

Abstract
In a surprisingly short time, electron cryo-microscopy (cryoEM) has developed from a niche technique in structural biology to a mainstream method practiced in a rapidly growing number of laboratories around the world. From its beginnings about 40 years ago, cryoEM has had a major impact on the study of membrane proteins, in particular the energy-converting systems from bacterial, mitochondrial and chloroplast membranes. Early work on two-dimensional crystals attained resolutions ∼3.5 Å, but at present, single-particle cryoEM delivers much more detailed structures without crystals. Electron cryo-tomography of membranes and membrane-associated proteins adds valuable context, usually at lower resolution. The review ends with a brief outlook on future prospects.
Keywords: electron cryo-microscopy, electron cryo-tomography, photosynthesis, respiratory chain complexes, membrane structure, ATP synthase
Introduction
Over the past 40 years, electron cryo-microscopy (cryoEM) has advanced from a rarified, avant-garde technique pursued by a small number of dedicated laboratories, into one of the most powerful and sought-after methods in molecular cell biology. CryoEM can address a wider scope of biological questions than any other method in structural biology, on samples ranging from tens or hundreds of nanometers in electron cryo-tomography (cryoET) to truly atomic resolution in single-particle work. CryoEM is having a major impact on our understanding of membrane systems, in particular the large energy-converting complexes of mitochondria and chloroplasts, but also ion channels, membrane transport proteins, protein translocases and membrane-perforating toxins. I present this breathtaking development from the perspective of someone who witnessed it at first hand from its beginnings in the early 1980s to the present day. Please note that this short paper is intended as a personal account rather than a comprehensive overview, focusing on how our own projects have progressed and benefitted from cryoEM, without wishing to detract in any way from the outstanding contributions of numerous colleagues.
Early days
When I completed my PhD with Nigel Unwin at the MRC-LMB in 1981, biological electron microscopy was mostly concerned with negatively stained samples, usually in uranyl acetate. Negative stain was able to produce useful projection views, and in the right hands even three-dimensional (3D) maps of objects such as ribosomes or viruses, at resolutions up to ∼20 Å, limited by the grain size of the stain. These images and maps showed the overall outer shape, but no internal details of the structures were visible. Looking inside the molecules to find out how they work was not possible. The most notable and by that time already famous exception was Richard Henderson’s and Nigel Unwin’s 7 Å map of bacteriorhodopsin, the first high-resolution EM structure of any biological macromolecule and the first structure of a membrane protein to reveal trans-membrane α-helices [1]. Richard and Nigel had collected data of unstained purple membrane, protected against dehydration in the high vacuum of the microscope column in the quasi-aqueous environment of an air-dried glucose solution. Purple membranes are near-perfect two-dimensional (2D) crystals of bacteriorhodopsin that form naturally in the cell membrane of some halophilic archaea. My PhD project was to apply the same approach to 2D crystals of cytoplasmic ribosomes that formed naturally on the surface of endoplasmic reticulum membranes of hibernating lizards. The resolution we achieved was only ∼60 Å, due to the poor inherent order of the ribosome arrays, even though they were lightly crosslinked with glutaraldehyde, and air-drying in glucose probably did not help. Still, since glucose and protein scatter electrons roughly to the same extent, the contrast in the micrographs came mainly from the denser, more strongly scattering ribosome components, and therefore, the resulting structure gave us a first low-resolution map of the ribosomal RNA [2,3]. The membrane was nowhere to be seen.
Data for both the bacteriorhodopsin and the ribosome maps had been collected at room temperature, because at the time the development of electron cryo-microscopes had barely begun. The pioneering groups were at the Lawrence Berkeley National Laboratory in the USA (Glaeser), EMBL Heidelberg (Dubochet), Osaka University (Fujiyoshi) and the Fritz-Haber-Institute in Berlin (Zemlin). Jacques Dubochet at EMBL [4] and Robert Glaeser in Berkeley [5] were experimenting with frozen-hydrated specimens. At the MRC-LMB in Cambridge, Richard Henderson was perfecting computer programs for processing high-resolution images of 2D crystals. In 1990, he and his colleagues published the first cryoEM structure, again bacteriorhodopsin, at near-atomic resolution [6]. The 3.5 Å map was the culmination of a 15-year effort, combining electron diffraction data that provided very accurate amplitudes of Fourier terms, with precise structure factor phases obtained by processing of high-resolution images. The majority of images had been recorded at liquid-helium temperature with a prototype 100-kV instrument, called Suleika, at the Fritz Haber Institute of the Max Planck Society in Berlin, in the research group of Fritz Zemlin [7].
Recording a single image on photographic film with the Suleika required no fewer than 70 steps of manual operation, not counting film development, quality assessment by optical diffraction and digitization on a flat-bed microdensitometer. Somewhere in the middle of this bafflingly complex procedure, Fritz would call out ‘Freuen!’ (rejoice!) to those around him, who looked on in amazement. Rejoicing was certainly in order if nothing had gone wrong up to that point, which did happen occasionally. The Suleika had a helium-cooled superconducting objective lens, designed and built by Isolde Dietrich at the company Siemens in Munich [8]. The superconducting lens was extremely stable, which had its advantages, but the objective lens current could not be changed without warming it up, and therefore, each image had to be focused by adjusting the acceleration voltage. Many of the film images at high tilt angles for the 3.5 Å map came from Ken Downing in Berkeley. They were recorded with a liquid-nitrogen-cooled 100-kV JEOL 100B, converted into the world’s first electron cryo-microscope by Ken Taylor and Robert Glaeser [9,10]. The Berkeley 100B was far ahead of its time in other respects as well: it could be operated in spotscan mode, which largely avoided beam-induced movement, and it was equipped with a field emission electron gun, which became standard in cryoEM only 15–20 years later.
For my postdoc, I was hoping to combine my two passions in science—membrane proteins and high-resolution cryoEM. I was therefore looking for a membrane protein amenable to structure determination by the methods that had been devised by Richard Henderson and Nigel Unwin for purple membrane. Fortunately, I found such a protein in the light-harvesting chlorophyll–protein complex from the photosynthetic membranes of chloroplasts, LHC-II for short. I was introduced to it by Ernst Wehrli at the ETH Zürich in Switzerland. Ernst had attended the first-ever EMBO course on EM and image processing (now known as the EMBO cryo-course, but at the time there was no cryo). The course was held at the MRC-LMB in 1978 by Linda Amos, Richard Henderson, Nigel Unwin, Aaron Klug, Tony Crowther and others, with first-year PhD students like me as instructors and luminaries such as Michael Rossmann, the world-famous protein crystallographer, as participants.
LHC-II has a natural, fortuitous (but unphysiological) tendency to form crystalline arrays when the isolated, detergent-solubilized complex is precipitated from solution with monovalent or divalent cations. That much was known from freeze-fracture electron microscopy when I started my postdoc at the ETH in Ernst Wehrli’s group, but it took more than two years to persuade the complex to form large, well-ordered, purple-membrane-like 2D crystals that would lend themselves to electron diffraction and high-resolution image processing [11]. It took another year to work out an initial structure of negatively stained LHC-II membrane crystals by electron crystallography [12]. The map resolution was 16 Å. None of this was cryoEM, but the stage was set.
LHC-II at liquid-helium temperature
Since there were no instruments for high-resolution cryoEM anywhere nearby, I took the 2D crystals to Berkeley, where Ken Downing and I were able to record images for a 3.7 Å projection map of LHC-II with their JEOL 100B [13]. The visit proved that the crystals had potential, but large-scale data collection with this ageing machine (bits of the mu metal magnetic shielding sometimes dropped off the ceiling during a recording session) was out of the question. The temperamental home-built cryo-stage did not go much below −120°C. Occasionally, the column vacuum would collapse for no apparent reason, and on the fluorescent screen, I could watch my precious sample freeze over with a thick layer of contaminating ice. Fixing the microscope and getting it ready for another cryo-transfer took Ken and his team most of the day. Because I was not able to help and there was nothing else to do, I had ample time to read and explore the beautiful UC Berkeley campus. I also took the 2D crystals to Berlin, but Suleika’s long-winded and idiosyncratic image recording routine would not have made large-scale data collection easy either. In any case, the Berlin group seemed none too keen to accommodate a young whipper-snapper like me, who was not even a professor. A perfect solution offered itself when I visited Yoshinori Fujiyoshi, then at the Protein Engineering Research Laboratory (PERI) in Osaka. Yoshi had designed and built a top-entry, helium-cooled cold stage for his 400-kV JEOL electron microscope [14]. The stage was particularly suitable for high-resolution cryoEM of 2D crystals, and Yoshi welcomed me with open arms. The lens was not superconducting, and therefore, it took only ∼10 manual operations to record one film image, which made data collection feasible.
I used my Heisenberg Fellowship from the DFG for repeated visits to Yoshi’s laboratory. I took the developed negatives back to EMBL Heidelberg, where in the meantime I had taken up a research group leader position. Together with my first postdoc Da Neng Wang, now Professor of Structural Biology at New York University, we scanned and processed the films. Within a year or two, we had gathered a sufficient number of images for a 3.4 Å structure of LHC-II [15] (Fig. 1). The complementary high-resolution electron diffraction patterns required no more than a standard electron microscope with a liquid-nitrogen-cooled Gatan cryo-transfer holder, and Da Neng and I collected them with the JEOL 2000 at EMBL [16]. The 3.4 Å map of LHC-II was the second high-resolution structure determined by cryoEM (again a membrane protein) and proved that the method worked for specimens other than bacteriorhodopsin. The map afforded fascinating insights into how chlorophylls and carotenoids were held and oriented by the polypeptide scaffold in an optimal position for harvesting solar energy and photoprotection.
Fig. 1.
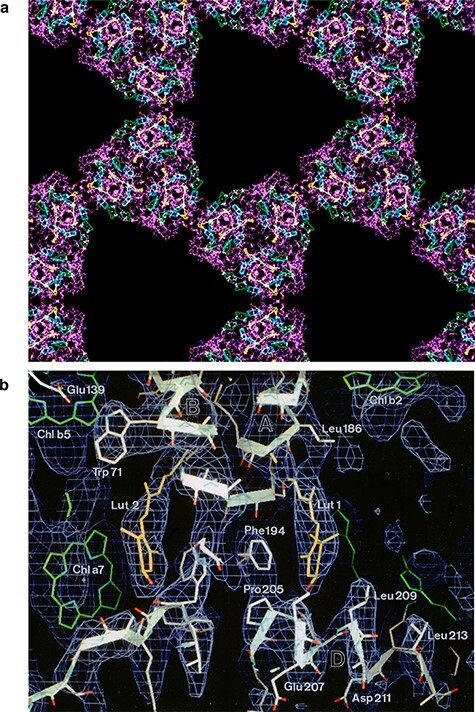
Structure of plant light-harvesting complex LHC-II determined by electron crystallography of two-dimensional crystals at liquid-helium temperature. (a) Atomic model of LHC-II trimers on the 2D crystal lattice. Polypeptide (purple) with bound chlorophylls (green and cyan) and carotenoids (yellow). (b) Details of the 3.4 Å map with fitted polypeptide (white), chlorophylls (green) and carotenoids (yellow). From [15].
The LHC-II structure was the overture to a number of highly successful collaborations for which colleagues from other laboratories teamed up with Yoshi to collect data with his superb instrument. Tom Walz came with his 2D crystals of aquaporin he had grown with Andreas Engel in Basel (Switzerland) [17] and Inga Schmidt-Krey and Hans Hebert with theirs of microsomal glutathione transferase [18]. An enduring partnership ensued with Nigel Unwin. Nigel recorded countless images of his tubular crystals of nicotinic acetyl-choline receptor at increasingly higher resolution with Yoshi’s microscopes [19,20], culminating in a 4 Å map of the receptor in the membrane that resolved sidechains [21]. Yoshi was accumulating a growing number of helium-cooled 400-kV and later 300-kV instruments, each of them filling a fairly large room, which all moved with him from Nara to Kyoto and then to Nagoya. When Nigel was awarded the Jeantet Prize in recognition of his groundbreaking work on the acetyl-choline receptor, he bought an apartment in Kyoto for his frequent stays, and Yoshi kept a JEOL 4000 specially for him that the Kyoto team called Janet. When I was appointed as a director at the Max Planck Institute of Biophysics in Frankfurt in 1997, it was my greatest wish to set up my own Yoshi-style microscope, and I was fortunate in that the Max Planck Society afforded me the means to do so. Like Yoshi’s latest model it was a 300-kV instrument with a liquid-helium-cooled top-entry stage, and it worked very reliably until 2012. Liquid helium was roughly twice as effective in protecting the specimen against radiation damage as liquid nitrogen [22,23], but also several times more expensive, and supplies were dwindling. Fortunately, it was possible to compensate for the lower signal-to-noise ratio by collecting and processing a correspondingly larger number of images at liquid-nitrogen temperature. When the opportunity arose, we replaced the JEOL 3000 SFF with a similar model, the JEOL 3200 FSC, which had a side-entry stage that could be cooled with either liquid nitrogen or liquid helium. Liquid nitrogen was less demanding and less costly, and we used it for cooling this new instrument almost exclusively.
Years later, we discovered another and—at least for me—completely unexpected disadvantage of liquid-helium cooling. It turned out that beam-induced movement of unsupported cryo-specimens in vitreous water is up to four times worse at liquid-helium than at liquid-nitrogen temperature [24]. This seemed to matter less for 2D crystals, which were routinely deposited on a continuous carbon support (even so, at least 80% of the film negatives showed signs of severe beam-induced movement, which made them useless). However, it does matter greatly for single-particle cryoEM. In retrospect, this finding explained why we had never been able to record any good single-particle images with the helium-cooled JEOL 3000. The reasons for the excessive beam-induced movement of vitrified solutions at liquid-helium temperature are not understood and need to be investigated further, if the superior cryo-protection qualities of liquid helium are ever to be taken advantage of for single particles. The causes may be related to the poor conductivity and viscous fluidity of vitreous water at very low temperatures, where carbon or graphene films are both highly conductive and mechanically stable.
2D and 3D crystals of membrane proteins
The liquid-helium-cooled top entry stage of the JEOL 3000 SFF was particularly appropriate for 2D crystals, but suitable specimens were hard to come by. No more than around 10 2D protein crystals are known to diffract electrons to better than 4 Å: purple membrane; LHC-II; the bacterial porins OmpF and PhoE; aquaporins-0, 1 and 4; microsomal glutathione transferase and its relatives (for reviews, see [25,26]); and zinc-induced sheets of tubulin [27], the only non-membrane protein amongst them. All are naturally abundant and readily available from natural sources in milligram quantities and thus amenable to extensive 2D crystallization screens. In the late 1990s, it became routine to produce—at first mostly bacterial—membrane proteins in similar amounts by heterologous expression. Quite a few of these formed 2D arrays, mostly in the shape of wide, flattened tubular vesicles. In our JEOL 3000, some yielded useful projection maps at 4–7 Å or 3D structures at 6–8 Å resolution by electron crystallography, notably the sodium-proton antiporters NhaA [28,29] and NhaP [30,31], the osmoregulatory transporter BetP [32], the bacterial protein translocase SecYEG [33–35] and the light-gated ion channel channelrhodopsin [36,37]. However, the 2D crystals remained small, and none of them were ever as good as LHC-II or purple membrane. At the same time, heterologously expressed membrane transporters invariably produced good 3D crystals, which meant that we [38–40] or others [41,42] were able to solve their high-resolution structures by X-ray crystallography. With a few rare exceptions, the—mostly hydrophobic—interactions that hold 2D crystals together are weaker and less numerous than the—mostly polar—crystal contacts in a 3D protein lattice, and therefore, the 2D crystals remained small and tended to be poorly ordered. Whatever the reason, for anyone interested in understanding the structure and mechanisms of membrane proteins rather than optimizing the growth of 2D crystals, X-ray crystallography was clearly the way to go. So for the next 10 years, this was what we did, until, very recently, single-particle cryoEM caught up. Structures of even fairly small membrane proteins can now be determined without any crystals at all, at considerably higher resolution.
Cryo-tomography of mitochondrial membranes
As a method in structural biology, cryoEM has the great advantage of not being restricted to crystals or pure protein solutions. cryoET can examine objects from any biological source of any shape and—within limits—size, provided that they are thin enough to be considered as weak phase objects, so that multiple electron scattering is negligible. In principle, the same microscopes can be used equally well for the three forms of cryoEM—electron crystallography, single-particle analysis and cryoET—as long as the sample can be tilted to about ±60°, which is a prerequisite for tomography and electron crystallography.
One of the first non-crystalline specimens we investigated by single-particle cryoEM was the mitochondrial respiratory supercomplex, also called the respirasome. The respirasome is a 1.7-MDa assembly of three mitochondrial respiratory chain complexes, complex I, III and IV, the latter two also known as cytochrome c reductase and cytochrome c oxidase. The supercomplex was first identified on blue-native gels of detergent-solubilized mitochondrial membranes by the Frankfurt membrane biochemist Hermann Schägger [43]. Initially, we extracted it from gels and examined it in negative stain [44]. Later, we purified it for cryoEM by gradient centrifugation [45]. The resolution of ∼20–25 Å we achieved with film images was low by today’s standards, but good enough to dock X-ray structures of the component complexes, which indicated where they interacted. In the meantime, with better preparation techniques but mainly thanks to better electron detectors and image processing programs (see below), several cryoEM structures of respiratory chain supercomplexes at higher resolution have been published by us [46] and others [47,48]. The precise function of these large assemblies and how they work in fine-tuning cellular metabolism and organismal fitness are not known and remain controversial.
At the time, there was a debate as to whether the respiratory chain supercomplexes actually existed in mitochondria or whether they were artefacts of detergent solubilization. It occurred to me that, if they existed, they should be visible in tomographic volumes of mitochondrial membranes. The membranes were easy enough to prepare, but we had never done cryo-tomography. Mike Strauss, who was then in the middle of his PhD and now runs the cryoEM facility at McGill University in Montreal, visited the laboratory of Grant Jensen at CalTech to learn the technique. He stayed for 2 weeks and on his return to Frankfurt established the procedure on our recently acquired FEI Polara electron microscope. Compared to 2D crystallography with the JEOL 3000 or single-particle cryoEM, cryo-tomography with the Polara turned out to be remarkably straightforward.
We collected tilt series of isolated inner mitochondrial membranes and generated 3D tomographic volumes, as Mike had learned at CalTech. But when we examined the volumes in the computer, the first thing that struck us was not the respirasome, but long rows of lollipop-shaped 10-nm particles that were clearly the F1 heads of the mitochondrial F1Fo ATPase [49]. We were intrigued that seemingly identical rows showed up in consecutive parallel slices through the tomographic volumes. At first, we suspected a problem with the program that had generated the tomograms, but when we discovered parallel rows of the same particles in perpendicular views, it dawned on us that they were rows of ATP synthase dimers. Dimeric forms of the mitochondrial ATP synthase had been known for some time from negative-stain single-particle EM [50,51], but because they all looked different, I thought they were bound to be artefacts. Also, it was difficult to see how a V-shaped dimer of two such large assemblies could sit stably in a planar lipid bilayer.
Mitochondrial ATP synthase dimers
Our tomographic volumes showed that the membranes were of course far from planar. The edges of the inner membrane cristae were tightly bent, and the dimer rows always sat in the regions with the smallest, ∼17-nm curvature radius. The dimer rows actually induce high local membrane curvature and indeed cristae formation, as we now know and were able to demonstrate by reconstituting purified ATP synthase dimers into liposomes [52]. Tomographic volumes of the reconstituted proteoliposomes resembled those of mitochondrial inner membranes closely and the membrane curvature at the position of the dimer rows was the same [53]. The rows run along the tightly bent cristae rims or ridges, but they are absent from the remaining regions of the inner membrane that are indeed roughly planar. Long rows of ATP synthase dimers are a defining feature of the mitochondrial inner membrane, and the high local curvature of the ridges is characteristic of metazoan and fungal mitochondria. Dimer rows are ubiquitous in mitochondria of all of the roughly 15 species we investigated, even though the dimers themselves vary greatly in terms of overall structure and subunit composition [54] (Fig. 2). ATP synthase dimers of metazoans and fungi differ significantly from those of ciliates such as Paramecium [55], which in turn differ from those of trypanosomes and Euglena [56]. Dimers of unicellular green algae including Chlamydomonas and Polytomella are strikingly different from all the above [57,58], and green-plant dimers are again different (unpublished). Yet, mitochondria of all these groups have dimer rows and their inner membranes form cristae, even though their appearance varies from lamellar (metazoans, fungi, green plants) to tubular (ciliates) to anywhere in between (Polytomella, trypanosomes). The dimer rows are often >1 µm long [49] and conspicuous in tomographic volumes of all unfixed, flash-frozen mitochondria. The reason why they had escaped notice for so long is that they do not survive the preparation steps (chemical fixation, dehydration, plastic embedding and heavy-metal staining) for conventional room-temperature EM. They are difficult to see by any technique other than cryoET, although they have been spotted once by rapid-freeze deep-etch electron microscopy of Paramecium mitochondria [59].
Fig. 2.
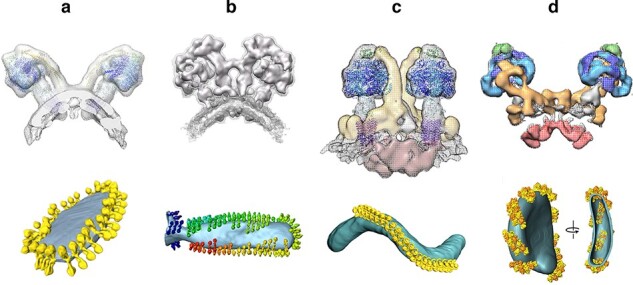
Cryo-ET of mitochondrial ATP synthase dimers. Subtomogram averages (above) and rendered tomographic volumes of inner mitochondrial membranes (below) from (a) Saccharomyces cerevisiae, (b) Polytomella sp., (c) Paramecium tetraurelia and (d) Euglena gracilis. From [54].
ATP synthase dimers, dimer rows and inner membrane cristae seem to play a central part in cellular and organismal physiology, as do the mitochondria themselves. The tight curvature at the cristae ridges is a prerequisite for close lamellar stacking, e.g. in cardiac muscle [60] (Fig. 3a). Cristae stacking increases the inner membrane surface area, and therefore, the potential for ATP production, by a factor of ∼10 [54], but this is most likely not the main reason why the dimers rows are so important. Yeast mutants lacking the dimer-specific ATP synthase subunits e and g that stabilize the dimers do not have dimer rows and do not develop cristae [61]. The mutants are less fit than wild type, grow more slowly and, according to one study, their membrane potential is reduced [62]. Even more strikingly, Drosophila fruit flies in which subunits equivalent to e and g are knocked down are deficient in germline stem cell maturation [63]. Mitochondria of these knockdowns do not have cristae, which points to a profound role of the cristae in Drosophila development. At present, we cannot even guess what this role might be.
Fig. 3.
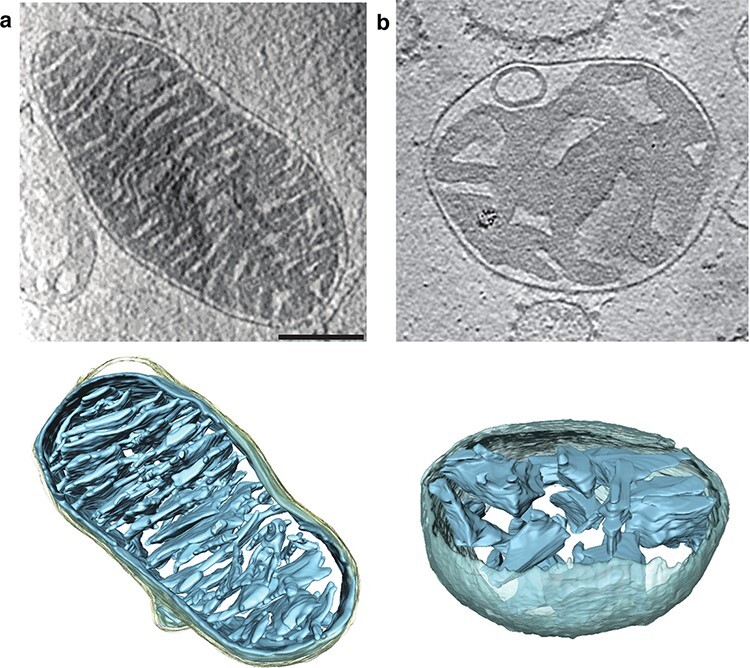
Cryo-ET of whole mitochondria from (a) mouse heart and (b) mouse liver. Upper panels: slices through tomographic volumes (scale bar, 250 nm). Lower panels: segmented 3D volumes of two typical mitochondria. Heart mitochondria have closely stacked, roughly parallel cristae (blue). Cristae of liver mitochondria are more loosely arranged and not stacked. Grey, inter-membrane space; light yellow, outer membrane. From [60].
After the dimer rows had sidetracked us for some time, we collected more tomograms of mitochondrial membranes to look for respiratory chain complexes and respirasomes. Indeed, they were there, in the flat membrane regions on either side of the ATP synthase rows. We detected the L-shaped density of the 1-MDa complex I next to ATP synthase dimers in four different species and were able to confirm its identity by antibody labelling [53]. The resolution and quality of the tomograms, recorded in our Polara with a 2k charge-coupled device (CCD) camera and Tridiem filter, was not sufficient to tell reliably whether or not they contained complex III or IV as well, which would have meant that they were respirasomes. A few years later, after we had installed our first Titan Krios electron microscope with a Quantum energy filter and Gatan K2 detector, we re-examined isolated inner membranes of bovine, fungal (Yarrowia lipolytica) and plant (Asparagus officinalis) mitochondria by cryoET. The tomographic volumes were very much clearer and enabled us to generate sub-tomogram averages of respirasomes from the three species at about 25 Å resolution [64] (Fig. 4). Like the dimer rows, the supercomplexes were ubiquitous. They looked similar in mammalian, yeast and plant mitochondria, which told us that interactions between the two major components, complexes I and III, were conserved.
Fig. 4.
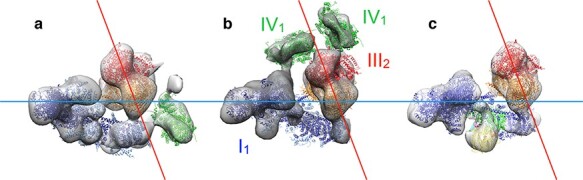
Subtomogram averages of respiratory chain supercomplexes in inner membranes of (a) bovine heart, (b) fungal (Y. lipolytica) and (c) plant mitochondria. The arrangement of complex I (blue) and complex III dimer (orange) is conserved, as indicated by the blue and red guidelines. Adapted from [64].
Cryo-tomography of whole mitochondria
Having examined membrane protein complexes in mitochondrial membrane fragments, we were curious to see the dimer rows in intact organelles. We were especially interested in mitochondria of the filamentous fungus Podospora anserina, which has an exceptionally short lifespan of 20 days, and we were wondering how the mitochondria and dimer rows would change during ageing. To our surprise, we found that the inner membrane of mitochondria from aged Podospora underwent a profound transformation within a few days [65]. The cristae disappeared, the dimer rows became increasingly disrupted, the dimers broke up into monomers and finally the mitochondrial matrix vesiculated. The double rows along the cristae ridges, or what was left of them in ageing cultures, were easily visible in small organelles [65] (Fig. 5), but in larger mitochondria, molecular detail was obscured by the dense mitochondrial matrix.
Fig. 5.
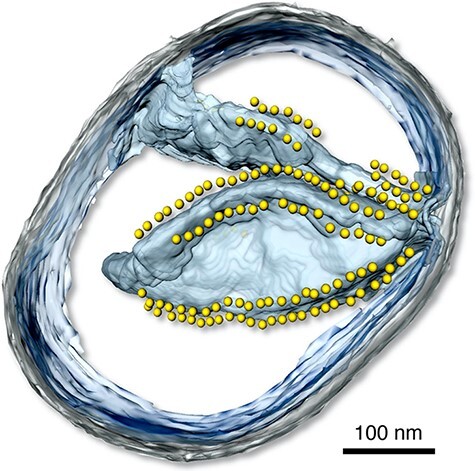
Three-dimensional tomographic volume of a small mitochondrion from P. anserina. The inner membrane is light blue, the outer membrane is grey. Yellow spheres indicate the catalytic F1 heads of ATP synthase dimers arranged in rows on the cristae ridges. Scale bar, 100 nm (B. Daum and W. Kühlbrandt, MPI of Biophysics, unpublished).
In collaboration with the MPI for the Biology of Ageing, we examined age-related changes in mitochondria of mouse and Drosopohila melanogaster, two popular, longer-lived model organisms. The differences were less drastic than in Podospora, but nonetheless clear [60]. In mouse, changes in inner membrane morphology were strongly organ-dependent. Heart mitochondria were particularly resilient to ageing, such that mitochondria from young and old mice were indistinguishable and equally tightly packed with lamellar cristae (Fig. 3a). We found that liver mitochondria have fewer cristae, which are not stacked (Fig. 3b). In old mice, around 20% of liver mitochondria developed a central vacuole that was contiguous with the cristae. In kidney, a similar proportion of mitochondria lost their inner membrane morphology. These changes presumably go along with reduced respiratory activity, ATP production and organellar fitness with increasing age. The picture was similar for Drosophila.
As a postdoc in the group, Vicki Gold used cryoET to investigate protein import into mitochondria. She generated a pre-protein construct with a mitochondrial import signal sequence and conjugated it with fluorescent quantum dots by biotin/streptavidin biochemistry. When import-competent mitochondria were incubated with this construct, the quantum-dot-labelled pre-protein got stuck in the translocase of the outer mitochondrial membrane, known as the Translocase of the Outer Membrane (TOM) complex. Her approach ensured that only complexes active in protein translocation were labelled, and that she could keep track of them by fluorescence during cryoEM grid preparation. In the tomographic volumes, the dense quantum dots showed up beautifully on the mitochondrial outer membrane, highlighting the location of active import complexes [66]. By subtomogram averaging, she also managed to visualize the import receptor protein subunits protruding on the cytoplasmic side at low resolution. Apparently, the receptor subunits are flexible, which explains why they have so far eluded us in our single-particle cryoEM studies of the isolated TOM complex (see below).
Cryo-tomography of reconstituted membranes
CryoET also works well for membrane assemblies reconstituted in vitro. Oli Daumke and Katja Faelber at the Max Delbrück Centre in Berlin were determining the X-ray structure of the mitochondrial membrane-remodelling GTPase Mgm1 and wanted to know how it interacts with the lipid bilayer. In collaboration with them, Lea Dietrich was able to assemble a lattice of helical Mgm1 filaments on pre-formed liposomes. The helical arrays shaped the liposomes into thin long tubes, and by image processing and sub-tomogram averaging, she generated a volume that indicated how the GTPase in the X-ray structure interacts with the membrane [67]. To our surprise, Lea discovered similar but different helical arrays inside the membrane tubes after some of them had ruptured, so that the protein could diffuse to the inside. The inside Mgm1 lattice generated wider tubes, presumably resembling the situation at tubular cristae junctions, which are most likely stabilized by Mgm1. The topography of the wider tubes would resemble the inner surface of the mitochondrial inner membrane, where Mgm1 is thought to promote membrane fission or fusion. The structures of the inside and outside helical lattices suggested mechanisms by which GTP hydrolysis generates a power stroke that would constrict or dilate the membrane tube [67].
Pore-forming proteins
Other membrane systems that lend themselves well to cryoET are bacterial and other pore-forming proteins. Membrane-perforating bacterial toxins are produced, or can be expressed, as soluble monomers which insert into the target membranes to form large circular pores. In collaboration with Trinad Charkaborty at the University of Giessen (Germany), we expressed, crystallized and determined the X-ray structure of the membrane-perforating toxin listeriolysin (LLO) from Listeria monocytogenes [68]. Later, we turned to pneumolysin (PLY), the virulence factor of the human pathogen Streptococcus pneumoniae. Both toxins insert into cholesterol-containing membranes of animal cells, assembling into ring-like structures of 40 or more monomers [69]. We obtained the structure of the closed PLY prepore in the membrane by cryoET and subtomogram averaging, and that of the fully open, 42-membered ring of the isolated, detergent-solubilized pore complex by single-particle cryoEM [70] (Fig. 6). Comparison of the prepore and pore structures indicated how the four-domain monomer inserts into the lipid bilayer and then, upon conversion of one of its domains into 80 Å beta hairpins, forms ∼400 Å beta barrels that perforate the bilayer and kill the cell. Molecular dynamics simulation in collaboration with Gerhard Hummer at the MPI of Biophysics revealed the detailed mechanism of pore formation and extrusion of the lipid plug to open the pore [71].
Fig. 6.
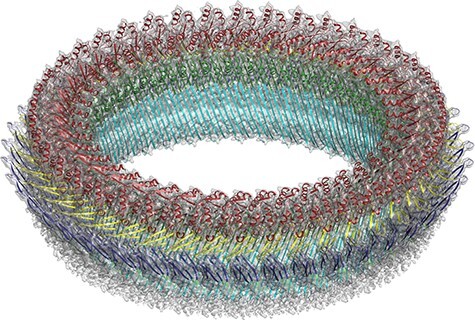
Single-particle cryoEM structure of the pneumolysin (PLY) toxin pore complex at 4.5 Å resolution. The four fitted domains in the 2.4 Å X-ray structure of the PLY monomer are shown in colour. One domain has rearranged into ∼80 Å beta hairpins (cyan) that form a 42-membered ring with an inner diameter of ∼400 Å perforating the membrane. From [70].
A very different way in which a protein can perforate a membrane, this time from the inside to allow a virus to escape from an infected cell, was brought to our attention by David Prangishvili at the Institut Pasteur in Paris, France. David was studying phages that infect certain thermophilic archaea, inducing them to produce large numbers of a small, 10-kDa membrane protein encoded in the viral genome. The gene product inserts spontaneously into the archaeal membrane, where it self-assembles into ∼1000 Å pyramids of 7-fold symmetry that pierce through the S-layer. The S-layer forms a tough protective lattice around archaea living in extreme environments and would otherwise trap the virus in the cell. Once the S-layer is punctured, the sides of the pyramids open like the petals of a flower to create ∼200 nm holes, through which the new virions emerge [72].
Chloroplast tomography
Ever since our LHC-II work in the 1990s, we had kept a close interest in chloroplasts, the characteristic membrane organelles of green plants and algae, and their photosynthetic membrane proteins, including the photosystem II reaction centre [73,74]. Chloroplasts are much larger than mitochondria, measuring around 5 µm across. They are too bulky and dense for direct tomographic imaging of plunge-frozen samples, the approach we had taken with whole mitochondria. At the beginning of his PhD, Bertram Daum (now a PI at the University of Exeter, UK) found out that chloroplasts ruptured by mechanical blotting on the cryoEM grid maintained the intricate molecular organization of their membrane networks at least for the few seconds it takes to flash-freeze them [75,76]. The protein-dense chloroplast stroma had leaked out, which improved the contrast and resolution of the tomograms. By cryoET of such samples, Bertram examined the arrangement and distribution of large membrane protein complexes in chloroplast thylakoids. We were especially interested in the chloroplast ATP synthase, to see whether it formed dimers and dimer rows as in mitochondria. The tomographic volumes of chloroplast grana stacks and unstacked stromal lamellae showed unambiguously that this was not the case. The chloroplast ATP synthase was homogenously distributed over the stroma thylakoids and grana end membranes, and all copies were monomers (Fig. 7). In particular, there were no dimers or dimer rows around the tightly curved edges of the thylakoid grana, where one might have expected to find them. In retrospect, this is not surprising since the chloroplast ATP synthase lacks the subunits that are required for dimer formation [54]. The same is true of the related bacterial ATP synthases, which are likewise all monomers.
Fig. 7.
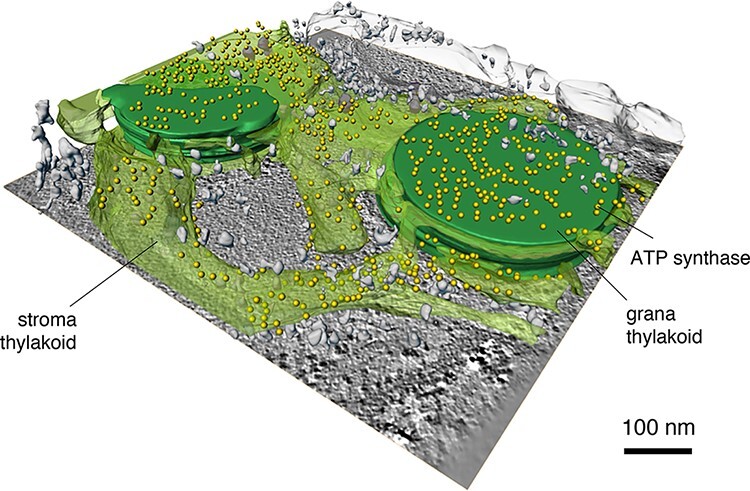
CryoET of chloroplast thylakoid membranes. F1 heads (yellow) of the chloroplast F1Fo ATP synthase are confined to stroma and grana end membranes. The chloroplast ATP synthase is monomeric and not found at the highly curved grana edges. Grana stacks (dark green); stroma membranes (transparent green). Scale bar, 100 nm. Courtesy of Bertram Daum, MPI of Biophysics.
More recently, when he joined the laboratory as a PhD student, Davide Floris adopted the same approach of specimen preparation to examine the molecular landscape of etioplasts, the precursor organelles of chloroplasts in the leaves of greening plants. The etioplast inner membrane is special because it forms the pro-lamellar body, a meshwork of tubular membranes arranged in the lattice of a lipidic cubic sponge phase. This had been known since the 1970s from conventional EM of plastic sections, but no one knew how or why the membranes were arranged in this peculiar way that appears to have no parallel in biology. By cryoET, Davide discovered that the membrane tubes were surrounded, and indeed formed, by helical arrays of large numbers of a small, membrane-associated protein. By sub-tomogram averaging, he obtained a map at 9 Å resolution and was able to show that the protein is the 37-kDa light-dependent protochlorophyllide oxidoreductase (LPOR) [77]. LPOR is the most abundant enzyme in etioplasts. It converts the pale yellow protochlorophyllide into chlorophyllide, which in turn is converted into the deep-green chlorophyll by chlorophyll synthase. Both enzymatic reactions are key steps in the biogenesis of photosynthetic reaction centres and light-harvesting complexes of chloroplasts, including LHC-II that consists of 33% chlorophyll by weight.
Direct electron detectors
Before 2012, only very few cryoEM structures reached a resolution of 3.5–4 Å, where large sidechains become visible. All were of 2D crystals, mostly of membrane proteins (see above), or of large icosahedral viruses (see [78]). For assemblies smaller than ∼20 MDa that did not consist of large numbers of identical units related by strict crystallographic, helical or point-group symmetry, high resolution was out of the question. The reason was the poor signal-to-noise ratio of individual electron micrographs, which meant that smaller particles could not be aligned and averaged with sufficient accuracy to warrant high resolution. Until that point, photographic film was by far the best recording medium for cryoEM. Although tedious to use, it was very much better than the solid-state CCD electron detectors available at the time, which were quick and convenient, but their modulation transfer function (MTF), and hence, detective quantum efficiency (DQE) at high resolution was poor. Richard Henderson’s incisive 1995 review [79] had shown beyond any doubt that of the three forms of radiation which can in principle deliver images of individual biological macromolecules—X-rays, neutrons and electrons—only the latter held the promise of producing such images at 3 Å resolution or better. Therefore, if cryoEM was ever to realize this promise and come close to the success of X-ray crystallography, electronic detectors with a signal-to-noise ratio and DQE at least as good as film would be mandatory.
In 2004, Richard set up a consortium of engineers and members of the cryoEM community to develop such a detector. Apart from himself and his colleagues Greg McMullan and Wasi Faruqi at the MRC-LMB, core members of the team were Renato Turchetta and Nicola Guerrini at the Rutherford Appleton Laboratories (RAL) outside Oxford, UK. Both were brilliant electronics engineers with expertise in detector chip design. Together, Renato and Nicola embarked on designing and building a solid-state device with all the desirable properties of an optimal electron detector: a large active area (at least 4000 × 4000 pixels), suitable pixel size (the consortium decided on 14 µm), a DQE as close as possible to 1 (where 1.0 would be that of a perfect detector), high sensitivity, rapid readout, low readout noise, high resistance to electron irradiation and reasonable production cost. These very demanding specifications were defined in preliminary consortium meetings. The new generation of detectors would be based on complementary metal oxide semiconductor technology that was already in widespread use in digital cameras and mobile phones, but needed to be adapted to the stringent requirements of high-resolution cryoEM. Although Richard could easily have shouldered the whole effort by himself, he was intent on putting the endeavour on a broader scientific base and invited me and Jürgen Plitzko of the MPI for Biochemistry in Martinsried (Germany) to join the consortium. The FEI company in Eindhoven (The Netherlands; acquired by Thermo Fisher in 2017) joined with a view of producing and marketing the detector in due course. The LMB, the two MPIs, FEI and RAL each contributed 20% to the considerable cost of getting this project off the ground. It was clear to us that if these new direct electron detectors could be made to work, everyone in the cryoEM world would want to have one. We now know they work far better than we could have imagined in our wildest dreams and everyone does want one. Indeed, most cryoEM groups have at least one already.
Initially, test chips with different pixel topologies were designed and produced at RAL and evaluated by the LMB detector development team. Criteria included signal-to-noise ratio, radiation hardness and DQE. Results exceeded expectations [80,81]. The most promising pixel design was then selected for further development. Before the camera chip went into production, the consortium decided that the silicon wafer of the detector should be back-thinned to ∼30–50 µm, which improved the signal-to-noise ratio and MTF substantially [82]. Eventually, FEI produced a back-thinned detector chip for a camera called the Falcon-II, and each consortium member obtained one prototype.
Of course, our consortium was not the only initiative in the world to develop direct electron detectors. The US company Gatan Inc., who had long experience in producing CCD cameras and energy filters for cryoEM, came out with their own design more or less simultaneously, as did the smaller Direct Electron company in San Diego, CA. Gatan’s direct electron detector camera was called the K2 Summit, and the one from Direct Electron was the DE-20. The sensor areas of all three cameras were similar (roughly 4000 by 4000 pixels), but the pixels of both the K2 Summit and the DE20 were very much smaller (5 and 6.4 µm). All three performed well (see [83]), but in practical terms, the high readout speed of the K2 (400 frames per second) meant that up to 20 times more images could be recorded per hour. Primarily for this reason, the K2 became particularly popular with cryoEM labs. It was also the camera of choice for cryoET, not least because the Falcon-II (produced by FEI) was not on offer in combination with an energy filter (essential for tomography), and post-column energy filters were at the time only produced by FEI’s competitor Gatan. On the other hand, the larger pixel of the Falcon-II meant that it had a better DQE at high spatial frequencies, which made it the best choice for high-resolution cryoEM of large single particles. Since then, both the Gatan K2 and the Falcon-II have been upgraded to the Gatan K3 and Falcon-III (and now the Falcon-IV), each with significantly improved performance, although the K3 has the same pixel as the K2. Even better, faster and more sensitive direct electron detectors are bound to be developed in the years to come.
The resolution revolution
We had our Falcon-II camera installed on our Polara electron microscope, and by October 2012, it performed to specification. With Greg McMullan’s help, we recorded our first high-resolution images of the Frh complex a few months later. Frh is a rigid, soluble, dodecameric assembly with a mass of 1.2 MDa that was isolated and purified from methanogenic archaea by our colleagues Seigo Shima and Stella Vitt at the MPI of Terrestrial Microbiology in Marburg (Germany). We had just published a nice cryoEM map of Frh from images recorded on Kodak SO163 film [84]. The nominal resolution was 5.5 Å, although β-strands were fully resolved and the map therefore looked much better. With the Falcon-II, we were able to improve the nominal resolution to 3.4 Å within a few weeks [85]. Since then, we have improved the map further to just better than 2 Å with the Falcon-III and K3 cameras (Fig. 8). The Frh map was one of the first single-particle cryoEM structures in this resolution range. The other three were the 20S proteasome from Thermoplasma thermophilum at 3.3 Å [86], the TRPV1 ion cannel from rats at 3.4 Å [87] and the large subunit of the yeast mitochondrial ribosome at 3.2 Å [88]. Two of them (Frh and the mitoribosome large subunit) were done with the Falcon-II, the other two with the K2 Summit. Together, these four structures marked the beginning of what has become known as the ‘resolution revolution’ in cryoEM [89], which is still gathering momentum.
Fig. 8.
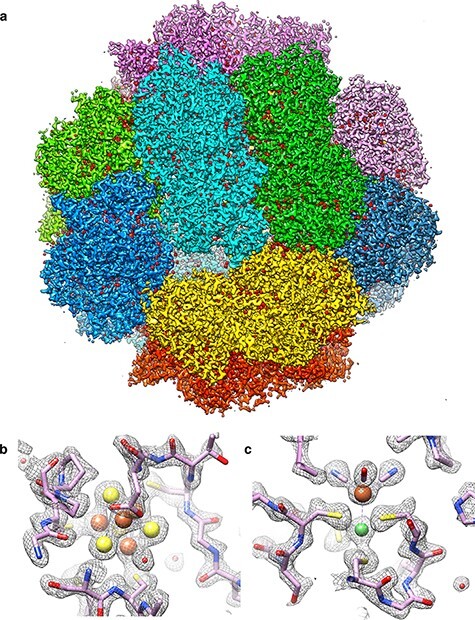
(a) CryoEM structure of Frh complex at 1.97 Å resolution, with each of the 12 heterotrimers in a different colour. Small red densities on the protein surface are water molecules. (b, c) Map details with fitted polypeptide chain and metal ions (Fe, orange; Ni, green). Sulphur is yellow. The iron in (c) coordinates one carbon monoxide and two cyanide ligands (J. Vonck and D. J. Mills, unpublished).
Although the resolution revolution was triggered by the direct electron detectors, it could not have progressed with the same force and speed without a set of powerful image processing programs that fortuitously became available around the same time. These programs enabled the cryoEM community to take full advantage of the superior image quality delivered by the new detectors. The high sensitivity and fast readout speed of the new cameras meant that the electron dose of ∼20 e/Å2 for a high-resolution micrograph could be spread over a series of movie frames. This made it possible to measure [90] and correct for beam-induced movement [91] that had been a persistent and intractable problem with film images. With the new cameras, it became convenient and quick to determine and correct for the contrast transfer function at the stage of image recording, e.g. by the programs ctffind4 [92] or Gctf [93]. Well-known and much used image program suites include EMAN2 [94], Frealign [95] and CryoSparc [96]. The most popular and perhaps most incisive program suite is RELION, developed by Sjors Scheres at the MRC-LMB [97,98], on the basis of the Xmipp programs from José-Maria Carazo in Madrid [99]. RELION applies rigorous Bayesian maximum-likelihood statistics and an intuitively obvious and generally accepted ‘gold standard’ resolution criterion based on an objective comparison of maps generated from two random halves of a data set. As the resolution revolution progresses, top software developers in protein crystallography and structural biology are joining the fray, devising powerful programs, e.g. phenix_resolve [100], to improve high-resolution cryoEM maps further.
Single-particle cryoEM of membrane proteins
Even though most single-chain membrane proteins are at present too small for high-resolution cryoEM, they often form dimers or higher oligomers, or associate with other subunits into complexes, which brings them into a suitable size range for single-particle analysis. When Arne Möller joined the department as a group leader in 2015, he focused on two such membrane proteins. His long-standing interest in ABC transporters came to fruition in the cryoEM structure of TmrAB, a 137-kDa heterodimeric ABC transporter [101]. A bound camelid nanobody added ∼15 kDa to the mass of the heterodimer, breaking its near-2-fold symmetry and helping alignment. The membrane environment of a lipid nanodisc stabilized the protein without restricting functionally important conformational changes. Arne’s team was able to resolve no fewer than eight different conformations of the transporter under turnover conditions at resolutions between 2.8 and 4.2 Å (Fig. 9). Together, the different conformations provided detailed snapshots of the complete transport cycle, as an impressive demonstration of the post-revolutionary power of cryoEM. The other moderately sized membrane protein studied by Arne and his team was a ∼210-kDa P-type ATPase from yeast that works as a lipid flippase [102]. The project was a collaboration with Poul Nissen in Aarhus (Denmark), who has a long and distinguished track record in protein crystallography of P-type ATPases. Arne and colleagues were able to resolve three different states, which together illustrated the molecular mechanism by which lipids are moved from one membrane leaflet to the other.
Fig. 9.
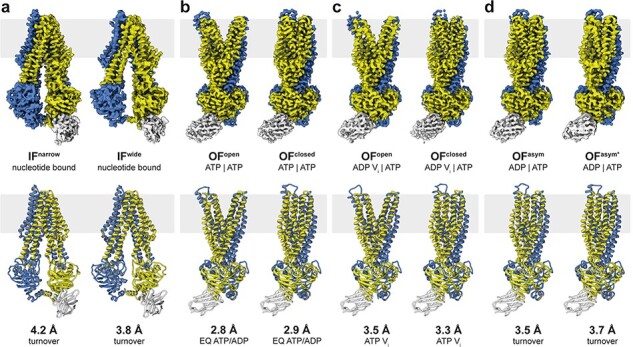
CryoEM structures of the ABC transporter TmrAB under turnover conditions. (a) Narrow and wide inward-facing conformations. (b) In the ATP-bound state, the nucleotide-binding domains associate closely and the exporter is either in an outward-facing open or occluded conformation. (c) Outward-facing open and occluded conformations also dominate the vanadate-trapped state. (d) Asymmetric states during TmrAB turnover resemble an outward-facing occluded conformation with slightly separated nucleotide-binding domains. TmrA is blue, TmrB is yellow and the bound nanobody is light grey. From [101].
Protein translocases
For years, we had been fascinated by membrane protein complexes that translocate pre-proteins into or out of cells or into cell organelles. The first of these was the protein translocase SecYEG from E. coli. As a postdoc in the group, Ian Collinson had grown 2D crystals of the complex, which Cécile Breyton analysed by electron crystallography, yielding a ∼8 Å 3D map [33,34]. The collaboration continued when Ian, by then Professor of Biochemistry at the University of Bristol (UK), was able to bind a pre-protein mimic to SecY. This gave rise to a 3D map at similar resolution, which indicated how the substrate unlocks the complex for protein export [35].
Another of our long-standing interests was the mitochondrial TOM complex. An initial collaboration with Nikolaus Pfanner at the University of Freiburg (Germany) resulted in the cryoEM structure of a heterotrimeric form from yeast mitochondria at ∼18 Å resolution. Data were recorded on film, first with an FEI Technai F20, and then with the Polara [103]. We revisited the project after the arrival of the direct detectors, this time together with Stephan Nussberger, who had been my first PhD student at EMBL and was now a professor at the University of Stuttgart (Germany). Stephan had tried for 10 years to crystallize the TOM complex from the filamentous fungus Neurospora crassa, but the crystals never diffracted to better than ∼8 Å. He had however fine-tuned the preparation of the complex from N. crassa mitochondria, which put us in a position to make rapid progress by cryoEM. As a PhD student, Thomas Bausewein collected single-particle images of the TOM complex in detergent or reconstituted into amphipols with our JEOL 3200 FSC at liquid-nitrogen temperature, which he processed to generate a map at 6.8 Å resolution [104]. The map showed how the two Tom40 β-barrels in the dimeric complex form a twin pore. The α-helical Tom20 preprotein receptor and the small Tom7, 6 and 5 subunits were assigned on the basis of the shape and size of their map density. Subunit assignment was helped by mass spectrometry indicating the size and sequence of subunits present in the complex. Since then, two higher-resolution cryoEM structures of the S. cerevisiae TOM complex have both confirmed our subunit assignments and orientations [105,106].
Respiratory chain complexes
An interesting side product of the TOM project was the cryoEM structure of the N. crassa cytochrome c oxidase, which co-purified accidentally with the TOM complex by Ni affinity chromatography. Its identity was revealed by the shape of the 5.5 Å cryoEM map [107]. As far as we know, this is the first time that an unknown protein contaminant has been identified solely on the basis of its cryoEM structure, but it will almost certainly not be the last.
In collaboration with Manuela Pereira at the ITQB in Lisbon (Portugal), Joana Sousa worked out the 3.9 Å cryoEM structure of a bacterial Alternative Complex III [108]. More recently, Alex Hahn in my group and Schara Safarian in Hartmut Michel’s department at our institute obtained a superb 2.7 Å map of a bacterial cytochrome bd oxidase that resolved a number of water molecules in its two proton channels [109]. Hartmut Michel, who pioneered X-ray crystallography of membrane proteins in the 1980s, is perhaps the most prominent recent convert in the growing number of protein crystallographers who now use cryoEM almost exclusively.
Complex I
For ∼20 years, we had collaborated closely with the group of Uli Brandt and Volker Zickermann at the Frankfurt University Medical School to study the structure of complex I from the aerobic yeast Yarrowia lipolytica by electron microscopy, initially in negative stain [110]. Complex I from Y. lipolytica has an important advantage over the mammalian complex in that it is readily accessible to directed mutagenesis. Brandt and Zickermann had established a genetic system to modify the complex of this particular organism, since the more established bakers’ yeast S. cerevisiae lacks complex I. This long-term investment is now paying off. The group had been quite successful in growing 3D crystals and determining the X-ray structure of this ∼1-MDa membrane complex, first at 6.3 Å [111] and then at 3.6–3.9 Å resolution [112]. They were thus reluctant to jettison crystallography and EM of negatively stained samples in favour of high-resolution cryoEM. Only when K.R. Vinothkumar (who had been my PhD student in Frankfurt and now runs the first Indian cryoEM facility in Bangalore) together with Judy Hirst in Cambridge published the structure of the bovine complex at 5 Å resolution [113] did they see the point of the new method, but by that time we had almost missed the boat.
Our first cryoEM structure of the Yarrowia complex [114] was not at very high resolution (7.9 Å), but did resolve the 8 FeS clusters in the peripheral arm and most of the roughly 80 trans-membrane helices in the membrane arm. It also revealed a new accessory subunit in the peripheral arm, which we identified as a sulfur transferase that links the respiratory chain to mitochondrial sulfur metabolism. The project took off when Kristian Parey joined Volker Zickermann and me as a postdoc. With the help of Deryck Mills, and taking advantage of our best microscopes and cameras, Kristian tirelessly collected large numbers of particle images of increasingly high quality. He generated 3D maps, first at 4.3 Å [115], then at 3.2 Å [116], and, most recently, at 2.1 Å resolution [117]. Janet Vonck built the atomic models. Of the recent high-resolution cryoEM structures of complex I [118,119], this latest map is in some ways the most astonishing in that it resolves ∼1600 water molecules, of which roughly 100 were found in the complex interior. The inner water molecules (Fig. 10) define a horizontal proton path that runs from the quinone reduction site at the junction between the peripheral and membrane arms, to the outlet of the proton pump at the far end of the membrane arm. Notwithstanding all this great progress, we still do not understand the mechanistic details of how the electron transfer reactions at the quinone reduction site are energetically coupled to proton transport by the three proton translocation modules in the membrane arm.
Fig. 10.
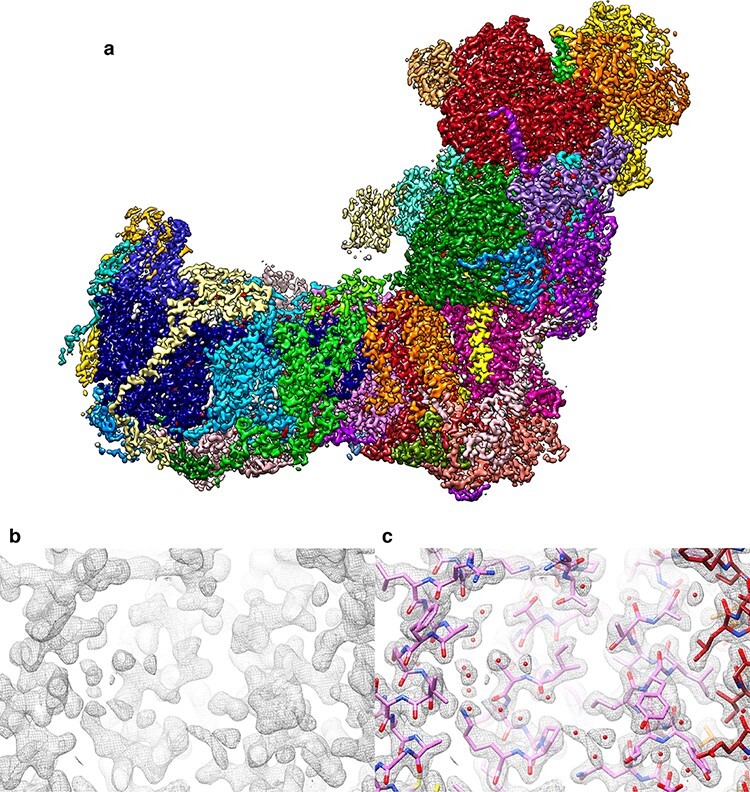
Structure of mitochondrial complex I from Y. lipolytica at 2.1 Å resolution. (a) Map coloured by subunit. Small red densities are water molecules. Map section without (b) and with (c) fitted atomic model. Small red spheres in (c) indicate a subset of the ∼100 internal water molecules along the proton transfer path. From [117].
Following on from his work on the Polytomella ATP synthase dimer (see below), Niklas Klusch took it upon himself to isolate and determine the cryoEM structure of complex I from Polytomella mitochondria. We joined forces with Hans-Peter Braun at the University of Hannover (Germany), who had spent years in purifying mitochondrial complex I of the model plant Arabidopsis thaliana and in characterizing its many subunits by mass spectrometry. In a tour-de-force of data collection and image processing, Niklas was able to obtain the cryoEM structures of both the Polytomella and the Arabidopsis complex at ∼2.9 Å resolution in minimum time, the latter in two different conformations [120]. Mitochondrial complex I from plants and green algae (including Polytomella) distinguishes itself from that of fungi and mammals by a large external domain of three carbonic anhydrase subunits (Fig. 11a,b). A metal ion (most likely zinc) resolved in the active site of one of the Arabidopsis subunits was absent in Polytomella. From this observation, we concluded that the carbonic anhydrase in the plant complex is active, whereas that of Polytomella is not.
Fig. 11.
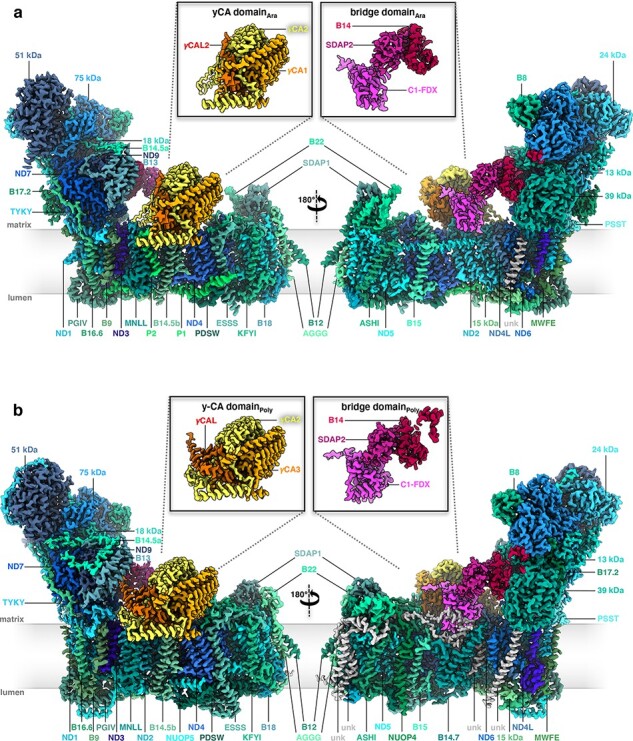
CryoEM structures of mitochondrial complex I from (a) Arabidopsis thaliana and (b) Polytomella sp., both at ∼2.9 Å resolution, coloured by subunit. The 14 core subunits are drawn in shades of blue; accessory subunits in shades of green. The three subunits of the carbonic anhydrase domain are yellow, light orange and orange; subunits of the bridge domain are pink, purple and red. Insets: γ-carbonic anhydrase (γCA) and bridge domain. Unknown subunits in the membrane arm (unk) are grey. From [120].
Another striking difference between complex I from plants and algae to that of fungi and mammals is a bridge of three accessory subunits, joining the peripheral and membrane arms. By structural homology, Niklas discovered that one of the bridge subunits was a ferredoxin (Fig. 11a,b). Again, a metal ion (in this case most likely Fe) in the Arabidopsis ferredoxin was missing in Polytomella, suggesting that the ferredoxin is active in Arabidopsis but not in Polytomella. We proposed that the ferredoxin plays a key role in redox-coupling the activity of the mitochondrial respiratory chain to photosynthesis [120].
Chloroplast and mitochondrial ATP synthases
One of our main aims with the new direct electron detectors had been to obtain a high-resolution structure of a mitochondrial or chloroplast ATP synthase. The F1Fo ATPases consist of two rotors, both of which can stall in distinct but structurally similar states that are simultaneously present, e.g. in a crystallization drop. Partly for this reason and because of the long-term instability of the complex in detergent, decades of intense effort had failed to produce a high-resolution X-ray structure of any intact, functionally competent ATP synthase. Only structures of some key components, notably an F1 subcomplex [121] and the c-ring rotor [122], were available from X-ray crystallography—the latter preceded by a cryoEM structure from 2D crystals at intermediate resolution [123]. Yet for an understanding of the fundamental mechanisms of ATP production in all forms of life, detailed structures of the whole, functional assembly were essential, ideally in several different states. CryoEM, which examines ATP synthase structures particle-by-particle within hours or minutes of isolation rather than in crystals of detergent-protein complexes that take weeks to grow, promised to overcome the problem.
Niklas Klusch, at that point an MSc student, isolated the dimers from Polytomella, and Matteo Allegretti, who had worked on the Frh structure during the first part of his PhD, proceeded to collect data with the Falcon-II camera on our Polara. Together with Karen Davies, an experienced postdoc who up to that point had done mostly cryoET, they obtained a map at 6.2 Å resolution, which revealed something entirely unexpected: a set of α-helices in the membrane-embedded Fo subcomplex that were >80 Å long and included a shallow angle of ∼20° with the membrane plane [124]. The paper was rejected twice, because one reviewer was sure that our structure had to be wrong. Everybody knew, the reviewer maintained, that α-helices in membrane proteins were ∼40 Å long and ran perpendicular to the membrane, which had been common knowledge since the first bacteriorhodopsin structure [1]. Eventually, we managed to convince the editor that our structure was not wrong. We found the long Fo helices again in the 7 Å map of the yeast ATP synthase dimer. This time we had collected the data in our JEOL 3200 that was now equipped with a K2 Summit camera [125]. Subsequently, the long, membrane-intrinsic helices were seen in a number of similar structures and are now known to be a hallmark of all rotary ATPases [54,126].
Of course, the different but similar ATP synthase conformations are also simultaneously present on a cryoEM grid. Our first structures of mitochondrial ATP synthase dimers from Polytomella [124] and yeast [125] appeared to be averages of several such states, and this explains why the resolution was limited to 6 or 7 Å. Once we had learned to apply yet more powerful image processing programs and had collected sufficient numbers of images to ensure that the various conformations in the particle set were well enough populated, it became possible to tease them apart by image processing. The first to succeed in this endeavour in our laboratory was Alexander Hahn, as a postdoc in the independent group of Thomas Meier (now Professor of Structural Biology at Imperial College London). Alex and Thomas had isolated the chloroplast ATP synthase from kilograms of spinach bought at a local supermarket—incidentally by ammonium sulphate precipitation, an old, brute-force method which I would never have thought would yield structurally and functionally intact preparations of such an intricate and fragile assembly. Alex collected a large number of movies with the faster and better Falcon-III camera installed on our new FEI Titan Krios electron microscope. He generated a very beautiful map that extended to ∼2.9 Å resolution in the catalytic F1 assembly of the most populated of three rotary states [127] (Fig. 12). In the catalytic sites of the F1 head, bound nucleotides, Mg ions and even water molecules were resolved. A comparison of the three rotary states indicated that the long and thin peripheral stalk, which connects the F1 head to the Fo rotor, is flexible, whereas the γ-subunit of the central stalk works as a rigid, non-flexible driveshaft. Without the benefit of high-resolution cryoEM, earlier biophysical work and computer simulations had come to the opposite conclusion, i.e. that the central stalk was flexible and the peripheral stalk was rigid [128].
Fig. 12.
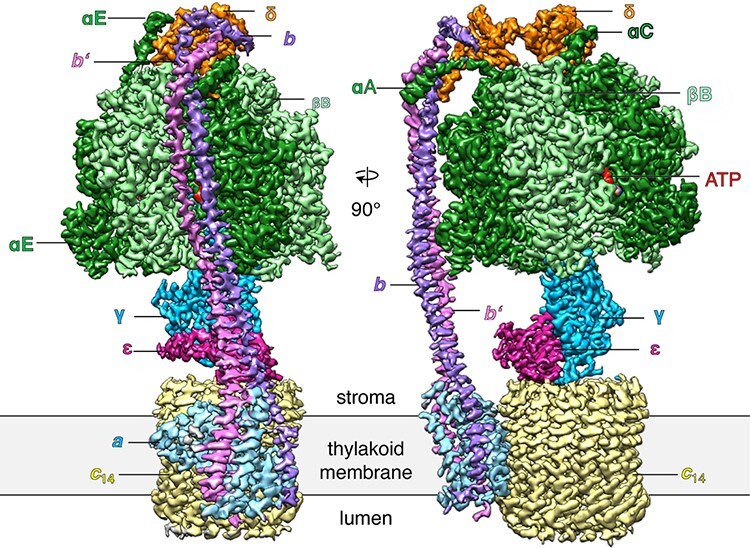
Cryo-EM structure of the spinach chloroplast ATP synthase. Surface representation of the map density with subunits α, dark green; β, light green; γ, blue; δ, orange; ε, purple; a, light blue; b, violet; b’, pink; 14-membered c-ring rotor c14, pale yellow; ATP, red. The Fo subcomplex (abb’c14) is embedded in the thylakoid membrane (grey), while the catalytic F1 subcomplex (α3β3γεδ) extends into the stroma. N-terminal helices of the αA, αC and αE subunits are indicated. The resolution of the F1 subcomplex is 2.9 Å. From [127].
Encouraged by Alex Hahn’s and Thomas Meier’s chloroplast ATP synthase structure, Bonnie Murphy, a new postdoc, together with Niklas Klusch set out to tackle the Polytomella dimer. They progressed rapidly to a 4.1 Å map that refined locally to 3.7 Å in the most rigid parts of the peripheral stalk and Fo subcomplex. In this region, all sidechains were resolved [129]. The map revealed two aqueous channels, one each extending from the middle of the membrane to the lumenal or matrix membrane surface. Each channel was lined by charged and polar residues of the a-subunit stator. In the hydrophobic core of the Fo subcomplex, the two half-channels were separated by a conserved arginine sidechain and approached one another to within ∼6 Å. The steep potential gradient at the channel end points would exert a strong force on the charge of the critical glutamates in the c-ring rotor, generating torque and resulting in net directional rotation [54].
In the 3.7 Å map, the static parts of the stator assembly were well-defined, but rotary states were not resolved. Data for the 3.7 Å map had been collected with a K2 Summit detector and the in-column energy filter of the JEOL 3200 FSC. Collecting a much larger number of particle images (∼730 000) with a more modern, fully automated microscope (Titan Krios G2) and a considerably better detector (Falcon-III), Niklas and Bonnie produced a map of the Polytomella ATP synthase dimer at an overall resolution of 2.7–2.8 Å. In an image processing marathon, Bonnie was able to separate 13 well-defined rotary substates that together provided a detailed picture of the molecular motions that accompany c-ring rotation and ATP synthesis [58] (Fig. 13). We found that the F1 head rotates along with the central stalk and c-ring rotor for the first ∼30° of each 120° primary rotary step to facilitate flexible coupling of the stoichiometrically mismatched F1 and Fo subcomplexes. Flexibility is mediated by the interdomain hinge of the OSCP subunit (known as δ in the chloroplast ATP synthase) on top of the F1 head. Since this subunit is highly conserved, as are all crucial elements of rotary catalysis, we can be confident that our mechanistic conclusions apply in principle to all rotary ATP synthases. With the high-resolution structures of the ATP synthase dimer and complex I from Polytomella and the known distribution of complex I and ATP synthase dimer rows in the membrane [53], we can now draw a model of a crista that contains all the known molecular details and summarizes how they are arranged and work together in the mitochondrial inner membrane (Fig. 14).
Fig. 13.
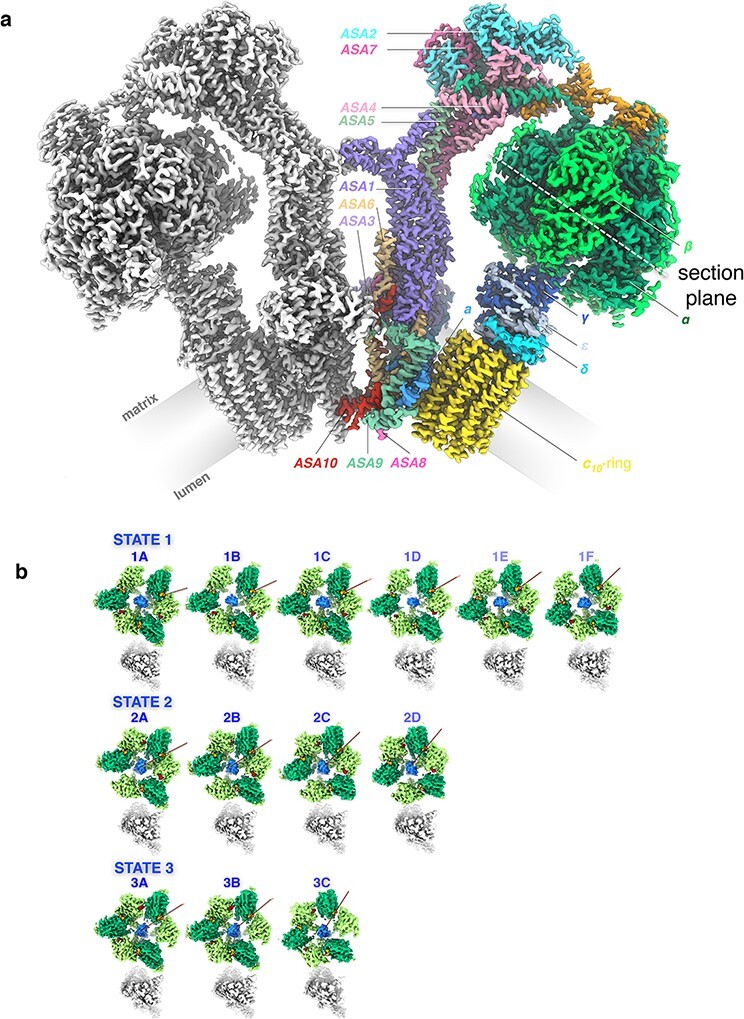
(a) Cryo-EM map density of the 62-subunit 1.58 MDa Polytomella ATP synthase dimer. The monomer map on the left is shown in grey. The map on the right is coloured by subunit. The F1 head consists of three catalytic β subunits (bright green) and three structural α subunits (dark green). The dashed white line indicates the section plane in (b). The long C-terminal extensions of β wrap around the α subunits on the outside of the F1 head. The central stalk subunits γ (dark blue), δ (cyan) and ε (light blue) connect F1 to the Fo motor in the membrane (grey). The c10 rotor ring (yellow) and subunit a (bright blue) are the main components of the Fo motor complex. The two-domain OSCP subunit (orange) links the three α subunits to the peripheral stalk subunits ASA 1–10. The local map resolution is 2.7 Å for the peripheral stalk, Fo, and c-ring, and 3.0–3.2 Å for the F1 head and central stalk. (b) Rotary substates. Sections through cryo‐EM maps of the Polytomella ATP synthase F1 head at the level of nucleotide‐binding sites (dashed white line in a). Top row: Six substates of primary state 1. Middle row: Four substates of primary state 2. Bottom row: Three substates of primary state 3. Bright green, β‐subunits; dark green, α‐subunits; blue, γ‐subunit; red, ADP in catalytic sites; orange, structural ATP; grey, peripheral stalk. Red lines indicate the rotary positions of consecutive substates. From [58].
Fig. 14.
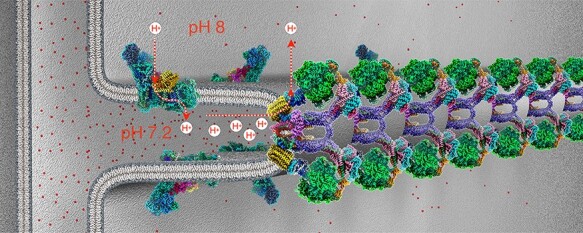
Molecular organization of mitochondrial inner membrane cristae. Cryo-EM structures of the F1Fo ATP synthase dimers [58] and complex I [120], both from Polytomella sp. in the membrane. Three-dimensional density maps at 2.7 Å (ATP synthase) and 2.9 Å resolution (complex I) are coloured by subunit. ATP synthase dimers assemble into long rows that impose a tight local curvature on the inner membrane, giving rise to the formation of cristae as mitochondrial microcompartments. Complex I and other respiratory chain complexes (not shown) occupy the flat membrane regions on either side of the dimer rows. Complex I generates most of the proton gradient across the inner membrane, pumping protons (dashed red arrows) into the cristae, which work as proton traps. Protons (red dots) flow through channels in the Fo subcomplex of the ATP synthase from the cristae lumen (∼pH 7.2) to the mitochondrial matrix (pH 8), driving the rotor (yellow) in the membrane. The torque generated by the electrochemical gradient is transmitted via the central stalk (blue) to the catalytic F1 subcomplex (green), where ATP is produced from ADP and phosphate by rotary catalysis.
Outlook
In the past 40 years, cryoEM of membrane proteins has come further than most of us would have predicted, and it has done so more rapidly than any of us could have hoped. Yet we are by no means at the end, but right in the middle, or perhaps even still at the beginning of this exhilarating and fascinating journey.
For single-particle cryoEM, a few technical challenges are left. Beam-induced movement, for many years the bane of cryoEM, may have been overcome once and for all by the new HexAuFoil grids developed by Chris Russo and colleagues [130], although they have not yet been widely tested. Alternative approaches, such as controlled devitrification of vitrified samples [131], may help in special circumstances. Protein adsorption and denaturation at the air–water interface is a fundamental and ubiquitous problem in single-particle cryoEM, which means that only a small percentage (typically ∼5–20%) of a particle set contribute to a high-resolution structure. The remaining 80–95% are in some way damaged or distorted at the air–water interface. It may be possible to overcome these problems with hydrophilized graphene support films, which the complexes adhere to in random orientations in preference to the air–water interface [132,133].
Ultimately, the success of single-particle cryoEM depends on the quality of the protein sample that goes on the grid. There is no substitute for optimizing the conditions a protein is exposed to during isolation. Therefore, when all other technical and computational issues are settled, protein biochemistry is likely to remain as the hardest part of single-particle cryoEM, because each protein is different, details matter, and there are no shortcuts.
With the most recent detectors and a matching battery of image processing software, structures of virtually any macromolecular assembly above a certain size can now be tackled by single-particle cryoEM. The current lower limit with soluble proteins is ∼60 kDa [134]. The recent 3.3 Å structure of the 55-kDa proton-coupled folate transporter in complex with a nanobody [135] suggests that the size limit for membrane proteins is similar, even though single-particle cryoEM of small membrane proteins is inherently more difficult. The detergent micelles or lipid nanodiscs that keep them in solution do not have a defined structure and reduce the contrast in the trans-membrane region, which makes the particles harder to align. The theoretical size limit has been estimated by Richard Henderson for perfect images recorded with 300-kV electrons (see Table 2 in [79]) to be ∼38 kDa. This limit is now almost within reach and likely to be approached asymptotically. With 100 -kV microscopes [136] and detectors, slightly smaller molecules should become accessible, because the scattering cross section is higher and the ratio of elastically and inelastically scattered electrons more favourable at lower acceleration voltage (see Fig. 2 in [79]).
Very accurate high-resolution structures can be obtained by electron diffraction of microscopic 3D crystals, a technique known as MicroED. MicroED has been applied mostly to small or very small proteins or protein fragments, in particular to a number of fibre-forming amyloid proteins that are recalcitrant to other techniques [137]. As such, it is a welcome addition to the cryoEM methods spectrum. It is however unlikely to become as popular as single-particle cryoEM or cryoET, because it still requires 3D protein crystals, which removes one of the key advantages of cryoEM over X-ray crystallography.
At present, structural biology is undergoing a profound transformation. After years of futile efforts, computational methods for solving the protein folding problem have become so powerful that the structure of virtually any single protein can now be instantly predicted with near-atomic accuracy from its genomic sequence. A new deep-learning algorithm called AlphaFold2 [138,139] has been shown to produce correct protein structures even if there are no related templates in the protein data bank (pdb). We tried it out with a new membrane protein without known homologues in the pdb, and the result was shockingly similar to our unreleased experimental structure. In the short to medium term, AlphaFold2 will thus shift the emphasis and take some of the pain and hard work (but also fun!) out of structure determination by cryoEM and X-ray crystallography. However, it will not make the experimental methods redundant.
A single structure is rarely enough to understand how a protein works. Most if not all proteins in the cell undergo conformational changes while performing their tasks, and cryoEM in particular is good at resolving different, simultaneously present functional states. Such conformational ensembles would be difficult to predict. More often than not, the function of a protein depends on interactions with other proteins or small molecules, and some of the interaction partners may not be known. Similarly, the structures of protein subunits that assemble into large macromolecular complexes may not be predicted reliably at present. Computational methods are unlikely to outperform cryoEM in determining the structures of large multi-subunit complexes, such as complex I or ATP synthase in the short term. It will be some time before AlphaFold can generate the correct structures of the 13 different rotary states of the Polytomella ATP synthase [58], the 8 different conformations of an ABC transporter [101] or even the two different states of plant complex I [120] as parts of a molecular mechanism. There is however no principal reason why it should not work, and therefore, it is likely to happen sooner or later. In any event, single-particle cryoEM and crystallography will be critical for validating structure predictions.
For cryoET, the best times are still ahead. As more and more high-resolution structures of proteins and complexes are done or predicted, cryoEM labs will increasingly turn from crystallography and single-particle work to cryoET, which is not subject to the same constraints of sample purity and homogeneity. At the same time, it will become increasingly urgent to investigate and understand the context of macromolecular components in the cell. CryoET in particular stands to gain from protein structure prediction, because it generates lower-resolution volumes quite readily, to which the predicted structures can then be fitted. CryoEM instrumentation can in principle be used equally well for single-particle work and cryoET (although an acceleration voltage of 300 kV is preferable for tomography), which makes the transition easier. CryoET does have its own constraints, but in terms of the molecular systems and biomedical questions it can address, it is essentially unlimited. Every cell contains a universe of molecular landscapes that is best explored by cryoET.
Constraints include sample thickness and tilt geometry. Most samples are too thick, but they can be thinned into thin lamellae by focused ion beam (FIB) milling at low temperature. Cryo-FIB milling is a demanding technique but it is practiced successfully in a growing number of cryoEM laboratories around the world [140,141]. Constraints of tilt geometry can be overcome by FIB-milling cylindrical pencils instead of planar lamellae, and several laboratories are experimenting in this direction.
A major unresolved challenge in cryoET is the lack of a universal cloneable label equivalent to green fluorescent protein (GFP) in light microscopy that is fused with a protein so it can be identified and studied in detail in its native context. Correlative light/electron microscopy alleviates the problem by identifying regions of interest in a larger volume, such as a cell, but within that region individual assemblies still have to be recognized by their shape and position, e.g. relative to a membrane. Because phosphorus scatters electrons more strongly than carbon, nitrogen or oxygen, phospholipid bilayers show up well in tomographic volumes, and therefore, cryoET works well for protein complexes in or at a membrane. CryoET of HIV capsids in vitrified aqueous buffer has achieved 3.4 Å resolution [142]. There is no fundamental reason why cryoET, just as single-particle cryoEM, should not go to true atomic resolution, given a sufficiently large number of tomographic volumes to average. In cellular tomography, the target complexes are usually surrounded by other macromolecules of similar density, which makes the targets more difficult to detect and average than single particles in dilute aqueous buffers. The ultimate limitation in both cases is the signal-to-noise ratio of the electron micrographs.
The signal-to-noise ratio of electron micrographs can be enhanced by a phase plate. It is fair to say that current phase plate designs, including the Volta phase plate [143] or our own electrostatic Boersch phase plate [144], have not kept their promise. Even though they can improve low-resolution image contrast, potential gains at high resolution are more than offset by charging problems and a lack of reproducibility associated with introducing an external device into the electron beam [143]. These problems may be overcome by a new elegant phase plate design that shifts the phase of the electron beam by the ponderomotive force of a strong laser [145]. The laser phase plate enhances contrast in the entire resolution range and would be particularly useful for cryoET. However, its implementation is technically demanding, the electron microscope needs an extra lens and a tremendously powerful laser is necessary for shifting the phase of a 300 kV beam by 90° for optimum contrast. For single-particle cryoEM, recent improvements in electron microscope hardware, in particular cold field emitters, better image filters and direct electron detectors, have increased phase contrast to an extent that makes the available phase plates less of a help than a hindrance for high resolution.
Even though the present generation of direct electron detectors is already superb, further improvements in their design and production are likely and will benefit both single-particle cryoEM and cryoET. At present, the DQE of the best available detectors (Falcon-IV) is 1.0 at low resolution and close to 0.4 at Nyquist frequency [146]. The Nyquist frequency defines the maximum resolution possible with the given pixel size of the detector. For a perfect detector, the DQE would be 1.0 at Nyquist, but this may be hard to achieve. Still, any improvement will boost the signal-to-noise ratio at high resolution and translate into better maps from fewer particle images or tomograms.
So far, the high cost of cryoEM hardware does not seem to have taken away much of its popularity. The number of new cryoEM labs around the globe is still growing, and research councils and agencies are willing to support them, because of the expected gains in mechanistic insight for basic biomedical research, which will ultimately translate into benefits for human health. But this trend cannot go on forever. Cost scales roughly with acceleration voltage. It is possible to produce less expensive 100-kV electron microscopes that perform equally well, and in some ways better for single-particle cryoEM than 300-kV instruments [136]. Bringing the cost of EM hardware down substantially will be essential if cryoEM is to be as widely accessible to biomedical research as protein crystallography has been in the past. An alternative would be to create more of the national cryoEM facilities such as eBIC at the Diamond synchrotron in the UK or the National Centers in the USA, to cumulate resources and provide cryoEM as a service. However, sample preparation for cryoEM is still very much a skill and a craft. Automation is in its infancy [147,148] and reproducibly thin ice layers, a key factor for high resolution, are hard to achieve. Similarly, collecting top-quality high-resolution images requires close familiarity with the sample. In the long run, it may therefore be preferable not to separate researchers from their microscopes and cryoEM samples.
Within the past year, two cryoEM structures of a standard soluble test specimen, recombinant mouse apoferritin, have been reported at ∼1.25 Å, i.e. true atomic resolution [146,149]. This proves that cryoEM is capable of delivering structures of equal quality as atomic-resolution X-ray crystallography, which not many of us would have imagined a few years ago. For most biological questions, a resolution ∼2 Å is probably more than adequate, but higher resolution is always nice, and it is hard to predict what it might reveal. Further improvements in electron microscopes, detectors and image processing software are bound to result in more and more atomic-resolution structures of interesting membrane proteins. Already with the present generation of cryoEM hardware and software, water molecules are resolved in the cytochrome bd oxidase [109], complex I [117,119] and the GABAA receptor [146]. At very high resolution, cryoEM may be able to visualize ionization states of bound ions and protonation states of sidechains and water molecules, which would provide more detailed insights into molecular mechanisms. The future for studying membrane proteins by cryoEM gets brighter and more fascinating all the time!
Acknowledgements
I thank Richard Henderson, Nigel Unwin, Janet Vonck and Hester Robinson for their critical reading and helpful comments on the manuscript.
Funding
Most of the work described in this review was funded by the Max Planck Society. Some of the earlier work at EMBL was funded by the European Molecular Biology Laboratory.
References
- 1. Henderson R and Unwin P N (1975) Three-dimensional model of purple membrane obtained by electron microscopy. Nature 257: 28–32. [DOI] [PubMed] [Google Scholar]
- 2. Kühlbrandt W and Unwin P N T (1982) Distribution of RNA and protein in crystalline eukaryotic ribosomes. J. Mol. Biol. 156: 431–448. [DOI] [PubMed] [Google Scholar]
- 3. Kühlbrandt W (1982) Discrimination of protein and nucleic-acids by electron-microscopy using contrast variation. Ultramicroscopy 7: 221–232. [DOI] [PubMed] [Google Scholar]
- 4. Dubochet J et al. (1982) Frozen aqueous suspensions. Ultramicroscopy 10: 55–61. [Google Scholar]
- 5. Jaffe J S and Glaeser R M (1984) Preparation of frozen-hydrated specimens for high-resolution electron-microscopy. Ultramicroscopy 13: 373–377. [DOI] [PubMed] [Google Scholar]
- 6. Henderson R et al. (1990) Model for the structure of bacteriorhodopsin based on high resolution electron cryo-microscopy. J. Mol. Biol. 213: 899–929. [DOI] [PubMed] [Google Scholar]
- 7. Baldwin J M et al. (1988) Images of purple membrane at 2.8 Å resolution obtained by cryo-electron microscopy. J. Mol. Biol. 202: 585–591. [DOI] [PubMed] [Google Scholar]
- 8. Dietrich I et al. (1977) Improvements in electron microscopy by application of superconductivity. Ultramicroscopy 2: 241–249. [DOI] [PubMed] [Google Scholar]
- 9. Taylor K A and Glaeser R M (1974) Electron diffraction of frozen, hydrated protein crystals. Science 186: 1036–1037. [DOI] [PubMed] [Google Scholar]
- 10. Hayward S B and Glaeser R M (1980) High resolution cold stage for the JEOL 100B and 100C electron microscopes. Ultramicroscopy 5: 3–8. [DOI] [PubMed] [Google Scholar]
- 11. Kühlbrandt W, Thaler T, and Wehrli E (1983) The structure of membrane crystals of the light-harvesting chlorophyll a/b protein complex. J. Cell Biol. 96: 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kühlbrandt W (1984) Three-dimensional structure of the light-harvesting chlorophyll a/b-protein complex. Nature 307: 478–480. [Google Scholar]
- 13. Kühlbrandt W and Downing K H (1989) Two-dimensional structure of plant light-harvesting complex at 3.7 Å resolution by electron crystallography. J. Mol. Biol. 207(4): 823–828. [DOI] [PubMed] [Google Scholar]
- 14. Fujiyoshi Y et al. (1991) Development of a superfluid helium stage for high-resolution electron microscopy. Ultramicroscopy 38: 241–251. [Google Scholar]
- 15. Kühlbrandt W, Wang D N, and Fujiyoshi Y (1994) Atomic model of plant light-harvesting complex by electron crystallography. Nature 367: 614–621. [DOI] [PubMed] [Google Scholar]
- 16. Wang D N and Kühlbrandt W (1992) Three-dimensional electron diffraction of plant light-harvesting complex. Biophys. J. 61: 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walz T et al. (1997) The three-dimensional structure of aquaporin-1. Nature 387: 624–627. [DOI] [PubMed] [Google Scholar]
- 18. Hebert H et al. (1997) The 3.0 Å projection structure of microsomal glutathione transferase as determined by electron crystallography of p2(1)2(1)2 two-dimensional crystals. J. Mol. Biol. 271: 751–758. [DOI] [PubMed] [Google Scholar]
- 19. Miyazawa A et al. (1999) Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel. J. Mol. Biol. 288: 765–786. [DOI] [PubMed] [Google Scholar]
- 20. Miyazawa A, Fujiyoshi Y, and Unwin N (2003) Structure and gating mechanism of the acetylcholine receptor pore. Nature 423: 949–955. [DOI] [PubMed] [Google Scholar]
- 21. Unwin N (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346: 967–989. [DOI] [PubMed] [Google Scholar]
- 22. Stark H, Zemlin F, and Boettcher C (1996) Electron radiation damage to protein crystals of bacteriorhodopsin at different temperatures. Ultramicroscopy 63: 75–79. [Google Scholar]
- 23. Fujiyoshi Y (1998) The structural study of membrane proteins by electron crystallography. Adv. Biophys. 35: 25–80. [DOI] [PubMed] [Google Scholar]
- 24. Pfeil-Gardiner O et al. (2019) A comparative study of single-particle cryo-EM with liquid-nitrogen and liquid-helium cooling. IUCrJ 6: 1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kühlbrandt W (1992) Two-dimensional crystallization of membrane proteins. Q. Rev. Biophys. 25: 1–49. [DOI] [PubMed] [Google Scholar]
- 26. Abeyrathne P D et al. (2012) Analysis of 2-D crystals of membrane proteins by electron microscopy (Chap. 1.15). In: Egelman E H (ed.), Comprehensive Biophysics 1, Biophysical Techniques for Structural Characterization of Macromolecules, pp 277–310 (Academic Press, Oxford: ). [Google Scholar]
- 27. Nogales E, Wolf S G, and Downing K H (1998) Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391(6663): 199–203. [DOI] [PubMed] [Google Scholar]
- 28. Williams K A et al. (1999) Projection structure of NhaA, a secondary transporter from Escherichia coli, at 4.0 Å resolution. EMBO J. 18: 3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams K A (2000) Three-dimensional structure of the ion-coupled transport protein NhaA. Nature 403: 112–115. [DOI] [PubMed] [Google Scholar]
- 30. Paulino C and Kühlbrandt W (2014) pH- and sodium-induced changes in a sodium/proton antiporter. eLife 3: e01412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paulino C et al. (2014) Structure and transport mechanism of the sodium/proton antiporter MjNhaP1. eLife 3: e03583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ziegler C et al. (2004) Projection structure and oligomeric state of the osmoregulated sodium/glycine betaine symporter BetP of Corynebacterium glutamicum. J. Mol. Biol. 337: 1137–1147. [DOI] [PubMed] [Google Scholar]
- 33. Breyton C et al. (2002) Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature 418: 662–665. [DOI] [PubMed] [Google Scholar]
- 34. Bostina M et al. (2005) Atomic model of the E-coli membrane-bound protein translocation complex SecYEG. J. Mol. Biol. 352: 1035–1043. [DOI] [PubMed] [Google Scholar]
- 35. Hizlan D et al. (2012) Structure of the SecY complex unlocked by a preprotein mimic. Cell Rep 1: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Müller M et al. (2011) Projection structure of channelrhodopsin-2 at 6 Å resolution by electron crystallography. J. Mol. Biol. 414: 86–95. [DOI] [PubMed] [Google Scholar]
- 37. Müller M et al. (2015) Light-induced helix movements in channelrhodopsin-2. J. Mol. Biol. 427: 341–349. [DOI] [PubMed] [Google Scholar]
- 38. Ressl S et al. (2009) Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature 458: 47–52. [DOI] [PubMed] [Google Scholar]
- 39. Wöhlert D, Kühlbrandt W, and Yildiz Ö (2014) Structure and substrate ion binding in the sodium/proton antiporter PaNhaP. eLife 3: e03579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wöhlert D et al. (2015) Mechanism of Na(+)-dependent citrate transport from the structure of an asymmetrical CitS dimer. eLife 4: e09375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van den Berg B et al. (2004) X-ray structure of a protein-conducting channel. Nature 427: 36–44. [DOI] [PubMed] [Google Scholar]
- 42. Kato H E et al. (2012) Crystal structure of the channelrhodopsin light-gated cation channel. Nature 482: 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schägger H and Pfeiffer K (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19: 1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schäfer E et al. (2006) Architecture of active mammalian respiratory chain supercomplexes. J. Biol. Chem. 281: 15370–15375. [DOI] [PubMed] [Google Scholar]
- 45. Althoff T et al. (2011) Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 30: 4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sousa J S et al. (2016) Functional asymmetry and electron flow in the bovine respirasome. eLife 5: e21290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Letts J A, Fiedorczuk K, and Sazanov L A (2016) The architecture of respiratory supercomplexes. Nature 537: 644–648. [DOI] [PubMed] [Google Scholar]
- 48. Wu M et al. (2016) Structure of mammalian respiratory supercomplex I1III2IV1. Cell 167: 1598–1609. [DOI] [PubMed] [Google Scholar]
- 49. Strauss M et al. (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27: 1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minauro-Sanmiguel F, Wilkens S, and Garcia J J (2005) Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc. Natl. Acad. Sci. U.S.A. 102: 12356–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dudkina N V et al. (2006) Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett. 580: 3427–3432. [DOI] [PubMed] [Google Scholar]
- 52. Blum T B et al. (2019) Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc. Natl. Acad. Sci. U.S.A. 116: 4250–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davies K M et al. (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108: 14121–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kühlbrandt W (2019) Structure and mechanisms of F-type ATP synthases. Annu. Rev. Biochem. 88: 515–549. [DOI] [PubMed] [Google Scholar]
- 55. Mühleip A W et al. (2016) Helical arrays of U-shaped ATP synthase dimers form tubular cristae in ciliate mitochondria. Proc. Natl. Acad. Sci. U.S.A. 113: 8442–8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mühleip A W et al. (2017) In situ structure of trypanosomal ATP synthase dimer reveals a unique arrangement of catalytic subunits. Proc. Natl. Acad. Sci. U.S.A. 114: 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dudkina N V et al. (2010) Row-like organization of ATP synthase in intact mitochondria determined by cryo-electron tomography. Biochim. Biophys. Acta 1797: 272–277. [DOI] [PubMed] [Google Scholar]
- 58. Murphy B J et al. (2019) Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1-Fo coupling. Science 364: eaaw9128. [DOI] [PubMed] [Google Scholar]
- 59. Allen R D, Schroeder C C, and Fok A K (1989) An investigation of mitochondrial inner membranes by rapid-freeze deep-etch techniques. J. Cell Biol. 108: 2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brandt T et al. (2017) Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. eLife 6: e24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Davies K M et al. (2012) Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 109: 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bornhövd C et al. (2006) Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J. Biol. Chem. 281: 13990–13998. [DOI] [PubMed] [Google Scholar]
- 63. Teixeira F K et al. (2015) ATP synthase promotes germ cell differentiation independent of oxidative phosphorylation. Nat. Cell Biol. 17: 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davies K M, Blum T B, and Kühlbrandt W (2018) Conserved in situ arrangement of complex I and III2 in mitochondrial respiratory chain supercomplexes of mammals, yeast, and plants. Proc. Natl. Acad. Sci. U.S.A. 115: 3024–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Daum B et al. (2013) Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 110: 15301–15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gold V A et al. (2014) Visualizing active membrane protein complexes by electron cryotomography. Nat. Commun. 5: 4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Faelber K et al. (2019) Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature 571: 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Köster S et al. (2014) Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 5: 3690. [DOI] [PubMed] [Google Scholar]
- 69. van Pee K et al. (2016) Unraveling the pore-forming steps of pneumolysin from streptococcus pneumoniae. Nano Lett. 16: 7915–7924. [DOI] [PubMed] [Google Scholar]
- 70. van Pee K et al. (2017) CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin. eLife 6: e23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vögele M et al. (2019) Membrane perforation by the pore-forming toxin pneumolysin. Proc. Natl. Acad. Sci. U.S.A. 116: 13352–13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Daum B et al. (2014) Self-assembly of the general membrane-remodeling protein PVAP into sevenfold virus-associated pyramids. Proc. Natl. Acad. Sci. U.S.A. 111: 3829–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rhee K-H et al. (1997) Two-dimensional structure of plant photosystem II at 8-Å resolution. Nature 389: 522–526. [DOI] [PubMed] [Google Scholar]
- 74. Rhee K-H et al. (1998) Three-dimensional structure of the plant photosystem II reaction centre at 8 Å resolution. Nature 396: 283–286. [DOI] [PubMed] [Google Scholar]
- 75. Daum B et al. (2010) Arrangement of photosystem II and ATP synthase in chloroplast membranes of spinach and pea. The Plant Cell 22: 1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Daum B and Kühlbrandt W (2011) Electron tomography of plant thylakoid membranes. J. Exp. Bot. 62: 2393–2402. [DOI] [PubMed] [Google Scholar]
- 77. Floris D and Kühlbrandt W (2021) Molecular landscape of etioplast inner membranes in higher plants. Nat. Plants 7: 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Grigorieff N and Harrison S C (2011) Near-atomic resolution reconstructions of icosahedral viruses from electron cryo-microscopy. Curr. Opin. Struct. Biol. 21(2): 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Henderson R (1995) The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 28: 171–193. [DOI] [PubMed] [Google Scholar]
- 80. McMullan G et al. (2009) Detective quantum efficiency of electron area detectors in electron microscopy. Ultramicroscopy 109: 1126–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Faruqi A R and McMullan G (2011) Electronic detectors for electron microscopy. Q. Rev. Biophys. 44: 357–390. [DOI] [PubMed] [Google Scholar]
- 82. McMullan G et al. (2009) Experimental observation of the improvement in MTF from backthinning a CMOS direct electron detector. Ultramicroscopy 109: 1144–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. McMullan G et al. (2014) Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy 147: 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mills D J et al. (2013) De novo modeling of the F420-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. eLife 2: e00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Allegretti M et al. (2014) Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. eLife 3: e01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li X et al. (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Meth. 10: 584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Liao M et al. (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504: 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Amunts A et al. (2014) Structure of the yeast mitochondrial large ribosomal subunit. Science 343: 1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kühlbrandt W (2014) The resolution revolution. Science 343: 1443–1444. [DOI] [PubMed] [Google Scholar]
- 90. Brilot A F et al. (2012) Beam-induced motion of vitrified specimen on holey carbon film. J. Struct. Biol. 177: 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zheng S Q et al. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Meth. 14: 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rohou A and Grigorieff N (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192: 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang K (2016) Gctf: real-time CTF determination and correction. J. Struct. Biol. 193: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tang G et al. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157: 38–46. [DOI] [PubMed] [Google Scholar]
- 95. Grigorieff N (2007) FREALIGN: high-resolution refinement of single particle structures. J. Struct. Biol. 157: 117–125. [DOI] [PubMed] [Google Scholar]
- 96. Punjani A et al. (2017) cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Meth. 14: 290–296. [DOI] [PubMed] [Google Scholar]
- 97. Scheres S H (2012) A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415: 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zivanov J et al. (2018) New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7: e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Scheres S H et al. (2008) Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3: 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Terwilliger T C et al. (2020) Improvement of cryo-EM maps by density modification. Nat. Meth. 17: 923–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hofmann S et al. (2019) Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571: 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Timcenko M et al. (2019) Structure and autoregulation of a P4-ATPase lipid flippase. Nature 571: 366–370. [DOI] [PubMed] [Google Scholar]
- 103. Model K, Meisinger C, and Kühlbrandt W (2008) Cryo-electron microscopy structure of a yeast mitochondrial preprotein translocase. J. Mol. Biol. 383: 1049–1057. [DOI] [PubMed] [Google Scholar]
- 104. Bausewein T et al. (2017) Cryo-EM structure of the TOM core complex from Neurospora crassa. Cell 170: 693–700. [DOI] [PubMed] [Google Scholar]
- 105. Araiso Y et al. (2019) Structure of the mitochondrial import gate reveals distinct preprotein paths. Nature 575: 395–401. [DOI] [PubMed] [Google Scholar]
- 106. Tucker K and Park E (2019) Cryo-EM structure of the mitochondrial protein-import channel TOM complex at near-atomic resolution. Nat. Struct. Mol. Biol. 26: 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bausewein T, Nussberger S, and Kühlbrandt W (2019) Cryo-EM structure of Neurospora crassa respiratory complex IV. IUCrJ 6: 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sousa J S et al. (2018) Structural basis for energy transduction by respiratory alternative complex III. Nat. Commun. 9: 1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Safarian S et al. (2019) Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 366: 100–104. [DOI] [PubMed] [Google Scholar]
- 110. Zickermann V et al. (2003) Functional implications from an unexpected position of the 49-kDa subunit of NADH: ubiquinoneoxidoreductase. J. Biol. Chem. 278: 29072–29078. [DOI] [PubMed] [Google Scholar]
- 111. Hunte C, Zickermann V, and Brandt U (2010) Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science 329: 448–451. [DOI] [PubMed] [Google Scholar]
- 112. Zickermann V et al. (2015) Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347: 44–49. [DOI] [PubMed] [Google Scholar]
- 113. Vinothkumar K R, Zhu J, and Hirst J (2014) Architecture of mammalian respiratory complex I. Nature 515: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. D’Imprima E et al. (2016) Cryo-EM structure of respiratory complex I reveals a link to mitochondrial sulfur metabolism. Biochim. Biophys. Acta. 1857: 1935–1942. [DOI] [PubMed] [Google Scholar]
- 115. Parey K et al. (2018) Cryo-EM structure of respiratory complex I at work. eLife 7: e39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Parey K et al. (2019) High-resolution cryo-EM structures of respiratory complex I: mechanism, assembly, and disease. Sci. Adv. 5: eaax9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Parey K et al. (2021) High-resolution structure and dynamics of mitochondrial complex I – insights into the proton pumping mechanism. Sci. Adv. 7: eabj3221.doi: 10.1126/sciadv.abj3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Agip A N A et al. (2018) Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25: 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kampjut D and Sazanov L A (2020) The coupling mechanism of mammalian respiratory complex I. Science 370: eabc4209. [DOI] [PubMed] [Google Scholar]
- 120. Klusch N et al. (2021) A ferredoxin bridge connects the two arms of plant mitochondrial complex I. The Plant Cell 33: 2072–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Abrahams J P et al. (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370: 621–628. [DOI] [PubMed] [Google Scholar]
- 122. Meier T et al. (2005) Structure of the rotor ring of F-type Na+-ATPase from Ilyobacter tartaricus. Science 308: 659–662. [DOI] [PubMed] [Google Scholar]
- 123. Vonck J et al. (2002) Molecular architecture of the undecameric rotor of a bacterial Na+-ATP synthase. J. Mol. Biol. 321: 307–316. [DOI] [PubMed] [Google Scholar]
- 124. Allegretti M et al. (2015) Horizontal membrane-intrinsic alpha-helices in the stator a-subunit of an F-type ATP synthase. Nature 521: 237–240. [DOI] [PubMed] [Google Scholar]
- 125. Hahn A et al. (2016) Structure of a complete ATP synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol. Cell 63: 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kühlbrandt W and Davies K M (2016) Rotary ATPases: a new twist to an ancient machine. Trends Biochem. Sci. 41: 106–116. [DOI] [PubMed] [Google Scholar]
- 127. Hahn A et al. (2018) Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 360: eaat4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wächter A et al. (2011) Two rotary motors in F-ATP synthase are elastically coupled by a flexible rotor and a stiff stator stalk. Proc. Natl. Acad. Sci. U.S.A. 108: 3924–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Klusch N et al. (2017) Structural basis of proton translocation and force generation in mitochondrial ATP synthase. eLife 6: e33274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Naydenova K, Jia P, and Russo C J (2020) Cryo-EM with sub-1 Å specimen movement. Science 370: 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wieferig J P, Mills D J, and Kühlbrandt W (2021) Devitrification reduces beam-induced movement in cryo-EM. IUCrJ 8: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. D’Imprima E et al. (2019) Protein denaturation at the air-water interface and how to prevent it. eLife 8: e42747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. D’Imprima E and Kühlbrandt W (2021) Current limitations to high-resolution structure determination by single-particle cryoEM. Q. Rev. Biophys. 54: e4, 1–15. [DOI] [PubMed] [Google Scholar]
- 134. Herzik M A, Wu M, and Lander G C (2019) High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Nat. Comm. 10(1): 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Parker J L et al. (2021) Structural basis of antifolate recognition and transport by PCFT. Nature 595: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Naydenova K et al. (2019) CryoEM at 100 keV: a demonstration and prospects. IUCrJ 6: 1086–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Rodriguez J A, Eisenberg D S, and Gonen T (2017) Taking the measure of MicroED. Curr. Opin. Struct. Biol. 46: 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tunyasuvunakool K et al. (2021) Highly accurate protein structure prediction for the human proteome. Nature 596: 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wagner F R et al. (2020) Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nat. Protoc. 15: 2041–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zachs T et al. (2020) Fully automated, sequential focused ion beam milling for cryo-electron tomography. eLife 9: e52286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Turonova B et al. (2017) Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4 angstrom. J. Struct. Biol. 199: 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Buijsse B et al. (2020) Spectral DQE of the Volta phase plate. Ultramicroscopy 218: 113079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Walter A et al. (2015) Towards an optimum design for electrostatic phase plates. Ultramicroscopy 153: 22–31. [DOI] [PubMed] [Google Scholar]
- 145. Schwartz O et al. (2019) Laser phase plate for transmission electron microscopy. Nat. Meth. 36: 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Nakane T et al. (2020) Single-particle cryo-EM at atomic resolution. Nature 587: 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Budell W C et al. (2021) Cryo-electron microscopic grid preparation for time-resolved studies using a novel robotic system, spotiton. J. Vis. Exp..doi: 10.3791/62271. [DOI] [PubMed] [Google Scholar]
- 148. Weissenberger G, Henderikx R J M, and Peters P J (2021) Understanding the invisible hands of sample preparation for cryo-EM. Nat. Meth. 18: 463–471. [DOI] [PubMed] [Google Scholar]
- 149. Yip K M et al. (2020) Atomic-resolution protein structure determination by cryo-EM. Nature 587: 157–161. [DOI] [PubMed] [Google Scholar]