

Abstract
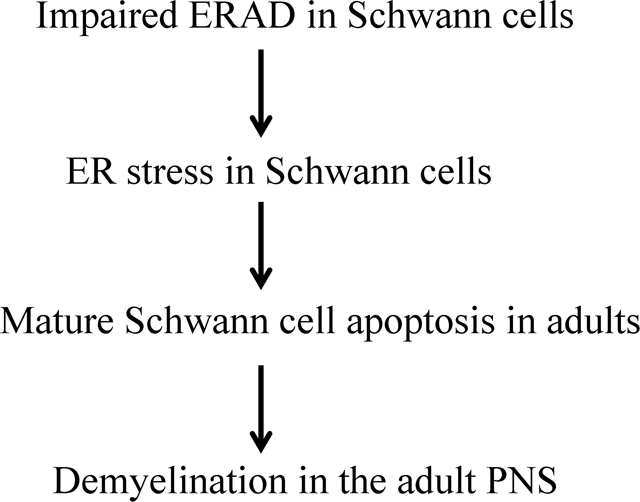
The integrated unfolded protein response (UPR) and endoplasmic reticulum associated degradation (ERAD) is the principle mechanisms that maintain endoplasmic reticulum (ER) homeostasis. Schwann cells (SCs) must produce an enormous amount of myelin proteins via the ER to assemble and maintain myelin structure; however, it is unclear how SCs maintain ER homeostasis. It is known that Suppressor/Enhancer of Lin-12-like (Sel1L) is necessary for the ERAD activity of the Sel1L- hydroxymethylglutaryl reductase degradation protein 1(Hrd1) complex. Herein, we showed that Sel1L deficiency in SCs impaired the ERAD activity of the Sel1L-Hrd1 complex and led to ER stress and activation of the UPR. Interestingly, Sel1L deficiency had no effect on actively myelinating SCs during development, but led to later-onset mature SC apoptosis and demyelination in the adult PNS. Moreover, inactivation of the pancreatic ER kinase (PERK) branch of the UPR did not influence the viability and function of actively myelinating SCs, but resulted in exacerbation of ER stress and apoptosis of mature SCs in SC-specific Sel1L deficient mice. These findings suggest that the integrated UPR and ERAD is dispensable to actively myelinating SCs during development, but is necessary for maintaining ER homeostasis and the viability and function of mature SCs in adults.
Keywords: UPR, ERAD, Schwann cells, Myelin, Sel1L, PERK
Graphical Abstract
Introduction
Membrane proteins are modified and folded in the endoplasmic reticulum (ER) (Kaufman, 1999; W. Lin & Popko, 2009). Protein folding is an error-prone process. Unfolded or misfolded proteins are recognized, retrotranslocated to the cytosol, and degraded by the ubiquitin-proteasome system, a process referred to as ER associated degradation (ERAD)(Nakatsukasa, Kamura, & Brodsky, 2014; Qi, Tsai, & Arvan, 2017; Ruggiano, Foresti, & Carvalho, 2014). Accumulation of unfolded or misfolded proteins in the ER lumen disrupts ER homeostasis and results in ER stress and activation of the unfolded protein response (UPR). The UPR comprises three parallel branches, pancreatic ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6α (ATF6α), which restores ER homeostasis in ER-stress cells and preserves their viability and function (Han & Kaufman, 2017; Marciniak & Ron, 2006; Walter & Ron, 2011). PERK activation inhibits global protein translation but stimulates the expression of cytoprotective genes and genes related with ERAD by phosphorylating eukaryotic translation initiation factor 2α (eIF2α). IRE1 activation upregulates the expression of genes that enhances protein folding and protein degradation by splicing X-box binding protein 1 (XBP1) mRNA. ATF6α undergoes cleavage and the cleaved ATF6α (cATF6α) increases the expression of ER chaperones and genes related to ERAD. The UPR restores ER homeostasis by facilitating protein folding, attenuating protein translation, and enhancing ERAD. Moreover, evidence suggests that ERAD can influence the UPR by degrading ER-resident UPR effectors (Horimoto et al., 2013; Sugimoto et al., 2017; S. Y. Sun et al., 2015). The integrated UPR and ERAD is the principle ER quality control mechanism that maintains ER homeostasis (Hwang & Qi, 2018; McCaffrey & Braakman, 2016; Qi et al., 2017). Nevertheless, if the integrated UPR and ERAD cannot restore ER homeostasis, persistent or severe ER stress triggers apoptotic programs to eliminate cells (Han & Kaufman, 2017; Marciniak & Ron, 2006; Walter & Ron, 2011).
Myelin is an enormous plasma membrane that wraps around axons to facilitate action potential propagation. Schwann cells (SCs) are the myelin-producing cells in the peripheral nervous system (PNS). During developmental myelination, each actively myelinating SC must produce an enormous amount of myelin proteins via the ER to assemble myelin sheath. Mature SCs in adults need to produce a substantial amount of myelin proteins via the ER, although far less than actively myelinating SCs, to maintain myelin structure homeostasis (Anitei & Pfeiffer, 2006; Clayton & Popko, 2016; W. Lin & Popko, 2009; Volpi, Touvier, & D’Antonio, 2017). It is generally believed that maintaining ER homeostasis is necessary for myelin protein biosynthesis and the myelinating function of SCs, and that actively myelinating SCs are more susceptible to disruption of ER homeostasis than mature SCs, due to the rate of myelin protein production (Clayton & Popko, 2016; W. Lin & Popko, 2009; W. Lin & Stone, 2020; Volpi et al., 2017). Nevertheless, the mechanisms by which SCs maintain ER homeostasis remain largely unknown.
A number of studies showed that ER stress and the UPR in SCs contributes to the development of myelin disorders in the PNS (D’Antonio et al., 2013; Pennuto et al., 2008; Sidoli et al., 2016). Conversely, a recent study reported that impaired UPR in SCs via double deletion of PERK and ATF6α does not affect their viability or function in young mice under normal conditions (Stone, Wu, Nave, & Lin, 2020). Interestingly, a study showed that deletion of Derlin-2 (an ERAD effector) in SCs compromises myelin maintenance in the PNS of naïve adult mice and exacerbates myelin abnormalities in a mouse model of Charcot-Marie-Tooth 1B (CMT1B) neuropathy (Volpi et al., 2019). Thus, we sought to determine the role of ERAD in maintaining ER homeostasis in SCs under physiological conditions. The Suppressor/Enhancer of Lin-12-like (Sel1L) - hydroxymethylglutaryl reductase degradation protein 1(Hrd1) complex is highly conserved ERAD machinery (Nakatsukasa et al., 2014; Qi et al., 2017; Wu & Rapoport, 2018). Importantly, it is known that Sel1L is necessary for the ERAD activity of the Sel1L-Hrd1 complex (Qi et al., 2017; S. Sun et al., 2014; Wu & Rapoport, 2018). We generated SC-specific Sel1L deficient mice and demonstrated the essential role of the ERAD activity of the Sel1L-Hrd1 complex in mature SCs in adult mice, but not in actively myelinating SCs during development. Moreover, we showed that PERK inactivation had a minimal effect on actively myelinating SCs, but exacerbated ER stress and apoptosis of mature SCs in SC-specific Sel1L deficient mice. These findings imply the paradoxical role of the integrated UPR and ERAD in maintaining ER homeostasis in actively myelinating and mature SCs and their viability.
Materials and Methods
Mice.
CNP/Cre mice (Lappe-Siefke et al., 2003), Sel1Lloxp mice (S. Sun et al., 2014), and PERKloxp mice (Zhang et al., 2002) were on the C57BL/6J background. Sel1LloxP mice were crossed with CNP/Cre mice, and the resulting progeny were further crossed with Sel1LloxP mice to obtain Sel1LloxP/loxP; CNP/Cre mice and control mice, including CNP/Cre mice, Sel1LloxP/loxP mice and wild type mice. Moreover, Sel1LloxP mice were crossed with CNP/Cre mice, and the resulting progeny were crossed with PERKloxP mice to obtain Sel1LloxP; PERKloxP; CNP/Cre mice and Sel1LloxP; PERKloxP mice. Sel1LloxP; PERKloxP; CNP/Cre mice were further crossed with Sel1LloxP; PERKloxP mice to obtain Sel1LloxP/loxP; PERKloxP/loxP; CNP/Cre mice, Sel1LloxP/loxP; CNP/Cre mice, PERKloxP/loxP; CNP/Cre mice, and control mice, including Sel1LloxP/loxP mice, PERKloxP/loxP mice, and CNP/Cre mice. Genotypes were determined by PCR from DNA extracted from tail tips as described in previous papers (Lappe-Siefke et al., 2003; S. Sun et al., 2014; Zhang et al., 2002). To assess Cre-mediated recombination of floxed alleles in myelin-producing cells of these mice, genomic DNA was isolated from the indicated tissues and PCR was performed as described in previous papers (S. Sun et al., 2014; Zhang et al., 2002). We monitored mice daily to detect neurological phenotypes. All animal procedures were conducted in complete compliance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Minnesota.
Immunohistochemistry (IHC).
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in PBS. The tissues were removed, postfixed with paraformaldehyde for 2 h, cryopreserved in 30% sucrose for 72 h, embedded in optimal cutting temperature compound, and frozen on dry ice. Frozen sections were cut using a cryostat at a thickness of 10 μm. The sections were treated with −20°C acetone, blocked with PBS containing 10% goat serum and 0.1% Triton X-100, and incubated overnight with the primary antibody diluted in blocking solution. Fluorescein (Vector Laboratories, Burlingame, CA, anti-rabbit, RRID:AB_2336197) or Cy3 (Millipore, Temecula, CA, anti-mouse, RRID:AB_11213281, anti-rabbit, RRID:AB_92489) were used for detection. Immunohistochemical detection of S100 (1:200, Abcam, Cambridge, MA, RRID:AB_304258), Hrd1(1:200, Thermo Fisher Scientific, RRID:AB_2199832), cleaved caspase-3 (1:200, Cell signaling technology, RRID:AB_2070042), Krox-20 (1:200, Abcam, Cambridge, MA, RRID:AB_942051), c-Jun (1:200, Santa Cruz Biotechnology, RRID:AB_1121646), Proliferating cell nuclear antigen (PCNA,1:200, Santa Cruz Biotechnology, RRID:AB_628110), and P0 (1:200, Abcam, Cambridge, MA, RRID:AB_2144668) were performed. Fluorescent stained sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories) and visualized with a Zeiss Axioskop 2 fluorescence microscope or an Olympus FV1000 confocal microscope.
Toluidine blue staining and electron microscopy (EM) analysis.
Mice were anaesthetized and perfused with PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde. The sciatic nerve was processed and embedded. Thin sections were cut, stained with toluidine blue, and visualized with a Zeiss Axioskop 2 microscope as described in our previous papers (Y. Lin, Pang, et al., 2014; Stone et al., 2017). Moreover, ultrathin sections were cut, stained with uranyl acetate and lead citrate, and visualized with a Phillips CM12 transmission EM as described in our previous papers (Y. Lin, Pang, et al., 2014; Stone et al., 2018). We counted the total number of axons, the number of myelinated axons, and the number of demyelinated axons, and measured the diameter of axons and the thickness of myelin using the NIH ImageJ software (http://rsb.info.nih.gov/ij/; RRID:SCR_003070) as described in our previous papers (Y. Lin, Pang, et al., 2014; Stone et al., 2018). The luminal width of the rough ER sheets was measured by using the NIH ImageJ software.
Western blot analysis.
Sciatic nerves harvested from mice were rinsed in ice-cold PBS and were homogenized using a motorized homogenizer as previously described (W. Lin, Harding, Ron, & Popko, 2005; Stone et al., 2018). After incubating on ice for 15 min, the extracts were cleared by centrifugation at 14,000 rpm for 30 min twice. The protein content of each extract was determined by DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). The extracts (40 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with a primary antibody against Sel1L (1:500, Santa Cruz Biotechnology, catalog no. sc-377351), Hrd1(1:2000, Thermo Fisher Scientific, RRID:AB_2199832); CCAAT/enhancer-binding protein homologous protein (CHOP, 1:1000, Santa Cruz Biotechnology, RRID:AB_783507), eIF2α (1:1000, Santa Cruz Biotechnology, RRID:AB_640075); phosphorylated eIF2α (p-eIF2α, 1:1000, Cell Signaling Technology, Danvers, MA, RRID:AB_330951), ATF6α (1:500; B-Bridge, Santa Clara, CA, RRID:AB_10709801), ), c-Jun (1:500, Santa Cruz Biotechnology, RRID:AB_1121646), or β-actin (1:5000, Sigma-Aldrich, St. Louis, MO, USA, RRID:AB_476694), followed by an HRP-conjugated secondary antibody (1:1000, Vector Laboratories anti-mouse, RRID:AB_2336177; anti-rabbit, RIDD:AB_2336198). The chemiluminescent signal was detected using the ECL Detection Reagents (GE Healthcare Biosciences, Pittsburgh, PA). The intensity of the chemiluminescence signals of p-eIF2α, eIF2α, CHOP, cATF6α, and β-actin were quantified using the NIH ImageJ software.
Real-time PCR.
Sciatic nerves harvested from mice were rinsed in ice-cold PBS and snap frozen with dry ice. RNA was isolated from the Sciatic nerve using Trizol reagent (Invitrogen) and treated with DNaseI (Invitrogen) to eliminate genomic DNA. Reverse transcription was performed using the iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Real-time PCR for spliced XBP1 mRNA (XBP1s) was performed with LightCycler® 480 SYBR Green I Master (Roche Diagnostics Corporation, Indianapolis, IN) on the LightCycler 480 System (Roche Diagnostics Corporation) as described in a previous paper (Yoon et al., 2019).
Statistical analysis.
The sample size for each individual experiment was listed in the corresponding figure legend. All data were expressed as mean ± standard deviation (SD). Comparison between two groups was statistically evaluated by t-test using GraphPad Prism 6 (GraphPad Software, RRID:SCR_002798). Multiple comparisons were statistically evaluated by a 1-way ANOVA with a Tukeys posttest using GraphPad Prism 6. P values less than 0.05 were considered significant.
Results
Sel1L deficiency impaired ERAD and led to ER stress and activation of the UPR in SCs.
In mammals, there are over 16 ER membrane-associated E3 ligase complexes involved in the ERAD. Among them, the Sel1L-Hrd1 complex is highly conserved and the best-characterized ERAD machinery (Nakatsukasa et al., 2014; Qi et al., 2017; Wu & Rapoport, 2018). Hrd1 is an ER-resident E3 ligase, its transmembrane regions form a retrotranslocation channel to export the ER proteins and its cytoplasmic RING finger domain ubiquitinizes the proteins. The ERAD activity of Hrd1 is regulated by autoubiquitination of its RING finger domain (Baldridge & Rapoport, 2016; Schoebel et al., 2017; Wu & Rapoport, 2018). Sel1L, an ER transmembrane protein, interacts with Hrd1 and blocks Hrd1 autoubiquitination (Jeong et al., 2016; Qi et al., 2017; Vashistha, Neal, Singh, Carroll, & Hampton, 2016). A large number of studies have demonstrated that Sel1L is essential for Hrd1 stability and the ERAD activity of the SelL-Hrd1 complex (Ji et al., 2016; Kim et al., 2018; Qi et al., 2017; S. Sun et al., 2014). Thus, to explore the role of the ERAD in SCs, we generated a mouse model that allows for deletion of Sel1L specifically in myelin-producing cells, SCs in the PNS and oligodendrocytes in the central nervous system (CNS). Sel1LloxP mice (S. Sun et al., 2014) were crossed with CNP/Cre mice (Lappe-Siefke et al., 2003), and the resulting progeny were further crossed with Sel1LloxP mice to obtain Sel1LloxP/loxP; CNP/Cre mice (Sel1L cKO mice) and control mice, including CNP/Cre mice, Sel1LloxP/loxP mice and wild type mice. We found that the exon 6 of the Sel1L gene was deleted selectively in the CNS and PNS of Sel1L cKO mice, but not in other organs of Sel1L cKO mice or in any organs of control mice. We also found that young developing Sel1L cKO mice were asymptomatic; however, adult Sel1L cKO mice exhibited tremor, ataxia, and signs of muscle weakness starting around the age of 8 weeks (w). Moreover, rotarod tests showed that young developing Sel1L cKO mice displayed normal motor activity, but adult Sel1L cKO mice showed impairment of motor activity starting around the age of 8 w. Additionally, we showed that Sel1L deficiency caused the impairment of the ERAD activity of the Sel1L-Hrd1 complex and the UPR activation in oligodendrocytes, and led to adult-onset, progressive myelin thinning in the CNS starting around the age of 6 w (These results are reported in the parallel manuscript entitled “The integrated UPR and ERAD in oligodendrocytes maintains myelin thickness in adults by regulating myelin protein translation”).
Western blot analysis showed that the protein levels of Sel1L and Hrd1 were significantly decreased in the sciatic nerve of 3-w-old Sel1L cKO mice compared to control mice (Figure 1A, B). Moreover, Hrd1 and S100 (a marker for SCs) double immunostaining showed that the immunoreactivity of Hrd1 was dramatically decreased in SCs in the sciatic nerve of 3-w-old Sel1L cKO mice compared to control mice (Figure 1C). These data demonstrate that Sel1L deficiency in SCs leads to elimination of Hrd1 protein. Moreover, we determined whether elimination of the Sel1L-Hrd1 complex disturbs ER homeostasis and leads to ER stress and activation of the UPR in SCs. Western blot analysis showed the levels of the UPR effectors p-eIF2α, CHOP, and cATF6α were significantly increased in the sciatic nerve of 3, 6 and 16-w-old Sel1L cKO mice compared to control mice (Figure 1D, E, Figure 6A, B, D, E). Real-time PCR analysis showed that the level of spliced XBP1 (XBP1s) mRNA was significantly increased in the sciatic nerve of 3, 6 and 16-w-old Sel1L cKO mice compared to control mice (Figure 1F, Figure 6 C, F ). Thus, these data demonstrate that Sel1L deficiency in SCs diminishes the ERAD activity of the Sel1L-Hrd1 complex, and results in ER stress and activation of the UPR in SCs.
Figure 1. Sel1L deficiency led to elimination of Hrd1 and activation of the UPR in SCs.
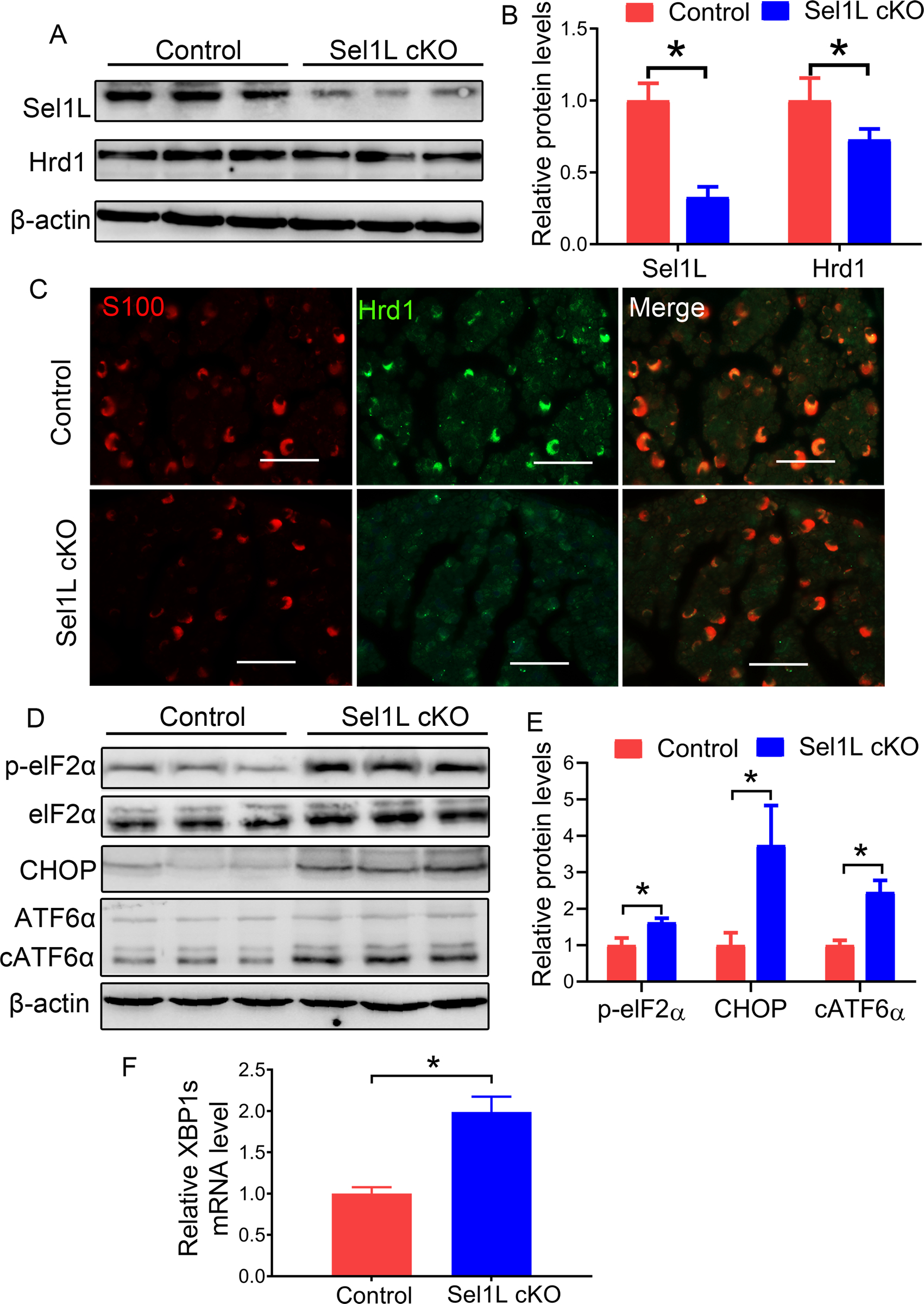
A, B. Western blot analysis showed that the levels of Sel1L and Hrd1 were significantly decreased in the sciatic nerve of 3-w-old Sel1L cKO mice compared to control mice. C. S100 and Hrd1 double labeling revealed a dramatic reduction of Hrd1 immunoreactivity in SCs in the sciatic nerve of 3-w-old Sel1L cKO mice compared to control mice. D, E. Western blot analysis showed the elevated levels of p-eIF2α, CHOP, and cATF6α in the sciatic nerve of 3-w-old Sel1L cKO mice compared to control mice. F. Real time-PCR analysis revealed that the level of XBP1s mRNA was significantly increased in the sciatic nerve of 3-w-old Sel1L cKO mice compared to control mice. Scale bars: 50 μm. N = 4 animals. Statistical analyses were done with a t-test. Error bars represent SD, P < 0.05.
Figure 6. The activity of the PERK pathway was abrogated in Double cKO mice.
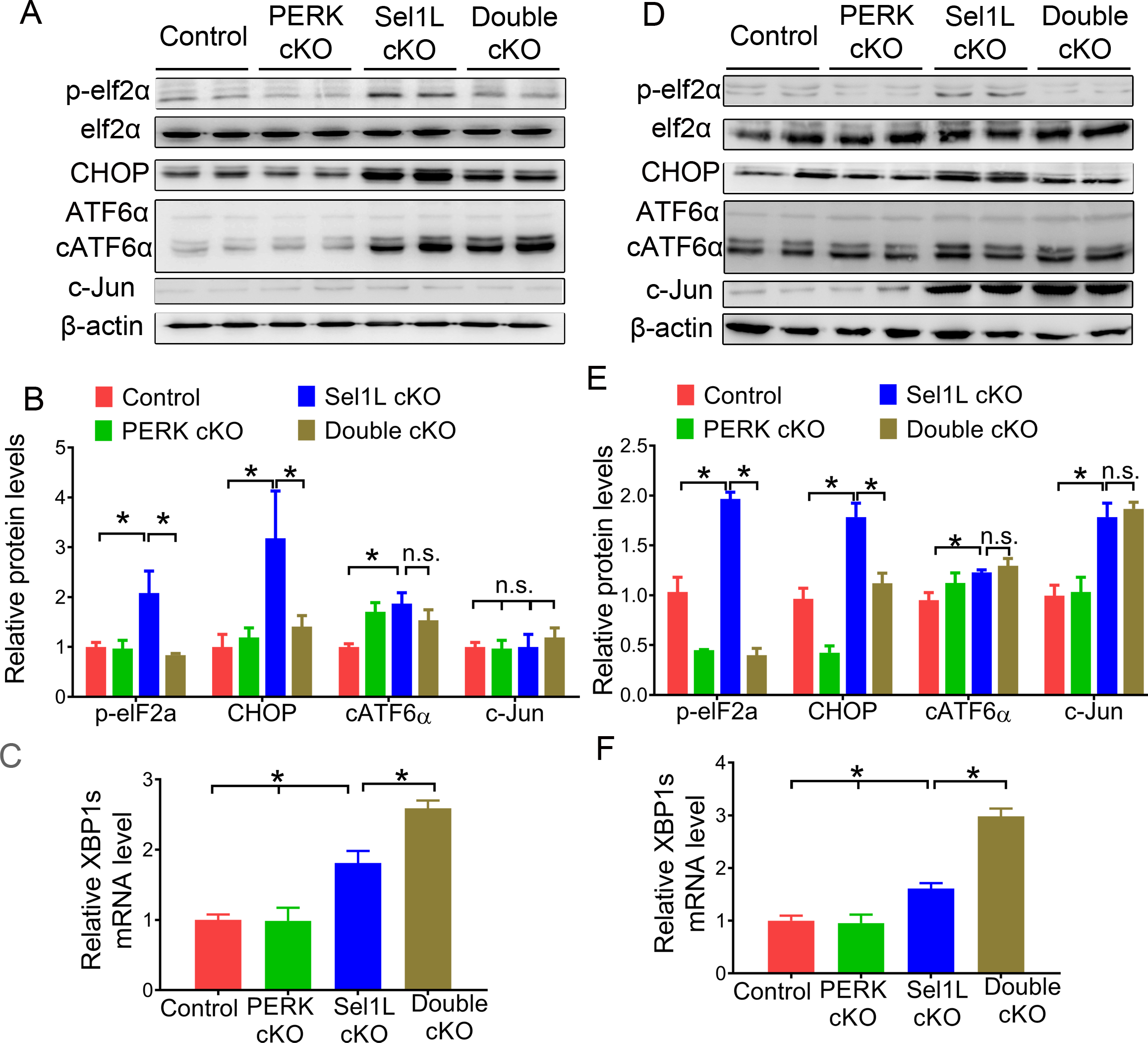
A, B. Western blot analysis for p-eIF2α, eIF2α, CHOP, ATF6α, cATF6α, and c-Jun in the sciatic nerve of 6-w-old Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice. C. Real time-PCR analysis for XBP1s mRNA in the sciatic nerve of 6-w-old Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice. D, E. Western blot analysis for p-eIF2α, eIF2α, CHOP, ATF6α, cATF6α, and c-Jun in the sciatic nerve of 16-w-old Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice. F. Real time-PCR analysis for XBP1s mRNA in the sciatic nerve of 16-w-old Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice. N = 4 animals. Statistical analyses were done with a 1-way ANOVA with a Tukeys posttest. Error bars represent SD, n.s. not significant, P < 0.05.
Sel1L deficiency did not affect the viability or function of actively myelinating SCs in young developing mice.
We determined the effects of Sel1L deficiency on SCs during development. P0 immunostaining showed that the immunoreactivity of P0 was comparable in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice (Figure 2A, B, E, F, I, J, M, N). Toluidine blue staining showed a comparable degree of myelination in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice (Figure 2C, D, G, H, K, L, O, P). EM analysis showed that Sel1L deficiency in SCs did not change the percentage of myelinated axons, the thickness of myelin, or the diameter of axons in the sciatic nerve of 3, 5, 6, and 10-w-old mice (Figure 3A–L, S, T). S100 immunostaining showed a comparable number of SCs in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice (Figure 4A–D, G–J, Y). Similarly, Krox-20 (another maker for SCs) and c-Jun double staining showed that both Krox-20 positive SC numbers and c-Jun positive cell numbers were comparable in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice (Figure 4M–P, S–V, Z, AA). Western blot analysis showed that the level of c-Jun was comparable in the sciatic nerve of 6-w-old Sel1L cKO mice and control mice (Figure 6A, B). Moreover, PCNA (a marker for proliferating cells) and S100 double immunostaining showed that PCNA and S100 double positive cells were undetectable in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice (supplemental Fig 1 A–D, G–J, M). Interestingly, EM analysis showed that Sel1L deficiency did not alter the morphology of SCs or the morphology of the ER in SCs in the sciatic nerve of 3, 5, 6, and 10-w-old mice (Figure 5A–D, G–J, M). Thus, these results demonstrate that both SCs and myelin appear normal in the PNS of Sel1L cKO mice until the age of 10 w. Taken together, these data suggest that the impaired ERAD activity of the Sel1L-Hrd1 complex causes modest ER stress in actively myelinating SCs, but does not affect their viability or function in young developing mice.
Figure 2. Sel1L cKO mice displayed adult-onset demyelination in the PNS.
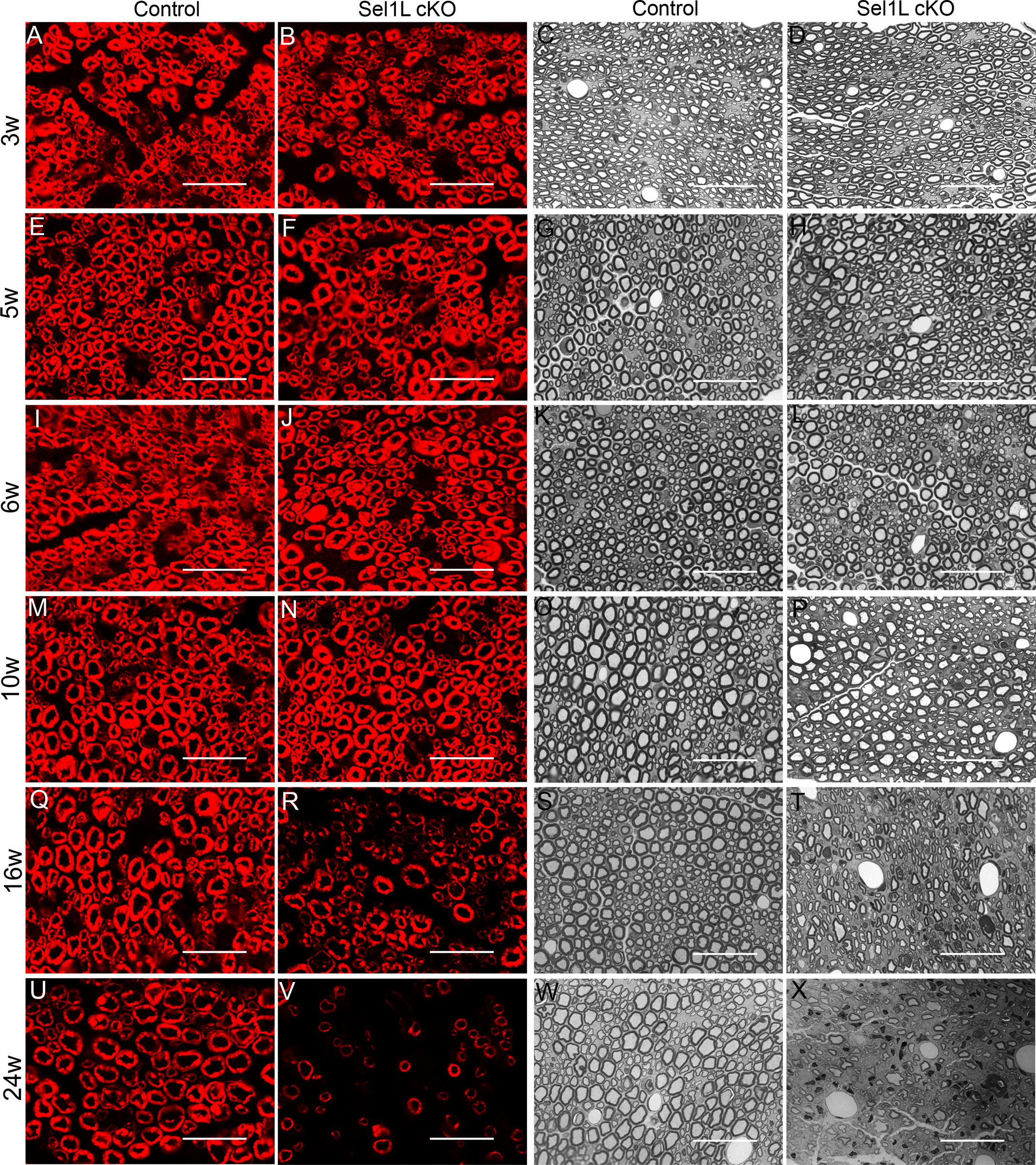
A, B, E, F, I, J, M, N. P0 immunostaining showed comparable P0 immunoreactivity in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice. Q, R, U, V. P0 immunostaining showed that P0 immunoreactivity was noticeably reduced in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice. C, D, G, H, K, L, O, P. Toluidine blue staining showed that the degree of myelination was comparable in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice. S, T, W, X. Toluidine blue staining showed noticeable myelin loss in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice. Scale bars: 20 μm. N = 4 animals.
Figure 3. EM analysis revealed adult-onset demyelination in the PNS of Sel1L cKO mice.
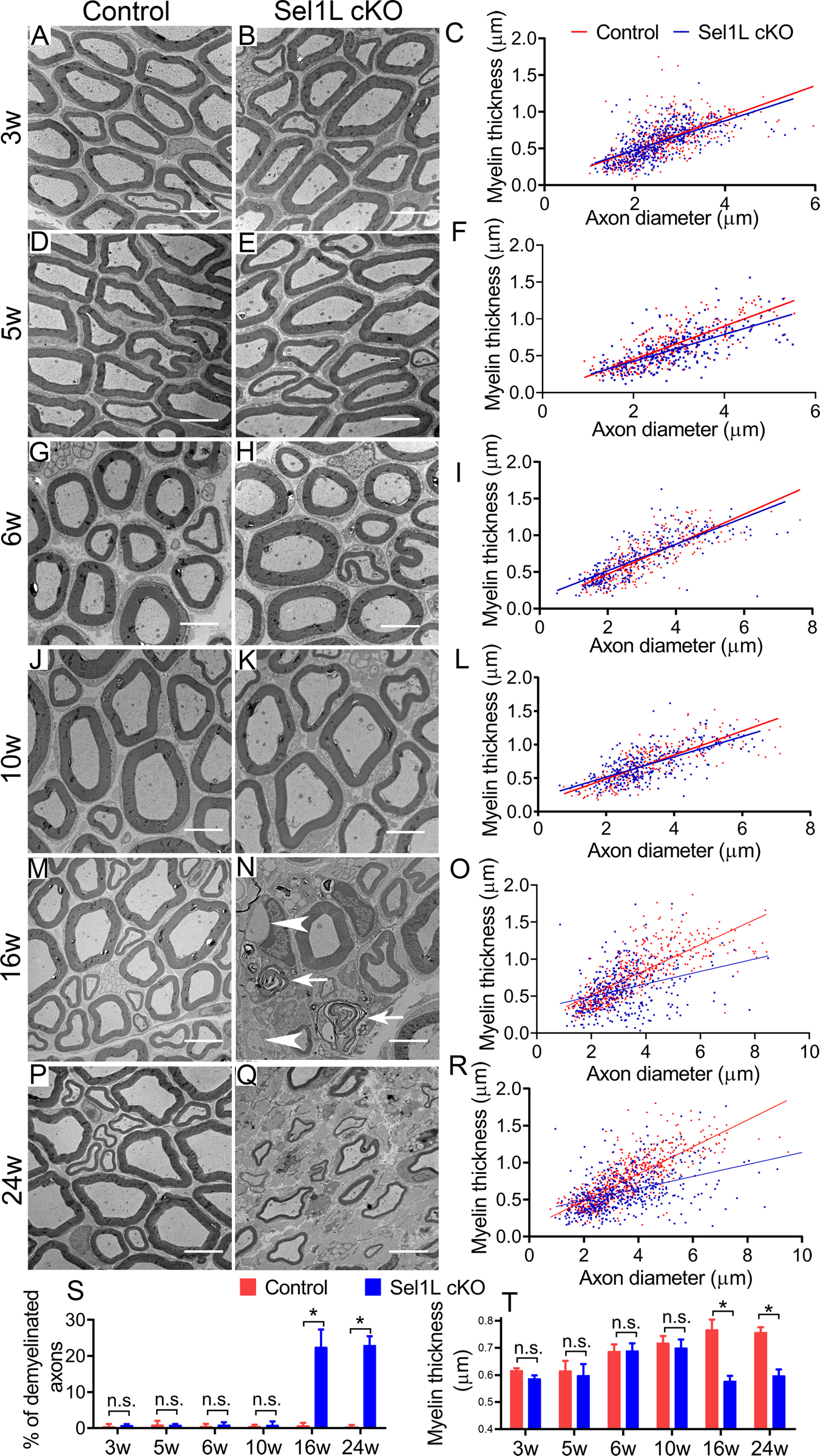
A-L, S, T. EM analysis showed that the percentage of myelinated axons, the thickness of myelin, and the diameter of axons were comparable in the sciatic nerve of 3, 5, 6, 10-w-old Sel1L cKO mice and control mice. M-R, S, T. EM analysis showed that there were a significant increase in the percentage of demyelinated axons (arrowhead, naked axon; arrow, loose myelin) and a significant decrease in the thickness of myelin in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice. Scale bars: 5 μm. N = 4 animals. Statistical analyses were done with a t-test. Error bars represent SD, n.s. not significant, P < 0.05.
Figure 4. Sel1L cKO mice displayed adult-onset SC loss in the PNS. A-D, G-J, Y.
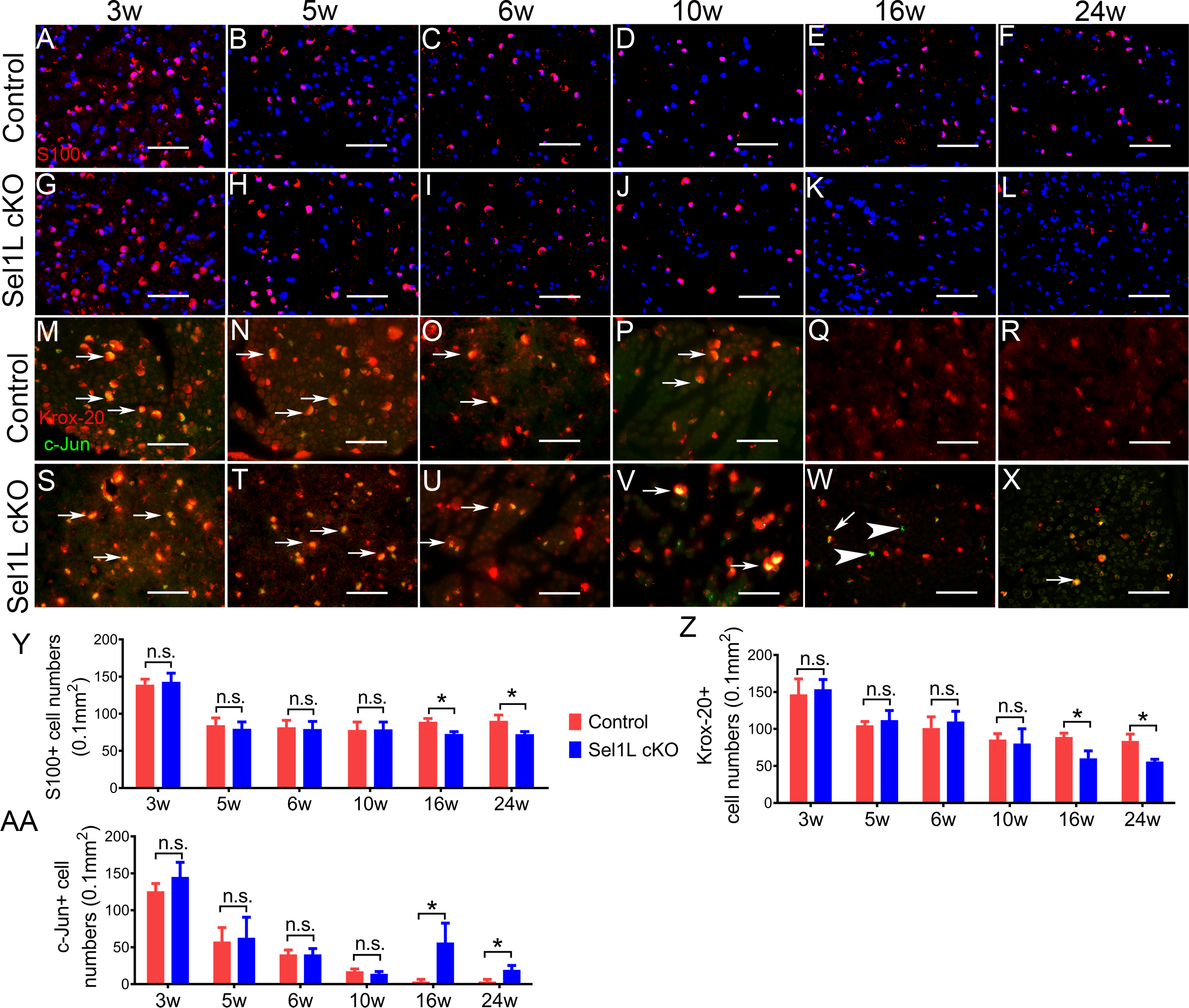
S100 immunostaining showed a comparable number of SCs in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice. E, F, K, L, Y S100 immunostaining showed that the number of SCs was significantly reduced in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice. M-P, S-V, Z, AA. Krox-20 and c-Jun double immunostaining showed that both Krox-20 positive cell numbers and c-Jun Krox-20 positive cell numbers were comparable in the sciatic nerve of 3, 5, 6, and 10-w-old Sel1L cKO mice and control mice. Arrow, Krox-20 and c-Jun double positive cells. Q, R, W, X, Z, AA. Krox-20 and c-Jun double immunostaining showed that the number of Krox-20 positive was significantly decreased in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice, and that the number of c-Jun positive cells was significantly increased in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice. Arrow, Krox-20 and c-Jun double positive cells; arrowhead, c-Jun positive cells. Scale bars: A-L, 50 μm. N = 4 animals. Statistical analyses were done with a t-test. Error bars represent SD, n.s. not significant, P < 0.05.
Figure 5. Sel1L deficiency did not alter the ER morphology in SCs in the PNS.
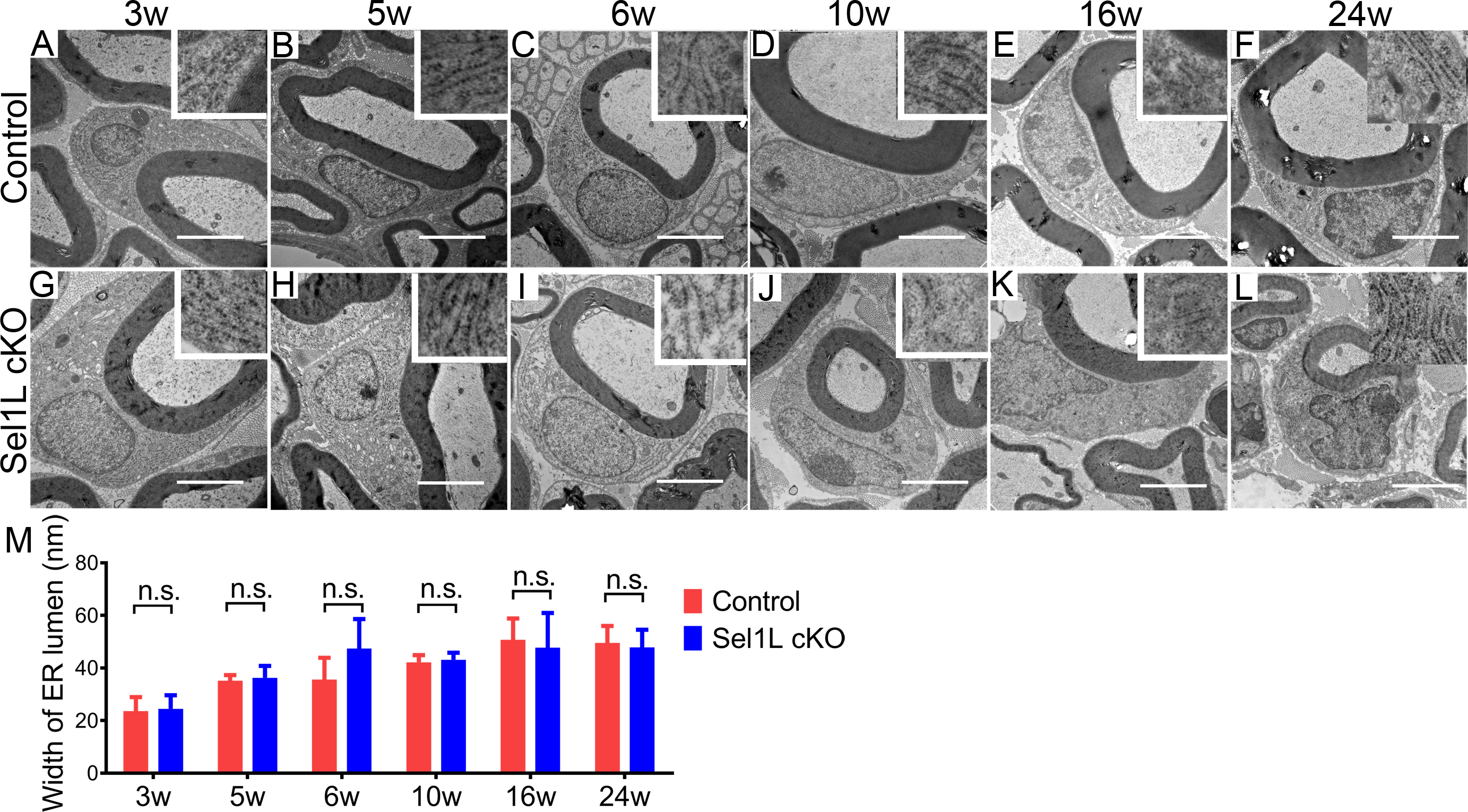
A-M. EM analysis showed that Sel1L deficiency did not alter the morphology of SCs or the morphology of the ER (insets) in SCs in the sciatic nerve of 3, 5, 6, 10, 16 and 24-w-old mice. Scale bars: 1.0 μm. N = 4 animals. Statistical analyses were done with a t-test. Error bars represent SD, n.s. not significant.
Sel1L deficiency impaired the viability and function of mature SCs in adult mice.
We further determined the effects of Sel1L deficiency on SCs in adult mice. P0 immunostaining showed that the immunoreactivity of P0 was markedly decreased in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice (Figure 2Q, R, U, V). Toluidine blue staining showed noticeable myelin loss in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice (Figure 2S, T, W, X). EM analysis showed that Sel1L deficiency in SCs significantly increased the percentage of demyelinated axons and reduced the thickness of myelin in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice (Figure 3M–T). S100 immunostaining showed that the number of SCs was significantly reduced in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice (Figure 4E, F, K, L, Y). Importantly, S100 and cleaved-caspase 3 (a marker of apoptosis) double immunostaining showed that none of S100 positive SCs was positive for cleaved-caspase 3 in the sciatic nerve of 16-w-old control mice; however, there were a few cleaved-caspase 3 positive SCs in Sel1L cKO mice (Fig 10 A, C, N). Similarly, Krox-20 and c-Jun double immunostaining showed that the number of Krox-20 positive SCs was significantly reduced in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice, and that the number of c-Jun positive cells was significantly increased in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice (Figure 4Q, R, W, X, Z, AA). Western blot analysis showed that the level of c-Jun was significantly increased in the sciatic nerve of 16-w-old Sel1L cKO mice compared to control mice (Figure 6D, E). Moreover, PCNA and S100 double immunostaining showed that the number of PCNA and S100 double positive cells was significantly increased in the sciatic nerve of 16 and 24-w-old Sel1L cKO mice compared to control mice (Supplemental Fig 1 E, F, K, L, M). Nevertheless, EM analysis showed that Sel1L deficiency did not alter the morphology of SCs or the morphology of the ER in SCs in the sciatic nerve of 16 and 24-w-old mice (Figure 5E, F, K–M). Collectively, these data suggest that the impaired ERAD activity of the Sel1L-Hrd1 complex causes ER stress in mature SCs and leads to cell apoptosis and subsequent demyelination in the adult PNS.
Figure 10. PERK inactivation exacerbated SC loss in the PNS of adult Sel1L cKO mice.
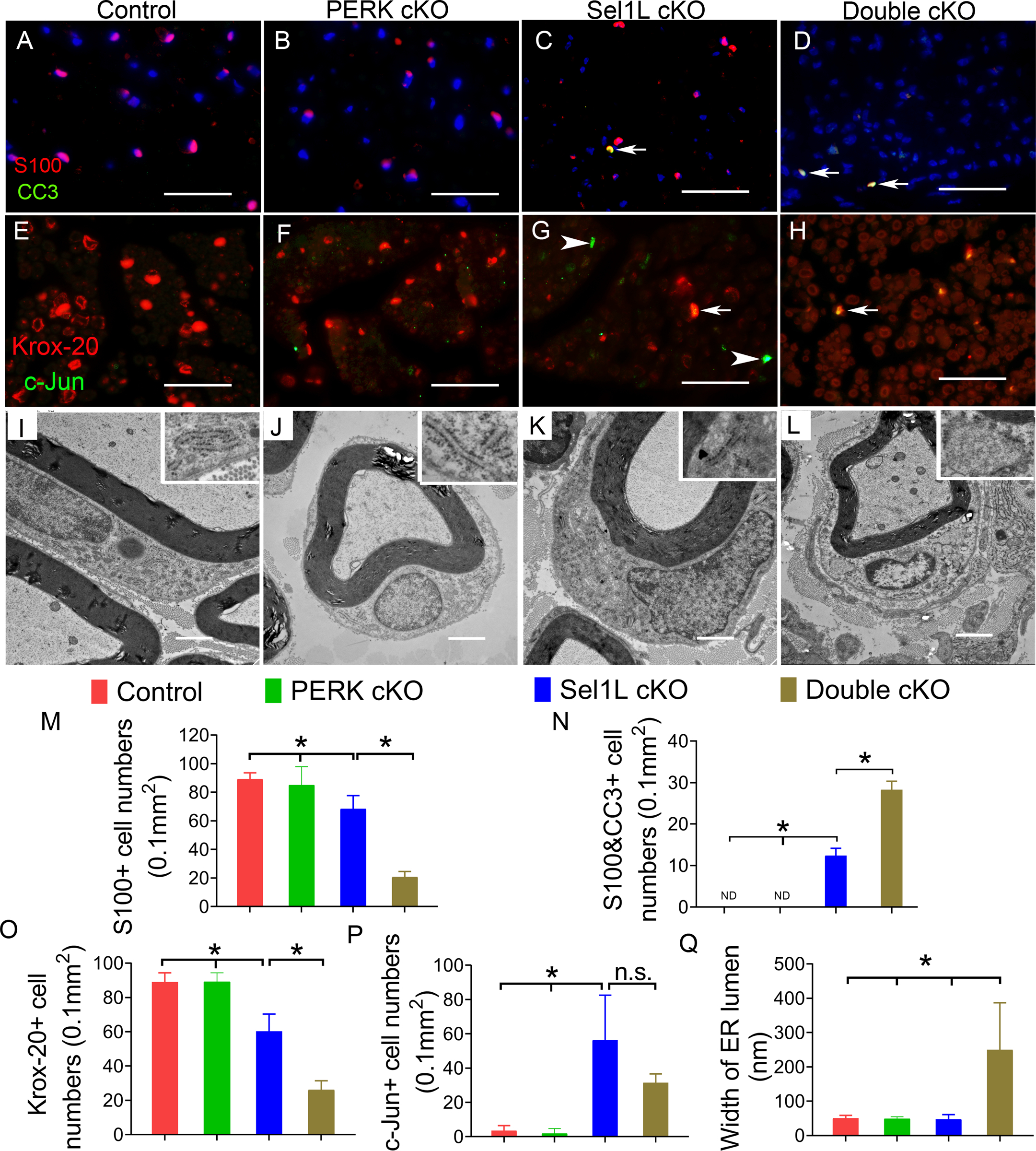
A-D, M, N. S100 and cleaved caspase-3 (CC3) double immunostaining showed that the number of SCs was significantly reduced in the sciatic nerve of 16-w-old Sel1L cKO mice compared to PERK cKO mice and control mice, and was further reduced in Double cKO mice. Importantly, the number of cleaved-caspase 3 positive SCs (arrow) was significantly increased in Sel1L cKO mice compared to PERK cKO mice and control mice, and was further increased in Double cKO mice. ND, undetectable. E-H, O, P. Krox-20 and c-Jun double immunostaining showed that the number of Krox-20 positive cells was significantly decreased in the sciatic nerve of 16-w-old Sel1L cKO mice compared to PERK cKO mice and control mice, and was further decreased in Double cKO mice. The number of c-Jun positive cells in the sciatic nerve of 16-w-old Sel1L cKO mice was significantly increased compared to PERK cKO mice and control mice, but was not significantly changed compared to Double cKO mice. Arrow, Krox-20 and c-Jun double positive cells; arrowhead, c-Jun positive cells. I-L, Q. EM analysis showed that neither Sel1L deficiency nor PERK inactivation alter the morphology of SCs or the morphology of the ER (insets) in SCs in the sciatic nerve of 16-w-old mice. Importantly, double deficiency of Sel1L and PERK led to significantly enlarged ER lumen (insets) in SCs in the sciatic nerve of 16-w-old mice. Scale bars: A-D, 50 μm; G-J, 1.0 μm. N = 4 animals. Statistical analyses were done with a 1-way ANOVA with a Tukeys posttest. Error bars represent SD, P < 0.05.
PERK inactivation in SCs did not influence developmental myelination, but exacerbated demyelination in the adult PNS in Sel1L cKO mice.
The elevated levels of p-eIF2α and CHOP in the PNS suggest activation of the PERK-eIF2α pathway in SCs of Sel1L cKO mice. It is known that activation of the PERK-eIF2α pathway restores ER homeostasis in ER-stressed cells by inhibiting protein translation and facilitating protein degradation, and promotes cell survival by stimulating certain cytoprotective gene expression (Han & Kaufman, 2017; Marciniak & Ron, 2006; Walter & Ron, 2011). Moreover, a number of studies show that the PERK-eIF2α pathway modulates the viability and function of SCs in animal models of PNS myelin disorders (D’Antonio et al., 2013; Musner et al., 2016; Pennuto et al., 2008; Sidoli et al., 2016). Thus, we exploited mice with PERK inactivation specifically in myelin-producing cells to determine whether Sel1L deficiency causes mature SC apoptotic by inducing ER stress. A recent study has demonstrated PERK inactivation specifically in oligodendrocytes in the CNS and SCs in the PNS in PERKloxP/loxP; CNP/Cre mice (PERK cKO mice), which are phenotypically normal and do not display any abnormalities of myelin in the CNS and PNS (Stone et al., 2020). PERK cKO mice were crossed with Sel1L cKO mice to generate Sel1LloxP/loxP; PERKloxP/loxP; CNP/Cre mice (Double cKO mice), Sel1L cKO mice, PERK cKO mice, and control mice. We found that young developing Double cKO mice were asymptomatic, and that adult Double cKO mice exhibited noticeably milder neurological phenotypes than Sel1L cKO mice. Rotarod test showed that motor activity in young developing Double cKO mice appeared normal, and that the decline of motor coordination in adult Double cKO mice was significantly milder than in age-matched Sel1L cKO mice. Moreover, we found that PERK inactivation in oligodendrocytes rescued progressive myelin thinning and oligodendrocyte death in the CNS of adult Sel1L cKO mice (These results are reported in the parallel manuscript entitled “The integrated UPR and ERAD in oligodendrocytes maintains myelin thickness in adults by regulating myelin protein translation”).
As expected, western blot analysis showed that the elevated levels of p-eIF2α and CHOP in the sciatic nerve of 6 and 16-w-old Sel1L cKO mice were abrogated by PERK inactivation (Figure 6A, B, D, E). However, PERK inactivation did not alter the elevated level of cATF6α in the sciatic nerve of 6 and 16-w-old Sel1L cKO mice (Figure 6A, B, D, E). Interestingly, real-time PCR analysis showed that PERK inactivation significantly increased the level of XBP1s mRNA in the sciatic nerve of 6 and 16-w-old Sel1L cKO mice (Figure 6C, F). These data demonstrate that PERK deficiency abrogates activation of the PERK-eIF2α pathway in the ER-stressed SCs in the PNS of Sel1L cKO mice.
We examined the histopathology of the sciatic nerve of these four groups of mice at the age of 6 w. In accordance with previous studies (Musner et al., 2016; Sidoli et al., 2016; Stone et al., 2020), we found that PERK inactivation in SCs alone did not change SC numbers or myelin integrity in the PNS (Figure 7, 8, 9, 10). P0 immunostaining showed that the immunoreactivity of P0 was comparable in the sciatic nerve of Double cKO mice, Sel1L cKO, PERK cKO mice, and control mice (Figure 7A–D). Toluidine blue staining showed a comparable degree of myelination in the sciatic nerve of Double cKO mice, Sel1L cKO, PERK cKO mice, and control mice (Figure 7E–H). EM analysis showed that the percentage of myelinated axons, the thickness of myelin, and the diameter of axons were comparable in the sciatic nerve of Double cKO mice, Sel1L cKO, PERK cKO mice, and control mice (Figure 7I–O). S100 immunostaining showed a comparable number of SCs in the sciatic nerve of Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice (Figure 8A–D, M). Similarly, Krox-20 and c-Jun double staining showed that both Krox-20 positive SC numbers and c-Jun positive cell numbers were comparable in the sciatic nerve of Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice (Figure 8E–H, N, O). Western blot analysis showed a comparable level of c-Jun in the sciatic nerve of Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice (Figure 6A, B). Moreover, PCNA and S100 double immunostaining showed that PCNA and S100 double positive cells were undetectable in the sciatic nerve of Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice (Supplemental Figure 2). Interestingly, EM analysis showed normal morphology of SCs in the sciatic nerve of Double cKO mice, Sel1L cKO mice, PERK cKO mice, and control mice (Figure 8I–L). Additionally, the morphology of the ER in SCs in the sciatic nerve of Double cKO mice appeared normal compared to Sel1L cKO mice, PERK cKO mice, and control mice (Figure 8I–L, P). These results suggest that PERK inactivation does not affect viability or function of actively myelinating SCs in Sel1L cKO mice during development. Thus, these data demonstrate the minimal role of the integrated UPR and ERAD in maintaining ER homeostasis and the viability and function of active myelinating SCs during development.
Figure 7. PERK inactivation in SCs did not affect developmental myelination in the PNS of Sel1L cKO mice. A-D.
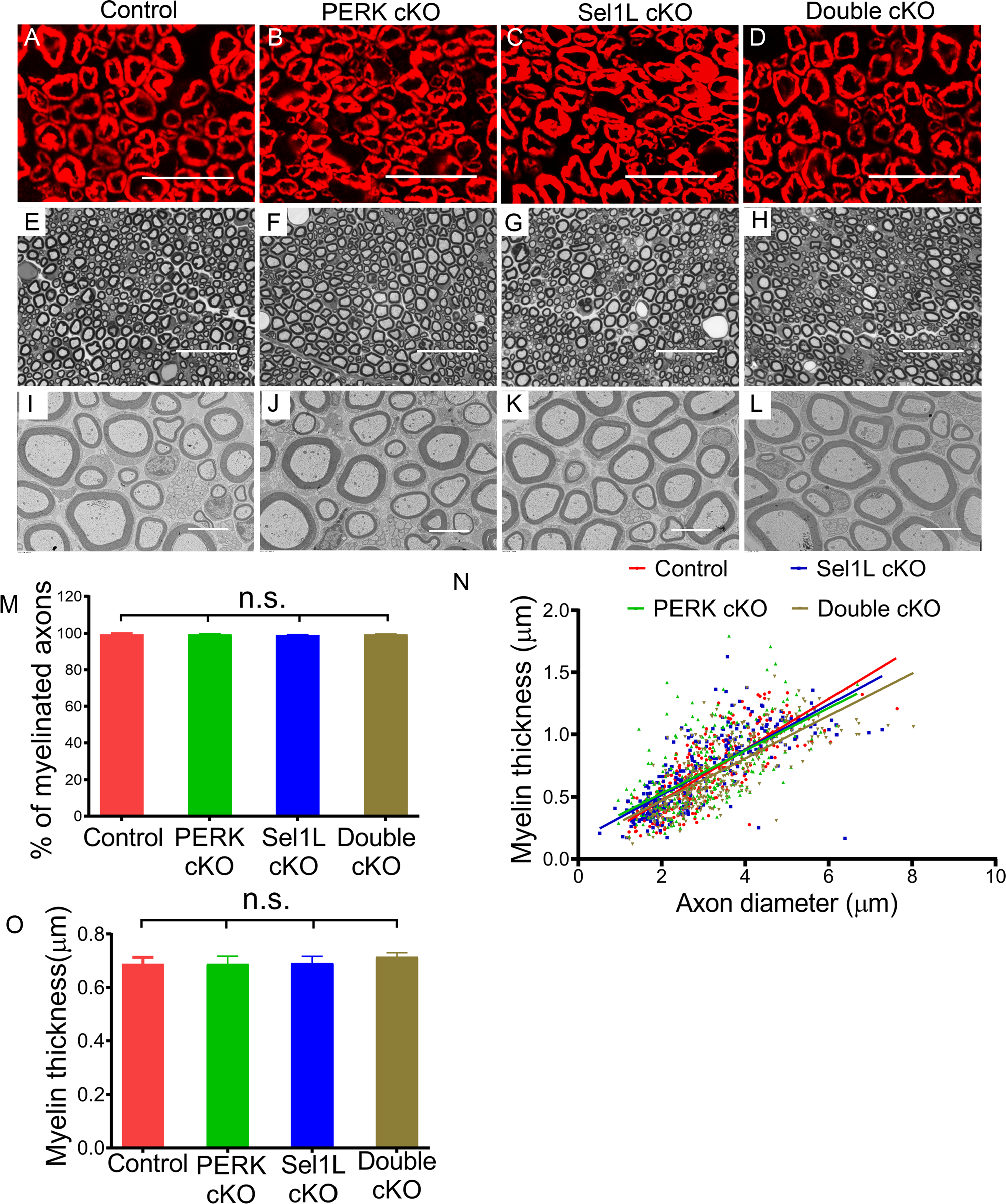
P0 immunostaining showed a comparable P0 immunoreactivity in the sciatic nerve of 6-w-old control mice, PERK cKO mice, Sel1L cKO mice, and Double cKO mice. E-H. Toluidine blue staining showed a comparable degree of myelination in the sciatic nerve of 6-w-old control mice, PERK cKO mice, Sel1L cKO mice, and Double cKO mice. I-O. EM analysis showed that the percentage of myelinated axons and the thickness of myelin were comparable in the sciatic nerve of 6-w-old control mice, PERK cKO mice, Sel1L cKO mice, and Double cKO mice. Scale bars: A-H, 20 μm; I-L, 5 μm. N = 4 animals. Statistical analyses were done with a 1-way ANOVA with a Tukeys posttest. Error bars represent SD, n.s. not significant.
Figure 8. PERK inactivation had no effect on SCs in the PNS of Sel1L cKO mice during development.
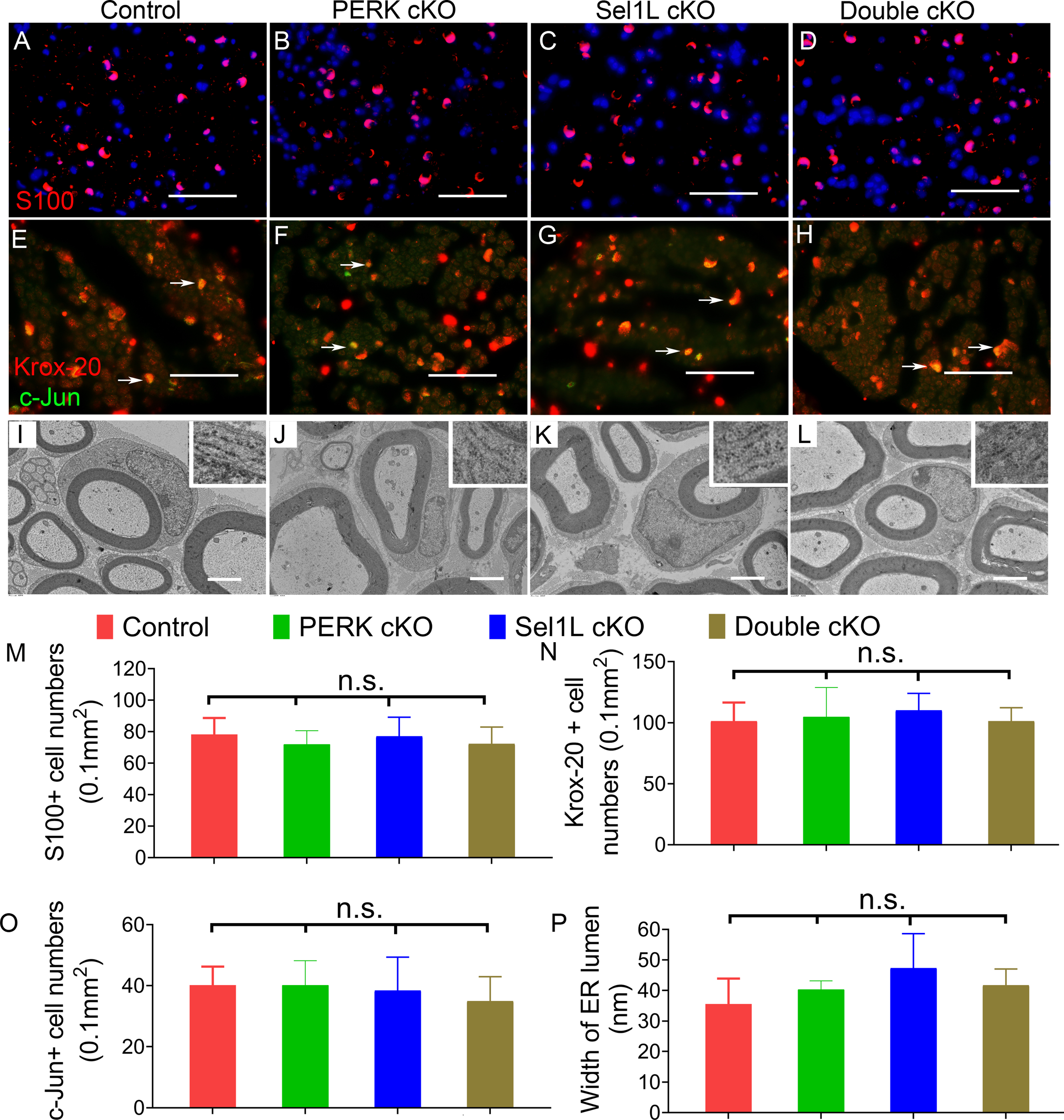
A-D, M. S100 immunostaining showed a comparable number of SCs in the sciatic nerve of 6-w-old control mice, PERK cKO mice, Sel1L cKO mice, and Double cKO mice. E-H, N, O. Krox-20 and c-Jun double immunostaining showed that both Krox-20 positive cell numbers and c-Jun positive cell numbers were comparable in the sciatic nerve of 6-w-old control mice, PERK cKO mice, Sel1L cKO mice, and Double cKO mice. Arrow, Krox-20 and c-Jun double positive cells. I-L, P. EM analysis showed normal morphology of SCs and normal morphology of the ER (insets) in SCs in the sciatic nerve of 6-w-old control mice, PERK cKO mice, Sel1L cKO mice, and Double cKO mice. Scale bars: A-D, 50 μm; F-I, 2.0 μm. N = 4 animals. Statistical analyses were done with a 1-way ANOVA with a Tukeys posttest. Error bars represent SD, n.s. not significant.
Figure 9. PERK inactivation exacerbated demyelination in the PNS of adult Sel1L cKO mice.
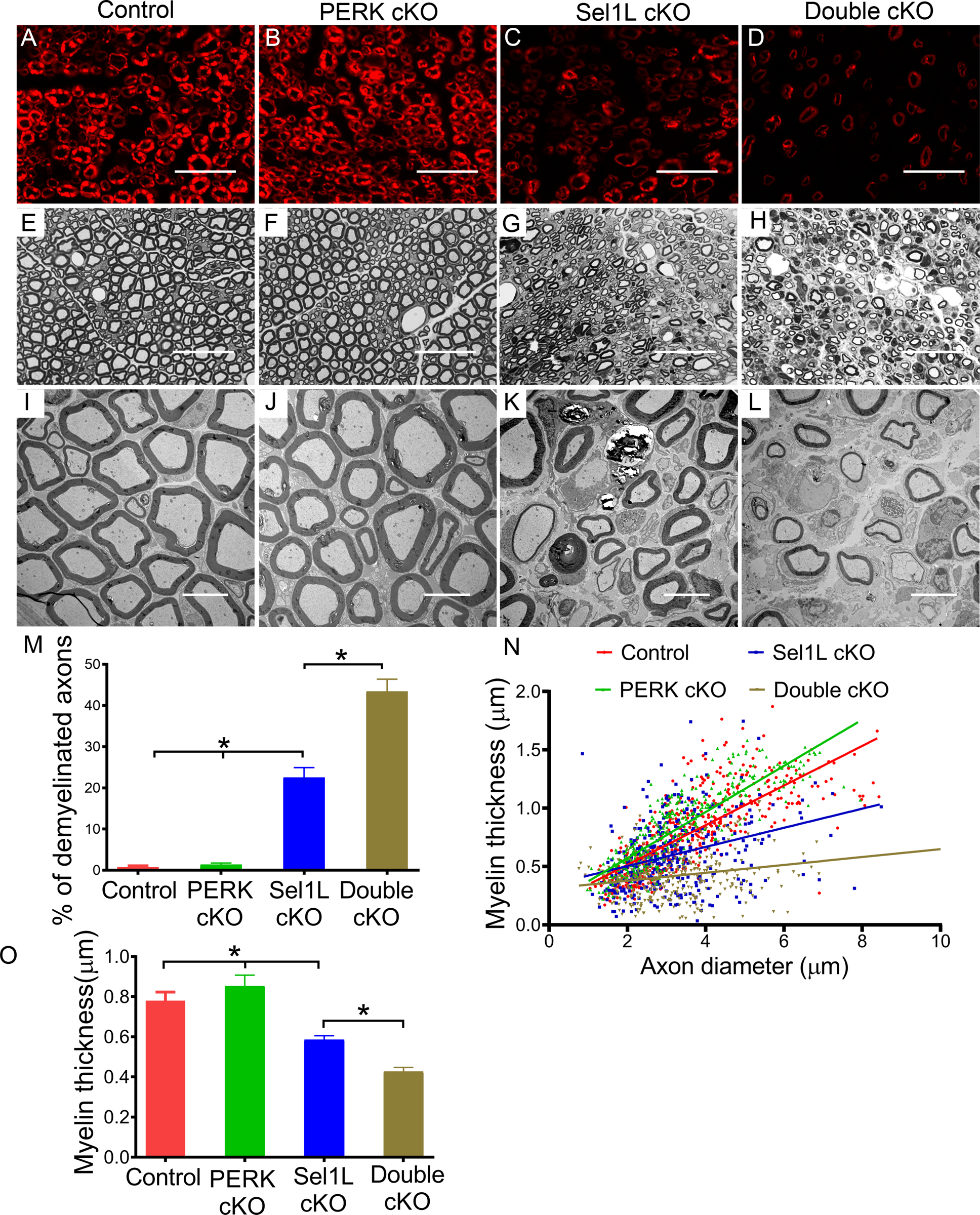
A-D. P0 immunostaining showed that P0 immunoreactivity was noticeably reduced in the sciatic nerve of 16-w-old Sel1L cKO mice compared to PERK cKO mice and control mice, and was further reduced in Double cKO mice. E-H. Toluidine blue staining showed noticeable myelin loss in the sciatic nerve of 16-w-old Sel1L cKO mice compared to PERK cKO mice and control mice, which was exacerbated in Double cKO mice. I-O. EM analysis showed that Sel1L deficiency in SCs significantly increased the number of demyelinated axons and decreased the thickness of myelin in the sciatic nerve of 16-w-old mice, which was exacerbated by PERK inactivation. Scale bars: A-H, 20 μm; I-L, 5 μm. N = 4 animals. Statistical analyses were done with a 1-way ANOVA with a Tukeys posttest. Error bars represent SD, P < 0.05.
Next, we examined the histopathology of the sciatic nerve of these four groups of mice at the age of 16 w. P0 immunostaining showed that the immunoreactivity of P0 was further decreased in the sciatic nerve of Double cKO mice compared to Sel1L cKO mice (Figure 9A–D). Toluidine blue staining showed that PERK inactivation noticeably exacerbated myelin loss in the sciatic nerve of Sel1L cKO mice (Figure 9E–H). EM analysis showed that PERK inactivation further increased the percentage of demyelinated axons and decreased the thickness of myelin in the sciatic nerve of Sel1L cKO mice (Figure 9I–O). S100 immunostaining showed that PERK inactivation further reduced the number of SCs in the sciatic nerve of Sel1L cKO mice (Figure 10A–D, M). Accordingly, S100 and cleaved-caspase 3 double immunostaining showed that the number of cleaved-caspase 3 positive SCs was significantly increased in Double cKO mice compared to Sel1L cKO mice (Figure 10A–D, N). Krox-20 and c-Jun double immunostaining showed that the number of Krox-20 positive SCs was further reduced in the sciatic nerve of Double cKO mice compared to Sel1L cKO mice; however, the number of c-Jun positive cells was not significantly changed in the sciatic nerve of Double cKO mice compared to Sel1L cKO mice (Figure 10E–H, O, P). Western blot analysis showed that the level of c-Jun was not significantly changed in the sciatic nerve of Double cKO mice compared to Sel1L cKO mice (Fig 6D, E). PCNA and S100 double immunostaining showed that PERK inactivation in SCs did not significantly change the number of PCNA and S100 double positive cells in the sciatic nerve of Sel1L cKO mice (Supplemental Figure 3). Importantly, EM analysis revealed that the ER lumen were significantly enlarged in SCs in the sciatic nerve of Double cKO mice compared to Sel1L cKO mice, PERK cKO mice, and control mice (Figure 10I–L, Q). These results suggest that PERK inactivation aggravates ER stress in mature SCs and results in exacerbated SC apoptosis and demyelination in the PNS of adult Sel1L cKO mice. Thus, these data demonstrate the essential role of the integrated UPR and ERAD in maintaining ER homeostasis and the viability and function of mature SCs in adults. On the other hand, the minimal alteration of c-Jun level in the sciatic nerve of 16-w-old Double cKO mice vs Sel1L cKO mice suggests that c-Jun is not a major player involved in ER stress-induced SC apoptosis and demyelination in the adult PNS of these mice.
Discussion
Schwann cells (SCs) must produce an enormous amount of myelin proteins via the ER to assemble myelin sheath or maintain myelin structure homeostasis. Maintaining ER homeostasis is necessary for the production of myelin proteins and the myelinating function of SCs (Clayton & Popko, 2016; W. Lin & Popko, 2009; Volpi et al., 2017). However, to date, attempts to understand the mechanisms by which SCs maintain ER homeostasis have been unsuccessful. ERAD is responsible for recognition and degradation of unfolded or misfolded proteins in the ER. Interestingly, a recent study showed that deletion of Derlin-2 (a component of the Sel1L-Hrd1 complex) in SCs moderately reduces myelin thickness in the PNS of naïve adult mice and significantly increases demyelinated axons in the PNS of mice carrying P0 deleted for S63 (P0S63del mice), an animal model of CMT1B (Volpi et al., 2019). These data raise the possibility that ERAD is major player in maintaining ER homeostasis in SCs. The SelL-Hrd1 complex is highly conserved and the best-characterized ERAD machinery (Nakatsukasa et al., 2014; Qi et al., 2017; Wu & Rapoport, 2018). Importantly, it is known that Sel1L is necessary for the ERAD activity of the Sel1L-Hrd1 complex. Thus, to explore the role of ERAD in maintaining ER homeostasis in SCs, we generated Sel1L cKO mice that allowed for deletion of Sel1L specifically in myelin-producing cells. As expected, we showed that Sel1L deficiency led to elimination of Hrd1 in SCs. Interestingly, we found that the impaired ERAD activity of the SelL-Hrd1 complex resulted in ER stress and activation of the UPR in SCs.
It is known that actively myelinating SCs during development produce a far larger amount of myelin proteins than adult mature SCs. The dogma is that actively myelinating SCs are more sensitive to disruption of ER homeostasis than mature SCs (Clayton & Popko, 2016; W. Lin & Popko, 2009; W. Lin & Stone, 2020; Volpi et al., 2017). Although Sel1L deficiency disrupted ER homeostasis and led to ER stress in actively myelinating SCs during development, young developing Sel1L cKO mice did not display any evidence of myelin abnormalities in the PNS, including no neurological phenotypes, no behavioral abnormalities, and normal appearance of SCs and myelin. More dramatically, young developing Double cKO mice with double deletion of Sel1L and PERK in SCs also did not show any abnormalities of SCs or myelin in the PNS. Furthermore, our previous study showed that the impaired UPR in SCs via double deletion of PERK and ATF6α has no effect on actively myelinating SCs during development (Stone et al., 2020). Additionally, a recent study showed that deletion of Derlin-2 in SCs does not affect developmental myelination in the PNS (Volpi et al., 2019). Collectively, these data suggest that the integrated UPR and ERAD is not a major player in regulating ER homeostasis or the viability and function of actively myelinating SCs during development. The mechanisms by which actively myelinating SCs in young developing mice maintain ER homeostasis and their myelinating function appear unique and deserve further investigation.
Unexpectedly, Sel1L cKO mice displayed adult-onset neurological phenotypes and behavioral abnormalities, and exhibited mature SC apoptosis and demyelination in the adult PNS. Interestingly, a previous study showed that mice with Derlin-2 deletion in SCs display a moderate reduction of myelin thickness in the adult PNS. Evidence suggests that Derlin-2, a component of the Sel1L-Hrd1 complex, is required for retrotranslocation of several ERAD substrates into the cytosol (Huang, Hsiao, Chu, Ye, & Chen, 2013; Lilley & Ploegh, 2005); however, Sel1L is essential for the ERAD activity of the Sel1L-Hrd1 complex. Therefore, these data likely reflect that ERAD impairment in SCs induces myelin abnormalities in the adult PNS in a dose-dependent manner. We further found that Sel1L deficiency caused ER stress in mature SCs of adult mice, and that PERK inactivation exacerbated ER stress in mature SCs of adult Sel1L cKO mice, as evidenced by the increased XBP1 splicing and the enlarged ER lumen in mature SCs of adult Double cKO mice. Importantly, PERK inactivation exacerbated mature SC apoptosis and demyelination in the PNS of adult Sel1L cKO mice. The PERK branch of the UPR restores ER homeostasis in ER-stressed cells by inhibiting protein translation and facilitating protein degradation, and promotes cell survival by stimulating certain cytoprotective gene expression (Han & Kaufman, 2017; Marciniak & Ron, 2006; Walter & Ron, 2011). Thus, our results suggest that ER stress induced by Sel1L deficiency causes mature SC apoptosis in the adult PNS, and that PERK inactivation exacerbates the ER stress induced by Sel1L deficiency in mature SCs, resulting in exacerbated cell apoptosis. Collectively, these data demonstrate the essential role of the integrated UPR and ERAD in maintaining ER homeostasis and the viability and function of adult mature SCs under physiological conditions. On the other hand, our recent study showed that mice with double deletion of PERK and ATF6α in myelinating cells display a tremoring phenotype starting at postnatal day (P) 28, and die by P65. Although the impaired UPR in myelinating cells via double deletion of PERK and ATF6α leads to severe oligodendrocyte loss and demyelination in the CNS at P45, there is no evidence of SC dysfunction or death in the PNS of these mice at P45 (Stone et al., 2020). In fact, in accordance to the study, we showed here that double deletion of PERK and Sel1L did not attenuate the viability or function of SCs in the PNS at the age of 6 w, and that Sel1L cKO mice did not display evidence of SC dysfunction or death at the age of 10 w. Therefore, the possibility that the impaired UPR via double deletion of PERK and ATF6α causes dysfunction and death of mature SCs in adult mice (after 10 w) cannot be ruled out and deserves further investigation.
Although the cytoprotective effects of the PERK-eIF2α is well documented in ER-stressed cells, PERK activation is not exclusively beneficial (Hetz & Papa, 2018; Tabas & Ron, 2011). A number of studies have suggested that the effects of PERK activation, beneficial or detrimental, on cells are activity-dependent and/or context-dependent (Hetz & Saxena, 2017; W. Lin et al., 2013; Y. Lin, Huang, et al., 2014; Y. Lin, Pang, et al., 2014; Ranganathan, Ojha, Kourtidis, Conklin, & Aguirre-Ghiso, 2008; Stone et al., 2016). The results presented in this study suggest that PERK activation restores ER homeostasis in mature SCs and preserves their viability and function in adult Sel1L cKO mice. Conversely, the data concerning the role of the PERK-eIF2α-CHOP pathway in SCs and myelin abnormalities in the PNS of P0S63del mice are somehow contradictory (D’Antonio et al., 2013; Das et al., 2015; Musner et al., 2016; Pennuto et al., 2008; Sidoli et al., 2016). One report showed that CHOP deletion does not influences hypomyelination but attenuates demyelination in the PNS of P0S63del mice (Pennuto et al., 2008). Other reports showed that inhibition of GADD34 (a regulatory subunit of a phosphatase complex that dephosphorylates p-eIF2α), via genetic and pharmacological approaches, elevates the level of p-eIF2α and ameliorates myelin abnormalities in the PNS of P0S63del mice (D’Antonio et al., 2013; Das et al., 2015). In contrast, using global PERK heterozygous deficient mice, a study showed that heterozygous PERK deficiency attenuates demyelination, but does not alter hypomyelination in the PNS of P0S63del mice (Musner et al., 2016). Additionally, using Schwann cell-specific PERK deficient mice, another study showed that PERK deficiency specifically in Schwann cells attenuates hypomyelination in the PNS of P0S63del nerves (Sidoli et al., 2016). It is unclear how the PERK-eIF2α-CHOP pathway has the paradoxical effects on myelin abnormalities in the PNS of P0S63del mice, possibly due to the degree of PERK activation or the pathological contexts.
In summary, using mice with Sel1L deletion specifically in myelin-producing cells, we showed here that Sel1L deficiency induced ER stress in SCs and led to adult-onset SC apoptosis and subsequent demyelination in the PNS. Moreover, we showed that PERK inactivation aggravated ER stress in mature SCs and results in exacerbated mature SC apoptosis and demyelination in the adult PNS of SC-specific Sel1L deficient mice. In contrast, double deletion of Sel1L and PERK in SCs did not affect the viability or function of actively myelinating SCs in the PNS during development. These data suggest that ERAD is dispensable for actively myelinating SCs during development, but is necessary for maintaining ER homeostasis and the viability and function of mature SCs in adults.
Supplementary Material
Main Points.
The integrated UPR and ERAD is dispensable to actively myelinating Schwann cells during development.
The integrated UPR and ERAD is necessary for maintaining ER homeostasis and the viability and function of mature Schwann cells in adults.
Acknowledgements:
We thank Dr. Ling Qi (University of Michigan, Ann Arbor, Michigan) for providing the Sel1LloxP/loxP mice. We thank Dr. Klaus-Armin Nave (Max Planck Institute of Experimental Medicine, Göttingen, Germany) for providing the CNP/Cre mice. This study was supported by grants from the National Institutes of Health (NS094151 and NS105689) to WL.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- Anitei M, & Pfeiffer SE (2006). Myelin biogenesis: sorting out protein trafficking. Curr Biol, 16(11), R418–421. doi: 10.1016/j.cub.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Baldridge RD, & Rapoport TA (2016). Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell, 166(2), 394–407. doi: 10.1016/j.cell.2016.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton BLL, & Popko B (2016). Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res, 1648, 594–602. doi: 10.1016/j.brainres.2016.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, … Wrabetz L (2013). Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med, 210(4), 821–838. doi: 10.1084/jem.20122005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, … Bertolotti A (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science, 348(6231), 239–242. doi: 10.1126/science.aaa4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, & Kaufman RJ (2017). Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev, 31(14), 1417–1438. doi: 10.1101/gad.297374.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, & Papa FR (2018). The Unfolded Protein Response and Cell Fate Control. Mol Cell, 69(2), 169–181. doi: 10.1016/j.molcel.2017.06.017 [DOI] [PubMed] [Google Scholar]
- Hetz C, & Saxena S (2017). ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol, 13(8), 477–491. doi: 10.1038/nrneurol.2017.99 [DOI] [PubMed] [Google Scholar]
- Horimoto S, Ninagawa S, Okada T, Koba H, Sugimoto T, Kamiya Y, … Mori K (2013). The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J Biol Chem, 288(44), 31517–31527. doi: 10.1074/jbc.M113.476010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Hsiao HT, Chu YR, Ye Y, & Chen X (2013). Derlin2 protein facilitates HRD1-mediated retro-translocation of sonic hedgehog at the endoplasmic reticulum. J Biol Chem, 288(35), 25330–25339. doi: 10.1074/jbc.M113.455212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, & Qi L (2018). Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci, 43(8), 593–605. doi: 10.1016/j.tibs.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Sim HJ, Song EK, Lee H, Ha SC, Jun Y, … Lee C (2016). Crystal structure of SEL1L: Insight into the roles of SLR motifs in ERAD pathway. Sci Rep, 6, 20261. doi: 10.1038/srep20261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Kim H, Yang L, Sha H, Roman CA, Long Q, & Qi L (2016). The Sel1L-Hrd1 Endoplasmic Reticulum-Associated Degradation Complex Manages a Key Checkpoint in B Cell Development. Cell Rep, 16(10), 2630–2640. doi: 10.1016/j.celrep.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ (1999). Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev, 13(10), 1211–1233. doi: 10.1101/gad.13.10.1211 [DOI] [PubMed] [Google Scholar]
- Kim GH, Shi G, Somlo DR, Haataja L, Song S, Long Q, … Qi L (2018). Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J Clin Invest, 128(3), 1125–1140. doi: 10.1172/JCI96420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, … Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet, 33(3), 366–374. doi: 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Lilley BN, & Ploegh HL (2005). Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A, 102(40), 14296–14301. doi: 10.1073/pnas.0505014102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Harding HP, Ron D, & Popko B (2005). Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol, 169(4), 603–612. doi: 10.1083/jcb.200502086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, … Popko B (2013). Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci, 33(14), 5980–5991. doi: 10.1523/JNEUROSCI.1636-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, & Popko B (2009). Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci, 12(4), 379–385. doi: 10.1038/nn.2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, & Stone S (2020). Unfolded protein response in myelin disorders. Neural Regen Res, 15(4), 636–645. doi: 10.4103/1673-5374.266903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Huang G, Jamison S, Li J, Harding HP, Ron D, & Lin W (2014). PERK activation preserves the viability and function of remyelinating oligodendrocytes in immune-mediated demyelinating diseases. Am J Pathol, 184(2), 507–519. doi: 10.1016/j.ajpath.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Pang X, Huang G, Jamison S, Fang J, Harding HP, … Lin W (2014). Impaired eukaryotic translation initiation factor 2B activity specifically in oligodendrocytes reproduces the pathology of vanishing white matter disease in mice. J Neurosci, 34(36), 12182–12191. doi: 10.1523/JNEUROSCI.1373-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, & Ron D (2006). Endoplasmic reticulum stress signaling in disease. Physiol Rev, 86(4), 1133–1149. doi: 10.1152/physrev.00015.2006 [DOI] [PubMed] [Google Scholar]
- McCaffrey K, & Braakman I (2016). Protein quality control at the endoplasmic reticulum. Proteostasis, 60(2), 227–235. doi: 10.1042/Ebc20160003 [DOI] [PubMed] [Google Scholar]
- Musner N, Sidoli M, Zambroni D, Del Carro U, Ungaro D, D’Antonio M, … Wrabetz L (2016). Perk Ablation Ameliorates Myelination in S63del-Charcot-Marie-Tooth 1B Neuropathy. Asn Neuro, 8(2). doi: 10.1177/1759091416642351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Kamura T, & Brodsky JL (2014). Recent technical developments in the study of ER-associated degradation. Current Opinion in Cell Biology, 29, 82–91. doi: 10.1016/j.ceb.2014.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M, Tinelli E, Malaguti M, Del Carro U, D’Antonio M, Ron D, … Wrabetz L (2008). Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron, 57(3), 393–405. doi: 10.1016/j.neuron.2007.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Tsai B, & Arvan P (2017). New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends in Cell Biology, 27(6), 430–440. doi: 10.1016/j.tcb.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, & Aguirre-Ghiso JA (2008). Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Research, 68(9), 3260–3268. doi: 10.1158/0008-5472.CAN-07-6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, & Carvalho P (2014). ER-associated degradation: Protein quality control and beyond. Journal of Cell Biology, 204(6), 868–878. doi: 10.1083/jcb.201312042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoebel S, Mi W, Stein A, Ovchinnikov S, Pavlovicz R, DiMaio F, … Liao MF (2017). Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature, 548(7667), 352–355. doi: 10.1038/nature23314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli M, Musner N, Silvestri N, Ungaro D, D’Antonio M, Cavener DR, … Wrabetz L (2016). Ablation of Perk in Schwann Cells Improves Myelination in the S63del Charcot-Marie-Tooth 1B Mouse. Journal of Neuroscience, 36(44), 11350–11361. doi: 10.1523/Jneurosci.1637-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Ho Y, Li XT, Jamison S, Harding HP, Ron D, & Lin WS (2016). Dual role of the integrated stress response in medulloblastoma tumorigenesis. Oncotarget, 7(39), 64124–64135. doi: 10.18632/oncotarget.11873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Jamison S, Yue Y, Durose W, Schmidt-Ullrich R, & Lin WS (2017). NF-kappa B Activation Protects Oligodendrocytes against Inflammation. Journal of Neuroscience, 37(38), 9332–9344. doi: 10.1523/Jneurosci.1608-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Wu SC, Jamison S, Durose W, Pallais JP, & Lin WS (2018). Activating transcription factor 6 alpha deficiency exacerbates oligodendrocyte death and myelin damage in immune-mediated demyelinating diseases. Glia, 66(7), 1331–1345. doi: 10.1002/glia.23307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Wu SC, Nave KA, & Lin WS (2020). The UPR preserves mature oligodendrocyte viability and function in adults by regulating autophagy of PLP. Jci Insight, 5(5). doi:ARTN e132364 10.1172/jci.insight.132364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto T, Ninagawa S, Yamano S, Ishikawa T, Okada T, Takeda S, & Mori K (2017). SEL1L-dependent Substrates Require Derlin2/3 and Herp1/2 for Endoplasmic Reticulum-associated Degradation. Cell Structure and Function, 42(2), 81–94. doi:Doi 10.1247/Csf.17007 [DOI] [PubMed] [Google Scholar]
- Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonca N, … Qi L (2014). Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A, 111(5), E582–591. doi: 10.1073/pnas.1318114111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY, Shi GJ, Sha HB, Ji YW, Han XM, Shu X, … Qi L (2015). IRE1 alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nature Cell Biology, 17(12), 1546–1555. doi: 10.1038/ncb3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, & Ron D (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nature Cell Biology, 13(3), 184–190. doi: 10.1038/ncb0311-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashistha N, Neal SE, Singh A, Carroll SM, & Hampton RY (2016). Direct and essential function for Hrd3 in ER-associated degradation. Proc Natl Acad Sci U S A, 113(21), 5934–5939. doi: 10.1073/pnas.1603079113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi VG, Ferri C, Fregno I, Del Carro U, Bianchi F, Scapin C, … D’Antonio M (2019). Schwann cells ER-associated degradation contributes to myelin maintenance in adult nerves and limits demyelination in CMT1B mice. Plos Genetics, 15(4). doi:ARTN e1008069 10.1371/journal.pgen.1008069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi VG, Touvier T, & D’Antonio M (2017). Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Frontiers in Molecular Neuroscience, 9. doi:Artn 162 10.3389/Fnmol.2016.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, & Ron D (2011). The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science, 334(6059), 1081–1086. doi: 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wu XD, & Rapoport TA (2018). Mechanistic insights into ER-associated protein degradation. Current Opinion in Cell Biology, 53, 22–28. doi: 10.1016/j.ceb.2018.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SB, Park YH, Choi SA, Yang HJ, Jeong PS, Cha JJ, … Kim SU (2019). Real-time PCR quantification of spliced X-box binding protein 1 (XBP1) using a universal primer method. Plos One, 14(7). doi:ARTN e0219978 10.1371/journal.pone.0219978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PC, McGrath B, Li SA, Frank A, Zambito F, Reinert J, … Cavener DR (2002). The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Molecular and Cellular Biology, 22(11), 3864–3874. doi: 10.1128/Mcb.22.11.3864-3874.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.