

Abstract
The etiology of idiopathic Parkinson’s disease (iPD) is multifactorial, and both genetics and environmental exposures are risk factors. While mutations in the LRRK2 gene that are associated with increased kinase activity are the most common cause of autosomal dominant PD, the role of LRRK2 in iPD, independent of mutations, remains uncertain. This review discusses how the architecture of LRRK2 influences kinase activation and how enhanced LRRK2 substrate phosphorylation may contribute to pathogenesis. We describe how oxidative stress and endolysosomal dysfunction, both of which occur in iPD, can activate non-mutated LRRK2 to a similar degree as pathogenic mutations. Similarly, environmental toxicants that are linked epidemiologically to iPD risk, can also activate LRRK2. In aggregate, current evidence suggests LRRK2 may play an important role in iPD.
Keywords: Autophagy-lysosomal pathway, endogenous protein expression, environmental toxicants, kinase, oxidative stress
LRRK2: A potential link between genetic and idiopathic forms of Parkinson’s disease
Parkinson’s disease (PD) is a complex, multifactorial, progressive disease characterized by motor and non-motor neurologic symptoms. The motor symptoms are driven, in part, by the loss of dopaminergic neurons in the substantia nigra (SN) and, in most cases, are accompanied by proteinaceous inclusions, referred to as Lewy bodies and neurites. The age of diagnosis, clinical manifestation, disease progression and neuropathology vary widely between individuals. Approximately 10% of PD cases are attributed to inherited genetic mutations, while the rest are considered idiopathic or sporadic. Mutations in risk genes elevate the chances of developing PD; however, the phenotypic penetrance (see Glossary) of risk genes is incomplete. Likelihood of developing idiopathic PD (iPD) depends on several factors including age, the presence of risk alleles, and environmental factors (e.g. toxicants and lifestyle) [1–4]. The G2019S mutation in the Leucine-rich repeat kinase-2 (LRRK2) protein is the most common pathogenic mutation, accounting for 1–6% of sporadic and 3–19% of familial PD [5]. Additionally, common variants in the LRRK2 locus are known risk factors for iPD. Indeed, it has been argued that LRRK2 provides a link between genetic and idiopathic forms of PD [6].
Despite the fact that LRRK2 is a low-abundance protein in neurons, its kinase activity is significantly increased in SN dopamine (DA) neurons and microglia of iPD patients [7]. Sustained elevation in LRRK2 kinase activity likely contributes to the pathogenesis of iPD: using LRRK2 kinase inhibitors or LRRK2 silencing, several investigators have shown prevention of brain pathologies in rodent models of PD [8–11]. While there is a growing consensus that LRRK2 kinase activity is increased in iPD, how it gets activated and its roles in disease pathogenesis are still not clear.
Clinically, LRRK2 mutation carriers who develop PD are largely indistinguishable from late-onset iPD. Despite the clinical similarity, the neuropathology is extremely heterogenous in LRRK2 mutation carriers, even among family members with the same mutation [12]. It is clear that LRRK2 mutation carriers exhibit profound nigrostriatal and locus coeruleus degeneration. However, the presence of Lewy pathology is variable: while most G2019S mutation carriers exhibit Lewy pathology, a minority of these carriers do not [13]. This variability, even among those with the same mutation, suggests that α-synuclein pathology may not be necessary for the typical clinical manifestations of the disease, and disease progression may occur through divergent mechanisms. In this context, it seems likely that the manifestations and progression of PD are influenced by gene-environment interactions – in which environmental factors play a role in both the development of clinical symptoms and the type of neuropathology observed at autopsy. This review will discuss the role LRRK2 plays in iPD, the mechanisms and toxicants that lead to LRRK2 activation, the cellular consequences of prolonged LRRK2 kinase activity and potential ways to reduce it therapeutically.
LRRK2: Conformational dynamics and kinase activation
LRRK2 is a 2527 amino acid protein that possesses two enzymatic domains: (i) a Ras-of-complex (ROC), C-terminal of the Roc domain (COR) (ROC-COR) GTPase domain and (ii) a serine/threonine kinase domain (Figure 1). The enzymatic domains are flanked by an N-terminal armadillo domain, ankyrin domain, leucine rich repeat domain, and a C-terminal WD40 domain (Figure 1). The most frequent pathogenic mutations reside in the ROC-COR GTPase (R1441G/C/H, Y1669C) and kinase domains (G2019S, I2020T) [14], indicating important roles for both enzymatic domains in the pathogenicity of LRRK2-driven PD. Each of the pathogenic mutations results in a toxic gain-of-function: elevated kinase activity [15]. These facts, together with evidence of elevated LRRK2 kinase activity in iPD [7, 16], suggest that elucidating the mechanisms that drive LRRK2 kinase activation will be crucial for understanding the pathogenesis of PD.
Figure 1. Architecture of the LRRK2 protein and effects of Parkinson’s disease-associated mutations.
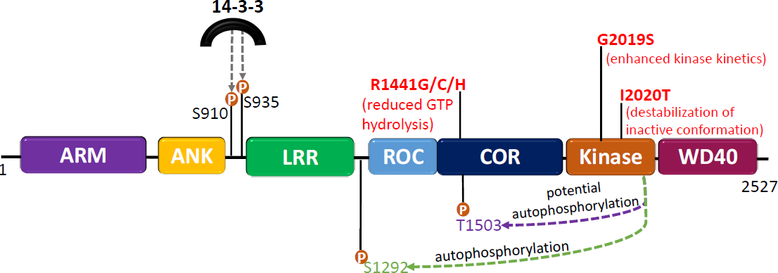
LRRK2 consists of 2527 amino acids and has 7 distinct domains: ARM, armadillo; ANK, ankyrin; LRR, leucine-rich repeat; ROC, Ras of complex; COR C-terminal of ROC; Kinase; and WD40, WD40 domain. LRRK2’s enzymatic domains are a ROC-COR GTPase domain and a serine/threonine kinase domain. The most common pathogenic mutations (in red) arise in the ROC-COR GTPase or kinase domain of the protein. The proposed mechanism of how each mutation affects the dynamics of LRRK2 is described below each mutation. Also depicted are the autophosphorylation sites, pSer1292 (green) and pThr1503 (purple). The autophosphorylation site pSer1292 is the most widely studied and is commonly used as a readout of kinase activity. The Thr1503 autophosphorylation site has been used in several studies as an indicator of kinase activity, however, this site has yet to be validated in an endogenous system. Phosphorylation sites in the N-terminal (pSer910 and pSer935) mediate 14–3-3 binding to LRRK2.
Embedded in all kinases is a DFGψ motif which controls its catalytic activity [17]. LRRK2 is unique in that it instead possesses a DYGI sequence (residues 2017–2020), the functional consequence being that a hydroxyl moiety on tyrosine2018 forms a hydrogen bonding node, keeping LRRK2 locked in its inactive conformation [18]. There are two common pathogenic LRRK2 mutations that lie within this DYGI sequence, G2019S and I2020T, which both result in enhanced kinase activity, presumably because they unleash the inactive conformation of the protein [18]. A recent study demonstrated that the G2019S mutation results in a more flexible kinase domain that enhances its dynamics and likely contributes to elevated kinase activity [19]. This idea was supported by an independent group that reported the structure of full length inactive human LRRK2 via cryo-electron microscopy [20]. Using full length human LRRK2, the authors showed that the structure of monomeric inactive wildtype LRRK2 and G2019S LRRK2 are almost identical [20], suggesting that the G2019S mutation may not result in elevated activity by a structural change in LRRK2, but rather through altered kinase kinetics [20]. Thus, the dynamics of the kinase domain may drive LRRK2 activation [19].
Emerging evidence suggests that GTP binding and hydrolysis affects LRRK2 kinase activity, and G2019S mutation-driven neurodegeneration relies on both GTPase and kinase activity [21]. This points to an intricate relationship between the two enzymatic domains of the LRRK2 protein. Mutations that arise in the ROC-COR GTPase domain affect GTP hydrolysis and result in a prolonged active state [22]. These mutations do not increase the catalytic activity in vitro, but they do result in increased substrate phosphorylation [15] and increased autophosphorylation at serine1292 (Ser1292) in vivo [23]. Both the ROC-COR GTPase and kinase domains interact with each other, suggesting that GDP and GTP binding can influence the conformation or activity of LRRK2 [24]. A 3.5Å structure of full length inactive LRRK2 revealed interdomain interactions between the ROC and COR domains [20]. Notably, this interface between the Roc-Cor GTPase domain is the site for many of the LRRK2 mutations that increase the risk for PD [20] suggesting that the interface of the ROC-COR GTPase domain is central to LRRK2 regulation.
Those mutations arising in different enzymatic domains result in elevated kinase activity, through unique mechanisms, which points to the complexity of LRRK2 kinase activation. As LRRK2 is a highly dynamic protein, elucidating the crucial regulatory interactions that stabilize LRRK2 in different conformations will lead to a deeper understanding of kinase activation and, possibly, more effective LRRK2 therapeutics to treat PD.
LRRK2: Substrates and kinase regulation
The functions of LRRK2 and its paralog LRRK1 are not fully understood. However, it is clear that a subset of Rab small GTPases, (e.g., Rab8A and Rab10) are physiological substrates of LRRK2 [25], and are distinct from those that are phosphorylated by LRRK1 [26]. It should be noted that only a subset of LRRK2 substrates, including Rab GTPases, have been validated in endogenous expression systems [25, 27]. As Rab GTPases regulate vesicular trafficking, LRRK2 appears to function as a master regulator of vesicular trafficking. Upon LRRK2-dependent phosphorylation of a Rab, the Rab’s interaction with its GDP dissociation inhibitor (GDI) is prevented, leading to accumulation of the Rab protein in membranes [28]. Heightened accumulation of membrane Rab proteins can lead to inefficient Rab cycling, which can result in both deficits in the endolysosomal system and vesicular trafficking. For example, overexpression of G2019S LRRK2 can disrupt endolysosomal trafficking by impairing Rab8A and Rab10, resulting in accumulation of Rab4-postive recycling endosomes and lysosomal dysfunction [29, 30]. To help maintain and facilitate efficient Rab cycling, the phosphatase, PPM1H, counteracts LRRK2 mediated Rab phosphorylation by dephosphorylating Rab8A and Rab10 [31]. Elevated phosphorylated Rab10 has been observed in post-mortem human brain tissue [7], peripheral immune cells [32], and blood from iPD patientsi. Additionally, preliminary results indicate elevated phosphorylated Rab10 is present in peripheral blood mononuclear cells (PBMCs) in two cohorts of subjects with iPD [33]. These results suggest there is increased LRRK2 kinase activity or decreased phosphatase activity in iPD. However, the status of PPM1H (expression, localization, activity) in iPD remains to be investigated.
Most of the LRRK2 protein is believed to reside in the cytosol in an inactive state [34] bound to members of the ubiquitously expressed 14–3-3 family of proteins. LRRK2 binding to 14–3-3 proteins at phosphorylation sites in the N-terminal domain (pSer910 and pSer935) of LRRK2 decrease kinase activity. Presently, these phosphorylation sites are considered biomarkers for on-target Type I LRRK2 kinase inhibitors, as these inhibitors result in dephosphorylation of Ser910 and Ser935 [35]. This has been validated by several independent groups using kinase inhibitors [21, 36–39]. Additionally, using overexpression systems and purified LRRK2 phospho-peptides, more recent data have suggested that LRRK2 binding to 14–3-3 proteins may also occur in the ROC-COR GTPase domain at pSer1444 [40, 41]. However, it will be important to validate this phosphorylation site using endogenous expression systems. Loss of 14–3-3:LRRK2 interaction leads to enhanced kinase activity, presumably by de-stabilization of the inactive conformation [41], and it has been reported that some PD-related pathogenic LRRK2 mutations (e.g., R1441G/C/H or Y1699C) that result in increased kinase activity, have less LRRK2:14–3-3 binding [42]. It is unclear if the G2019S mutation also results in a reduction in LRRK2:14–3-3 binding, as conflicting results have been reported [7, 42], possibly because of the use of overexpression versus endogenous expression systems. Nevertheless, the combination of the loss of this regulatory interaction and altered dynamics of the enzymatic domains of LRRK2 may help explain why these mutations result in enhanced kinase activity. In addition, overexpression of 14–3-3 protein can prevent G2019S LRRK2 kinase activity-driven toxicity [43]. Together, these data indicate that 14–3-3 proteins play a major role in LRRK2 kinase regulation and activation. In the context of iPD, phosphorylation of Ser232 on 14–3-3, which leads to its dysfunction, was reported in neuroblastoma cells treated with rotenone or overexpressing α-synuclein [44]. This was also observed in iPD brain tissue [44]. Dissociation of 14–3-3 from LRRK2 has been observed in iPD post-mortem human brain tissue [7], indicating that impaired 14–3-3:LRRK2 binding might contribute to the elevated LRRK2 activity observed in iPD.
LRRK2 not only resides in the cytoplasm but also localizes to many membrane-bound organelles including endosomes, lysosomes, and mitochondria, as well as microtubules [45–50]. Understanding LRRK2’s subcellular localization and its interacting proteins will help define its function and how it contributes to PD pathogenesis. At the trans-Golgi network (TGN), overexpression of Rab29 recruits and activates LRRK2 by promoting LRRK2 autophosphorylation at Ser1292, which then increases phosphorylation of Rab10 [49–51]. Interestingly, the PARK16 locus, which contains the gene for Rab29, has been identified as a risk site for iPD [52]. As such, Rab29 recruitment and activation of LRRK2 at the TGN might be a mechanism that contributes to PD pathogenesis. As an important caveat, however, a recent study reported that endogenous (as opposed to overexpressed) Rab29 does not recruit LRRK2 or stimulate LRRK2 kinase activity [53]. Therefore, Rab29-mediated LRRK2 recruitment and activation, if it occurs, may depend on cell type and/or subcellular microdomains with elevated endogenous Rab29 levels. That being said, under some conditions, Rab29 can recruit LRRK2 to stressed lysosomes and stabilizes Rab8 and Rab10 at the lysosomal membrane preventing lysosomal enlargement [47]. This LRRK2-Rab machinery can also upregulate lysosomal secretion through the Rab effectors, EH Domain-Binding Protein 1 (EHBP1) and EHBP1-like 1 (EHBP1L1) [47, 54]. This LRRK2-dependent recruitment of EHBP1 and EHBP1L1 to the lysosome promotes vesicle formation and budding from lysosomal membranes to maintain lysosomal homeostasis [55, 56]. It has also been reported that LRRK2 can recruit c-Jun-N-Terminal Kinase (JNK)-Interacting Protein-4 (JIP4) to stressed lysosomes via Rab35 and Rab10, which causes budding of vesicles from stressed lysosomes [46]. Nevertheless, it is clear that LRRK2 is a regulator of endolysosomal function.
LRRK2: Endolysosomal dysfunction
The genetic architecture of PD strongly implicates impairments of vesicular trafficking, endolysosomal function and autophagy in PD pathogenesis [6, 57]. Aberrant LRRK2 kinase activity is associated with pharmacologically stressed lysosomes. It is possible that genetic mutations that result in lysosomal dysfunction might also result in increased LRRK2 kinase activity. In addition, a number of LRRK2-interacting proteins (e.g. Auxilin, ATP13A2, LAMP2a, and ArfGAP1) are associated with lysosomal dysfunction [58–62]. Moreover, the D290N VSP35 mutation and certain GBA1 mutations (i.e., N370S and E326K), which impair endolysosomal function, result in increased LRRK2 kinase activity [63, 64]. Together, these data suggest that aberrant LRRK2 activity is associated with endolysosomal dysfunction.
Early endosomes are the first cargo vesicle in the endolysosomal pathway (Box 1), and any disruption in maturation or trafficking of early endosomes to late endosomes will likely affect overall lysosomal function. In sporadic Alzheimer’s disease, swollen Rab5-positive early endosomes accumulate in neurons early in the disease progression, even before there is deposition of amyloid beta [65], and a similar buildup of early endosomes is seen in SN DA neurons of iPD-patients [9]. Because LRRK2 colocalizes with EEA1-positive early endosomes [66], it is possible that the observed increase in LRRK2 kinase activity in iPD is responsible for the accrual of Rab5- and transferrin-positive endosomes iPD [9]. Consistent with this hypothesis, blocking LRRK2 activity with PF-360 prevented the accumulation of Rab5-positive early endosomes in the rotenone model of iPD [9]. While Rab5b, and not Rab5a has been nominated as a potential substrate for LRRK2 in overexpression studies [25, 67, 68], available antibodies cannot distinguish between different Rab5 isoforms, or their phosphorylation state. Nevertheless, if Rab5b is a LRRK2 substrate, this could explain the observed accumulation of Rab5 in iPD DA neurons [9]. It should be noted, however, that Rab5b has yet to be validated as a bona fide LRRK2 substrate in an endogenous context. It is possible that prolonged LRRK2 kinase activity leads to general disruption of early to late endosomal maturation by preventing recruitment of the Rab5-specific GDI. Supporting this interpretation, the number of late endosomes and lysosomes was normalized by LRRK2 kinase inhibition in a rodent model of iPD [9]. Further investigation is necessary to determine if early increases in LRRK2 activity coincide temporally with accumulation of Rab5 (endosomes) and whether this might serve as an early indicator for iPD pathology.
Box 1. The endolysosomal pathway.
As shown in Figure I, the endolysosomal pathway uses a series of cargo vesicles to internalize nutrients and recycle or degrade cargo (via lysosomes). Newly internalized cargo from the plasma membrane is rapidly targeted to early endosomes and shuttled to either the distinct thin tubular or the large luminal microdomain of the early endosome. The tubular microdomain of the early endosome is involved in recycling cargo either back to the plasma membrane via recycling endosomes or shuttling cargo to the TGN via retrograde transport. These two distinct pathways sometimes involve the retromer complex. Cargo in the large luminal microdomain of the early endosome eventually matures into the late endosome (also known as multivesicular bodies). This maturation process is marked by Rab5 to Rab7 conversion, in which Rab5-enriched early endosomes become Rab7-enriched (acidic) late endosomes. As cargo is trafficked toward lysosomes, the luminal pH of endosomes gradually decreases. The ESCRT machinery facilitates late endosomal trafficking and fusion of these mature acidic late endosomes with lysosomes, where proteolytic degradation of cargo occurs. As recycling or degrading endosomes are regulated by distinct Rab GTPases, and a subset of these Rab GTPases are substrates of LRRK2, LRRK2 is believed to regulate aspects of the endolysosomal pathway.
Figure I (for Box 1).

Schematic diagram of endolysosomal pathways. (1) From plasma membrane endocytosis to the formation of early endosomes. This is subsequently followed by two divergent pathways: (2) formation of recycling endosomes and (3) maturation to late endosome and eventual (4) lysosomes. (5) The retromer complex can traffic cargo to the TGN and (6) support anterograde vesicular trafficking from the TGN.
The balance between lysosomal repair and clearance may be dysregulated in PD. This imbalance is functionally significant because extensive damage to the outer lysosomal membrane is highly detrimental to cell health. To circumvent harmful consequences that may arise from membrane permeabilization, damaged lysosomes may be tagged for autophagic removal in a process called lysophagy. Lysophagy is initiated by recognition of cytosolic lectins, such as Galectin-3, at the lysosome membrane. However, Endosomal Sorting Complexes Required Transport (ESCRT) machinery can also be recruited to damaged or permeabilized lysosomes prior to lysophagy machinery – in an attempt to seal and repair damaged lysosomal membranes [69, 70]. New evidence suggests that LRRK2 can recruit Rab8A and the ESCRT-III component, Charged Multivesicular Body Protein 4B (CHMP4B) to damaged lysosomes and regulate the decision between membrane repair and lysophagy [48]. Any disruption of ESCRT recruitment can interrupt lysosome repair and result in irreversible lysosome damage, which can be lethal to the cell [69]. Interestingly, macrophages from LRRK2-PD mutation carriers (G2019S or R1441C) accumulate vesicles that are positive for Rab8A and Galectin-3, suggesting increased LRRK2 activity may disrupt the balance between lysosomal membrane repair and lysophagy [48].
It is apparent that pathological LRRK2 kinase activity can disrupt membrane and vesicle trafficking and may affect lysosomal function deleteriously (Figure 2). Therefore, a deeper understanding of the factors that can activate aberrant LRRK2 kinase activity – and the mechanisms thereof – is essential.
Figure 2. The role of LRRK2 in endolysosomal trafficking.
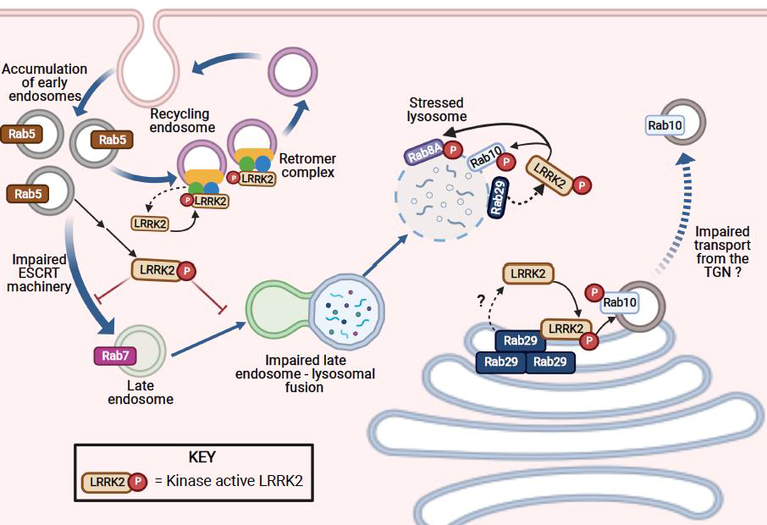
In PD, elevated LRRK2 activity can lead to accumulation of Rab5-positive early endosomes as well as impairment in the ESCRT machinery. These deficits can in turn prevent early to late endosomal maturation; as a result, there are fewer Rab7-positive late endosomes. These deficits in vesicle trafficking can, in turn, cause an increase in LRRK2 activity, albeit through an undefined mechanism(s). Impaired late endosomal–lysosomal fusion caused by aberrant LRRK2 kinase activity can result in stressed lysosomes, which then recruit and activate LRRK2, possibly via Rab29. At a stressed lysosome, active LRRK2 can recruit and phosphorylate Rab8A and Rab10. The biological significance of lysosomal recruitment of Rab10 and Rab8A has yet to be fully characterized but likely involves lysosomal vesicle formation / budding and exocytosis. At the recycling endosome, LRRK2 can be activated by the retromer complex. Although the mechanism responsible for this recruitment is currently unknown, it is clear that LRRK2 recruitment to the recycling endosome can result in improper retrograde trafficking from the recycling endosome to the trans-Golgi network (TGN). Moreover, at the TGN, high levels of Rab29 (in overexpression systems) can recruit and activate LRRK2, which results in accumulation of Rab10 positive vesicles at the TGN, which in turn, can lead to impaired anterograde trafficking.
LRRK2: Activation by oxidative and endolysosomal stress
Dopaminergic neurons are highly vulnerable to oxidant stress. This enhanced sensitivity to oxidative stress may be due, in part, to DA itself, which can auto-oxidize and lead to the accumulation of free radicals and the highly reactive DA-quinone. Accumulation of these toxic free radicals and quinones can lead to mitochondrial dysfunction, reactive oxygen species (ROS) production and oxidatively damaged proteins [71]. Therefore, nigrostriatal DA neurons rely heavily on surveillance mechanisms (e.g., autophagy) and the endolysosomal pathway to identify and remove dysfunctional proteins, organelles, and unwanted material to maintain homeostasis. Disruption in these surveillance pathways may occur preferentially in DA neurons, as DA and its oxidation products can cause lysosomal and mitochondrial dysfunction [64, 71–73].
LRRK2 is a reduction/oxidation (redox)-sensitive protein and elevated oxidative stress may exert its effects, in part, through enhanced LRRK2 kinase activity (Figure 3, Key Figure). Several reports indicate that LRRK2 kinase activity is increased by H2O2 [7, 74, 75] or decreased by the antioxidant curcumin [74]. The kinase activity of wildtype endogenous LRRK2 can be elevated dose-dependently by physiological concentrations of H2O2, and this can be blocked by the ROS scavenger α-tocopherol [7]. Others have reported that H2O2 increases wildtype LRRK2 kinase activity to the same extent as the G2019S mutation [75]. Moreover, there is strong evidence that LRRK2 can be modulated by (i) the major ROS producer, NADPH oxidase 2, (ii) exposure to the toxicant rotenone, and (iii) overexpression α-synuclein, all of which promote the formation of ROS [7]. Thus, redox state may be an important regulator of LRRK2 kinase activity. In this regard, the kinase domain shows a high degree of homology with other redox-sensitive MAP-kinases [76]. Cysteine residues on proteins can readily undergo oxidative modifications which, in turn, can influence function and enzymatic activity. The kinase activation loop of LRRK2 contains two solvent-exposed vicinal cysteine residues. These residues are conserved across many species [77] and are not present in LRRK1, suggesting they may play a specific role in LRRK2 activation. It is plausible that these cysteine residues are important for sensing the redox state of the local subcellular environment and function to activate LRRK2 [18]. If so, it may be possible to design LRRK2 inhibitors that target them [78]. Going forward, exploration of the redox sensitivity of LRRK2 may yield novel insights into the molecular mechanisms that drive its kinase activity.
Figure 3, Key Figure: Activation of wildtype LRRK2 kinase.
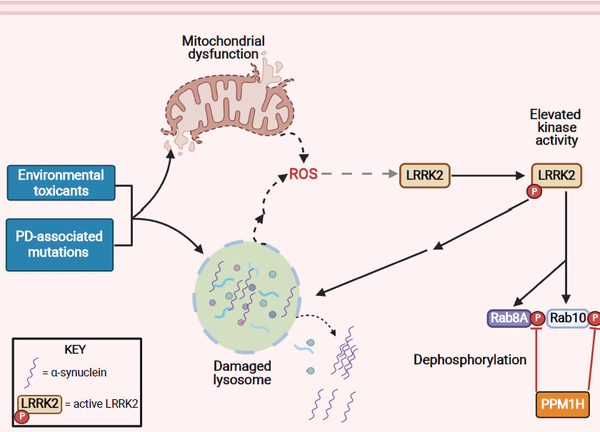
Environmental contaminants linked to PD [82] and PD-associated risk genes [2] affect mitochondrial function and the endolysosomal system, either directly or indirectly. Interestingly, mitochondrial dysfunction and endolysosomal stress are key mediators of LRRK2 kinase activation. A convergent event of both mitochondrial and lysosomal dysfunction is the generation and accumulation of ROS, which in turn can activate LRRK2. Whether the ROS buildup from damaged lysosomes is released directly from these lysosomes or is a downstream consequence is currently undefined. Enhanced LRRK2 kinase activity, as a result of either mitochondrial dysfunction or stressed lysosomes, can further damage lysosomes and result in membrane permeabilization and leakage of damaged cargo and proteins, including α-synuclein into the cytoplasm. This, in turn, results in the toxic accumulation and aggregation of α-synuclein. The phosphatase PPM1H, which is known to counteract LRRK2 activity by dephosphorylating specific Rab proteins (e.g. Rab8A, Rab10) may normalize aberrant LRRK2 kinase activity. However, this signaling axis remains to be explored.
While LRRK2 is regulated by oxidative stress, it may also enhance oxidative stress [7, 79], and it is possible that increased LRRK2 kinase activity may indirectly damage components of the endolysosomal pathway. In addition to oxidative stress, endolysosomal and autophagic stressors also activate LRRK2. For example, pleiotropic trafficking deficits induced by monensin, and lysosomal membrane rupture caused by chloroquine or L-Leucyl-L-Leucine-Methyl Ester can induce LRRK2 activation in several different cell types [46, 48, 53, 54]. Whether these seemingly disparate stimuli share similar molecular mechanisms of LRRK2 activation is currently unknown, but it should be noted that interfering with endolysosomal or autophagic function can lead to secondary oxidative stress [80–82]. Conversely, oxidative stress and ROS can cause impairment of endolysosomal function [81], which highlights the reciprocal interplay between redox signaling and autophagy.
Conceivably, LRRK2-mediated lysosomal stress may cause mitochondrial deficits, perhaps via impaired mitophagy, thereby leading to increased mitochondria-derived ROS, but the sequence and directionality of this signaling remain to be defined. Interestingly, blocking LRRK2 kinase activity in vivo using PF-360 prevented SNc DA neuron endolysosomal deficits caused by rotenone, an oxidative stressor [9]. Thus, endolysosomal impairment can activate LRRK2 and LRRK2 kinase activity contributes to endolysosomal dysfunction.
LRRK2: A target of environmental toxicants
The mechanisms underlying aberrant LRRK2 activity and its downstream effects, may be influenced by exogenous factors (Figure 3). In this context, an obvious – but often overlooked factor involved in LRRK2 kinase activation is environmental contaminants linked to PD [83], which are increasingly reported to enhance LRRK2 kinase activity in the brain [7, 84–87]. Epidemiological data shows increased risk for iPD among pesticide-exposed farmers and residents of agricultural or rural communities [88, 89], as well as industrial workers exposed to chlorinated solvents [90, 91]. The pesticides paraquat and rotenone are both associated with a 2.5-fold increased risk for PD [92]. Similarly, twin studies show the organic solvent trichloroethylene (TCE) increases PD risk (Odds Ratio 6.1) [91]. Thus far, the mechanisms that enhance the risk for developing iPD following toxicant exposure appear to involve accumulation of ROS and/or mitochondrial dysfunction.
Emerging evidence indicates that LRRK2 may link mitochondrial toxicant-driven oxidative stress with PD-related pathologies. For example, when rats were chronically exposed to the mitochondrial complex I inhibitor, rotenone, or the industrial solvent, TCE, there was oxidative damage in the SN, increased LRRK2 kinase activity, phosphorylated Rab10 accumulation, and endolysosomal dysfunction, with resultant selective nigrostriatal DA neuron degeneration [9, 84]. Co-treatment with the selective LRRK2 kinase inhibitor, PF-360, prevented all rotenone-induced pathologies, including DA neuron degeneration in rats [9].
Peripheral inflammation may be important in PD pathogenesis and LRRK2 is highly expressed in immune cells [93], so exposure to exogenous toxicants that influence LRRK2’s kinase activity may also contribute to excessive neuroinflammation in PD. Support for this idea comes from a study using macrophages treated with manganese, and which suggests that non-neuronal cells may also be an important target of toxicant-induced LRRK2 kinase activity [94, 95].
LRRK2: Therapeutic Strategies
Therapeutic approaches for LRRK2 activation stemming from environmental toxicant exposure
The molecular mechanisms by which LRRK2 is activated by environmental contaminants remain to be delineated. Clearer understanding of these mechanisms may lead to new therapeutic and prophylactic measures. Inhibition of LRRK2 by small molecule kinase inhibitors or antisense oligonucleotides may protect against LRRK2-mediated pathology induced by environmental contaminants as seen experimentally in the rotenone and pre-formed fibril α-synuclein rodent models of PD [9, 10]. Thus, individuals who (1) have had a known exposure to an environmental toxicant linked to increased PD risk, and (2) display elevated LRRK2 kinase activity in a biological sample (e.g., blood neutrophils, PBMCs, or urinary exosomes) may represent a population in which to attempt to prevent the onset, or slow the progression, of PD. Thus, LRRK2 inhibitors may be a unique therapeutic avenue for individuals exposed to PD-linked environmental toxicants. However, at a minimum, this approach would require integration of environmental monitoring, validation of LRRK2 biomarkers, and off-label usage of approved LRRK2 therapeutics. Accordingly, the bar to mitigate neuropathology caused by environmental toxicant-induced LRRK2 kinase activity is high and will require further characterization and validation in human subjects.
Therapeutic approaches targeting LRRK2 for iPD
In contrast to the toxicant exposure mitigation discussed previously, the use of LRRK2 therapeutics for iPD disease modification will likely require long-term, possibly life-long treatment after initiation of therapy. Given the expression of LRRK2 in tissues other than the brain, especially the lungs, there is justified concern about the potential for “on-target” (LRRK2-specific) toxicity, such as pulmonary toxicity. There are a number of lines of evidence which, at least in our view, argue against the possibility of severe on-target toxicity. A recent study examined in non-human primates the pulmonary effects of 3 structurally distinct LRRK2 kinase inhibitors and found only mild histopathological lung changes that were reversable and without noticeable effect on lung function [96]. Studies in mice and rats also yielded reassuring results [38, 97–99]. Importantly, two recent genetic studies demonstrated that LRRK2 loss-of-function variants in humans do not seem to have negative effect on health or survival [100, 101], which further argues in favor of the potential safety of LRRK2-targeted therapeutic approaches.
As of this writing, a Phase 1b clinical trial to evaluate the safety, target engagement and effects on putative biomarkers of a LRRK2 kinase inhibitor (BIIB122/DNL151) in PD subjects with or without LRRK2 mutations has recently completedi. Additionally, an antisense oligonucleotide therapeutic targeting LRRK2 is entering Phase 1 clinical studies (NCT03976349)ii and is open to subjects with LRRK2 mutations as well as those with iPD. A consideration to keep in mind, however, is that only a fraction of LRRK2 is believed to be active at a given time and it remains uncertain that reducing overall LRRK2 levels will reduce the amount of LRRK2 kinase activity. Going forward, as more is learned about the structure, function and biology of LRRK2, it is likely that additional approaches to target LRRK2 therapeutically will emerge.
Concluding Remarks
Despite current uncertainties and knowledge gaps (see Outstanding Questions), it is becoming increasingly clear that LRRK2 plays a central role in the pathogenesis of iPD. Common variants in the LRRK2 locus are known risk factors for iPD. Oxidative stress and endolysosomal impairment, both of which play critical roles in PD, activate LRRK2 kinase activity. Similarly, environmental toxicants, implicated epidemiologically in PD risk, activate LRRK2. Moreover, in vivo blockade of LRRK2 activity is protective in genetic and toxicant models of PD. As noted, early-stage clinical trials of LRRK2-based therapeutics are already underway. Given that (i) PD is the fastest growing neurological disease in the world [102] and (ii) that LRRK2 is central to PD pathogenesis, a deeper understanding of the cellular biology of LRRK2 and its activation mechanisms is essential.
Outstanding questions.
What are the biological functions of LRRK2 – and how do they differ by cell type?
The phosphatase PPM1H counters the effects LRRK2 kinase. Does dysregulated PPM1H contribute to LRRK2-driven pathologies?
What are the molecular mechanisms by which oxidative stress and endolysosomal/autophagic stress activate wildtype LRRK2 – and are they the same?
Does dopamine or its oxidation products activate LRRK2 in SNc neurons?
What is the most efficacious and safe way to mitigate aberrant LRRK2 kinase activity in PD – small molecule kinase inhibitors, RNAi or other?
Inhibition of LRRK2 kinase activity appears to prevent neurodegeneration in vivo – but can it slow or stop degeneration after the pathogenic cascade of PD has been initiated?
Given that PD-associated environmental toxicants (e.g., rotenone, paraquat and TCE) activate LRRK2 kinase activity, can LRRK2 therapeutics be used to mitigate post-exposure risk of developing PD?
Are individuals with LRRK2 loss of function mutations less likely to develop PD – and are these individuals relatively resistant to PD-associated environmental toxicants?
Could assessment of peripheral LRRK2 kinase activity provide the basis for a reliable biomarker for disease state, patient stratification or target engagement?
Highlights.
Mutations in the LRRK2 gene that are associated with aberrantly enhanced kinase activity are the most common cause of genetic Parkinson’s disease (PD). Emerging evidence now points to a role of elevated LRRK2 kinase activity without LRRK2 mutations in non-genetic (idiopathic) forms of the disease.
The architecture of LRRK2 influences kinase activation, and enhanced LRRK2 substrate phosphorylation may contribute to PD pathogenesis.
Evidence indicates that LRRK2 kinase activity may be enhanced in idiopathic PD by oxidative stress and/or endolysosomal stress.
Exposure to environmental toxicants linked to idiopathic PD by epidemiological studies is also reported to activate LRRK2 kinase activity.
Two therapeutic approaches targeting LRRK2 – an antisense oligonucleotide and a kinase inhibitor – are in clinical trials for both LRRK2 mutation carriers and those with idiopathic PD.
Acknowledgements
This work was supported by research grants from the National Institutes of Health (R00ES029986-04 to BRD, and NS100744, NS095387 to JTG), the American Parkinson’s disease Association (JTG), the Parkinson’s Foundation (BRD), the Michael J Fox Foundation (MJFF-006910 to JTG and MJFF-008011 and MJFF-000849 to EMR), Claude D. Pepper Older Americans Independence Center (P30AG024827 to EMR), the Blechman Foundation, and the friends and family of Sean Logan (JTG). All schematic figures were created with BioRender.com.
Glossary
- Anti-sense oligonucleotide (ASO)
Synthetic, short single-stranded oligodeoxynucleotides that are complementary to an mRNA target and thereby can lead to transcript knockdown and, consequently, a reduction in protein expression
- Autophagy
Lysosome-dependent degradation system that allows for recycling of macromolecules. It is responsible for removal of unnecessary and/or dysfunctional components of the cell
- Chloroquine
A lysosomotropic drug that becomes protonated in the acidic environment of the lysosome and causes an influx of water into the lysosome, a process referred to as the osmotic effect
- L-leucyl-L-leucine methyl ester (LLOME)
A lysosomotropic agent that results in lysosomal membrane permeabilization and rupture. After endocytosis, it reaches the lysosome where it interacts with Cathepsin C and causes lysosomal membrane rupture
- Monensin
An antibiotic derived from Streptomyces cinnamonensis. It is a monovalent cation ionophore that causes disruption in vesicular transport from the trans-Golgi network
- Oxidative stress
The pathophysiological event that occurs when the redox state of the cell shifts towards being more oxidized. This shift is the consequence of the accumulation of free radicals and/or loss of antioxidant defense systems
- Penetrance
In the context of human genetics, penetrance refers to the proportion of individuals carrying a particular genetic variant (allele) who also express the phenotype associated with that variant. LRRK2 mutations have incomplete penetrance, meaning that not all individuals carrying a mutation develop Parkinson’s disease
- Quinone
A class of cyclic organic compounds that contains two carbonyl groups in a six-member unsaturated ring. They are oxidized derivatives of aromatic compounds and are electrophilic molecules that can readily form covalent bonds with nucleophilic moieties such as sulfhydryl groups of cysteine residues
- Rab GTPase
Members of the Ras superfamily of small G proteins. About 70 Rab proteins have been identified to date and are involved in membrane trafficking, including vesicle formation and movement along tubules. A subset of Rab GTPases are LRRK2 substrates
- Reduction/oxidation (redox) state
The cellular or subcellular state of balance between oxidants and antioxidants in a cell
- Toxicant
A substance of natural or synthetic origin that produces transient or permanent damage (toxicity) to cells, tissues, and organs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ball N, et al. (2019) Parkinson’s Disease and the Environment. Front Neurol 10, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blauwendraat C, et al. (2020) The genetic architecture of Parkinson’s disease. Lancet Neurol 19, 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leonard H, et al. (2020) Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson’s disease. J Med Genet 57, 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reeve A, et al. (2014) Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res Rev 14, 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Artzi M, et al. (2017) DaT-SPECT assessment depicts dopamine depletion among asymptomatic G2019S LRRK2 mutation carriers. PLoS One 12, e0175424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kluss JH, et al. (2019) LRRK2 links genetic and sporadic Parkinson’s disease. Biochem Soc Trans 47, 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Maio R, et al. (2018) LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim C, et al. (2020) LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci Transl Med 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rocha EM, et al. (2020) LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol Dis 134, 104626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao HT, et al. (2017) LRRK2 Antisense Oligonucleotides Ameliorate alpha-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol Ther Nucleic Acids 8, 508–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daher JP, et al. (2015) Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates alpha-Synuclein Gene-induced Neurodegeneration. J Biol Chem 290, 19433–19444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Fonzo A, et al. (2005) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365, 412–415 [DOI] [PubMed] [Google Scholar]
- 13.Kalia LV, et al. (2015) Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor M and Alessi DR (2020) Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr Opin Cell Biol 63, 102–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leandrou E, et al. (2019) Kinase activity of mutant LRRK2 manifests differently in heterodimeric vs. homo-dimeric complexes. Biochem J 476, 559–579 [DOI] [PubMed] [Google Scholar]
- 16.Fraser KB, et al. (2016) Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov Disord 31, 1543–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor SS, et al. (2020) Kinase Domain Is a Dynamic Hub for Driving LRRK2 Allostery. Front Mol Neurosci 13, 538219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt SH, et al. (2019) The dynamic switch mechanism that leads to activation of LRRK2 is embedded in the DFGpsi motif in the kinase domain. Proc Natl Acad Sci U S A 116, 14979–14988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt SH, et al. (2021) Conformation and dynamics of the kinase domain drive subcellular location and activation of LRRK2. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myasnikov A, et al. (2021) Structural analysis of the full-length human LRRK2. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen APT, et al. (2020) Dopaminergic neurodegeneration induced by Parkinson’s disease-linked G2019S LRRK2 is dependent on kinase and GTPase activity. Proc Natl Acad Sci U S A 117, 17296–17307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu CX, et al. (2019) Parkinson’s disease-associated mutations in the GTPase domain of LRRK2 impair its nucleotide-dependent conformational dynamics. J Biol Chem 294, 5907–5913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheng Z, et al. (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 4, 164ra161. [DOI] [PubMed] [Google Scholar]
- 24.Deniston CK, et al. (2020) Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature 588, 344–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steger M, et al. (2016) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malik AU, et al. (2021) Deciphering the LRRK code: LRRK1 and LRRK2 phosphorylate distinct Rab proteins and are regulated by diverse mechanisms. Biochem J 478, 553–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lis P, et al. (2018) Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. Biochem J 475, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez RC, et al. (2019) Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J Cell Biol 218, 4157–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivero-Rios P, et al. (2020) Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations. Cells 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivero-Rios P, et al. (2019) The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J Biol Chem 294, 4738–4758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berndsen K, et al. (2019) PPM1H phosphatase counteracts LRRK2 signaling by selectively dephosphorylating Rab proteins. Elife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan Y, et al. (2018) Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem J 475, 23–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petropoulou-Vathi L, et al. (2021) Distinct profiles of LRRK2 activation and Rab GTPase phosphorylation in clinical samples from different PD cohorts. bioRxiv, 10.1101/2021.11.05.465894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schapansky J, et al. (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet 23, 4201–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasegian A, et al. (2021) Impact of Type II LRRK2 inhibitors on signaling and mitophagy. Biochem J 478, 3555–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dzamko N, et al. (2010) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14–3-3 binding and altered cytoplasmic localization. Biochem J 430, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez B, et al. (2019) Centrosomal cohesion deficits as cellular biomarker in lymphoblastoid cell lines from LRRK2 Parkinson’s disease patients. Biochem J 476, 2797–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kluss JH, et al. (2021) Preclinical modeling of chronic inhibition of the Parkinson’s disease associated kinase LRRK2 reveals altered function of the endolysosomal system in vivo. Molecular Neurodegeneration 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S, et al. (2020) Exosome markers of LRRK2 kinase inhibition. NPJ Parkinsons Dis 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manschwetus JT, et al. (2020) Binding of the Human 14–3-3 Isoforms to Distinct Sites in the Leucine-Rich Repeat Kinase 2. Front Neurosci 14, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muda K, et al. (2014) Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14–3-3. Proc Natl Acad Sci U S A 111, E34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, et al. (2011) Phosphorylation-dependent 14–3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS One 6, e17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavalley NJ, et al. (2016) 14–3-3 Proteins regulate mutant LRRK2 kinase activity and neurite shortening. Hum Mol Genet 25, 109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slone SR, et al. (2015) Increased 14–3-3 phosphorylation observed in Parkinson’s disease reduces neuroprotective potential of 14–3-3 proteins. Neurobiol Dis 79, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biskup S, et al. (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol 60, 557–569 [DOI] [PubMed] [Google Scholar]
- 46.Bonet-Ponce L, et al. (2020) LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eguchi T, et al. (2018) LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A 115, E9115–E9124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herbst S, et al. (2020) LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J 39, e104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purlyte E, et al. (2019) Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beilina A, et al. (2014) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A 111, 2626–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Z, et al. (2018) LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum Mol Genet 27, 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon-Sanchez J, et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41, 1308–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalogeropulou AF, et al. (2020) Endogenous Rab29 does not impact basal or stimulated LRRK2 pathway activity. Biochem J 477, 4397–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuwahara T, et al. (2020) Roles of lysosomotropic agents on LRRK2 activation and Rab10 phosphorylation. Neurobiol Dis 145, 105081. [DOI] [PubMed] [Google Scholar]
- 55.Nakajo A, et al. (2016) EHBP1L1 coordinates Rab8 and Bin1 to regulate apical-directed transport in polarized epithelial cells. J Cell Biol 212, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang P, et al. (2016) RAB-10 Promotes EHBP-1 Bridging of Filamentous Actin and Tubular Recycling Endosomes. PLoS Genet 12, e1006093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vazquez-Velez GE and Zoghbi HY (2021) Parkinson’s Disease Genetics and Pathophysiology. Annu Rev Neurosci 44, 87–108 [DOI] [PubMed] [Google Scholar]
- 58.Henry AG, et al. (2015) Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet 24, 6013–6028 [DOI] [PubMed] [Google Scholar]
- 59.Meng D, et al. (2021) ArfGAP1 inhibits mTORC1 lysosomal localization and activation. EMBO J 40, e106412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen M and Krainc D (2018) LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A 115, 5576–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Orenstein SJ, et al. (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16, 394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stafa K, et al. (2012) GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet 8, e1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mir R, et al. (2018) The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J 475, 1861–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ysselstein D, et al. (2019) LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun 10, 5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cataldo AM, et al. (2004) Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging 25, 1263–1272 [DOI] [PubMed] [Google Scholar]
- 66.Schreij AM, et al. (2015) LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep 16, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yun HJ, et al. (2015) An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J Biochem 157, 485–495 [DOI] [PubMed] [Google Scholar]
- 68.Steger M, et al. (2017) Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Radulovic M, et al. (2018) ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Skowyra ML, et al. (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hastings TG (2009) The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J Bioenerg Biomembr 41, 469–472 [DOI] [PubMed] [Google Scholar]
- 72.Burbulla LF, et al. (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hastings TG, et al. (1996) Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci U S A 93, 1956–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang D, et al. (2012) LRRK2 kinase activity mediates toxic interactions between genetic mutation and oxidative stress in a Drosophila model: suppression by curcumin. Neurobiol Dis 47, 385–392 [DOI] [PubMed] [Google Scholar]
- 75.Li X, et al. (2010) Reevaluation of phosphorylation sites in the Parkinson disease-associated leucine-rich repeat kinase 2. J Biol Chem 285, 29569–29576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson WM, et al. (2015) The roles of redox enzymes in Parkinson’s disease: Focus on glutaredoxin. Ther Targets Neurol Dis 2 [PMC free article] [PubMed] [Google Scholar]
- 77.Fan TS, et al. (2016) Clinical heterogeneity of LRRK2 p.I2012T mutation. Parkinsonism Relat Disord 33, 36–43 [DOI] [PubMed] [Google Scholar]
- 78.Ray S, et al. (2014) The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J Biol Chem 289, 13042–13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heo HY, et al. (2010) LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res 316, 649–656 [DOI] [PubMed] [Google Scholar]
- 80.Ketola K, et al. (2010) Monensin is a potent inducer of oxidative stress and inhibitor of androgen signaling leading to apoptosis in prostate cancer cells. Mol Cancer Ther 9, 3175–3185 [DOI] [PubMed] [Google Scholar]
- 81.Lee J, et al. (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441, 523–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu JJ, et al. (2009) Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 1, 425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Miranda BR, et al. (2021) Preventing Parkinson’s Disease: An Environmental Agenda. J Parkinsons Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De Miranda BR, et al. (2021) The industrial solvent trichloroethylene induces LRRK2 kinase activity and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. Neurobiol Dis 153, 105312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deshpande P, et al. (2020) Protein synthesis is suppressed in sporadic and familial Parkinson’s disease by LRRK2. FASEB J 34, 14217–14233 [DOI] [PubMed] [Google Scholar]
- 86.Ho DH, et al. (2021) LRRK2 Kinase Inhibitor Rejuvenates Oxidative Stress-Induced Cellular Senescence in Neuronal Cells. Oxid Med Cell Longev 2021, 9969842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mendivil-Perez M, et al. (2016) Neuroprotective Effect of the LRRK2 Kinase Inhibitor PF-06447475 in Human Nerve-Like Differentiated Cells Exposed to Oxidative Stress Stimuli: Implications for Parkinson’s Disease. Neurochem Res 41, 2675–2692 [DOI] [PubMed] [Google Scholar]
- 88.Ascherio A, et al. (2006) Pesticide exposure and risk for Parkinson’s disease. Ann Neurol 60, 197–203 [DOI] [PubMed] [Google Scholar]
- 89.Gorell JM, et al. (1998) The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 50, 1346–1350 [DOI] [PubMed] [Google Scholar]
- 90.Gash DM, et al. (2008) Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol 63, 184–192 [DOI] [PubMed] [Google Scholar]
- 91.Goldman SM, et al. (2012) Solvent exposures and Parkinson disease risk in twins. Ann Neurol 71, 776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanner CM, et al. (2011) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119, 866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cook DA, et al. (2017) LRRK2 levels in immune cells are increased in Parkinson’s disease. npj Parkinson’s Disease 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim J, et al. (2019) LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS One 14, e0210248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen J, et al. (2018) Role of LRRK2 in manganese-induced neuroinflammation and microglial autophagy. Biochem Biophys Res Commun 498, 171–177 [DOI] [PubMed] [Google Scholar]
- 96.Baptista MAS, et al. (2020) LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci Transl Med 12 [DOI] [PubMed] [Google Scholar]
- 97.Herzig MC, et al. (2011) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Human Molecular Genetics 20, 4209–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Andersen MA, et al. (2018) PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology 395, 15–22 [DOI] [PubMed] [Google Scholar]
- 99.Bryce DK, et al. (2021) Characterization of the Onset, Progression, and Reversibility of Morphological Changes in Mouse Lung after Pharmacological Inhibition of Leucine-Rich Kinase 2 Kinase Activity. J Pharmacol Exp Ther 377, 11–19 [DOI] [PubMed] [Google Scholar]
- 100.Blauwendraat C, et al. (2018) Frequency of Loss of Function Variants in LRRK2 in Parkinson Disease. JAMA Neurol 75, 1416–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Whiffin N, et al. (2020) The effect of LRRK2 loss-of-function variants in humans. Nat Med 26, 869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dorsey ER and Bloem BR (2018) The Parkinson Pandemic-A Call to Action. JAMA Neurol 75, 9–10 [DOI] [PubMed] [Google Scholar]