors for the Brain. Trends Pharmacol Sci 2018; 39(12): 1004–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Q, Prior M, Dargusch R, et al. A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease. PLoS One 2011; 6(12): e27865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prior M, Dargusch R, Ehren JL, Chiruta C, Schubert D. The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimers Res Ther 2013; 5(3): 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prior M, Goldberg J, Chiruta C, et al. Selecting for neurogenic potential as an alternative for Alzheimer’s disease drug discovery. Alzheimers Dement 2016; 12(6): 678–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Currais A, Goldberg J, Farrokhi C, et al. A comprehensive multiomics approach toward understanding the relationship between aging and dementia. Aging (Albany NY) 2015; 7(11): 937–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Currais A, Huang L, Goldberg J, et al. Elevating acetyl-CoA levels reduces aspects of brain aging. Elife 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda T Senescence-accelerated mouse (SAM) with special references to neurodegeneration models, SAMP8 and SAMP10 mice. Neurochem Res 2009; 34(4): 639–59. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA, Poon HF. The senescence-accelerated prone mouse (SAMP8): a model of age-related cognitive decline with relevance to alterations of the gene expression and protein abnormalities in Alzheimer’s disease. Exp Gerontol 2005; 40(10): 774–83. [DOI] [PubMed] [Google Scholar]
- 10.Akiguchi I, Pallas M, Budka H, et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology 2017; 37(4): 293–305. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg J, Currais A, Prior M, et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin RM, Fu X, Pai MY, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014; 510(7505): 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon GM, Niphakis MJ, Cravatt BF. Determining target engagement in living systems. Nat Chem Biol 2013; 9(4): 200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durham TB, Blanco MJ. Target engagement in lead generation. Bioorg Med Chem Lett 2015; 25(5): 998–1008. [DOI] [PubMed] [Google Scholar]
- 15.Daugherty D, Goldberg J, Fischer W, Dargusch R, Maher P, Schubert D. A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimers Res Ther 2017; 9(1): 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Currais A, Prior M, Lo D, Jolivalt C, Schubert D, Maher P. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging Cell 2012; 11(6): 1017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin SY, Fauman EB, Petersen AK, et al. An atlas of genetic influences on human blood metabolites. Nat Genet 2014; 46(6): 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29(1): 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38(4): 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4(1): 44–57. [DOI] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979; 282(5739): 615–6. [DOI] [PubMed] [Google Scholar]
- 24.Huang L, McClatchy DB, Maher P, et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis 2020; 11(10): 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodson L, Gunn PJ. The regulation of hepatic fatty acid synthesis and partitioning: the effect of nutritional state. Nat Rev Endocrinol 2019; 15(12): 689–700. [DOI] [PubMed] [Google Scholar]
- 26.Parola M, Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med 2019; 65: 37–55. [DOI] [PubMed] [Google Scholar]
- 27.Ye X, Meeker HC, Kozlowski PB, et al. Pathological changes in the liver of a senescence accelerated mouse strain (SAMP8): a mouse model for the study of liver diseases. Histol Histopathol 2004; 19(4): 1141–51. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, He J, Ji S, et al. Comparative studies of early liver dysfunction in senescence-accelerated mouse using mitochondrial proteomics approaches. Mol Cell Proteomics 2008; 7(9): 1737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dashti N, Wolfbauer G. Secretion of lipids, apolipoproteins, and lipoproteins by human hepatoma cell line, HepG2: effects of oleic acid and insulin. J Lipid Res 1987; 28(4): 423–36. [PubMed] [Google Scholar]
- 30.Maher P, Currais A, Schubert D. Using the Oxytosis/Ferroptosis Pathway to Understand and Treat Age-Associated Neurodegenerative Diseases. Cell Chem Biol 2020; 27(12): 1456–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kepchia D, Currais A, Dargusch R, Finley K, Schubert D, Maher P. Geroprotective effects of Alzheimer’s disease drug candidates. Aging (Albany NY) 2021; 13(3): 3269–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panwar P, Butler GS, Jamroz A, Azizi P, Overall CM, Bromme D. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. Matrix Biol 2018; 65: 30–44. [DOI] [PubMed] [Google Scholar]
- 33.Gan L, Chitturi S, Farrell GC. Mechanisms and implications of age-related changes in the liver: nonalcoholic Fatty liver disease in the elderly. Curr Gerontol Geriatr Res 2011; 2011: 831536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertolotti M, Lonardo A, Mussi C, et al. Nonalcoholic fatty liver disease and aging: epidemiology to management. World J Gastroenterol 2014; 20(39): 14185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu HW, Chan YC, Wang MF, Wei CC, Chang SJ. Dietary (−)-Epigallocatechin-3-gallate Supplementation Counteracts Aging-Associated Skeletal Muscle Insulin Resistance and Fatty Liver in Senescence-Accelerated Mouse. J Agric Food Chem 2015; 63(38): 8407–17. [DOI] [PubMed] [Google Scholar]
- 36.Carling D AMPK signalling in health and disease. Curr Opin Cell Biol 2017; 45: 31–7. [DOI] [PubMed] [Google Scholar]
- 37.Hardie DG, Schaffer BE, Brunet A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol 2016; 26(3): 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30(1): 207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Changes in plasma and liver lipid metabolites 1. (a) Metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual metabolites. (b) Correlation plots of metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Figure S2: Changes in plasma and liver lipid metabolites 2. (a) Metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual metabolites. (b) Correlation plots of metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Figure S3: Changes in plasma and liver lipid metabolites 3. (a) Metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice or 13-month-old J147-treated mice to 9-month-old untreated mice (n = 6/group). Results were compared by Wilcoxon matched-pairs signed rank test. Dots represent the log fold change of individual metabolites. (b) Correlation plots of metabolite levels in the liver and plasma of female SAMP8 mice. Data are presented as the log fold change of 13-month-old to 9-month-old mice (top row) or log fold change of 13-month-old J147-treated mice to 9-month-old untreated mice (bottom row) (n = 6/group).
Figure S4: J147 treatment activates the AMPK/ACC1 pathway in SAMP8 liver. Raw Western blot images of liver from 9-month-old, 13-month-old, and J147-treated 13-month-old SAMP8 mice (n = 6/group).
Figure S5: J147 treatment activates the AMPK/ACC1 pathway in HepG2 cells. Raw Western blot images of a time course of 1 μM J147 activation of the AMPK/ACC1 signaling pathway. Increasing phosphorylation of AMPK (α-Thr172) and ACC1 (Ser79) in HepG2 cells (n = 3/group).
Table S1: List of plasma FFAs in female and male Wistar rats treated with J147.
Table S2: Lipidomics with the liver of 9-month-old, 13-month-old, and J147-treated 13-month-old female SAMP8 mice.
Table S3: List of reversed age-related gene expression changes in the liver by J147 treatment.