

Abstract
Trialkylamines are widely found in naturally-occurring alkaloids, synthetic agrochemicals, biological probes, and especially pharmaceuticals agents and pre-clinical candidates. Despite the recent breakthrough of catalytic alkylation of dialkylamines, the selective α-C(sp3)–H bond functionalization of widely available trialkylamine scaffolds holds promise to streamline complex trialkylamine synthesis, accelerate drug discovery and execute late-stage pharmaceutical modification with complementary reactivity. However, the canonical methods always result in functionalization at the less-crowded site. Herein, we describe a solution to switch the reaction site through fundamentally overcoming the steric control that dominates such processes. By rapidly establishing an equilibrium between α-amino C(sp3)–H bonds and a highly electrophilic thiol radical via reversible hydrogen atom transfer, we leverage a slower radical-trapping step with electron-deficient olefins to selectively forge a C(sp3)–C(sp3) bond with the more-crowded α-amino radical, with the overall selectivity guided by Curtin-Hammett principle. This subtle reaction profile has unlocked a new strategic concept in direct C–H functionalization arena for forging C–C bonds from a diverse set of trialkylamines with high levels of site-selectivity and preparative utility. Simple correlation of site-selectivity and 13C NMR shift serves as a qualitative predictive guide. The broad consequences of this dynamic system, together with the ability to forge N-substituted quaternary carbon centers and implement late-stage functionalization techniques, holds potential to streamline complex trialkylamine synthesis and accelerate small-molecule drug discovery.
Graphical Abstract
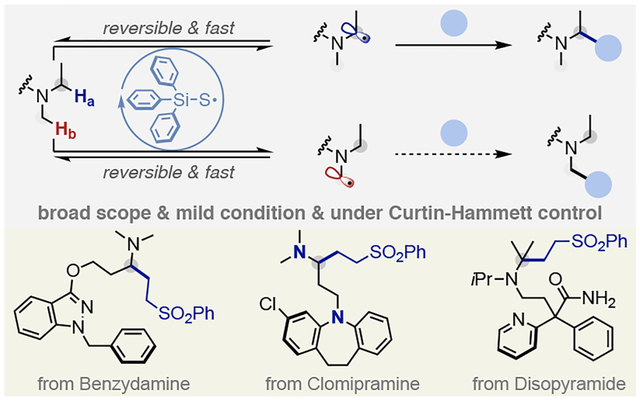
INTRODUCTION
Trialkylamine-containing pharmaceuticals are ubiquitous, with the amine functionality incorporated to mimic the multiple functions of biogenic amines or to provide desirable pharmacokinetic properties.1 Against this back-drop, there is a constant synthetic interest in the development and improvement of methods to catalytically construct complex trialkylamine-scaffolds in synthetic and medicinal chemistry.2 Traditional approaches to trialkylamines use alkylation of secondary amines, with some noteworthy catalytic examples recently disclosed.3 Alternatively, the direct α-C–H functionalization of widely available trialkylamines would enable facile access to functionally diverse libraries, and consequently allow for streamlined routes for the catalytic synthesis of structurally complex trialkylamines with complementary reactivity, amenable to late-stage functionalization strategies.4 However, α-C–H bonds of unsymmetrical unfunctionalized trialkylamines often have nearly identical bond dissociation energies (for example, N-methyl piperidine: 91 kcal/mol for the secondary vs 92 kcal/mol for the primary site).5 Thus, their selective activation on the basis of enthalpic differences will be difficult. Currently, the vast majority of strategies based on fundamentally different reactive intermediates, including carbene insertion, direct hydride or hydrogen abstraction or single electron oxidation/deprotonation processes, predominantly rely on steric control to activate the more accessible and least substituted position selectively (Fig.1, A).6 One type of exception that has recently appeared involves benzylic amines where selectivity is apparently governed by obvious differences in acidity or bond dissociation energies.7 The other exception involves diaryl ketone as catalyst under irradiation with high energy UV light in presence of large excess of the trialkylamines (20 equiv or more),8 which largely limits the synthetic potential and makes late-stage applications nearly impossible.9 Thus, a general approach to functionalize the more substituted and less accessible α-C–H bond of a wide range of trialkylamines with enhanced structural complexity therefore constitutes an elusive and long-standing synthetic challenge.
Figure 1.
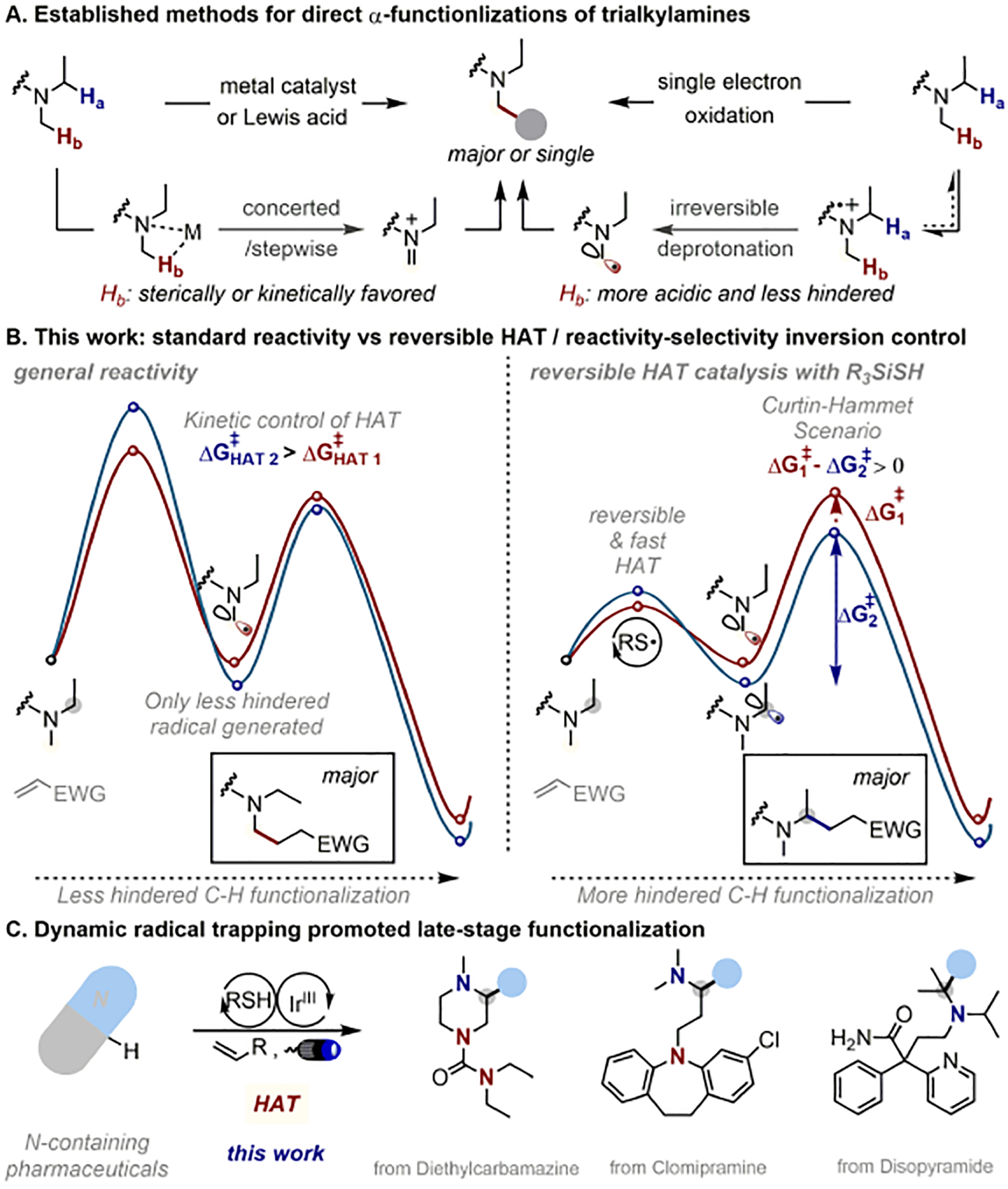
(A) Canonical methods for trialkylamine α-functionalization. (B) Energy profile of trialkylamine α-functionalization enabled by standard photoredox catalysis vs. reversible HAT catalysis. (C) Late-stage modification of trialkylamine-containing pharmaceuticals.
Thiyl radicals have had a transformative impact in modern synthetic chemistry.10 The electrophilicity of thiyl radicals make them preeminent and efficient species for a broad range of biological and chemical processes, especially those involving hydrogen atom transfer (HAT).11 Recently, MacMillan and co-workers demonstrated the isotope labelling utility of silyl thiyl radicals as HAT catalysts to perform photoredox-catalyzed α-deuteration and tritiation of trialkylamine-containing pharmaceuticals.12 Diagnostically, this strategy results in exhaustive α-C–H isotopic labeling. Our continuing interest in photoredox promoted amine functionalization led us to examine the feasibility of selective C(sp3)–C(sp3) bond formation at the more-substituted position of trialkylamines.13,16f If accomplishable, this strategy would offer a pathway complementary to existing methods for complex trialkylamine construction and late-stage functionalization of pharmaceuticals.
RESULTS AND DISCUSSION
Specifically, under photoredox conditions,14 we envisioned using thiol catalyst to generate a highly electrophilic thiyl radical via a single electron transfer (SET) and subsequent deprotonation process. The resultant thiyl radical would then ideally participate in an unselective, fast and reversible HAT of the C(sp3)–H bond adjacent to the basic nitrogen atoms,15 an equilibrium reminiscent of the racemization step in dynamic kinetic resolution and other Curtin-Hammett controlled processes.16 However, under equilibrium conditions, the primary radical might be expected to react preferentially as it is kinetically more accessible. Thus, the key to this strategyis to find a suitable electrophilic coupling partner that inverts that natural trend and reacts preferentially at the more-substituted (and more-nucleophilic) position of the α-amino radical. To do so, fine tuning of reaction barriers is required since the initial HAT should be fully reversible and the subsequent radical trapping should be slow and irreversible to kinetically control the selectivity in a Curtin-Hammett mechanistic scenario (Fig.1, B right).
In line with this notion, we considered that radical trapping with electron-deficient olefins— the Giese reaction — would meet the requirements for the inversion of selectivity, (Fig.1, C).17 To achieve the proposed mechanism, we sought a thiol catalyst capable of unselective, fast and reversible HAT. After testing a series of thiol catalysts, we found that commercially available triphenyl silanethiol (TPS-SH, E1/2ox = 0.43 V vs Ag/AgCl in MeCN) and triisopropyl silanethiol (TIPS-SH, E1/2ox = 0.28 V vs Ag/AgCl in MeCN), were particularly suited for selective alkylation of N-methyl piperidine with tert-butyl acrylate (see SI section II for details). By contrast, omitting TPS-SH leads to mixtures favouring C–H functionalization at the less hindered position as previously reported.6e,6f
After optimization, a combination of 1 mol% Ir(ppy)2(dtbbpy)PF6 and 20 mol% TPS-SH in toluene (0.1 M) in presence of vinyl phenyl sulfone, under blue light-emitting diode (LED) irradiation provides the best results, giving rise to compound 20 in 86% isolated yield with excellent site-selectivity.18 Next, we aimed to study the generality of our protocol. As shown in Fig. 2, a wide variety of trialkylamines undergo the targeted sp3 C–H alkylation with vinyl sulfone, acrylate, acrylonitrile, 1,1-diarylethylene and vinyl ketone. Strikingly, our catalytic system selectively alkylates N-ethyl piperidine on the ring, albeit with moderate selectivity (5, 11). Indeed, the method generally displays an excellent site-selectivity profile, and is tolerant of multiple functional groups, such as alcohol (7), ester (8, 13), imide (14), ketone (15) and saturated N-heterocycles such as piperazine (16), pyrrolidine (17) and piperidine (20-24). As expected, the reactivity can be smoothly extended to aniline derivatives (25). We found that the selectivity is affected by the steric bulk of both olefin acceptor and amines; in an extreme case, diisopropyl methyl amine affords tertiary-alkylation with unsubstituted Michael acceptors but methyl-alkylation when the olefin bears a β-substituent (3 vs. 26), indicating the fast reversibility of HAT step is tempered by increased steric crowding in the selectivity-determining alkylation event.
Figure 2. Site-selective alkylation of simple trialkylamines and novel disconnection.
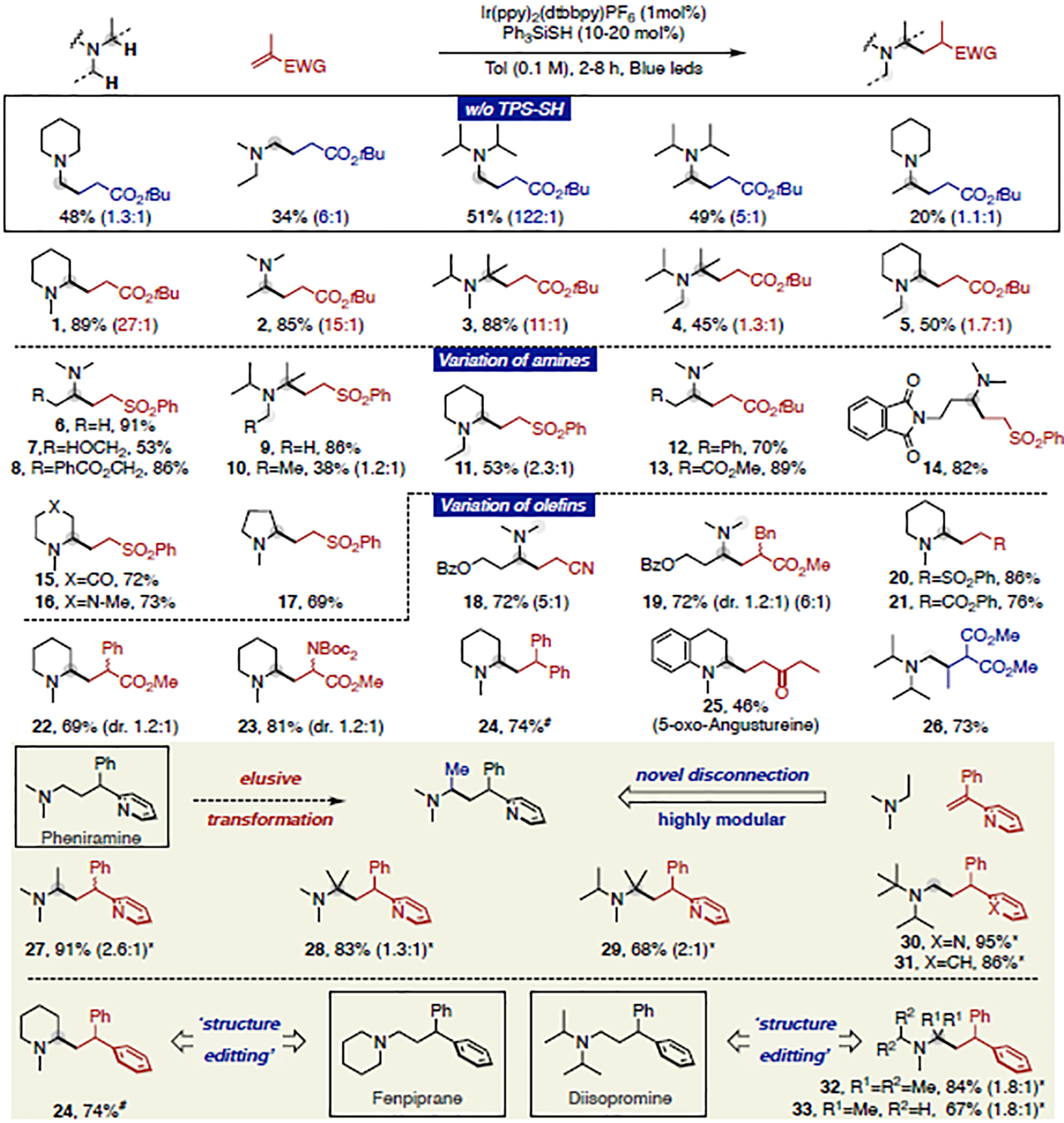
Reaction conditions: trialkylamine (3.0 eq.), olefin (1.0 eq.), Ir(ppy)2(dtbby)PF6 (1 mol%), Ph3SiSH (20 mol%), Tol (0.1 M), Blue LED (32W), 28 °C, 2–8 h. #2-tert-butyl-1,1,3,3-tetramethylguanidine (50 mol%) used as additive.*TPS-SH (5 mol%) and Dioxane (0.1 M) were used instead.
With our approach, we considered whether it also may provide an orthogonal solution to strategic derivatization efforts of trialkylamine-containing biologically active compounds. The biological activity of 3,3-Diarylpropyl-amines, critical units widely found in H1-antihistamines, can vary from antiallergic to antispasmodic, antipyretic, and choleretic by fine-tuning of the structure.19 Editing the structure by selective introduction of methyl groups via C–H functionalization remains a daunting challenge,20 with several recent noteworthy solutions.21 We considered a retrosynthetic disconnection involving the corresponding trialkylamine motif alkylated with dia-rylethylenes (Figure 2). The strategy provides facile access to methylated (27-31) versions of pheniramine, albeit with moderate selectivity. Even more noteworthy is the structural reorganization that is enabled by this approach – a pheniramine analogue is easily assembled by reacting N-methyl piperidine with diphenyl ethylene (24) while an isostructural diisopromine is formed from diisopropyl methyl amine (32). From a chemical perspective, these reactions are distinguished by a superb reaction profile with simple precursors being converted to products of significantly increased complexity that are difficult to access with existing methods, thus showcasing the preparative potential of this transformation.
MECHANISTIC STUDIES
To gain insight into the reaction mechanism and understand the origin of the high levels of selectivity attained with our system, we performed a series of computational and experimental studies. Our control study indicates that a radical chain mechanism is operative, as both photoredox and traditional radical initiation with azobisisobu-tyronitrile (AIBN) give essentially the same results for N-methyl piperidine. Further support for the chain process under photoredox conditions was gained from the observed quantum yield (Φ=23 - see SI section III for details). Under thiol-free conditions (Figure 3), the radical is generated (and later regenerated) via an irreversible HAT to a carbon-centered radical, which is of relatively high activation barrier (ΔG‡= 16.4–17.6 kcal/mol) and kinetically favors the least substituted site (by ΔΔG‡ = 1.2 kcal/mol). This is followed by rapid radical addition (C-C bond formation).
Figure 3.
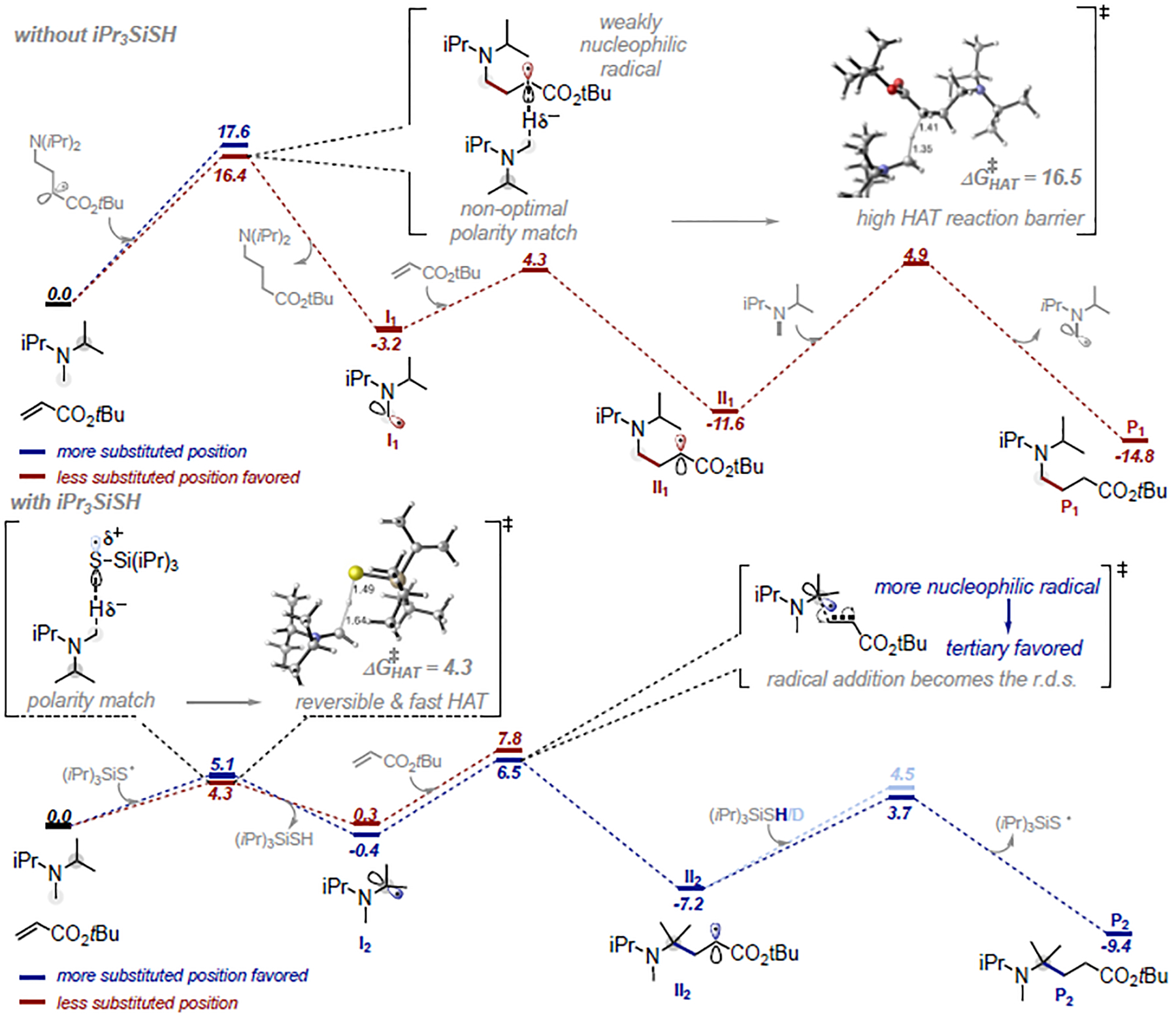
DFT computational studies of reaction mechanism.
In stark contrast, when the thiol is present, initial formation of a thiyl radical is favored, which triggers H-atom abstraction with roughly a third of the activation barrier (ΔG‡ = 4.3–5.1 kcal/mol) as compared to the carbon centered radical, and essentially without driving force (ΔGrxn = −0.4 (tertiary) / 0.3 (primary) kcal/mol), rendering the process fully reversible. As such, the calculated profile fully supports that the HAT process is unselective, fast and reversible under thiol-conditions, in line with the deuteration experiments, in which α-amino C(sp3)–H deuteration of both recovered amine and products only occurs in the presence of thiol catalyst (Figure 4) (see SI section III for details). Our calculations suggest that this different behaviour in HAT is due to an excellent polarity match of the electrophilic thiyl radical with the relatively electronrich α-N C–H bond. On the contrary, the α-ester radical II1 under thiol-free conditions is also weakly nucleophilic22 in character and there is hence a barrier-enhancing polarity mismatch (see SI section IV for details).
Figure 4.
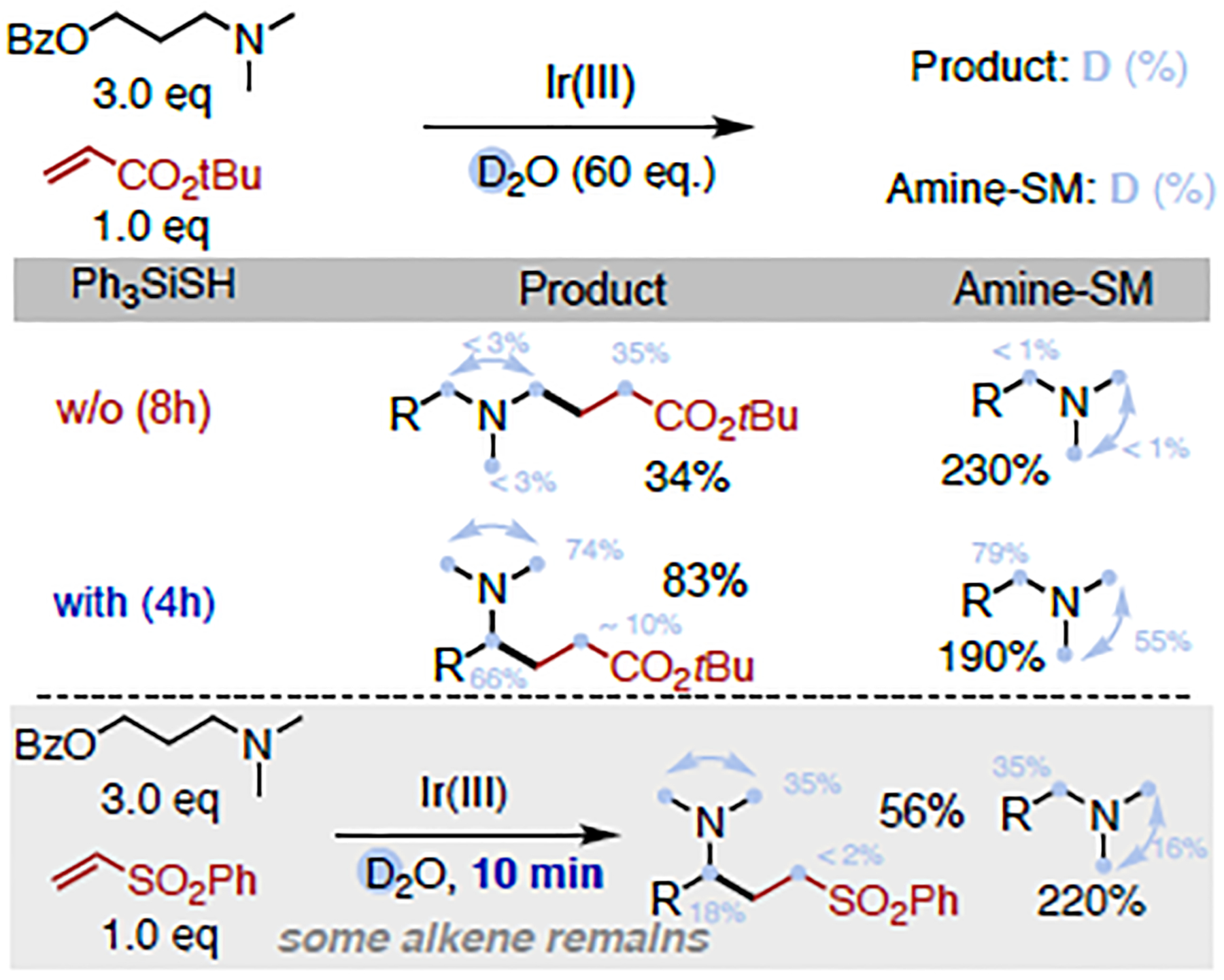
Deuteration studies.
Thus, while HAT is the selectivity-determining step under thiol-free conditions, in the presence of R3Si-SH, the later slower C–C bond-forming step becomes the rate determining step and determines the product outcome. Interestingly, the more substituted radical (tertiary) reacts via a slightly lower activation barrier (ΔΔG‡ = 1.3 kcal/mol). Our data indicate that this is primarily due to the electronic impact of the adjacent nitrogen in these radicals and the resulting pyramidalization. Additionally, our calculation of the corresponding nitrogen-free system predicts inverted reactivities (i.e. primary radical reacts with lower barrier than tertiary, see SI section IV for details).
To further corroborate this point, we calculated the corresponding barrier of a series of α-C, α-N and α-O tertiary radicals (Figure 5), which follow the trend of the more pyramidal the radical, the lower the barrier (see Figure S14–15 for details). Finally, regeneration of the thiyl radical occurs irreversibly through HAT from the resulting α-ester radical. This process is influenced by the deuteration of silanethiol: the free energy barrier of deuterium transfer is 0.8 kcal/mol higher in energy than HAT from R3Si-S(H/D), explaining the low deuterium incorporation in the α-ester position of the acrylate.
Figure 5.
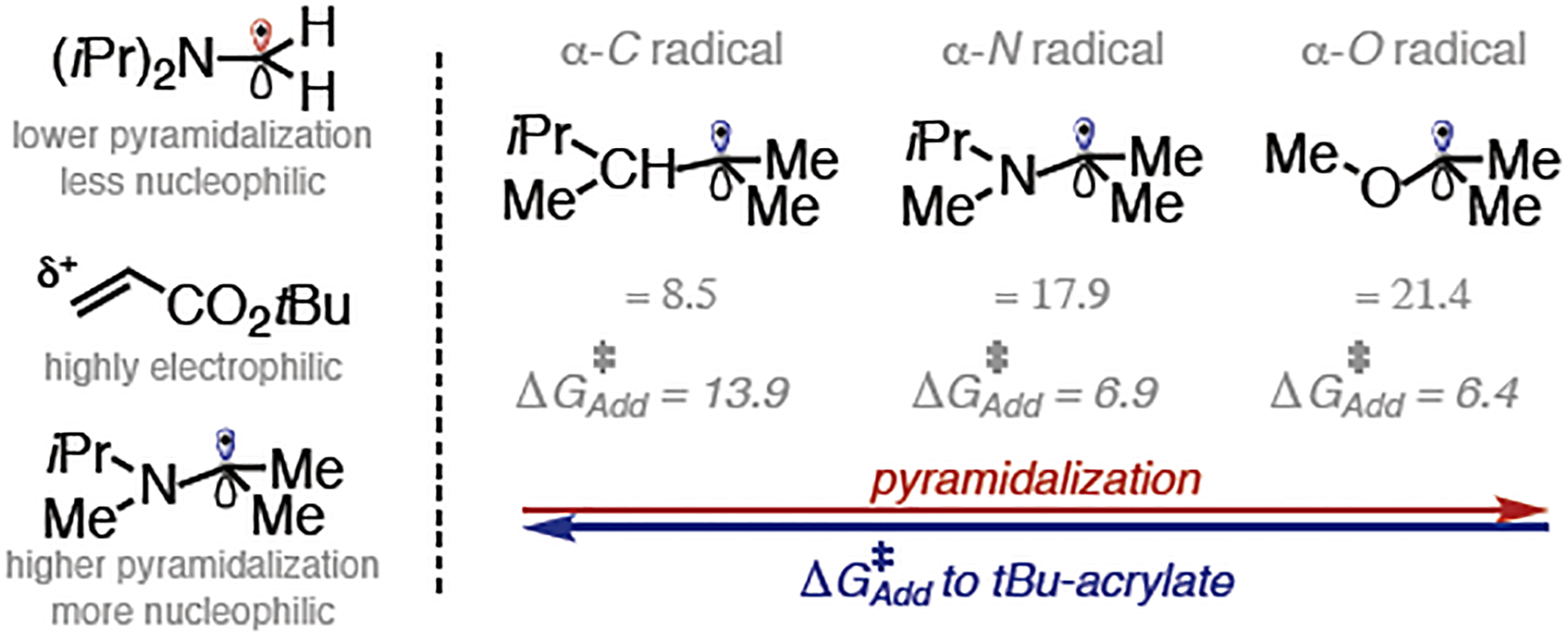
Correlation between radical pyramidalization and free energy barrier (kcal/mol) in radical addition.
In order to show that the HAT catalyst reacts as an electrophilic radical, we calculated the LUMO energy of the HAT catalyst radical and its correlation with the free energy barrier for the HAT at the primary position of (iPr)2NMe. Interestingly, there is a linear correlation of both parameters (Figure 6): the higher the electrophilicity of the radical, the lower the barrier of the corresponding HAT process. The two best catalysts (Ph3SiSH and iPr3SiSH) promote the HAT with a very low free energy barrier due to the polarity match in the transition states. In contrast, less electrophilic but weakly nucleophilic α-ester radical, shows a significant increase of the activation barrier for HAT process.
Figure 6.
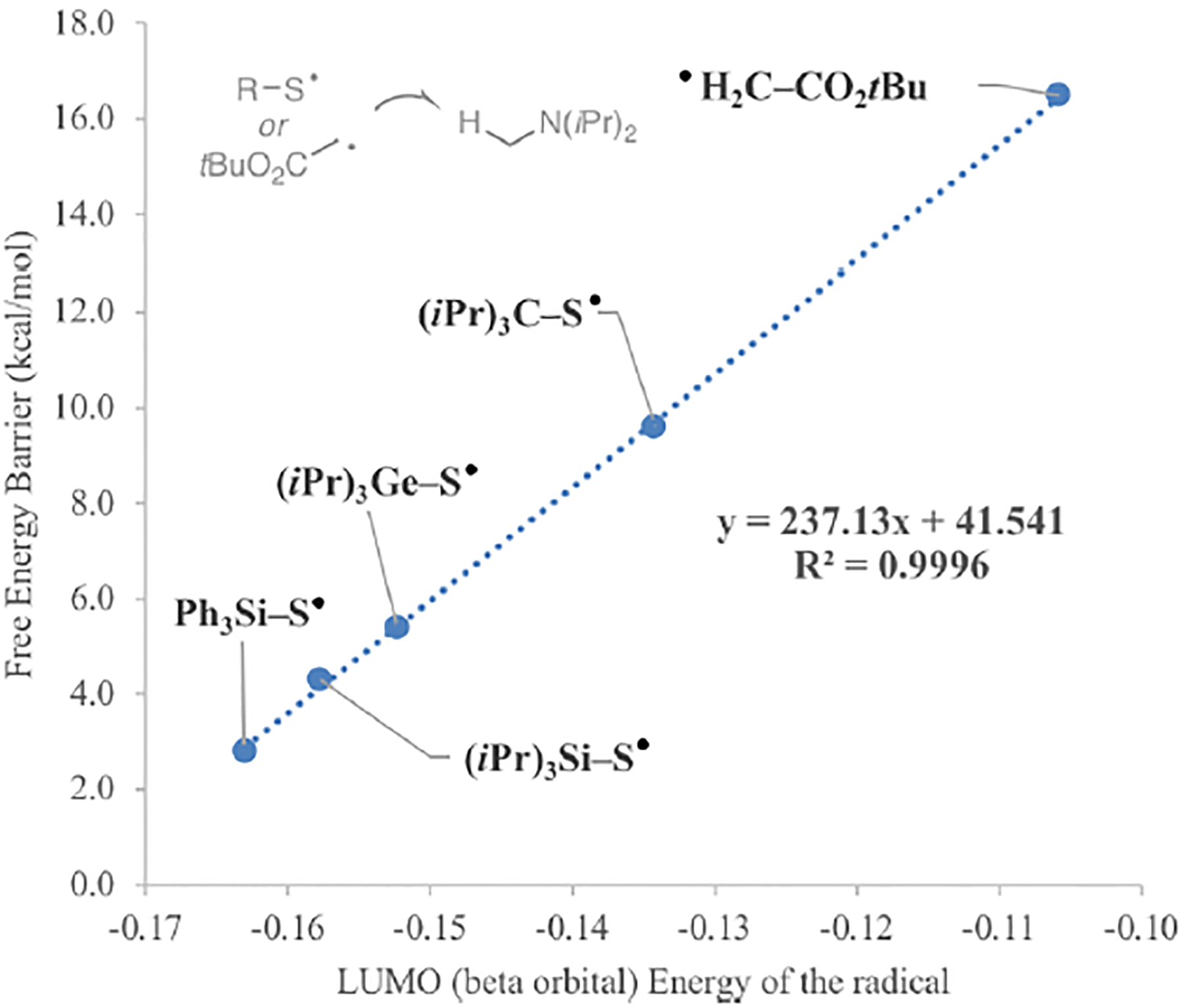
Correlation between LUMO orbital of the radical and HAT barrier (kcal/mol).
With a rationalization of the underlying reactivity and selectivity in hand, we wondered whether certain simple molecular parameters could potentially correlate with the overall site-selectivity even in the context of late-stage functionalization of complex pharmaceuticals. As expected, bond dissociation energies (BDE) do not correlate with the observed reactivity (Figure 7). However, our calculations of a variety of parameters for simple amines revealed that the site-selectivity has a qualitative relationship with the 13C NMR shift of the α-amino carbon signal; alkylation occurs selectively at the carbon bearing the higher 13C NMR shift.
Figure 7. Qualitative reaction site prediction and late-stage alkylation of pharmaceuticals.
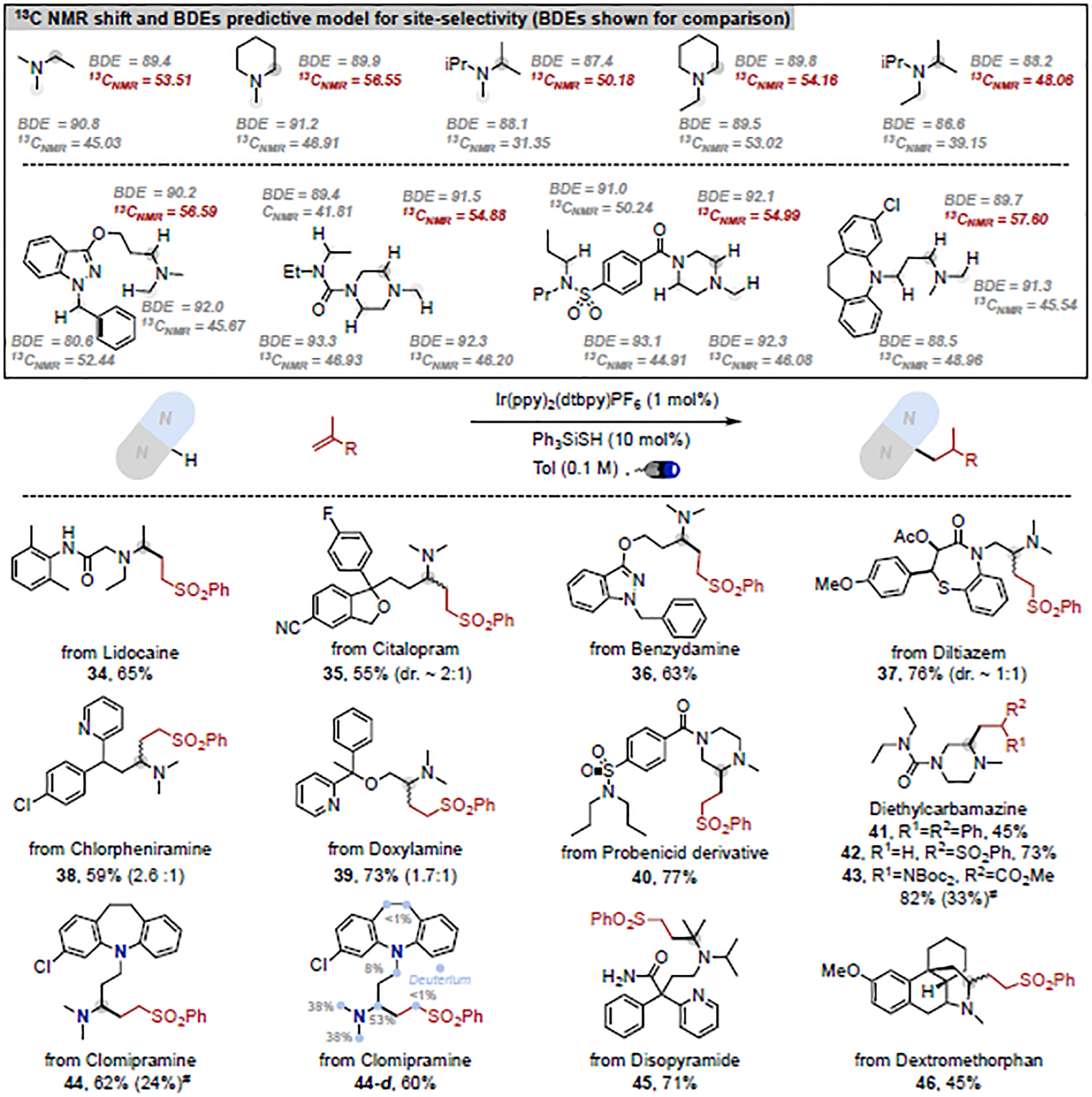
BDEs listed are in kcal/mol. 13C NMR shifts are in ppm. Reaction conditions: trialkylamine (3.0 eq.), olefin (1.0 eq.), Ir(ppy)2(dtbby)PF6 (1 mol%), Ph3SiSH (10 mol%), Tol (0.1 M), Blue LED (32W), 28 °C, 8 h. # 1H NMR yield, amine (1.0 eq.) used instead.
We then extrapolated this predictive model to a library of commercially available drugs containing trialkylamine scaffolds and other potential reactive sites. As evident from the results compiled for 34–46 in Figure 7 and as predicted by our simple model, all these trialkylamine-containing complex molecules underwent effective site-selective alkylation under our conditions. Importantly, the α-amino C–H bonds of aniline (44), amide (37, 40, 45), sulfonamide (40) and urea (41-43), α-C–H bonds to oxygen (35, 36, 39) and benzylic C–H bonds (35, 36, 38, 44) all remained untouched under the reaction conditions, highlighting the potential of the current transformation in the rapid and efficient late-stage modification of medicinally relevant molecules. Trialkyl N- substituted quaternary carbon centers, which are challenging to form with conventional methods,23 are readily accessible even in the context of late-stage functionalization (9, 45). Strikingly, selective alkylation of secondary α-amino C–H bonds in dextromethorphan was observed with good yield (46). Isotope labelling is a paramount diagnostic tool in pharmacokinetics,24 and in the presence of D2O, almost all alkylated drug molecules presented in Fig. 4 should undergo decent isotope incorporation.12 As a testament, clomipramine was selected to perform the desired alkylation/D-labelling sequence with good yield, and deuterium incorporation (44-d), a result that underscores our mechanistic conclusions – HAT is rapid and equilibrating leading to per-deuteration of the substrate while the alkylation event is selective and product-determining leading to a monoalkylation.
CONCLUSION
In summary, we have developed a general regioselective α-C(sp3)–H functionalization of the least accessible site of trialkylamines via reversible HAT catalysis. Using silanethiols as reversible HAT catalysts under visible light photoredox conditions, this transformation has proven to be mild, efficient, and tolerant to a wide variety of functionalities present in complex amines. The key to success is the rapid reversibility of the HAT process and subsequent selective trapping of the more substituted and nucleophilic α-amino radical under Curtin-Hammett control. Conceptually, we expect that the broader consequences of the inversion of canonical selectivity in trialkylamine functionalization will foster systematic investigations for more challenging site-selective transformations involving radical species.
Supplementary Material
ACKNOWLEDGMENT
We thank NIGMS (GM125206) for support. I.F.A. thanks the Alexander von Humboldt Foundation for a scholarship. Calculations were performed with computing resources granted by JARA HPC from RWTH Aachen University under project jara0091.
Funding Sources
The authors declare no competing financial interest.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Reaction optimization, detailed experimental procedures, computational studies, and compound characterization (PDF)
REFERENCES
- (1).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nature Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (2).(a) Trowbridge A; Walton SM; Gaunt MJ New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev 2020, 120, 2613–2692; [DOI] [PubMed] [Google Scholar]; (b) Matheau-Raven D; Gabriel P; Leitch JA; Almehmadi YA; Yamazaki K; Dixon DJ Catalytic Reductive Functionalization of Tertiary Amides Using Vaska’s Complex: Synthesis of Complex Tertiary Amine Building Blocks and Natural Products. ACS Catal. 2020, 10, 8880–8897; [Google Scholar]; (c) Liu RY; Buchwald SL CuH-Catalyzed Olefin Functionalziation: From Hydroamination to Carbonyl Addition. Acc. Chem. Res 2020, 53, 1229–1243; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ganley JM; Murray PRD; Knowles RR Photocatalytic Generation of Aminium Radical Cations for C–N Bond Formation. ACS Catal. 2020, 10, 11712–11738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Yang Y; Shi S-L; Niu D; Liu P; Buchwald SL Catalytic Asymmetric Hydroamination of Unactivated Internal Olefins to Aliphatic Amines. Science 2015, 349, 62–66; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Catalytic Intermolecular Hydroaminations of Unactivated Olefins with Secondary Alkyl Amines. Science 2017, 355, 727–730; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trowbridge A; Reich D; Gaunt MJ Multicomponent Synthesis of Tertiary Alkylamines by Photocatalytic Olefin-Hydroaminoalkylation. Nature 2018, 561, 522–527; [DOI] [PubMed] [Google Scholar]; (d) Li M-L; Yu J-H; Li Y-H; Zhu S-F; Zhou Q-L Highly Enantioselective Carbene Insertion into N–H Bonds of Aliphatic Amines. Science 2019, 366, 990–994; [DOI] [PubMed] [Google Scholar]; (e) Kumar R; Flodén NJ; Whitehurst WG; Gaunt MJ A General Carbonyl Alkylative Amination for Tertiary Amine Synthesis. Nature 2020, 581, 415–420; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shi S; Qiu W; Miao P; Li R; Lin X; Sun Z Three-Component Radical Homo Mannich Reaction. Nat Commun 2021, 12, 1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res 2015, 48, 1474–1484; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu J; Wang J; Nguyen TH; Zheng N The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem 2013, 9, 1977–2001; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nakajima K; Miyake Y; Nishibayashi Y Synthetic Utilization of α-Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res 2016, 49, 1946–1956. [DOI] [PubMed] [Google Scholar]
- (5).Wayner DDM; Clark KB; Rauk A; Yu D; Armstrong DA C–H Bond Dissociation Energies of Alkyl Amines: Radical Structures and Stabilization Energies. J. Am. Chem. Soc 1997, 119, 8925–8932. [Google Scholar]; For recent site-selective functionalization of nearly identical allylic C–H bonds, see:; (b) Li J; Zhang Z; Wu L; Zhang W; Chen P; Lin Z; Liu G Site-specific Allylic C–H Bond Functionalization with A Copper-bond N-centred Radical. Nature 2019, 574, 516–521; [DOI] [PubMed] [Google Scholar]; (c) Lei H; Rovis T A Site-selective Amination Catalyst Discriminates between Nearly Identical C–H Bonds of Unsymmetrical Disubstituted Alkenes. Nat. Chem 2020, 12, 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) He J; Hamann LG; Davies HML; Beckwith REJ Late-Stage C–H Functionalization of Complex Alkaloids and Drug Molecules via Intermolecular Rhodium-Carbenoid Insertion. Nat Commun 2015, 6, 5943; [DOI] [PubMed] [Google Scholar]; (b) Chan JZ; Chang Y; Wasa M B(C6F5)3-Catalyzed C–H Alkylation of N-Alkylamines Using Silicon Enolates without External Oxidant. Org. Lett 2019, 2, 984–988; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Catino AJ; Nichols JM; Nettles BJ; Doyle MP The Oxidative Mannich Reaction Catalyzed by Dirhodium Caprolactamate. J. Am. Chem. Soc 2006, 128, 5648–5649; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Barham JP; John MP; Murphy JA Contra-Thermodynamic Hydrogen Atom Abstraction in the Selective C–H Functionalization of Trialkylamine N-CH3 Groups. J. Am. Chem. Soc 2016, 138, 15482–15487; [DOI] [PubMed] [Google Scholar]; (e) Aycock RA; Pratt CJ; Jui NT Aminoalkyl Radicals as Powerful Intermediates for the Synthesis of Unnatural Amino Acids and Peptides. ACS Catal. 2018, 8, 9115–9119; [Google Scholar]; (f) Thullen SM; Rovis T A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System. J. Am. Chem. Soc 2017, 139, 15504–15508. [DOI] [PubMed] [Google Scholar]
- (7).(a) Ide T; Barham JP; Fujita M; Kawato Y; Egami H; Hamashima Y Regio- and Chemoselective Csp3–H Arylation of Benzylamines by Single Electron Transfer/Hydrogen Atom Transfer Synergistic Catalysis. Chem. Sci 2018, 9, 8453–8460; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leng L; Fu Y; Liu P; Ready JM Regioselective, Photocatalytic α-Functionalization of Amines. J. Am. Chem. Soc 2020, 142, 11972–11977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Bertrand S; Hoffmann N; Pete J-P Highly Efficient and Stereoselective Radical Addition of Tertiary Amines to Electron-Deficient Alkenes – Application to the Enantioselective Synthesis of Necine Bases. Eur. J. Org. Chem 2000, 12; [Google Scholar]; (b) Hoffmann N; Bertrand S; Marinković S; Pesch J Efficient Radical Addition of Tertiary Amines to Alkenes Using Photochemical Electron Transfer. Pure and Applied Chemistry 2006, 78, 2227–2246. [Google Scholar]
- (9). As mentioned in ref. 21a, the use of use of simple acrylate or acrylonitrile as coupling partner resulted in polymerization, which could be avoided by conducting the reaction in neat amine (0.1 M).
- (10).Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (11).(a) Jin J; MacMillan DWC Alcohols as Alkylating Agents in Heteroarene C–H Functionalization. Nature 2015, 525, 87–90; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y; Carder HM; Wendlandt AE Synthesis of Rare Sugar Isomers through Site-selective Epimerization. Nature 2020, 578, 403–408. [DOI] [PubMed] [Google Scholar]
- (12).Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW; MacMillan DWC Photoredox-Catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 358, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).DiRocco DA; Rovis T Catalytic Asymmetric α-Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis. J. Am. Chem. Soc 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For selective reviews on photoredox catalysis, see:; (a) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]; (c) Marzo L; Pagire SK; Reiser O; König B Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; (d) Reed NL; Yoon TP Oxidase Reactions in Photoredox Catalysis. Chem. Soc. Rev, 2021, 50, 2954–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Escoubet S; Gastaldi S; Vanthuyne N; Gil G; Siri D; Bertrand MP Thiyl Radical Mediated Racemization of Benzylic Amines. Eur. J. Org. Chem 2006, 2006, 3242–3250. [DOI] [PubMed] [Google Scholar]
- (16).(a) Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-Driven Deracemization Enabled by Excited-State Electron Transfer. Science 2019, 366, 364–369; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeHovitz JS; Loh YY; Kautzky JA; Nagao K; Meichan AJ; Yamauchi M; MacMillan DWC; Hyster TK Static to Inducibly Dynamic Stereocontrol: The Convergent Use of Racemic β-Substituted Ketones. Science 2020, 369, 1113–1118; [DOI] [PubMed] [Google Scholar]; (c) Li X-T; Lv L; Wang T; Gu Q-S; Xu G-X; Li Z-L; Ye L; Zhang X; Cheng G-J; Liu X-Y Diastereo- and Enantioselective Catalytic Radical Oxysulfonylation of Alkenes in β,γ-Unsaturated Ketoximes. Chem 2020, 6, 1692–1706; [Google Scholar]; (d) Verho O; Bäckvall J -E. Chemoenzymatic Dynamic Kinetic Resolution: A Powerful Tool for the Preparation of Enantiomerically Pure Alcohols and Amines. J. Am. Chem. Soc 2015, 137, 3996–4009; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bhat V; Welin ER; Guo X; Stoltz BM Advances in Stereoconvergent Catalysis from 2005 to 2015: Transition-Metal-Mediated Stereoablative Reactions, Dynamic Kinetic Resolutions, and Dynamic Kinetic Asymmetric Transformations. Chem. Rev 2017, 117, 4528–4561; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Seeman JI Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations, Applications, and Extensions of Curtin-Hammett Winstein-Holness Kinetics. Chem. Rev 1983, 83, 83–134. [Google Scholar]
- (17).Giese B Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew. Chem. Int. Ed. Engl 1983, 22, 753–764. [Google Scholar]
- (18).For selective examples of α-functionalization of anilines and amides via Giese addition, see:; (a) Miyake Y; Nakajima K; Nishibayashi Y Visible-Light-Mediated Utilization of α-Aminoalkyl Radicals: Addition to Electron-Deficient Alkenes Using Photoredox Catalysts. J. Am. Chem. Soc 2012, 134, 3338–3341; [DOI] [PubMed] [Google Scholar]; (b) McManus JB; Onuska NPR; Nicewicz DA Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 9056–9060. [DOI] [PubMed] [Google Scholar]
- (19).Ahmed M; Buch C; Routaboul L; Jackstell R; Klein H; Spannenberg A; Beller M Hydroaminomethylation with Novel Rhodium–Carbene Complexes: An Efficient Catalytic Approach to Pharmaceuticals. Chem. Eur. J 2007, 13, 1594–1601. [DOI] [PubMed] [Google Scholar]
- (20).(a) Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed 2013, 52, 12256–12267; [DOI] [PubMed] [Google Scholar]; (b) Aynetdinova D; Callens MC; Hicks HB; Poh CYX; Shennan BDA; Boyd AM; Lim ZH; Leitch JA; Dixon DJ Installing the “Magic Methyl” – C–H Methylation in Synthesis. Chem. Soc. Rev 2021, 50, 5517–5563. [DOI] [PubMed] [Google Scholar]
- (21).(a) Feng K; Quevedo RE; Kohrt JT; Oderinde MS; Reilly U; White MC Late-Stage Oxidative C(sp3)–H Methylation. Nature 2020, 580, 621–627; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Friis SD; Johansson MJ; Ackermann L Cobalt-Catalysed C–H Methylation for Late-Stage Drug Diversification. Nat. Chem 2020, 12, 511–519; [DOI] [PubMed] [Google Scholar]; (c) Vasilopoulos A; Krska SW; Stahl SS C(sp3)–H Methylation Enabled by Peroxide Photosensitization and Ni-Mediated Radical Coupling. Science 2021, 372, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) De Vleeschouwer F; Van Speybroeck V; Waroquier M; Geerlings P; De Proft F Electrophilicity and Nucleo-philicity Index for Radicals. Org. Lett 2007, 9, 2721–2724; [DOI] [PubMed] [Google Scholar]; (b) Héberger K; Lopata A; Jászberényi J Cs. Separation of Polar and Enthalpy Effects in Radical Addition Reactions Using Polar (σ) and Radical (σ ·) Sigma Scales. J. Phys. Org. Chem 2000, 13, 151–156. [Google Scholar]
- (23).Hager A; Vrielink N; Hager D; Lefranc J; Trauner D Synthetic Approaches towards Alkaloids Bearing α-Tertiary Amines. Nat. Prod. Rep 2016, 33, 491–522. [DOI] [PubMed] [Google Scholar]
- (24).Elmore CS; Bragg RA Isotope Chemistry; a Useful Tool in the Drug Discovery Arsenal. Bioorganic & Medicinal Chemistry Letters 2015, 25, 167–171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.