

Abstract
The main protease (Mpro) and papain‐like protease (PLpro) play critical roles in SARS‐CoV‐2 replication and are promising targets for antiviral inhibitors. The simultaneous visualization of Mpro and PLpro is extremely valuable for SARS‐CoV‐2 detection and rapid inhibitor screening. However, such a crucial investigation has remained challenging because of the lack of suitable probes. We have now developed a dual‐color probe (3MBP5) for the simultaneous detection of Mpro and PLpro by fluorescence (or Förster) resonance energy transfer (FRET). This probe produces fluorescence from both the Cy3 and Cy5 fluorophores that are cleaved by Mpro and PLpro. 3MBP5‐activatable specificity was demonstrated with recombinant proteins, inhibitors, plasmid‐transfected HEK 293T cells, and SARS‐CoV‐2‐infected TMPRSS2‐Vero cells. Results from the dual‐color probe first verified the simultaneous detection and intracellular distribution of SARS‐CoV‐2 Mpro and PLpro. This is a powerful tool for the simultaneous detection of different proteases with value for the rapid screening of inhibitors.
Keywords: Inhibitor Screening, Main Protease, Papain-Like Protease, SARS-CoV-2, Simultaneous Visualization
The simultaneous visualization of crucial proteases within SARS‐CoV‐2‐infected cells can be a significant benefit for the accurate detection of virus and rapid screening of inhibitors as well as for understanding the viral lifecycle. The catalytic efficiency, intracellular enzyme activity, and distribution of SARS‐CoV‐2 main protease and papain‐like protease have now been exploited using peptide‐based dual‐color FRET probes and their inhibitors.
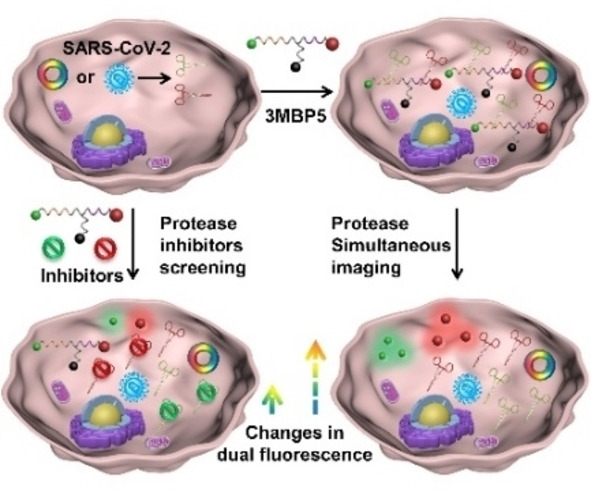
Introduction
The coronavirus disease 2019 (COVID‐19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) continues to threaten global health.[ 1 , 2 ] The detection of SARS‐CoV‐2 RNAs and antibodies has been widely explored using the quantitative reverse transcription polymerase chain reaction, isothermal nucleic acid amplification methods, lateral flow assays, and electrochemical chips. [3] Understanding the viral lifecycle and distribution of relevant proteases are critical to controlling disease propagation, diagnosing infection, and screening therapies. The main protease (Mpro, also known as 3CLpro) and papain‐like protease (PLpro) are specifically encoded from the open‐reading frames (ORFs) of the coronavirus RNA genome.[ 4 , 5 ] Mpro and PLpro are essential nonstructural proteins (NSPs) for processing the viral precursor polyprotein to form functional proteins during viral replication. Their amino acid sequences are highly conserved in SARS‐CoV, SARS‐CoV‐2, and Middle East respiratory syndrome (MERS). [6] Importantly, these enzymes are not closely related to any human protease, thus making them selective biomarkers for virus detection and drug development, since targeting Mpro and PLpro are unlikely to target the host proteases. However, the potential spatial and temporal distribution as well as the functionalities of Mpro and PLpro in SARS‐CoV‐2‐infected cells remain unclear. [7]
Numerous methods have been applied to simultaneously detect multiple biomarkers in complex biological samples, such as recombinant fluorescent proteins, and nanoparticle‐based optical and electrochemical sensors. [8] Among them, fluorescence (or Förster) resonance energy transfer (FRET) probes that report biochemical processes in living cells have generated great interest because of their rapid signal acquisition, high detection sensitivity, low background, and non‐invasiveness. [9] Furthermore, multicolor FRET (mFRET) has been broadly used to determine distances, structures, interactions, and dynamics of multiple biomolecules. [10] Mpro cleaves viral polyproteins after the Leu‐Gln (LQ) sequence, and PLpro cleaves viral polyproteins after the Leu‐Xaa‐Gly‐Gly (LXGG, where Xaa represents any amino acid) sequence. [11] PLpro can remove the ubiquitin‐like ISG15 protein modifications in addition to Lys48‐linked polyubiquitin through domain interaction to regulate viral spread and innate immunity. [12] Thus, Mpro and PLpro are suitable targets for FRET‐based detection.
There are a variety of commercially available and research‐grade substrate‐screening tools available, including several for Mpro and PLpro. Recently reported substrates show high specificity and good selectivity. [13] However, these are all single‐channel screens and it can be difficult to confirm if the two different probes are internalized in the same cell at similar concentrations for precise cell imaging analysis.[ 12a , 14 ] It could be more accurate and effective to image both proteases with a dual‐color FRET probe. However, no such probe is available for the simultaneous detection of Mpro and PLpro, despite the potential value in understanding the virus lifecycle and treatment impact arising from the simultaneous imaging of Mpro and PLpro in SARS‐CoV‐2‐infected cells.
Here we designed and synthesized a dual‐color fluorescent probe named 3MBP5 for the simultaneous visualization of Mpro and PLpro in SARS‐CoV‐2‐infected cells. 3MBP5 comprises five segments (Scheme 1a): 1) a fluorophore, Cyanine3 (Cy3, 3), 2) a Mpro‐responsive peptide (SAVLQ/SGFRKMA, M), 3) a black hole quencher‐2 (BHQ‐2, B), 4) a PLpro‐responsive peptide (RLRGG/K, P), and 5) another fluorophore, Cyanine5 (Cy5, 5).
Scheme 1.
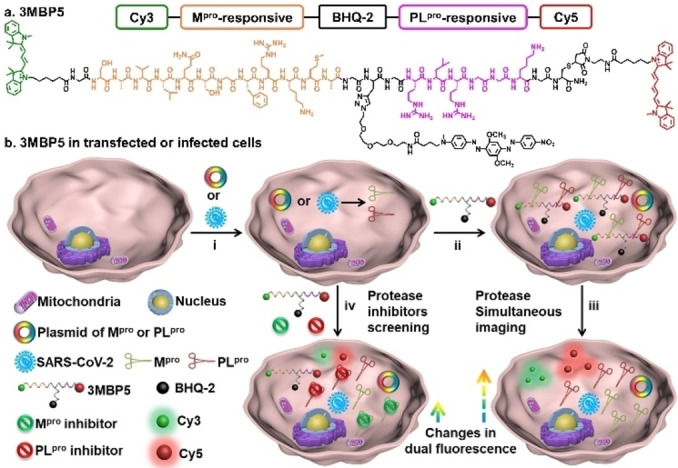
Structure and function of 3MBP5. a) Molecular structure of 3MBP5. b) 3MBP5 is used for the simultaneous visualization of SARS‐CoV‐2 Mpro and PLpro, and rapid inhibitor screening. i) Mpro and PLpro plasmid transfected or SARS‐CoV‐2‐infected cells can produce Mpro and PLpro; ii) 3MBP5 was incubated with the transfected or infected cells; iii) after being cleaved by Mpro and PLpro, the fluorescence of the Cy3 and Cy5 fragments recovered without spatial proximity to the BHQ‐2 quencher; iv) 3MBP5 fluorescence remains quenched when the cells were incubated with Mpro and PLpro inhibitors.
The combined segments are abbreviated as 3MBP5. These components were covalently linked using a combination of Fmoc‐based solid‐phase peptide synthesis, thiol‐maleimide Michael addition, and a copper‐catalyzed azide‐alkyne click reaction. The fluorescence of 3MBP5 in both the Cy3 and Cy5 channels is effectively quenched by BHQ‐2 when the peptide is intact (Scheme 1b). After cleavage by Mpro and PLpro, the fluorescence intensities of Cy3 and Cy5 will significantly increase since they are no longer quenched by BHQ‐2. Upon addition of Mpro and PLpro inhibitors, the protease activities of Mpro and PLpro are inhibited, and the fluorescence signals of Cy3 and Cy5 will decrease as the inhibitor concentrations increase. Therefore, the activities of Mpro, PLpro, and their inhibitors can be monitored by fluorescence changes. This well‐designed and robust dual‐color probe provides a novel opportunity for the simultaneous detection of protease and the rapid screening of inhibitors.
Results and Discussion
To simultaneously detect Mpro and PLpro in vitro and in cells, the commercial fluorescent dyes Cy3, Cy5, and BHQ‐2 were chosen as optimal FRET pairs as a result of the sizable overlap of their absorption and emission spectra. [15] Cy3 and Cy5 showed strong absorption bands at 450–580 nm and 550–700 nm, respectively, in a mixed solution of DMSO/water (v/v=1 : 99) at room temperature (Supporting Information Figure S1). To avoid the overlap of the emission spectra of Cy3 and Cy5, 500 nm and 600 nm were selected as the optimal excitation wavelengths for Cy3 and Cy5, respectively. The fluorescence bands of Cy3 and Cy5 were evident at 530–700 nm and 630–800 nm, respectively. Taking the spatial distance between the donor and acceptor into consideration, we designed 3MBP5 with the two donors (Cy3 and Cy5) at the two ends of a protease‐cleavable peptide, and one acceptor (BHQ‐2) in the center. [16] We then chose the Cy3‐BHQ‐2 pair for Mpro detection and the Cy5‐BHQ‐2 pair for PLpro detection based on FRET efficiency (EFRET) and substrate length (Scheme S1). The experimental calculated Förster radius (rDA) is smaller than the estimated Förster radius (RDA), thus indicating FRET in Cy3–Cy5 (4.40–<6.20 nm), Cy3‐BHQ‐2 (2.73–<5.09 nm), and Cy5‐BHQ‐2 (3.48–<4.75 nm; Table S1). [17] Furthermore, the lowest energy structure and center‐to‐center separation distance were analyzed by Molecular Operating Environment (MOE) software (Figure S2). The extracted donor–acceptor distances agreed well with the molecular simulations, and indicated that Cy3, Cy5, and BHQ‐2 can be recruited for a dual‐color FRET probe.
Azide‐functionalized BHQ‐2 was first synthesized (Scheme S2). Cysteine (Cys) and propargylglycine (Pra) linkers were reacted with maleimide‐ and azide‐functionalized dyes, respectively. Glycine (Gly) spacers were used to enhance the molecular flexibility and reduce steric hindrance (Scheme S3). [18] Before cleavage off the resin, Cy3 was linked to Gly on the N‐terminus through an amidation reaction. Then, maleimide‐functionalized Cy5 was coupled to Cys by a thiol‐maleimide Michael addition. Finally, azide‐functionalized BHQ‐2 was covalently bound to Pra through a copper‐catalyzed azide‐alkyne click reaction. [19]
Control probes verified the substrate specificity and enzyme activity of the Cy5‐BHQ‐2 pair: BRM5 (R denotes the abbreviation of tetra‐polyarginine) for Mpro detection, BRP5 for PLpro detection, and 3EBP5. Here, E is the abbreviation for the Mpro‐responsive sequence (with a Gln to Glu mutation to prevent the Mpro‐substrate interactions) for PLpro detection (Figure 1a, Schemes S4–S6). A hydrophilic tetra‐polyarginine was incorporated into BRM5 and BRP5 to enhance their solubility and cell permeability. [20] This motif can promote probe entry into cells and reduce probe degradation in the cell microenvironment.
Figure 1.
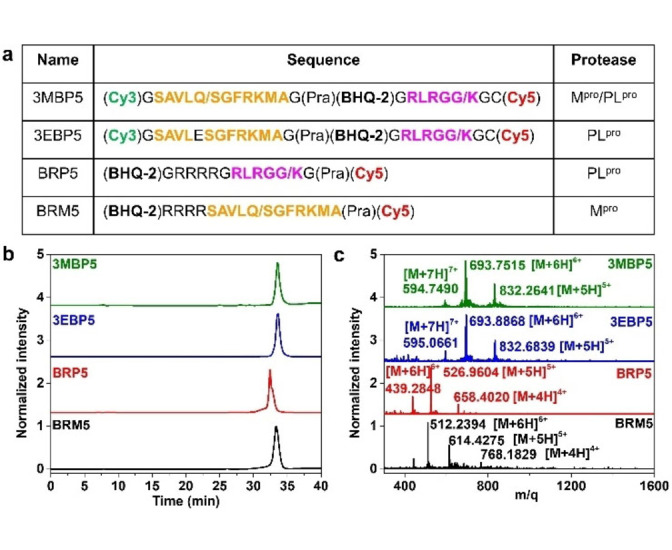
Characteristics of 3MBP5 and its derivatives. a) Molecular structures and peptide sequences, b) high‐performance liquid chromatography (HPLC), and c) electrospray ionization mass spectrometry (ESI‐MS) analysis of 3MBP5, 3EBP5, BRP5, and BRM5 confirm that all the probes were prepared with at least 95 % purity. The orange peptide sequence represents Mpro binding and the cleavage fraction. The pink peptide sequence represents PLpro binding and the cleavage fraction. The slash represents the protease cleavage site.
All products were purified by high‐performance liquid chromatography (HPLC) (Figure 1b). Their chemical structures were characterized by electrospray ionization mass spectrometry (ESI‐MS) or nuclear magnetic resonance (NMR) spectroscopy (Figures 1c, S3–S9). Taking 3MBP5 as an example, as shown in Figure 1c, a strong signal was evident at 832.2641 attributed to the [M+5H]5+ ion of 3MBP5 (calculated, 832.0489), a strong signal at 693.7515 attributed to the [M+6H]6+ ion of 3MBP5 (calculated, 693.5421), and a strong signal at 594.7490 attributed to the [M+7H]7+ ion of 3MBP5 (calculated, 594.6086). Importantly, the exact molecular weights of hydrolyzed five‐membered rings of maleimide‐functionalized Cy5 can be observed in the mass spectra of 3EP5, 3MP5, 3EBP5, and 3MBP5 as a result of the reversible thiol‐maleimide conjugation reaction (Figures S10, S11). [21] These results confirmed that 3MBP5, 3EBP5, BRM5, and BRP5 were successfully synthesized.
We evaluated whether these probes can be specifically cleaved by recombinant Mpro and PLpro. HPLC and ESI‐MS results showed that the BRM5 and 3MBP5 probes were cleaved by Mpro after the AVLQ site on incubation in 20 mM Tri‐HCl buffer (pH 8.0) for 1 h at 37 °C. The cleavage efficiency of BRM5 and 3MBP5 by Mpro was 85.52 % and 93.39 %, respectively (Figures S12–S14, S18). Under the same assay conditions, the BRP5 and 3MBP5 probes were cleaved by PLpro after the RLRGG site, but the cleavage efficiency was only 54.34 % and 12.63 %, respectively (Figures S15–S17, S19–S22). These results demonstrated that 3MBP5 can be cleaved by both Mpro and PLpro at engineered sites with different efficiencies, and the PLpro activity is greatly affected by the chemical structure.
We then studied the potential optical spectral properties of 3MBP5 with Mpro, PLpro, and their inhibitors (Figure 2a). BHQ‐2 showed a complex absorption profile at 400–720 nm (Figure 2b). The fluorescence intensity of Cy5 but not Cy3 in 3MP5 was clearly enhanced on excitation at 500 nm, thus revealing a significant FRET effect between Cy3 and Cy5 (yellow line, Figure 2c). After being modified with BHQ‐2, the fluorescence intensities of Cy3 and Cy5 in 3MBP5 clearly decreased as a result of the effective quenching efficiency of BHQ‐2 (pink lines, Figure 2c,d). According to the fluorescence changes of Cy5 before and after modification with peptide and BHQ‐2 in BRM5, 2 μM of the probe was considered the optimal concentration for protease detection (Figure S23). In addition, BRM5, BRP5, 3EBP5, and 3MBP5 were incubated with different concentrations of Mpro and PLpro to validate the probe sensitivity. As seen in Figures 2e,f, S24, and S25, the fluorescence intensities of BRM5 and 3MBP5 but not 3EBP5 were gradually enhanced as the concentration of Mpro increased. BRP5, 3EBP5, and 3MBP5 were all activated by PLpro.
Figure 2.

Photophysical properties and probe activation by proteases. a) Schematic representation of the FRET effect between Cy3, Cy5, and BHQ‐2 in 3MBP5 with Mpro, PLpro, and inhibitors. b–d) The UV/Vis absorption and fluorescence spectra of 10 μM Cy3, BHQ‐2, Cy5, 3MP5, and 3MBP5 showed good spectral overlap for FRET. e,f) The fluorescence spectra and g,h) fluorescent kinetics of 2.0 μM 3MBP5 with Mpro and PLpro showed a dual‐color increase in the fluorescence signal. i,j) Probe specificity to the target Mpro and PLpro as shown by the fluorescence response of 2.0 μM 3MBP5 with 100 nM Mpro, PLpro, Mpro and PLpro, thrombin (TB), hemoglobin (HGB), and bovine serum albumin (BSA). k,l) Impact of protease inhibitors as studied with 2.0 μM 3MBP5 and 100 nM Mpro, 1.0 μM PLpro, GC376, and GRL0617; the inhibitor decreased the 3MBP5 signal. In (k) and (l), the turquoise line denotes no inhibitor, and the taupe line denotes probe only (no protease, no inhibitor). All other colors represent different inhibitor concentrations. For all panels, the probes were dissolved in Tris‐HCl buffer with 1 % DMSO. The excitation wavelength for Cy3 is 500 nm in (c, e, g, i, and k). The excitation wavelength for Cy5 is 600 nm in (d, f, h, j, and l).
The kinetics of incubating 3MBP5 with Mpro and PLpro were evaluated over time. The fluorescence intensity of 3MBP5 at 560 nm (500 nm excitation) reached a maximum within 20 min of incubation with Mpro (Figure 2g), but it took more than 3 h to achieve maximum fluorescence at 660 nm (600 nm excitation) on incubation with PLpro (Figure 2h). We evaluated three different sources of PLpro protease and noted similar results (Figures S26, S27). Next, 3MBP5, BRM5, and BRP5 were treated with different control proteins under identical conditions to investigate the selectivity: thrombin (TB), hemoglobin (HGB), and bovine serum albumin (BSA; Figures 2i,j, S28). The fluorescence intensities of Cy3 and Cy5 in 3MBP5 were clearly enhanced only on incubation with Mpro and PLpro. In the Cy3 channel, co‐incubation of BRM5 and 3MBP5 with both Mpro and PLpro led to a much stronger fluorescence intensity than Mpro incubation alone, with a fluorescence enhancement of 33.6 % and 40.4 %. In the Cy5 channel, however, the fluorescence intensity showed negligible changes after BRP5, 3EBP5, and 3MBP5 were co‐incubated with Mpro and PLpro compared to incubation with PLpro alone. Clear color changes were observed after 3MBP5 was incubated with different concentrations of Mpro for 1 h at room temperature (25 °C, Figure S29). Thin layer chromatography (TLC) showed more cleavage of the probe when 3MBP5 was co‐incubated with Mpro and PLpro than with Mpro incubation alone. These results confirmed that 3MBP5 can quantitatively detect both Mpro and PLpro together, and PLpro can promote Mpro cleavage efficiency when co‐incubated.
Commercial inhibitors GC376 for Mpro and GRL0617 for PLpro were used to confirm whether the probes can be used for screening Mpro and PLpro inhibitors.[ 5 , 12 , 13b ] Different concentrations of inhibitors were incubated with a fixed concentration of Mpro (100 nM) and PLpro (1.0 μM) and 2.0 μM BRM5, 2.0 μM BRP5, and 2.0 μM 3MBP5 for 1 h at 37 °C in 20 mM Tri‐HCl buffer (pH 8.0). The changes in the fluorescence were recorded (Figures 2k,l and S30); both the Cy3 and Cy5 signals were significantly reduced as the inhibitor concentrations increased. The results demonstrated that 3MBP5 can simultaneously detect Mpro and PLpro inhibitors and thus holds potential for rapid inhibitor screening.
We next used 3MBP5 for cell imaging. HEK 293T cells were transfected with three different plasmids: A SARS‐CoV‐2 Mpro plasmid, a SARS‐CoV‐2 PLpro plasmid, and a control influenza virus protein (A/PR8/1834 NP, referred to as PR8) plasmid. [13b] The plasmid‐transfected HEK 293T cells were subsequently incubated with different probes, and Mpro and PLpro inhibitors for confocal laser scanning microscopy (CLSM) analysis (Figure 3a). The cell imaging incubation time, probe concentration, and optical parameters of Cy3 and Cy5 were optimized first. Different concentrations (1.0 μM, 2.0 μM, 4.0 μM, and 8.0 μM) of Cy3 and Cy5 were co‐incubated with HEK 293T cells for 3 h under standard cell culture conditions (Figure S31). The commercial dye Hoechst 33258 was chosen to stain the nuclei of cells. According to the CLSM images and cytotoxicity analysis, 2.0 μM Cy5 was sufficient and suitable for cell imaging. Higher concentrations of Cy5 may result in high background and cytotoxicity (Figure S32). Additionally, to validate nonspecific cleavage and avoid false positive signals, 2.0 μM 3MBP5 was incubated with Dulbecco's modified eagle medium (DMEM; Figure S33). DMEM alone does not activate the probe. The Cy3 but not the Cy5 fluorescence signal of 3MBP5 rapidly increased within 15 min in DMEM with Mpro. 10 % FBS in DMEM cleaves the peptide after 30 min incubation, and its maximum fluorescence was reached after 95 min incubation. This cleavage is significantly slower (80 min) than that detected for Mpro. The results indicate that i) Mpro is more robust and active than PLpro in cell culture media and ii) prolonged incubation times will lead to the nonspecific activation of 3MBP5 in complex cell culture media. Therefore, the incubation of 2.0 μM of each probe for 30 min is optimal for cell imaging experiments.
Figure 3.

Validation of 3MBP5 by plasmids and inhibitors. Confocal laser scanning microscopy (CLSM) images and mean fluorescence intensity of HEK 293T cells incubated with PR8, Mpro, and PLpro plasmids as well as the impact of inhibitors. a) Experimental scheme of HEK 293T cells incubated with plasmids, inhibitors, and probes in DMEM without FBS. b) 3MBP5 and PBS show that there is no background activation of 3MBP5. c) 3MBP5 is activated in the green channel when the cells are cultured with the Mpro plasmid. d) 3MBP5 is activated in the red channel when cells are incubated with the PLpro plasmid. e) 3MBP5 produces signal in both the red and green channels when cells are incubated with both Mpro and PLpro plasmids. f) 3MBP5 was added to cells treated with a control plasmid (PR8)—no signal activation was observed. g) The GC376 Mpro inhibitor decreased the activity of Mpro and led to a decrease in the green emission of 3MBP5. h) The GRL0617 PLpro inhibitor decreased the activity of PLpro and led to a decrease in the red emission of 3MBP5. i) The use of both inhibitors caused both signal channels to decrease. All scale bars are 20 μm.
To assess the Mpro and PLpro activity in plasmid‐transfected HEK 293T cells, CLSM cell imaging experiments were performed after incubation with 2.0 μM BRM5 and 2.0 μM BRP5 for 30 min (Figures S34 and S35). The red fluorescence of BRM5 and BRP5 were observed in Mpro or PLpro plasmid transfected cells but not in PR8 plasmid‐transfected cells. Their fluorescence intensities clearly increased as the plasmid concentration increased. Furthermore, Mpro and PLpro plasmid transfected cells were incubated with 2.0 μM 3MBP5 for 30 min. Compared with the weak fluorescence in naked cells or PR8‐transfected cells (Figure 3b,f), Mpro plasmid transfected cells showed clear green fluorescence (Figure 3c), and PLpro plasmid transfected cells showed strong red fluorescence (Figure 3d). Only the Mpro and PLpro plasmid co‐transfected cells showed both green and red fluorescence, leading to a yellow overlap (Figure 3e). Moreover, the red fluorescence intensity was enhanced as the ratio of plasmids between Mpro and PLpro increased (Figure S36). To validate the use of 3MBP5 for cell‐based inhibitor screening, 10 μM Mpro inhibitor (GC376) and 10 μM PLpro inhibitor (GRL0617) was added to the plasmid‐transfected cells (Figure 3g–i). The green and red fluorescence decreased, which corresponds to inhibition of both viral proteases. These data demonstrated that 3MBP5 can be used for imaging the intracellular activity of Mpro and PLpro in plasmid‐transfected cells and identifying protease inhibition.
After confirming that 3MBP5 could detect Mpro and PLpro in plasmid‐transfected HEK 293T cells, we next determined if 3MBP5 could be used for imaging Mpro and PLpro as well as screening their inhibitors in SARS‐CoV‐2‐infected cells. Thus, TMPRSS2‐Vero cells were infected with SARS‐CoV‐2 (USA‐WA1/2020) at a multiplicity of infection (MOI) of 0.02 for 24 h before adding the probes (Figure 4a). [13b] At 24 h post‐infection, 3MBP5 and Hoechst 33258 were added into the cells through another 30 min incubation. In terms of the fluorescence of the non‐infected cells and infected cells, strong green and red fluorescence were observed in infected cells but not in un‐infected cells when incubated with 2.0 μM 3MBP5 (Figure S37). The 1.0 μM 3MBP5 could not produce fluorescence in infected cells, but 4.0 μM 3MBP5 led to strong nonspecific green and red fluorescence in un‐infected cells. Therefore, 2.0 μM 3MBP5 was chosen for subsequent cell imaging studies.
Figure 4.

Simultaneous visualization of Mpro, PLpro, and inhibitors in SARS‐CoV‐2‐infected TMPRSS2‐Vero cells. a) Experimental scheme of SARS‐CoV‐2‐infected cells incubated with different probes and inhibitors. Enlarged view shows the imaging mechanism with Alexa488. b) Un‐infected cells and c) infected cells were incubated with 3MBP5, anti‐SARS‐CoV‐2 nucleocapsid (Capsid) primary antibodies, and Alexa488‐labeled secondary antibodies. Panel (b) shows no false positive signal activation. Panel (c) confirms that the 3MBP5 can image Mpro and PLpro from actual SARS‐CoV‐2 viruses and that the virus is present (by capsid immunostaining). d) The immunostaining of the Mpro protein independently confirms that the target is present. e–h) The impact of inhibitors in SARS‐CoV‐2‐infected cells: e) no inhibitors, f) GC376 Mpro inhibitor, g) GRL0617 PLpro inhibitor, and h) both inhibitors. The results confirm that the signal decreases when the target increases. i) Western Blotting of Mpro, PLpro, capsid, and GAPDH in un‐infected and infected cells after 24 h of infection, again confirming the presence/absence of target. All the nuclei were stained with 5 μM Hoechst33258. The yellow scale bar is 20 μm. The white scale bar is 50 μm. The vertical dotted line denotes the YZ plane cutting line. The transverse dotted line denotes the XZ plane cutting line. The pink arrow shows clear fluorescence only from Alexa488.
SARS‐CoV‐2 infection of cells was confirmed by staining with anti‐SARS‐CoV‐2 nucleocapsid (Capsid) and anti‐SARS‐CoV‐2 Mpro primary antibodies as well as AlexaFluor 488‐labeled secondary antibodies (Alexa488). There was clear green (Mpro), red (PLpro), and cyan (Capsid) fluorescence in the infected cells but not in un‐infected cells (Figure 4b,c, Videos S1 and S2). These data confirmed that the virus was present and producing proteases of interest. Interestingly, some infected cells with most Alexa488‐labeled Capsid staining had little Mpro and PLpro signal, and not all infected cells had the same degree of signal for each protease. This staining indicated that there were differences in intracellular protein expression levels between Mpro, PLpro and Capsid.
When the infected cells were incubated with Mpro primary antibodies, Alexa488, and 3MBP5, the green (Mpro), red (PLpro), and cyan (Mpro) fluorescence had good overlap, and their fluorescence intensities varied between cells (Figure 4d and Video S3). In addition, Pearson‘s correlation coefficients of co‐localization between green (Mpro), red (PLpro), and cyan (Capsid or Mpro) fluorescence increased after infection with SARS‐CoV‐2 (Figure S38). We can also observe some cyan fluorescence without overlap of the green and red fluorescence near the cell membrane (Figure 4d, pink arrow). This difference was caused because 3MBP5 represented the protease activity, and the Alexa488‐labeled Mpro antibody showed the protease location. Moreover, the expression of PLpro, Mpro, Capsid, and GAPDH were confirmed by Western Blot analysis 24 h post‐infection (Figure 4i). To further evaluate the detection of protease inhibitors by 3MBP5, infected cells were incubated with different concentrations of Mpro inhibitor and PLpro inhibitor before adding the 3MBP5. The green and red fluorescence of 3MBP5 decreased in infected cells after incubation with GC376 (Figure 4f), and the red fluorescence of 3MBP5 decreased upon incubation with GRL0617 (Figure 4g), thereby demonstrating that a reduction in the protease activity by the presence of Mpro inhibitor and PLpro inhibitor can be detected by 3MBP5 (Figures 4f–h, and S39). These results confirmed that 3MBP5 could be used for the simultaneous screening of Mpro and PLpro as well as their inhibitors in SARS‐CoV‐2‐infected cells.
Conclusion
In conclusion, we report the simultaneous visualization of SARS‐CoV‐2 Mpro and PLpro activity in vitro and in cells with a dual‐color FRET probe. The FRET efficiencies in Cy3‐BHQ‐2, Cy3‐Cy5, and Cy5‐BHQ‐2 were simulated by theoretical calculations and found to agree well with experimental results. We then successfully synthesized four different probes named 3MBP5, 3EBP5, BRM5, and BRP5—fluorogenic substrates for these proteases—through Fmoc‐based solid‐phase peptide synthesis and click reactions. It should be possible, in theory, to apply this approach to link three different cargoes in the same peptide and selectively deliver them into targeted sites. The LC‐MS and optical results all confirmed that the substrates could be specifically cleaved by Mpro and PLpro. Importantly, their catalytic efficiency was first verified together with 3MBP5. This probe design could also be applied to simultaneously detect different proteases, thus having significant value in the rapid screening of inhibitors to other emerging diseases by changing only the peptide cleavage sites. We also found that the intracellular distribution and content of SARS‐CoV‐2 nucleocapsid, Mpro, and PLpro were different between cells. As a research tool, 3MBP5 may allow rapid identification of infected cells for sorting or verification of infection without the need to modify the virus or the host cell, as with reporter genes. This method would assist future studies to accurately understand the complex biological process involving multiple proteases.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
We thank Dr. Nicholas S. Heaton for SARS‐CoV‐2 Mpro plasmid and influenza virus protein (A/PR8/1834 NP) plasmid, Dr. Rolf Hilgenfeld for the SARS‐CoV‐2 Mpro plasmid used for E.coli expression, Dr. Shaun K. Olsen and Dr. Tony T. Huang for SARS‐CoV‐2 PLpro, and Dr. Liangfang Zhang for HEK 293T cells. We thank internal funding from the UC Office of the President (R00RG2515) and the National Institutes of Health (R01 DE031114, R21 AI157957, and R21 AG065776‐01S1) for financial support. This work was supported in part by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE‐1650112. C.M. acknowledges support from the Achievement Reward for College Scientists (ARCS) Foundation. R.M.B. acknowledges support from the National Cancer Institute of the National Institutes of Health under the award number T32CA153915. This research was supported by NIH grant (K08 AI130381) and Career Award for Medical Scientists from the Burroughs Wellcome Fund to AFC. The confocal imaging work was performed at the school of medicine microscopy and histology core of the University of California San Diego, a member of the National Institute of Neurological Disorders and Stroke (NINDS), which is supported by the National Science Foundation (Grant NS047101). The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS‐Related Coronavirus 2, Isolate USA‐WA1/2020, NR‐52281.
Y. Cheng, R. M. Borum, A. E. Clark, Z. Jin, C. Moore, P. Fajtová, A. J. O'Donoghue, A. F. Carlin, J. V. Jokerst, Angew. Chem. Int. Ed. 2022, 61, e202113617; Angew. Chem. 2022, 134, e202113617.
References
- 1. Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W. J., Wang D., Xu W., Holmes E. C., Gao G. F., Wu G., Chen W., Shi W., Tan W., Lancet 2020, 395, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., Zhang Y. Z., Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Xiong E., Jiang L., Tian T., Hu M., Yue H., Huang M., Lin W., Jiang Y., Zhu D., Zhou X., Angew. Chem. Int. Ed. 2021, 60, 5307–5315; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 5367–5375; [Google Scholar]
- 3b. Yousefi H., Mahmud A., Chang D., Das J., Gomis S., Chen J. B., Wang H., Been T., Yip L., Coomes E., Li Z., Mubareka S., McGeer A., Christie N., Gray-Owen S., Cochrane A., Rini J. M., Sargent E. H., Kelley S. O., J. Am. Chem. Soc. 2021, 143, 1722–1727; [DOI] [PubMed] [Google Scholar]
- 3c. Kevadiya B. D., Machhi J., Herskovitz J., Oleynikov M. D., Blomberg W. R., Bajwa N., Soni D., Das S., Hasan M., Patel M., Senan A. M., Gorantla S., McMillan J., Edagwa B., Eisenberg R., Gurumurthy C. B., Reid S. P. M., Punyadeera C., Chang L., Gendelman H. E., Nat. Mater. 2021, 20, 593–605; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Talebian S., Wallace G. G., Schroeder A., Stellacci F., Conde J., Nat. Nanotechnol. 2020, 15, 618–621; [DOI] [PubMed] [Google Scholar]
- 3e. Liu T., Hsiung J., Zhao S., Kost J., Sreedhar D., Hanson C. V., Olson K., Keare D., Chang S. T., Bliden K. P., Gurbel P. A., Tantry U. S., Roche J., Press C., Boggs J., Rodriguez-Soto J. P., Montoya J. G., Tang M., Dai H., Nat. Biomed. Eng. 2020, 4, 1188–1196. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Nature 2020, 582, 289–293; [DOI] [PubMed] [Google Scholar]
- 4b. Muramatsu T., Takemoto C., Kim Y. T., Wang H., Nishii W., Terada T., Shirouzu M., Yokoyama S., Proc. Natl. Acad. Sci. USA 2016, 113, 12997; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., Hilgenfeld R., Science 2003, 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 5. Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S. J., El Oualid F., Huang T. T., Bekes M., Drag M., Olsen S. K., Sci. Adv. 2020, 6, eabd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Dömling A., Gao L., Chem 2020, 6, 1283–1295; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S. H., J. Med. Chem. 2016, 59, 6595–6628; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Báez-Santos Y. M., St John S. E., Mesecar A. D., Antiviral Res. 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Yan S., Wu G., FASEB J. 2021, 35, e21197; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Zohar T., Loos C., Fischinger S., Atyeo C., Wang C., Slein M. D., Burke J., Yu J., Feldman J., Hauser B. M., Caradonna T., Schmidt A. G., Cai Y., Streeck H., Ryan E. T., Barouch D. H., Charles R. C., Lauffenburger D. A., Alter G., Cell 2020, 183, 1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Frei A. P., Bava F. A., Zunder E. R., Hsieh E. W., Chen S. Y., Nolan G. P., Gherardini P. F., Nat. Methods 2016, 13, 269–275; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Canales A., Jia X., Froriep U. P., Koppes R. A., Tringides C. M., Selvidge J., Lu C., Hou C., Wei L., Fink Y., Anikeeva P., Nat. Biotechnol. 2015, 33, 277–284; [DOI] [PubMed] [Google Scholar]
- 8c. Zhou L., Wang R., Yao C., Li X., Wang C., Zhang X., Xu C., Zeng A., Zhao D., Zhang F., Nat. Commun. 2015, 6, 6938; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Lam A. J., St-Pierre F., Gong Y., Marshall J. D., Cranfill P. J., Baird M. A., McKeown M. R., Wiedenmann J., Davidson M. W., Schnitzer M. J., Tsien R. Y., Lin M. Z., Nat. Methods 2012, 9, 1005–1012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Mi L. Z., Lu C., Li Z., Nishida N., Walz T., Springer T. A., Nat. Struct. Mol. Biol. 2011, 18, 984–989; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8f. Andresen M., Stiel A. C., Fölling J., Wenzel D., Schönle A., Egner A., Eggeling C., Hell S. W., Jakobs S., Nat. Biotechnol. 2008, 26, 1035–1040. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Li J., Pu K., Chem. Soc. Rev. 2019, 48, 38–71; [DOI] [PubMed] [Google Scholar]
- 9b. Ye D., Shuhendler A. J., Cui L., Tong L., Tee S. S., Tikhomirrov G., Felsher D. W., Rao J., Nat. Chem. 2014, 6, 519–526; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Pu K., Shuhendler A. J., Rao J., Angew. Chem. Int. Ed. 2013, 52, 10325–10329; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10515–10519; [Google Scholar]
- 9d. Razgulin A., Ma N., Rao J., Chem. Soc. Rev. 2011, 40, 4186–4216; [DOI] [PubMed] [Google Scholar]
- 9e. Lovell J. F., Liu T. W. B., Chen J., Zheng G., Chem. Rev. 2010, 110, 2839–2857. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Widen J. C., Tholen M., Yim J. J., Antaris A., Casey K. M., Rogalla S., Klaassen A., Sorger J., Bogyo M., Nat. Biomed. Eng. 2021, 5, 264–277; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Dagher M., Kleinman M., Ng A., Juncker D., Nat. Nanotechnol. 2018, 13, 925–932; [DOI] [PubMed] [Google Scholar]
- 10c. Ma Y., Yamamoto Y., Nicovich P. R., Goyette J., Rossy J., Gooding J. J., Gaus K., Nat. Biotechnol. 2017, 35, 363–370. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. El-Baba T. J., Lutomski C. A., Kantsadi A. L., Malla T. R., John T., Mikhailov V., Bolla J. R., Schofield C. J., Zitzmann N., Vakonakis I., Robinson C. V., Angew. Chem. Int. Ed. 2020, 59, 23544–23548; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23750–23754; [Google Scholar]
- 11b. Ma-Lauer Y., Carbajo-Lozoya J., Hein M. Y., Muller M. A., Deng W., Lei J., Meyer B., Kusov Y., von Brunn B., Bairad D. R., Hunten S., Drosten C., Hermeking H., Leonhardt H., Mann M., Hilgenfeld R., von Brunn A., Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Békés M., van der Heden van Noort G. J., Ekkebus R., Ovaa H., Huang T. T., Lima C. D., Mol. Cell 2016, 62, 572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A. R., Tascher G., Geurink P. P., Wilhelm A., van der Heden van Noort G. J., Ovaa H., Muller S., Knobeloch K. P., Rajalingam K., Schulman B. A., Cinatl J., Hummer G., Ciesek S., Dikic I., Nature 2020, 587, 657–662; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B. S., Johnson M. E., Baker S. C., Ghosh A. K., Mesecar A. D., Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Pawlik B., Wang X., Jochmans D., Neyts J., Mlynarski W., Hilgenfeld R., Drag M., Nat. Chem. Biol. 2021, 17, 222–228; [DOI] [PubMed] [Google Scholar]
- 13b. Froggatt H. M., Heaton B. E., Heaton N. S., J. Virol. 2020, 94, e01265-20;32843534 [Google Scholar]
- 13c. Wang L., Hu W., Fan C., Protein Sci. 2020, 29, 1228–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Peñalver L., Schmid P., Szamosvári D., Schildknecht S., Globisch C., Sawade K., Peter C., Böttcher T., Angew. Chem. Int. Ed. 2021, 60, 6799–6806; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 6874–6881; [Google Scholar]
- 14b. Steuten K., Kim H., Widen J. C., Babin B. M., Onguka O., Lovell S., Bolgi O., Cerikan B., Neufeldt C. J., Cortese M., Muir R. K., Bennett J. M., Geiss-Friedlander R., Peters C., Bartenschlager R., Bogyo M., ACS Infect. Dis. 2021, 7, 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Cho Y., An H. J., Kim T., Lee C., Lee N. K., J. Am. Chem. Soc. 2021, 143, 14125; [DOI] [PubMed] [Google Scholar]
- 15b. Sun W., Guo S., Hu C., Fan J., Peng X., Chem. Rev. 2016, 116, 7768–7817; [DOI] [PubMed] [Google Scholar]
- 15c. Lu C. H., Cecconello A., Elbaz J., Credi A., Willner I., Nano Lett. 2013, 13, 2303–2308. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Zhang C., Dong X., Ong S. Y., Yao S. Q., Anal. Chem. 2021, 93, 12081–12089; [DOI] [PubMed] [Google Scholar]
- 16b. Xue L., Prifti E., Johnsson K., J. Am. Chem. Soc. 2016, 138, 5258–5261; [DOI] [PubMed] [Google Scholar]
- 16c. Jia X., Chen Q., Yang Y., Tang Y., Wang R., Xu Y., Zhu W., Qian X., J. Am. Chem. Soc. 2016, 138, 10778–10781. [DOI] [PubMed] [Google Scholar]
- 17. Clapp A. R., Medintz I. L., Mauro J. M., Fisher B. R., Bawendi M. G., Mattoussi H., J. Am. Chem. Soc. 2004, 126, 301–310. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Chen X., Zaro J. L., Shen W. C., Adv. Drug Delivery Rev. 2013, 65, 1357–1369; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Reddy Chichili V. P., Kumar V., Sivaraman J., Protein Sci. 2013, 22, 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Cheng Y., Sun C., Liu R., Yang J., Dai J., Zhai T., Lou X., Xia F., Angew. Chem. Int. Ed. 2019, 58, 5049–5053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5103–5107; [Google Scholar]
- 19b. Cheng Y., Sun C., Ou X., Liu B., Lou X., Xia F., Chem. Sci. 2017, 8, 4571–4578; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Cheng Y., Huang F., Min X., Gao P., Zhang T., Li X., Liu B., Hong Y., Lou X., Xia F., Anal. Chem. 2016, 88, 8913–8919. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Zhang P., Cui Y., Anderson C. F., Zhang C., Li Y., Wang R., Cui H., Chem. Soc. Rev. 2018, 47, 3490–3529; [DOI] [PubMed] [Google Scholar]
- 20b. Peraro L., Kritzer J. A., Angew. Chem. Int. Ed. 2018, 57, 11868–11881; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12042–12057; [Google Scholar]
- 20c. Stewart M. P., Sharei A., Ding X., Sahay G., Langer R., Jensen K. F., Nature Nature. 2016, 538, 183–192. [DOI] [PubMed] [Google Scholar]
- 21. Lyon R. P., Setter J. R., Bovee T. D., Doronina S. O., Hunter J. H., Anderson M. E., Balasubramanian C. L., Duniho S. M., Leiske C. I., Li F., Senter P. D., Nat. Biotechnol. 2014, 32, 1059–1062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information