

Abstract
The understanding of lysosomes has come a long way since the initial discovery of their role in degrading cellular waste. The lysosome is now recognized to be a highly dynamic organelle positioned at the crossroads of cell signaling, transcription, and metabolism. Underscoring its importance is the observation that in addition to rare monogenetic lysosomal storage disorders, genes regulating lysosomal function are implicated in common sporadic neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Developing therapies for these disorders is particularly challenging, largely due to gaps in knowledge of the underlying molecular and cellular processes. In this review, we discuss technological advances that have propelled deeper understanding of the lysosome in neurodegeneration, from elucidating the functions of lysosome-related disease risk variants at the level of the organelle, cell and tissue, to the development of disease-specific biological models that recapitulate disease manifestations. Finally, we identify key questions that remain to be addressed to successfully bridge the gap to the clinic.
Keywords: Parkinson’s disease, Alzheimer’s disease, frontotemporal dementia, amyotrophic lateral sclerosis, lysosomes
Lysosomes and neurodegeneration
The lysosome is a key organelle found in eukaryotic cells. Initially recognized for its function in intracellular degradation, the lysosome is now understood to have far-reaching roles in the maintenance of cell homeostasis and viability. A large body of evidence indicates that lysosomes serve as a major signaling hub in the cell, affecting fundamental processes such as membrane repair, energy metabolism, and inflammatory pathways [1–4] Pathogenic variants in over 50 lysosomal genes result in lysosomal storage disorders (LSDs, see Glossary), the most common neurodegenerative diseases in children, highlighting the importance of proper lysosomal functioning in maintenance of the nervous system [5]. A plethora of risk variants in lysosomal genes have also been identified in individuals with neurodegenerative diseases such as Parkinson’s disease (PD), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) [6–12] (Figure 1, Table 1). The critical role of lysosomes in the autophagy pathway places them at the crossroads of several intracellular processes including the regulation of proteotoxicity and inflammation. Indeed, variants in risk alleles associated with late-onset Alzheimer’s disease (AD) such as APOE, SORL1, BIN1, CD2AP, PICALM, TREM2 and PLD3, as well as genes related to APP processing, a pathologically and genetically relevant pathway in AD, are associated with lysosomal-autophagy pathways [13–19]. Many of these genes, however, do not impact neurons alone. In AD, analysis of post-mortem brain sections from patients carrying risk variants in TREM2, a gene expressed in myeloid cells, reveals a significant increase in the percentage of microglia containing autophagic vesicles [20]. Other studies have shown that lysosomal dysfunction in both glial and neuronal cells contribute to the spreading of the pathologic hallmarks amyloid-beta and tau in AD, and alpha-synuclein in PD [21–25]. Taken together, these findings highlight the pervasive influence of lysosomal dysfunction across multiple cell types within the nervous system.
Figure 1: Genes implicated in lysosomal function and lysosomal processes are at the crossroads of several neurodegenerative diseases.
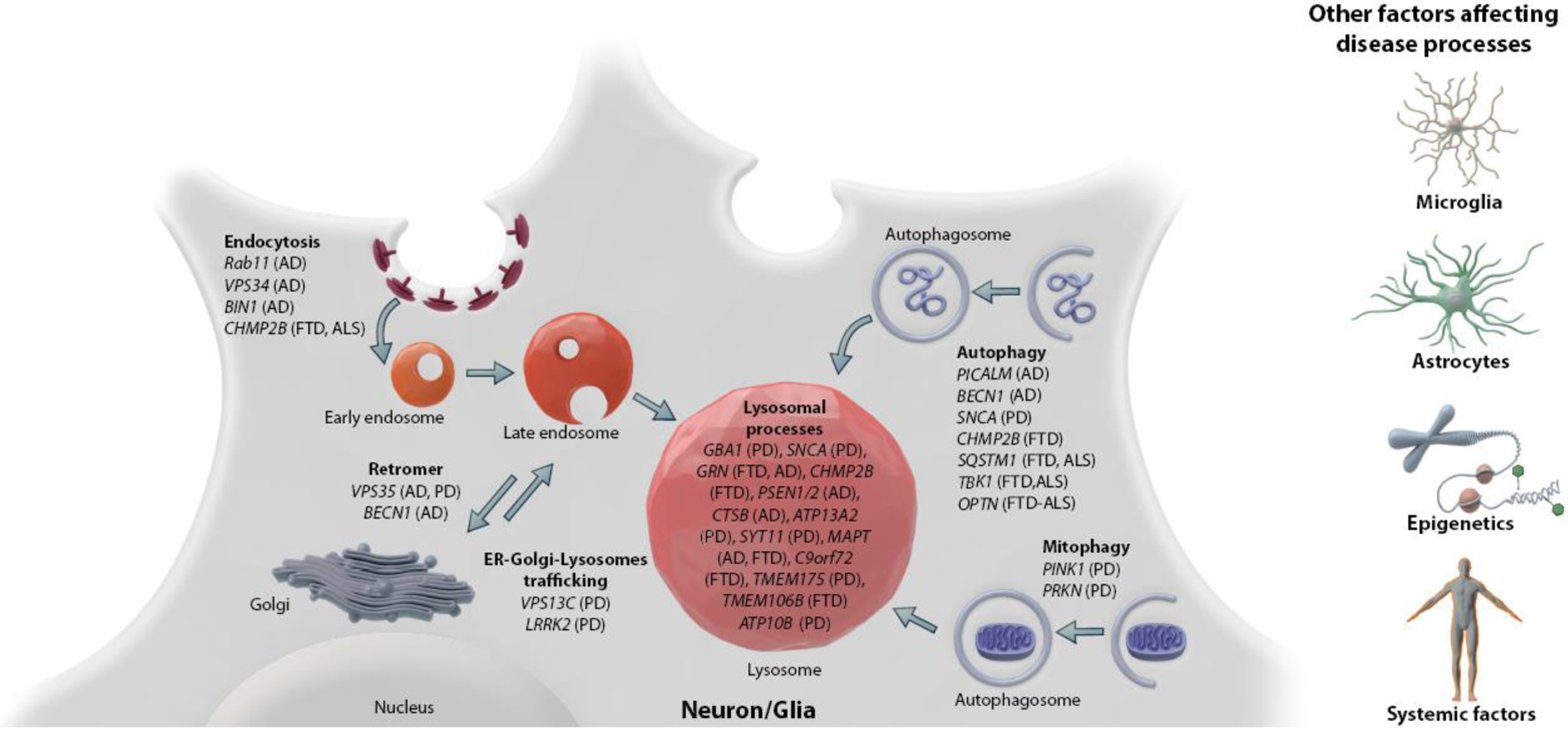
Risk variants in lysosomal genes have been identified in several neurodegenerative diseases, including AD, PD, ALS and FTD. The implicated genes are involved in a broad range of lysosome-related pathways, such as acidification, endocytosis, autophagy, mitophagy and ER-golgi-lysosome trafficking. The risk variants may mediate their pathogenicity in/through various cell types, including neurons, microglia and astrocytes. Epigenetic pathways and systemic factors likely modulate the risk of these variants for the disease and thus contribute to the heterogeneity in disease progression.
Table 1:
Summary of genes associated with PD, ALS and FTD risk and their proposed roles in lysosomal physiologya
| Gene | Protein | Proposed lysosomal function | Mutation/Variant | Proposed disease mechanism | Frequency | Clinical phenotypes | References |
|---|---|---|---|---|---|---|---|
| GBA1 | Glucocerebrosidase | Hydrolysis of GluCer and GluSph | Missense or PTC | Likely LOF | Common | PD, an earlier age at onset | [163,164] |
| LRRK2 | Leucine rich repeat kinase 2 | Recruitment to lysosome and phosphorylation of Rab proteins | Missense | GOF | Common | LOPD | [83,165,166] |
| TMEM175 | Transmembrane protein 175 | Lysosomal K+ channel | Missense | LOF, GOF (protective) | Common | Increased or decreased PD risk | [71,74] |
| ATP13A2 | ATPase cation transporting 13A2 | Exporter of polyamines such as spermine | Missense or PTC | LOF | Rare | Atypical PD, also called Kufor-Rakeb syndrome | [9,167] |
| ATP10B | ATPase phospholipid transporting 10B | Flippase of GlcCer/PC | Missense and splice site mutation | LOF | Rare | PD | [168] |
| VPS13C | Vacuolar protein sorting 13C | Lipid transport from ER to lysosomes | Deletions and PTC mutations | LOF | Rare | PD, an earlier age at onset | [169,170] |
| SNCA | Alpha-synuclein | No clear function | Multiplication & Missense | GOF | Rare | PD, an earlier age at onset LOPD | [171,172] |
| C9orf72 | Chromosome 9 open reading frame 72 | GTPase-activating protein (GAP) complex with SMCR8-WD41; lysosome trafficking | G4C2-repeat expansion | GOF, LOF, possibly both LOF and GOF | Common | FTD/ALS | [10,33,101,173,174] |
| GRN | Progranulin | Granulin precursor, lipid metabolism and hydrolase trafficking | Missense or PTC | LOF | Common | FTD/ALS | [11,62,64] |
| CHMP2B | Charged multivesicular body protein 2B | Endocytic multivesicular body formation | C-terminal truncation | LOF | Rare | FTD/ALS | [175,176] |
| TMEM106B | Transmembrane protein 106B | Regulation of lysosomal trafficking and morphology | Intron variant | Increased protein expression | Common | FTD/ALS | [12,177,178] |
Abbreviations: GluCer: glucosylceramide; GlcCer: Galactosylceramide; PC: phosphatidylcholine; LOF: loss-of-function; GOF: gain-of-function; PTC: premature termination codon; LOPD: late-onset PD
Immune cells are also exquisitely sensitive to lysosomal perturbations. In immune cells, lysosomal pathways regulate key functions including phagocytosis, processing and secretion of inflammatory cytokines and antigen presentation [26,27]. In ALS and FTD, the most prevalent causally-linked risk gene, C9orf72, plays an important role in lysosomal homeostasis, and when disrupted, results in dysfunctional immune cells in the periphery and brain [28–33]. GRN, another gene important in lysosomal function and the second most common cause of inherited FTD (interestingly not associated with ALS), has been linked to microglial dysfunction and neuroinflammation [32,34–36]. Notably, homozygous loss-of-function mutations in GRN lead to a LSD subtype known as neuronal ceroid lipofuscinoses (NCL) affecting children and young adults. In NCL, the progressive neurological deterioration is accompanied by dementia, motor disturbances, epilepsy, loss of vision, and early death [37,38]. Altogether, these lines of evidence highlight lysosomal dysfunction as a critical driver of disease pathology involving both neuroinflammation and neurodegeneration.
Understanding of the genetic architecture and pathological mechanisms associated with lysosomal disorders has progressed significantly over the past decade. The growing body of knowledge on the physiological functions of neurodegeneration-related lysosomal genes such as GBA1, LRRK2, C9orf72, TMEM175, ATP13A2, TMEM106B and GRN (Table 1) has paved the way for ongoing clinical trials in GBA1- and LRRK2-associated PD, C9orf72-associated ALS or FTD and GRN-associated FTD [39,40]. Nevertheless, major gaps in the understanding remain, hampering the identification of biomarkers and the development of targeted disease-modifying therapeutics. Current challenges include: a. Defining the specific lysosomal alterations resulting from lysosomal risk variants and identifying the underlying pathogenic mechanisms at the organelle level, b. Determining the impact of risk variants on non-neuronal cell types and their specific contribution to pathogenicity, c. Identifying genetic and cellular modifiers of disease risk, and d. Translating these discoveries into therapy for neurodegenerative diseases. In the following four sections, we systematically address each of these challenges and discuss how emerging technologies can be harnessed to advance the understanding of lysosome-related neurodegenerative diseases and bridge the existing knowledge gaps.
Pathophysiological analysis of lysosomes at the level of the organelle
The application of single-cell transcriptomic, proteomic and metabolomic approaches, as well as next-generation sequencing in neurodegenerative diseases have shed light on disease-specific genetic and cellular changes with increased granularity [41–44]. However, the complexity of lysosome function presents a key barrier in unravelling its specific roles in neurodegenerative disease. An example is the identification of variants in GBA1, a common genetic risk factor for PD and the associated disorders dementia with Lewy bodies (DLB) and rapid eye movement sleep behavioral disorder (RBD) [45,46]. Despite more than a decade of studies, precisely how these GBA1 variants contribute to PD remains unresolved [47–49]. GBA1 encodes the enzyme glucocerebrosidase, a lysosomal hydrolase deficient in the autosomal recessively inherited LSD Gaucher disease (GD). In GD there is a clear accumulation of the lipid substrates glucosylceramide and glucosylsphingosine in macrophages, and this is causally linked to aspects of the disease pathology [50]. Curiously, this does not appear to be the case in GBA1-associated PD, where scant substrate accumulation has been reported [51–53]. One explanation could be that glucocerebrosidase dysfunction specifically results in intra-lysosomal lipid accumulation, leading to biochemical, functional and structural changes that are not detected using conventional methodologies focusing on whole cell/tissue analysis. Of note, glucocerebrosidase also appears to play a role in sporadic PD, for there is an inverse pathological correlation between glucocerebrosidase protein levels and alpha-synuclein pathology, proposed to be further propagated by a bidirectional pathological loop [53–55].
While lysosomes constitute only a minute fraction of the intracellular volume (under 5%), lysosomal proteins, lipids and metabolites are also present elsewhere in the cell [56]. Lysosome isolation combined with downstream analyses such as proteomics and metabolomics are essential to more accurately evaluate lysosome-specific alterations in disease. Such an approach could help delineate the physiological and pathological roles of genes that are implicated in PD, such as GBA1, TMEM175, LRRK2 and SNCA, and enable the identification of converging and/or diverging disease mechanisms in these different forms of PD.
Until recently, isolation of intact lysosomes for functional and biochemical analyses has been challenging, mostly due to the time-consuming and cumbersome methods available. Recently a novel lysosomal immunoprecipitation method (Lyso-IP) has enabled the rapid isolation of intact and sufficiently pure lysosomes [57]. This method involves labeling of lysosomes with a Lyso-tag, a 3xHA-tagged version of the lysosomal protein, Transmembrane protein 192 (TMEM192), followed by immunoprecipitation of the labeled lysosomes using anti-HA antibody-bound resins. This fast and efficient approach has been used to facilitate metabolomic, proteomic and lipidomic analyses of lysosomes from various cellular models [57–60].
Analysis of lysosomes isolated using the Lyso-IP method have been critical in demonstrating the mechanisms of amino acid and lipid exchange between lysosomes and the cytosol, highlighting processes that could be relevant in disease. For example, metabolite profiling of isolated lysosomes revealed that lysosomal arginine sensing serves as a trigger for interactions between SLC38A9 and Rag-Ragulator, thereby coupling mTORC1 signaling to lysosomal metabolism [58]. In the LSD Niemann Pick C (NPC), Lyso-IP technique was used to understand how cholesterol accumulation caused by loss of NPC1 leads to lysosomal dysfunction and other cellular abnormalities. Proteomic analysis of lysosomes isolated from NPC1-null cells showed marked accumulation of numerous substrates ordinarily degraded in lysosomes, thus indicating broad proteolytic defects [59]. Furthermore, immunoblotting of these isolated lysosomes indicated ongoing lysosomal membrane damage, as evidenced by the significant enrichment of ESCRT components. This pathological signaling mechanism was not detectable with conventional immunofluorescence staining, further highlighting the advantages of the Lyso-IP method [59]. In another study, by performing quantitative proteomic analysis of lysosomes isolated by Lyso-IP the authors identified a key process by which cells survive conditions of nutrient starvation [61]. The study demonstrated that under nutrient-limitation/starvation conditions cells utilize ribosomes as a critical source of nutrients by actively delivering ribosomes to lysosomes (via autophagy) for degradation and thereby nutrient recycling [61].
The findings discussed above illustrate the potential of Lyso-IP-based studies in helping resolve pressing outstanding questions. For example, the method could help to better clarify the precise lysosomal functions of progranulin, the growth factor protein encoded by GRN. Recent studies indicate a role for progranulin in lysosomal lipid metabolism and as a potential regulator of glucocerebrosidase and cathepsin D activity [62–65]. Both the full-length progranulin protein and the ~6 kDa granulin peptides formed by progranulin cleavage in lysosomes have important roles. However, how progranulin or the distinct granulins act in the lysosome, and whether there is functional heterogeneity amongst the distinct granulins in a cell-type- and context-dependent manner is still being explored. Full-length progranulin is also present in other cellular organelles where its function is unclear. Comprehensive proteomic and lipidomic profiling of lysosomes versus whole cells from cellular models of GRN knock-out/variants before and after therapeutic rescue with recombinant progranulin, for example, could illuminate the precise mechanistic link between progranulin, lysosomal lipid metabolism and proteolytic defects. When modelling human disease, the Lyso-tag concept can also be applied to iPSC-based organoids and regionalized brain organoids called assembloids. For example, one can compare isolated lysosomes from GBA1-PD versus non-GBA1-PD organoids to identify converging and diverging features in these different forms of PD. In the future, Lyso-tagged mice expressing the 3xHA-TMEM192 tag in a cell- and tissue-specific manner may be exploited to further the understanding of disease pathophysiology, for example, by delineating the glial versus neuronal effects of a lysosomal risk allele in vivo. A limitation of the Lyso-tag approach is that it cannot be applied towards human post-mortem tissues and hence here, conventional lysosome isolation methods must be optimized to analyze intact lysosomes.
Lysosomes have the ability to transport ions across the membrane in a bi-directional manner, for the maintenance of their internal homeostasis (pH, calcium levels, potassium levels etc.), as well as for triggering downstream signaling cascades. Lysosomes are studded with different types of ion channels, many of which are directly relevant to human disease. Thus, beyond purifying lysosomes, organelle electrophysiology and imaging are emerging as essential tools for clarifying the mechanisms of lysosomal disease, and for identifying lysosomal therapeutic targets such as TRPML1 and mTORC1 [66–70]. In patients with PD, as well as in the related disorders DLB and RBD, genetics studies have identified both risk and protective variants in TMEM175 [8,71,72]. Lysosomal patch clamp revealed that TMEM175 encodes a lysosomal potassium channel, and that protective and risk variants in PD either activate or inactivate the channel, respectively [73,74]. Interestingly the protective variant not only increased channel opening, but also enhanced neuronal resistance to damage [74]. Accordingly, activating TMEM175 could offer a potential therapeutic strategy for PD. Targeted studies to determine the precise effect of TMEM175 variants on lysosomal physiology and cell survival are needed. Beyond the potential role in maintaining lysosomal pH, there is very limited understanding of how an increase or decrease in TMEM175-mediated lysosomal potassium currents affect lysosome function and neuronal survival. TMEM175 knock-out cells display impaired regulation of lysosomal pH, decreased lysosomal catalytic activity (Cathepsin B and D), decreased glucocerebrosidase activity and impaired autophagy [75]. Most patients with TMEM175 variants carry one copy of the wild-type protein, so it is also important to assess these phenotypes in cells heterozygous for the variant.
While LRRK2 variants are implicated in both familial PD and in the risk for sporadic PD, how LRRK2 contributes to the clinical phenotypes remains unresolved. Studies at the level of the lysosome are shedding new light on underlying pathomechanisms, demonstrating that lysosomal rupture plays a role in the aggregation of pathological proteins including tau and alpha-synuclein and in NLRP3 inflammasome activation [76–82]. Interestingly, LRRK2 is recruited to damaged lysosomes, a process which is exacerbated by PD-related LRRK2 mutations [83]. However, the signaling mechanism that recruits LRRK2 onto a subset of damaged lysosomes, the functional consequence of this recruitment and its relevance in PD remain unclear. High- and super-resolution microscopy methodologies such as focused ion beam scanning electron microscopy [83], correlative light electron microscopy, three-dimensional super-resolution structured illumination microscopy, combined with dyes that report lysosomal pH and lysosomal hydrolytic function are being employed to better understand these processes. As a case-in-point, a study using such imaging techniques identified stable mitochondria-lysosome contact sites that facilitated bidirectional mitochondrial and lysosomal interaction in the cell [84]. Interestingly, PD patient-derived neurons carrying mutant GBA1 displayed prolonged mitochondria-lysosome contacts leading to mitochondrial dysfunction, which could be rescued by increasing glucocerebrosidase activity [85].
Establishing the non-neuronal contribution to lysosome-related neurodegeneration using human cellular models
Neuroinflammation is an important component of neurodegeneration, though it is still debated whether it represents a cause, consequence or both. Microglia, astrocytes and other non-neuronal cell types in the brain are involved in many essential physiological processes, including neurotrophic support, protection against pathogens, removal of cellular debris, physical and metabolic support, and transport of cargoes across the blood-brain barrier [86–88]. Several lysosomal genes implicated in neurodegenerative disorders are known to be crucial for the proper functioning of non-neuronal cell types. For example, in LSDs such as GD and NPC, disease-relevant phenotypes have been reported in astrocytes and microglia [89–91]. In GD, patient iPSC-derived astrocytes carrying GBA1 mutations demonstrated increased proliferation, severe cytoskeletal hypertrophy and impaired clearance of neuronally-released alpha-synuclein [89]. In a murine model of NPC, brains of NPC1 knock-out mice showed pronounced and early reactive gliosis, and microglia in NPC1 knock-out mice displayed enhanced phagocytosis and impaired turnover of myelin [90,91]. In another example, deficits in lysosomal degradation caused by the loss of NPC1 has been proposed to drive stimulator of interferon genes (STING) activation in neurons and microglia [92,93]. Of note, STING is a transmembrane protein which functions as part of the normal interferon-based immune response against DNA pathogens; dysregulation of this pathway is involved in various inflammatory and autoimmune diseases [92,93]. Overall, these findings suggest that pathogenicity of risk variants in lysosomal genes may involve both neuronal and non-neuronal cells.
Another lysosome-related disease risk gene that connects lysosomes, neuroinflammation and neurodegeneration is C9orf72. C9orf72 repeat expansions are the most common genetic link associated with ALS and FTD. C9orf72 is functionally linked to multiple cellular processes, including vesicle trafficking, lysosome homeostasis and mTORC1 signaling [31,94–96]. C9orf72 repeat expansions are bidirectionally transcribed into repetitive RNA species which can then be translated into dipeptide-repeat (DPR) proteins. Studies focusing on the pathogenic roles of C9orf72 repeat expansions have identified three distinct disease mechanisms that may not be mutually exclusive: i. Loss-of-function of the C9orf72 protein, ii. Toxic gain-of-function from DPR proteins, and iii. Toxic gain-of-function from RNA repeats [97–101]. Varying involvement of these mechanisms in different brain cell types may be partially responsible for the clinical and pathological heterogeneity observed in C9orf72-associated FTD and ALS. Evidence obtained from patients with C9orf72 mutations as well as from C9orf72 knock-out mice suggest that both develop a neuroinflammatory phenotype [29,102]. Mice with a selective deletion of C9orf72 in myeloid cells (primarily monocytes, tissue macrophages and dendritic cells) recapitulate phenotypes such as lymphoid hypertrophy and the systemic inflammation observed in mice with a generalized knock-out of C9orf72. These observed phenotypes were attributed to decreased lysosomal degradation of STING in myeloid cells and subsequent upregulation of the type I interferon response and inflammation [102]. In another study, microglia in C9orf72 knock-out mice were shown to exhibit an aberrant increase in lysosomes, a heightened inflammatory state and increased complement-mediated pruning of synaptic terminals in the motor cortex, thereby contributing to the cognitive deficits observed [103].
In glial cells, the interplay between lysosomal dysfunction and inflammatory mechanisms can exacerbate the aggregation of proteins associated with pathology. For example, astrocytes with the G2019S LRRK2 mutation exhibit a decreased capacity to take up and degrade fibrillary alpha-synuclein via the endo-lysosomal pathway [104]. In a GRN knock-out mouse model of NCL, single-nucleus RNA sequencing of thalamic samples showed that upon progranulin loss, microglia switch from a homeostatic to a disease-specific state that may lead to endo-lysosomal dysfunction and neurodegeneration. In fact, GRN knock-out microglia retain this phenotype in ex vivo cultures, and conditioned medium from GRN knock-out microglia was sufficient to promote TDP-43 granule formation, nuclear pore defects and cell death in neurons via the complement activation pathway [105]. Taken together, these findings highlight the importance of understanding the impact of lysosomal disease variants on glial function.
Human disease-relevant cellular models that sufficiently recapitulate neuron-glial interactions can be used to study lysosomal gene variants in physiological settings. In-vitro models based on immortalized cell lines have limited utility when characterizing neuron-glia interactions, neuron-neuron interactions, and spatially organized microenvironments. Although murine in-vivo models of neurodegenerative diseases help overcome some of these challenges, they often do not sufficiently recapitulate the analogous disease phenotype at the cellular and molecular level [106–108]. Human iPSC-derived neurons, microglia, macrophages and astrocytes from patient fibroblasts can be used to circumvent some of these problems, and their application has advanced the mechanistic understanding of PD, AD, ALS and FTD [109–117]. Recent studies of human 3D cerebral organoid models of disease, generated using hiPSC and CRISPR technologies, have recapitulated some of the hallmarks of AD and PD pathology, such as amyloid aggregation, hyperphosphorylated tau and the reduced number and complexity of dopaminergic neurons [118–122]. The use of such organoid models, including those incorporating neuron-glia interactions, will help address questions on how disease variants in glial cells affect neuronal physiology, stress, and survival, as well as the impact of neurons on glial biology. A major open question is whether, as with C9orf72 carriers, risk alleles in genes such as SNCA, GBA1, or TDP43 contribute to the disease process via loss-of-function, gain-of-function or both, and whether this is cell-type specific. The use of neurons and glia derived from CRISPR-edited human iPSCs can help to determine whether disease variants confer pathogenicity to different cell types via common or distinct pathways. Determining the relative contribution of neurons and glia in mediating toxicity associated with lysosomal risk variants is essential for the design of improved therapeutic strategies for lysosome-related neurodegenerative diseases.
Screens to identify genetic modifiers of disease
While establishing that a subset of neurodegenerative diseases is lysosome-related based on genetic and clinical evidence can narrow down the potential disease etiology, the factors that modify the risk conferred by pathological variants in lysosomal genes in a spatio-temporal manner are largely unknown. These potential genetic or epigenetic modifiers are largely undetected by conventional disease-association studies [123,124]. In fact, even in monogenic LSDs, genetic and epigenetic modifiers are thought to contribute towards the observed heterogeneity in associated phenotypes, age-of-onset and disease course [125,126]. Considering the much wider heterogeneity associated with sporadic forms of neurodegenerative diseases, it is likely that additional disease modifiers play a significant role in determining the ultimate impact of risk alleles in lysosomal genes on disease etiology [127,128].
The identification of genetic modifiers in sporadic forms of neurodegenerative diseases is challenging. Approaches employed have included family studies, large-scale association studies or genomic sequencing focusing on patients with broadly divergent phenotypes. Recent developments in large-scale functional genomic analyses allow performing large-scale CRISPR-based genetic screens to identify modifiers that impact lysosomal function or that modulate the activity of specific lysosomal enzymes. In addition to well-established CRISPR knock-out screens using conventional active Cas9, catalytically inactive versions of Cas9, termed dead Cas9 (dCas9), can be used in CRISPR interference (CRISPRi) screens to recruit transcriptional repressor domains to transcription start sites in order to repress gene transcription, or in CRISPR activation (CRISPRa) screens, in order to recruit transcriptional activator domains to induce the target gene [129]. The ability of CRISPRi and CRISPRa to inhibit and activate genes of interest will further expedite the mapping of directional interactions between disease variants and modifiers.
Nevertheless, the application of CRISPR-based genetic screens to identify novel regulators of neurodegenerative disease is only the first step. A key component of any CRISPR screen is the biological readout upon which the screening is based, such as endocytosis, organelle biogenesis, or protein function at the organelle level. In lysosome-related neurodegenerative diseases, screens aimed at identifying disease modifiers should include readouts that are disease-relevant and in-line with newly appreciated functions attributed to lysosomes in neurons and glia, such as neuronal axon maintenance, phagocytosis and exocytosis [130,131]. For example, in highly polarized cells like neurons, lysosomal trafficking along axons is considered to be an important mechanism for proper cellular functioning and survival [132–134]. Nevertheless, little is known about the regulation of axonal lysosome trafficking and its impact on neurodegeneration. There is growing interest in studying the processes that regulate spatial distribution of lysosomes in cells and its role in neurodegenerative disease. A recent study, for instance, showed that elevated cholesterol in lysosomes in NPC1 knock-out neurons leads to impaired axonal trafficking, while reducing cholesterol reverses the defect, reducing autophagic stress and neuronal death in these cells [135].
In addition to analyzing disease risk variants, another promising application of CRISPR-based genetic screens is at the intersection of aging-associated lysosomal dysfunction and disease. For example, lipofuscin is a fluorescent substance composed of oxidized macromolecules, thought to be the byproduct of incomplete lysosomal degradation. Once considered merely a bystander and biomarker of brain aging, recent evidence shows that these accumulations are intimately associated with neurodegeneration [136]. At autopsy, brains from patients with NCL demonstrate lipofuscinosis associated with substantial neuronal death [137]. In patients with PD, there is a correlation between increased lipofuscin aggregates and midbrain neurodegeneration [138–140]. A combination of disease-relevant cell models and targeted CRISPR screens may be leveraged to clarify both the etiology of lipofuscin in the aging brain and its potential contribution to pathogenicity [14].
One major aspect of CRISPR screens that needs careful consideration is the selection of the appropriate cellular model. At present, most genetic screens have been performed in non-neuronal cancer cell lines, which are not ideal for studying CNS-specific pathways. Though technically challenging, the use of hiPSCs has now enabled CRISPR screening in CNS cell types such as neurons, microglia and astrocytes. A recent screen performed to identify genetic interactors of C9orf72 further emphasizes the value of screening with the appropriate cellular model [141]. Since C9orf72 regulates macrophage function, the genome-wide CRISPR screen was performed in myeloid cells. This synthetic lethal screen in C9orf72 knock-out cells identified a strong genetic interaction between C9orf72 and FIS1. The findings suggest that FIS1 and C9orf72 work in parallel pathways to repress inflammation, and that FIS1 has a compensatory role in suppressing inflammation in the absence of C9orf72 [141]. In another demonstration of the merits of screening in CNS-relevant models, a genome-wide CRISPRi and CRISPRa screen using hiPSC-derived neurons resulted in the identification of the lysosomal protein prosaposin (PSAP) and other proteins in the same pathway (Cathepsin D and GM2 ganglioside activator) as modifiers of a stress-induced neuronal death process known as ferroptosis [142]. PSAP variants are associated with both LSDs and PD [143,144]. Interestingly, PSAP knock-out in HEK cells did not recapitulate phenotypes observed specifically in neurons, including lipofuscin accumulation, further highlighting the need to screen in hiPSC-derived CNS cellular models. Advances in high-throughput imaging and machine learning have made it possible to perform previously untractable image-based CRISPR-screens for subcellular phenotypes. As a case-in-point, an image-based whole-genome CRISPRi screen was recently performed to identify regulators of TFEB nuclear translocation [145] The TFEB CRISPRi screen identified regulators such as TGFBR1, TMEM184B and a phosphatase PPP1R1B, which is particularly interesting since phosphorylation status of TFEB is a critical determinant of its activity and subcellular location.
Isogenic hiPSC lines harboring disease variants of interest represent a powerful tool for recapitulating neuronal disease phenotypes such as alpha synuclein aggregation, lewy body/neurite-like pathology and oxidized dopamine accumulation [110,146–148]. CRISPR screens can now be designed using CNS-relevant cellular models for disease-relevant attributes such as microglial phagocytosis, neuronal lipofuscin levels, lysosomal and mitochondrial stress, and/or studying lysosomal variant functions such glucocerebrosidase activity and TMEM175-dependent pH regulation. The use of disease-relevant hiPSC in CRISPR screening platforms that employ disease-specific readouts holds tremendous potential for unravelling the function of disease variants that are associated with complex clinical phenotypes.
Next-generation translational research in lysosome-associated neurodegenerative diseases
Several lysosomal-targeted therapeutics are in clinical or late pre-clinical development for neurological LSDs and disorders associated with lysosomal variants such as ALS, FTD and PD. The strategic rationale for these therapies consist of targeting either the pathways associated with the genetic variants or general lysosomal mechanisms (i.e. mTORC1 inhibition, TRPML1 activation). There is a growing consensus that an effective strategy to tackle neurodegenerative diseases is to stratify patients based on shared genetic, cellular/biochemical, pathological and clinical phenotypes, so as to create sub-groups of patients that are most likely to respond to a specific therapeutic strategy. Exemplifying this is an ongoing Phase II trial in FTD-GRN patients (INFRONT-2i) using AL001, an antibody designed to elevate progranulin levels by blocking its degradation, resulting in normalization of CSF levels of lysosomal (LAMP1, Cathepsin D) and immune (C1QB) biomarkers of progranulin deficiencies. This normalization of disease-relevant biomarkers was shown to occur concomitantly with an elevation of CSF progranulin levelsii.
An important open question is whether patients carrying different lysosome-related PD risk genes share common or distinct disease mechanisms. Interestingly, in a PD patient cohort, the TMEM175 p.M393T risk variant, seen in over 20% of PD patients, was associated with reduced glucocerebrosidase activity in blood [8]. In iPSC-derived dopaminergic neurons from patients with LRRK2-associated PD, LRRK2, through its substrate Rab10, negatively regulated lysosomal glucocerebrosidase activity [149]. Unexpectedly, patients carrying the LRRK2 p.M1646T PD risk variant had increased glucocerebrosidase activity in peripheral blood [150]. Additional studies including analysis of post-mortem brain and CSF from PD patients carrying LRRK2 variants are needed to clarify whether LRRK2 impacts glucocerebrosidase activity, and if so, how. Nevertheless, glucocerebrosidase appears to play a role in PD pathogenesis beyond GBA1-PD, suggesting that glucocerebrosidase-based therapeutics and biomarkers may have a broader application.
Neurodegenerative diseases are complex, as exemplified by their regional and cell-subtype specific vulnerability [151,152]. Single-cell RNA sequencing and single-cell proteomics, which profile a multitude of individual cells, enable a focus on specific cell types in the brain. Several comprehensive single-cell omics studies performed in AD have led to novel insights into pathogenesis, including cell-type specific ApoE repression (in oligodendrocytes and astrocytes), ApoE upregulation (in microglia) and sex-specific transcriptional responses in multiple cell types [42,153,154]. A large-scale proteomic analysis performed in AD brains and cerebrospinal fluid, showed that changes in cellular energy metabolism occur early in the disease process, identifying novel potential therapeutic targets and biomarkers [155]. In AD, single-cell transcriptomics have been extended to genotype-specific analyses. For example, the effects of pathologic variants in TREM2 were assessed in different cell types in AD brains, leading to the identification of glial-type specific phenotypes in mutation carriers, including an IRF8-driven reactive phenotype in microglia and weakened metabolic coordination with neurons in astrocytes [156]. Similar comprehensive omics studies in patients with PD, FTD, ALS or AD carrying risk alleles in lysosomal genes can now be performed to address outstanding questions, including whether the molecular trajectories of these cases are similar to those seen in sporadic forms of the disease, and whether risk alleles such as GRN, C9orf72, GBA1, LRRK2 and TMEM175 lead to cell-specific phenotypes in patients.
Another pertinent issue common to lysosome-related neurodegenerative disease is the lack of functional biomarkers that accurately report brain target engagement and track the efficacy of therapeutic interventions. An emerging putative lysosomal biomarker that is relevant to neurodegenerative disease is BMP, di-docohexaenoyl (22:6) bis(monoacylglycerol)phosphate (di-22:6-BMP). BMP, is an endo-lysosomal inner membrane lipid that is increased in the urine of LRRK2 G2019S mutation carriers, and is reported to correlate with cognitive decline [157]. LRRK2 knock-out mice and nonhuman primates treated with LRRK2 kinase inhibitors have reduced levels of urinary di-22:6-BMP [158]. While this lipid might have potential as a biomarker, the biological significance of altered di-22:6-BMP levels is poorly understood. The accumulation of di-22:6-BMP is also seen with the loss of VPS13C [159]. VPS13C is a gene associated with early-onset PD, recently implicated in the transfer of lipids between the ER and endosomes/lysosomes. Interestingly, a panel of LRKK2 biomarkers has been developed for LRRK2-targeted clinical trials, including the lysosomal lipid BMP [160,161]. However, further work is needed to validate this lipid as a disease-relevant biomarker.
The complexity of the molecular interactions surrounding each potential therapeutic target is posing a tremendous challenge for identifying relevant biomarkers to support optimal assessment of clinical interventions. The recently unsuccessful Phase II trial with venglustat, a glucosylceramide synthase inhibitor (MOVES-PDiii) is a case-in-point. The aim of this trial was to evaluate the safety and efficacy of venglustat in PD patients who were heterozygous for a GBA1 mutation; however, the trial failed to meet its primary endpoint (change from baseline in the Unified Parkinson’s Disease Rating Scale Part II and III after one year). Despite the lack of cinical improvement, venglustat was reported to show target engagement at the molecular level, resulting in decreased levels of glucosylceramide in patient plasma and cerebrospinal fluid of up to 75%.
The discrepancy between robust target engagement and the lack of stablilization or improvement of symptoms recapitulate the translational gap between preclinical research and clinical development. Among the lessons learned are the following: First, there is a crucial need for a better mechanistic understanding of the key players involved in the pathogenic process. Clearly, glucocerebrosidase interacts within a larger biological framework much of which still remains to be elucidated. Second, great care must be exercised when selecting a biomarker. It is unclear whether the lowering of CSF glucosylceramide levels was a target engagement proxy for the reduction of glucosylceramide levels in the lysosomal membrane of brain cells, since glucosylceramide is present in other cellular compartments. In addition to measuring CSF levels of glucosylceramide, investigation of lysosomal glucosylceramide levels in PBMCs using the Lyso-tag approach or using lysosomal-specific glucocerebrosidase substrates are warranted [162]. Lastly, to accurately report targeting of lysosomes in the brain for glucocerebrosidase-augmenting therapies such as gene therapy or chaperones, the development of lysosome-specific PET ligands will be critical. The existence of ongoing trials, including the Phase I trial for GBA1 gene therapy PR001 (PROPELiv) and the Phase II trial for the glucocerebrosidase chaperone Ambroxolv, underscores the urgent need for disease-relevant functional biomarkers that can drive the transition from the bench to the clinic.
Concluding Remarks
A common theme that emerges from studying disorders of the lysosome is that multiple processes likely contribute to the pathophysiology observed. This is mirrored by the heterogeneous clinical manifestations of neurodegenerative disorders such as AD, PD, and the LSDs. Indeed, the complexity of lysosome function across many cell types poses a major challenge in translational research: the road from preclinical data to new therapies is fraught with obstacles to be overcome (see Outstanding Questions). Two aspects of these challenges, we would argue, require particular attention. First, it should be emphasized that lysosomes play important roles beyond cellular waste disposal, and are vital for a wide range of cellular functions. Second, it is important to appreciate that in most cases, neurodegeneration is an outcome of physiological processes gone awry in multiple cell types in the brain, rather than in neurons alone.
Outstanding questions.
What alterations in lysosome physiology are caused by the risk alleles identified, and how do they relate to disease etiology? For example, while in Gaucher disease there is a clear accumulation of lipid substrates, this is not evident in GBA1 heterozygotes, suggesting that substrate accumulation is unlikely to be the basis of pathology in GBA1-PD. What then mediates the risk for GBA1? Furthermore, how does alpha-synuclein alter lysosomal function in normal and diseased states?
What is the contribution of different cell types to the disease pathology? Specifically, what is the contribution of neurons vs glial vs peripheral cells to neuropathology in neurodegenerative diseases? Many lysosomal genes are widely expressed in different cell and tissue types. Do pathological variants in these genes confer their pathogenicity through a specific cell type or is it a cumulative effect on multiple cells and organ systems?
What makes different sets of neurons more vulnerable than others in neurodegenerative disorders? Cell-type and regional-specific vulnerability is well established in most neurodegenerative disorders and corresponds to clinical manifestations, although the pathophysiological basis is not well delineated. For example, why are dopaminergic neurons from the substantia nigra pars compacta highly vulnerable in PD, when similar dopaminergic neurons in the ventral tegmental area are spared? What makes the pyramidal neurons particularly vulnerable to disturbances in proteostasis?
Are there converging molecular mechanisms across neurodegenerative diseases, and if so, could these pathways explain the overlap in pathology and clinical manifestations? For example, one feature that might suggest a shared mechanism could be lipofuscin accumulation. Do the various implicated lysosomal genes somehow impact lipofuscin levels, ultimately resulting in pathology?
Major efforts to delineate the specific role of neurodegeneration-related lysosomal genes and aging mechanisms should be undertaken using relevant cell types at the level of the organelle. Recent advancements in deep omics analyses should be leveraged to analyze post-mortem brain samples from genotype-curated cohorts to catalogue cell-specific signatures. Novel insights and hypotheses from such analyses should then be tested and validated in patient hiPSC-derived models using CRISPR-Cas9 techniques. Such comprehensive analyses have the potential to uncover novel pathways and expand the knowledge of disease processes (Figure 2).
Figure 2: Blueprint for a better understanding of lysosomal dysfunction across neurodegenerative disorders.
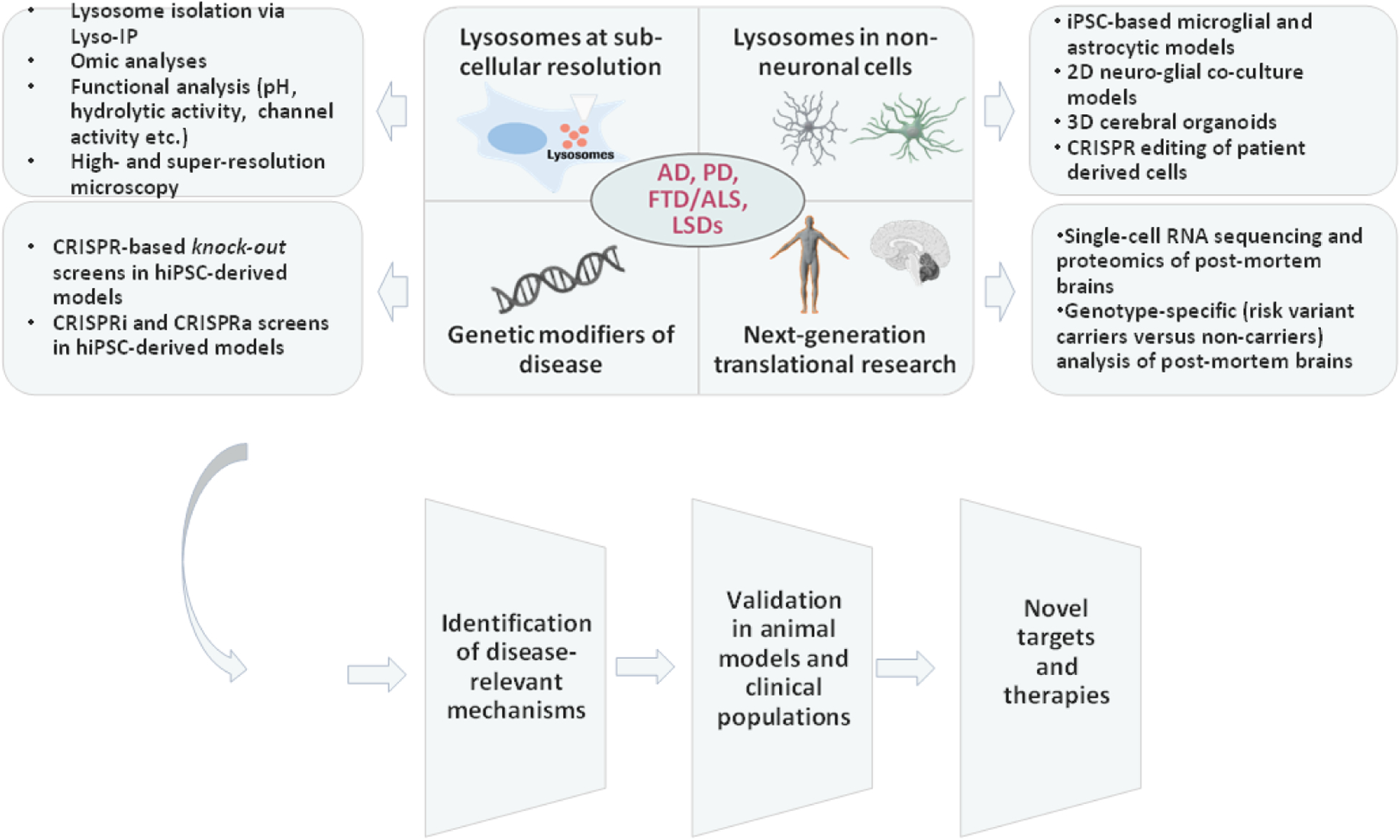
In-depth analysis of lysosomal roles in neurodegenerative diseases such as AD, PD,ALS, FTD and LSDs should include the following approaches: 1. Studying lysosomes in physiology and disease at the subcellular resolution, 2. Assessing the impact of lysosomal disease risk variants on non-neuronal cells and its relation to pathology, 3. Screening for genetic modifiers of disease risk conferred by lysosomal variants, and 4. Next-generation translational research involving analysis of genotype-specific post-mortem brains from patients. Such an approach will help identify disease-relevant mechanisms that can be investigated for development of novel disease-modifying therapies. Illustrations of human brain and DNA double-helix were exported from BioRender.com.
Finally, it should be acknowledged that lysosomal genes and the pathways they impact represent only one element of the underlying pathophysiological processes in neurodegenerative diseases. Trans-disciplinary efforts will be needed to synthesize the existing wealth of information and to leverage mechanistic understanding for guiding clinical development. Hopefully, these efforts will support a transition from the current focus on symptomatic treatments to the discovery of therapeutic agents that can truly alter the course of these complex disorders.
Highlights.
Genetic and clinical studies have implicated the lysosomal pathway as a key contributor to pathology in several neurodegenerative diseases, including, AD, PD and FTD/ALS.
Lysosome-related disease risk variants in neurodegeneration affect different aspects of the lysosomal pathway (e.g. pH regulation, enzymatic activity and levels, cargo-delivery), potentially contributing to the differences in pathology observed in different neurodegenerative diseases.
Recent studies have shown that lysosomes are sophisticated organelles that regulate and interact with key disease-relevant pathways such as nutrient-sensing, neuroinflammation, and ferroptosis.
Human disease-relevant iPSC models and advanced methodologies such as CRISPR, single-cell omics, and deep-sequencing are leading to novel insights into lysosome-related mechanisms of disease and the identification of new therapeutic targets.
Acknowledgements
We would like to thank Max Iglesias for designing Figure 1 and for his inputs on Figure 2; Dr. Luka Kulic for critical inputs in shaping the manuscript and the Roche postdoctoral fellowship program for funding V.U. E.S. and Y.C are supported by the Intramural Research Programs of the National Human Genome Institute and the National Institutes of Health.
Declaration of interests
V.U. and R.J. are employed by F. Hoffmann-La Roche.
The Sidransky lab has received funding from the Michael J Fox Foundation, the Aligning Science Across Parkinson’s Initiative and from F.Hoffmann-La Roche Ltd. under a Cooperative Research and Development Agreement with the NHGRI and NCATS.
Glossary
- Alpha-synuclein
A predominantly neuronal protein that is linked genetically and neuropathologically to Parkinson’s disease.
- Alzheimer’s disease (AD)
Common neurodegenerative disease characterized by amyloid plaques and tau tangles in brain. In AD, memory loss is often the initial symptom, followed by a range of cognitive and behavioral impairments.
- Amyotrophic lateral sclerosis (ALS)
A progressive neurodegenerative disorder affecting nerves in the brain and spinal cord, leading to loss of muscle control.
- APP processing
A sequential protein cleavage process where amyloid precursor protein (APP) is cleaved by specific proteases to produce amyloid-beta (Aβ), a peptide that is the main component of AD-related amyloid plaques.
- Assembloids
Organoid generated by spatially organizing multiple cell types.
- Autophagy pathway
A physiological intracellular pathway wherein damaged or unnecessary cellular components are delivered to lysosomes for degradation.
- Cerebral organoid
3D brain models derived from pluripotent stem cells that show structural organization reminiscent of the brain. Their organization and composition can be modulated by exogenous patterning factors.
- Correlative light electron microscopy
A combination of fluorescence microscopy with high-resolution electron microscopy that allows study of cellular events (reported by multi-color labels) at ultrastructural resolution.
- CRISPR technology
A technology characterized by DNA elements termed CRISPRs (clustered regularly interspaced short palindromic repeats) and CRISPR-associated (Cas) proteins. It has two components, a Cas protein and a single guide RNA (sgRNA) that targets the Cas endonucleases to genes of interest in a sequence-specific manner. Depending on the Cas protein, it can induce gene knock-out, inactivation or activation.
- Deep omics
Deep omics refers to the application of deep neural networks and machine-learning for multi-level and multi-factorial analysis of ‘omic’ data such as those from proteomic, lipidomic, and transcriptomic analyses.
- Epigenetic modifiers
Epigenetic modifiers refer to processes (or factors regulating these processes) such as DNA methylation and histone modification. Epigenetic modifiers change the risk for a certain disease by changing gene expresion without altering the underlying DNA sequence.
- Endosomal sorting complexes required for transport (ESCRT)
ESCRT refers to a protein machinery composed of cytosolic protein complexes, known as ESCRT-0, I, II and III. The complexes are involved in membrane remodeling resulting in alterations such as membrane bending and budding. ESCRT complexes are also known to be recruited to damaged lysosomal membrane for its repair.
- Focused ion beam scanning electron microscopy
A technique which combines serial etching of a resin block (tissue sample) by focused ion beam with the scanning of the exposed surface in a repetitive manner, resulting in 3D-representation of ultrastructural features.
- Frontotemporal dementia (FTD)
A group of disorders caused by progressive nerve cell loss in the brain’s frontal or temporal lobe, associated with changes in personality, behavior and language.
- Gaucher disease (GD)
Lysosomal storage disorder resulting from the deficiency of the enzyme glucocerebrosidase, associated with both neuronopathic and non-neuronopathic phenotypes.
- Glucocerebrosidase
Lysosomal enzyme that cleaves the substrates glucocerebroside and glucosylsphingosine.
- Human induced pluripotent stem cells (hiPSC)
Pluripotent cells derived from somatic cells such as fibroblasts and peripheral blood mononuclear cells that can be differentiated into different cell types.
- Lipofuscin
Electron-dense autofluorescent material that accumulates over time predominantly in lysosomes of post-mitotic cells.
- Lysosomal storage disorders (LSDs)
Group of inherited metabolic disorders that affect lysosomal function, causing accumulation of toxic materials in various cells. LSDs may affect different organs, including the skeleton, skin, liver, heart, and central nervous system.
- mTORC1 signaling
A signaling network mediated by protein complex known as mechanistic target of rapamycin complex 1 (mTORC1) that integrates intracellular and extra-cellular growth signals with metabolic processes in the cell.
- Neuronal ceroid lipofuscinoses (NCL)
A group of LSDs comprising of 14 distinct forms that together are the most common degenerative brain diseases in children.
- Niemann Pick C (NPC)
A rare, progressive, neurodegenerative disease caused by autosomal recessive mutations in either NPC1 or NPC2 gene. The disease is associated with neurovisceral symptoms resulting from lysosomal dysfunction and lipid accumulation.
- Parkinson’s disease (PD)
A progressive neurodegenerative disorder characterized by loss of predominantly dopaminergic neurons in the brain, leading to motor and non-motor symptoms.
- Phagocytosis
Cellular process for uptake and elimination of large particles, including microorganisms, foreign substances, and apoptotic cells.
- SLC38A9
A lysosomal membrane protein belonging to the solute carrier (SLC) group of membrane transport proteins. It transports many amino acids out of the lysosomes into the cytosol in an arginine-dependent manner.
- Tau
Microtubule-associated proteins that are predominantly expressed in neurons and dynamically regulate key processes such as axonal transport and neurite outgrowth.
- Three-dimensional super-resolution structured illumination microscopy
A 3D imaging technique that increases the resolution of conventional optical microscope to about 100 nm lateral and 250 nm axial by combining it with structure illumination microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
References
- 1.Settembre C et al. (2013) Signals for the lysosome: a control center for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 14, 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawrence RE and Zoncu R (2019) The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol 21, 133–142 [DOI] [PubMed] [Google Scholar]
- 3.Tsunemi T et al. (2019) Increased lysosomal exocytosis induced by lysosomal Ca2+ channel agonists protects human dopaminergic neurons from α-Synuclein toxicity. J. Neurosci 39, 5760–5772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campagno KE and Mitchell CH (2021) The P2X7 receptor in microglial cells modulates the endolysosomal axis, autophagy, and phagocytosis. Front. Cell. Neurosci 15, 645244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parenti G et al. (2021) The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med 13, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallings RL et al. (2019) Lysosomal dysfunction at the centre of Parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci. 42, 899–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alcalay RN et al. (2019) SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov. Disord 34, 526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krohn L et al. (2020) Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann. Neurol 87, 139–153 [DOI] [PubMed] [Google Scholar]
- 9.van Veen S et al. (2020) ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578, 419–424 [DOI] [PubMed] [Google Scholar]
- 10.Renton AE et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker M et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 [DOI] [PubMed] [Google Scholar]
- 12.Van Deerlin VM et al. (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet 42, 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Acker ZP et al. (2019) Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol. Neurodegener 14, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bourdenx M et al. (2021) Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 184, 2696–2714.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Q et al. (2020) MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy 16, 641–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finkbeiner S (2020) The autophagy lysosomal pathway and neurodegeneration. Cold Spring Harb. Perspect. Biol 12, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung C et al. (2021) SORL1 deficiency in human excitatory neurons causes APP-dependent defects in the endolysosome-autophagy network. Cell Rep. 35, 109259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH et al. (2015) Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fedeli C et al. (2019) PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 15, 2044–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulland TK et al. (2017) TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu H et al. (2021) In vitro amplification of pathogenic tau conserves disease-specific bioactive characteristics. Acta Neuropathol. 141, 193–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi I et al. (2020) Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun 11, 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Domert J et al. (2014) Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis 65, 82–92 [DOI] [PubMed] [Google Scholar]
- 24.Cho MH et al. (2014) Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10, 1761–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minakaki G et al. (2018) Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14, 98–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Y et al. (2011) Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci. Signal 4, ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ge W et al. (2015) The Roles of Lysosomes in inflammation and autoimmune diseases. Int. Rev. Immunol 34, 415–431 [DOI] [PubMed] [Google Scholar]
- 28.Amick J et al. (2016) C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell 72, 3040–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Rourke JG et al. (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mccauley ME and Baloh RH (2019) Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 137, 715–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M et al. (2020) C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell 19, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Root J et al. (2021) Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis 154, 105360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeJesus-Hernandez M et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka Y et al. (2013) Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 250, 8–19 [DOI] [PubMed] [Google Scholar]
- 35.Bossù P et al. (2011) Loss of function mutations in the progranulin gene are related to pro-inflammatory cytokine dysregulation in frontotemporal lobar degeneration patients. J. Neuroinflammation 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galimberti D et al. (2015) Inflammatory molecules in frontotemporal dementia: cerebrospinal fluid signature of progranulin mutation carriers. Brain. Behav. Immun 49, 182–187 [DOI] [PubMed] [Google Scholar]
- 37.Martens LH et al. (2012) Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest 122, 3955–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward ME et al. (2017) Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med 9, 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonam SR et al. (2019) Lysosomes as a therapeutic target. Nat. Rev. Drug Discov 18, 923–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiernan MC et al. (2021) Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol 17, 104–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lodato MA et al. (2018) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 559, 555–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grubman A et al. (2019) A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 22, 2087–2097 [DOI] [PubMed] [Google Scholar]
- 43.Hook PW et al. (2018) Single-cell RNA-Seq of mouse dopaminergic neurons informs candidate gene selection for sporadic Parkinson disease. Am. J. Hum. Genet 102, 427–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffman JM et al. (2017) Proteomics and metabolomics in ageing research: from biomarkers to systems biology. Essays Biochem. 61, 379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goker-Alpan O et al. (2006) Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 67, [DOI] [PubMed] [Google Scholar]
- 46.Gan-Or Z et al. (2015) GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann. Clin. Transl. Neurol 2, 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanz J and Saftig P (2016) Parkinson’s disease: acid-glucocerebrosidase activity and alpha-synuclein clearance. J. Neurochem 139, 198–215 [DOI] [PubMed] [Google Scholar]
- 48.Aflaki E et al. (2017) The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron 93, 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Do J et al. (2019) Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener 14, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aflaki E et al. (2016) A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J. Neurosci 36, 7441–7452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schöndorf DC et al. (2014) IPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun 5, 4028. [DOI] [PubMed] [Google Scholar]
- 52.Gegg ME et al. (2015) No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord 30, 1085–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gündner AL et al. (2019) Path mediation analysis reveals GBA impacts Lewy body disease status by increasing α-synuclein levels. Neurobiol. Dis 121, 205–213 [DOI] [PubMed] [Google Scholar]
- 54.Mazzulli JR et al. (2011) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazzulli JR et al. (2016) Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci 36, 7693–7706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klumperman J and Raposo G (2014) The complex ultrastructure of the endolysosomal system. Cold Spring Harb. Perspect. Biol 6, a016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abu-Remaileh M et al. (2017) Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wyant GA et al. (2017) mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–647.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis OB et al. (2021) NPC1-mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in Niemann-Pick Type C. Dev. Cell 56, 260–276.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh J et al. (2020) Systematic comparison of strategies for the enrichment of lysosomes by data independent acquisition. J. Proteome Res 19, 371–381 [DOI] [PubMed] [Google Scholar]
- 61.Wyant GA et al. (2018) NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Logan T et al. (2021) Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 184, 4651–4668.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evers BM et al. (2017) Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Rep. 20, 2565–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holler CJ et al. (2017) Intracellular proteolysis of progranulin generates stable, lysosomal granulins that are haploinsufficient in patients with frontotemporal dementia caused by GRN mutations. eNeuro 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X et al. (2017) The interaction between progranulin and prosaposin is mediated by granulins and the linker region between saposin B and C. J. Neurochem 143, 236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dong XP et al. (2008) The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cang C et al. (2013) mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 152, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W et al. (2015) Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. U. S. A 112, E1373–E1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li G et al. (2019) Control of lysosomal TRPML1 channel activity and exosome release by acid ceramidase in mouse podocytes. Am. J. Physiol. - Cell Physiol 317, C481–C494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chin MY et al. (2021) Genetically encoded, pH-sensitive mTFP1 biosensor for probing lysosomal pH. ACS Sensors 6, 2168–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jinn S et al. (2019) Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum. Mol. Genet 28, 3244–3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chia R et al. (2021) Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet 53, 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cang C et al. (2015) TMEM175 is an organelle K+ channel regulating lysosomal function. Cell 162, 1101–1112 [DOI] [PubMed] [Google Scholar]
- 74.Wie J et al. (2021) A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 591, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jinn S et al. (2017) TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A 114, 2389–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Freeman D et al. (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8, e62143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang P et al. (2017) Impaired endo-lysosomal membrane integrity accelerates the seeding progression of α-synuclein aggregates. Sci. Rep 7, 7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen JJ et al. (2019) Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J. Biol. Chem 294, 18952–18966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.See SK et al. (2021) PIKfyve inhibition blocks endolysosomal escape of α-synuclein fibrils and spread of α-synuclein aggregation. Bioarxiv DOI: 10.1101/2021.01.21.427704 [DOI] [Google Scholar]
- 80.Polanco JC et al. (2021) Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta Neuropathol. 141, 235–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Okada M et al. (2014) The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J. Biol. Chem 289, 32926–32936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Manna S et al. (2018) Immunomodulation of the NLRP3 inflammasome through structure-based activator design and functional regulation via lysosomal rupture. ACS Cent. Sci 4, 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonet-Ponce L et al. (2020) LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv 6, eabb2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wong YC et al. (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via Rab7 GTP hydrolysis. Nature 554, 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim S et al. (2021) Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease. Nat. Commun 12, 1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Obermeier B et al. (2016) The blood – brain barrier. Handb. Clin. Neurol 133, 39–59 [DOI] [PubMed] [Google Scholar]
- 87.Colonna M and Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Physiol 35, 441–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verkhratsky A (2020) Untangling complexities of glial-neuronal communications : astroglial metabolic cascades orchestrate tonic inhibition in the thalamus. Neuron 108, 585–587 [DOI] [PubMed] [Google Scholar]
- 89.Aflaki E et al. (2020) A characterization of Gaucher iPS-derived astrocytes: potential implications for Parkinson disease. Neurobiol. Dis 134, 10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pressey SNR et al. (2012) Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis 45, 1086–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Colombo A et al. (2021) Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun 12, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gonugunta VK et al. (2017) Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. 21, 3234–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chu TT et al. (2021) Tonic prime-boost of STING signalling mediates Niemann–Pick disease type C. Nature DOI: 10.1038/s41586-021-03762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aoki Y et al. (2017) C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897 [DOI] [PubMed] [Google Scholar]
- 95.Shi Y et al. (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corrionero A and Horvitz HR (2018) A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr. Biol 28, 1522–1535.e5 [DOI] [PubMed] [Google Scholar]
- 97.Burberry A et al. (2016) Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med 8, 347ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mori K et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1339 [DOI] [PubMed] [Google Scholar]
- 99.Ash PEA et al. (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Donnelly CJ et al. (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhu Q et al. (2020) Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci 23, 615–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McCauley ME et al. (2020) C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 585, 96–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lall D et al. (2021) C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 109, 2275–2291.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Streubel-Gallasch L et al. (2021) Parkinson’s disease–associated LRRK2 interferes with astrocyte-mediated alpha-synuclein clearance. Mol. Neurobiol DOI: 10.1007/s12035-021-02327-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang J et al. (2020) Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature 588, 459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.LaFerla FM and Green KN (2012) Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med 2, a006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Volta M and Melrose H (2017) LRRK2 mouse models : dissecting the behavior, striatal neurochemistry and neurophysiology of PD pathogenesis. Biochem. Soc. Trans 45, 113–122 [DOI] [PubMed] [Google Scholar]
- 108.Dawson TM et al. (2018) Animal models of neurodegenerative diseases. Nat. Neurosci 21, 1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fernandes HJR et al. (2016) ER stress and autophagic perturbations lead to elevated extracellular a-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports 6, 342–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Burbulla LF et al. (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’ s disease. Science 357, 1255–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Verheyen A et al. (2018) Genetically engineered iPSC-derived FTDP-17 MAPT neurons display mutation-specific neurodegenerative and neurodevelopmental phenotypes. Stem Cell Reports 11, 363–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.di Domenico A et al. (2019) Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Reports 12, 213–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lang C et al. (2019) Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies HDAC4 as a regulator of Parkinson cell phenotypes. Cell Stem Cell 24, 93–106.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Okarmus J et al. (2020) Lysosomal perturbations in human dopaminergic neurons derived from induced pluripotent stem cells with PARK2 mutation. Sci. Rep 10, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gowrishankar S et al. (2021) Overlapping roles of JIP3 and JIP4 in promoting axonal transport of lysosomes in human iPSC-derived neurons. Mol Biol Cell. 32, 1094–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haenseler W et al. (2017) Excess α-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia-Reitboeck P et al. (2018) Human induced pluripotent stem cell-derived microglia-ike cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep. 24, 2300–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raja WK et al. (2016) Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One 11, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smits LM et al. (2019) Modeling Parkinson’s disease in midbrain-like organoids. npj Parkinson’s Disease 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim H et al. (2019) Modeling G2019S-LRRK2 sporadic parkinson’s disease in 3D midbrain organoids. Stem Cell Reports 12, 518–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao J et al. (2020) APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun 11, 5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wulansari N et al. (2021) Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci. Adv 7, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Do C et al. (2017) Genetic-epigenetic interactions in cis: A major focus in the post-GWAS era. Genome Biol. 18, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Alcalà-Vida R et al. (2021) Epigenetic mechanisms underlying enhancer modulation of neuronal identity, neuronal activity and neurodegeneration. Neurobiol. Dis 147, 105155. [DOI] [PubMed] [Google Scholar]
- 125.Hassan S et al. (2017) The role of epigenetics in lysosomal storage disorders: uncharted territory. Mol. Genet. Metab 122, 10–18 [DOI] [PubMed] [Google Scholar]
- 126.Platt FM et al. (2018) Lysosomal storage diseases. Nat. Rev. Disease Primers 4, 27. [DOI] [PubMed] [Google Scholar]
- 127.van Heesbeen HJ and Smidt MP (2019) Entanglement of genetics and epigenetics in Parkinson’s disease. Front. Neurosci 13, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jain N and Chen-Plotkin AS (2018) Genetic modifiers in neurodegeneration. Curr. Genet. Med. Rep 6, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gilbert LA et al. (2014) Resource genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ferguson SM (2019) Neuronal lysosomes. Neurosci. Lett 697, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kreher C et al. (2021) Lysosomal functions in glia associated with neurodegeneration. Biomolecules 11, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gowrishankar S et al. (2017) Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol 216, 3291–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Farías GG et al. (2017) BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. U. S. A 114, E2955–E2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Farfel-Becker T et al. (2019) Neuronal soma-derived degradative lysosomes are continuously delivered to distal axons to maintain local degradation capacity. Cell Rep. 28, 51–64.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Roney JC et al. (2021) Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev. Cell 56, 1452–1468.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moreno-García A et al. (2018) An overview of the role of lipofuscin in age-related neurodegeneration. Front. Neurosci 12, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Geraets RD et al. (2016) Moving towards effective therapeutic strategies for Neuronal Ceroid Lipofuscinosis. Orphanet J. Rare Dis 11, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Braak E et al. (2001) α-Synuclein immunopositive Parkinson’s disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathol. 101, 195–201 [DOI] [PubMed] [Google Scholar]
- 139.Lv Z et al. (2011) Increased iron levels correlate with the selective nigral dopaminergic neuron degeneration in Parkinson’s disease. J. Neural Transm 118, 361–369 [DOI] [PubMed] [Google Scholar]
- 140.Wellings TP et al. (2017) Altered neurofilament protein expression in the lateral vestibular nucleus in Parkinson’s disease. Exp. Brain Res 235, 3695–3708 [DOI] [PubMed] [Google Scholar]
- 141.Chai N et al. (2020) Genome-wide synthetic lethal CRISPR screen identifies FIS1 as a genetic interactor of ALS-linked C9ORF72. Brain Res. 1728, 146601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tian R et al. (2021) Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci 24, 1020–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Diaz-font SA et al. (2005) A mutation within the saposin D domain in a Gaucher disease patient with normal glucocerebrosidase activity. Hum Genet DOI: 10.1007/s00439-005-1288-x [DOI] [PubMed] [Google Scholar]
- 144.Oji Y et al. (2020) Variants in saposin D domain of prosaposin gene linked to Parkinson ’ s disease. Brain 143, 1190–1205 [DOI] [PubMed] [Google Scholar]
- 145.Kanfer G et al. (2021) Image-based pooled whole-genome CRISPRi screening for subcellular phenotypes. J. Cell Biol 220, e202006180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ryan SD et al. (2013) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155, 1351–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Reinhardt P et al. (2013) Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367 [DOI] [PubMed] [Google Scholar]
- 148.Nickels SL et al. (2019) Impaired serine metabolism complements LRRK2-G2019S pathogenicity in PD patients. Park. Relat. Disord 67, 48–55 [DOI] [PubMed] [Google Scholar]
- 149.Ysselstein D et al. (2019) LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun 10, 5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sosero YL et al. (2021) LRRK2 p.M1646T is associated with glucocerebrosidase activity and with Parkinson’s disease. Neurobiol. Aging 103, 142.e1–142.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Saxena S and Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71, 35–48 [DOI] [PubMed] [Google Scholar]
- 152.Pandya VA and Patani R (2021) Region-specific vulnerability in neurodegeneration: lessons from normal ageing. Ageing Res. Rev 67, 101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Del-aguila JL et al. (2019) A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimer’s Res. Ther 11, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mathys H et al. (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nat. Neurosci 570, 2087–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Johnson ECB et al. (2020) Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26, 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhou Y et al. (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and - independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Alcalay RN et al. (2020) Higher urine bis(monoacylglycerol)phosphate levels in LRRK2 G2019S mutation carriers: implications for therapeutic development. Mov. Disord 35, 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fuji RN et al. (2015) Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med 7, 273ra15. [DOI] [PubMed] [Google Scholar]
- 159.Hancock-Cerutti W et al. (2021) ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA- dependent STING signaling. Bioarxiv DOI: 10.1101/2021.06.08.447593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kelly K and West AB (2020) Pharmacodynamic biomarkers for emerging LRRK2 therapeutics. Front. Neurosci 14, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang S et al. (2020) Exosome markers of LRRK2 kinase inhibition. npj Parkinson’s Disease 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ashmus RA et al. (2018) Fluorescence-quenched substrates for quantitative live cell imaging of glucocerebrosidase activity. Methods Enzymol. 598, 199–215 [DOI] [PubMed] [Google Scholar]
- 163.Sidransky E et al. (2009) Multi-center analysis of glucocerebrosidase mutations in Parkinson disease. N. Engl. J. Med 361, 1651–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Alcalay RN et al. (2014) Comparison of Parkinson risk in Ashkenazi jewish Gaucher patients and GBA heterozygotes. JAMA Neurol. 71, 752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Healy DG et al. (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zimprich A et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 [DOI] [PubMed] [Google Scholar]
- 167.Ramirez A et al. (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet 38, 1184–1191 [DOI] [PubMed] [Google Scholar]
- 168.Martin S et al. (2020) Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 139, 1001–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kumar N et al. (2018) VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol 217, 3625–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Darvish H et al. (2019) Identification of a large homozygous VPS13C deletion in a patient with early-onset parkinsonism. Mov. Disord 33, 1968–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bussi C et al. (2018) Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J. Cell Sci 131, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rosborough K et al. (2017) α-Synuclein and parkinsonism: updates and future perspectives. Curr. Neurol. Neurosci. Rep 17, [DOI] [PubMed] [Google Scholar]
- 173.Su MY et al. (2021) Structural basis for the ARF GAP activity and specificity of the C9orf72 complex. Nat. Commun 12, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tang D et al. (2020) Cryo-EM structure of C9ORF72–SMCR8–WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc. Natl. Acad. Sci. U. S. A 117, 9876–9883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lindquist SG et al. (2008) Frontotemporal dementia linked to chromosome 3 (FTD-3) - current concepts and the detection of a previously unknown branch of the Danish FTD-3 family. Eur. J. Neurol 15, 667–670 [DOI] [PubMed] [Google Scholar]
- 176.Skibinski G et al. (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet 37, 806–808 [DOI] [PubMed] [Google Scholar]
- 177.Stagi Massimiliano et al. (2014) Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol. Cell. Neurosci 61, 226–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Schwenk BM et al. (2014) The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 33, 450–467 [DOI] [PMC free article] [PubMed] [Google Scholar]