

Abstract
The COVID-19 pandemic sparked rapid development of SARS-CoV-2 diagnostics. However, emerging variants pose the risk for target dropout and false-negative results secondary to primer/probe binding site (PBS) mismatches. The Agena MassARRAY® SARS-CoV-2 Panel combines RT-PCR and MALDI-TOF mass-spectrometry to probe for five targets across N and ORF1ab genes, which provides a robust platform to accommodate PBS mismatches in divergent viruses. Herein, we utilize a deidentified dataset of 1,262 SARS-CoV-2-positive specimens from Mount Sinai Health System (New York City) from December 2020 through April 2021 to evaluate target results and corresponding sequencing data. Overall, the level of PBS mismatches was greater in specimens with target dropout. Of specimens with N3 target dropout, 57% harbored an A28095T substitution that is highly-specific for the alpha (B.1.1.7) variant of concern. These data highlight the benefit of redundancy in target design and the potential for target performance to illuminate the dynamics of circulating SARS-CoV-2 variants.
Keywords: RT-PCR, MALDI-TOF, SARS-CoV-2, B.1.1.7, variants, diagnostic, dropout
Introduction
Molecular diagnostic assays for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), the etiologic agent of coronavirus disease 2019 (COVID-19), utilize nucleic acid amplification test (NAAT) methods to assess for presence of viral nucleic acids in clinical specimens. These assays rely on primers and probes targeting one or more viral gene regions including open reading frame 1ab (ORF1ab), open reading frame 8 (ORF 8), nucleocapsid (N), spike (S), and envelope (E) 1. These targets have been designed primarily based on sequences from virus strains that circulated early in the pandemic, including the reference genome collected from Wuhan, China in January, 2020 2–4.
In addition to the quantity of viral nucleic acids in a clinical specimen, the diagnostic and analytic capabilities of NAATs depend on the complementarity of primers and probes to viral genome sequences to reliably amplify targets of interest. As a result, binding of primers and probes can be impacted by progressive accumulation of changes in the viral genomes at primer binding sites (PBSs). Indeed, mismatches in PBSs – particularly the 2-3 nucleotides at the 3’ end of the oligonucleotide – can result in reduced binding and subsequent failure to amplify (termed “dropout” in diagnostic NAAT assays) 5–8. In fact, SARS-CoV-2 has diversified over the past 18 months, and mutations in the N, S, and E genes have been reported in viruses from specimens with corresponding target dropout during testing on commercial NAAT-based diagnostic platforms 9–15. Moreover, in silico analyses have utilized publicly-available SARS-CoV-2 genome sequences to identify mutations in circulating viral variants that have the potential to interfere with diagnostic targets 1,5,16–18. These findings highlight the potential diagnostic challenge as increasingly diverse SARS-CoV-2 lineages (e.g., B.1.1.7) continue to emerge globally 19,20.
To limit the risk of false-negative results, most NAAT assays for SARS-CoV-2 that currently have emergency use authorization (EUA) from the US Food and Drug Administration (FDA) utilize two or more diagnostic targets 21,22. We recently reported the analytic performance of the Agena MassARRAY® SARS-CoV-2 Panel which combines RT-PCR and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) technologies to detect SARS-CoV-2 23. The Agena MassARRAY® platform probes for five distinct targets in the ORF1ab and N viral genes 24, providing a robust platform for diagnosis of SARS-CoV-2 in clinical specimens despite the emergence of virus strains that have accumulated mutations that can interfere with some diagnostic targets. We evaluated the pattern of target detections for SARS-CoV-2-positive specimens collected at the Mount Sinai Health System (MSHS) to interrogate the impact of viral genetic variation on this diagnostic platform.
To do this, we compared detection of Agena diagnostic targets and genomic sequence data for SARS-CoV-2-positive specimens that were deidentified and banked as part of our Pathogen Surveillance Program (MSHS PSP) at the Icahn School of Medicine at Mount Sinai (ISMMS), which has been previously described 25. Complete viral genomes underwent phylogenetic analyses to characterize emergent evolutionary lineages among the SARS-CoV-2-positive specimens at MSHS (manuscript in preparation). For this analysis, we utilized a dataset comprised of 1,262 viral genomes recovered from deidentified clinical specimens collected from patients seeking care at the Mount Sinai Health System from December 1, 2020 through April 24, 2021. We identified PBS mismatches associated with lineage-specific substitutions in SARS-CoV-2 variants of concern (VOC) that resulted in Agena MassARRAY® platform target dropout.
Materials and Methods
Ethics statement
This study was reviewed and approved by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai (HS#13-00981).
SARS-CoV-2 specimen collection and testing
Upper respiratory tract (e.g., nasopharyngeal, anterior nares) and saliva specimens collected for SARS-CoV-2 testing underwent diagnostic testing in the MSHS Clinical Microbiology Laboratory (CML), which is certified under Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a and meets requirements to perform high-complexity tests. For this study, we retrospectively utilized deidentified data available for diagnostic specimens tested on the Agena MassARRAY® SARS-CoV-2 Panel and MassARRAY® System (Agena, CPM384) platform during the study period.
As previously described, prior to SARS-CoV-2 testing, saliva specimens underwent an initial processing step involving a 15 minute incubation at 55°C prior to RNA extraction 23. Upper respiratory specimens did not undergo any pre-processing prior to testing. RNA was extracted from 300μL of each specimen using the chemagic™ Viral DNA/RNA 300 Kit H96 (PerkinElmer, CMG-1033-S) on the automated chemagic™ 360 instrument (PerkinElmer, 2024-0020) per the manufacturer’s protocol. The MS2 phage RNA internal control (IC) was included in all extraction steps. The extracted RNA underwent RT-PCR with iPLEX® Pro chemistry to amplify different Agena targets, per the manufacturer’s protocol. After inactivation of unincorporated dNTPs by treatment with shrimp alkaline phosphatase (SAP), a sequence-specific primer extension step was performed, in which a mass-modified terminator nucleotide was added to the probe, using supplied extension primers and iPLEX® Pro reagents.
Extension products (analytes) were desalted, transferred to a SpectroCHIP® Array (silicon chip with pre-spotted matrix crystal) and loaded into the MassARRAY® Analyzer (a MALDI-TOF mass spectrometer). The analyte/matrix co-crystals were irradiated by a laser inducing desorption and ionization, and positively charged molecules accelerated into a flight tube towards a detector. Separation occurred by time-of-flight, which is proportional to molecular mass. After data processing, a spectral fingerprint was generated for each analyte that characterizes the mass/charge ratio and relative intensity of the molecules. Data acquired by the MassARRAY® Analyzer was processed with the MassARRAY® Typer software and SARS-CoV-2 Report software. The assay detects five viral targets: three in the nucleocapsid (N) gene (N1, N2, N3) and two in the ORF1ab gene (ORF1A, ORF1AB). If the IC was detected, results were interpreted as positive if ≥ 2 targets were detected or negative if < 2 targets were detected. If no IC and no targets were detected, the result was invalid and required repeat testing of the specimen before reporting. If IC was detected and no targets were detected, the sample was interpreted as negative.
Overall, 86,781 upper respiratory and saliva specimens underwent clinical testing in the CML at MSHS, during the period from December 1, 2020 through April 24, 2021. Of those specimens, 2,062 tested positive for SARS-CoV-2. A subset of 1,262 specimens were deidentified, related data was entered in to the MSHS PSP database, and underwent SARS-CoV-2 next-generation sequencing as previously described 25,26.
SARS-CoV-2 sequencing, assembly and phylogenetic analyses
SARS-CoV-2 viral RNA underwent reverse transcription, PCR amplification and next-generation sequencing followed by genome assembly and lineage assignment using a phylogenetic-based nomenclature as described by Rambaut et al. 27 using the PANGOLIN tool, version 2021-04-28 28 as previously described 25,26. Ultimately, this yielded 1,176 complete genomes (≥95% completeness) and 86 partial genomes (<95% completeness).
Agena target sequence alignment
Agena MassARRAY® target detection results were matched to the corresponding genome sequences. Primer and probe sequences for each Agena target were obtained from published FDA EUA documentation for the Agena MassARRAY® SARS-CoV-2 Panel (Table S1) 24. We generated reverse-complement sequences for reverse primers for all five targets and probes that are designed in the reverse orientation (e.g., N1-N3). An unaligned FASTA file including sequence data for the clinical specimens and the Wuhan-Hu-1 reference sequence (NCBI nucleotide: NC_045512.2 (Genbank: MN908947.3)) was generated for each of the fifteen primers/probes. The Multiple Alignment using Fast Fourier Transform (MAFFT) platform 29,30 which is publicly available for use online (https://mafft.cbrc.jp/alignment/server/add_fragments.html?frommanualnov6) was used to align each file. To enable inclusion of incomplete genomes that had intact regions sequenced at PBSs, we did not remove uninformative sequences (e.g., with ambiguous letters). Otherwise, the default settings were used to align all sequences to the reference genome, which generated a resulting FASTA alignment file for each primer and probe sequence.
Sequence variation in primer/probe target regions
To identify mismatches in the primer and probe regions of the viral genomes, FASTA alignment files were processed locally in a Bash environment. Custom Unix-code (https://github.com/AceM1188/SACOV_primer-probe_analyses) was used to identify mismatches at each nucleotide position within each primer and probe sequence 31. A tab-delimited output file that identified mismatches by primer/probe nucleotide position across the viral genome sequences was generated for each alignment.
Note that for the viral genome sequences with stretches of Ns that corresponded with the PBS, mismatches could not be called, and these sequences were excluded from the mismatch counting for the given primer/probe. In addition, genomes with gaps that spanned the entire region of a PBS were excluded from the analyses for the given primer/probe.
Mismatches by position in PBS regions of forward/reverse primer and probe sequences were manually counted on Microsoft Excel v16.48. To account for differences in completeness of consensus genomes, the number of PBS mismatches was normalized to the number of nucleotides in the PBS of each specimen consensus sequence.
Statistical analyses
For statistical comparison of fraction of PBS with mismatches in genomes with detected targets versus those with dropout targets, normality was assessed by D’Agostino and Pearson test (GraphPad Prism 9.1.0), which indicated that all distributions were non-parametric; thus, a Mann-Whitney test (two-tailed) was performed (GraphPad). To determine if specific mismatches were associated with specific target dropout results, specimens were grouped by (1) presence or absence of the mismatch of interest (in the setting of no other mismatches) and (2) detection or dropout of the target of interest – which resulted in a 2x2 contingency table which underwent association testing by Fisher’s exact test.
Display Items
All figures are original and were generated using the GraphPad Prism software 9.1.0, R software package ggplot2, NCBI Multiple Sequence Alignment Viewer v.1.17.0 (https://www.ncbi.nlm.nih.gov/tools/msaviewer/), and finished in Adobe Illustrator 2021 (v.25.2.1).
Results
Overall, of the 2,062 SARS-CoV-2-positive specimens, 1,274 (62%) had all five targets detected with the remaining having one (n = 419) or more (n = 369) targets dropout. For the subset of 1,262 SARS-CoV-2-positive specimens sequenced in our study, all five diagnostic targets were detected in 943 (75%), with the remaining having one (n = 227) or more (n = 92) target dropout (Table S2). When we calculated the target detection rate among these SARS-CoV-2-positive specimens by week, the ORF1AB target had the lowest average detection rate per week (0.87) followed by the N3 target (0.88) and the N2 target (0.94) (Figure 1). Notably, the N3 detection rate declined over time with the lowest detection occurring during the last four weeks of the timeframe studied (week ending April 3 (0.75) – April 24, 2021 (0.79)). Given these observations, we used the diagnostic data and corresponding genome sequences to identify mismatches to each primer/probe utilized by the Agena MassARRAY® platform to determine the impact on target detection results.
Figure 1. Agena target detection rate in SARS-CoV-2-positive specimens by week.
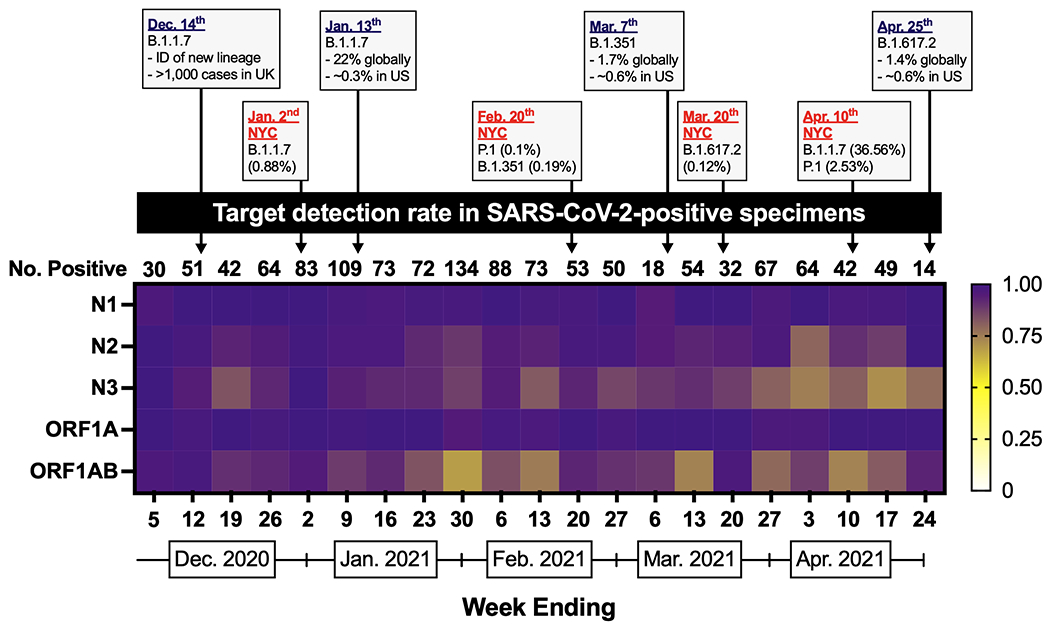
Heatmap depicting the proportion of sequenced SARS-CoV-2-positive specimens that have detectable N1, N2, N3, ORF1A, or ORF1AB targets by week from December 1, 2020 through April 24, 2021. International, national, and global statistics are indicated by dates in purple font. NYC statistics are indicated by dates in red font. Data for epidemiologic events obtained from 38,39,41,44,46. The number of sequenced SARS-CoV-2-positive specimens per week is indicated above each week (column).
Nucleotide mismatches across diagnostic targets
We aligned each forward primer, reverse primer, and probe sequence of the Agena MassARRAY® SARS-CoV-2 Panel to the set of 1,262 SARS-CoV-2 genome sequences.
To examine the impact of mismatches on target results, we measured the number of mismatches (normalized to the number of nucleotides in the PBS; see methods) in specimens with detected and undetected target results (Figure 2). Detection of each of four targets (N1, N3, ORF1A, ORF1AB) was associated with perfect complementarity (0 mismatches) between the genome sequence and the respective target PBSs. Specifically, > 96% of specimens with either detectable N1 or N3 targets had perfect complementarity to the respective forward/reverse/probe PBS, and > 95% of specimens with detectable ORF1A or ORF1AB targets had perfect complementarity to the respective forward/reverse/probe PBS. The remaining specimens had – at most – only one mismatch to each of the target PBSs. The exception to this was the N2 target, for which, more specimens with detectable N2 target had mismatches to N2 forward (43%) and N2 reverse (39%) PBSs (Figure 2B). Indeed, up to four mismatches to the N2 forward and up to two mismatches to the N2 reverse PBSs were found in the specimens for which the N2 target was detected.
Figure 2. Impact of SARS-CoV-2 primer/probe binding site mismatches on Agena target detection results.
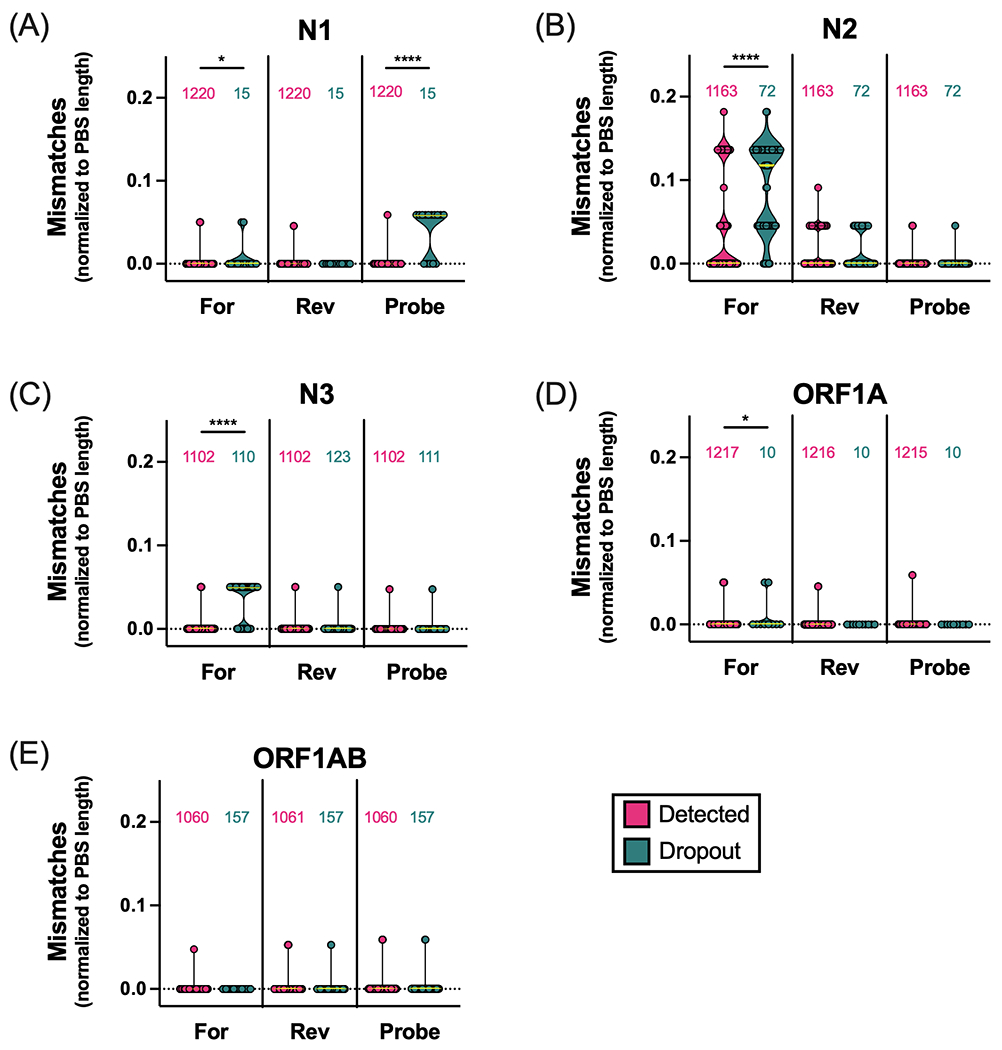
Number of mismatches normalized to the number of nucleotides in primer/probe binding sites (PBS length) across five Agena MassARRAY® diagnostic targets: (A) N1, (B) N2, (C) N3, (D) ORF1A, (E) ORF1AB. Each point represents the calculated mismatches per specimen consensus genome for each target PBS. Violin plots represent the distribution as density of the points grouped by primer/probe sequence (forward (For), reverse (Rev), Probe) and by target detection result (detected (magenta), dropout (turquoise)). The number of genome sequences analyzed for mismatches are depicted above each violin plot. Medians are depicted as yellow lines. Bars above distributions reflect statistical comparison of underlying distributions by Mann-Whitney test. Asterisks reflect p-values (*, p < 0.05; ****, p < 0.0001).
When compared across target result groups, the number of mismatches was significantly higher in specimens with N1, N2, N3 and ORF1A target dropout (Figure 2A–D). In addition, we found the fraction of N1 probe PBS with mismatches was significantly higher in specimens with N1 target dropout than in those with detectable N1 (Figure 2A).
Because the position of mismatches within PBSs affect primer binding capabilities 1,6–8, we characterized the mismatch frequency by position of each primer/probe. Specifically, we measured the proportion of specimen genomes with a mismatch at each independent position along the full length of each target’s primer/probe (Figure 3, Figure S1). From 5’ to 3’ direction, we found that among 15 specimens with N1 target dropout, 10 harbored single mismatches to the 4th – 14th basepair (bp) (SARS-CoV-2 genome positions 28714 – 28704) of the 17-bp-long N1 probe PBS (Figure 3A). Specifically, these mismatches reflected the following substitutions: G28714A (n = 1 specimens), G28713A (n = 2), C28709T (n = 2), C28706T (n = 1), G28704C (n = 3), G28704T (n = 1).
Figure 3. SARS-CoV-2 positional mismatches at target primer/probe binding sites.
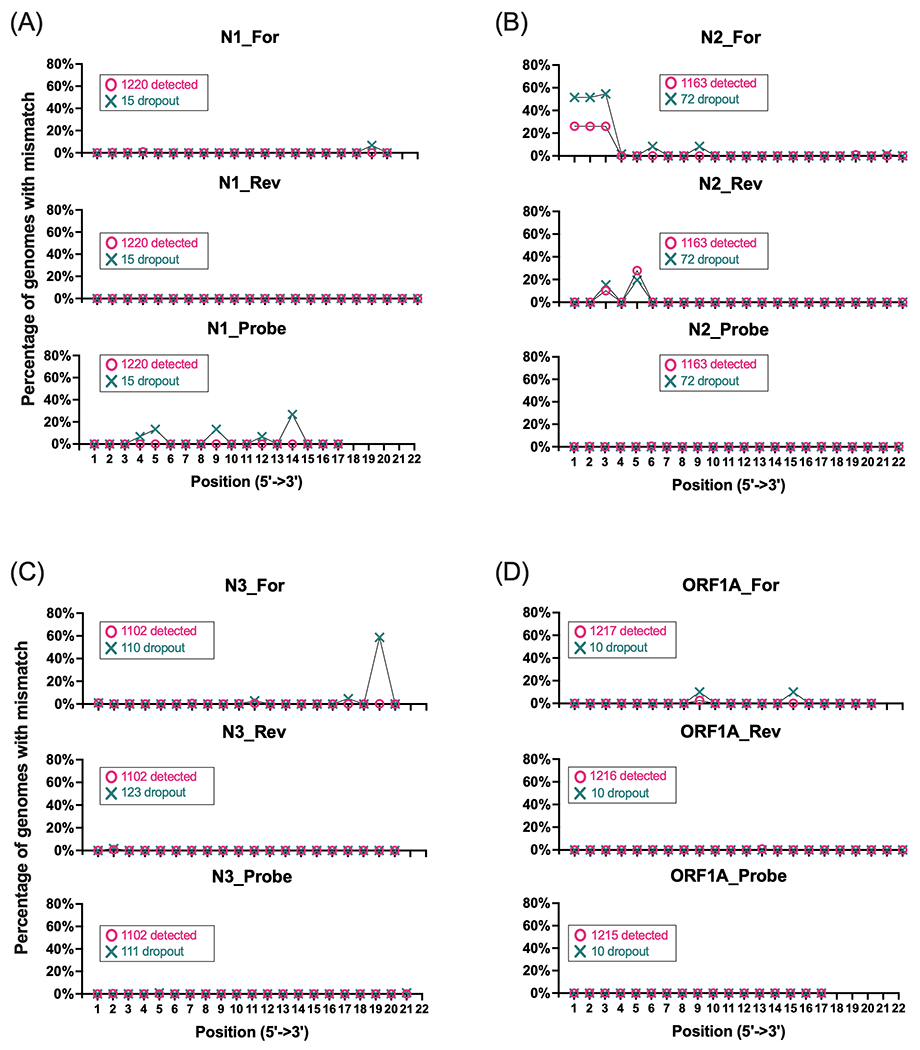
Line graphs depict the percentage of specimen genomes with mismatches at individual basepair positions across Agena MassARRAY® target PBSs: (A) N1, (B) N2, (C) N3, (D) ORF1A. There are three plots for each target that correspond with the forward (For), reverse (Rev), and Probe binding sites. Two line plots are depicted for each binding site to depict mismatches in genomes from specimens that yielded a detected target result (magenta) or target dropout (turquoise). The percentage represents the number of genomes with mismatches at each position relative to the number of genome sequences detected or not detected by each target (annotated in each graph).
By contrast, mismatches in the 5’ end of the 22-bp-long N2 primer PBSs (forward, 1st – 3rd bp (28881 – 28883); reverse, 3rd bp (28977) and 5th bp (28975)) were identified in sequences that yielded both N2 target detection and dropout (Figure 3B). In 340 specimens with any one mismatch to the first 3 bp of the N2 forward primer, 336 (99%) harbored the concurrent substitutions G28881A, G28882A, and G28883C in the N gene. Of the 72 specimen genomes with N2 target dropout, 34 (47%) had this substitution trio. Although, this polymorphism was found in 304 (26%) of the 1,163 specimen genomes with N2 target detection, statistically, this represents a significant association of the GGG-to-AAC substitution with N2 target dropout (Fisher’s exact, p = 0.0002).
In addition, specimens that harbor mismatches to the 5’ end of the N2 reverse primer are the result of the C28977T or G28975A substitutions. However, of 466 specimens that harbor either substitution, only 1 had both suggesting these substitutions occur independently of one another. When grouped by N2 target detection result, neither substitution was significantly associated with N2 target dropout (Fisher’s exact, p ≥ 0.1351).
Interestingly, we found that of the 110 specimen genomes with N3 target dropout, 63 (57%) had a mismatch at the penultimate nucleotide towards the 3’ end in the 20-bp-long N3 forward primer (Figure 3C). All mismatches at this position are the result of a specific adenine-to-thymine substitution in ORF8 (A28095T) of the SARS-CoV-2 genome. Of the 1,102 genomes with detected N3 target, only two harbored this mismatch; overall, this represents a statistically significant association of this positional mismatch with N3 target dropout (Fisher’s exact, p < 0.0001).
We also assessed whether the association of these mismatches with target dropout is maintained when the quantity of virus in the specimen is controlled. Although the Agena platform yields a qualitative diagnostic result, we have demonstrated previously that the number of detected targets is proportional to the quantity of virus in a given specimen 23. When we limit our dataset only to specimens for which all other (e.g., non-N3) targets are detected, the association of the A28095T substitution with N3 target dropout remains statistically significant (Fisher’s exact, p < 0.0001), indicating that N3 target dropout due to the A28095T substitution is independent of differences in virus concentration.
Lineage-specific variation and target dropout
In order to assess whether target dropout was due to lineage-specific variation, we examined the phylogenetic lineages of genomes harboring distinct substitutions in our dataset. Among the 34 specimens with the concurrent GGG-to-AAC tri-nucleotide substitution and N2 target dropout, the earliest was from December 29, 2020 (PV24926) which belonged to the B.1.1.434 lineage. This polymorphism did not demonstrate bias to any one lineage in specimens that yielded N2 target dropout as it was found in specimens that mapped to 15 different lineages including B.1.1.7 (alpha, n = 11), B.1.1.434 (n = 6), and B.1.1 (n = 4) lineages.
We next examined the phylogenetic lineage of genomes harboring the A28095T substitution in our dataset to assess whether N3 target dropout was due to lineage-specific variation. We found that the earliest specimen with this substitution was from January 8, 2021 (specimen PV25263) and belonged to the B.1.1.7 lineage. Indeed, the substitution appeared in a subset of genomes of the B.1.1.7 lineage. Interestingly, of the 127 B.1.1.7 genomes, approximately half (n = 65) harbored the A28095T substitution while the remaining maintained the adenine at the position (Figure 4). Ninety-seven percent (63/65) of the B.1.1.7 specimens with the A20895T substitution demonstrated N3 target dropout. Furthermore, the converse was also true as almost all B.1.1.7 specimens with N3 target dropout (63/64 (98%)) had the A28095T substitution (Figure 4A).
Figure 4. Lineage-specific substitution interferes with SARS-CoV-2 diagnostic target detection.
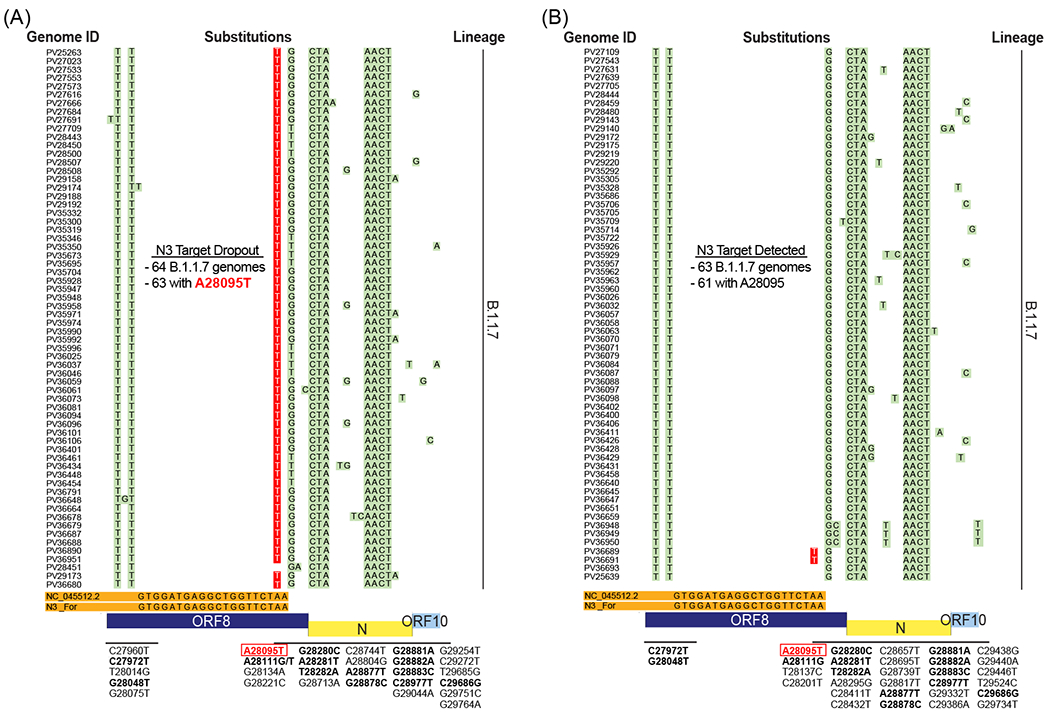
(A) Alignment of B.1.1.7 genomes associated with N3 target dropout. View is magnified to display mismatches across the N3 forward (For) primer binding site. Sixty-four specimen genomes are indicated by laboratory identifiers (Genome ID) and mismatches to the Wuhan-Hu-1 reference sequence (NC_045512.2) and the N3 For primer (orange) are highlighted in green. Substitutions that correspond with each of the mismatches are annotated below each panel. The lineage specific A28095T substitution that is associated with N3 target dropout is highlighted in red with white typeface font. (B) Alignment of B.1.1.7 genomes associated with N3 target detection. Sixty-three individual genomes are indicated by laboratory identifiers and mismatches are highlighted and annotated as in (A). Note for substitutions that are shared across both target results (e.g., dropout and detected), annotations are in boldface font.
Among the other 45 specimens with N3 target dropout, 10 harbored mismatches in the N3 PBS (Figure S2). Of the 10 genomes, only one (specimen PV36946) had the A28095T substitution, but the sequence recovered was incomplete (42% completeness) and a lineage could not be assigned. The other 9 genomes represented B.1.2 (n = 3), B.1.36.18 (n = 1), B.1.427 (n = 2), B.1.575 (n = 2), and B.1.621 (n = 1) lineages. Among these non-B.1.1.7 genomes, three mismatches were identified at the 1st (G28077T; n = 1), 11th (C28087T; n = 3) and 17th (C28093T; n = 5) bp of the N3 forward PBS which were encoded by viruses from multiple lineages. Of note, these non-B.1.1.7 genomes did not have any other mismatches in the N3 reverse or probe PBS. Furthermore, the two mismatches closest to the 3’ end of the N3 forward primer – C28087T and C28093T – were significantly associated with N3 target dropout (Fisher’s exact, p = 0.0407 and p < 0.0001, respectively), but the mismatch at the first position was not significantly associated with N3 target dropout (Fisher’s exact, p = 0.0914).
Discussion
Molecular assays for the diagnosis of COVID-19 developed early in the pandemic utilize primers and probes based on conserved regions in the then-available SARS-CoV-2 genome sequences. Now, more than 18 months later, circulating SARS-CoV-2 variants have accumulated numerous nucleotide substitutions in response to evolutionary pressures. These genomic variations can be associated with increased infectivity, transmissibility, and disease pathogenesis 32–36, warranting accurate and quick surveillance efforts. However, genome variation can also be a challenge for detection of these variants of interest (VOIs) or VOCs if mismatches to PBSs in diagnostic targets are present. A number of studies have described substitutions in the ORF1ab, S, E, and N genes that may interfere with specific RT-PCR targets 1,5,9,11,12,14–16,18, but these studies have inherent limitations. Several are in silico analyses that do not reflect diagnostic performance in the clinical setting 1,5,16,18, whereas others do not definitively demonstrate target dropout due to substitutions as they utilize platforms for which primer/probe sequence information is not publicly available 9,11,14,15. In addition, the later studies are based on assays that interrogate up to 3 diagnostic targets, and are limited by the number and diversity of viral sequences surveyed over finite timeframes, some prior to the emergence and expansion of VOCs.
In the current study, we describe a robust evaluation of the impact of PBS mismatches on Agena MassARRAY® SARS-CoV-2 Panel target results for over 1,200 specimens over a five-month time period that corresponds with the rapid emergence of viral VOIs/VOCs (December 2020 through April 2021). This large dataset enabled direct correlation of detection of each of five different Agena diagnostic viral targets with genomic sequence data
Additionally, by using publicly available primer/probe sequences to map lineage-specific substitutions, we were able to further evaluate the impact of mismatches on target results and to demonstrate an association between variation in SARS-CoV-2 PBSs and target dropout. Our analysis revealed that several mutations result in N1 and N3 target dropout. Interestingly, although specimens from other lineages harbor mismatches in the N3 target region, we identified a distinct association between the B.1.1.7-associated A28095T substitution and dropout of the N3 diagnostic target on the Agena MassARRAY® SARS-CoV-2 Panel. This finding represents the first description of a lineage-specific substitution that introduces a mismatch to a publicly available primer sequence and yields diagnostic target dropout. This underscores the utility of publicly available sequences to further monitor their diagnostic ability as SARS-CoV-2 continues to evolve and new lineages emerge.
The B.1.1.7 lineage (alpha) has been designated as a variant of concern by the World Health Organization and the US Centers for Diseases Control and Prevention due to its increased transmissibility 20,33–35,37. This lineage was first reported in > 1,100 cases in the United Kingdom (UK) on December 14, 2020 38, but it has been estimated to have emerged in September 2020 20,35. Since then, the B.1.1.7 lineage spread rapidly, comprising > 90% of new SARS-CoV-2 infections in the UK by March 2021 35. This lineage also spread globally, including in the US 39,40, where recent epidemiological reports indicate B.1.1.7 variants caused > 60% of the new infections as of May 6, 2021 41.
These characteristics further underscore the urgency to update and continually develop robust screening modalities to capture VOCs like B.1.1.7. Sequencing of these variants remains the ‘gold standard’ of surveillance, but not all diagnostic laboratories have the infrastructure or capacity to readily utilize this technology. S gene target failure (SGTF) on commercial RT-PCR platforms has been proposed as a screening alternative to detect the S protein deletion H69-V70 (ΔH69/ΔV70) 14,15,42,43 which is found in B.1.1.7 and, to a lesser extent, in other circulating lineages (e.g., B.1.525, B.1.620 (NextStrain, build June 23, 2021)) 15.
In this study, we describe diagnostic target dropout that can be utilized to promptly identify specimens of interest for whole genome sequencing and variant classification. Notably detection of the B.1.1.7 variant containing the A28095T substitution is associated with the Agena N3 target dropout. This substitution is characteristic of 50% of the circulating B.1.1.7 specimens in our dataset and reflects a unique snapshot of genomic variation occurring within the circulating B.1.1.7 variants in New York City. The A28095T substitution introduces a stop codon in the ORF8 gene producing a truncated version with potential functional changes in encoded protein (K68stop). Among a dataset of >2 million publicly available viral genomes from global surveillance efforts (GISAID, June 23, 2021), 309,050 genomes harbor this substitution. Nearly all (99.9%) of these A28095T-genomes belong to the B.1.1.7 lineage which, in turn, represent a sub-population (33.9%) of all B.1.1.7 genomes. Therefore, continued diagnostic surveillance of the Agena N3 target dropout and subsequent genomic surveillance can be exploited to monitor the spread of the B.1.1.7 variant as well as other VOCs. Indeed, based on publicly available genomes, other VOCs harbor substitutions that result in mismatches to Agena target PBSs. For example, 73% of delta (B.1.617.2) variant genomes have the non-synonymous substitution, G28916T in the N gene (amino acid change, G215C) which introduces a mismatch to the terminal bp of the N2 probe (GISAID, June 23, 2021). Given its position in the probe, this mismatch likely impacts N2 target performance. This warrants further study, particularly as the B.1.617.2 VOC continues to expand globally 44–46 since its parent lineage was first identified in India in October 2020 47–49.
An important potential limitation of our study is that target performance can be affected when the quantity of viral nucleic acids in diagnostic specimens is at or near the assay limit of detection, and that the limit of detection varies for different targets. We have demonstrated previously that Agena MassARRAY® target detection is proportional to quantity of viral nucleic acids 23. Thus, detection of other targets can be used as a control to evaluate the performance of an individual target, such as N3 target dropout in the setting of the B.1.1.7 A28095T variant. In addition, we have also identified other mutations in this study that are associated with N1 and N3 target dropout. However, unlike the B.1.1.7 A28095T/A genomes in our dataset, these are fewer in number and further comprehensive evaluation is needed to determine the definitive impact on target performance.
Assay platforms that incorporate testing of multiple targets within the virus genome are more likely to retain diagnostic sensitivity as SARS-CoV-2 continues to diversify and new variants emerge. Diagnostic target performance patterns on these redundant platforms have the potential to accommodate unfolding genomic variation in a timely manner, and highlight the potential of diagnostic results to serve as a robust system for detection of these emergent SARS-CoV-2 variants. These qualities demonstrate the importance of these platforms to capture the evolutionary consequences of the ongoing pandemic to inform public health and infection prevention measures.
Supplementary Material
Acknowledgments
We thank the members of MSHS CML, Simon, and van Bakel laboratories for providing any assistance when needed throughout this study. We are grateful for the continuous expert guidance provided by the ISMMS Program for the Protection of Human Subjects (PPHS).
The Research reported in this paper was supported by the National Institutes of Health (NIH) contract number HHSN272201400008C, the NIH Office of Research Infrastructure under award numbers S10OD018522 and S10OD026880, institutional and philanthropic funds (Open Philanthropy Project, #2020-215611), as well as a Robin Chemers Neustein Postdoctoral Fellowship Award (to Dr. Gonzalez-Reiche).
Footnotes
Code availability
To generate genome sequences, sequencing data were analyzed using a custom reference-based (MN908947.3) pipeline, https://github.com/mjsull/COVID_pipe 50. To analyze mismatches to diagnostic target PBSs, genome sequences were analyzed using a custom Unix-code https://github.com/AceM1188/SACOV_primer-probe_analyses 31.
Competing Interests
Robert Sebra is VP of Technology Development and a stockholder at Sema4, a Mount Sinai Venture. This work, however, was conducted solely at Icahn School of Medicine at Mount Sinai. Otherwise, the authors declare no competing interests.
Data availability
SARS-CoV-2 sequencing read data for all study genomes were deposited in GISAID [www.gisaid.org] (EPI_ISL_882805 – EPI_ISL_5336552) and GenBank [https://www.ncbi.nlm.nih.gov/genbank/] databases. Specimen and corresponding database accession identifiers are annotated in Table S2.
References
- 1.Wang R, Hozumi Y, Yin C, Wei G-W. Mutations on COVID-19 diagnostic targets. Genomics. 2020;112(6):5204–5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan JF-W, Yip CC-Y, To KK-W, et al. Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens. J Clin Microbiol. 2020;58(5). doi: 10.1128/JCM.00310-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Udugama B, Kadhiresan P, Kozlowski HN, et al. Diagnosing COVID-19: The disease and tools for detection. ACS Nano. 2020;14(4):3822–3835. [DOI] [PubMed] [Google Scholar]
- 5.Gand M, Vanneste K, Thomas I, et al. Deepening of In Silico Evaluation of SARS-CoV-2 Detection RT-qPCR Assays in the Context of New Variants. Genes . 2021;12(4). doi: 10.3390/genes12040565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwok S, Kellogg DE, McKinney N, et al. Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990;18(4):999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefever S, Pattyn F, Hellemans J, Vandesompele J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 2013;59(10):1470–1480. [DOI] [PubMed] [Google Scholar]
- 8.Whiley DM, Sloots TP. Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J Clin Virol. 2005;34(2):104–107. [DOI] [PubMed] [Google Scholar]
- 9.Tahan S, Parikh BA, Droit L, Wallace MA, Burnham CA, Wang D. SARS-CoV-2 E gene variant alters analytical sensitivity characteristics of viral detection using a commercial RT-PCR assay. J Clin Microbiol. Published online April 26, 2021. doi: 10.1128/JCM.00075-21 [DOI] [PMC free article] [PubMed]
- 10.Vanaerschot M, Mann SA, Webber JT, et al. Identification of a polymorphism in the N gene of SARS-CoV-2 that adversely impacts detection by reverse transcription-PCR. J Clin Microbiol. 2020;59(1):e02369–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Artesi M, Bontems S, Göbbels P, et al. A Recurrent Mutation at Position 26340 of SARS-CoV-2 Is Associated with Failure of the E Gene Quantitative Reverse Transcription-PCR Utilized in a Commercial Dual-Target Diagnostic Assay. J Clin Microbiol. 2020;58(10). doi: 10.1128/JCM.01598-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogels CBF, Brito AF, Wyllie AL, et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat Microbiol. 2020;5(10):1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziegler K, Steininger P, Ziegler R, Steinmann J, Korn K, Ensser A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Euro Surveill. 2020;25(39). doi: 10.2807/1560-7917.ES.2020.25.39.2001650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown KA, Gubbay J, Hopkins J, et al. S-Gene Target Failure as a Marker of Variant B.1.1.7 Among SARS-CoV-2 Isolates in the Greater Toronto Area, December 2020 to March 2021. JAMA. 2021;325(20):2115–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bal A, Destras G, Gaymard A, et al. Two-step strategy for the identification of SARS-CoV-2 variant of concern 202012/01 and other variants with spike deletion H69-V70, France, August to December 2020. Euro Surveill. 2021;26(3). doi: 10.2807/1560-7917.ES.2021.26.3.2100008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gand M, Vanneste K, Thomas I, et al. Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection. Int J Mol Sci. 2020;21(15). doi: 10.3390/ijms21155585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahamtan A, Ardebili A. Real-time RT-PCR in COVID-19 detection: issues affecting the results. Expert Rev Mol Diagn. 2020;20(5):453–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan KA, Cheung P. Presence of mismatches between diagnostic PCR assays and coronavirus SARS-CoV-2 genome. R Soc Open Sci. 2020;7(6):200636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercer TR, Salit M. Testing at scale during the COVID-19 pandemic. Nat Rev Genet. Published online May 4, 2021. doi: 10.1038/s41576-021-00360-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Published December 18, 2020. Accessed May 21, 2021. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- 21.In Vitro Diagnostics EUAs. U.S. Food and Drug Administration. Published Febraury 17, 2021. Accessed February 21, 2021. https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas#individual-molecular
- 22.Rybicka M, Miłosz E, Bielawski KP. Superiority of MALDI-TOF mass spectrometry over real-time PCR for SARS-CoV-2 RNA detection. Viruses. 2021;13(5). doi: 10.3390/v13050730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez MM, Banu R, Shrestha P, et al. RT-PCR/MALDI-TOF mass spectrometry-based detection of SARS-CoV-2 in saliva specimens. J Med Virol. Published online May 8, 2021. doi: 10.1002/jmv.27069 [DOI] [PMC free article] [PubMed]
- 24.Agena Bioscience, Inc. MassARRAY® SARS-CoV-2 Panel Instructions for Use.; 2021. https://agenabio.com/wp-content/uploads/2020/04/GEN0027-02-CoV2-EUA-Product-Sheet-WEB.pdf
- 25.Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science. 2020;369(6501):297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez MM, Gonzalez-Reiche AS, Alshammary H, et al. Molecular evidence of SARS-CoV-2 in New York before the first pandemic wave. Nat Commun. 2021;12(1):3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rambaut A, Holmes EC, O’Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Toole Á, Scher E, Underwood A, et al. pangolin: lineage assignment in an emerging pandemic as an epidemiological tool. Github. https://github.com/cov-lineages/pangolin [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katoh K, Misawa K, Kuma K-I, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu T, Li J, Zhou H, Li C, Holmes EC, Shi W. Bioinformatics resources for SARS-CoV-2 discovery and surveillance. Brief Bioinform. 2021;22(2):631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez (AceM1188) MM. AceM1188/SACOV_primer-probe_analyses. Zenodo. Published May 22, 2021. Accessed June 9, 2021. https://10.5281/zenodo.4920818 [Google Scholar]
- 32.Sanders RW, de Jong MD. Pandemic moves and countermoves: vaccines and viral variants. Lancet. 2021;397(10282):1326–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caniels TG, Bontjer I, van der Straten K, et al. Emerging SARS-CoV-2 variants of concern evade humoral immune responses from infection and vaccination. medRxiv. Published online June 1, 2021. doi: 10.1101/2021.05.26.21257441 [DOI] [PMC free article] [PubMed]
- 34.Volz E, Mishra S, Chand M, et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. bioRxiv. Published online January 4, 2021. doi: 10.1101/2020.12.30.20249034 [DOI]
- 35.Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372(6538). doi: 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kupferschmidt K Danish scientists see tough times ahead as variant rises. Science. 2021;371(6529):549–550. [DOI] [PubMed] [Google Scholar]
- 37.Kemp SA, Meng B, Ferriera IA, et al. Recurrent emergence and transmission of a SARS-CoV-2 spike deletion H69/V70. bioRxiv. Published online March 8, 2021:2020.12.14.422555. doi: 10.1101/2020.12.14.422555 [DOI]
- 38.Public Health England. PHE investigating a novel variant of COVID-19.https://www.gov.uk/government/news/phe-investigating-a-novel-variant-of-covid-19. Published December 14, 2020.
- 39.Galloway SE, Paul P, MacCannell DR, et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage - United States, December 29, 2020-January 12, 2021. MMWR Morb Mortal Wkly Rep. 2021;70(3):95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Firestone MJ, Lorentz AJ, Wang X, et al. First Identified Cases of SARS-CoV-2 Variant B.1.1.7 in Minnesota - December 2020-January 2021. MMWR Morb Mortal Wkly Rep. 2021;70(8):278–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paul P, France AM, Aoki Y, et al. Genomic Surveillance for SARS-CoV-2 Variants Circulating in the United States, December 2020-May 2021. MMWR Morb Mortal Wkly Rep. 2021;70(23):846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Methods for the detection and identification of SARS-CoV-2 variants. Published March 3, 2021. Accessed June 23, 2021. https://www.ecdc.europa.eu/en/publications-data/methods-detection-and-identification-sars-cov-2-variants
- 43.Braybrook E, Pandey S, Vryonis E, Anderson NR, Young L, Grammatopoulos D. Screening for the alpha variant of SARS-CoV-2 (B.1.1.7) and the impact of this variant on circulating biomarkers in hospitalised patients. bioRxiv. Published online June 21, 2021. doi: 10.1101/2021.06.18.21258699 [DOI] [Google Scholar]
- 44.Centers for Disease Control and Prevention, US Department of Health and Human Services. CDC COVID Data Tracker. Accessed July 4, 2021. https://covid.cdc.gov/covid-data-tracker/#variant-proportions
- 45.Coronavirus Disease (COVID-19): Weekly Epidemiological Update (29 June 2021) - World. Accessed July 4, 2021. https://reliefweb.int/report/world/coronavirus-disease-covid-19-weekly-epidemiological-update-29-june-2021
- 46.New York City Department of Health. COVID-19: Data. NYC Health COVID-19: Data on Variants. Accessed July 4, 2021. https://www1.nyc.gov/site/doh/covid/covid-19-data-variants.page
- 47.The Delta Variant: What Scientists Know. The New York Times. Published June 22, 2021. Accessed July 4, 2021. https://www.nytimes.com/2021/06/22/health/delta-variant-covid.html [Google Scholar]
- 48.WHO EMRO Weekly Epidemiological Monitor: Volume 14; Issue no 19; 9 May 2021. Accessed July 4, 2021. https://reliefweb.int/report/afghanistan/who-emro-weekly-epidemiological-monitor-volume-14-issue-no-19-9-may-2021 [Google Scholar]
- 49.GISAID - hCov19 Variants. Tracking of Variants. Accessed July 4, 2021. https://www.gisaid.org/hcov19-variants/ [Google Scholar]
- 50.mjsull. mjsull/COVID_pipe: initial release (Version v0.1.0). Zenodo. Published 2020, April 29. 10.5281/zenodo.3775031 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SARS-CoV-2 sequencing read data for all study genomes were deposited in GISAID [www.gisaid.org] (EPI_ISL_882805 – EPI_ISL_5336552) and GenBank [https://www.ncbi.nlm.nih.gov/genbank/] databases. Specimen and corresponding database accession identifiers are annotated in Table S2.