

Abstract
Using reported glutathione S-transferase omega 1 (GSTO1–1) cocrystal structures, we designed and synthesized acrylamide-containing compounds that covalently bind to Cys32 on the catalytic site. Starting from a thiazole derivative 10 (GSTO1–1 IC50 = 0.6 μM), compound 18 was synthesized and cocrystallized with GSTO1. Modification on the amide moiety of hit compound 10 significantly increased the GSTO1–1 inhibitory potency. We solved the cocrystal structures of new derivatives, 37 and 44, bearing an amide side chain bound to GSTO1. These new structures showed a reorientation of the phenyl thiazole core of inhibitors, 37 and 44, when compared to 18. Guided by the cocrystal structure of GSTO1:44, analogue 49 was designed, resulting in the most potent GSTO1–1 inhibitor (IC50 = 0.22 ± 0.02 nM) known to date. We believe that our data will form the basis for future studies of developing GSTO1–1 as a new drug target for cancer therapy.
Graphical Abstract

INTRODUCTION
Glutathione S-transferases (GSTs) are a diverse family of cytosolic, mitochondrial, and microsomal enzymes that conjugate glutathione to a variety of exogenous compounds including drugs, industrial intermediates, pesticides, herbicides, environmental pollutants, and carcinogens, which mainly act as part of an integrated defense strategy.1,2 In addition, GSTs also play an important role in biotransformation of endogenous substrates and other biological functions.3 The mammalian GSTs are classified into seven different classes, alpha, mu, pi, theta, sigma, zeta, and omega. Among them, the omega GSTs (GSTOs) belong to an atypical cytosolic class and two isozymes, GSTO1–1 and GSTO2–2, have been identified in humans that share low sequence identity with other GST classes.4 GSTO1–1 possesses S-phenacyl glutathione reductase and monomethylarsonate reductase activity, whereas GSTO2–2 catalyzes dehydroascorbate reduction.5 S-(4-Nitrophenacyl)-glutathione (4-NPG) is also reported as a good substrate for GSTO1–1.6 They are also unique among GSTs in terms of their catalytic amino acid residues. Rather than a catalytic serine or tyrosine, GSTOs possess a reactive cysteine nucleophile in the active site.7 Therefore, their enzymatic activities are sensitive to molecules that are able to conjugate with the thiol group, such as N-ethylmaleimides or haloacetamides.8–10 The crystal structure of GSTO1 revealed two important active sites (PDB ID: 1EEM).7 One is the hydrophobic or electrophile binding site (H-site) that can easily accommodate lipophilic electrophiles and the other is a narrow strip consisting of a series of hydrophilic residues that can selectively bind with glutathione (G-site).7
GSTO1–1 is overexpressed in select human cancers such as pancreatic cancer,11 head and neck cancer,12 urinary bladder cancer,13 and colorectal cancer,14 which leads to an accelerated detoxification of drug substrates and thus is implicated in drug resistance.15–18 We previously reported that silencing GSTO1–1 with siRNA impairs cancer cell viability, suggesting that GSTO1–1 is a potential target for cancer therapy.4 It is also reported that GSTO1–1 plays a proinflammatory role in models of inflammation, colitis, and obesity.19 Only a limited number of GSTO1–1 inhibitors have been reported, and there is a strong correlation between thiol-alkylating groups in these compounds and their GSTO1–1 inhibitory activities (Figure 1). The epoxide-bearing cytokine release inhibitory drug (CRID) has affinity toward GSTO1–1, which may be related to their inhibition of ATP-induced IL-1β posttranslational processing.20 Previous screen of the Molecular Libraries Small Molecule Repository (MLSMR) library uncovered a class of selective α-chloroacetamide GSTO1–1 inhibitors. After further optimization, KT53 was selected as a potent GSTO1–1 inhibitor that enhanced cisplatin-induced cytotoxicity.21 However, KT53 showed instability in phosphate-buffered saline (PBS) within 6 h at room temperature because of its intramolecular cyclization reaction. A GSTO1 cocrystal with ML175 was subsequently reported (PDB ID: 5V3Q).22 Additionally, an aryl fluorosulfate compound was identified as a GSTO1–1 probe by an “inverse drug discovery” strategy and its X-ray cocrystal structure was also solved (PDB ID:5UEH).23 Previously, we developed a series of chloroaceta-mide-containing GSTO1–1 inhibitors by high-throughput screening of our in-house compound library.4 The lead compound C1–27 was discovered as a low-molecular-weight chloroacetamide derivative with good potency. The cocrystal structure of the GSTO1-C1–27 complex (PDB ID: 4YQM) was also obtained, showing C1–27 binding in the H-site of GSTO1. C1–27 exhibited cytotoxicity in several cancer cell lines, but knockdown of GSTO1–1 did not significantly affect C1–27 cytotoxicity, suggesting that C1–27 has off-target effects at higher concentrations than those required for GSTO1–1 inhibition. All of GSTO1–1 inhibitors discussed above display similar structural features to match the GSTO1 protein structure. Their hydrophobic cores (usually electron-deficient aromatic rings) allow them to enter the H-site easily. Most importantly, they all contain electrophilic reactive groups (epoxide, chloroacetamide, benzyl chloride, aryl fluorosulfate) that can covalently bind with the Cys32 thiol group of GSTO1. We noticed that α,β-unsaturated carbonyls, such as acrylamides, acrylic esters, and chalcones, can also be served as covalent modifiers. Drugs or compounds with α,β-unsaturated carbonyls are mostly designed to undergo an irreversible hetero-Michael addition reaction with a specific residue (typically, unsubstituted cysteines) of the target protein. Compared with other covalent binding groups, α,β-unsaturated carbonyls, especially acrylamides, are considered to be mild and present in select Food Drug and Administration-approved drugs.24–26 Previous reports showed that a series of artemisinin derivatives containing 1,2,4-trioxane acrylates displayed GSTP1 inhibition activity, indicating that α,β-unsaturated carbonyl modifiers might be applicable for GST inhibitors. 27 Currently, there is no reported GSTO1–1 inhibitor containing an α,β-unsaturated carbonyl moiety. Herein, we describe a structure-guided approach of developing novel GSTO1–1 inhibitors based on our previously published GSTO1 crystal structures.
Figure 1.

Examples of reported GSTO1–1 inhibitors.
RESULTS AND DISCUSSION
Chemistry.
The synthesis of compounds 1–12 is shown in Scheme 1. Compounds 1–3 were synthesized from a C1–27 intermediate 5-amino-2-chloro-N,N-dimethylbenzenesulfonamide 4i conjugated with acryloyl chloride, 2-chloroethylsulfonyl chloride, and methyl 4-bromocrotonate, respectively, as described in our previous study.4 Commercially available heterocycle amines were treated with acryloyl chloride in dichloromethane (DCM) to give 5–12. Scheme 2 describes the synthesis of compounds 16–27 with different substituents. Bromination of commercially available acetophenones 13a–13l using copper bromide in ethyl acetate gave 2-bromoacetophenones 14a–14l.28 Treatment of 2-bromoacetophenones with thiourea in ethanol yielded 4-phenyl-2-aminothiazole derivatives 15a–15l.29 The amines were then conjugated with acryloyl chloride to give compounds 16–27.
Scheme 1. Synthesis of Compounds 1–12a.

aReagents and conditions: (a) acryloyl chloride, DCM, DIEA, rt, 2 h; (b) 2-chloroethylsulfonyl chloride, DCM, DIEA, rt, 2 h; (c) methyl (E)-4-bromocronate, potassium fluoride, CH3CN, 100 °C, 4 h.
Scheme 2. Synthesis of Thiazole Acrylamide Analogues 16–27a.

aReagents and conditions: (a) CuBr2, EtOAc, reflux, 3 h; (b) thiourea, EtOH, reflux, 2 h; (c) acryloyl chloride, DCM, DIEA, rt, 2 h.
Compounds 29–34 were synthesized as described in Scheme 3 to investigate the influence of installing substituents on the thiazole ring and the amide nitrogen. Compounds with 5-substituents on the thiazole ring 29 and 30 were obtained by treating commercially available α-bromoketones 14o and 14p with thiourea, followed by coupling with acryloyl chloride. Compounds with N-substitutions on the acrylamide moiety were synthesized under two different conditions. Intermediates 28c and 28d were obtained by 4f and corresponding bromides. N-substituted 4-phenylthiazol-2-amines 28c–28d were heated with acryloyl chloride in toluene to give 33–34. The total yield of 33–34 was relatively low because the bromination process resulted in bisubstituted side products. In order to overcome this limitation and generate more derivatives with an amide side chain efficiently, we developed another synthetic route for compounds 37–45 (Scheme 4). The commercially available starting material 2-bromoacetophenone 14m was treated with potassium thiocyanate followed by primary amines to give the intermediate 28e–28h.30 Acid hydrolysis of methyl ester intermediate 28e provided the free carboxylic acid 35, which was then conjugated with amines to give amides 36a–36d. Compounds 28e–28h, 36a–36d were treated with acryloyl chloride to give 37–45.
Scheme 3. Synthesis of Thiazole Acrylamide Analogues 29–34a.

aReagents and conditions: (a) corresponding N-substituted thiourea, EtOH, reflux, 2 h; (b) K2CO3, CH3CN, reflux, 8 h; (c) acryloyl chloride, DCM, rt, 2 h; (d) acryloyl chloride, toluene, 70 °C, 0.5 h.
Scheme 4. Synthesis of Thiozale Acrylamide Analogues 37–45a.

aReagents and conditions: (a) KSCN, EtOH, reflux, 3 h; (b) corresponding primary amines, EtOH, reflux, overnight; (c) HCl, THF/H2O, reflux, 2 h; (d) EDC, HOBt, DIEA, DMF, rt, overnight; (e) acryloyl chloride, toluene, 70 °C, 0.5 h; (f) TFA, DCM, rt, 2 h.
Compounds 47–52 were synthesized as described in Scheme 5. Hydroxyl protection of 13p and 13q gave intermediate 13r and 13s. Bromination and cyclization processes were similar with the protocols presented in Scheme 4 to give 28i–28l from 14e, 14r, 14s, and 14l, respectively. Compound 46c was obtained by conjugating 28l with 2-chloroacetyl chloride. Methyl ether deprotection of 46a–46c and 48 gave the hydroxyl derivatives 49–52.
Scheme 5. Synthesis of Thiazole Acrylamide Analogues 49–52a.

aReagents and conditions: (a) CH3I, acetone, reflux, 8 h; (b) CuBr2, EtOAc, reflux, 3 h; (c) KSCN, EtOH, reflux, 3 h; (d) (3-methylisoxazol-5-yl)methanamine, EtOH, reflux, 32 h; (d) acryloyl chloride, toluene, 70 °C, 0.5 h; (e) 2-chloroacetyl chloride, DIEA, toluene, 70 °C, 1 h; (f) BBr3, DCM, −78 °C to rt, 0.5–2 h, THF, TEA, reflux, 1 h.
The synthesis of compound 53 started with tert-butyl (4-bromothiophen-2-yl)carbamate 54 conjugated with 5-(bromomethyl)-3-methylisoxazole in the presence of NaH (Scheme 6). The Boc group of intermediate 55 was cleaved before conjugating with acryloyl chloride. A Suzuki coupling with (2-hydroxyphenyl)boronic acid offered the final product 53.
Scheme 6. Synthesis of Compound 53a.

aReagents and conditions: (a) 5-(bromomethyl)-3-methylisoxazole, NaH, DMF, 0 °C to rt, 2 h; (b) TFA, DCM, rt, 2 h; (c) acryloyl chloride, toluene, 70 °C, 0.5 h; (d) Pd(PPh3)4, Na2CO3, dioxane, H2O, 80 °C, 8 h.
Design and SAR Studies.
GSTO1–1 is considered a relatively new target for early stage drug discovery. Various assays can be used to identify GSTO1 inhibitors.31 We used several assays to evaluate the GSTO1–1 inhibitor C1–27, including the 4-NPG substrate assay and in-gel competitive fluorescence assay. Because C1–27 and all other GSTO1–1 inhibitors bind with GSTO1–1 on the catalytic active site residues, we tested the GSTO1–1 inhibitory activity of all compounds by a competitive fluorescence assay. Using 5-chloromethylfluorescein diacetate (CMFDA), which has been reported to be a potent and irreversible GSTO1–1 inhibitor (Figure 1),32 we optimized the gel-based binding assay that efficiently measures the competitive inhibition of CMFDA binding to GSTO1; a representative example of this assay is shown in Figure S1. Recombinant GSTO1–1 was used to reduce assay interference caused by limitation of gel separation. It is of interest to note that for irreversible covalent inhibitor ideal measurement is Kinact because it considers the rate of inactivation and the binding affinity.26 The limitation of our assay is that it cannot measure the rate of inactivation and the binding affinity. Thus, in this study, we determined % inhibition of the binding of CMFDA to GSTO1 by the compound, where compounds are preincubated with GSTO1 for 30 min, followed by addition of CMFDA for 30 min. Preincubation at various time points did not show notable difference in the IC50 values (Figure S2). In this study, we determined IC50 for all compounds preincubated for 30 min.
Initially, we modified the chloroacetamide group of C1–27 with other electrophilic covalent binding groups. Its replacement with an acrylamide or a vinyl sulfonamide moiety caused a significant decrease in either GSTO1–1 potency or cytotoxicity (Table 1). By analyzing the cocrystal structure of C1–27 and GSTO1–1, we observed an important H-bond interaction between the sulfonamide moiety and Trp180 that restricts compound’s conformation in the binding pocket (Figure 2A). The chloroacetamide moiety and acrylamide moiety have similar molecular shapes but different binding positions. The cysteine thiol group binds to the alpha-carbonyl carbon atom of C1–27 but with the beta-carbonyl carbon of the acrylamide analogue 1. When we superimposed these two structures, the distance between the two active sites was measured as approximately 1.34 Å (Figure 2B). Direct replacement of chloroacetamide on C1–27 with an acrylamide moiety might result in the loss of H-bond interaction because of the different length of the covalent binding groups. We further synthesized methyl crotonate derivative 3 where the distance from the sulfonamide group to the covalent binding site was retained. We expected that the ester moiety of 3 could interact with any residues on the G-site of GSTO1. However, this modification resulted in a loss of activity, perhaps due to the bulky methyl crotonate motif decreasing the chance of Michael acceptor reaction with the thiol group of Cys32. The unsatisfying result indicated that the C1–27 core was not suitable for acrylamide replacement, and new cores or scaffolds needed to be developed.
Table 1.
GSTO1–1 Binding Assay Activity of C1–27 Analogues 1–3

| ||||
|---|---|---|---|---|
|
| ||||
| Cpd | R | aGSTO 1–1 IC50(μM) | bIC50-MTT (μM) | |
| HCT116 | HT29 | |||
|
| ||||
| Cl-27 |

|
0.021 | 1.2 ±0.6 | 4.3 ±0.6 |
| 1 |

|
3.14 ±3.32 | 24.6; 25.0 | >30 |
| 2 |

|
1.29 ±1.56 | 9.9; 11.0 | 10.2; 16.8 |
| 3 |

|
>10 | >30 | >30 |
GSTO IC50 values were determined using the GSTO binding assay. Values are the mean ± SD of at least three independent experiments.
MTT IC50 values are reported from two independent experiments.
Figure 2.

(A) Cocrystal structure of GSTO1:C1–27 (pink stick, PDB ID: 4YQM). (B) Structural differences of 1 (cyan) and C1–27 (orange) under nucleophilic attack. 3D structures were generated, overlapped, and measured in Maestro. (C) Compound 5 (cyan) displayed similar binding pose with C4–10 (orange) at the GSTO1 H-site in a docking study perform by Glide. (D) Alignment of 5 (yellow green) and 10 (pink) showed the advantage of the thiazole core structure, and 3D structures were generated and overlapped in Maestro.
We previously built a chloroacetamide-containing compound library to screen potential GSTO1–1 inhibitors.4 On this basis of this library, a GSTO1 covalent docking study was conducted to search for novel inhibitors. The chloroacetamide moieties of active compounds were replaced with arylamide, and the new library generated was docked on the GSTO1 active site. Taking into consideration the difference in physical and chemical properties between chloroacetamide and acrylamide, we selected C4–10, a phenylpyrazole derivative, that showed modest potency on the GSTO1–1 binding assay in our previous study as a model compound (Figure 1), and the cocrystal structure of C4–10 with GSTO1 was also solved (PDB ID: 4YQV).4 We observed that its acrylamide analogue 5 exhibited a very similar pose with C4–10 (Figure 2C) in our docking study. The acrylamide moiety was bound to Cys32, the N-phenylpyrazole moiety occupied the H-site, and the benzene ring was located deep into the hydrophobic pocket. Additionally, some other phenyl heterocyclic derivatives were docked onto the GSTO1 active site and showed similar binding poses. Thus, a series of analogues were synthesized and investigated in the GSTO1–1 binding assay to verify our docking results. The compounds with the pyrazole core were inactive (5 and 6). Compounds 7 and 8 with a nonaromatic piperidine core also did not possess significant GSTO1–1 binding activity. However, compounds with a thiazole core displayed potency and 4-phenyl (10) was more potent than 5-phenyl derivatives (11). Most of these heterocyclic compounds are rigid molecules, and their binding conformation and low energy conformation are likely to be quite similar. Therefore, a three-dimensional (3D) structure superimposition was conducted to analyze the structure–activity relationship (SAR). We observed that the benzene ring of the thiazole compounds displayed a shift compared with the pyrazole compound 5, while other motifs were perfectly aligned (Figure 2D), which is probably due to the unique sulfur–carbon bond angle in the thiazole ring (88.6°).33 This special angle allows the benzene ring to insert into the narrow hydrophobic cleft to improve potency. Two other compounds with a thiophene core structure (11 and 12) also showed good potencies, suggesting that the sulfur atom in the heterocyclic core displays positive contribution to potency. Thiophene analogue 11 showed slightly decreased potency as compared to 10, perhaps due to the conformational restriction imposed by its intramolecular H-bond. Endogenous GSTO1–1 binding activity was also observed on 9 and 10 in a cell-based assay. Compound 10 exhibits most desirable ligand efficiency (LE)34 and ligand-lipophilicity efficiency (LLE)35 among these analogues. Therefore, we chose compound 10 as a new GSTO1–1 inhibitor template for further studies and optimization. Compound 10 also displayed cytotoxicity on HCT116 and HT29 cell lines, but whether the cytotoxicity was resulted from GSTO1–1 inhibition needs further investigation (Table 2).
Table 2.
GSTO1–1 Binding Assay Activity, Calculated Molecule Properties, and Cytotoxicity of Phenyl Heterocyclic Arylamides 5–12a

| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Cpd | Het | aGST01–1 IC50(μM) | bcLogP | cLE | dLLE | eIC50-MTT (μM) | |
| HCT116 | HT29 | ||||||
|
| |||||||
| 5 |

|
>10 | 1.87 | - | - | >30 | >30 |
| 6 |

|
>10 | 1.89 | - | - | >30 | >30 |
| 7 |
|
>10 | 2.43 | - | - | >30 | >30 |
| 8 |

|
>10 | 2.88 | - | - | >30 | >30 |
| 9 |

|
1.03 ±0.81 | 2.75 | 0.50 | 3.05 | >30 | 29.3; 24.3 |
| 10 |

|
0.47 ±0.17 | 2.75 | 0.54 | 3.47 | 18.6; 29.0 | 6.6; 6.6 |
| 11 |

|
1.01 ±0.97 | 4.00 | 0.38 | 1.76 | >30 | >30 |
| 12 |

|
0.22 ±0.12 | 4.21 | 0.43 | 2.31 | >30 | >30 |
GSTO IC50 values were determined using the GSTO binding assay. Values represent the mean ± SD of at least three independent experiments.
clog P values were calculated using ChemBioDraw Professional 16.
Calculated LE = −1.4 log IC50/N, where N is the number of nonhydrogen atom. LE > 0.25 means a good hit.
Calculated LLE = pIC50 (M) – clog P. LLE > 3 means a good hit.
MTT IC50 values are reported from two independent experiments.
The binding pose of compound 10 in our docking study showed that the benzene ring does not fully occupy the H-site and additional lipophilic groups can be accommodated. We investigated whether modifications on the phenyl group can improve potency. All compounds were tested in the GSTO1–1 binding assay and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide (MTT) cytotoxicity assay. Because the major interaction between GSTO1 and our compounds is hydrophobic interaction, the LLE of these compounds was also investigated. Substitution of the benzene ring with chlorine atoms showed a preference at 4-position (18, IC50 = 0.17 μM) over other positions. This may be due to the fact that 4-chloro can fill additional space in the hydrophobic pocket. The classic bioisosteric replacement of chlorine with the methyl group resulted in a slightly decreased potency. We hypothesized that the activity change was related to the different electron inductive effects between chlorine and the methyl group on the benzene ring. When bound to GSTO1, the benzene ring located in the hydrophobic pocket is surrounded by Trp180, Phe31, Phe225, and Tyr229. These residues can form π–π interaction with the benzene ring, and the interaction intensity is significantly influenced by the electron density of the aromatic ring. In order to test the feasibility of this hypothesis, additional analogues with substitutions at 4-position were synthesized. As anticipated, analogues with strong electron-withdrawing groups (e.g., trifluoromethyl analogue 20, IC50 = 0.30 μM) displayed improved potency, whereas analogues with 2,3-dioxole, a strong electron-donating substituent, showed decreased potencies (IC50 = 1.22 μM).
To validate our design strategy and to guide future analogue design, we obtained the cocrystal structure of 18 bound to GSTO1 (PDB ID: 6MHB, Figure 3A). The 2.75 Å structure of GSTO1:18 demonstrates similarity to the cocrystal structure of GSTO1 with a chloroacetamide inhibitor C4–10 (PDB ID: 4YQV)4 in both the overall structure of GSTO1 [an average root mean square deviation (RMSD) between two structures of 0.49 Å] as well as the orientation of the ligand. Compound 18 occupies the same deep hydrophobic cleft of the H-site aligned by aromatic residues including Trp180, Phe31, Phe225, and Tyr229 (Figure 3B). The 4-chlorophenyl moiety makes van der Waals contacts with the side chains of Val127, Arg183, Met187, Trp222, Phe225, and Leu226 as well as the backbone amide of Phe31 (Figure 3A). The propionamide and thiazole ring make hydrophobic contacts with Met29, Pro33, Phe34, and Tyr229. Although Tyr229 appears to interact with the thiazole, its orientation varies between the six chains of GSTO1 modeled as part of this structure, and a lack of electron density in some chains disallows modeling of the Tyr229 side chain altogether. In conjunction with the poor electron density of the thiazole ring, this seems to suggest that binding of 18 is likely driven by interactions of the 4-chlorophenyl moiety with GSTO1. The most significant difference between GSTO1:18 and GSTO1:C4–10 is a change in conformation of the Leu226 side chain, which reorients to interact with the chlorine of the 4-chlorophenyl substituent of 18 (Table 3).
Figure 3.

Cocrystal structure of GSTO1:18 (PDB ID: 6MHB, cyan ribbons:teal sticks). (A) Hydrophobic interactions of 18 within the H-site of GSTO1. (B) Structural comparison of GSTO1:18 and GSTO1:C4–10 (PDB ID: 4YQV, gray ribbons:black sticks) demonstrates that these inhibitors bind with similar orientations within a hydrophobic cavity (gray surface) of the GSTO1 H-site. Average RMSD = 0.49 Å
Table 3.
GSTO1–1 Binding Assay, clog P, LLE, and Cytotoxicity of Phenyl Thiazole Arylamides 16–27a

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cpd | R |
aGST01-l IC50(μM) |
bcLogP | cLLE | dIC50-MTT (μM) | |
| HCT116 | HT29 | |||||
|
| ||||||
| 16 |
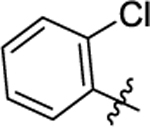
|
0.88 ±1.17 | 3.11 | 2.66 | 20.1; 12.3 | 10.2; 19.5 |
| 17 |

|
0.60 ± 0.00 | 3.47 | 2.75 | 14.3; 11.9 | 11.4; 11.0 |
| 18 |

|
0.17 ±0.01 | 3.47 | 3.23 | 20.6; 12.6 | 17.3; 23.7 |
| 19 |

|
0.81 ±0.41 | 3.29 | 2.80 | 18.6; 15.3 | 16.7; 16.1 |
| 20 |

|
0.30 ± 0.29 | 3.64 | 2.88 | 5.7; 4.0 | 5.7; 4.0 |
| 21 |

|
4.49 ±0.13 | 2.77 | 2.58 | 12.9; 8.0 | 12.2; 12.0 |
| 22 |

|
1.22 ±0.78 | 2.79 | 3.13 | 17.3; 16.8 | 20.1; 17.7 |
| 23 |

|
0.4 ± 0.07 | 3.92 | 2.48 | 17.4; 11.8 | 12.5; 13.4 |
| 24 |

|
0.86 ±0.51 | 1.98 | 4.09 | >30 | 16.2; 24.9 |
| 25 |

|
>10 | 2.28 | - | 12.5; 7.1 | 13.5; 6.1 |
| 26 |

|
>10 | 2.31 | - | >30 | >30 |
| 27 |

|
1.58 ±0.97 | 2.90 | 2.90 | 21.3; 20.4 | 19.1; 18.7 |
GSTO IC50 values were determined using the GSTO binding assay. Values represent the mean ± SD of at least three independent experiments.
clog P values were calculated using ChemBioDraw Professional 16.
Calculated LLE = pIC50 (M) – clog P.
MTT IC50 values are reported from two independent experiments.
Interestingly, we found that the benzene ring was close to the Phe31 backbone amide (3.50 Å), which could be treated as a potential binding site. Therefore, we generated 2-hydroxyl analogue 24. The potency was improved although not that significantly. Meanwhile, the other 2-position substitutions or 4-hydroxyl substitution led to a potency reduction. The 2-methoxyl analogue 26 was inactive. We speculated that the 2-hydroxyl group probably formed an H-bond with the backbone oxygen of Phe31, but adding ortho-position substituents altered the binding energy and offset the potency gained from the H-bonding. We also tested the cytotoxicity of these compounds in the MTT assay. Most of these analogues showed inhibition of cell viability, but their cytotoxicities did not show an obvious correlation with their GSTO1 binding potency.
In order to extend our SAR study and improve potency, we analyzed the cocrystal structure of 18 bound to GSTO1 looking for other potential interaction sites. Two regions on the binding pocket can be identified as exploitable sites. One is a hydrophobic area near the “T cave” surrounded by aromatic ring residues such as Tyr229, Phe31, and Phe225. The other one is Trp180 close to the thiazole ring (distance = 5.46 Å), which plays an important role as an H-bond donor when bound with C1–27. Therefore, we designed analogues with substituents on the 5-position of the thiazole ring or adding side chain on amide nitrogen.
Inserting a methyl on the 5-position of the thiazole ring led to a potency reduction (29, 3.1 ± 1.5 μM), whereas methyl 5-carboxylate analogue 30 exhibited slightly increased potency. We assumed that 5-substitutions generally affected the low energy conformation, which accounted for the potency decrease for 29, whereas the carboxylate group in 30 improved the binding free energy by forming another H-bond with Trp180. Compounds 33 and 34 displayed increased potencies, suggesting that more area occupied in the H-site helps exclude “unfavorable water molecules” and contribute to potency. Therefore, additional compounds with amide side chains were synthesized to improve potency based on this theory. Because ethyl acetate derivative 34 showed improved potency, we further examined if its homologues 37, 38, and 39 could be accommodated within the cavity. A bulky tert-butyl group can be tolerated, whereas the free carboxylic acid replacement resulted in a significant activity loss, suggesting that lipophilic groups may be beneficial to improve potency as we expected. Additional derivatives were synthesized to investigate the tolerance of other isosteres. Replacement of the methyl ester group with more lipophilic cyclohexyl amides led to increased potency, which also validated our hypothesis. Considering that there are still polar residues around the molecule, we sought to design amide derivatives with potential H-bond acceptors. Compounds with a terminal oxygen, including 43 with a bulky N-(2-morpholinoethyl) group, exhibited moderate to good potencies. Compound 42 displayed the best potency up to this stage (IC50 = 0.03 ± 0.02 μM). All analogues showed improved LLE, suggesting that our optimization was reasonable. In addition, we also synthesized compounds based on 33. We hypothesized that the additional benzyl group forms edge-to-face π-stacking with Tyr229. However, the benzyl group installation made the compound greasy (clog P = 4.48). In order to reduce lipophilicity, the pyridine derivative was synthesized. Unfortunately, compound 45 showed decreased potency probably due to the electron-deficient pyridine ring impairing the π-interaction. Hence, we designed a molecule with the 3-methyl-isoxazole group, which was regarded as a preferred druglike and electron-rich heterocycle group. This modification resulted in fairly good activity (44, IC50 = 0.051 μM) and improved clog P (2.96). When the potency of the GSTO1–1 binding activity improved, these derivatives, however, lost their cytotoxicity, indicating that inhibiting GSTO1–1 might not influence cell viability.
The cocrystal structures of GSTO1 with 37 and 44 were solved to resolutions of 2.0 (PDB ID: 6MHC) and 2.15 Å (PDB ID: 6MHD), respectively. The two structures reveal very little difference in the overall structure of GSTO1 (average RMSD = 0.33 Å) when compared to one another but demonstrate more substantial differences when compared with the structure of GSTO:18 (average RMSD = 0.78 Å for GSTO1:37 and 0.80 Å for GSTO1:44) (Figure 4A). In particular, the structures of GSTO1 with 37 and 44 show a rearrangement of the H-site as a result of a shift in the α9 helix when compared to the GSTO1:18 structure (Figure 4A). In this state, the side chain of Trp222 is reoriented to occupy the cavity that accommodates the 4-chlorophenyl ring of 18. In both structures, the electron density of inhibitors 37 and 44 suggests modeling the thiazole ring such that it engages in π–π stacking interactions with the side chain of Tyr229, which appears well ordered in all chains of both structures, and to a lesser extent Phe225 (Figure 4B). The electron density associated with the phenyl ring of both inhibitors is less definitive, suggesting that there is likely a degree of free rotation about the bond connecting the two ring systems. The phenyl substituent is poised to make weak hydrophobic contacts with α4 helix residues Gly128, Ile131, Arg132, as well as Leu226 and Tyr229, which form a shallow pocket in the H-site. In the GSTO1:37 structure, the electron density did not support modeling the ester, which is projected into bulk solvent and consequently disordered (Figure 4B). In contrast, an interaction with the side chain of Phe34 orders the isoxazole ring of 44, which extends this portion of the ligand toward the G-site (Figure 5A). This additional interaction likely contributes to the improvement in potency of 44 when compared to 37 because it may block glutathione to enter the G-site. This discovery led us to further optimization by adding more flexible substituents attached to functional groups to interact with residues at the G-site that may further improve potency (Table 4).
Figure 4.

Structural comparison of GSTO1:37 (PDB ID: 6MHC, 37 is shown in pink sticks), GSTO1:44 (PDB ID: 6MHD, 44 is shown in green sticks) and GSTO1:18 (PDB ID: 6MHA, 18 is shown in teal sticks). (A) Structural differences are a result of the N-substituted inhibitors 37 (magenta sticks) and 44 (dark green sticks) adopting a different orientation within the GSTO1 H-site compared to 18 (teal sticks). Trp222 makes pi-stacking with the chlorophenyl group of 18 but has no contact with the phenyl groups of 37 and 44. (B) Hydrophobic interactions of GSTO1 with inhibitors 37 and 44.
Figure 5.

(A) N-substitution of thiazole acrylamide inhibitors, as demonstrated by the cocrystal structure of GSTO1:44 (PDB ID: 6MHD, dark green sticks), may provide a handle to access hydrogen-bonding or water-mediated interactions with residues in the glutathione binding site (glutathione from PDB ID: 1EEM shown in black sticks). Residues that interact with both glutathione and 44 are highlighted in light blue, whereas sites of potential enthalpic interactions with extended N-substituted thiazole inhibitors are highlighted in yellow. (B) 2-Position of the phenyl ring of inhibitor 44 is directed into a pocket with a strong overall positive charge. The 2-hydroxyl substituent of 49, bearing a partial negative charge, is likely stabilized through electrostatic interactions with the inhibitor binding site. Electrostatic maps were generated using APBS from −5.0 eV (red) to +5.0 eV (blue).
Table 4.
GSTO1–1 Binding Assay, clog P, LLE, and Cytotoxicity of Thiazole Arylamides 37–45a

| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Cpd | R1 | R2 | aGSTO1–1IC50(μM) | bcLogP | cLLE | dIC50-MTT (μM) | |
| HCT116 | HT29 | ||||||
|
| |||||||
| 29 | H | Me | 3.11±1.54 | 2.94 | 2.57 | 21.7; 20.1 | 22.8; 22,0 |
| 30 | H | COOEt | 0.27±0.13 | 3.38 | 3.14 | 24.7; 19.6 | 25.3; 18.0 |
| 33 |

|
H | 0.72±0.14 | 4.48 | 2.44 | >30 | >30 |
| 34 |

|
H | 0.57±0.07 | 3.05 | 3.91 | >30 | 19.6; 19.0 |
| 37 |

|
H | 0.37 ± 0.01 | 2.52 | 3.91 | >30 | >30 |
| 38 |
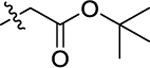
|
H | 0.15 ± 0.03 | 3.75 | 3.07 | 20.9 | >30 |
| 39 |

|
H | 2.04 ± 0.34 | 2.25 | 3.44 | >30 | >30 |
| 40 |

|
H | 0.08 ± 0.04 | 3.69 | 3.41 | 29.3; >30 | >30 |
| 41 |

|
H | 0.53 ± 0.77 | 2.21 | 4.07 | 19.8; 14.6 | 14.8; 13.3 |
| 42 |
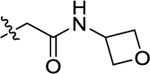
|
H | 0.03 ± 0.02 | 2.22 | 5.30 | 20.8; 23.2 | >30 |
| 43 |

|
H | 0.23 ± 0.09 | 2.26 | 4.38 | 15.6; 7.3 | 14.5; 12.2 |
| 44 |

|
H | 0.051 ± 0.001 | 2.96 | 4.33 | >30 | >30 |
| 45 |

|
H | 0.74 ± 0.02 | 2.98 | 3.15 | >30 | >30 |
GSTO IC50 values were determined using the GSTO binding assay. Values represent the mean ± SD of at least three independent experiments.
clog P values were calculated using ChemBioDraw Professional 16.
Calculated LLE = pIC50 (M) – clog P.
MTT IC50 values are reported from two independent experiments.
The reorientation of the phenyl thiazole core of inhibitors 37 and 44 when compared to 18 seems to suggest that the deep cleft in the H-site occupied by inhibitors C4–10 and 18 cannot accommodate much steric bulk associated with N-substitution of the acrylamide. This ultimately forces N-substituted ligands to occupy a more solvent-exposed pocket of the H-site, which is driven mostly by the hydrophobic packing of the thiazole, whereas the deep hydrophobic cleft occupied by inhibitors C4–10 and 18 locks the phenyl ring into a more ordered state.
On the basis of the above preliminary SAR results, we continued to design analogues based on our new optimized compound 44. We first combined the 4-trifluromethylphenyl group and 3-methyl-isoxazole side chain on acrylamide nitrogen to provide a hybrid compound 47. This modification showed a little improvement in potency as compared to 44. We then examined the isoxazole compound with the 2-hydroxylphenyl group to match the potential protein–ligand interaction requirement, and this molecule exhibits a remarkable picomole level potency (49, IC50 = 0.00022 μM). Meanwhile, its 2-methoxylphenyl analogue 48 was 200-fold less potent than 49, validating the hypothesis that the hydroxyl group did play an important role on binding interaction. From the cocrystal structure of GSTO1: 44 and docking studies, it was not apparent that the hydroxyl group would form H-bonds with any protein residues. We believe that the increase in potency is due to charge interaction. The partial negative charge of the oxygen atom would be highly attracted to the positive charge of the drug-binding pocket. Thus, compound 49 with the 2′-hydroxyl substitution would bind tighter than compound 44 (Figure 5B). We also noticed that additional space can be occupied at the 4-position on the benzene ring; therefore, analogues with 4-chloro or 4-fluoro were designed. Compound 50 also displayed excellent potency, whereas decreased potency was observed with the fluorinated analogue 51. High LLE values and optimal clog P of these compounds also illustrated that they also have good druglike properties. These compounds showed remarkable potency in the GSTO1–1 assay but did not show significant cytotoxicity, indicating that the selectivity of these compounds can be further improved (Table 5).
Table 5.
GSTO1–1 Binding Assay, clog P, LLE, and Cytotoxicity of Thiazole Arylamides 47–51

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cpd | R | aIC50(μM) | bcLogP | cLLE | dIC50-MTT (μM) | |
| HCT116 | HT29 | |||||
|
| ||||||
| 47 |

|
0.025 ± 0.02 | 3.85 | 4.04 | 20.3; 19.7 | 25.9; 22.3 |
| 48 |

|
0.36 ± 0.22 | 2.42 | 4.02 | >30 | >30 |
| 49 |
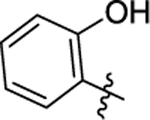
|
0.00022 ± 2.43E-05 | 2.19 | 7.51 | >30 | >30 |
| 50 |

|
0.00032 ± 2.57E-05 | 3.07 | 6.45 | >30 | >30 |
| 51 |

|
0.04 ± 0.04 | 2.50 | 4.90 | >30 | >30 |
GSTO IC50 values were determined using the GSTO binding assay. Values represent the mean ± SD of at least three independent experiments.
clog P values were calculated using ChemBioDraw Professional 16.
Calculated LLE = pIC50 (M) – clog P.
MTT IC50 values are reported from two independent experiments.
Compound 49, designed based on the GSTO1:44 co-crystal structure, is the most potent GSTO1 inhibitor (IC50 = 0.22 ± 0.02 nM) known to date and did not show off-target-induced cytotoxicity. There is a possibility that 2-OH of this molecule may make the overall molecule a potential chelator. We tested the compound in the presence or absence of various metals to examine whether a chelation could give rise to assay interference. We did not see a significant effect by various metals in the activity of the compound (Figure S3). This compound also potently inhibited the GSTO1 enzyme activity in the 4-nitrophenacyl glutathione (4NPG) reduction assay (Figure S4). Although not tested in this study, due to 64% sequence identity of GSTO1 and GSTO2, we expect these inhibitors may also bind to GSTO2.
Covalent inhibitors sometimes display instability in vitro or in vivo because of their reactivity.24 Compounds 18 and 49 were selected for stability studies. They showed good stability in 20% acetonitrile/PBS buffer solution at room temperature overnight as detected by liquid chromatography–mass spectrometry (LC–MS). An in vitro mouse and human liver microsome stability assay was used to evaluate their metabolic properties (Table 6). The results showed that compounds were rapidly metabolized in both mouse and human microsomes perhaps due to the acrylamide moiety being the main metabolic liability.
Table 6.
Metabolic Stability and Half-Life of Compounds 18 and 49 in Mouse and Human Liver Microsomes
| percentage remaining (%) |
|||||
|---|---|---|---|---|---|
| time point (min) | a18 in MLM | b18 in HLM | a49 in MLM | b49 in HLM | cVerapamil in MLM |
| 0 | 100 | 100 | 100 | 100 | 100 |
| 5 | 33.93 | 66.67 | 16.00 | 14.81 | 26.51 |
| 10 | 17.86 | 48.28 | 12.00 | 7.41 | 10.11 |
| 15 | 12.5 | 39.08 | 12.00 | 3.70 | 4.73 |
| 30 | 7.14 | 19.54 | 8.00 | 3.70 | |
| 45 | 5.36 | 8.04 | 4.00 | 3.70 | |
| 60 | 5.36 | 5.75 | 4.00 | 3.70 | |
| half-life (min) | 3.21 | 9.52 | 1.81 | 1.89 | 2.70 |
MLM: mouse liver microsome
HLM: human liver microsome.
Verapamil was set as a positive control.
Additional optimizations were conducted to improve the metabolic stability without major decrease in potency. Two more analogues were synthesized (Table 7). Chloroacetamide, the most commonly used covalent reactive group of GSTO1–1 inhibitors, was conjugated with the scaffold of compound 49 to obtain compound 52. However, the replacement displayed respectable potency loss, indicating that the scaffold is more suitable for acrylamide. Concerning the thiazole nitrogen of compound 49 may be attacked by its acrylamide structure forming an inactive cyclic product like KT53, we synthesized 53 by replacing the thiazole ring by thiophene but that resulted in a 1000-fold loss in potency. Future studies will focus on seeking analogues with better PK properties.
Table 7.
GSTO1–1 Binding Assay, clog P, LLE, and Cytotoxicity of Compounds 52 and 53
| Cpd | Structure |
aGST01–1 IC50(μM) |
bcLogP | cLLE | dIC50-MTT (μM) | |
|---|---|---|---|---|---|---|
| HCT116 | HT29 | |||||
|
| ||||||
| 52 |

|
0.0056±0.01 | 1.65 | 6.60 | >30 | >30 |
| 53 |

|
0.19±0.12 | 3.12 | 3.60 | 20.7; 19.3 | 24.4; 29.5 |
GSTO IC50 values were determined using the GSTO binding assay. Values represent the mean ± SD of at least three independent experiments.
clog P values were calculated using ChemBioDraw Professional 16.
Calculated LLE = pIC50 (M) – clog P.
MTT IC50 values are reported from two independent experiments.
CONCLUSIONS
This study reports our structure-based design efforts toward the development of novel GSTO1 inhibitors with better potency and druglike properties. Our efforts toward discovering new analogues with an acrylamide structure led to a potent phenylthiazole compound 10 (IC50 = 0.5 ± 0.2 μM) possessing desirable clog P, LE, and LLE. Compound 10 was identified as a lead, and a series of analogues were synthesized based on a new cocrystal structure with GSTO1. A cocrystal structure of 18 and GSTO1 revealed that the compound inserted into a hydrophobic cave with the GSTO1 H-site. Derivatives with an amide side chain showed improved potency as compared to those without substitutions. Cocrystal structures of 37 and 44 with GSTO1 showed slightly different binding poses than with 18, but the core structures were still located in the electrophilic pocket. The introduction of an amide side chain caused a shift in the α9 helix and resulted in a reorientation of the H-site. We also found that the isoxazole ring of compound 44 occupies a small portion of the G-site and contributed to improved potency.
The inhibitors disclosed in this study present suitable lead compounds for further development. Future studies will focus on further optimizations of 49 to acquire potent inhibitors with improved PK and pharmaceutical properties. Detailed in vitro and in vivo studies will also be performed to evaluate their efficacy and safety in cancer therapy.
EXPERIMENTAL SECTION
Chemistry.
Reagents and anhydrous solvents were purchased from commercial sources and used without further purification. Reaction progress was monitored by UV absorbance using thin-layer chromatography (TLC) on aluminum-backed precoated silica plates from Silicycle (SiliaPlate, 200 μm thickness, F254). Purifications using flash chromatography were performed using a Biotage Isolera chromatography system equipped with 10 and 25 g Ultra-SNAP Cartridge columns (25 μM spherical silica). Glassware for reactions was oven-dried in reactions performed using nitrogen or argon atmosphere using standard inert conditions. 1H NMR spectra were obtained using a Bruker NMR instrument (300 or 400 MHz), and spectral data are reported using the following abbreviations: (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets); coupling constants are reported in hertz, followed by integration. A Shimadzu LCMS 2020 system was utilized for generating high-performance liquid chromatography (HPLC) traces and obtaining MS data. The system was equipped with a photodiode array (PDA) UV detector and Kinetex 2.6 μm, XB-C18 100 Å, 75 × 4.6 mm column, which was used at room temperature. The HPLC gradient method utilized was 10–95% MeCN in H2O with 0.1% formic acid over 15 min with a 0.50 mL/min flow rate. Reverse-phase preparative purifications were performed on a Shimadzu LC20 modular HPLC system utilizing a PDA detector and a Kinetex 5 μm XB-C18 100 Å, 150 × 21.2 mm column. The purification method used was 25 min gradient of 10–90% MeCN in H2O with 0.05% trifluoroacetic acid as the additive. The purity of final compounds (≥95%) was assessed at 254 nm using the described column and method.
N-(4-Chloro-3-(N,N-dimethylsulfamoyl)phenyl)acrylamide (1).
To a solution of 5-amino-2-chloro-N,N-dimethylbenzenesulfonamide 4i (50 mg, 0.21 mmol) in DCM (10 mL) was added N,N-diisopropylethylamine (DIEA) (89 μL, 0.63 mmol) and stirred at 0 °C for 20 min. Acryloyl chloride (16 μL, 0.60 mmol) in DCM (5 mL) was added slowly. The reaction mixture was stirred at room temperature for 2 h and then partitioned between DCM and saturated NaHCO3. The aqueous layer was extracted with DCM for another two times. The combined organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 2–50% EtOAc in hexane to give a white solid (48 mg, 80% yield). 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 2.6 Hz, 1H), 7.55–7.35 (m, 2H), 7.28 (s, 1H), 7.02 (s, 1H), 6.60 (dd, J = 16.5, 9.8 Hz, 1H), 6.35 (d, J = 16.5 Hz, 1H), 6.07 (d, J = 9.8 Hz, 1H), 2.93 (s, 6H). MS (ESI): 289.0 [M + H]+. Purity: 95.8%.
2-Chloro-N,N-dimethyl-5-(vinylsulfonamido)benzenesulfonamide (2).
To a solution of 5-amino-2-chloro-N,N-dimethylbenzenesulfonamide 4i (50 mg, 0.21 mmol) in DCM (10 mL) was added DIEA (89 μL, 0.63 mmol) and stirred at 0 °C for 20 min. 2-Chloroethylsulfonyl chloride (34 mg, 0.21 mmol) in 5 mL DCM was added slowly. The reaction mixture was stirred at room temperature for 2 h and then partitioned between DCM and saturated NaHCO3. The aqueous layer was extracted with DCM for another two times. The combined organic layer was dried with MgSO4, filtered, concentrated, and purified via flash chromatography using a gradient method of 2–50% EtOAc in hexane to give a white solid (25 mg, 36% yield). 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 2.6 Hz, 1H), 7.55–7.35 (m, 2H), 7.28 (s, 1H), 7.02 (s, 1H), 6.60 (dd, J = 16.5, 9.8 Hz, 1H), 6.35 (d, J = 16.5 Hz, 1H), 6.07 (d, J = 9.8 Hz, 1H), 2.93 (s, 6H). MS (ESI): 325.0 [M + H]+. Purity: 95.7%.
Methyl (E)-4-((4-Chloro-3-(N,N-dimethylsulfamoyl)phenyl)amino)but-2-enoate (3).
To a solution of 5-amino-2-chloro-N,N-dimethylbenzenesulfonamide 4i (100 mg, 0.42 mmol) in acetonitrile (10 mL) were added potassium fluoride (73 mg, 12.8 mmol) and methyl (E)-4-bromocronate (150 mg, 0.84 mmol). The solution was heated at 100 °C for 4 h using a microwave reactor and concentrated in vacuo. The residue was purified with flash chromatography using DCM as the eluent to give 3 as a white solid (73 mg, 53% yield). 1H NMR (300 MHz, CDCl3) δ 7.36–7.25 (m, 2H), 6.93 (dt, J = 15.7, 4.3 Hz, 1H), 6.70 (dd, J = 8.8, 3.1 Hz, 1H), 5.87 (dt, J = 15.8, 2.0 Hz, 1H), 4.12 (dd, J = 4.6, 1.9 Hz, 2H), 3.74 (d, J = 0.8 Hz, 3H), 2.88 (d, J = 0.8 Hz, 6H). MS (ESI): 332.9 [M + H]+. Purity: 96.4%.
General Procedure A: Preparation of Acrylamides 5–12.
To a solution of phenyl heterocyclic amines (0.50 mmol) in DCM (10 mL) was added DIEA (1.50 mmol) and stirred at 0 °C for 20 min. Acryloyl chloride (0.60 mmol) in DCM (10 mL) was added slowly. The reaction mixture was stirred at room temperature for 2 h and then partitioned between DCM and saturated NaHCO3. The aqueous layer was extracted with DCM for two more times. The combined organic layer was dried with MgSO4, filtered, concentrated, and purified via flash chromatography using a gradient method of 2–50% EtOAc in hexane.
N-(1-Phenyl-1H-pyrazol-4-yl)acrylamide (5).
Compound 5 was prepared from 1-phenyl-1H-pyrazol-4-amine 4a (100 mg, 0.62 mmol), DIEA (210 μL, 1.26 mmol), and acryloyl chloride (56 mg, 0.62 mmol) according to the general procedure A as a yellow solid (100 mg, 75% yield). 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 1.4 Hz, 1H), 7.69 (d, J = 1.7 Hz, 1H), 7.39–7.21 (m, 3H), 6.43 (dt, J = 16.9, 1.6 Hz, 1H), 6.34–6.18 (m, 1H), 5.74 (ddd, J = 10.1, 2.7, 1.4 Hz, 1H). MS (ESI): 214.1 [M + H]+. Purity: 98.5%.
N-(1-Benzyl-1H-pyrazol-4-yl)acrylamide (6).
Compound 6 was prepared from 1-benzyl-1H-pyrazol-4-amine 4b (100 mg, 0.58 mmol), DIEA (193 μL, 1.16 mmol), and acryloyl chloride (52 mg, 0.58 mmol) according to the general procedure A as a yellow solid (107 mg, 82% yield). 1H NMR (300 MHz, CDCl3) δ 8.45 (s, 1H), 8.02 (s, 1H), 7.45 (s, 1H), 7.37–7.24 (m, 1H), 7.29 (s, 2H), 7.20 (dd, J = 7.4, 2.2 Hz, 2H), 6.37 (dd, J = 17.0, 1.6 Hz, 1H), 6.21 (dd, J = 16.9, 10.0 Hz, 1H), 5.67 (dd, J = 10.0, 1.6 Hz, 1H), 5.23 (s, 2H). MS (ESI): 227.9 [M + H]+. Purity: 98.5%.
1-(4-Phenylpiperidin-1-yl)prop-2-en-1-one (7).
Compound 7 was prepared from 4-phenylpiperidine 4c (100 mg, 0.62 mmol), DIEA (207 μL, 1.24 mmol), and acryloyl chloride (56 mg, 0.62 mmol) according to the general procedure A as a colorless oil (117 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ 7.40–7.17 (m, 5H), 6.65 (dd, J = 16.8, 10.5 Hz, 1H), 6.32 (dd, J = 16.8, 2.0 Hz, 1H), 5.72 (dd, J = 10.5, 2.0 Hz, 1H), 4.86 (d, J = 13.2 Hz, 1H), 4.21–4.10 (m, 1H), 3.20 (t, J = 13.0 Hz, 1H), 2.93–2.75 (m, 1H), 2.87–2.55 (m, 3H), 1.69 (td, J = 16.3, 12.6, 7.5 Hz, 2H). MS (ESI): 215.9 [M + H]+. Purity: 100.0%.
1-(4-Benzylpiperidin-1-yl)prop-2-en-1-one (8).
Compound 8 was prepared from 4-benzylpiperidine 4d (100 mg, 0.57 mmol), DIEA (190 μL, 1.14 mmol), and acryloyl chloride (51 mg, 0.57 mmol) according to the general procedure A as a colorless oil (117 mg, 90% yield). 1H NMR (300 MHz, CDCl3) δ 7.37–7.19 (m, 2H), 7.16 (dd, J = 5.5, 3.3 Hz, 3H), 6.59 (dd, J = 16.8, 10.5 Hz, 1H), 6.27 (dd, J = 16.8, 2.0 Hz, 1H), 5.67 (dd, J = 10.6, 2.0 Hz, 1H), 4.14 (q, J = 7.1 Hz, 1H), 2.67–2.47 (m, 4H), 2.05 (d, J = 11.3 Hz, 2H), 1.71 (s, 4H). MS (ESI): 230.1 [M + H]+. Purity: 100.0%.
N-(5-Phenylthiazol-2-yl)acrylamide (9).
Compound 9 was prepared from 5-phenylthiazol-2-amine 4e (100 mg, 0.57 mmol), DIEA (190 μL, 1.14 mmol), and acryloyl chloride (51 mg, 0.57 mmol) according to the general procedure A as a white solid (58 mg, 45% yield). 1H NMR (300 MHz, CDCl3) δ 7.71–7.55 (m, 3H), 7.51–7.30 (m, 3H), 6.68 (dd, J = 17.0, 1.3 Hz, 1H), 6.50 (dd, J = 17.0, 10.1 Hz, 1H), 6.00 (dd, J = 10.1, 1.3 Hz, 1H). MS (ESI): 231.0 [M + H]+. Purity: 97.7%.
N-(4-Phenylthiazol-2-yl)acrylamide (10).
Compound 10 was prepared from 4-phenylthiazol-2-amine 4f (100 mg, 0.57 mmol), DIEA (190 μL, 1.14 mmol), and acryloyl chloride (51 mg, 0.57 mmol) according to the general procedure A as a white solid (52 mg, 40% yield). 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 7.6 Hz, 2H), 7.41 (dt, J = 23.4, 7.1 Hz, 3H), 7.21 (s, 1H), 6.56–6.44 (m, 1H), 6.06 (ddd, J = 16.8, 10.4, 6.2 Hz, 1H), 5.75 (dd, J = 10.4, 5.5 Hz, 1H). MS (ESI): 231.0 [M + H]+. Purity: 98.0%.
Ethyl 2-Acrylamido-4-phenylthiophene-3-carboxylate (11).
Compound 11 was prepared from ethyl 4-phenylthiophene-2-amino-3-carboxylate 4g (100 mg, 0.40 mmol), DIEA (135 μL, 0.82 mmol), and acryloyl chloride (36 mg, 0.40 mmol) according to the general procedure A as a yellow solid (93 mg, 77% yield). 1H NMR (300 MHz, CDCl3) δ 8.91 (s, 1H), 7.49 (d, J = 1.7 Hz, 5H), 6.46 (dd, J = 17.0, 1.5 Hz, 1H), 6.34 (dd, J = 17.0, 9.8 Hz, 1H), 5.85 (dd, J = 9.9, 1.6 Hz, 1H), 4.26 (q, J = 7.1 Hz, 2H), 1.14 (t, J = 7.1 Hz, 3H), 0.09 (s, 4H). MS (ESI): 302.1 [M + H]+. Purity: 95.0%.
Ethyl 2-Acrylamido-5-phenylthiophene-3-carboxylate (12).
Compound 11 was prepared from ethyl 5-phenylthiophene-2-amino-3-carboxylate 4h (100 mg, 0.40 mmol), DIEA (135 μL, 0.82 mmol), and acryloyl chloride (36 mg, 0.40 mmol) according to the general procedure A as a yellow solid (97 mg, 80% yield). 1H NMR (300 MHz, CDCl3) δ 11.23 (s, 1H), 7.63 (dd, J = 7.3, 1.7 Hz, 2H), 7.50–7.25 (m, 5H), 6.56 (dd, J = 17.0, 1.1 Hz, 1H), 6.39 (dd, J = 17.0, 10.1 Hz, 1H), 5.93 (dd, J = 10.2, 1.2 Hz, 1H), 4.40 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H). MS (ESI): 302.1 [M + H]+. Purity: 100.0%.
4-Chloro-2-methoxyacetophenone (13r).
To a solution of 4-chloro-2-hydroxylacetophenone 13p (1.0 g, 5.86 mmol) in acetonitrile (20 mL) was added anhydrous K2CO3 (1.61 g, 11.7 mmol) and stirred at room temperature for 10 min. Methyl iodine (0.98 g, 7.03 mmol) was then added, and the reaction mixture was heated to reflux overnight and concentrated. The residue was suspended in H2O and extracted with EtOAc (3×). The organic layer was dried with MgSO4, filtered, and concentrated to give 4-chloro-2-methoxyacetophenone 13r as a white solid (1.04 g, 97% yield). 1H NMR (300 MHz, CDCl3) δ 7.72 (d, J = 8.2 Hz, 1H), 7.05–6.94 (m, 2H), 3.93 (s, 3H), 2.61 (d, J = 0.6 Hz, 3H). MS (ESI): 185.1 [M + H]+.
4-Fluoro-2-methoxyacetophenone (13s).
To a solution of 4-fluoro-2-hydroxylacetophenone 13p (1.0 g, 6.49 mmol) in acetonitrile (20 mL) was added anhydrous K2CO3 (1.79 g, 13.0 mmol) and stirred at room temperature for 10 min. Methyl iodine (1.10 g, 7.78 mmol) was then added, and the reaction mixture was heated to reflux overnight and concentrated. The residue was suspended in H2O and extracted with EtOAc (3×). The organic layer was dried with MgSO4, filtered, and concentrated to give 4-chloro-2-methoxyacetophenone 13s as a white solid (1.05 g, 94% yield). 1H NMR (300 MHz, CDCl3) δ 7.88–7.77 (m, 1H), 6.77–6.63 (m, 2H), 3.92 (s, 3H), 2.60 (s, 3H). MS (ESI): 169.1 [M + H]+.
General Procedure B: Preparation of Compounds 14a–14t.
To a solution of 2-bromoacetophenones (3.00 mmol) in EtOAc (25 mL) was added copper bromide (6.00 mmol), and the suspension was heated to reflux under vigorous stirring for 3 h. Then the resulting white suspension was cooled to room temperature, and the solid was removed by filtration. The filtrate was washed with water, dried with MgSO4, filtered, and concentrated to give compounds 14a–14t (55–97%)
2-Bromo-2′-chloroacetophenone (14a).
Intermediate 14a was prepared from 2-chloroacetophenone 13a according to the general procedure B as a colorless oil (539 mg, 77% yield). 1H NMR (300 MHz, CDCl3) δ 7.62–7.54 (m, 1H), 7.46 (d, J = 4.9 Hz, 2H), 7.39 (dq, J = 7.7, 4.6, 3.6 Hz, 1H), 4.54 (d, J = 0.9 Hz, 2H). MS (ESI): 232.9, 234.9 [M + H]+.
2-Bromo-3′-chloroacetophenone (14b).
Intermediate 14b was prepared from 3-chloroacetophenone 13b according to the general procedure B as a white solid (616 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ 7.98 (t, J = 1.9 Hz, 1H), 7.93–7.83 (m, 1H), 7.66–7.56 (m, 1H), 7.47 (t, J = 7.9 Hz, 1H), 4.44 (s, 2H). MS (ESI): 232.9, 234.9 [M + H]+.
2-Bromo-4′-chloroacetophenone (14c).
Intermediate 14c was prepared from 4-chloroacetophenone 13c according to the general procedure B as a white solid (658 mg, 95% yield). 1H NMR (300 MHz, CDCl3) δ 7.95 (d, J = 8.2 Hz, 2H), 7.50 (d, J = 8.6 Hz, 2H), 4.43 (s, 2H). MS (ESI): 232.9, 234.9 [M + H]+.
2-Bromo-4′-methylacetophenone (14d).
Intermediate 14d was prepared from 4-chloroacetophenone 13d according to the general procedure B as a white solid (658 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 4.46 (d, J = 0.6 Hz, 2H), 2.45 (s, 3H). MS (ESI): 212.9, 214.9 [M + H]+.
2-Bromo-4′-trifluoromethylacetophenone (14e).
Intermediate 14e was prepared from 4-trifluoromethylacetophenone 13e according to the general procedure B as a white solid (440 mg, 55% yield). 1H NMR (300 MHz, CDCl3) δ 8.13 (d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.2 Hz, 2H), 4.48 (s, 2H). MS (ESI): 266.9, 268.9 [M + H]+.
2-Bromo-4′-methoxyacetophenone (14f).
Intermediate 14f was prepared from 4-methoxyacetophenone 13f according to the general procedure B as a gray solid (612 mg, 90% yield). 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 9.1 Hz, 1H), 6.98 (d, J = 8.7 Hz, 1H), 4.42 (s, 1H), 3.91 (s, 2H). MS (ESI): 228.2, 230.2 [M + H]+.
2-Bromo-3′,4′-dioxolacetophenone (14g).
Intermediate 14g was prepared from 3,4-dioxolacetophenone 13g according to the general procedure B as a yellow solid (568 mg, 79% yield). 1H NMR (300 MHz, CDCl3) δ 7.66–7.54 (m, 2H), 7.51–7.34 (m, 2H), 6.90 (d, J = 8.2 Hz, 1H), 6.09 (s, 2H), 4.40 (s, 2H). MS (ESI): 242.9, 244.9 [M + H]+.
2-Bromo-2′-acetonaphthone (14h).
Intermediate 14h was prepared from 2-acetonaphthone 13h according to the general procedure B as a white solid (720 mg, 97% yield). 1H NMR (300 MHz, CDCl3) δ 8.53 (d, J = 1.6 Hz, 1H), 8.05 (dd, J = 8.6, 1.6 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.97–7.88 (m, 3H), 7.72–7.54 (m, 3H), 4.61 (s, 2H). MS (ESI): 248.9, 250.9 [M + H]+.
2-Bromo-2′-hydroxylacetophenone (14i).
Intermediate 14i was prepared from 2-hydroxylacetophenone 13i according to the general procedure B as a yellow solid (460 mg, 72% yield). 1H NMR (300 MHz, CDCl3) δ 11.76 (s, 1H), 7.77 (dd, J = 8.1, 1.6 Hz, 1H), 7.55 (ddd, J = 8.7, 7.2, 1.6 Hz, 1H), 7.05 (d, J = 8.5 Hz, 1H), 7.03–6.90 (m, 1H), 4.47 (s, 2H). MS (ESI): 214.9, 216.9 [M + H]+.
2-Bromo-4′-hydroxylacetophenone (14j).
Intermediate 14j was prepared from 4-hydroxylacetophenone 13j according to the general procedure B as a yellow solid (496 mg, 73% yield). 1H NMR (300 MHz, acetone-d6): δ 9.40 (s, 1H), 7.98 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 4.65 (s, 2H). MS (ESI): 214.9, 216.9 [M + H]+.
2-Bromo-2′-methoxyacetophenone (14k).
Intermediate 14k was prepared from 2-methoxyacetophenone 13k according to the general procedure B as a yellow solid (612 mg, 91% yield). 1H NMR (300 MHz, CDCl3) δ 7.85 (dd, J = 7.7, 1.9 Hz, 1H), 7.61–7.48 (m, 1H), 7.12–6.97 (m, 2H), 4.64 (s, 2H), 3.97 (s, 3H). MS (ESI): 228.2, 230.2 [M + H]+.
2-Bromo-2′-fluoroacetophenone (14l).
Intermediate 14l was prepared from 2-fluoroacetophenone 13l according to the general procedure B as a colorless oil (611 mg, 94% yield). 1H NMR (300 MHz, CDCl3) δ 7.96 (td, J = 7.6, 2.1 Hz, 1H), 7.61 (q, J = 7.1 Hz, 1H), 7.36–7.23 (m, 1H), 7.19 (dd, J = 11.0, 8.0 Hz, 1H), 4.55 (dt, J = 2.5, 1.3 Hz, 2H). MS (ESI): 216.9, 218.9 [M + H]+.
2-Bromo-2′-methoxy-4′-chloroacetophenone (14r).
Intermediate 14r was prepared from 2-methoxy-4-chloroacetophenone 13r according to the general procedure B as a yellow solid (687 mg, 87% yield). 1H NMR (300 MHz, CDCl3) δ 7.73 (dd, J = 8.3, 6.2 Hz, 1H), 7.02–6.97 (m, 2H), 4.56 (s, 2H), 3.95 (s, 3H). MS (ESI): 262.9, 264.9 [M + H]+.
2-Bromo-2′-methoxy-4′-fluoroacetophenone (14s).
Intermediate 14s was prepared from 2-methoxy-4-fluoroacetophenone 13s according to the general procedure B as a yellow solid (666 mg, 90% yield). 1H NMR (300 MHz, CDCl3) δ 7.89 (dd, J = 8.6, 6.9 Hz, 1H), 6.85–6.58 (m, 2H), 4.57 (s, 2H), 3.95 (s, 3H). MS (ESI): 246.9, 248.9 [M + H]+.
General Procedure C: Preparation of Compounds 15a–15o and 28a–28b.
To a solution of 2-bromoacetophenones (1.0 equiv) in EtOH (20 mL) was added corresponding thiourea (1.2 equiv). The reaction mixture was heated to reflux for 2 h. Then the resulting solution was cooled to room temperature and poured into 2 M ammonium hydroxide aqueous solution (40 mL). The solid was collected by filtration, washed with water, and dried to give compounds 15a–15p and 28a–28b.
4-(2-Chlorophenyl)thiazol-2-amine (15a).
Intermediate 15a was prepared from 2-bromo-2′-chloroacetophenone 14a (200 mg, 0.85 mmol) and thiourea (64 mg, 0.85 mmol) according to the general procedure C as a white solid (164 mg, 92% yield). 1H NMR (300 MHz, acetone-d6): δ 7.97 (dd, J = 7.7, 1.9 Hz, 1H), 7.47 (dd, J = 7.8, 1.5 Hz, 1H), 7.33 (dtd, J = 21.8, 7.4, 1.7 Hz, 3H), 7.15 (s, 1H), 6.50 (s, 2H). MS (ESI): 211.0 [M + H]+.
4-(3-Chlorophenyl)thiazol-2-amine (15b).
Intermediate 15b was prepared from 2-bromo-3′-chloroacetophenone 14b (200 mg, 0.85 mmol) and thiourea (64 mg, 0.85 mmol) according to the general procedure C as a white solid (173 mg, 97% yield). 1H NMR (300 MHz, acetone-d6): δ 7.90 (t, J = 1.9 Hz, 1H), 7.85–7.76 (m, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.33–7.24 (m, 1H), 7.09 (s, 1H), 6.52 (s, 2H). MS (ESI): 211.0 [M + H]+.
4-(4-Chlorophenyl)thiazol-2-amine (15c).
Intermediate 15c was prepared from 2-bromo-4′-chloroacetophenone 14c (200 mg, 0.85 mmol) and thiourea (64 mg, 0.85 mmol) according to the general procedure C as a white solid (171 mg, 96% yield). 1H NMR (300 MHz, acetone-d6): δ 7.87 (t, J = 8.3 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.01 (s, 1H), 6.51 (s, 2H). MS (ESI): 211.0 [M + H]+.
4-(4-Methylphenyl)thiazol-2-amine (15d).
Intermediate 15d was prepared from 2-bromo-4′-methylacetophenone 14d (200 mg, 0.86 mmol) and thiourea (65 mg, 0.86 mmol) according to the general procedure C as a white solid (161 mg, 99% yield). 1H NMR (300 MHz, acetone-d6): δ 7.75 (d, J = 8.2 Hz, 2H), 7.17 (t, J = 7.9 Hz, 2H), 6.87 (s, 1H), 6.42 (s, 2H), 2.33 (s, 3H). MS (ESI): 191.1 [M + H]+.
4-(4-Trifluoromethylphenyl)thiazol-2-amine (15e).
Intermediate 15e was prepared from 2-bromo-4′-trifluoromethylacetophenone 14e (200 mg, 0.75 mmol) and thiourea (57 mg, 0.75 mmol) according to the general procedure C as a white solid (177 mg, 97% yield). 1H NMR (300 MHz, DMSO-d6): δ 7.81 (d, J = 8.5 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.11 (s, 2H), 7.07 (s, 1H). MS (ESI): 245.0 [M + H]+.
4-(4-Methoxyphenyl)thiazol-2-amine (15f).
Intermediate 15f was prepared from 2-bromo-4′-methoxyacetophenone 14f (200 mg, 0.88 mmol) and thiourea (71 mg, 0.88 mmol) according to the general procedure C as a white solid (161 mg, 89% yield). 1H NMR (300 MHz, acetone-d6): δ 7.79 (d, J = 8.8 Hz, 2H), 6.90 (dd, J = 14.7, 8.6 Hz, 2H), 6.78 (s, 1H), 6.37 (s, 1H), 3.83 (d, J = 5.9 Hz, 3H). MS (ESI): 207.0 [M + H]+.
4-(3′,4′-Dioxolphenyl)thiazol-2-amine (15g).
Intermediate 15g was prepared from 2-bromo-3′,4′-dioxolacetophenone 14g (200 mg, 0.82 mmol) and thiourea (62 mg, 0.85 mmol) according to the general procedure C as a white solid (142 mg, 79% yield). 1H NMR (300 MHz, acetone-d6): δ 7.41 (dd, J = 8.1, 1.7 Hz, 1H), 7.35 (d, J = 1.6 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.81 (s, 1H), 6.41 (s, 2H), 6.01 (s, 2H). MS (ESI): 221.0 [M + H]+.
4-(Naphthalen-2-yl)thiazol-2-amine (15h).
Intermediate 15h was prepared form 2-bromo-2′-acetonaphthone 14h (200 mg, 0.81 mmol) and thiourea (61 mg, 0.81 mmol) according to the general procedure C as a white solid (144 mg, 79% yield). 1H NMR (300 MHz, acetone-d6): δ 8.41 (s, 1H), 8.00 (dd, J = 8.6, 1.7 Hz, 1H), 7.90 (t, J = 8.9 Hz, 3H), 7.49 (tt, J = 7.4, 5.6 Hz, 2H), 7.11 (s, 1H), 6.53 (s, 2H). MS (ESI): 227.1 [M + H]+.
4-(2-Hydroxylphenyl)thiazol-2-amine (15i).
Intermediate 15i was prepared from 2-bromo-2′-hydroxylacetophenone 14i (200 mg, 0.93 mmol) and thiourea (71 mg, 0.93 mmol) according to the general procedure C as a white solid (119 mg, 67% yield). 1H NMR (300 MHz, acetone-d6): δ 11.93 (s, 1H), 7.67 (dd, J = 7.8, 1.6 Hz, 1H), 7.22–7.09 (m, 1H), 7.00 (s, 1H), 6.99–6.92 (m, 2H), 6.88–6.74 (m, 2H). MS (ESI): 193.0 [M + H]+.
4-(4-Hydroxylphenyl)thiazol-2-amine (15j).
Intermediate 15j was prepared from 2-bromo-4′-hydroxylacetophenone 14j (200 mg, 0.93 mmol) and thiourea (71 mg, 0.93 mmol) according to the general procedure C as a white solid (131 mg, 74% yield). 1H NMR (300 MHz, acetone-d6): δ 8.87 (s, 1H), 7.70 (d, J = 8.9 Hz, 2H), 6.83 (d, J = 8.9 Hz, 2H), 6.71 (s, 1H). MS (ESI): 193.0 [M + H]+.
4-(2-Methoxyphenyl)thiazol-2-amine (15k).
Intermediate 15k was prepared from 2-bromo-2′-methoxyacetophenone 14k (200 mg, 0.88 mmol) and thiourea (71 mg, 0.88 mmol) according to the general procedure C as a white solid (148 mg, 82% yield). 1H NMR (300 MHz, acetone-d6): δ 8.17 (dd, J = 7.8, 1.8 Hz, 1H), 7.24 (dd, J = 15.5, 1.8 Hz, 1H), 7.24 (s, 2H), 7.10–6.92 (m, 2H), 6.47 (s, 2H), 3.94 (s, 3H). MS (ESI): 207.0 [M + H]+.
4-(2-Fluorophenyl)thiazol-2-amine (15l).
Intermediate 15l was prepared from 2-bromo-2′-fluoroacetophenone 14l (200 mg, 0.92 mmol) and thiourea (71 mg, 0.92 mmol) according to the general procedure C as a white solid (171 mg, 96% yield). 1H NMR (300 MHz, acetone-d6): δ 8.11 (td, J = 1.5, 7.7 Hz, 1H), 7.38–7.11 (m, 3H), 7.02 (d, J = 2.5 Hz, 1H), 6.48 (s, 2H). MS (ESI): 195.0 [M + H]+.
5-Methyl-4-phenylthiazol-2-amine (15o).
Intermediate 15o was prepared from 2-bromo-1-phenylpropan-1-one 14o (200 mg, 0.86 mmol) and thiourea (64 mg, 0.86 mmol) according to the general procedure C as a white solid (83 mg, 51% yield). 1H NMR (300 MHz, CDCl3) δ 7.62–7.52 (m, 2H), 7.48–7.26 (m, 4H), 5.34 (s, 2H), 2.40 (s, 3H). MS (ESI): 191.1 [M + H]+.
General Procedure D: Preparation of Acrylamides 16–27, 29, 30.
To a solution of thiazol-2-amines (1.0 equiv) in DCM (10 mL) was added DIEA (2.0 equiv) and stirred at 0 °C for 20 min. Acryloyl chloride (1.0 equiv) in DCM (10 mL) was added slowly. The reaction mixture was stirred at room temperature for 2 h and then partitioned between DCM and saturated NaHCO3. The aqueous layer was extracted with DCM for two more times. The combined organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 2–50% EtOAc in hexane.
N-(4-(2-Chlorophenyl)thiazol-2-yl)acrylamide (16).
Compound 16 was prepared from 4-(2-chlorophenyl)thiazol-2-amine 15a (50 mg, 0.24 mmol), DIEA (79 μL, 0.48 mmol), and acryloyl chloride (21 mg, 0.24 mmol) according to the general procedure D as a white solid (32 mg, 52% yield). 1H NMR (300 MHz, CDCl3) δ 7.76 (dd, J = 7.2, 2.3 Hz, 1H), 7.55–7.41 (m, 2H), 7.40–7.25 (m, 3H), 6.43 (d, J = 16.9 Hz, 1H), 5.92 (dd, J = 16.9, 10.4 Hz, 1H), 5.66 (d, J = 10.3 Hz, 1H). MS (ESI): 265.0 [M + H]+. Purity: 95.6%.
N-(4-(3-Chlorophenyl)thiazol-2-yl)acrylamide (17).
Compound 17 was prepared from 4-(3-chlorophenyl)thiazol-2-amine 15b (50 mg, 0.24 mmol), DIEA (79 μL, 0.48 mmol), and acryloyl chloride (21 mg, 0.24 mmol) according to the general procedure D as a white solid (30 mg, 48% yield). 1H NMR (300 MHz, CDCl3) δ 7.82 (s, 1H), 7.71 (d, J = 7.4 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.36 (s, 1H), 7.22 (s, 1H), 6.60 (d, J = 16.8 Hz, 1H), 6.29 (dd, J = 17.3, 10.2 Hz, 1H), 5.94 (d, J = 10.4 Hz, 1H). MS (ESI): 265.0 [M + H]+. Purity: 99.1%
N-(4-(4-Chlorophenyl)thiazol-2-yl)acrylamide (18).
Compound 18 was prepared from 4-(4-chlorophenyl)thiazol-2-amine 15c (50 mg, 0.24 mmol), DIEA (79 μL, 0.48 mmol), and acryloyl chloride (21 mg, 0.24 mmol) according to the general procedure D as a white solid (31 mg, 50% yield). 1H NMR (300 MHz, CDCl3) δ 10.10 (s, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.20 (s, 1H), 6.53 (d, J = 16.9 Hz, 1H), 6.12 (dd, J = 17.0, 10.4 Hz, 1H), 5.82 (d, J = 10.3 Hz, 1H). MS (ESI): 265.0 [M + H]+. Purity: 100.0%.
N-(4-(4-Methylphenyl)thiazol-2-yl)acrylamide (19).
Compound 19 was prepared from 4-(4-methylphenyl)thiazol-2-amine 15d (50 mg, 0.26 mmol), DIEA (87 μL, 0.52 mmol), and acryloyl chloride (23 mg, 0.26 mmol) according to the general procedure D as a white solid (39 mg, 61% yield). 1H NMR (300 MHz, CDCl3) δ 7.71 (d, J = 7.9 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.14 (s, 1H), 6.51 (d, J = 16.9 Hz, 1H), 6.08 (dd, J = 16.9, 10.4 Hz, 1H), 5.77 (d, J = 10.4 Hz, 1H), 2.40 (s, 3H). MS (ESI): 245.2 [M + H]+. Purity: 100.0%.
N-(4-(4-Trifluoromethylphenyl)thiazol-2-yl)acrylamide (20).
Compound 20 was prepared from 4-(4-trifluoromethylphenyl)thiazol-2-amine 15e (50 mg, 0.20 mmol), DIEA (68 μL, 0.40 mmol), and acryloyl chloride (18 mg, 0.20 mmol) according to the general procedure D as a white solid (20 mg, 33% yield). 1H NMR (300 MHz, CDCl3) δ 7.95 (d, J = 8.2 Hz, 2H), 7.70 (d, J = 8.1 Hz, 2H), 7.28 (s, 2H), 6.58 (d, J = 16.9 Hz, 1H), 6.24 (dd, J = 16.9, 10.4 Hz, 1H), 5.90 (d, J = 10.4 Hz, 1H), 1.27 (s, 1H), 0.09 (s, 5H). MS (ESI): 299.0 [M + H]+. Purity: 100.0%.
N-(4-(4-Methoxyphenyl)thiazol-2-yl)acrylamide (21).
Compound 21 was prepared from 4-(4-methoxyphenyl)thiazol-2-amine 15f (50 mg, 0.24 mmol), DIEA (79 μL, 0.48 mmol), and acryloyl chloride (22 mg, 0.24 mmol) according to the general procedure D as a white solid (43 mg, 68% yield). 1H NMR (300 MHz, CDCl3) δ 7.76 (d, J = 8.6 Hz, 2H), 7.07 (s, 1H), 6.97 (d, J = 8.6 Hz, 2H), 6.48 (d, J = 16.9 Hz, 1H), 6.05 (dd, J = 16.9, 10.4 Hz, 1H), 5.72 (d, J = 10.4 Hz, 1H), 3.86 (s, 3H). MS (ESI): 261.0 [M + H]+. Purity: 100.0%.
N-(4-(3,4-Dioxylphenyl)thiazol-2-yl)acrylamide (22).
Compound 22 was prepared from 4-(3,4-dioxylphenyl)thiazol-2-amine 15g (50 mg, 0.23 mmol), DIEA (76 μL, 0.45 mmol), and acryloyl chloride (21 mg, 0.23 mmol) according to the general procedure D as a white solid (41 mg, 66% yield). 1H NMR (300 MHz, acetone-d6): δ 11.29 (s, 1H), 7.52–7.35 (m, 3H), 6.88 (d, J = 8.1 Hz, 1H), 6.71 (dd, J = 17.0, 10.1 Hz, 1H), 6.53 (dd, J = 17.0, 1.9 Hz, 1H), 6.04 (s, 2H), 5.92 (dd, J = 10.1, 1.9 Hz, 1H). MS (ESI): 275.0 [M + H]+. Purity: 98.0%.
N-(4-(Naphthalen-2-yl)thiazol-2-yl)acrylamide (23).
Compound 23 was prepared from 4-(naphthalen-2-yl)thiazol-2-amine 15h (50 mg, 0.22 mmol), DIEA (74 μL, 0.44 mmol), and acryloyl chloride (20 mg, 0.22 mmol) according to the general procedure D as a white solid (49 mg, 79% yield). 1H NMR (300 MHz, acetone-d6): δ 11.40 (s, 1H), 8.46 (s, 1H), 8.07 (dd, J = 8.6, 1.7 Hz, 1H), 7.99–7.86 (m, 3H), 7.67 (s, 1H), 7.51 (hept, J = 5.0 Hz, 2H), 6.76 (dd, J = 17.0, 10.1 Hz, 1H), 6.56 (dd, J = 17.0, 1.8 Hz, 1H), 5.95 (dd, J = 10.1, 1.8 Hz, 1H). MS (ESI): 281.1 [M + H]+. Purity: 96.8%.
N-(4-(2-Hydroxylphenyl)thiazol-2-yl)acrylamide (24).
Compound 24 was prepared from 4-(2-hydroxylphenyl)thiazol-2-amine 15i (50 mg, 0.26 mmol), DIEA (87 μL, 0.52 mmol), and acryloyl chloride (24 mg, 0.26 mmol) according to the general procedure D as a white solid (9 mg, 15% yield). 1H NMR (300 MHz, CDCl3) δ 9.55 (s, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.25 (d, J = 4.2 Hz, 1H), 7.10–6.87 (m, 2H), 6.64 (d, J = 16.8 Hz, 1H), 6.38 (dd, J = 17.0, 10.3 Hz, 1H), 5.99 (d, J = 10.3 Hz, 1H). MS (ESI): 246.1 [M + H]+.
N-(4-(4-Hydroxylphenyl)thiazol-2-yl)acrylamide (25).
Compound 25 was prepared from 4-(4-hydroxylphenyl)thiazol-2-amine 15j (50 mg, 0.26 mmol), DIEA (87 μL, 0.52 mmol), and acryloyl chloride (24 mg, 0.26 mmol) according to the general procedure D as a white solid (14 mg, 22% yield). 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J = 8.7 Hz, 3H), 7.48 (s, 1H), 7.03–6.88 (m, 3H), 6.68 (d, J = 17.3 Hz, 1H), 6.55–6.40 (m, 1H), 6.01 (d, J = 10.2 Hz, 1H). MS (ESI): 246.1 [M + H]+. Purity: 98.2%.
N-(4-(2-Methoxyphenyl)thiazol-2-yl)acrylamide (26).
Compound 26 was prepared from 4-(2-methoxyphenyl)thiazol-2-amine 15k (50 mg, 0.26 mmol), DIEA (88 μL, 0.52 mmol), and acryloyl chloride (21 mg, 0.26 mmol) according to the general procedure D as a white solid (30 mg, 48% yield). 1H NMR (300 MHz, acetone-d6): δ 11.25 (s, 1H), 8.15 (dd, J = 7.8, 1.8 Hz, 1H), 7.76 (s, 1H), 7.37–7.24 (m, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.01 (t, J = 7.5 Hz, 1H), 6.72 (dd, J = 17.0, 10.1 Hz, 1H), 6.53 (dd, J = 17.1, 1.8 Hz, 1H), 5.92 (dd, J = 10.1, 1.9 Hz, 1H), 4.00 (s, 3H). MS (ESI): 261.0 [M + H]+. Purity: 97.2%.
N-(4-(2-Fluorophenyl)thiazol-2-yl)acrylamide (27).
Compound 27 was prepared from 4-(2-fluorophenyl)thiazol-2-amine 15l (50 mg, 0.25 mmol), DIEA (86 μL, 0.50 mmol), and acryloyl chloride (23 mg, 0.25 mmol) according to the general procedure D as a white solid (38 mg, 60% yield). 1H NMR (300 MHz, CDCl3) δ 10.29 (s, 1H), 8.07–7.95 (m, 1H), 7.48 (d, J = 2.1 Hz, 1H), 7.37–7.28 (m, 1H), 7.32–7.11 (m, 3H), 6.51 (d, J = 16.9 Hz, 1H), 6.11 (dd, J = 17.0, 10.3 Hz, 1H), 5.78 (d, J = 10.3 Hz, 1H). MS (ESI): 248.0 [M + H]+. Purity: 100.0%.
N-Benzyl-4-phenylthiazol-2-amine (28c).
To a solution of 4-phenylthiazol-2-amine 4f (200 mg, 1.13 mmol) in acetonitrile (20 mL) was added anhydrous K2CO3 (470 mg, 3.34 mmol) and stirred at room temperature for 30 min. Benzyl bromide (193 mg, 1.13 mmol) was added, and the reaction mixture was heated at reflux for 8 h before concentrated. The residue was suspended in saturated NaHCO3 aqueous solution before being extracted with EtOAc (3×). The organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 2–30% EtOAc in hexane to give N-benzyl-4-phenylthiazol-2-amine 28c as a white solid (52 mg, 17% yield). 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 7.5 Hz, 2H), 7.43–7.27 (m, 8H), 6.72 (s, 1H), 6.14 (s, 1H), 4.52 (s, 2H). MS (ESI): 267.1 [M + H]+.
Ethyl (4-Phenylthiazol-2-yl)glycinate (28d).
To a solution of 4-phenylthiazol-2-amine 4f (200 mg, 1.13 mmol) in acetonitrile (20 mL) was added anhydrous K2CO3 (470 mg, 3.34 mmol) and stirred at room temperature for 30 min. Ethyl bromoacetate (188 mg, 1.13 mmol) was added, and the reaction mixture was heated to reflux for 8 h. Acetonitrile was removed in vacuo after the reaction completed. The residue was suspended in saturated NaHCO3 aqueous solution before being extracted with EtOAc (3×). The organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 2 to 35% EtOAc in hexane to give N-benzyl-4-phenylthiazol-2-amine 28d as a white solid (77 mg, 26% yield). 1H NMR (300 MHz, CDCl3) δ 7.88–7.77 (m, 2H), 7.47–7.17 (m, 3H), 6.77 (d, J = 14.4 Hz, 1H), 4.39 (s, 2H), 4.28 (dq, J = 11.9, 7.3 Hz, 2H), 1.33 (q, J = 7.2 Hz, 3H). MS (ESI): 263.1 [M + H]+.
General Procedure E: Preparation of Compounds 28e–28l.
To a solution of 2-bromoacetophenones (1.0 equiv) in 15 mL EtOH was added potassium thiocyanate (1.0 equiv) and was heated under reflux for 2 h. Then to the resulting solution were added corresponding primary amines (1.2 equiv) in EtOH (10 mL), and it was heated to reflux overnight before being concentrated. The residue was suspended in H2O and neutralized before extracting with EtOAc (3×). The organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 2 to 35% EtOAc in hexane.
Methyl (4-phenylthiazol-2-yl)glycinate (28e).
Compound 28e was prepared from 2-bromoacetophenone 14m (1.0 g, 5.03 mmol), potassium thiocyanate (488 mg, 5.03 mmol), and methyl glycinate hydrochloride (757 mg, 6.03 mmol) according to the general procedure E as a yellow solid (910 mg, 73% yield). 1H NMR (300 MHz, CDCl3) δ 7.88–7.78 (m, 2H), 7.46–7.31 (m, 3H), 7.28 (s, 1H), 6.75 (s, 1H), 5.84 (s, 1H), 4.25 (s, 2H), 3.83 (s, 3H). MS (ESI): 249.1 [M + H]+.
tert-Butyl(4-phenylthiazol-2-yl)glycinate (28f).
Compound 28f was prepared from 2-bromoacetophenone 14m (300 mg, 1.51 mmol), potassium thiocyanate (146 mg, 1.51 mmol), and tert-butyl glycinate hydrogen chloride (303 mg, 1.81 mmol) according to the general procedure E as a yellow solid (380 mg, 87% yield). 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 7.6 Hz, 2H), 7.46–7.30 (m, 3H), 7.28 (s, 1H), 6.74 (s, 1H), 5.91 (s, 1H), 4.11 (s, 2H), 1.52 (s, 9H). MS (ESI): 291.1 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-4-Phenylthiazol-2-amine (28g).
Compound 28g was prepared from 2-bromoacetophenone 14m (300 mg, 1.51 mmol), potassium thiocyanate (146 mg, 1.51 mmol), and (3-methylisoxazol-5-yl)methanamine (203 mg, 1.81 mmol) according to the general procedure E as a yellow solid (229 mg, 56% yield). 1H NMR (300 MHz, CDCl3) δ 7.88–7.78 (m, 2H), 7.46–7.32 (m, 13H), 7.28 (s, 1H), 6.75 (s, 1H), 6.07 (s, 1H), 4.63 (s, 2H), 2.42 (s, 3H). MS (ESI): 272.1 [M + H]+.
4-Phenyl-N-(pyridin-3-ylmethyl)thiazol-2-amine (28h).
Compound 28h was prepared from 2-bromoacetophenone 14m (300 mg, 1.51 mmol), potassium thiocyanate (146 mg, 1.51 mmol), and pyridin-3-ylmethanamine (194 mg, 1.81 mmol) according to the general procedure E as a yellow solid (133 mg, 33% yield). 1H NMR (300 MHz, CDCl3) δ 8.58 (d, J = 2.3 Hz, 1H), 8.50 (dd, J = 4.9, 1.6 Hz, 1H), 7.74 (ddt, J = 21.5, 7.9, 1.8 Hz, 3H), 7.50–7.19 (m, 5H), 6.95 (s, 1H), 6.68 (s, 1H), 4.49 (s, 2H). MS (ESI): 268.1 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-4-(4-trifluoromethylphenyl)thiazol-2-amine (28i).
Compound 28i was prepared from 2-bromo-4′-trifluoromethylacetophenone 14e (300 mg, 1.12 mmol), potassium thiocyanate (109 mg, 1.12 mmol), and (3-methylisoxazol-5-yl)methanamine (151 mg, 1.35 mmol) according to the general procedure E as a yellow solid (255 mg, 67% yield). 1H NMR (300 MHz, CDCl3) δ 7.93 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 8.1 Hz, 3H), 6.87 (s, 1H), 6.08 (s, 1H), 4.67 (s, 2H), 2.43 (d, J = 0.9 Hz, 3H). MS (ESI): 340.1 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-4-(4-chloro-2-methoxyphenyl)thiazol-2-amine (28j).
Compound 28j was prepared from 2-bromo-4′-chloro-2′-methoxyacetophenone 14r (300 mg, 1.14 mmol), potassium thiocyanate (111 mg, 1.14 mmol), and (3-methylisoxazol-5-yl)methanamine (153 mg, 1.37 mmol) according to the general procedure E as a yellow solid (233 mg, 61% yield). 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1H), 7.21 (s, 1H), 7.05–6.91 (m, 2H), 6.09 (s, 1H), 4.66 (s, 2H), 3.94 (s, 3H), 2.28 (s, 3H). MS (ESI): 336.1 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-4-(4-fluoro-2-methoxyphenyl)thiazol-2-amine (28k).
Compound 28k was prepared from 2-bromo-4′-fluoro-2′-methoxyacetophenone 14s (300 mg, 1.21 mmol), potassium thiocyanate (118 mg, 1.21 mmol), and (3-methylisoxazol-5-yl)methanamine (163 mg, 1.46 mmol) according to the general procedure E as a yellow solid (213 mg, 55% yield). 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 8.4 Hz, 1H), 7.12 (s, 1H), 6.91 (dd, J = 8.4, 2.1 Hz, 1H), 6.85 (d, J = 2.0 Hz, 1H), 6.00 (s, 1H), 4.56 (s, 2H), 3.84 (s, 3H), 2.18 (s, 3H). MS (ESI): 320.1 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-4-(2-methoxylphenyl)thiazol-2-amine (28l).
Compound 28l was prepared from 2-bromo-2′-methoxyacetophenone 14l (300 mg, 1.31 mmol), potassium thiocyanate (127 mg, 1.31 mmol), and (3-methylisoxazol-5-yl)methanamine (176 mg, 1.57 mmol) according to the general procedure E as a yellow solid (236 mg, 60% yield). 1H NMR (300 MHz, CDCl3) δ 8.15 (d, J = 7.7 Hz, 1H), 7.34–7.21 (m, 1H), 7.15–6.93 (m, 2H), 6.06 (d, J = 4.1 Hz, 1H), 4.63 (s, 2H), 3.96 (d, J = 1.5 Hz, 3H), 2.40 (d, J = 2.0 Hz, 3H). MS (ESI): 302.1 [M + H]+.
N-(4-Phenyl-5-methyl-thiazol-2-yl)acrylamide (29).
Compound 29 was prepared from 5-methyl-4-phenylthiazol-2-amine 15o (50 mg, 0.26 mmol), DIEA (88 μL, 0.52 mmol), and acryloyl chloride (23 mg, 0.26 mmol) according to the general procedure D as a white solid (35 mg, 55% yield). 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 7.4 Hz, 2H), 7.38 (dt, J = 27.7, 7.3 Hz, 3H), 6.23 (dd, J = 16.5, 1.5 Hz, 1H), 5.46 (dd, J = 16.7, 10.3 Hz, 1H), 5.31 (dd, J = 10.3, 1.6 Hz, 1H), 2.54 (s, 3H). MS (ESI): 245.2 [M + H]+.
Ethyl 2-Acrylamido-4-phenylthiazole-5-carboxylate (30).
Compound 16 was prepared from commercially available ethyl 2-amino-4-phenylthiazole-5-carboxylate 15p (50 mg, 0.18 mmol), DIEA (67 μL, 0.36 mmol), and acryloyl chloride (18 mg, 0.18 mmol) according to the general procedure D as a white solid (37 mg, 61% yield). 1H NMR (300 MHz, CDCl3) δ 7.75 (dd, J = 6.6, 2.9 Hz, 2H), 7.44 (dd, J = 5.3, 3.6 Hz, 2H), 6.29 (dd, J = 15.9, 2.2 Hz, 1H), 5.48–5.37 (m, 1H), 5.35 (dd, J = 15.9, 10.3 Hz, 1H), 4.31 (q, J = 7.1 Hz, 2H), 1.33 (t, J = 7.1 Hz, 3H). MS (ESI): 303.1 [M + H]+. Purity: 95.6%
General Procedure G: Preparation of N-Substituted Acrylamides 33–34, 37–45, 46a–46b, 47–48.
To a solution of 2-aminethiazoles (0.25 mmol) in toluene (15 mL) was added DIEA (0.75 mmol) and stirred at room temperature for 10 min. Acryloyl chloride (0.37 mmol) was then added slowly to the solution. The reaction mixture was heated at 70 °C for 0.5 h, and the solvent was removed in vacuo. The residue was suspended in saturated NaHCO3 aqueous solution and extracted with EtOAc (3×). The organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 2–35% EtOAc in hexane.
N-Benzyl-N-(4-phenylthiazol-2-yl)acrylamide (33).
Compound 32 was prepared from N-benzyl-4-phenylthiazol-2-amine 28c (50 mg, 0.17 mmol), DIEA (84 μL, 0.50 mmol), and acryloyl chloride (23 mg, 0.25 mmol) according to the general procedure G as a white solid (19 mg, 32% yield). 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 1.6 Hz, 1H), 7.46–7.25 (m, 10H), 6.80–6.57 (m, 2H), 5.89 (dd, J = 9.4, 2.6 Hz, 1H), 5.71 (s, 2H). MS (ESI): 321.1 [M + H]+. Purity: 98.9%.
Ethyl N-acryloyl-N-(4-phenylthiazol-2-yl)glycinate (34).
Compound 34 was prepared from ethyl (4-phenylthiazol-2-yl) glycinate 28d (50 mg, 0.19 mmol), DIEA (96 μL, 0.57 mmol), and acryloyl chloride (26 mg, 0.29 mmol) according to the general procedure G as a white solid (21 mg, 34% yield). 1H NMR (300 MHz, CDCl3) δ 7.93–7.83 (m, 2H), 7.48–7.25 (m, 3H), 7.26 (s, 1H), 6.65 (d, J = 5.9 Hz, 2H), 5.98 (t, J = 5.9 Hz, 1H), 5.11 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H). MS (ESI): 317.1 [M + H]+. Purity: 96.5%.
(4-Phenylthiazol-2-yl)glycine Hydrogen Chloride (35).
To a solution of intermediate 28e (1.0 g, 4.03 mmol) in tetrahydrofuran (THF) (20 mL) was added 1 M HCl aqueous solution (5 mL). The reaction mixture was heated under reflux for 2 h, and the solvent was removed to give (4-phenylthiazol-2-yl)glycine hydrogen chloride 35 as a light yellow solid (890 mg, 94%). 1H NMR (300 MHz, acetone-d6): δ 7.99–7.80 (m, 2H), 7.44–7.32 (m, 3H), 7.01 (s, 1H), 4.48 (s, 1H), 4.27 (s, 2H). MS (ESI): 235.0 [M + H]+.
General Procedure F: Preparation of Compounds 36a–36d.
To a solution of intermediate 35 (100 mg, 0.37 mmol) in DMF (10 mL) were added 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDCI) (105 mg, 0.55 mmol), hydroxybenzotriazole (HOBt) (74 mg, 0.55 mmol), amines (0.44 mmol), and DIEA (185 μL, 1.11 mmol). The reaction mixture was stirred at room temperature overnight. After the reaction was completed, the solution was poured into saturated NaHCO3 aqueous solution (25 mL) and extracted with EtOAc (3×). The organic layer was dried with MgSO4, filtered, concentrated, and purified with flash chromatography using a gradient method of 0–10% methanol in DCM.
N-Cyclohexyl-2-((4-phenylthiazol-2-yl)amino)acetamide (36a).
Compound 36a was prepared from 35 and cyclohexylamine (43 mg, 0.44 mmol) according to the general procedure F as a yellow solid (92 mg, 67% yield). 1H NMR (300 MHz, CDCl3) δ 7.80 (dd, J = 7.1, 1.7 Hz, 2H), 7.39 (dd, J = 8.2, 6.5 Hz, 3H), 7.37–7.25 (m, 2H), 6.75 (s, 1H), 4.04 (s, 2H), 1.90 (dd, J = 12.6, 4.0 Hz, 3H), 1.74–1.54 (m, 4H), 1.46–1.24 (m, 3H), 1.29–1.05 (m, 4H). MS (ESI): 316.1 [M + H]+.
1-Morpholino-2-((4-phenylthiazol-2-yl)amino)ethan-1-one (36b).
Compound 36b was prepared from 35 and morpholine (38 mg, 0.44 mmol) according to the general procedure F as a yellow oil (103 mg, 78% yield). 1H NMR (300 MHz, CDCl3) δ 7.75–7.65 (m, 2H), 7.49 (d, J = 7.2 Hz, 3H), 6.61 (s, 1H), 4.26 (s, 2H), 3.70 (dd, J = 18.7, 6.8 Hz, 6H), 3.52 (d, J = 5.1 Hz, 2H). MS (ESI): 304.1 [M + H]+.
N-(Oxetan-3-yl)-2-((4-phenylthiazol-2-yl)amino)acetamide (36c).
Compound 36c was prepared from 35 and oxetan-3-amine hydrogen chloride (48 mg, 0.44 mmol) according to the general procedure F as a yellow solid (126 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ 7.88–7.78 (m, 2H), 7.46–7.32 (m, 3H), 7.28 (s, 1H), 6.75 (s, 1H), 6.07 (s, 1H), 4.63 (s, 2H), 2.42 (s, 3H). MS (ESI): 289.9 [M + H]+.
N-(2-Morpholinoethyl)-2-((4-phenylthiazol-2-yl)amino)acetamide (36d).
Compound 36d was prepared from 35 and 2-morpholinoethan-1-amine (57 mg, 0.44 mmol) according to the general procedure F as a yellow oil (89 mg, 59% yield). 1H NMR (300 MHz, CDCl3) δ 7.86–7.76 (m, 2H), 7.45–7.25 (m, 4H), 7.06 (s, 1H), 6.77 (s, 1H), 4.13 (s, 2H), 3.60 (t, J = 4.6 Hz, 4H), 3.43 (q, J = 5.6 Hz, 2H), 2.53 (t, J = 5.9 Hz, 2H). MS (ESI): 347.1 [M + H]+.
Methyl N-Acryloyl-N-(4-phenylthiazol-2-yl)glycinate (37).
Compound 37 was prepared from methyl (4-phenylthiazol-2-yl) glycinate 28e (50 mg, 0.20 mmol), DIEA (101 μL, 0.60 mmol), and acryloyl chloride (27 mg, 0.30 mmol) according to the general procedure G as a white solid (33 mg, 55% yield). 1H NMR (300 MHz, CDCl3) δ 7.92–7.82 (m, 2H), 7.48–7.23 (m, 5H), 6.69–6.60 (m, 2H), 6.03–5.93 (m, 1H), 5.15 (s, 2H), 3.83 (s, 3H). MS (ESI): 303.1 [M + H]+. Purity: 95.0%.
tert-Butyl N-Acryloyl-N-(4-phenylthiazol-2-yl)glycinate (38).
Compound 38 was prepared from t-butyl (4-phenylthiazol-2-yl) glycinate 28f (100 mg, 0.34 mmol), DIEA (173 μL, 1.03 mmol), and acryloyl chloride (47 mg, 0.52 mmol) according to the general procedure G as a white solid (71 mg, 60% yield). 1H NMR (300 MHz, CDCl3) δ 8.04–7.80 (m, 3H), 7.56–7.18 (m, 5H), 6.69–6.60 (m, 2H), 6.03–5.92 (m, 1H), 4.98 (d, J = 15.5 Hz, 2H). MS (ESI): 345.1 [M + H]+. Purity: 94.2%.
N-Acryloyl-N-(4-phenylthiazol-2-yl)glycine (39).
To a solution of tert-butyl N-acryloyl-N-(4-phenylthiazol-2-yl)glycinate 38 (50 mg, 0.14 mmol) in 10 mL DCM was added 10 mL trifluoroacetic acid. The solution was stirred at room temperature for 2 h before it was concentrated. The residue was purified with preparative HPLC to give a white solid (31 mg, 79% yield). 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 7.7 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.38–7.25 (m, 2H), 6.66 (d, J = 8.8 Hz, 2H), 5.97 (dd, J = 8.7, 3.4 Hz, 1H), 5.12 (s, 2H). MS (ESI): 289.06 [M + H]+. Purity: 95.6%.
N-(2-(Cyclohexylamino)-2-oxoethyl)-N-(4-phenylthiazol-2-yl)acrylamide (40).
Compound 40 was prepared from N-cyclohexyl-2-((4-phenylthiazol-2-yl)amino)acetamide 36a (50 mg, 0.16 mmol), DIEA (80 μL, 0.38 mmol), and acryloyl chloride (22 mg, 0.24 mmol) according to the general procedure G as a white solid (36 mg, 61% yield). 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 7.4 Hz, 2H), 7.50–7.25 (m, 4H), 7.00 (dd, J = 16.7, 10.4 Hz, 1H), 6.73–6.61 (m, 1H), 6.02 (d, J = 10.1 Hz, 1H), 4.92 (s, 2H), 3.81 (d, J = 8.0 Hz, 1H), 1.85 (d, J = 12.2 Hz, 2H), 1.63–1.44 (m, 3H), 1.41–1.24 (m, 3H), 1.11 (t, J = 11.0 Hz, 3H). MS (ESI): 370.1 [M + H]+. Purity: 100.0%.
N-(2-Morpholino-2-oxoethyl)-N-(4-phenylthiazol-2-yl)acrylamide (41).
Compound 41 was prepared from 1-morpholino-2-((4-phenylthiazol-2-yl)amino)ethan-1-one 36b (50 mg, 0.17 mmol), DIEA (83 μL, 0.50 mmol), and acryloyl chloride (22 mg, 0.25 mmol) according to the general procedure G as a white solid (35 mg, 60% yield). 1H NMR (300 MHz, CDCl3) δ 7.82 (d, J = 7.4 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.39–7.25 (m, 2H), 6.65 (d, J = 8.4 Hz, 2H), 5.94 (s, 1H), 5.20 (s, 2H), 3.85–3.67 (m, 8H). MS (ESI): 358.1 [M + H]+. Purity: 96.3%.
N-(2-(Oxetan-3-ylamino)-2-oxoethyl)-N-(4-phenylthiazol-2-yl)acrylamide (42).
Compound 42 was prepared from N-(oxetan-3-yl)-2-((4-phenylthiazol-2-yl)amino)acetamide 36c (50 mg, 0.15 mmol), DIEA (77 μL, 0.46 mmol), and acryloyl chloride (21 mg, 0.23 mmol) according to the general procedure G as a white solid (38 mg, 65% yield). 1H NMR (300 MHz, CDCl3) δ 7.93–7.83 (m, 2H), 7.79 (s, 1H), 7.42 (dt, J = 27.7, 7.4 Hz, 4H), 7.28 (s, 1H), 6.95 (dd, J = 16.6, 10.3 Hz, 1H), 6.68 (dd, J = 16.6, 1.6 Hz, 1H), 6.08–5.98 (m, 1H), 5.17–4.99 (m, 1H), 5.04–4.83 (m, 4H), 4.44 (t, J = 6.5 Hz, 2H). MS (ESI): 344.1 [M + H]+. Purity: 100.0%.
N-(2-((2-Morpholinoethyl)amino)-2-oxoethyl)-N-(4-phenylthiazol-2-yl)acrylamide (43).
Compound 43 was prepared from N-(2-morpholinoethyl)-2-((4-phenylthiazol-2-yl)amino)acetamide 36d (50 mg, 0.14 mmol), DIEA (72 μL, 0.43 mmol), and acryloyl chloride (20 mg, 0.22 mmol) according to the general procedure G as a white solid (26 mg, 45% yield). 1H NMR (300 MHz, CDCl3) δ 7.94–7.84 (m, 2H), 7.48–7.27 (m, 4H), 7.22 (s, 1H), 6.94 (dd, J = 16.6, 10.3 Hz, 1H), 6.68 (dd, J = 16.6, 1.7 Hz, 1H), 6.02 (dd, J = 10.4, 1.6 Hz, 1H), 5.04 (s, 2H), 3.37 (dd, J = 11.4, 5.6 Hz, 6H), 2.41 (t, J = 5.8 Hz, 2H). MS (ESI): 401.1 [M + H]+. Purity: 98.2%.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-phenylthiazol-2-yl)acrylamide (44).
Compound 44 was prepared from N-((3-methylisoxazol-5-yl)methyl)-4-phenylthiazol-2-amine 28g (50 mg, 0.18 mmol), DIEA (92 μL, 0.55 mmol), and acryloyl chloride (25 mg, 0.28 mmol) according to the general procedure G as a white solid (25 mg, 42% yield). 1H NMR (300 MHz, CDCl3) δ 7.99–7.83 (m, 2H), 7.51–7.25 (m, 4H), 7.03 (dd, J = 16.6, 10.3 Hz, 1H), 6.65 (dd, J = 16.6, 1.7 Hz, 1H), 6.08 (s, 1H), 5.99 (dd, J = 10.4, 1.7 Hz, 1H), 5.64 (s, 2H), 2.38 (d, J = 0.8 Hz, 3H). MS (ESI): 326.1 [M + H]+. Purity: 98.4%.
N-(4-Phenylthiazol-2-yl)-N-(pyridin-3-ylmethyl)acrylamide (45).
Compound 45 was prepared from 4-phenyl-N-(pyridin-3-ylmethyl)thiazol-2-amine 28h (50 mg, 0.19 mmol), DIEA (94 μL, 0.56 mmol), and acryloyl chloride (25 mg, 0.28 mmol) according to the general procedure G as a white solid (19 mg, 31% yield). 1H NMR (400 MHz, acetone-d6): δ 8.01–7.91 (m, 2H), 7.72 (s, 2H), 7.62 (s, 1H), 7.47–7.32 (m, 6H), 7.35–7.24 (m, 2H), 7.20 (d, J = 7.9 Hz, 1H), 6.97 (dd, J = 16.5, 10.4 Hz, 1H), 6.54 (dd, J = 16.6, 1.9 Hz, 1H), 5.93 (dd, J = 10.4, 2.0 Hz, 1H), 5.83 (s, 2H). MS (ESI): 322.1 [M + H]+. Purity: 97.2%.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(4-chloro-2-methoxyphenyl)thiazol-2-yl)acrylamide (46a).
Compound 46a was prepared from N-((3-methylisoxazol-5-yl)methyl)-4-(4-chloro-2-methoxyphenyl)thiazol-2-amine 28j (100 mg, 0.29 mmol), DIEA (148 μL, 0.88 m mol), and acryloyl chloride (40 mg, 0.45 mmol) according to the general procedure G as a white solid (51 mg, 44% yield). 1H NMR (300 MHz, CDCl3) δ 8.12 (d, J = 8.3 Hz, 1H), 7.72 (s, 1H), 7.09–6.91 (m, 2H), 6.94–6.82 (m, 1H), 6.68 (d, J = 16.5 Hz, 1H), 6.09 (s, 1H), 6.01 (d, J = 10.3 Hz, 1H), 5.65 (s, 2H), 3.98 (s, 3H), 2.26 (s, 3H). MS (ESI): 390.0 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(4-fluoro-2-methoxyphenyl)thiazol-2-yl)acrylamide (46b).
Compound 46b was prepared from N-((3-methylisoxazol-5-yl)methyl)-4-(4-fluoro-2-methoxyphenyl)thiazol-2-amine 28k (100 mg, 0.30 mmol), DIEA (149 μL, 0.89 mmol) and acryloyl chloride (40 mg, 0.45 mmol) according to the general procedure G as a white solid (38 mg, 44% yield). 1H NMR (300 MHz, CDCl3) δ 8.12 (d, J = 8.3 Hz, 1H), 7.72 (s, 1H), 7.09–6.82 (m, 3H), 6.68 (d, J = 16.5 Hz, 1H), 6.12–5.96 (m, 2H), 5.65 (s, 2H), 3.98 (s, 3H), 2.26 (s, 3H). MS (ESI): 374.1 [M + H]+.
2-Chloro-N-(4-(2-methoxyphenyl)thiazol-2-yl)-N-((3-methylisoxazol-5-yl)methyl)acetamide (46c).
A solution of 2-chloroacetyl chloride (57 mg, 0.50 mmol), 28l (100 mg, 0.33 mmol) and DIEA (129 mg, 1.00 mmol) in toluene (2 mL) was heated at 70 °C for 1 h. The mixture was concentrated, and the residue was purified with flash chromatography using DCM as the eluent to give 2 as a white solid (93 mg, 74% yield). 1H NMR (300 MHz, CDCl3) δ 8.10–8.04 (m, 1H), 7.80 (s, 1H), 7.31 (d, J = 8.8 Hz, 1H), 7.13 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 6.40 (s, 1H), 5.64 (s, 2H), 4.92 (s, 2H), 3.92 (s, 3H), 2.17 (s, 3H). MS (ESI): 377.9 [M + H]+.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(4-trifluoromethylphenyl)thiazol-2-yl)acrylamide (47).
Compound 47 was prepared from N-((3-methylisoxazol-5-yl)methyl)-4-(4-trifluoromethylphenyl)thiazol-2-amine 28i (50 mg, 0.16 mmol), DIEA (78 μL, 0.47 mmol), and acryloyl chloride (21 mg, 0.23 mmol) according to the general procedure G as a white solid (20 mg, 35% yield). 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 8.2 Hz, 2H), 7.41 (s, 1H), 7.03 (dd, J = 16.5, 10.4 Hz, 1H), 6.67 (d, J = 16.2 Hz, 1H), 6.08–5.96 (m, 2H), 5.64 (s, 2H), 2.39 (s, 3H). MS (ESI): 394.1 [M + H]+. Purity: 95.5%.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(2-methoxyphenyl)thiazol-2-yl)acrylamide (48).
Compound 48 was prepared from N-((3-methylisoxazol-5-yl)methyl)-4-(2-methoxylphenyl)thiazol-2-amine 28l (100 mg, 0.33 mmol), DIEA (166 μL, 1.00 mmol), and acryloyl chloride (45 mg, 0.50 mmol) according to the general procedure G as a white solid (59 mg, 50% yield). 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 7.9 Hz, 1H), 7.78 (s, 1H), 7.38–7.25 (m, 1H), 7.13–6.94 (m, 4H), 6.64 (d, J = 16.5 Hz, 1H), 6.07 (s, 1H), 5.97 (d, J = 10.4 Hz, 1H), 5.64 (s, 2H), 4.00 (s, 3H), 2.37 (s, 3H). MS (ESI): 356.1 [M + H]+.
General Procedure H: Preparation of Compounds 49–51.
A solution of methoxy-containing acrylamides (1.0 equiv) in anhydrous DCM (20 mL) was stirred at −78 °C for 30 min, and 1 M BBr3 solution in DCM (5.0 equiv) was added dropwise. The reaction mixture was slowly warmed to room temperature and stirred for 2 h. Methanol (1 mL) was slowly added at −30 °C to quench the reaction. The mixture was extracted with DCM three times. The combined organic layer was dried with MgSO4, filtered, and concentrated. The crude was refluxed in 5% DIEA THF solution (20 mL) for 1 h. The solvent was then removed in vacuo, and the residue was purified with preparative HPLC.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(2-hydroxylphenyl)thiazol-2-yl)acrylamide (49).
Compound 49 was prepared from N-((3-methylisoxazol-5-yl)methyl)-N-(4-(2-methoxyphenyl)thiazol-2-yl)acrylamide 48 (30 mg, 0.084 mmol) and 1 M BBr3 solution (0.25 mL, 0.25 mmol) according to the general procedure G as a white solid (10 mg, 35% yield). 1H NMR (300 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 1H), 7.33 (s, 1H), 7.08–6.88 (m, 3H), 6.70 (d, J = 16.5 Hz, 1H), 6.05 (d, J = 9.7 Hz, 2H), 5.49 (s, 2H), 2.42 (s, 3H). MS (ESI): 342.1 [M + H]+. Purity: 98.0%.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(4-chloro-2-hydroxylphenyl)thiazol-2-yl)acrylamide (50).
Compound 50 was prepared from N-((3-methylisoxazol-5-yl)methyl)-N-(4-(4-chloro-2-methoxyphenyl)thiazol-2-yl)acrylamide 46a (30 mg, 0.076 mmol) and 1 M BBr3 solution (0.22 mL, 0.22 mmol) according to the general procedure G as a white solid (10 mg, 36% yield). 1H NMR (300 MHz, CDCl3) δ 7.53 (d, J = 8.4 Hz, 1H), 7.05–6.85 (m, 3H), 6.73 (d, J = 16.4 Hz, 1H), 6.25 (s, 1H), 6.11 (d, J = 10.3 Hz, 1H), 5.49 (s, 2H), 2.31 (s, 3H). MS (ESI): 376.0 [M + H]+. Purity: 98.4%.
N-((3-Methylisoxazol-5-yl)methyl)-N-(4-(4-fluoro-2-hydroxylphenyl)thiazol-2-yl)acrylamide (51).
Compound 51 was prepared from N-((3-methylisoxazol-5-yl)methyl)-N-(4-(4-fluoro-2-methoxyphenyl)thiazol-2-yl)acrylamide 46b (30 mg, 0.077 mmol) and 1 M BBr3 solution (0.23 mL, 0.23 mmol) according to the general procedure G as a white solid (7 mg, 25% yield). 1H NMR (300 MHz, CDCl3) δ 7.57 (dd, J = 8.7, 6.4 Hz, 1H), 7.22 (s, 1H), 6.98 (dd, J = 16.3, 10.3 Hz, 1H), 6.82–6.59 (m, 3H), 6.24 (s, 1H), 6.11 (d, J = 10.3 Hz, 1H), 5.50 (s, 2H), 2.31 (s, 3H). MS (ESI): 374.1 [M + H]+. Purity: 95.0%.
2-Chloro-N-(4-(2-hydroxyphenyl)thiazol-2-yl)-N-((3-methylisoxazol-5-yl)methyl)acetamide (52).
Compound 52 was prepared from 46c (30 mg, 0.077 mmol) and 1 M BBr3 solution (0.23 mL, 0.23 mmol) according to the general procedure G as a white solid (14 mg, 24% yield). 1H NMR (300 MHz, CDCl3) δ 10.85 (s, 1H), 7.63 (dd, J = 7.8, 1.6 Hz, 1H), 7.34 (s, 1H), 7.31–7.25 (m, 1H), 7.02 (dd, J = 8.2, 1.2 Hz, 1H), 6.94 (ddd, J = 7.8, 7.2, 1.3 Hz, 1H), 6.34 (s, 1H), 5.54 (s, 2H), 4.63 (s, 2H), 2.31 (s, 3H). MS (ESI): 363.9 [M + H]+. Purity: 100.0%.
N-(4-(2-Hydroxyphenyl)thiophen-2-yl)-N-((3-methylisoxazol-5-yl)methyl)acrylamide (53).
To a solution of 57 (19 mg, 0.058 mmol), (2-hydroxyphenyl)boronic acid (12 mg, 0.087 mmol), and Na2CO3 (12 mg, 0.116 mmol) in dioxane (1.5 mL) and H2O (0.5 mL) was added Pd(PPh3)4 (6.7 mg, 0.0058 mmol) under argon. The mixture was heated at 80 °C under argon for 8 h. The reaction mixture was diluted with EtOAc, washed with saturated NaHCO3 and brine, dried over Na2SO4, filtered, and purified with preparative HPLC to give 53 as a white solid (8 mg, 41% yield). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 7.36 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.17 (s, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.54–6.37 (m, 2H), 6.16 (s, 1H), 5.72 (d, J = 9.9 Hz, 1H), 5.05 (s, 2H), 2.32 (s, 3H). MS (ESI): 341.1 [M + H]+. Purity: 100.0%.
tert-Butyl (4-bromothiophen-2-yl)((3-methylisoxazol-5-yl)methyl)carbamate (55).
To a solution of tert-butyl (4-bromothiophen-2-yl)carbamate (40 mg, 0.144 mmol) in DMF (1 mL) at 0 °C was added NaH (8.6 mg, 0.216 mmol, 60% in mineral oil). The mixture was stirred at room temperature for 15 min before 5-(bromomethyl)-3-methylisoxazole (32 mg, 0.188 mmol) was added. The mixture was stirred for another 4 h, quenched by H2O, and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, concentrated, and purified with flash chromatography (20% EtOAc in hexane) to give 55 as a white solid (43 mg, 80%). 1H NMR (400 MHz, CDCl3) δ 6.86 (s, 1H), 6.53 (s, 1H), 6.00 (s, 1H), 4.94 (s, 2H), 2.30 (s, 3H), 1.53 (s, 9H).
4-Bromo-N-((3-methylisoxazol-5-yl)methyl)thiophen-2-amine (56).
To a solution of 55 in DCM (1.5 mL) was added TFA (0.3 mL) dropwise. The mixture was stirred at room temperature for 3 h and concentrated. 56 was obtained as a colorless gel, which was directly used in the next step.
N-(4-Bromothiophen-2-yl)-N-((3-methylisoxazol-5-yl)methyl)acrylamide (57).
A solution of acryloyl chloride (21 mg, 0.23 mmol), 56 (31 mg, 0.12 mmol), and DIEA (44 mg, 0.35 mmol) in toluene (2 mL) was heated at 70 °C for 1 h. The mixture was concentrated, and the residue was purified with flash chromatography (20% EtOAc in hexane) to give 57 as a white solid (19 mg, 51% yield over two steps). 1H NMR (400 MHz, CDCl3) δ 7.19 (s, 1H), 6.82 (s, 1H), 6.49 (d, J = 16.7 Hz, 1H), 6.32 (d, J = 12.0 Hz, 1H), 6.11 (s, 1H), 5.73 (d, J = 10.3 Hz, 1H), 4.97 (s, 2H), 2.30 (s, 3H).
Molecular Modeling.
The published GSTO1 structures (PDB code: 4YQV and 4YQM) and in-house structures were used for the modeling of potential inhibitor binding modes from the fragment and previously screening results, respectively. All crystallographic water molecules were deleted for calculations, as they were not deemed to be possible to confidently predict important water molecules up front across the different series modeled. Protein alignment and hydrophobic surface calculations were conducted in Maestro.36 Molecular graphics and analyses were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.37 3D small molecule structures and distance measurements were generated in Maestro. Crystal structures were prepared for docking using the protein preparation wizard in Maestro, which optimizes hydrogen placements. The active site was defined by the bound ligand. Glide Covalent Docking Protocol38 was used for covalent ligand docking to target Cys32. Multiple tautomers and protonation states were enumerated for ligands using LigPrep.39 Docked binding modes were ranked using the docking score (GlideScore35) and visually inspected for the retention of key interactions along with other close contacts. Binding modes were deprioritized when the conformation of the docked molecule was considered to be unsatisfactory.
GSTO1–1 Expression and Purification.
GSTO1–1 [residues 1–241 bearing an N-terminal 6x histidine tag and a tobacco etch virus (TEV) protease cleavage site] was expressed and purified as described previously.4
In-Gel Fluorescence Binding Assay.
The inhibitory activity of the compounds was determined using a competitive inhibition of CMFDA binding to GSTO1. Briefly, an in vitro binding assay with purified GSTO1–1 was performed using 1 μM GSTO1–1 in a reaction buffer [100 mM Tris (pH 8.0), 1.5 mM ethylenediaminetetraacetic acid (EDTA), and 1 μM dithiothreitol (DTT)] incubated with compounds for 30 min at 37 °C, followed by addition of CMFDA (500 nM) for 30 min. The reaction was quenched with a Laemmli sample buffer and resolved on a 4–20% polyacrylamide gel. Gels were immediately scanned on an i-Bright imaging system (Thermo Fisher Scientific). Quantification of fluorescent band intensity was done using ImageJ software. % decrease in band intensity is equivalent to the % inhibition of CMFDA binding to the GSTO1 upon compound addition. Hence, % decrease in band intensity is the measurement of the inhibitory effect of GSTO1 because of binding of the compound to GSTO1.
4NPG Reductase Assay.4,6
Inhibition of GSTO1 enzyme activity was measured by monitoring the reduction of 4-NPG to 4-nitroacetophenone. Briefly, in a 200 mL reaction volume, 5 μg/mL purified GSTO1 in reaction buffer [100 mM Tris (pH 8.0), 1.5 mM EDTA, and 1 mM DTT] was incubated with DMSO or different concentrations of inhibitors for 30 min at 37 °C. A unit of 1 mM 4-NPG was added to the reaction, and decrease in absorbance at 305 nm was recorded on Synergy H1 plate reader (Biotek).
Cell Lines.
Colon cancer cell line HCT116 was generously provided by Dr. Bert Vogelstein, Johns Hopkins Medical Institutions, Baltimore, MD. HT29 cell line was purchased from the American Type Culture Collection (Manassas, VA). Cell lines were cultured in RPMI 1640 media supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere of 5% CO2. All cell lines were maintained in culture under 30 passages and tested regularly for Mycoplasma contamination using PlasmoTest (InvivoGen, San Diego, CA, USA).
Cell Viability Assays.
Cell proliferation was assessed by the MTT assay. Cancer cells were seeded in 96-well microtiter plates and, after overnight attachment, treated with synthesized compounds. After 72 h, MTT solution (3 mg/mL) was added to each well, and cells were incubated for 3 h at 37 °C. After incubation, media from each well were removed, and the dark blue formazan crystals formed by live cells were dissolved in DMSO (100 μL per well). The absorbance intensity was measured at 570 nm on a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Crystallization, Data Collection, and Structure Determination.
The GSTO1:18 complex was crystallized by mixing GSTO1–1 (2.66 mg/mL in 25 mM Tris pH 7.5, 60 mM NaCl, and 1 mM DTT) with 18 in a 1:1.5 molar ratio and incubating at 4 °C for 48 h. Protein was concentrated to 22 mg/mL and crystallized by sitting drop vapor diffusion at 20 °C by mixing protein in a 1:1 ratio with well solution containing 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.5, 25% poly(ethylene glycol) (PEG)-3350, and 3% methanol. Crystals were cryoprotected in a solution containing 100 mM MES pH 6.5 and 30% PEG-3350 and flash-frozen in liquid nitrogen.
The GSTO1:37 and GSTO1:44 complexes were crystallized by mixing concentrated GSTO1 (18.5–23 mg/mL in 25 mM Tris pH 7.5, 60 mM NaCl, and 1 mM DTT) in a 1:1.5 or 1:1.8 protein to inhibitor ratio with inhibitors 37 and 44, respectively. Protein was incubated with an inhibitor at 4 °C for 1 h prior to crystallization by sitting drop vapor diffusion at 16 °C. Crystals formed in drops containing protein in a 1:1 ratio with well solution containing 100 mM MES pH 6.5, 30% PEG-3350, and either 3% (+/−)-2-methyl-2,4-pentanediol for GSTO1:37 or 4% acetone for GSTO1:44. After 24 h, crystals were cryoprotected in a solution containing 100 mM MES pH 6.5, 30% PEG-3350, and 1.25 mM inhibitor and then supper-cooled in liquid nitrogen.
Diffraction data were collected on Advanced Photon Source LS-CAT beamlines 21-ID-G (GSTO-1 complexes with 18 and 37) or 21-ID-D (GSTO1:44) as described in Table 1. All data were processed using HKL2000,40 and all three structures were solved via molecular replacement using a published structure of a GSTO1/inhibitor complex sans ligand (PDB ID: 4YQU).4 Molecular replacement was performed in MOLREP41 (GSTO1:18) or Phaser42 (GSTO1:37 and GSTO1:44). Iterative model building and refinement were performed using COOT43 and BUSTER,44 respectively. Coordinates and geometric restraints for each ligand were generated in Grade.45 All structures were validated using Molprobity46 and the PDB validation server.47 All calculated RMSD values are based on SSM super-positioning in COOT.43
The GSTO1 structure bound to 18 was solved to 2.75 Å in the space group P1. The structure includes six chains of protein per asymmetric unit with an average RMSD of 0.47 Å between chains. Residues 6 through 241 were modeled in each chain with the exception of a disordered loop that varied in length between chains (133–135 in chains A, B, and D, 134–135 in chain C, and 133–136 in chains E and F). In chain E, a number of additional residues including 10–12, 57–58, and 234–235 were not modeled because of disorder. Difference electron density maps supported covalent modification of residue Cys32 by inhibitor 18 in all six chains (Figure S1A); however, weaker electron density in chain F allowed only partial modeling of the inhibitor. The electron density corresponding to MES, a component of the crystallization solution and cryoprotectant, was also observed in four of the six chains.
The GSTO1 structures with 37 and 44 were solved in the space group P1211 at resolutions of 2.0 and 2.15 Å, respectively. Both structures contained two molecules of protein per asymmetric unit with an RMSD of 0.48 Å between chains. In both structures, residues 1–241 are modeled in each chain, as well as three residues (SNA) associated with the N-terminal TEV cleavage site. Difference electron density maps supported covalent modification of the GSTO1 residue Cys32 by 37 or 44 in both chains of their corresponding cocrystal structures (Figure S1B,C). In the structure of GSTO1 with 37, the methyl ester substituent was not ordered. As with the GSTO1:18 structure, the electron density corresponding to MES was observed in both chains of the GSTO1 structures with 37 and 44. In the structure of GSTO1:44, the electron density on the surface of both protein chains supported modeling a monomer of PEG.
Microsome Stability.
The microsome incubation system was prepared as follows. Microsome (10 μL, 20 mg/mL) was diluted with 330 μL 0.1 M phosphate buffer (3.3 mM MgCl2), and 40 μL of 10 μM test compound phosphate buffer solution was added. The prepared master solution was prewarmed in 37 °C for 3 min. NADPH (3.5 mg) was dissolved in 210 μL of 0.1 M phosphate buffer (3.3 mM MgCl2). Then 20 μL NADPH was added to the above master solution to initiate the reaction for 0, 5, 10, 15, 30, 45, and 60 min. The final concentration of the test compound in the reaction system was 1 μM. Aliquot of 40 μL was pipetted from the reaction solution and stopped by the addition of 4 volumes of cold acetonitrile containing 50 nM of compounds 18 and 49 as an internal standard (IS) at the designated time points. The incubation solution was centrifuged at 3500g for 10 min to precipitate the protein. The supernatant was used for LC/MS/MS analysis. The natural log peak area ratio (compound peak area/IS peak area) was plotted against time, and the gradient of the line was determined.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported in part through a University of Michigan Developmental Research/GI SPORE (P50 CA130810) Pilot Award. N.N. and J.A.S. are grateful for the support of this work from NIH P30 CA046592. W.D. was supported by China Scholarship Council (201606240054). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
ABBREVIATIONS
- GSTO1
glutathione S-transferase omega 1
- CRID
cytokine release inhibitory drug
- SAR
structure–activity relationship
- CMFDA
5-chloromethylfluorescein diacetate
- LE
ligand efficiency
- LLE
lipophilic ligand efficiency
- TLC
thin-layer chromatography
- LC–MS
liquid chromatography–mass spectrometry
- EtOH
ethanol
- DCM
dichloromethane
- EtOAc
ethyl acetate
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- HOBt
hydroxybenzotriazole
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide
- DIEA
diisopropylethylamine
- BBr3
boron tribromide
- MgSO4
magnesium sulfate
- NaHCO3
sodium bicarbonate
- K2CO3
potassium carbonate
- HCl
hydrogen chloride
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01960.
Molecular formula strings including screening data, X-ray diffraction data, NMR spectra, and HPLC chromatograms (PDF)
PDB ID of new crystal (X-ray) structures/homology models: GSTO1:18 (PDB ID: 6MHB), GSTO1:37 (PDB ID: 6MHC), and GSTO1:44 (PDB ID: 6MHD) (CSV)
REFERENCES
- 1.Hayes JD; Flanagan JU; Jowsey IR Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [DOI] [PubMed] [Google Scholar]
- 2.Board PG; Menon D Glutathione transferases, regulators of cellular metabolism and physiology. BBA-Gen Subjects 2013, 1830, 3267–3288. [DOI] [PubMed] [Google Scholar]
- 3.Kensler TW; Wakabayashi N; Biswal S Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [DOI] [PubMed] [Google Scholar]
- 4.Ramkumar K; Samanta S; Kyani A; Yang S; Tamura S; Ziemke E; Stuckey JA; Li S; Chinnaswamy K; Otake H Mechanistic evaluation and transcriptional signature of a glutathione S-transferase omega 1 inhibitor. Nat. Commu. 2016, 7, 13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Board PG The omega-class glutathione transferases: structure, function, and genetics. Drug Metab. Rev. 2011, 43, 226–235. [DOI] [PubMed] [Google Scholar]
- 6.Board PG; Coggan M; Cappello J; Zhou H; Oakley AJ; Anders MW S-(4-Nitrophenacyl)glutathione is a specific substrate for glutathione transferase omega 1–1. Anal. Biochem. 2008, 374, 25–30. [DOI] [PubMed] [Google Scholar]
- 7.Board PG; Coggan M; Chelvanayagam G; Easteal S; Jermiin LS; Schulte GK; Danley DE; Hoth LR; Griffor MC; Kamath AV Identification, characterization, and crystal structure of the Omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [DOI] [PubMed] [Google Scholar]
- 8.Whitbread AK; Masoumi A; Tetlow N; Schmuck E; Coggan M; Board PG Characterization of the omega class of glutathione transferases. Methods Enzymol. 2005, 401, 78–99. [DOI] [PubMed] [Google Scholar]
- 9.Weerapana E; Simon GM; Cravatt BF Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol. 2008, 4, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MBD; Bachovchin DA; Mowen K; Baker D; Cravatt BF Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J-H; Ni R-Z; Xiao M-B; Guo J-G; Zhou J-W Comparative proteomic analysis of differentially expressed proteins in human pancreatic cancer tissue. Hepatobiliary Pancreat. Dis. Int. 2009, 8, 193–200. [PubMed] [Google Scholar]
- 12.Ginos MA; Page GP; Michalowicz BS; Patel KJ; Volker SE; Pambuccian SE; Ondrey FG; Adams GL; Gaffney PM Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004, 64, 55–63. [DOI] [PubMed] [Google Scholar]
- 13.Djukic T; Simic T; Pljesa-Ercegovac M; Matic M; Suvakov S; Coric V; Dragicevic D; Savic-Radojevic A Upregulated glutathione transferase omega-1 correlates with progression of urinary bladder carcinoma. Redox Rep. 2017, 22, 486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L; Zhao L; Zhang Y; Zhang Q; Ding Y Proteomic analysis of Tiam1-mediated metastasis in colorectal cancer. Cell Biol. Int. 2007, 31, 805–14. [DOI] [PubMed] [Google Scholar]
- 15.McIlwain CC; Townsend DM; Tew KD Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene 2006, 25, 1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bulus H; Oguztuzun S; Güler Simsek G; Kilic M; Ada AO; Göl S; Kocdogan AK; Kaygın P; Bozer B; Iscan M Expression of CYP and GST in human normal and colon tumor tissues. Biotech. Histochem. 2018, 1–9. [DOI] [PubMed] [Google Scholar]
- 17.Piaggi S; Raggi C; Corti A; Pitzalis E; Mascherpa MC; Saviozzi M; Pompella A; Casini AF Glutathione transferase omega 1–1 (GSTO1–1) plays an anti-apoptotic role in cell resistance to cisplatin toxicity. Carcinogenesis 2010, 31, 804–811. [DOI] [PubMed] [Google Scholar]
- 18.Yan X.-d.; Pan L.-y.; Yuan Y; Lang J.-h.; Mao N Identification of platinum-resistance associated proteins through proteomic analysis of human ovarian cancer cells and their platinum-resistant sublines. J. Proteome Res. 2007, 6, 772–780. [DOI] [PubMed] [Google Scholar]
- 19.Menon D; Innes A; Oakley AJ; Dahlstrom JE; Jensen LM; Brüstle A; Tummala P; Rooke M; Casarotto MG; Baell JB GSTO1–1 plays a pro-inflammatory role in models of inflammation, colitis and obesity. Sci. Rep. 2017, 7, 17832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laliberte RE; Perregaux DG; Hoth LR; Rosner PJ; Jordan CK; Peese KM; Eggler JF; Dombroski MA; Geoghegan KF; Gabel CA Glutathiones-transferase omega 1–1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1β posttranslational processing. J. Biol. Chem. 2003, 278, 16567–16578. [DOI] [PubMed] [Google Scholar]
- 21.Tsuboi K; Bachovchin DA; Speers AE; Spicer TP; Fernandez-Vega V; Hodder P; Rosen H; Cravatt BF Potent and Selective Inhibitors of GlutathioneS-Transferase Omega 1 That Impair Cancer Drug Resistance. J. Am. Chem. Soc. 2011, 133, 16605–16616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menon D; Coll R; Board PG Glutathione transferase omega 1 is required for the lipopolysaccharide-stimulated induction of NADPH oxidase 1 and the production of reactive oxygen species in macrophages. Free Radic. Biol. Med. 2014, 73, 318–327. [DOI] [PubMed] [Google Scholar]
- 23.Board DE; Brighty GJ; Plate L; Bare G; Chen W; Li S; Wang H; Cravatt BF; Forli S; Powers ET “Inverse drug discovery” strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J. Am. Chem. Soc. 2017, 140, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flanagan ME; Abramite JA; Anderson DP; Aulabaugh A; Dahal UP; Gilbert AM; Li C; Montgomery J; Oppenheimer SR; Ryder T Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- 25.Barf T; Kaptein A Irreversible protein kinase inhibitors: balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [DOI] [PubMed] [Google Scholar]
- 26.Jackson PA; Widen JC; Harki DA; Brummond KM Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-michael addition reactions. J. Med. Chem. 2016, 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bräutigam M; Teusch N; Schenk T; Sheikh M; Aricioglu RZ; Borowski SH; Neudörfl J-M; Baumann U; Griesbeck AG; Pietsch M Selective inhibitors of glutathione transferase P1 with trioxane structure as anticancer agents. ChemMedChem 2015, 10, 629–639. [DOI] [PubMed] [Google Scholar]
- 28.Aeluri R; Alla M; Polepalli S; Jain N Synthesis and antiproliferative activity of imidazo[1,2-a]pyrimidine Mannich bases. Eur. J. Med. Chem. 2015, 100, 18–23. [DOI] [PubMed] [Google Scholar]
- 29.Sravanthi TV; Manju SL Synthesis and Fluorescence Properties of Novel indol-3yl-thiazolo[3,2-a][1,3,5]triazines and indole-3-carbaldehyde Schiff Bases. J. Fluoresc. 2015, 25, 1727–1738. [DOI] [PubMed] [Google Scholar]
- 30.Strobel H; Wohlfart P; Zoller G; Will DW Heteroaryl-substituted amides comprising a saturated linker group, and their use as pharmaceuticals. U.S. Patent 851,897,6B2, 2013.
- 31.Xie Y; Dahlin JL; Oakley AJ; Casarotto MG; Board PG; Baell JB Reviewing hit discovery literature for difficult targets: glutathione transferase omega-1 as an example. J. Med. Chem. 2018, 61, 7448–7470. [DOI] [PubMed] [Google Scholar]
- 32.Son J; Lee J-J; Lee J-S; Schüller A; Chang Y-T Isozyme-specific fluorescent inhibitor of glutathione s-transferase omega 1. ACS Chem. Biol. 2010, 5, 449–453. [DOI] [PubMed] [Google Scholar]
- 33.Shaffer AA; Wierschke SG Comparison of computational methods applied to oxazole, thiazole, and other heterocyclic compounds. J. Comput. Chem. 1993, 14, 75–88. [Google Scholar]
- 34.Schultes S; de Graaf C; Haaksma EEJ; de Esch IJP; Leurs R; Krämer O Ligand efficiency as a guide in fragment hit selection and optimization. Discovery Today: Technol 2010, 7, e157–e162. [DOI] [PubMed] [Google Scholar]
- 35.Leeson PD; Springthorpe B The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Disco. 2007, 6, 881. [DOI] [PubMed] [Google Scholar]
- 36.Maestro, version 10.1; Schrödinger, LLC: New York, NY, 2015. [Google Scholar]
- 37.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 38.Glide, version 6.8; Schrödinger, LLC: New York, NY, 2015. [Google Scholar]
- 39.LigPrep, version 3.5; Schrödinger, LLC: New York, NY, 2015. [Google Scholar]
- 40.Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. In Methods Enzymol.; Elsevier, 1997; Vol. 276, pp 307–326. [DOI] [PubMed] [Google Scholar]
- 41.Vagin A; Teplyakov A MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar]
- 42.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development ofCoot. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bricogne G; Blanc E; Brandl M; Flensburg C; Keller P; Paciorek W; Roversi P; Sharff A; Smart OS; Vonrhein C; Womack TO BUSTER, 2.10.3; Global Phasing Ltd.: Cambridge, United Kingdom, 2017. [Google Scholar]
- 45.Smart OS; Womack TO; Sharff A; Flensburg C; Keller P; Paciorek W; Vonrhein C; Bricogne G GRADE, 1.2.13; Global Phasing Ltd.: Cambridge, United Kingdom, 2017. [Google Scholar]
- 46.Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2009, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. wwPDB Validation Server.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.