

Abstract
Next-generation sequencing (NGS) technology is used to evaluate hereditary cancer risks of patients worldwide; however, information concerning the germline multigene mutational spectrum among patients with breast cancer (BC) with consanguineous marriage (CM) is limited. Therefore, this prospective study aimed to determine the molecular characteristics of patients with BC who were tested with multigene hereditary cancer predisposition NGS panel and to show the effect of CM on cancer-related genes. Patients with BC with or without CM and family history (FH) of BC treated in our breast center were selected according to The National Comprehensive Cancer Network (NCCN) criteria for hereditary BC. In these patients, the analysis of a panel of 33 genes involved in hereditary cancer predisposition was performed after genetic counseling by using NGS. The pathogenic variant (PV) and the variant of uncertain significance (VUS) were found to be 15.8 and 47.4%, respectively. PVs were identified in 10/33 genes in 34 patients; 38.2% in BRCA1/2 genes; 6, 24, and 14% in other high, moderate and low-risk genes, respectively. The CM rate was 17.7% among the 215 patients with BC. The PV rate was 13.2% in patients with CM and 16.4% in patients without CM (P=0.80). When PV and VUS were evaluated together, the PV+VUS ratio was significantly higher in patients with CM and FH of BC than patients without CM and FH of BC (88.2 vs. 63.3%, P=0.045). Analysis of multigene panel provided 9.76% additional PVs in moderate/low-risk genes. The PV rate was similar in patients with BC with or without CM. A high PV+VUS ratio in patients with CM and FH of BC suggests that genes whose importance are unknown are likely to be pathogenic genes later.
Keywords: breast cancer, consanguineous marriage, multigene testing, pathogenic variant
Introduction
Breast cancer (BC) is the most frequently diagnosed cancer globally, with >2 million incident cases in 2020 (1). In Turkey, 25,345 new cases of BC were diagnosed in 2020, corresponding to 24.4% of all cancers diagnosed in women (2). Although hereditary factors contribute to 10–30% of BC pathogenesis, only a small fraction of BCs (5–10%) can be explained by germline mutations in cancer susceptibility genes (3,4), which means other susceptibility loci are likely to exist (5). Identifying unaffected disease-causing variant carriers and governing their risk has been shown to reduce BC and all-cause mortality (6). The selection of patients for BC risk assessment and counseling is mainly performed using information about the personal and/or family history (FH) of BC, usually following the guidelines and recommendations of The National Comprehensive Cancer Network (NCCN) (7). Although most patients with hereditary BC are found to have mutations in BRCA1/2 genes, it is estimated that as much as 70% of BC cases could be caused by mutations in other genes (8). Furthermore, recent studies have shown the importance of carefully selected patients who will benefit from genetic testing (9).
The use of multigene panel testing for detecting hereditary cancer risk has increased within the last few years, aiming to provide information and tailored management for more families in a convenient way regarding patients with a less typical presentation for a given cancer syndrome and/or missing FH data and to provide a cost-effective alternative to BRCA-only testing (10,11).
Consanguineous marriage (CM) is still common in various parts of the world (12) and mainly occurs as a first cousin union (13). For example, in Turkey, the prevalence of CMs is still relatively high at 20–25% (ranges from 11.5 to 46%) (14,15). However, the relationship between CM and BC-predisposing genes is not clear.
The present study aimed to analyze 33 genes implicated in hereditary cancer predisposition in Turkish patients with BC and investigate the impact of CM and FH of BC on carrying pathogenic mutations in genes associated with BC.
Materials and methods
Patients
In this prospective clinical study, patients who were treated with BC between 2018 and 2020 in our clinic were included. Patients who did not agree to participate in the study and did not have any genetic test indication were excluded. Patients were divided into two groups as CM [study group (SG)] and those without CM [control group (CG)] and were directed to genetic tests. All women received genetic counseling and psychological assistance and signed an informed consent form before molecular genetic testing and permission to use their data for research purposes. The Biruni University Ethical Committee approved the study (approval no. 2015-KAEK-43-18-11; 19/09/2018). Basic demographics, personal and family histories were noted, and pedigrees were drawn by ordering clinicians (Fig. 1).
Figure 1.
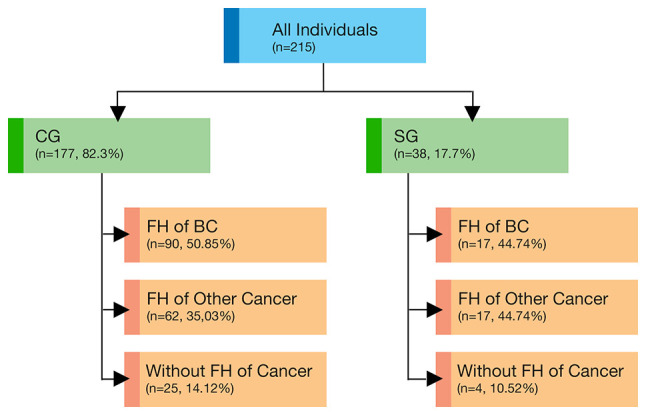
Study design and tested population groups and subgroups. FH, family history; BC, breast cancer; SG, study group; CG, control group.
Two next-generation sequencing (NGS) multigene panels, including 26 and 33 genes related to hereditary cancer predisposition, were performed in both the SG and CG as previously described (16). The panel genes were grouped according to the associated risk for BC [high-risk genes, BRCA1, BRCA2, CDH1, partner and localizer of BRCA2 (PALB2), PTEN, serine/threonine-protein kinase STK11 and TP53; moderate-risk genes, serine-protein kinase ATM (ATM), serine/threonine-protein kinase Chk2 (CHEK2) and nibrin (NBN); low/unknown-risk genes, APC, bone morphogenetic protein receptor type-1A, CDK4, CDKN2A, epithelial cell adhesion molecule (EPCAM), menin, DNA mismatch repair protein Mlh1 (MLH1), DNA mismatch repair protein Msh2 (MSH2), DNA mismatch repair protein Msh6 (MSH6), adenine DNA glycosylase (MUTYH), mismatch repair endonuclease PMS2 (PMS2), proto-oncogene tyrosine-protein kinase receptor Ret, SMAD4, von Hippel-Lindau disease tumor suppressor, Fanconi anemia group J protein (BRIP1), DNA repair protein RAD51 homolog 3 (RAD51C), DNA repair protein RAD51 homolog 4 (RAD51D), BRCA1-associated RING domain protein 1, serine/threonine-protein kinase Chk1, double-strand break repair protein MRE11, neurofibromin, DNA repair protein RAD50 (RAD50) and DNA repair protein RAD51 homolog 2] (16).
NGS and mutation confirmation
The genomic DNA was isolated from peripheral blood leukocytes according to manufacturers' protocol (Qiagen, Inc., and RBC Bioscience) and was quantified by using NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Inc.).
In the first protocol; a probe library (Roche NimbleGen SeqCap EZ Choice) targeting all coding exons and 50 bp of flanking intronic regions of the 33 genes was custom-designed, and sample preparation was performed following the SeqCap EZ Choice Library User's Guide (Roche NimbleGen, Inc.) as previously described (16). Briefly, after enzymatic fragmentation (Kappa Hyperplus kit), the library was prepared according to the manufacturer's protocol (Roche NimbleGen, Inc.). NGS was performed with MiSeq Reagent Kit v3 (600-cycle) on Illumina MiSeq machine (Illumina, Inc.). Alignment to the reference sequence (hg19) and variant calling was performed by the SeqNext module of the SeqPilot suite (JSI Medical Systems GmbH).
In the second protocol; a commercially designed panel, from SOPHiA Genetics™ was used: The SOPHiA Hereditary Cancer Solutions (HCS). The library preparation was performed according to manufacturer's protocol. Sequencing was performed on Illumina Next Seq machine (Illumina, Inc.) with manufacturer's appropriate NGS kits. Alignment to the reference sequence (hg19) and variant calling was performed by the SOPHiA DDM platform of the same manufacturer.
The annotation and interpretation of all identified variants were performed using an in-house local knowledge base and a proprietary bioinformatics pipeline. The clinical significance of variants was examined using the standards and guidelines for the interpretation of sequence variants recommended by the American College of Medical Genetics and Genomics (ACMG Laboratory Quality Assurance Committee) and the Association for Molecular Pathology (AMP) (17).
All pathogenic, likely pathogenic variants (PVs) obtained from the custom designed primers and VUS were confirmed by Sanger Sequencing for further segregation analysis using Applied Biosystems 3130 Genetic Analyzer (Thermo Fisher Scientific, Inc.). Sanger sequences are presented in Figs. S1-S6. Family segregations were performed by Sanger Sequencing for further segregation analysis also using Applied Biosystems 3130 Genetic Analyzer (Thermo Fisher Scientific, Inc.).
Analysis of large genomic rearrangements (LGRs)
The copy number variation (CNV) module of the software suite SeqPilot (JSI Medical Systems GmbH) and panels. MOPS (18) was used for the computational analysis of LGRs from NGS data for the following genes/gene regions, including BRCA1, BRCA2, CHEK2, EPCAM (Exons 8, 9), MLH1, MSH2, MSH6, MUTYH, PALB2, RAD50 (Exons 1, 2, 4, 10, 14, 21, 23 and 25), RAD51C, RAD51D, and TP53. All predicted LGRs detected with these algorithms were then experimentally studied using the Multiplex Ligation-dependent Probe Amplification technique as described previously (16,18).
Statistical analysis
Pearson correlation analysis with ‘N-1’ correction was performed to correlate two parameters. In addition, chi-square tests were conducted to compare the distribution of categorical variables. Fischer's exact test was used when chi-square tests assumptions did not hold due to low expected cell counts, All analyses were performed with R software version 3.4.4. All statistical tests were two-sided, P<0.05 was considered to indicate a statistically significant difference.
Results
Patient characteristics
A total of 38 patients with BC with CM (SG) and 177 patients with BC without CM (CG) were referred for testing. The detailed demographic and clinical features were summarized and compared in Table I. The median age of patients was 47 years (27–76 years). The median time from diagnosis to testing was 1 year, and 67% (144/215) of patients tested within 12 months following the diagnosis. A family history (FH) of cancer constituted a primary reason for referral, accounting for 86.4% (186/215). Almost half of women (49.7%, 107/215) reported FH of BC (44.7% in SG and 50.8% in CG). CM was reported in 17.7% (38/215) of the patients. There were no statistically significant differences in the patient characteristics between the two groups (Tables I–III).
Table I.
Overall characteristics and test results in two groups.
| Groups | ||||
|---|---|---|---|---|
|
|
||||
| Characteristic | Patients with BC | SG | CG | P-value |
| Total patients | 215a | 38 (17.7)b | 177 (82.3)b | |
| Age at testing, years | 0.3173 | |||
| Mean ± SD | 47.5±10.6 | 47.4±9.9 | 47.5±11.0 | |
| Median, range | 47 (27–76) | 47 (32–68) | 47 (27–76) | |
| FH of cancer, n (%) | ||||
| None | 29 (13.4) | 4 (10.5) | 25 (14.1) | 0.7900 |
| FH of BC | 107 (49.7) | 17 (44.7) | 90 (50.8) | 0.5900 |
| FH of other cancer(s) | 79 (36.7) | 17 (44.7) | 62 (35.0) | 0.2700 |
| Multigene test result, n (%) | ||||
| Negative | 79 (36.7) | 13 (34.2) | 66 (37.3) | 0.8500 |
| Positive | 34 (15.8) | 5 (13.2) | 29 (16.4) | 0.8000 |
| VUS only | 102 (47.4) | 20 (52.6) | 82 (46.3) | 0.5900 |
| Positive in gene categories, n (%) | ||||
| NCCN absolute risk category | 13 (37.1) | 1 (20.0) | 12 (41.4) | 0.6200 |
| >60% | ||||
| NCCN absolute risk category | 2 (0.5) | 0 | 2 (6.9) | 1.0000 |
| 41-60% | ||||
| NCCN absolute risk category | 19 (55.8) | 4 (80.0) | 15 (51.7) | 0.6200 |
| 15-40% and low/unknown risk | ||||
Data are presented as the n;
data are presented as the n (%). BC, breast cancer; FH, family history; SG, study group; CG, control group; VUS, variant of uncertain significance; NCCN, The National Comprehensive Cancer Network.
Table III.
Association between FH of BC and genetic test results.
| Groups | ||||
|---|---|---|---|---|
|
|
||||
| Genetic test results | FH status | CG | SG | P-value |
| Negative | FH of BC | 0.022 | ||
| No | 33 (50.0) | 11 (84.6) | ||
| Yes | 33 (50.0) | 2 (15.4) | ||
| FH of any cancer | 0.370 | |||
| No | 8 (12.0) | 3 (23.0) | ||
| Yes | 58 (88.0) | 10 (77.0) | ||
| VUS | FH of BC | 0.620 | ||
| No | 46 (56.1) | 10 (50.0) | ||
| Yes | 36 (43.9) | 10 (50.0) | ||
| FH of any cancer | 0.170 | |||
| No | 14 (17.0) | 1 (5.0) | ||
| Yes | 68 (83.0) | 19 (95.0) | ||
| Negative, PV+VUS | Only patients with FH of BC | 0.045 | ||
| Negative | 33 (36.7) | 2 (11.8) | ||
| PV+VUS | 57 (63.3) | 15 (88.2) | ||
Data are presented as the n (%). BC, breast cancer; FH, family history; SG, study group; CG, control group; VUS, variant of uncertain significance; PV, pathogenic variant.
Multigene panel testing results
Identified pathogenic variants (PVs) were in 10/33 genes in 34 patients; 38.2% in BRCA1/2 genes; 6, 24 and, 14.3% in other high, moderate, and low-risk genes, respectively (Table II). Specifically, the positive rate was 13.2 and 16.4% in the SG and the CG, respectively (P=0.80; Table I and Fig. 2).
Table II.
Multigene panel testing results among the categories of tested individuals.
| Positive in gene categories | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Individuals | N | Positive, % | BRCA1 and BRCA2, % | Other high-riska, % | Moderate riskb, % | Low/unknown V riskc, % | US, % |
| Total individuals | 215 | 15.8 | 6.0 | 0.9 | 3.7 | 2.2 | 47.4 |
| SG | 38 | 13.2 | 2.6 | 0.0 | 5.2 | 5.2 | 52.6 |
| CG | 177 | 16.4 | 6.7 | 1.1 | 3.3 | 5.0 | 46.3 |
| FH of BC | 107 | 24.3 | 10.2 | 0.9 | 4.6 | 8.4 | 21.4 |
| SG | 17 | 29.4 | 5.8 | 0.0 | 11.7 | 11.7 | 58.8 |
| CG | 90 | 23.3 | 12.2 | 1.1 | 3.3 | 7.7 | 40.0 |
| FH of any cancer | 187 | 17.1 | 6.9 | 1.0 | 3.7 | 5.3 | 46.5 |
| SG | 34 | 14.7 | 2.9 | 0.0 | 5.8 | 5.8 | 55.9 |
| CG | 153 | 17.6 | 7.8 | 1.3 | 3.2 | 5.2 | 44.6 |
CDH1, PALB2, PTEN, STK11, TP53;
ATM, CHEK2, NBN;
APC, BMPR1A, CDK4, CDKN2A, EPCAM, MEN1, MLH1, MSH2, MSH6, MUTYH, PMS2, RET, SMAD4, VHL, BRIP1, RAD51C, RAD51D, BARD1, CHEK1, MRE11 (MRE11A), NF1, RAD50, RAD51B. BC, breast cancer; FH, family history; SG, study group; CG, control group; VUS, variant of uncertain significance; PALB2, partner and localizer of BRCA2; STK11, serine/threonine-protein kinase STK11; ATM, serine-protein kinase ATM; CHEK2, serine/threonine-protein kinase Chk2; NBN, nibrin; BMPR1A, bone morphogenetic protein receptor type-1A; EPCAM, epithelial cell adhesion molecule; MEN1, menin; MLH1, DNA mismatch repair protein Mlh1; MSH2, DNA mismatch repair protein Msh2; MSH6, DNA mismatch repair protein Msh6; MUTYH, adenine DNA glycosylase; PMS2, mismatch repair endonuclease PMS2; RET, proto-oncogene tyrosine-protein kinase receptor Ret; VHL, von Hippel-Lindau disease tumor suppressor; BARD1, BRCA1-associated RING domain protein 1; CHEK1, serine/threonine-protein kinase Chk1; MRE11, double-strand break repair protein MRE11; NF1, neurofibromin; RAD50, DNA repair protein RAD50; RAD51B, DNA repair protein RAD51 homolog 2.
Figure 2.
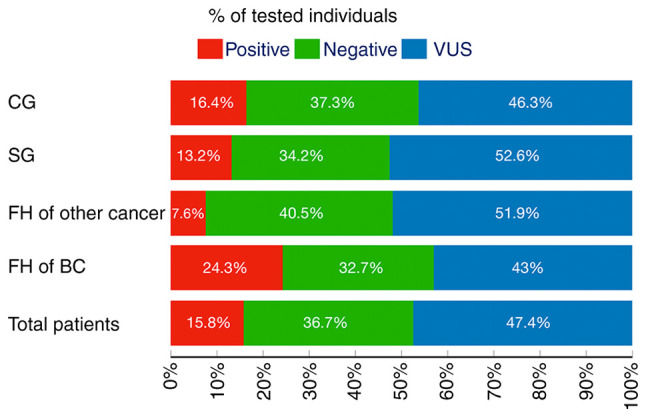
Multigene testing results for all groups and their subgroups according to FH of BC and other cancers and consanguineous marriage status. FH, family history; BC, breast cancer; SG, study group; CG, control group; VUS, variant of uncertain significance.
The distribution of PV in all patients was as follows: BRCA1 (14%), ATM (15%), MUTYH (15%), BRCA2 (23%), CHECK2 (9%), RAD51C (9%), PALBB2 (6%), RAD50 (3%), PMS2 (3%) and EPCAM (3%) (Table IV and Fig. 3A). There were five PVs in the SG (BRCA2, CHECK2, RAD51C, and MUTYH) and 29 PVs in the CG, respectively (Table IV and Fig. 3B and C). Variants classified as pathogenic or likely pathogenic were small deletions and insertions, missense, and splice site variants with decreasing frequency (Table IV).
Table IV.
List of PVs/LPVs identified in this study.
| Variant | Clinical significance | Personal history of cancer | FH of cancer | CM status |
|---|---|---|---|---|
| NM_007194(CHEK2): | LPV | Breast | Breast, colon | Yes |
| c.1427C>T, p. (Thr476Met) | ||||
| NM_007194(CHEK2): | LPV | Breast | Breast, colorectal, | |
| c.1427C>T, p. (Thr476Met) | kidney | |||
| NM_001128425(MUTYH): | PV (monoallelic) | Breast | Breast | Yes |
| c.1171C>T, p. (Gln391*) | ||||
| NM_007294(BRCA1): | PV | Breast | Breast, lung | Yes |
| c.4035delA, p. (Glu1346Lysfs*20) | ||||
| NM_000059(BRCA2): | PV | Breast | Breast, ovarian | No |
| c.9682delA, p. (Ser3228Valfs*21) | ||||
| NM_000059(BRCA2): | PV | Breast | Breast, prostate, lung, gastric | No |
| c.8087T>A, p. (Leu2696*) | ||||
| NM_024675(PALB2): | PV | Breast | Breast | No |
| c.3271C>T, p. (Gln1091*) | ||||
| NM_000059(BRCA2): | PV | Breast | Breast | No |
| c.5557dupT, p. (Cys1853Leufs*5) | ||||
| NM_007294(BRCA1): | PV | Breast | Breast, gastric, skin, lung | No |
| c.5266dupC, p. (Gln1756Profs*74) | ||||
| NM_058216(RAD51C): | LPV | Breast, ovarian | Breast, ovarian prostate | No |
| c.904+5G>T | ||||
| NM_000051(ATM):c.6527delT, p. | PV | Breast | Breast, lung, uterine | No |
| (Leu2176Cysfs*59) | ||||
| NM_007294(BRCA1): | PV | Breast | Breast, skin, melanoma, uterine | No |
| c.3700_3704delGTAAA, p. | ||||
| (Val1234Glnfs*8) | ||||
| NM_005732(RAD50): | PV | Breast | Breast, skin, melanoma, uterine | No |
| c.326_329delCAGA, p. (Thr109Asnfs*20) | ||||
| NM_002485(NBN): c.657_661delACAAA, p. (Lys219Asnfs*16) | PV | Breast | lymphoma | No |
| NM_007194(CHEK2): c.1427C>T, p. (Thr476Met) | LPV | Breast | None | No |
| NM_001128425(MUTYH): c.884C>T, p. (Pro295Leu) | PV (monoallelic) | None | Pancreatic | NA |
| NM_000059(BRCA2): c.4936_4939delGAAA, p.Glu1646Glnfs*23 | PV | None | Breast, ovarian | NA |
| NM_000179(MSH6):c.2764C>T, p.(Arg922*) | PV | None | Breast, intestine | NA |
| NM_007194(CHEK2):c.793-1G>A | PV | None | Breast, ovarian, lung | NA |
| NM_001128425.1(MUTYH):c.1187G>A | LPV | Breast | No | No |
| NM_002878.3(RAD51D):c.480+1G>A | PV | Breast | Breast | Yes |
| NM_000059.3(BRCA2):c.8940delA | PV | Breast | Prostate, lung | No |
| NM_007294.4(BRCA1):c.1621C>T | PV | Breast | Breast, ovarian | No |
| NM_000051.3(ATM):c.2922-2A>G | PV | Breast | Pancreas, lung | No |
| NM_000059.3(BRCA2): c.2307dupT | PV | Breast | Colon | No |
| NM_000059.3(BRCA2):c.5073dupA | PV | Breast | Breast, prostate | Yes |
| NM_001128425.1(MUTYH): | PV | Breast | Breast, colon | No |
| c.1437_1439delGGA | ||||
| NM_000051.3(ATM):c.2284_2285delCT | PV | Breast | Breast | No |
| NM_058216.3(RAD51C):c.181_182delCT | PV | Breast | No | No |
| NM_000051.3(ATM):c.6527delT | PV | Breast | Breast | No |
| NM_024675.4(PALB2):c.932_933insC | PV | Breast | Lung | No |
| NM_000535.7(PMS2):c.988+1del | PV | Breast | Breast | No |
| NM_000314.8(PTEN):c.93delC | LPV | Breast | Breast, gastric, prostate, lung | No |
| NM_005732.4(RAD50):c.1873delT | LPV | Breast | Breast, gastric, prostate, lung | No |
| NM_007294.4(BRCA1):c.5266dupC | PV | Breast | Breast, ovarian, lung | No |
| NM_000051.3(ATM):c.3576G>A | PV | Breast | NA | NA |
| NM_000059.3(BRCA2):c.4376dupA | LPV | Breast | Breast, ovarian, endometrium | No |
| NM_002354.2(EPCAM):c.556-14A>G | PV | Breast | Breast, gastric | No |
| NM_000059.3(BRCA2):rsa13q13.1 | LPV | Breast | Breast | No |
| (BRCA2exon3)x1 | ||||
| NM_007294.4(BRCA1):c.5176A>T | PV | Breast | Breast, ovarian, CNS | No |
| NM_001128425.1(MUTYH):c.1187G>A | PV | Breast | Colon, lung | No |
| NM_001128425.1(MUTYH):c.734G>A | PV | Breast | Breast | No |
LPV, likely pathogenic variants; PV, pathogenic variants; CNS, central nervous system; FH, family history; CM, consanguineous marriage; PV, pathogenic variant; NA, no available information.
Figure 3.

(A) Distribution of 34 PVs among the examined genes. (B) Distribution of five PVs in the SG. (C) Distribution of 29 PVs in the CG. PV, pathogenic variant.
The incidence of PV in FH of BC was slightly higher in the SG (29.4 vs. 23.3%, P=0.51; Tables II and III). Variants with uncertain significance (VUS) were identified in 47.4% of women. The VUS rates ranged from 21.4 to 55.9% (Table II). Among the women with a PV, 38.2% of them had BRCA1/2 gene mutations. The high mutation frequency of the BRCA1/2 genes was 19.6% in the SG and 40.8% in the CG, respectively (P=0.48). In addition, distributions of PVs in patients were identified in other high- (6%,), moderate (24%) or low risk (14.3%) genes apart from the BRCA1/2 genes, PVs were identified in CHEK2, ATM, NBN, PALB2, RAD50, RAD51C and MUTYH (monoallelic) (Fig. 3A and Table IV).
The negative genetic test result was significantly higher in those in the SG who did not have FH of BC (84.6 vs. 50%, P=0.022; Table III). However, when only patients with FH of BC were considered, the total PV and VUS rates were significantly higher in the SG compared with the CG (88.2 vs. 63.3%, P=0.045; Table III).
Discussion
Consanguineous marriage (CM) between biological relatives is a social custom with a long history in various parts of the world (12). Today, hundreds of millions of individuals live in consanguineous families (12–14). The offspring of consanguineous parents are more likely to have the same two alleles (homozygosity) by descent. The prevalence of CM is still relatively high at 20–25% (ranges from 11.5 to 46%) (14,15) in Turkey. However, the relationship between CM and BC-predisposing genes is not clear. In this prospective clinical study, the effect of CM on cancer-related genes in patients with BC was investigated.
The rate of pathogenic variants (PVs) identified with multigene NGS panel testing in Turkish patients with BC was 15.8% in our study. Most of these PVs were BRCA1 and BRCA2 genes (~39%), which is consistent with previous literature (16,19). Furthermore, the prevalence of BRCA1/2 mutations in the CG was 6.7% and in those with FH of BC was 14.1%, which was similar to the literature in which the prevalence ranges from 9 to 21% (20–22).
Analysis of positive gene mutations showed that more than half of the patients with BC (61.8%) having a PV would have been missed if genetic testing was restricted to BRCA1/2 or high-risk genes included in the NCCN guidelines for Genetic/Familiar high-risk assessment for breast and ovarian cancer and could enable personalized management decisions for these patients (23).
It is noteworthy that most of the variants classified as pathogenic or likely pathogenic in the current study were deleterious (variants with small deletions, insertions, and splice sites). This may be related to the fact that most missense variants remain in the VUS class upon compliance with the ACMG classification. A notable feature in the cohort of the present study was that no large deletions and duplications (CNVs) were observed. In cases where it is impossible to calculate CNVs by the NGS method, additional testing may be considered to detect pathological CNVs, especially in BRCA1 and BRCA2 genes. Thus, it is clear that VUS monitoring will continue to maintain its importance.
Our previous study analyzed genes involved in hereditary cancer predisposition using an NGS approach in 1,197 individuals from Greece, Romania, and Turkey (16). A PV was identified in 264 of the individuals (22.1%) analyzed, while a VUS was identified in 34.8% of cases. Nevertheless, the PV rate was lower (15.8%) and the VUS rate was higher (43.7%) in Turkish individuals. Moreover, as a PV, the BRCA1/2 ratio was 10.1%, while other high-risk, moderate-risk, and low/unknown risk ratios were 4.4, 0.6, and 1.9%, respectively, for these individuals. Similar to these findings, the PV and VUS rates were 12.8 and 47.4%, respectively, in the current study. This study also found similarities regarding BRCA1/2 and other high-risk, moderate-risk, and low/unknown risk ratios (Table I). There were no significant differences between the two groups regarding PV rates (Fig. 3A-C).
The Turkish public health system covered hereditary cancer predisposition gene tests on a routine basis, which has been covered by for several years. However, only two recent studies with multigene panels were performed in Turkey. The first was a multinational study, and the pathogenic/likely pathogenic mutation rate was found to be similar (15.8%) to our study (16). In the second study conducted by Akcay et al (19), cancer susceptibility genes were analyzed in breast and colorectal cancer patients. High BC risk genes were found in 17.2% of patients and, a low BC risk gene in 3.9%.
Due to the sample size of the present study, no LGRs were found by any of the bioinformatics approaches, which is essential to evaluate the full mutation spectrum of tested genes, especially for BRCA1/2. In Turkey, Large Genomic Rearrangements (LGRs). rates are between 1–4% (19,24,25) comparable with different populations around the world (0.1-12.7%) (16,26–30).
The likelihood of identifying variants of uncertain significance (VUS) increases with multigene panel testing as high as 40% (31). In the current study, the VUS rate was found to be 47.4%. Most VUS reclassifications involve a downgrade to a benign variant; a small proportion may be reclassified as pathogenic (32). Therefore, the VUS should not be used to guide medical management until the clinical significance of these findings is determined (33).
Multigene panel testing is a powerful tool that detects BRCA and non-BRCA germline mutations in individuals with a family history (FH) of BC. When FH of BC was used as a stratification factor among patients with BC, almost half of women (49.7%, 107/215) reported FH of BC (44.7% in SG and 50.8% in CG). The patients' suitability can explain this high FH of BC rate in this study for genetic testing.
In populations where CMs are prevalent, patients are at risk for homozygous/compound heterozygous germline mutations in BRCA2, BRIP1 and PALB2 result in Fanconi's anemia (34–36). In a previous study, the consanguinity rate in Southern Mediterranean populations was inversely correlated with families with BRCA1 germline deleterious mutations (37).
In a study by Medimegh et al (38), it was demonstrated that the parental consanguinity was protective against BC, especially over the age of 50. However, the results of the present study showed no significant relationships between patients with BC with or without CM and pathogenic mutations. Thus, although thousands of multigene panel tests have been performed worldwide (39), there is still an important fraction of BC cases that remain undiagnosed, underlying the predisposition to BC (40).
In a retrospective review of the multi-institutional tumor registry, including 2,237 patients with BC, 509 (60.7%) had negative results, 108 (12.8%) had deleterious mutations and 221 (26.3%) had VUS (41). In the present study, 15.8% of the patients had PVs, 47.4% had VUS and 36.7% had negative results. The PV rate did not differ between the two groups (13.2 and 16.4% in SG and CG, respectively), and the VUS rate was higher in the CM group (52.6 vs. 46.3% in the SG and CG, respectively, P=0.59). However, when the sum of PV and VUS results in patients with FH of BC in two groups were compared, it was found that the sum of PV and VUS was significantly higher in the SG (63.3 vs. 88.2% in the CG and SG, respectively, P=0.045). This significant result may suggest that genes of unknown importance in VUS may be important in the future.
In conclusion, the present study demonstrated the importance of using multigene panels in a multi-ethnic population and contributed to the knowledge of hereditary BC in Turkey. The pathogenic mutation rate was 24.3% in patients with FH of BC. The PV rate in patients with FH of BC reached 29.4% in the SG. Therefore, multigene panel testing should be considered for these patients. The CM did not significantly increase the pathogenic mutation rate, but the VUS rate was higher in this group. This observation should be confirmed with further prospective clinical trials with more extensive series.
Supplementary Material
Acknowledgements
Not applicable.
Funding Statement
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the ClinVar respiratory, ncbi.nlm.nih.gov/clinvar/?term=SUB10439877 and ncbi.nlm.nih.gov/clinvar/?term=SUB10447902
Authors' contributions
GNT, EP and AS drafted the manuscript. GNT performed bioinformatics and statistical analysis, analyzed and interpreted the variant data and prepared all supporting materials and figures. AOC, EP, KA, GP, SK and KY designed the study and experiments, collected the demographic data, and coordinated the study and all mutational analyses. GNT, EP, KA, GP, SK, GN, VO, TO, KY and AOC participated in the writing of the manuscript, performed DNA extraction, sequencing and Multiplex Ligation-dependent Probe Amplification experiments, and contributed to the analysis and interpretation of the variant data. VO, AOC, KY, CO, FA, TO, ASI, GS, GA, GNT, EP, KA, GP, SK, GN, ES and AS provided the patient material, diagnosis and management. VO, TO and AS conceived the study and participated in its design and coordination. GNT and KY confirm the authenticity of all raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
All tested individuals provided signed informed consent form before molecular genetic testing and permission for the anonymous use of their data for research purposes and/or scientific publications. The study was approved by The Biruni University Ethical Committee (approval no. 2015-KAEK-43-18-11; date, 19/09/2018).
Patient consent for publication
Present within informed consent forms.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, Bray F. Cancer statistics for the year 2020: An overview. Int J Cancer. 2021;149:778–789. doi: 10.1002/ijc.33588. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F. Global cancer observatory: Cancer today. https://gco.iarc.fr/today. [ May 28; 2019 ];Lyon, France: International Agency for Research on Cancer. (2018) Available from. [Google Scholar]
- 3.Newman B, Austin MA, Lee M, King MC. Inheritance of human breast cancer: evidence for autosomal dominant transmission in high-risk families. Proc Natl Acad Sci USA. 1988;85:3044–3048. doi: 10.1073/pnas.85.9.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claus EB, Risch N, Thompson WD. Genetic analysis of breast cancer in the cancer and steroid hormone study. Am J Hum Genet. 1991;48:232–242. [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–105. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, Garber JE, Neuhausen SL, Matloff E, Eeles R, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304:967–975. doi: 10.1001/jama.2010.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The National Comprehensive Cancer Network, corp-author. https://www.nccn.org. [ October 30; 2020 ]; [Google Scholar]
- 8.Valencia OM, Samuel SE, Viscusi RK, Riall TS, Neumayer LA, Aziz H. The role of genetic testing in patients with breast cancer: A review. JAMA Surg. 2017;152:589–94. doi: 10.1001/jamasurg.2017.0552. [DOI] [PubMed] [Google Scholar]
- 9.Beitsch PD, Whitworth PW, Hughes K, Patel R, Rosen B, Compagnoni G, Baron P, Simmons R, Smith LA, Grady I, et al. Underdiagnosis of hereditary breast cancer: Are genetic testing guidelines a tool or an obstacle? J Clin Oncol. 2019;37:453–460. doi: 10.1200/JCO.18.01631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asphaug L, Melberg HO. The cost-effectiveness of multigene panel testing for hereditary breast and ovarian cancer in Norway. MDM Policy Pract. 2019 Feb 1; doi: 10.1177/2381468318821103. (Epub ahead of print). doi: 10.1177/2381468318821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanislaw C, Xue Y, Wilcox WR. Genetic evaluation and testing for hereditary forms of cancer in the era of next-generation sequencing. Cancer Biol Med. 2016;13:55–67. doi: 10.20892/j.issn.2095-3941.2016.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romeo G, Bittles AH. Consanguinity in the contemporary world. Hum Hered. 2014;77:6–9. doi: 10.1159/000363352. [DOI] [PubMed] [Google Scholar]
- 13.Hamamy H. Consanguineous marriages: Preconception consultation in primary health care settings. J Community Genet. 2012;3:185–192. doi: 10.1007/s12687-011-0072-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alper OM, Erengin H, Manguoğlu AE, Bilgen T, Cetin Z, Dedeoğlu N, Lüleci G. Consanguineous marriages in the province of Antalya, Turkey. Annales de Genetique. 2004;47:129–138. doi: 10.1016/j.anngen.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Erdem Y, Tekşen F. Genetic screening services provided in Turkey. J Genet Couns. 2013;22:858–864. doi: 10.1007/s10897-013-9644-9. [DOI] [PubMed] [Google Scholar]
- 16.Tsaousis GN, Papadopoulou E, Apessos A, Agiannitopoulos K, Pepe G, Kampouri S, Diamantopoulos N, Floros T, Iosifidou R, Katopodi O, et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer. 2019;19:535. doi: 10.1186/s12885-019-5756-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Povysil G, Tzika A, Vogt J, Haunschmid V, Messiaen L, Zschocke J, Klambauer G, Hochreiter S, Wimmer K. panelcn.MOPS: Copy-number detection in targeted NGS panel data for clinical diagnostics. Hum Mutat. 2017;38:889–897. doi: 10.1002/humu.23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akcay IM, Celik E, Agaoglu NB, Alkurt G, Kizilboga Akgun T, Yildiz J, Enc F, Kir G, Canbek S, Kilic A, et al. Germline pathogenic variant spectrum in 25 cancer susceptibility genes in Turkish breast and colorectal cancer patients and elderly controls. Int J Cancer. 2020;148:285–295. doi: 10.1002/ijc.33199. [DOI] [PubMed] [Google Scholar]
- 20.Lang GT, Shi JX, Hu X, Zhang CH, Shan L, Song CG, Zhuang ZG, Cao AY, Ling H, Yu KD, et al. The spectrum of BRCA mutations and characteristics of BRCA-associated breast cancers in China: Screening of 2,991 patients and 1,043 controls by next-generation sequencing. Int J Cancer. 2017;141:129–142. doi: 10.1002/ijc.30692. [DOI] [PubMed] [Google Scholar]
- 21.Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006;295:1379–1388. doi: 10.1001/jama.295.12.1379. [DOI] [PubMed] [Google Scholar]
- 22.Han SA, Kim SW, Kang E, Park SK, Ahn SH, Lee MH, Nam SJ, Han W, Bae YT, Kim HA, et al. The prevalence of BRCA mutations among familial breast cancer patients in Korea: Results of the Korean hereditary breast cancer study. Fam Cancer. 2013;12:75–81. doi: 10.1007/s10689-012-9578-7. [DOI] [PubMed] [Google Scholar]
- 23.The National Comprehensive Cancer Network. Genetic/Familiar High-Risk Assessment, Breast and Ovarian (Version 3.2019), corp-author https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. [ October 30; ];2020 [Google Scholar]
- 24.Yazıcı H, Kılıç S, Akdeniz D, Şükrüoğlu Ö, Tuncer ŞB, Avşar M, Kuru G, Çelik B, Küçücük S, Saip P. Frequency of rearrangements versus small indels mutations in BRCA1 and BRCA2 Genes in Turkish patients with high risk breast and ovarian cancer. Eur J Breast Health. 2018;14:93–99. doi: 10.5152/ejbh.2017.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aktas D, Gultekin M, Kabacam S, Alikasifoglu M, Turan AT, Tulunay G, Kose MF, Ortac F, Yüce K, Tunçbilek E, Ayhan A. Identification of point mutations and large rearrangements in the BRCA1 gene in 667 Turkish unselected ovarian cancer patients. Gynecol Oncol. 2010;119:131–135. doi: 10.1016/j.ygyno.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Judkins T, Rosenthal E, Arnell C, Burbidge LA, Geary W, Barrus T, Schoenberger J, Trost J, Wenstrup RJ, Roa BB. Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer. 2012;118:5210–5216. doi: 10.1002/cncr.27556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold AG, Otegbeye E, Fleischut MH, Glogowski EA, Siegel B, Boyar SR, Salo-Mullen E, Amoroso K, Sheehan M, Berliner JL, et al. Assessment of individuals with BRCA1 and BRCA2 large rearrangements in high-risk breast and ovarian cancer families. Breast Cancer Res Treat. 2014;145:625–634. doi: 10.1007/s10549-014-2987-6. [DOI] [PubMed] [Google Scholar]
- 28.Casilli F, Tournier I, Sinilnikova OM, Coulet F, Soubrier F, Houdayer C, Hardouin A, Berthet P, Sobol H, Bourdon V, et al. The contribution of germline rearrangements to the spectrum of BRCA2 mutations. J Med Genet. 2006;43:e49. doi: 10.1136/jmg.2005.040212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Hormazabal P, Gutierrez-Enriquez S, Gaete D, Reyes JM, Peralta O, Waugh E, Gomez F, Margarit S, Bravo T, Blanco R, et al. Spectrum of BRCA1/2 point mutations and genomic rearrangements in high-risk breast/ovarian cancer Chilean families. Breast Cancer Res Treat. 2011;126:705–716. doi: 10.1007/s10549-010-1170-y. [DOI] [PubMed] [Google Scholar]
- 30.Pylkäs K, Erkko H, Nikkilä J, Sólyom S, Winqvist R. Analysis of large deletions in BRCA1, BRCA2 and PALB2 genes in Finnish breast and ovarian cancer families. BMC Cancer. 2008;8:146. doi: 10.1186/1471-2407-8-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, Roa BB, Wenstrup RJ, Hartman AR, Syngal S. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology. 2015;149:604–613.e20. doi: 10.1053/j.gastro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macklin S, Durand N, Atwal P, Hines S. Observed frequency and challenges of variant reclassification in a hereditary cancer clinic. Genet Med. 2018;20:346–350. doi: 10.1038/gim.2017.207. [DOI] [PubMed] [Google Scholar]
- 33.Moghadasi S, Eccles DM, Devilee P, Vreeswijk MP, van Asperen CJ. Classification and clinical management of variants of uncertain significance in high penetrance cancer predisposition genes. Hum Mutat. 2016;37:331–336. doi: 10.1002/humu.22956. [DOI] [PubMed] [Google Scholar]
- 34.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 35.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 36.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 37.Belaiba F, Medimegh I, Bidet Y, Boussetta S, Baroudi O, Mezlini A, Bignon YJ, Benammar El gaaied A. BRCA1/BRCA2 Mutations Shaped by ancient consanguinity practice in southern mediterranean populations. Asian Pac J Cancer Prev. 2018;19:2963–2972. doi: 10.22034/APJCP.2018.19.10.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medimegh I, Troudi W, Omrane I, Ayari H, Uhrhummer N, Majoul H, Benayed F, Mezlini A, Bignon YJ, Sibille C, Elgaaied AB. Consanguinity protecting effect against breast cancer among tunisian women: Analysis of BRCA1 haplotypes. Asian Pac J Cancer Prev. 2015;16:4051–4055. doi: 10.7314/APJCP.2015.16.9.4051. [DOI] [PubMed] [Google Scholar]
- 39.LaDuca H, Polley EC, Yussuf A, Hoang L, Gutierrez S, Hart SN, Yadav S, Hu C, Na J, Goldgar DE, et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet Med. 2019;22:407–415. doi: 10.1038/s41436-019-0633-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science. 2014;343:1466–1470. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bagwell AK, Sutton TL, Gardiner S, Johnson N. Outcomes of large panel genetic evaluation of breast cancer patients in a community-based cancer institute. Am J Surg. 2021;221:1159–1163. doi: 10.1016/j.amjsurg.2021.03.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the ClinVar respiratory, ncbi.nlm.nih.gov/clinvar/?term=SUB10439877 and ncbi.nlm.nih.gov/clinvar/?term=SUB10447902