

Abstract
Amyloid imaging by positron emission tomography (PET) is an important method for diagnosing neurodegenerative disorders such as Alzheimer’s disease. Many 11C and 18F labeled PET tracers show varying binding capacities, specificities, and affinities for their target proteins. The structural basis of these variations is poorly understood. Here we employ 19F and 13C solid-state NMR to investigate the binding sites of a PET ligand, flutemetamol, to the 40-residue Alzheimer’s β-amyloid peptide (Aβ40). Analytical high-performance liquid chromatography and 19F NMR spectra show that flutemetamol binds the current Aβ40 fibril with a stoichiometry of one ligand per four to five peptides. Half of the ligands are tightly bound while the other half are loosely bound. 13C and 15N chemical shifts indicate that this Aβ40 polymorph has an immobilized N-terminus, a non-β-sheet His14, and a non-β-sheet C-terminus. We measured the proximity of the ligand fluorine to peptide residues using 19F-13C and 19F-1H rotational-echo-double-resonance (REDOR) experiments. The spectra show that three segments in the peptide, 12VHH14, 18VFF20 and 39VV40, lie the closest to the ligand. REDOR-constrained docking simulations indicate that these three segments form multiple binding sites, and the ligand orientations and positions at these sites are similar across different Aβ polymorphs. Comparison of the flutemetamol-interacting residues in Aβ40 with the small-molecule binding sites in other amyloid proteins suggest that conjugated aromatic compounds preferentially bind β-sheet surface grooves lined by aromatic, polar, and charged residues. These motifs may explain the specificity of different PET tracers to different amyloid proteins.
Graphical Abstract
Introduction
The structural biology of neurodegenerative diseases has largely centered on the determination of the three-dimensional structures of the protein aggregates involved in these diseases 1–6. Because amyloid fibrils represent the thermodynamically stable structures at the end stage of disease, disaggregating these fibrils pose a significant challenge. To prevent, detect, and slow disease progression, one approach is to investigate the structure and dynamics of early-to-intermediate states of these proteins 7–8, while a second approach is to detect these amyloids in living human brains using imaging techniques. A widely used neuroimaging modality is positron emission tomography (PET), in which positive β-decay of radioactive nuclei such as 11C and 18F is detected as γ photons after the positron is annihilated by an electron. The location of positron annihilation is deduced from the positions of the detected γ photons, thus allowing the localization and quantification of the concentration of radiotracers in living tissues. 11C has a short half-life of 20 minutes while 18F has a longer half-life of 110 minutes. Thus many 18F-containing PET tracers 9 have been developed to diagnose diseases such as cancer, cardiovascular diseases, infectious diseases, and neurological disorders.
One of the most important applications of PET is the imaging of the two pathological hallmarks of Alzheimer’s disease (AD): the extracellular neuritic plaques formed by the β-amyloid peptide (Aβ) and the intracellular neurofibrillary tangles (NFTs) formed by the protein tau 10–11. Because aromatic conjugated dyes such as thioflavin T and Congo red bind cross-β amyloid fibrils, a series of 11C and 18F-containing aromatic compounds have been developed for PET imaging of Aβ deposits and tau inclusions 12–13. Among these, 11C-labeled Pittsburgh Compound B (11C-PiB) (Fig. 1), a benzothiazole derivative of thioflavin T, is the most extensively studied PET tracer for Aβ fibrils 14–15. PiB binds Aβ aggregates in AD cortical tissues with a high affinity (Kd of ~2 nM), favorable lipophilicity, two-fold higher uptake in AD brains than healthy controls, 9, 16–17 and negligible binding to tau NFTs and α-synuclein. A [18F]3’-F-derivative of PiB, 3’-F-PiB, has similar properties as the parent compound, and is known as flutemetamol or GE-067, with a tradename Vizamyl. 18F-flutemetamol PET is especially diagnostic of advanced phases of Aβ pathology that occurs in symptomatic AD cases 18.
Figure 1.
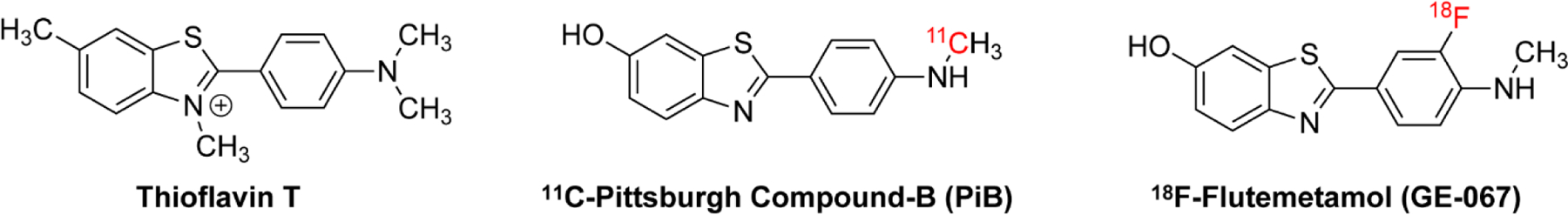
Chemical structures of thioflavin T, 11C-PiB, and 18F-flutemetamol.
Although amyloid PET imaging is important in the clinic, why multiple structurally similar compounds show different affinities, specificities, and stoichiometries to their target proteins is still poorly understood. Some PET tracers bind only Aβ fibrils while others target only tau NFTs 12. Even among Aβ-binding thioflavin derivatives, fluorescence experiments on synthetic Aβ fibrils found significantly different binding stoichiometries (Bmax) and binding constants 19. For PiB, radioligand assays indicate that Bmax and binding affinities differ widely among Aβ peptides obtained from different sources, including AD brains, healthy brains, transgenic mouse brains, and synthetic Aβ fibrils 15. The structural basis for these variations in binding characteristics is largely unknown. Determining the binding sites and binding geometries of PET ligands on the molecular level is necessary for designing more effective radiotracers, as well as small molecules that can potentially disaggregate these fibrils.
The presence of fluorine atoms in PET tracers provides an attractive opportunity for determining the binding sites of these PET ligands by NMR. The stable and NMR-active isotope of fluorine is 19F, which has favorable nuclear spin properties for structural studies. 19F is a 100% abundant spin-1/2 nucleus that possesses the second highest gyromagnetic ratio (γ) among all stable NMR-active isotopes. As a result, 19F NMR has high detection sensitivity, and nuclear dipole-dipole coupling involving 19F is stronger than couplings between low-γ nuclei such as 13C or 15N 20. This strong dipole coupling allows relatively long inter-atomic distances of 1–2 nm to be measured 21–31. In addition, the 19F chemical shift is sensitive to its molecular environment, which makes 19F an excellent probe of the conformation and dynamics of biomolecules 32–36. For these reasons, 19F solution and solid-state NMR has long been used to probe the structures of biological molecules. To speed up distance measurements, we recently developed two-dimensional (2D) solid-state NMR experiments that measure many 13C-19F, 1H-19F and 19F-19F distances simultaneously 20–24, 37. The heteronuclear 13C-19F and 1H-19F distances are measured using the dipolar recoupling technique REDOR under magic-angle spinning (MAS) 38. Each pair of 2D REDOR experiments yield multiple distance restraints, thus facilitating structure determination.
Here we apply 19F and 13C solid-state NMR experiments to investigate the residues in Aβ40 fibrils that interact with flutemetamol. We grew homogeneous fibrils without seeding using recombinant 13C, 15N-labeled Aβ40. HPLC analysis and 19F NMR indicate that flutemetamol binds these Aβ40 fibrils with a molar ratio of ~1 : 4.6, without about half of the ligands being tightly bound. 13C-19F and 1H-19F REDOR spectra indicate that three segments in the peptide lie the closest to the ligand. These segments include a double histidine (His) motif, a double phenylalanine (Phe) motif, and the double valine (Val) motif at the C-terminus of the peptide. Distance-constrained molecular docking yielded the binding orientations and positions of the ligand, which are similar for different Aβ structures. These results give insights into the residue-level interactions that stabilize this PET ligand in various Aβ structures.
Materials and Methods
Expression and purification of Aβ40
We expressed Aβ40 in E. coli as a soluble His-tagged fusion protein NT*-Aβ40 with a TEV recognition site. This construct produces a human Aβ40 peptide without additional residues after cleavage. The plasmid pT7His6NT* – TEV recognition site – Aβ(1–40) 39 was a gift from Professor Kronqvist and Professor Johansson at Karolinska Institute, Sweden. The plasmid was transformed into E. coli BL21(DE3) competent cells. An overnight culture was grown in a 250 mL unbaffled flask filled with 50 mL LB medium containing 50 µg/mL kanamycin at 37ºC with 225 rpm shaking (with an orbit diameter of 1.9 cm in this work). 5 mL of the overnight culture was inoculated into a 0.5 L LB medium in a baffled 2.8 L flask, and cells were grown at 37˚C with 250 rpm shaking until OD600 reached 0.8–0.9. Cells were centrifuged at 3,000 g and 4˚C for 5 min, and the pellet was resuspended in a baffled 2.8 L flask with 1 L of M9 medium containing 3 g/L 13C6-labeled D-glucose, 1 g/L 15N-labeled ammonium chloride, M9 salts (1 x), 2 mM MgSO4, 0.1 mM CaCl2, 50 mg/L kanamycin, and vitamin and mineral supplements. Cells were grown in this M9 medium at 37˚C and 250 rpm until an OD600 of 0.9, then protein expression was induced with 0.5 mM IPTG for 4 h.
After protein expression, cells from the M9 culture were centrifuged at 4,000 g and 4˚C for 10 min. Cell pellet was resuspended in 80 mL lysis buffer containing 20 mM Tris, 8 M urea, pH 8.0 and lysed using a tip sonicator (40% power, 5 s on, 5 s off, 10 min). The cell lysate was centrifuged at 15,000 g for 20 min to remove cell debris and protein aggregates. The supernatant containing the NT*-Aβ40 fusion protein was purified using a Ni-NTA affinity column. Fractions containing the fusion protein (Fig. S1a) were pooled and dialyzed against 1.5 L of 20 mM pH 8 Tris buffer overnight. After dialysis, TEV protease was added at a ratio of 1:10 TEV : substrate (w/w), and cleavage reaction was conducted in 50 mL cleavage buffer (20 mM pH 8 Tris, 0.5 mM EDTA and 1 mM DTT) at 4ºC without shaking overnight. After cleavage was assessed by SDS-PAGE to be complete (Fig. S1a), solid guanidinium chloride was added to the cleavage solution to a concentration of 6 M, and the reaction mixture was sonicated for 30 min. After filtration through a 0.45 μm syringe filter, Aβ40 was purified by reverse-phase HPLC using a semi-preparative C4 column (250 mm length, 10 mm I.D., 10 µm particle size, 300 Å pore size, Higgins analytical) using a linear gradient of 15–40% buffer B in 25 min (buffer A: water with 0.1% TFA, buffer B: acetonitrile with 0.1% TFA, column heated to 60 ºC). The purity of the peptide was verified by SDS-PAGE (Fig. S1b), analytical HPLC (Fig. S1c) and mass spectrometry (Fig. S1d). The pure fractions were combined and lyophilized to give the Aβ40 powders 40. The yields of purified Aβ peptides were 10 mg and 15 mg per liter of M9 culture for 13C, 15N-labeled protein and unlabeled Aβ40, respectively.
Fibrillization of Aβ40 peptide
The purified recombinant Aβ40 peptide was fibrillized using a previously published protocol 41–42. The lyophilized peptide was monomerized by sequential dissolution in HFIP and DMSO, then fibrils were grown at a low concentration of ~46 μM in pH 7.4 phosphate buffer at 37˚C for 3 days under shaking. Specifically, Aβ40 was first dissolved in HFIP (5 mg/mL) and sonicated for 5 min. Aliquots of 40 μl HFIP solution were transferred into 1.5 mL low-binding microcentrifuge tubes. 25–50 tubes of samples were prepared to obtain a sufficient quantity of fibrils for solid-state NMR experiments. The HFIP solution was evaporated, and 20 μl DMSO was added to each tube to dissolve the peptide film to 10 mg/mL. The solution was vortexed and sonicated for 30 min, after which 980 μL of 10 mM pH 7.4 sodium phosphate buffer was added to reach a peptide concentration of 0.2 mg/mL. First dissolving the peptide in DMSO led to fibrils with biological activity in mouse inoculation 43. This procedure was previously shown to reproduce the Osaka Aβ chemical shifts measured on recombinant protein 41, 44. The peptide solution was shaken at 37˚C and 900 rpm for 3 days for fibril growth. To collect the fibrils, we spun the tubes at 13,200 rpm and 4˚C for 20 min using a benchtop microcentrifuge, and combined all the pellets into one tube. No Aβ40 peak was detected from the supernatant by analytical HPLC (Fig. S1e), indicating that the fibrilization yield was more than 99%.
Binding of flutemetamol to Aβ40 fibrils
Like many PET tracers, the hydrophobic flutemetamol has very low solubility in aqueous solution 45, making it difficult to saturate the binding sites on the Aβ fibrils. To overcome this difficulty, we adopted a recently published procedure that uses bovine serum albumin (BSA) to increase the solubility of the ligand and facilitate its binding to Aβ40 45. BSA and flutemetamol were co-solubilized at equimolar concentrations of 50 μM in a 20 mM pH 7.4 sodium phosphate buffer containing 25% DMSO. We then removed DMSO by dialyzing 225 mL of this solution against 2 L of 20 mM sodium phosphate buffer for 4 h with fresh buffer change every hour. The dialyzed solution was centrifuged at 11,000 g for 20 min at 20˚C using a JA-10 rotor to remove insoluble flutemetamol. The supernatant was analyzed by HPLC to determine the flutemetamol concentration, which was 15 μM. We then added 75 mL suspension of 40 μM Aβ40 fibrils to 187 mL of the 15 μM flutemetamol-containing supernatant to reach a 1 : 1 molar ratio of flutemetamol to Aβ monomers. The solution was shaken at 250 rpm at 37˚C for an hour to facilitate mixing of the ligand to the fibril, then centrifuged at 18,000 rpm at room temperature for 40 min using a JA-20 rotor to collect the fibrils. The ligand-bound Aβ fibrils from six 50 mL centrifuge tubes were combined into two 1.5 mL tubes and either dried to ~50 wt% water or kept at the same hydration level before being spun down into MAS rotors using a benchtop centrifuge.
Measurement of flutemetamol binding affinity to Aβ40 fibrils
To measure the binding affinity of flutemetamol to Aβ fibrils, we titrated solubilized flutemetamol to preformed Aβ40 fibrils to reach 0–11 μM final concentrations of flutemetamol and 11 μM Aβ40. We also prepared a control sample by adding flutemetamol-containing solution to a peptide-free buffer. The solutions were shaken in an incubator at 250 rpm and 37˚C for an hour, then centrifuged at 13,000 rpm for 20 min. The supernatant was analyzed by HPLC at 358 nm to determine the concentration of free flutemetamol, [Tracer]free. The concentration of bound flutemetamol, [Tracer]bound, was calculated as the difference of the free flutemetamol concentration between the peptide-free and peptide-containing samples. The apparent dissociation constant KD and the maximum flutemetamol concentration (Bmax) bound to 11 μM Aβ40 fibrils were determined by fitting [Tracer]free and [Tracer]bound using the equation (Fig. S2)
We found a KD of 1.6 ±0.2 μM and a Bmax of 2.4±0.1 μM (Fig. S2). This Bmax value indicates a binding ratio of one flutemetamol to 4.6 peptides for the 11 μM Aβ40, or a binding mole fraction of 22%.
To determine whether flutemetamol is bound homogeneously to the fibrils, we treated both ligand-bound fibrils and ligand-free fibrils with 50 µL of 6 M guanidinium chloride in 20% DMSO solution. This procedure releases loosely bound flutemetamol in the ligand-bound fibrils. We then centrifuged the solution and measured the amount of flutemetamol in the supernatant by HPLC. About 12% of the total added flutemetamol was detected in the supernatant. Since the initially added flutemetamol concentration was equimolar to Aβ monomers, this means that ~12 mol% of flutemetamol relative to Aβ40 monomers is loosely bound to the peptide. Since the total bound flutemetamol is 22 mol% of the fibrils, this means ~10 mol% of flutemetamol relative to Aβ monomers is tightly bound.
To determine whether the flutemetamol-bound Aβ fibrils contain residual BSA after ultracentrifugation, we prepared another control sample in which BSA but not flutemetamol was added to the 25% DMSO/20 mM sodium phosphate buffer. The solution was mixed with either 11 μM Aβ40 or with buffer only. After centrifugation, we measured the BSA concentrations in the supernatant at 280 nm by HPLC. The difference in BSA concentrations between the supernatant without and with Aβ40 fibrils is the concentration of BSA bound to Aβ40 fibrils. Only 0.2 ± 0.4 μM of BSA was found to remain with the 11 μM Aβ fibrils. Given the uncertainty, essentially no BSA remained in the fibrils, while the bound flutemetamol has a molar ratio of 1 : 4.6 with respect to peptide monomers.
Transmission electron microscopy (TEM) experiments
Aβ fibrils were adsorbed onto freshly glow-discharged, 200-mesh formvar/carbon-coated copper grids (Ted Pella), washed with 0.1 M ammonium acetate followed by 0.01 M ammonium acetate, and stained with 2% (wt/vol) uranyl acetate. TEM images were taken on a JEOL JEM-1400 Transmission Electron Microscope at the University of Kansas Medical Center Electron Microscopy Research Laboratory.
Solid-state NMR experiments
Apo Aβ40 fibrils, flutemetamol-bound fibrils, and ligand-free BSA control fibrils were packed into 3.2 mm Revolution NMR rotors by benchtop centrifugation. Each rotor contained ~30 mg hydrated material, in which ~5 mg was the dry mass of the peptide. We also packed flutemetamol-bound Aβ40 fibrils into a Bruker 1.9 mm MAS rotor for 13C-19F and 1H-19F REDOR experiments. The hydrated mass of this rotor was ~17 mg, in which ~8.8 mg was the dry peptide mass.
All 13C and 15N solid-state NMR experiments were conducted on a Bruker Avance II 800 MHz (18.8 T) spectrometer using a BlackFox 3.2 mm HCN MAS probe. All 19F NMR experiments were carried out on a Bruker Avance III 600 MHz (14.1 T) spectrometer using a 1.9 mm HFX probe. 13C chemical shifts were referenced externally to the adamantane CH2 chemical shift at 38.48 ppm on the tetramethylsilane (TMS) scale. 15N chemical shifts were referenced to the 15N peak of 15N-acetylvaline at 122.0 ppm on the liquid ammonia scale. 19F chemical shifts were referenced to the 5F-Trp at −122.1 ppm on the CF3Cl scale. All experiments were conducted at an MAS frequency of 14 kHz. Sample temperature was kept at 287 K for most experiments. This was controlled by keeping the water 1H chemical shift (δH2O) at 4.89 ppm, using the equation Teff (K) = 96.9 × (7.83−δH2O) 46. Typical radiofrequency (rf) field strengths for the 3.2 mm MAS probe were 50–83 kHz for 1H, 15–62.5 kHz for 13C and 25–36 kHz for 15N. Typical rf field strengths for the 1.9 mm MAS experiments were 107.5 kHz for 1H, 62.5 kHz for 13C and 71.4 kHz for 19F.
2D 13C-13C and 15N-13C correlation experiments and 3D NCACX, NCOCX and CONCA experiments were used to assign the 13C and 15N chemical shifts of apo and flutemetamol-bound Aβ40 47–49 (Table S1). In these experiments, 15N-15C polarization transfer was conducted using the SPECIFICCP sequence 50 and 13C-13C polarization transfer was conducted using the CORD sequence 51. 1H TPPM decoupling was applied at an rf field strength of 83 kHz during the evolution and acquisition periods. To measure flutemetamol contact with Aβ40, we first carried out a 1D selective 13C-19F REDOR experiment 23 52. To obtain additional site resolution and extend the distance reach, we next implemented a 2D 13C-13C resolved 1H-19F REDOR experiment, whose pulse diagram is shown in Fig. S3a 24, 31. This experiment was conducted at an MAS frequency of 14 kHz, at which 1H homonuclear decoupling was necessary during the 1H-19F REDOR period. We used the frequency-switched Lee-Goldberg (FSLG) sequence for homonuclear decoupling 53. The transverse 1H rf field strength of the FSLG train was 107.5 kHz, which fit 75 FSLG cycles into a REDOR mixing time of 2.3 ms. The 19F-dephased 1H magnetization was then transferred to 13C and detected in a 2D 13C-13C correlation spectrum. More detailed experimental parameters are listed in Table S2.
One- and two-dimensional solid-state NMR spectra were processed and plotted in the Topspin software, and resonance assignment of the 3D correlation spectra were conducted in the NMRFAM-Sparky software 54. Backbone (ϕ, ψ) torsion angles were calculated from the assigned chemical shifts using the TALOS-N software 55 after adjusting the 13C chemical shift to the DSS scale. 19F spinning sideband patterns were fit using the solid lineshapes analysis tool in the Topspin software. The 19F chemical shift anisotropy (CSA) is reported in terms of the anisotropy parameter , asymmetry parameter 56–57, and the isotropic chemical shift , where δzz, δxx and δyy are the principal values of the chemical shift tensor in decreasing difference from δiso.
1H-19F REDOR simulations
REDOR simulations were conducted using the SIMPSON software 58 through NMRbox 59. Powder averaging used the REPULSION 168 scheme 60 with 32 γ angles. A 19F CSA δ of −73.7 ppm and η of 0.49 were used in the simulations, and the z-axis of the 19F CSA tensor was approximated as colinear with the 1H-19F vector. 19F pulse flip angle imperfection was taken into account by averaging the REDOR simulations with effective 19F flip angles of 180° to 150° in 5° increments, weighted by a Gaussian distribution centered at 180° with a standard deviation of 15° 23.
Molecular docking of flutemetamol to Aβ fibrils
A molecular structure model of flutemetamol was generated using ChemDraw and exported as a “.mol” file. This structure was energy-minimized in Chem3D using the MMFF94 force field 61 and a minimum gradient norm of 0.1. The optimized conformation shows a ~13 Å long and ~3 Å wide molecule. This structure was outputted as a “.pdb” file for docking simulations.
We used the HADDOCK software 62 to dock flutemetamol to four published Aβ40 fibril structural models 2–3, 63–64. These four structures were chosen to represent distinct fibril polymorphs and structure determination methods. A two-fold symmetric structure of in vitro fibrillized Aβ40 solved using solid-state NMR (PDB: 6TI5) 63 contains 2 × 8 monomers. Another two-fold symmetric structure was obtained from AD brain vasculature Aβ40 fibrils and was solved using cryoEM (PDB: 6SHS) 2. This structure model contains 2 × 6 monomers. Because flutemetamol spans approximately 3.5 β-strands when lying parallel to the fibril axis, we reduced these structural models to 2 × 4 monomers prior to input into HADDOCK to speed up the simulations. A three-fold symmetric structure of in vitro fibrilized Aβ40 (PDB: 2LMQ) contains 3 × 6 monomers 64. This model was not modified despite its large size because the structure is not translationally symmetric. A three-fold symmetric structure of brain-seeded fibrils (PDB: 2M4J) 3 contains 3 × 3 monomers and was used directly for docking.
These Aβ40 structural models and the flutemetamol structure were inputted into the HADDOCK software for peptide-ligand docking. Because V12, H13, H14, V18, F19, F20, V39, and V40 were found to be in close proximity to the ligand from the REDOR experiments, we ran eight separate simulations in which each of these residues was set as an “active” residue. HADDOCK uses the active residue list to generate Ambiguous Interaction Restraints, which impose an energy penalty for placing the ligand too far from the active residue. No explicit distances were inputted into the HADDOCK simulations.
In most simulations, we specified a single copy of the active residue and placed it in an interior strand. When all copies of a residue were set as active, control simulations showed very similar results, except that there was a stronger preference for the ligand to lie parallel to the fibril axis, contacting multiple copies of the active residue. In the single-active-residue simulations, the binding poses are more varied, and are sometimes tilted toward the β-strand backbone. We also included nonpolar hydrogens in the molecular structures and left all other settings at their default values. We left random patches and center of mass restraints off, and used 1000 structures for rigid-body docking, 200 for semi-flexible refinement, and 200 for the final refinement.
The HADDOCK software clusters and ranks the docking results according to an internal scoring function, which is a linear combination of various energy terms and buried surface area. We overlaid the top ten clusters for each simulation, or fewer for simulations that produced less than ten clusters in total. We discarded docking sites located on the first and last strands of the fibril. If these sites exist physically, they would account for only a small fraction of bound flutemetamol. The three-fold in vitro Aβ40 structure shows irregularity at the N-terminus, thus we discarded sites near the N-terminus. For each fibril structure, we combined results for all eight simulations for the eight active residues and discarded repeated instances of the same site for visual clarity. This yielded an ensemble of docked sites. The binding energy (kcal/mol) for each site was calculated using the PRODIGY-LIGAND software 65, a contact-based binding energy predictor compatible with HADDOCK output files.
Results
Flutemetamol binding to Aβ fibrils
The hydrophobic flutemetamol has very low solubility in aqueous solution. Even in the presence of 2% DMSO, the solubility is only ~0.2 μM, which makes it difficult to prepare ligand-bound Aβ fibrils for solid-state NMR experiments. Thus, we adopted a recently published procedure that uses BSA as a carrier protein to solubilize and titrate flutemetamol to preformed Aβ fibrils 45. BSA is an abundant blood plasma protein that binds a wide range of hydrophobic compounds 66–67, and is commonly used as a drug-delivery agent. In 25% DMSO solution, both flutemetamol and BSA can be dissolved to 50 μM. We dialyzed this flutemetamol-containing BSA/DMSO solution to remove DMSO, then centrifuged the solution to remove insoluble flutemetamol, giving a flutemetamol concentration of 15 μM. This flutemetamol-BSA solution was then titrated to preformed Aβ40 fibrils, and BSA was quantitatively removed by centrifugation. HPLC analysis shows that this BSA-mediated titration method results in a bound flutemetamol mole fraction of 22%, in which 12% is loosely bound, releasable after treatment of the fibrils with 6 M guanidinium chloride, while 10% is tightly bound.
Flutemetamol binding to Aβ40 fibrils assessed by 19F solid-state NMR
19F NMR spectra of fluorinated flutemetamol (Fig. 2a) allowed us to assess the structure and dynamics of the PET tracer when bound to the Aβ fibrils. Powder flutemetamol shows a 19F isotropic chemical shift of −134.8 ppm in the cross-polarization (CP) spectrum, and the spinning sideband intensity pattern fits to a rigid-limit CSA of −73.7 ppm (Fig. 2b). Binding to hydrated fibrils did not change the isotropic chemical shift nor the sideband intensity envelope (Fig. 2c), indicating that the bound flutemetamol is similarly immobilized as in the powder. However, the peptide-bound flutemetamol has much broader linewidths of 4.4 ppm compared to powder flutemetamol (1.7 ppm), indicating that the bound ligand is more disordered. Comparison of the CP intensities of the powder and the peptide-bound ligand indicate that the immobile fraction of the ligand represents ~10 mol% of the Aβ peptide. This 1:10 binding stoichiometry is in excellent agreement with the HPLC-determined fraction of tightly bound ligand.
Figure 2.
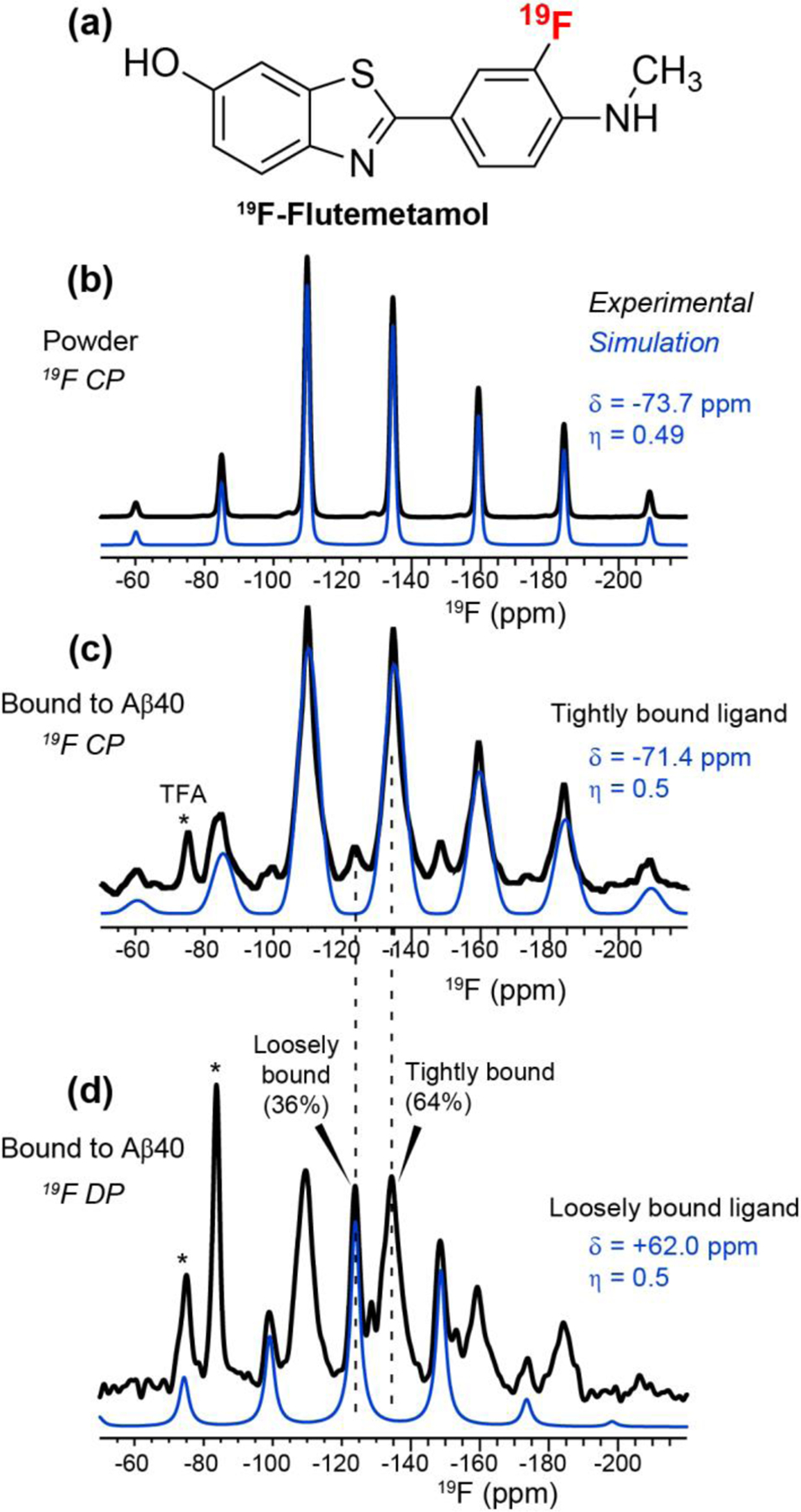
19F NMR spectra of flutemetamol. (a) Chemical structure of 19F-labeled flutemetamol. (b) 19F CP spectrum of powder flutemetamol, measured with a 40-s recycle delay. Spectral simulation yields a 19F CSA δ of −73.7 ppm and an asymmetry parameter η of 0.49. (c) 19F CP spectrum of Aβ-bound flutemetamol, measured with a 2-s recycle delay. Spectral simulations of the sideband pattern indicate very similar 19F CSA parameters as the powder sample. (d) 19F DP spectrum of Aβ-bound flutemetamol, measured with an 18-s recycle delay. A second component with an isotropic chemical shift of −124.2 ppm is observed, which has a reduced CSA of +62 ppm and narrower linewidth than the −134.8 ppm isotropic component. Asterisks denote residual peptide-bound TFA (−75 ppm) and free TFA (−85 ppm) in the samples. The latter is only detected in the DP spectrum while the former is detected in both the CP and DP spectra. All spectra were measured under 14 kHz MAS.
To obtain more quantitative information about the bound ligands, we measured a 19F direct-polarization (DP) spectrum of peptide-bound flutemetamol using a long recycle delay of 18 s (Fig. 2d). Interestingly, the DP spectrum exhibits a second 19F isotropic chemical shift of −124.2 ppm, which has narrower linewidths and a smaller CSA (+62 ppm) than the CP-detected component. Thus, a second population of ligand exists that undergoes small-amplitude motion. The intensity of this mobile component is 36% of the full spectrum. This mobile component also exhibits residual CP intensity, but only at ~20% that of the DP intensity. This CP intensity is much lower than the 1.3-fold enhancement of the CP intensity over DP for the immobilized fraction. Since the 19F CSA of the mobile fraction is still relatively large, 87% of the rigid-limit value, the low CP intensity of this component cannot be completely attributed to motion. Instead, it likely reflects the long distances of the mobile ligand from the peptides. The 1H T1 of powder flutemetamol is estimated to be ~6 s based on 1H-19F CP experiments with varying recycle delays, while the 1H T1 of the hydrated peptide fibrils is ~1 s. Thus, with the recycle delay of 2 s for the 1H-19F CP experiment, the loosely bound flutemetamol would have incompletely equilibrated intensities compared to the tightly bound ligand. These 19F NMR spectra, taken together with binding assays, indicate that two populations of flutemetamol coexist when bound to the fibrils: a tightly bound and immobile fraction that represents ~10 mol% of the peptide, and a loosely bound fraction that represents ~12 mol% of the peptide.
Secondary structure of apo Aβ40 fibrils from 13C and 15N chemical shifts
We assessed the morphology of the Aβ40 fibrils using TEM. The apo peptide produced straight and homogeneous filaments that are 17–20 nm wide (Fig. 3a). This apparent width is 2–3 fold larger than those of previously reported Aβ40 fibrils2–3, 45, 63–64, 68, suggesting lateral association of two or three protofilaments. One- and two-dimensional 13C and 15N spectra (Fig. 3b–d) of the apo peptide show narrow linewidths of 0.8 ppm for 15N and 0.6 ppm for 13C, indicating a homogeneous molecular conformation. In the Gly region of the 2D 15N-13Cα correlation spectrum (Fig. 3c), about nine Gly peaks are resolved. Since Aβ40 contains 6 glycine residues, this observation indicates the presence of more than one conformation for some residues.
Figure 3.
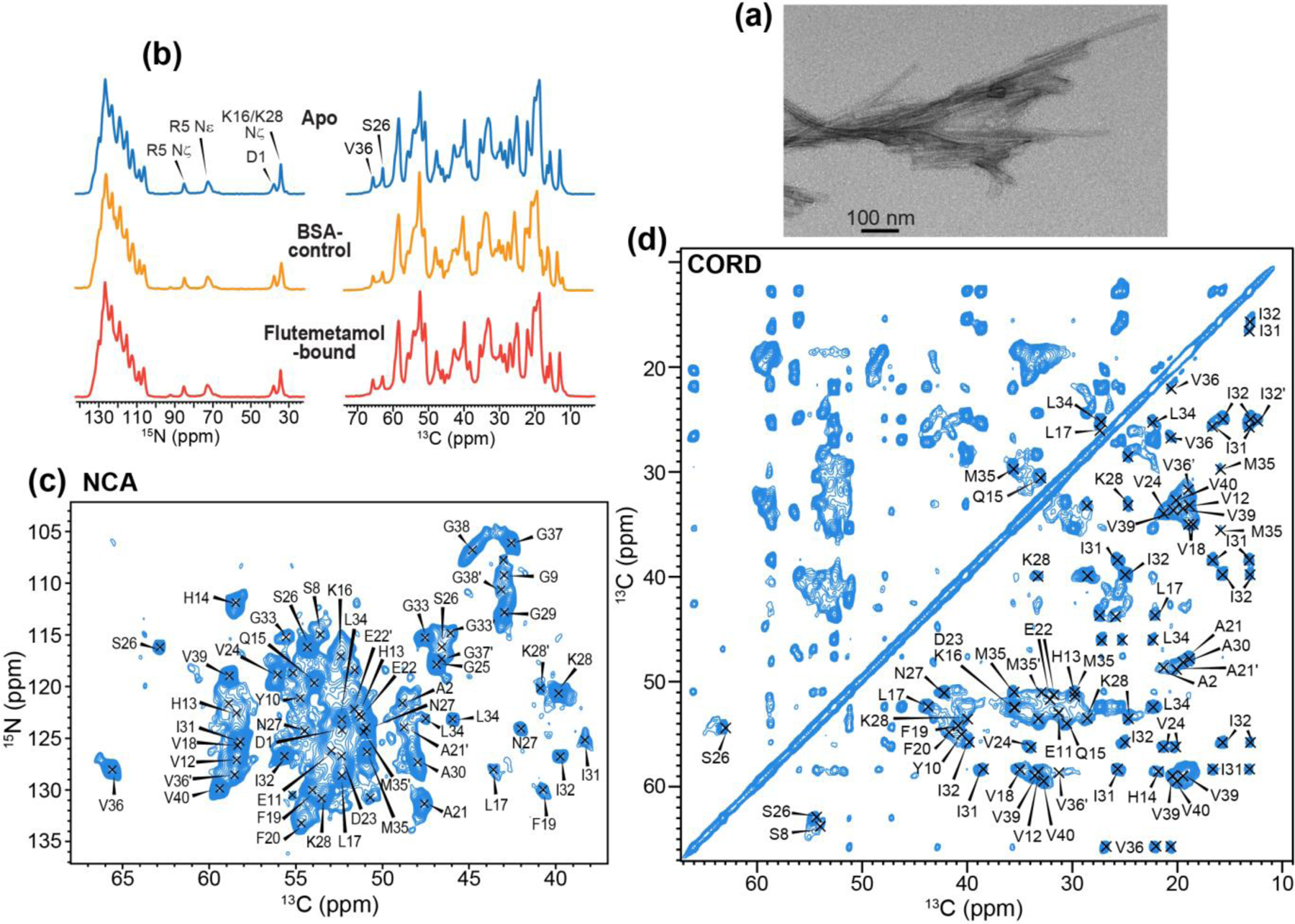
Characterization of apo, BSA control and flutemetamol-bound Aβ fibrils. (a) TEM image of apo Aβ40 fibrils. (b) 1D 13C and 15N MAS spectra of apo (blue), BSA control (orange) and flutemetamol-bound (red) Aβ40 fibrils. (c) 2D 15N-13Cα correlation spectrum of apo Aβ40 fibrils. Assignment is obtained from 3D correlation spectra. (d) 2D 13C-13C correlation spectrum of apo Aβ40.
We assigned the 13C and 15N chemical shifts of the apo and flutemetamol-bound peptide using 3D NCACX, NCOCX and CONCA correlation experiments. For the apo peptide, about 85% of the spectral intensities correspond to a major conformation whereas ~15% of the intensities result from a second conformation (Fig. 4). The latter is mostly found for residues 21AE22 and 32IGLMVGG38 (Fig. S4, S5). The flutemetamol-bound peptide has very similar chemical shifts and similar coexistence of a major conformer and a minor conformer. Therefore, ligand binding does not cause noticeable perturbation of the peptide conformation. In the major conformer, the N-terminal D1 and A2 exhibit rigid β-sheet signals, while the next five residues 3EFRHD7 lack intensities, indicating that they are dynamic 45, 64, 68. The rest of the peptide shows strong intensities, indicating it is immobilized. We detected clear intensities for 13HHQK16 (Fig. 4a), which is dynamic in many previously reported Aβ40 polymorphs 3, 45, 69–71. Moreover, H14 exhibits α-helical Cα and Cβ chemical shifts (Fig. 4a, Table S1), in contrast to all previously studied Aβ polymorphs that contain immobilized H13 and H14 63–64, 68. We also observed α-helical chemical shifts at V36 (Fig. 3d, Fig. 4b), in contrast to β-sheet chemical shifts in all other Aβ polymorphs so far studied by NMR. The root-mean-square deviation (RMSD) of CO, Cα and Cβ chemical shifts between the current Aβ40 fibrils and previously studied Aβ fibrils range from 2.5 ppm to 4.5 ppm (Fig. S6). TALOS-N (ϕ, ψ) torsion angles derived from these chemical shifts indicate three β-strands in the current polymorph, spanning residues S8-H13, Q15-E22, and V24-M35 (Fig. S7). The β-strand conformation of 24VGSNK28 differs from the turn structure adopted by this segment in previously reported polymorphs. Unexpectedly, the major breaks between β-strands in the current Aβ40 conformation are consistent with the β-strand breaks at H14, V24, and V36 found in the meningeal AD brain Aβ fibril structure 2. The non-β-strand character of the C-terminal 36VGGVV40 segment in the current polymorph is also qualitatively consistent with the C-terminal bend in an AD brain-seeded Aβ fibril sample 3. These observations suggest that the current Aβ polymorph might recapitulate some aspects of AD brain-derived Aβ aggregates.
Figure 4.
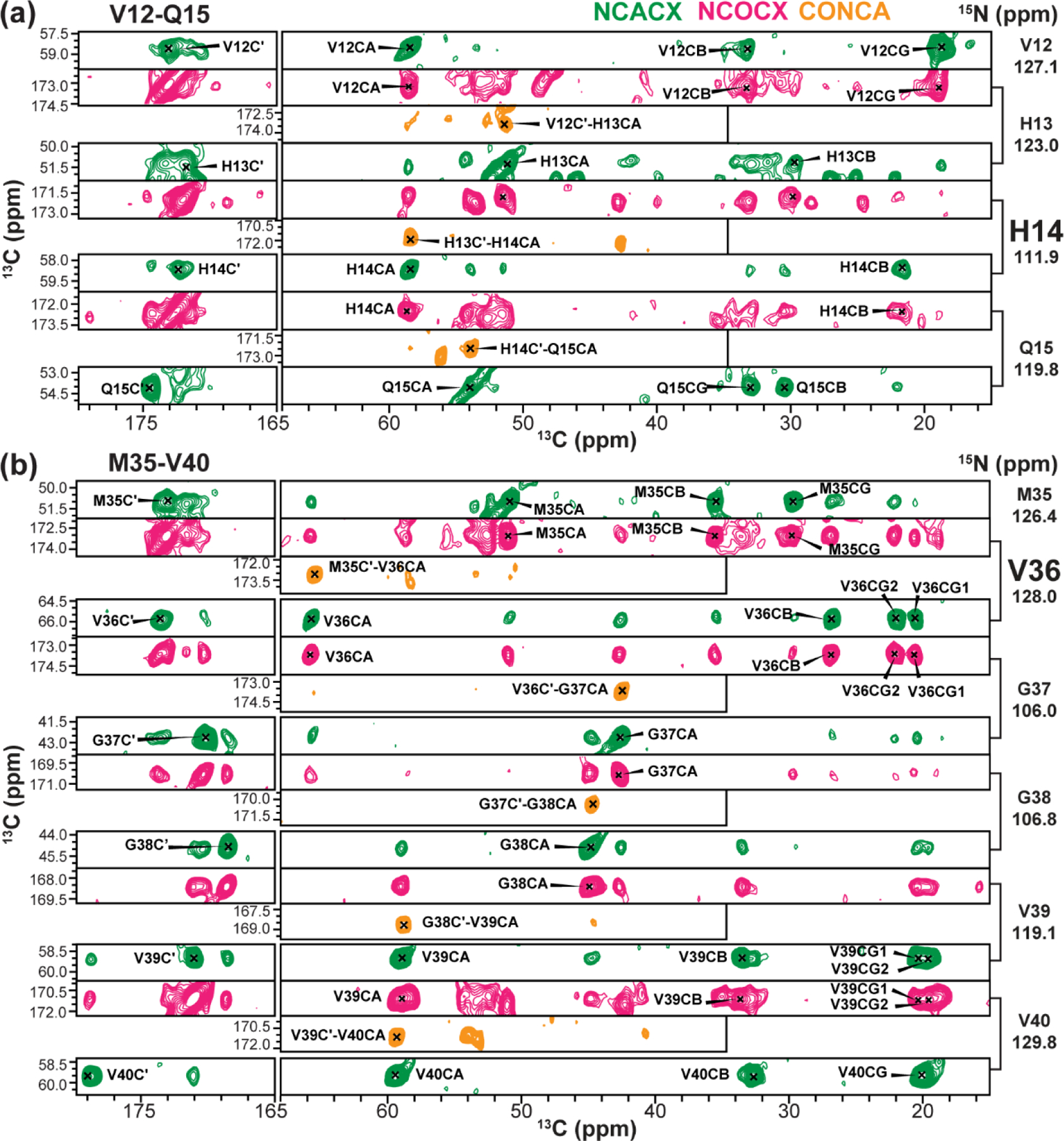
Representative 3D spectral strips for chemical shift assignment of apo Aβ40 fibrils. (a) 12VHHQ15 strip. (b) 35MVGGVV40 strip. Note the α-helical chemical shifts of H14 and V36. The NCACX spectrum provides intra-residue correlation peaks whereas the NCOCX and CONCA spectra provide inter-residue correlations.
Since flutemetamol-bound fibrils show similar chemical shifts and intensity distributions as the apo peptide (Fig. 3b, Fig. S4, S5), the PET tracer does not cause observable conformational changes to the fibrils. To evaluate whether incubating preformed Aβ40 fibrils with BSA solution during ligand titration might have perturbed the fibril conformation, we measured the 2D spectra of a BSA control sample produced by subjecting the fibrils to ligand-free BSA solution. This control sample exhibited the same major- and minor-conformer chemical shifts (Fig. S4c, S5c), with the major conformer peaks accounting for two thirds of the total intensities. The coexistence of the same two conformers in all three samples indicate that this structural polymorphism 72 results from the common sample preparation conditions used here, rather than from the use of BSA during ligand titration.
Flutemetamol-interacting residues in Aβ40 fibrils from 19F REDOR NMR
To measure the proximities of flutemetamol to peptide residues, we conducted 13C-detected 13C-19F (Fig. 5) and 1H-19F REDOR (Fig. 6) experiments. 13C spectra of the peptide were measured in the absence (S0, black) and presence of 19F pulses for various mixing times. Only carbons that are close to the fluorine will show intensity differences (ΔS, red). For the 13C-19F REDOR experiment, we first measured sidechain-selective and backbone-selective spectra, whose residue-type assignment is given in Fig. 5a. The sidechain-selective REDOR difference spectra show methyl signals of Leu, Ile and Val, whereas the Cα-selective REDOR difference spectra show the signals of Val, Ile and H14 (58.5 ppm) and Phe/Gln (54.5 ppm) (Fig. 5b). The 22-ppm peak for Leu/Val Cγ or H14 Cβ exhibits particularly high difference intensities, indicating the involvement of at least one of these sidechain carbons in ligand binding. To detect other sidechain signals, we also measured broadband 13C-detected 1H-19F REDOR spectra (Fig. 5c). We detected various Cβ difference peaks, for example at 40 ppm for Ile, Phe and Lys. The highest ΔS intensity is observed at 18.8 ppm, which contains signals from V12, V18, A21 or A30 sidechains. This peak shows a ΔS/S0 ratio of ~0.03. Since the tightly bound ligand has a stoichiometry of 10% relative to the Aβ monomers, this difference corresponds to a real ΔS/S0 of ~0.3, suggesting that some of the sidechains are very close to the fluorine.
Figure 5.
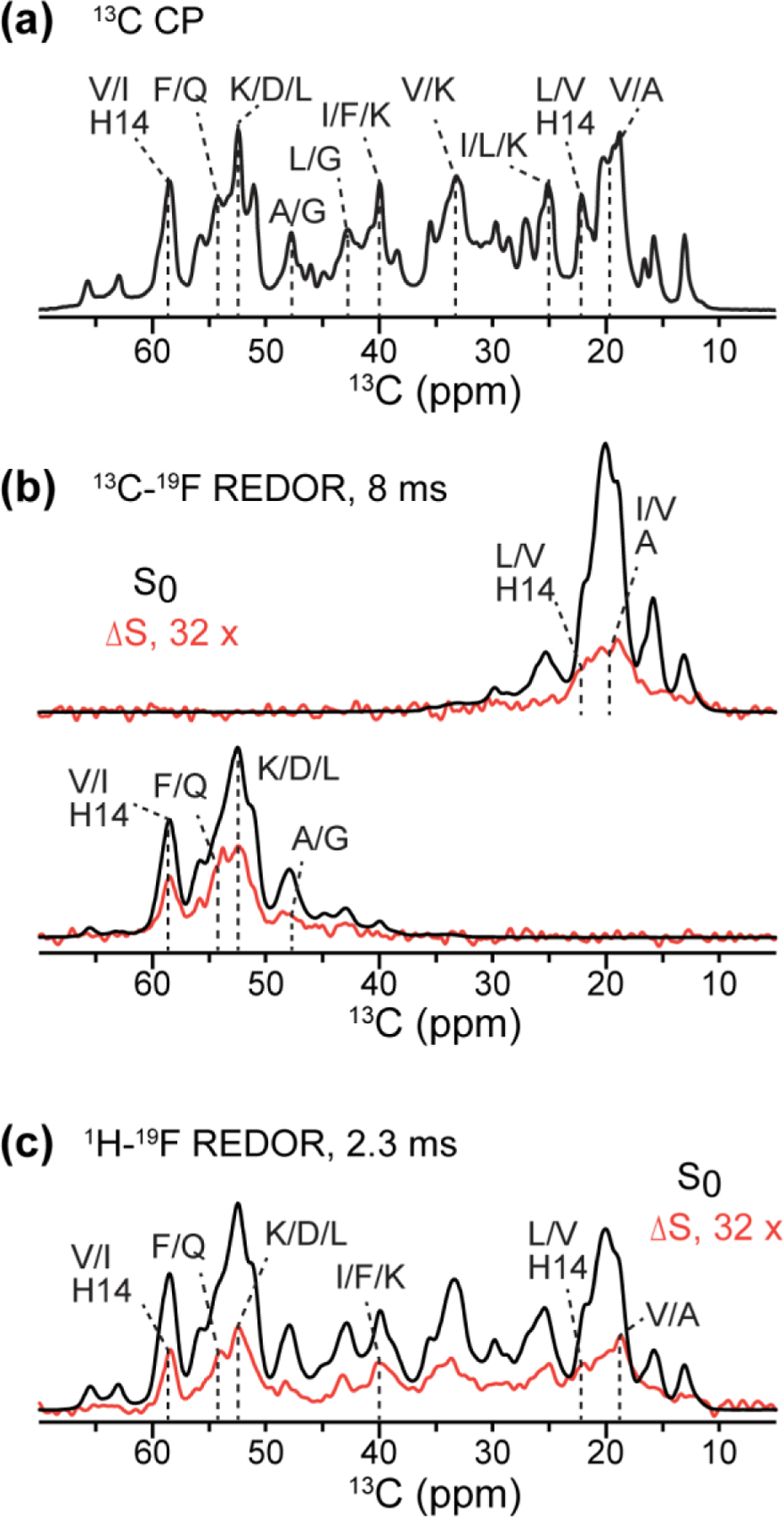
1D 13C-detected 19F-dephased REDOR spectra of flutemetamol-bound Aβ40. (a) 13C CP spectrum, showing amino acid type assignment. (b) Methyl- and Cα-selective 13C-19F REDOR spectra with 8 ms mixing. For clarity, the difference spectrum (ΔS) is scaled up 32-fold with respect to the control spectrum (S0). (c) 13C-detected 1H-19F REDOR spectra measured with 2.3 ms mixing. Both (b) and (c) indicate that multiple residues are dephased by the fluorine.
Figure 6.
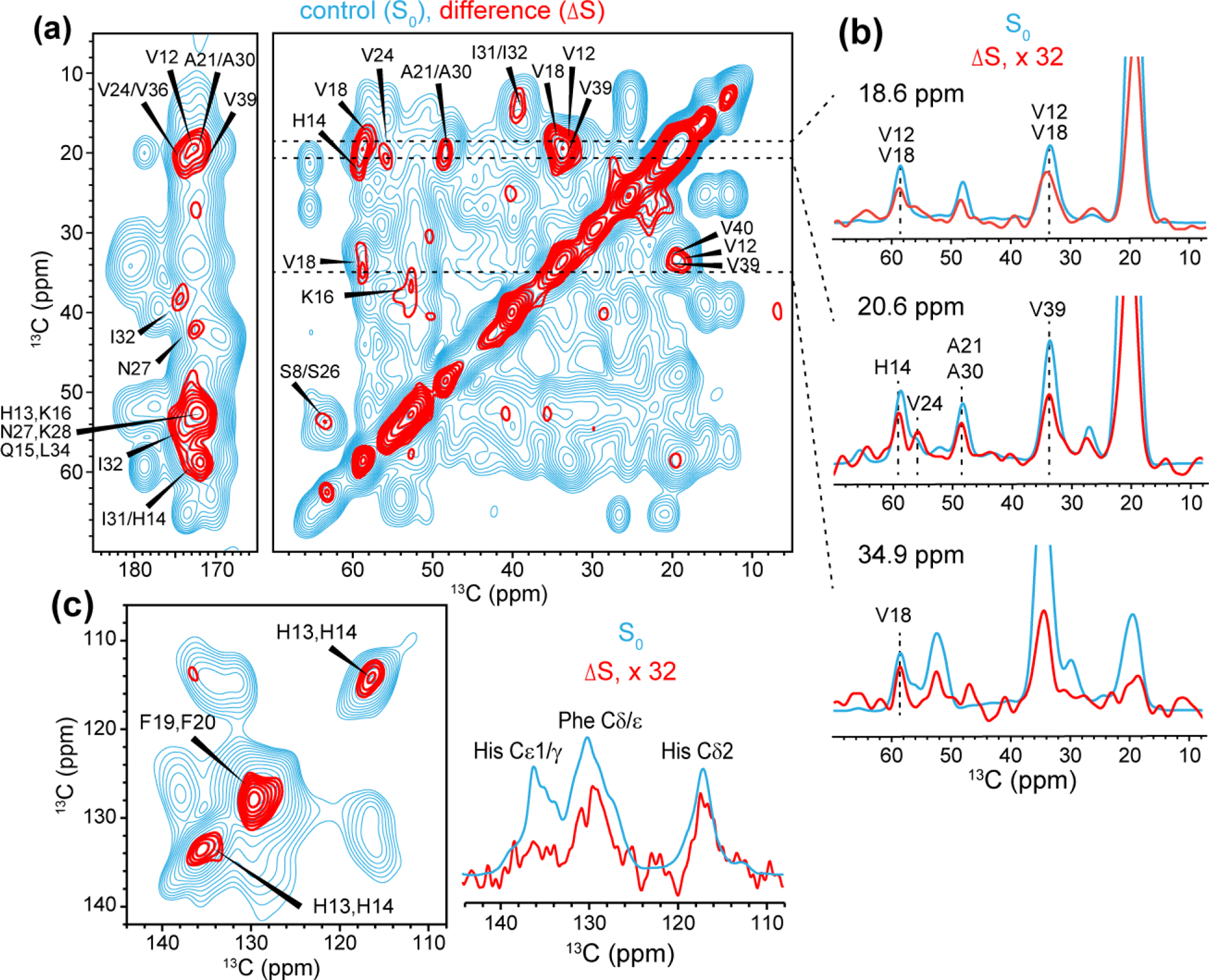
2D 13C-13C resolved 1H-19F REDOR S0 (cyan) and ΔS (red) spectra for determining residues in close proximity to flutemetamol. The REDOR mixing time was 2.3 ms. (a) Aliphatic and carbonyl regions. (b) Selected 1D cross sections. The ΔS spectrum is scaled up 32-fold to facilitate comparison with the S0 spectrum. These REDOR spectra are significantly line-broadened (LB = −30 and GB = 0.02) to maximize the signal-to-noise ratios. (c) Aromatic region of the 2D spectrum (left). The 1D 13C-detected 1H-19F REDOR spectra are shown on the right. Both spectra were measured using a mixing time of 2.3 ms and gave consistent results.
To better resolve the signals of flutemetamol-interacting residues, we next turned to 2D 13C-13C resolved 1H-19F REDOR experiment. Similar to the 1D REDOR experiments, the 2D spectra were measured in the absence (S0, cyan) and presence of 19F pulses, and their difference spectrum (ΔS, red) indicate the carbons whose directly bonded protons are in close proximity to the ligand fluorine. Because the 1H-19F REDOR period occurs before 13C t1 evolution (Fig. S3a), the ω1 frequency of the cross peaks indicates the fluorine-proximal protons that are bonded to a specific carbon. The 2D spectrum resolves a number of sites (Fig. 6, Table S3). In the 18.6 ppm cross section (Fig. 6a, b), cross peaks at 34.0 ppm and 58.5 ppm can be assigned to V12 and V18. In the 20.6-ppm cross section, cross peaks are observed at 59.1 ppm, 56.0 ppm, 48.6 ppm, and 33.8 ppm, which can be assigned to H14, V24, A21 or A30, and V39, respectively. In the 34.9-ppm cross section, correlations at 59.0 ppm and 19.0 ppm confirm that V18 is close to the ligand. A weak peak at (36.7, 52.8) ppm in the 2D difference spectrum can be assigned to K16. In the carbonyl region of the 2D spectrum, a correlation peak at (58.9, 172.0) ppm can be assigned to H14 or I31. Importantly, we observed strong difference intensities for the aromatic Phe and His residues along the diagonal of the 2D spectrum (Fig. 6c). Since there are only two Phe residues (F19 and F20) and three His residues (H6, H13 and H14) in the peptide, we assigned these Phe and His signals to F19/F20 and H13/H14. The 2D CC spectrum show ΔS/S0 values of 1.8–3.1%, which correspond to S/S0 values of 96.9 – 98.2% (Fig. S3b).
Based on the secondary structure predicted from the chemical shifts (Fig. S7), H13/H14 are located at the beginning of a β-strand, while F19/F20 are located at the other end of the same β-strand. Thus, these two sets of residues are unlikely to form a single binding site for flutemetamol. Thus, multiple binding sites must exist in the fibrils. Taking into account the 1 : 10 stoichiometry for the tightly bound flutemetamol, and assuming two equally populated binding sites, we broadly estimate a distance range of 6–7 Å for the nearest peptide protons to the ligand fluorine (Fig. S3b). These REDOR spectra, taken together, indicate that three clusters of residues, V12 and H13/H14, V18 and F19/F20, A21 or A30, and V39/V40, are in close proximity to the ligand fluorine. These three clusters of residues, 12VHH14, 18VFFA21, and the C-terminus, constitute at least two binding pockets for flutemetamol.
Experimentally constrained docking of flutemetamol to Aβ fibrils
To investigate whether the residues identified by REDOR experiments to be close to flutemetamol are energetically favorable for binding the ligand, we conducted docking analysis to several Aβ40 structures. In the Alzheimer brain, flutemetamol and PiB detect Aβ fibrils in cored plaques, diffuse plaques, and cerebral amyloid angiopathy 73. PiB also binds synthetic Aβ40 fibrils assembled in vitro 15. Therefore, these PET ligands bind multiple Aβ polymorphs 3, 74, which must share common binding sites. These binding sites should have similar local conformations and should stabilize the ligand by similar molecular interactions. We selected four Aβ40 structural models (Fig. 7, Fig. S8) for the docking simulations: a two-fold symmetric structure of in vitro fibrilized Aβ40 63, a two-fold symmetric structure of AD brain vasculature Aβ40 2, a three-fold symmetric structure of in vitro fibrilized Aβ40 75, and a three-fold symmetric structure of AD brain-seeded Aβ40 fibrils 3. The three structural models solved using solid-state NMR have a common β-arch fold with a U-turn at 22EDVGSNK28 in each protofilament, but differ in the packing between protofilaments. The AD vasculature Aβ fibrils have a distinct elongated C-shaped structure, with two turns at H14 and V36. Since our current Aβ sample has unusual α-helical chemical shifts at H14 and V36, our current Aβ polymorph may have structural similarities with this AD brain vasculature Aβ.
Figure 7.
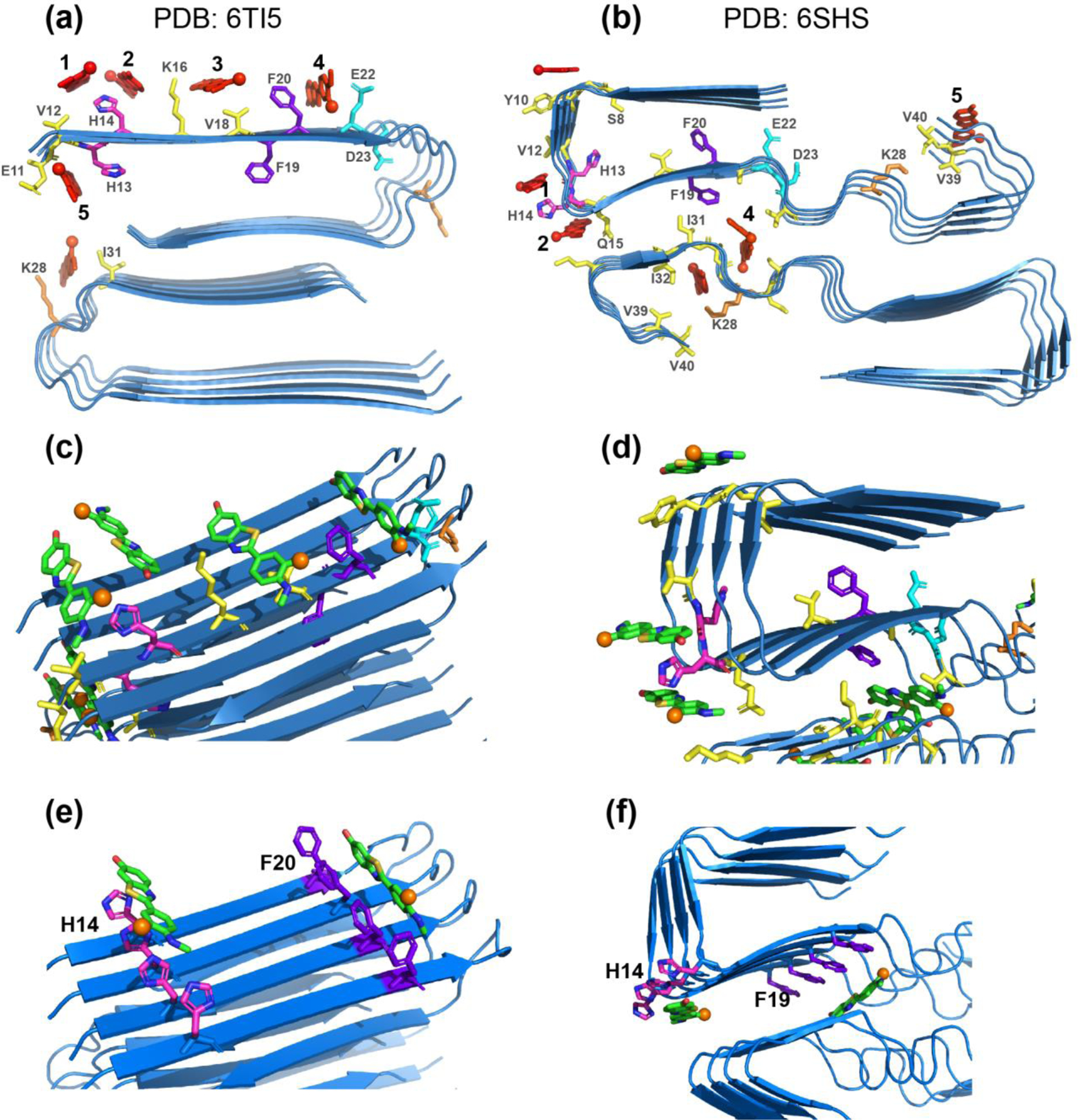
Experimentally constrained molecular docking of flutemetamol to two-fold symmetric Aβ40 structural models. The common docked sites are numbered as 1–5. H13 and H14 are shown in magenta, F19 and F20 in purple, E22 and D23 in cyan, K28 in orange, and all other residues in yellow. Flutemetamol is shown in red. (a) Two-fold symmetric in vitro Aβ40 structure from solid-state NMR 63 (PDB: 6TI5). (b) Two-fold symmetric meningeal AD brain Aβ40 structure 2 (PDB: 6SHS). (c-d) Additional views of flutemetamol binding sites in the solid-state NMR structure (c) and the cryo-EM structure (d). A single active residue was used in each simulation. Flutemetamol (green sticks) straddles several β-strands in most docked poses. The fluorine atom is shown in orange. (e-f) Simulations using one active residue per peptide and hence four active residues in each simulation. (e) Docking results at H14 and F20 for the two-fold symmetric solid-state NMR structure. (f) Docking results at H14 and F19 for the two-fold symmetric AD brain Aβ40 structure. The ligand binding pose is similar to the pose obtained when a single active residue was used in each simulation.
Based on the REDOR results that 12VHH14, 18VFF21 and 39VV40 are the closest to the ligand, we assigned these eight residues as active residues one at a time for HADDOCK simulations. Fig. 7 compares the lowest energy consensus binding sites for the two-fold symmetric structures. In both models, the surface-accessible 12VHH14 segment accommodated two flutemetamol molecules, sites 1 and 2, one on each side. The long axis of the ligand straddles 3.5 β-strands along the fibril axis. The aromatic plane of the ligand is slightly tilted from the local β-sheet plane in the in vitro structure model (Table 1). In the AD brain vasculature Aβ structure, 12VHH14 forms a severe turn between two β-strands, where the ligand plane is perpendicular to the local peptide plane at site 1 but parallel to it at site 2. Incorporating multiple active residues in each simulation did not change the ligand pose (Fig. 7e, f). The ligand at site 1 is significantly disordered in the three-fold symmetric in vitro Aβ model (Fig. S8c), which we attribute to the lack of strict translational symmetry and the uneven β-sheet surface of this structural model.
Table 1.
Docking results of flutemetamol to Aβ40 fibrils.
| Polymorph (PDB code) |
Properties | Site 1 V12, H14 |
Site 2 H14, K16 |
Site 3 V18 |
Site 4 F20 |
Site 5 V39, V40 |
|---|---|---|---|---|---|---|
| 2-fold in vitro (6TI5) |
Orientation | Tilted | Tilted | Parallel | Perpendicular | N/A |
| ΔG (kcal/mol) | −5.8 | −6.3 | −6.5 | −6.6 | ||
|
| ||||||
| 2-fold AD brain (6SHS) | Orientation | Perpendicular | Parallel | N/A | Tilted | Parallel |
| ΔG (kcal/mol) | −6.1 | −6.9 | −7.0 | −5.9 | ||
|
| ||||||
| 3-fold in vitro (2LMQ) | Orientation | Tilted | N/A | Parallel, tilted | Perpendicular | Tilted |
| ΔG (kcal/mol) | −6.5 | −7.6 to −6.4 | −7.8 | −6.7 | ||
|
| ||||||
| 3-fold brain-seeded in vitro (2M4J) | Orientation | Tilted | Tilted | Parallel, perpendicular | N/A | Tilted |
| ΔG (kcal/mol) | −5.9 | −6.0 | −6.2 | −7.3 to −7.0 | ||
The next common binding site, site 3, is located between K16 and V18 on the external β-sheet surface, and the ligand plane lies approximately parallel to the local β-sheet plane. The two-fold symmetric in vitro Aβ40 structure gives the clearest view of the stability of this binding site (Fig. 7c): the aromatic rings of flutemetamol engage in CH-π stacking interactions with the V18 methyl groups, π-π interactions with the F20 sidechain, and polar interactions with the K16 ammonium. Similar interactions are observed in the three-fold symmetric in vitro structure (Fig. S8c). The three-fold symmetric brain-seeded Aβ structure differs qualitatively from the other structures for these residues, because the F20 sidechain points into the fibril core rather than the fibril surface (Fig. S8d). Despite this difference, V18 and K16, together with E22, retain a binding pocket for flutemetamol, and allows more than one docked orientation. This suggests that polar interactions, provided by the cationic K16 on one side and the anionic E22 on the other side, might be more important for sequestering the ligand than hydrophobic interactions. Site 3 is missing in the two-fold AD brain vasculature Aβ structure (Fig. 7b). We attribute this to the fact that K16 and V18 face the N-terminal β-strand formed by 3EFRHD7, which may have prevented ligand entry into this confined space. Since this N-terminal segment is dynamically disordered in our current polymorph, we expect site 3 to be occupied by flutemetamol.
The fourth common binding site is associated with F20 and F19. In both the two-fold (Fig. 7a, c) and three-fold (Fig. S8c) in vitro structures, the ligand intercalates between F20 and E22 with the aromatic plane perpendicular to the β-sheet surface. In the three-fold symmetric brain-seeded Aβ structure, this site is absent due to the inward-facing direction of both F20 and F19 sidechains (Fig. S8d). In the two-fold AD brain vasculature Aβ structure (Fig. 7b, d), no binding site was found near F20 due to the same obstruction by the N-terminal latch, but a binding site was found on the other side, surrounded by F19, A21, and V24. We speculate that this internal site might be accessed from the ends of the long fibrils through molecular diffusion 76. This binding site was found without specifying an active residue, suggesting this site is energetically favorable.
In addition to the four sites centered around the two aromatic segments of the peptide, the cryoEM structure of brain-derived Aβ40 structure (Fig. 7b) and the three-fold symmetric in vitro structure (Fig. S8c) also displayed a binding site 5 at the C-terminal 38GVV40, consistent with the REDOR data. We also conducted free docking simulations, without the NMR constraints, for the two-fold symmetric in vitro Aβ40 structure and the two-fold symmetric meningeal AD brain Aβ40 structure (Fig. S8a, b). These unconstrained simulations reproduced some of the binding sites found in the REDOR constrained docking. For the in vitro Aβ40 structure, site 4 near F20 and site 5 near H13 and V40 are found, while in the meningeal Aβ40 structure, site 4 near F19 is found, but no histidine-binding sites nor C-terminal sites were observed. Therefore, experimental measurements are required for locating all the binding residues of flutemetamol in the Aβ fibrils.
Discussion
Determining the binding sites of PET tracers in amyloid fibrils is important for understanding the specificity and affinity of these ligands for diagnosis of neurodegenerative disorders. Here we identified the Aβ residues that are in close contact with flutemetamol using REDOR NMR, thus placing important constraints on the binding sites of this PET ligand. We found three sets of residues that are in close proximity with flutemetamol, 12VHH14, 18VFF20, and 39VV40. These residues do not cluster in a single location in Aβ structures, therefore they constitute at least two binding sites. The REDOR-constrained docking simulations indicate that as many as five binding sites can be formed by these residues, because the same cluster of residues can interact with the ligand on different sides (Fig. 7). These multiple binding sites are consistent with the much broader 19F linewidths of peptide-bound flutemetamol compared to the ligand by itself (Fig. 2c, d).
Although the peptide-ligand REDOR distances were measured necessarily on the specific Aβ fibril polymorph prepared here, we hypothesize that the ligand-proximal residues we detected may be common to many Aβ polymorphs, because it is the local residue-level molecular interaction that governs binding. The similar binding poses of flutemetamol in docking simulations of different Aβ structures support this notion. Our solid-state NMR data, biochemical analysis and molecular docking show that flutemetamol predominantly interacts with the two aromatic cores of the peptide, H13/H14 and F19/F20. In addition, the ligand also interacts with the C-terminal 39VV40. The two aromatic cores are surrounded by polar residues 12VHHQK16 and 18VFFAE22. Since H14 and F20 are separated into two grooves by the intervening K16 and V18 in all known Aβ40 structures, these two segments most likely form distinct binding sites. Docking simulations indicate that different Aβ polymorphs share these common binding sites. The V12/H14 pair and V18/F20 pair can both stabilize the ligand through CH-π and π-π interactions. In the simulations, the only Aβ polymorph in which V18/F20 does not interact with the ligand is the brain vasculature Aβ fibril, where an N-terminal β-strand restricted entry of the ligand to these residues. However, in this structure, another binding site is found near F19, facing the interface between the two protofilaments. The ligand is sandwiched between F19 on one monomer and A30 on the other monomer. In the 13C-detected 1H-19F REDOR spectra, both I31/I32 and A21/A30 signals are dephased by fluorine, indicating that these residues are close to flutemetamol in our samples. The fluorine-carbon distances in the various docked poses range from 4 Å to 7 Å, in qualitative agreement with the measured 1H-19F REDOR dephasing (Fig. S3b, c). In most docked poses, flutemetamol lies with its long axis parallel to the fibril axis, but the aromatic plane can be parallel, perpendicular to or tilted from the local β-sheet plane, depending on the sidechains interacting with the ligand. Fig. 7 and Fig. S8 summarize all the docked sites in each structural model. The multiple docked sites may be randomly populated in the fibrils, and the same site is expected to be occupied much more sparsely than every 3–4 strands because of the overall mole fraction of ~10% for the tightly bound ligand. This low stoichiometry is reasonable because occupation of one site over 3–4 β-strands may block diffusion of additional ligand molecules to adjacent strands for the internal binding sites 76–77.
Flutemetamol binds the current Aβ polymorph with a Bmax of 2.4 μM, which corresponds to ~22 mol% of the 11 μM Aβ fibrils (Fig. S2). Among the bound ligand, about 10% is tightly bound and immobilized while 12% is loosely bound and partly mobile. These two fractions are distinguished by their 19F CSAs, linewidths, and CP intensities (Fig. 2). Radioligand assays of brain homogenates suggest that 3H-labeled PiB binds to a high-affinity site with a KD of ~2.5 nM and a maximum specific binding (Bmax) of 565 pmol per 1 nmol of AD brain Aβ, i.e. 1 tracer per 2 Aβ peptide 15. However, the Bmax value drops by 1000-fold when the ligand is added to synthetic Aβ40 fibrils, suggesting that the polymorph structure of the fibrils has a significant impact on the binding capacity. Our ~22% binding stoichiometry is much closer to the Bmax of AD brain Aβ aggregates than to the Bmax of the previously reported synthetic in vitro fibrils, suggesting that the current Aβ polymorph may be a reasonable mimic of brain Aβ aggregates. In addition to the high-affinity site, AD brain Aβ aggregates show a low-affinity binding site with a Kd of ~250 nM 15, which is more comparable to our measured KD of 1.6 μM. It is worth noting that the radioligand binding assay 15 measured PiB binding to unpurified brain homogenate, while our assay was conducted on flutemetamol and purified Aβ40 fibrils. Moreover, we conducted our titration on micromolar concentrations of the ligand and Aβ, thus we are only able to detect binding sites with micromolar KD values. This does not preclude the existence of nanomolar binding sites in our samples. The fact that the REDOR spectra show multiple residues in contact with the ligand fluorine, and the 19F spectra resolve two signals, both indicate the existence of multiple binding sites. Some of these binding sites may have a high (nM) binding affinity while others may have weaker affinity, giving rise to more mobile ligands. Multiple binding sites have also been reported for thioflavin T analogs using fluorescence experiments 19, with evidence that the binding stoichiometry ranges from one ligand for ~4 Aβ peptides to one ligand for ~35 peptides.
It is of interest to compare the flutemetamol-interacting residues found here with the Aβ residues that interact with green tea flavonoid epigallocatechin-3-gallate (EGCG) 78 and the nonsteroidal anti-inflammatory drug (NSAID) sulindac sulfide 70. Reif and coworkers reported that EGCG increased the intensities of the aromatic sidechain 13C signals in the NMR spectra, indicating that this flavonoid immobilizes the H13/H14 and F19/F20 sidechains 78. EGCG binding also removed the β-strand conformation of residues 10–20, leading to the formation of non-toxic Aβ40 oligomers. Sulindac sulfide perturbed the chemical shifts of 18VFF20 as well as 33GLM35, and 13C-19F REDOR data pinpointed V18 and V39 as close to the fluorine on sulindac sulfide 70. Thus, sulindac sulfide shows a similar affinity to the pair of Phe residues as flutemetamol does to Aβ40. Interestingly, these authors did not detect significant interactions of two PET tracers, methylated-PiB and 4,4’-diaryl-2,2’-bithiazole (DABTA) 45, with Aβ. Using BSA, they titrated these two PET ligands at a 1 : 1 molar ratio to Aβ40, but the actual binding stoichiometry was not reported. 1H-detected NMR spectra of perdeuterated and 13C, 15N-labeled protein indicate that the largest chemical shift perturbations occur at Gly25 and Ser26, which lie in the loop connecting the two β-strands. Methylated PiB reduced the concentration of a minor peptide conformation, suggesting that this PiB derivative might increase the structural homogeneity of this Aβ40 polymorph by stabilizing the inter-strand loop. Thus, the binding site of methylated PIB appears to be qualitatively different from the binding sites of flutemetamol found here. Two possible reasons might explain this difference. First, the PiB analog used in the previous study methylates a phenol oxygen and an amine nitrogen at the two ends of the molecule, thus the compound is less polar than the parent compound. This non-polar nature may weaken the ligand interactions with the polar N-terminal region of Aβ. Second, the Aβ40 polymorph in the prior study differs from the polymorph obtained here, which might also cause different binding sites.
Other studies of small-molecule binding to amyloid proteins also gave insights into the molecular interactions that stabilize conjugated aromatic ligands to amyloid fibrils. Meier and coworkers investigated the binding of Congo red and luminescent conjugated polythiophenes (LCPs) 79–80 to the amyloid fibrils formed by the prion domain of the HET-s protein. Based on chemical shift perturbations, ligand-to-protein 1H-13C polarization transfer, and molecular docking, they identified a common high-affinity binding site that lies in a groove established by the cationic K229 and polar S265 on one side and S227 and S263 on the other. These cationic and polar sidechains interact electrostatically with the negatively charged sulfates and carboxylates on Congo red and LCPs, respectively. CryoEM maps of AD tau paired helical filaments bound to the PET tracer, APN-1607, showed two neighboring binding grooves separated by three polar residues, R349, Q351, and K353 81. The acidic carboxylated ligand tafamidis binds transthyretin at a position held by K16 and E54 82. In most of these binding sites, the elongated ligands straddle multiple β-strands in a groove of the β-sheet surface that is lined by polar and cationic residues such as Lys and Arg or by aromatic residues such as Phe and His. The multivalent stabilization may account for the selectivity of these ligands for certain proteins. Although Aβ has a high propensity to assemble into multiple different structures 83, the local conformations of the dual-histidine 12VHH14 motif and the dual-phenylalanine 18VFF20 motif are similar across multiple Aβ structures. Therefore, the flutemetamol binding sites obtained here are consistent with the specificity of PiB to Aβ but not to tau in AD brains.
The Aβ40 fibrils produced in this study exhibits chemical shifts that are distinct from all previous Aβ fibrils studied using solid-state NMR. The current polymorph is characterized by well-ordered and immobilized H13 and H14, non-β-sheet conformation at H14, and non β-sheet conformations at V36 and its C-terminal residues. The non-β-sheet conformations at H14 and V36 have qualitative resemblance to their conformations in the meningeal AD brain Aβ fibrils 2. The fibril growth protocol of the current study differs from the protocols in previous solid-state NMR studies in several important aspects. In our study, the peptides were first monomerized using HFIP and DMSO and then fibrilized at a monomer concentration of 46 μM 41–42. The sequential treatment by HFIP and DMSO is known to produce aggregate-free Aβ 84. In comparison, previous studies used guanidinium chloride or DMSO but not HFIP to solubilize the peptide and used significantly higher peptide concentrations of 100–210 μM for fibril formation 3, 63–64. The higher monomer concentrations and possibly incomplete solubilization in previous studies might have selectively amplified certain β-sheet conformations than others. A second difference in the fibrillization protocols is that the current study did not use seeding, whereas most previous fibril formation protocols used seeded growth either in a single step 3 or in multiple steps 45, 64, 68, which select for seeding-competent conformations. Third, the degree of agitation in the fibril growth protocols differ. We applied continuous shaking at 900 rpm during 3 days of fibril growth, whereas most previous studies either used gentle shaking 45, 68 or kept the solution quiescent 3, 64. To our knowledge, the only other study that employed a similar shaking speed of 950 rpm was that of Bertini and coworkers 63. These differences may all contribute to the distinct Aβ polymorphs found in this study.
In conclusion, the 19F and 13C NMR data shown here identified three sets of residues in Aβ40 that are in close contact with the PET tracer flutemetamol. These residues include the two aromatic motifs H13/H14 and F19/F20, and the C-terminal V39/V40. Together, these segments constitute at least two spatially distinct binding sites for the ligand. Experimentally constrained docking simulations indicate that flutemetamol binds with its long axis predominantly parallel to the fibril axis, and the ligand is stabilized by polar and aromatic residues at these sites. Flutemetamol binds the Aβ40 fibril polymorph studied here with a stoichiometry of 22%, in which 10% is tightly bound. Our results suggest that polar, charged, and aromatic amino acid residues on the β-sheet surface may be the most important residues for sequestering aromatic-rich PET ligands. This information might be useful for designing next-generation amyloid PET tracers with desired affinity and specificity.
Supplementary Material
Acknowledgements
This work is supported by NIH grant AG059661 to M.H. and startup funds provided by Wichita State University to H.W. A.J.D. is supported by an NIH Ruth L. Kirschstein Individual National Research Service Award (F31AG069418). This study made use of NMR spectrometers at the MIT-Harvard Center for Magnetic Resonance, which is supported by NIH grant P41 GM132079. We thank Pat St. John and Larysa Stroganova at the KUMC Electron Microscopy Research Laboratory for help with negative stain EM.
Footnotes
Supporting Information Available
Tables of NMR chemical shifts and experimental parameters, figures showing Aβ sample preparation details, 1H-19F REDOR pulse sequence and simulation, additional 2D NMR spectra, chemical shift comparison with other Aβ polymorphs, additional docking views, are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Data availability
NMR chemical shifts have been deposited in the Biological Magnetic Resonance Bank (BMRB) with ID numbers 51008 and 51009.
References
- 1.Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ, . . . Scheres SHW, Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kollmer M; Close W; Funk L; Rasmussen J; Bsoul A; Schierhorn A, . . . Fändrich M, Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun 2019, 10, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu JX; Qiang W; Yau WM; Schwieters CD; Meredith SC; Tycko R, Molecular Structure of b-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colvin MT; Silvers R; Ni QZ; Can TV; Sergeyev IV; Rosay M, . . . Griffin RG, Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wälti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Böckmann A, . . . Riek R, Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4976–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuttle MD; Comellas G; Nieuwkoop AJ; Covell DJ; Berthold DA; Kloepper KD, . . . Rienstra CM, Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol 2016, 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes CA; Robertson AJ; Louis JM; Anfinrud P; Bax A, Observation of β-Amyloid Peptide Oligomerization by Pressure-Jump NMR Spectroscopy. J. Am. Chem. Soc 2019, 141, 13762–13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceccon A; Tugarinov V; Ghirlando R; Clore GM, Abrogation of prenucleation, transient oligomerization of the Huntingtin exon 1 protein by human profilin I. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 5844–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng F; Goodman MM, Fluorine-18 radiolabeled heterocycles as PET tracers for imaging β-amyloid plaques in Alzheimer’s disease. Curr. Top. Med. Chem 2013, 13, 909–919. [DOI] [PubMed] [Google Scholar]
- 10.Querfurth HW; LaFerla FM, Alzheimer’s disease. N. Engl. J. Med 2010, 362, 329–344. [DOI] [PubMed] [Google Scholar]
- 11.Ballard C; Gauthier S; Corbett A; Brayne C; Aarsland D; Jones E, Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [DOI] [PubMed] [Google Scholar]
- 12.Villemagne VL; Fodero-Tavoletti MT; Masters CL; Rowe CC, Tau imaging: early progress and future directions. Lancet Neurol 2015, 14, 114–124. [DOI] [PubMed] [Google Scholar]
- 13.Hall B; Mak E; Cervenka S; Aigbirhio FI; Rowe JB; O’Brien JT, In vivo tau PET imaging in dementia: Pathophysiology, radiotracer quantification, and a systematic review of clinical findings. Ageing Res. Rev 2017, 36, 50–63. [DOI] [PubMed] [Google Scholar]
- 14.Klunk WE; Engler H; Nordberg A; Wang Y; Blomqvist G; Holt DP, . . . Långström B, Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol 2004, 55, 306–319. [DOI] [PubMed] [Google Scholar]
- 15.Klunk WE; Lopresti BJ; Ikonomovic MD; Lefterov IM; Koldamova RP; Abrahamson EE, . . . Mathis CA, Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J. Neurosci 2005, 25, 10598–10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koole M; Lewis DM; Buckley C; Nelissen N; Vandenbulcke M; Brooks DJ, . . . Van Laere K, Whole-body biodistribution and radiation dosimetry of 18F-GE067: a radioligand for in vivo brain amyloid imaging. J. Nucl. Med 2009, 50, 818–822. [DOI] [PubMed] [Google Scholar]
- 17.Vandenberghe R; Adamczuk K; Dupont P; Laere KV; Chételat G, Amyloid PET in clinical practice: Its place in the multidimensional space of Alzheimer’s disease. Neuroimage Clin 2013, 2, 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thal DR; Beach TG; Zanette M; Heurling K; Chakrabarty A; Ismail A, . . . Buckley C, [(18)F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer’s disease: specific detection of advanced phases of amyloid-β pathology. Alzheimers Dement 2015, 11, 975–985. [DOI] [PubMed] [Google Scholar]
- 19.Lockhart A; Ye L; Judd DB; Merritt AT; Lowe PN; Morgenstern JL, . . . Brown J, Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. J. Biol. Chem 2005, 280, 7677–7684. [DOI] [PubMed] [Google Scholar]
- 20.Shcherbakov AA; Medeiros-Silva J; Tran N; Gelenter MD; Hong M, From Angstroms to Nanometers: Measuring Interatomic Distances by Solid-State NMR. Chem. Rev 2021, 10.1021/acs.chemrev.1c00662. [DOI] [PMC free article] [PubMed]
- 21.Roos M; Mandala VS; Hong M, Determination of Long-Range Distances by Fast Magic-Angle-Spinning Radiofrequency-Driven (19)F-(19)F Dipolar Recoupling NMR. J. Phys. Chem. B 2018, 122, 9302–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roos M; Wang T; Shcherbakov AA; Hong M, Fast Magic-Angle-Spinning 19F Spin Exchange NMR for Determining Nanometer 19F–19F Distances in Proteins and Phamaceutical Compounds. J. Phys. Chem. B 2018, 122, 2900–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shcherbakov AA; Hong M, Rapid Measurement of Long-Range Distances in Proteins by Multidimensional 13C-19F REDOR NMR under Fast Magic-Angle Spinning. J. Biomol. NMR 2018, 71, 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shcherbakov AA; Mandala VS; Hong M, High-Sensitivity Detection of Nanometer 1H-19F Distances for Protein Structure Determination by 1H-Detected Fast MAS NMR. J. Phys. Chem. B 2019, 123, 4387–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M; Lu M; Fritz M; Quinn C; Byeon IJ; Byeon CH, . . . Polenova T, Fast Magic Angle Spinning ¹⁹F NMR of HIV-1 Capsid Protein Assemblies. Angew. Chem. Int. Ed. Engl 2018, 57, 16375–16379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu M; Wang M; Sergeyev IV; Quinn CM; Struppe J; Rosay M, . . . Polenova T, (19)F Dynamic Nuclear Polarization at Fast Magic Angle Spinning for NMR of HIV-1 Capsid Protein Assemblies. J. Am. Chem. Soc 2019, 141, 5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilchrist ML Jr; Monde K; Tomita Y; Iwashita T; Nakanishi K; McDermott AE, Measurement of interfluorine distances in solids. J. Magn. Reson 2001, 152, 1–6. [DOI] [PubMed] [Google Scholar]
- 28.Goetz JM; Poliks B; Studelska DR; Fischer M; Kugelbrey K; Bacher A, . . . Schaefer J, Investigation of the binding of fluorolumazines to the 1-MDa capsid of lumazine synthase by 15N{19F} REDOR NMR. J. Am. Chem. Soc 1999, 121, 7500–7508. [Google Scholar]
- 29.Yang H; Staveness D; Ryckbosch SM; Axtman AD; Loy BA; Barnes AB, . . . Cegelski L, REDOR NMR Reveals Multiple Conformers for a Protein Kinase C Ligand in a Membrane Environment. ACS Cent. Sci 2018, 4, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graesser DT; Wylie BJ; Nieuwkoop AJ; Franks WT; Rienstra CM, Long-range 19F-15N distance measurements in highly-13C, 15N-enriched solid proteins with 19F-dephased REDOR shift (FRESH) spectroscopy. Magn. Reson. Chem 2007, 45, S129–S134. [DOI] [PubMed] [Google Scholar]
- 31.Wi S; Sinha N; Hong M, Long range 1H-19F distance measurement in peptides by Solid-State NMR. J. Am. Chem. Soc 2004, 126, 12754–12755. [DOI] [PubMed] [Google Scholar]
- 32.Kitevski-LeBlanc JL; Prosser RS, Current applications of 19F NMR to studies of protein structure and dynamics. Prog. Nucl. Magn. Reson. Spectrosc 2012, 62, 1–33. [DOI] [PubMed] [Google Scholar]
- 33.Sharaf NG; Gronenborn AM, (19)F-Modified Proteins and (19)F-Containing Ligands as Tools in Solution NMR Studies of Protein Interactions. Methods Enzymol 2015, 565, 67–95. [DOI] [PubMed] [Google Scholar]
- 34.Grage SL; Xu X; Schmitt M; Wadhwani P; Ulrich AS, (19)F-Labeling of Peptides Revealing Long-Range NMR Distances in Fluid Membranes. J. Phys. Chem. Lett 2014, 5, 4256–4259. [DOI] [PubMed] [Google Scholar]
- 35.Matei E; Gronenborn AM, (19)F Paramagnetic Relaxation Enhancement: A Valuable Tool for Distance Measurements in Proteins. Angew. Chem. Int. Ed. Engl 2016, 55, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu JJ; Horst R; Katritch V; Stevens RC; Wüthrich K, Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shcherbakov AA; Roos M; Kwon B; Hong M, Two-dimensional 19F-13C correlation NMR for 19F resonance assignment of fluorinated proteins. J. Biomol. NMR 2020, 74, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gullion T; Schaefer J, Rotational-Echo Double-Resonance NMR. J. Magn. Reson 1989, 81, 196–200. [DOI] [PubMed] [Google Scholar]
- 39.Kronqvist N; Sarr M; Lindqvist A; Nordling K; Otikovs M; Venturi L, . . . Johansson J, Efficient protein production inspired by how spiders make silk. Nat. Commun 2017, 8, 15504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abelein A; Chen G; Kitoka K; Aleksis R; Oleskovs F; Sarr M, . . . Biverstål H, High-yield Production of Amyloid-β Peptide Enabled by a Customized Spider Silk Domain. Sci. Rep 2020, 10, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elkins MR; Wang T; Nick M; Jo H; Lemmin T; Prusiner SB, . . . Hong M, Structural Polymorphism of Alzheimer’s β-Amyloid Fibrils as Controlled by an E22 Switch: A Solid-State NMR Study. J. Am. Chem. Soc 2016, 138, 9840–9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang T; Jo H; DeGrado WF; Hong M, Water Distribution, Dynamics, and Interactions with Alzheimer’s β-Amyloid Fibrils Investigated by Solid-State NMR. J. Am. Chem. Soc 2017, 139, 6242–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stohr J; Condello C; Watts JC; Bloch L; Oehler A; Nick M, . . . Prusiner SB, Distinct synthetic Ab prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 10329–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schutz AK; Vagt T; Huber M; Ovchinnikova OY; Cadalbert R; Wall J, . . . Meier BH, Atomic-Resolution Three-Dimensional Structure of Amyloid b Fibrils Bearing the Osaka Mutation. Angew. Chem. Int. Ed. Engl 2015, 54, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niu Z; Sarkar R; Aichler M; Wester H-J; Yousefi BH; Reif B, Mapping the Binding Interface of PET Tracer Molecules and Alzheimer Disease Aβ Fibrils by Using MAS Solid-State NMR Spectroscopy. ChemBioChem 2020, 21, 2495–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Böckmann A; Gardiennet C; Verel R; Hunkeler A; Loquet A; Pintacuda G, . . . Lesage A, Characterization of different water pools in solid-state NMR protein samples. J. Biomol. NMR 2009, 45, 319–327. [DOI] [PubMed] [Google Scholar]
- 47.Hong M, Resonance Assignment of 13C/15N Labeled Proteins by Two- and Three-Dimensional Magic-Angle-Spinning NMR. J. Biomol. NMR 1999, 15, 1–14. [DOI] [PubMed] [Google Scholar]
- 48.Rienstra CM; Hohwy M; Hong M; Griffin RG, 2D and 3D 15N-13C-13C NMR chemical shift correlation spectroscopy of solids: assignment of MAS spectra of peptides. J. Am. Chem. Soc 2000, 122, 10979–10990. [Google Scholar]
- 49.Pauli J; Baldus M; vanRossum B; Groot H. d.; Oschkinat H, Backbone and side-chain 13C and 15N signal assignments of the alpha-spectrin SH3 domain by magic-angle spinning solid-state NMR at 17.6 Tesla. ChemBioChem 2001, 2, 272–281. [DOI] [PubMed] [Google Scholar]
- 50.Baldus M; Petkova AT; Herzfeld J; Griffin RG, Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys 1998, 95, 1197–1207. [Google Scholar]
- 51.Hou G; Yan S; Trebosc J; Amoureux JP; Polenova T, Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids. J. Magn. Reson 2013, 232, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaroniec CP; Tounge BA; Herzfeld J; Griffin RG, Frequency Selective Heteronuclear Dipolar Recoupling in Rotating Solids: Accurate 13C-15N Distance Measurements in Uniformly 13C,15N-labeled Peptides. J. Am. Chem. Soc 2001, 123, 3507–3519. [DOI] [PubMed] [Google Scholar]
- 53.Bielecki A; Kolbert AC; Levitt MH, Frequency-Switched Pulse Sequences - Homonuclear Decoupling and Dilute Spin NMR in Solids. Chem. Phys. Lett 1989, 155, 341–346. [Google Scholar]
- 54.Lee W; Tonelli M; Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen Y; Bax A, Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol 2015, 1260, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haeberlen U, High Resolution NMR in Solids: Selective Averaging Academic Press: 1976. [Google Scholar]
- 57.Schmidt-Rohr K; Spiess HW, Multidimensional Solid-State NMR and Polymers Academic Press: San Diego, 1994; p 478. [Google Scholar]
- 58.Bak M; Rasmussen JT; Nielsen NC, SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson 2011, 213, 366–400. [DOI] [PubMed] [Google Scholar]
- 59.Maciejewski MW; Schuyler AD; Gryk MR; Moraru II; Romero PR; Ulrich EL, . . . Hoch JC, NMRbox: A Resource for Biomolecular NMR Computation. Biophys. J 2017, 112, 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bak M; Nielsen NC, REPULSION A Novel Approach to Efficient Powder Averaging in Solid-State NMR. J. Magn. Reson 1997, 125, 132–139. [DOI] [PubMed] [Google Scholar]
- 61.Halgren TA, Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem 1996, 17, 490–519. [Google Scholar]
- 62.van Zundert GCP; Rodrigues JPGLM; Trellet M; Schmitz C; Kastritis PL; Karaca E, . . . Bonvin AMJJ, The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- 63.Bertini I; Gonnelli L; Luchinat C; Mao JF; Nesi A, A New Structural Model of Ab40 Fibrils. J. Am. Chem. Soc 2011, 133, 16013–16022. [DOI] [PubMed] [Google Scholar]
- 64.Paravastu AK; Leapman RD; Yau WM; Tycko R, Molecular structural basis for polymorphism in Alzheimer’s b-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vangone A; Schaarschmidt J; Koukos P; Geng C; Citro N; Trellet ME, . . . Bonvin, A., Large-scale prediction of binding affinity in protein-small ligand complexes: the PRODIGY-LIG web server. Bioinformatics 2019, 35, 1585–1587. [DOI] [PubMed] [Google Scholar]
- 66.Dockal M; Carter DC; Rüker F, The three recombinant domains of human serum albumin. Structural characterization and ligand binding properties. J. Biol. Chem 1999, 274, 29303–29310. [DOI] [PubMed] [Google Scholar]
- 67.Varshney A; Sen P; Ahmad E; Rehan M; Subbarao N; Khan RH, Ligand binding strategies of human serum albumin: how can the cargo be utilized? Chirality 2010, 22, 77–87. [DOI] [PubMed] [Google Scholar]
- 68.Petkova AT; Leapman RD; Guo Z; Yau WM; Mattson MP; Tycko R, Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 2005, 307, 262–265. [DOI] [PubMed] [Google Scholar]
- 69.Prade E; Barucker C; Sarkar R; Althoff-Ospelt G; Lopez del Amo JM; Hossain S, . . . Reif B, Sulindac Sulfide Induces the Formation of Large Oligomeric Aggregates of the Alzheimer’s Disease Amyloid-β Peptide Which Exhibit Reduced Neurotoxicity. Biochemistry 2016, 55, 1839–1849. [DOI] [PubMed] [Google Scholar]
- 70.Prade E; Bittner HJ; Sarkar R; Lopez Del Amo JM; Althoff-Ospelt G; Multhaup G, . . . Reif B, Structural Mechanism of the Interaction of Alzheimer Disease Aβ Fibrils with the Non-steroidal Anti-inflammatory Drug (NSAID) Sulindac Sulfide. J. Biol. Chem 2015, 290, 28737–28745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Petkova AT; Ishii Y; Balbach JJ; Antzutkin ON; Leapman RD; Delaglio F; Tycko R, A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tycko R; Wickner RB, Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc. Chem. Res 2013, 46, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikonomovic MD; Buckley CJ; Abrahamson EE; Kofler JK; Mathis CA; Klunk WE; Farrar G, Post-mortem analyses of PiB and flutemetamol in diffuse and cored amyloid-β plaques in Alzheimer’s disease. Acta Neuropathol 2020, 140, 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qiang W; Yau WM; Lu JX; Collinge J; Tycko R, Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paravastu AK; Qahwash I; Leapman RD; Meredith SC; Tycko R, Seeded growth of beta-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou R; Kuang G; Ågren H; Nordberg A; Långström B; Tu Y, Free Energy Profile for Penetration of Pittsburgh Compound-B into the Amyloid β Fibril. ACS Chem. Neurosci 2019, 10, 1783–1790. [DOI] [PubMed] [Google Scholar]
- 77.Kuang G; Murugan NA; Zhou Y; Nordberg A; Ågren H, Computational Insight into the Binding Profile of the Second-Generation PET Tracer PI2620 with Tau Fibrils. ACS Chem. Neurosci 2020, 11, 900–908. [DOI] [PubMed] [Google Scholar]
- 78.Lopez del Amo J-M; Dasari M; Fink U; Grelle G; Wanker EE; Bieschke J; Reif B, Structural Properties of EGCG induced, non-toxic Alzheimer’s disease Aβ oligomers J. Mol. Biol 2012, 421, 517–524. [DOI] [PubMed] [Google Scholar]
- 79.Schütz AK; Hornemann S; Wälti MA; Greuter L; Tiberi C; Cadalbert R, . . . Meier BH, Binding of Polythiophenes to Amyloids: Structural Mapping of the Pharmacophore. ACS Chem Neurosci 2018, 9, 475–481. [DOI] [PubMed] [Google Scholar]
- 80.Schütz AK; Soragni A; Hornemann S; Aguzzi A; Ernst M; Böckmann A; Meier BH, The amyloid-Congo red interface at atomic resolution. Angew. Chem. Int. Ed. Engl 2011, 50, 5956–5960. [DOI] [PubMed] [Google Scholar]
- 81.Shi Y; Murzin AG; Falcon B; Epstein A; Machin J; Tempest P, . . . Goedert M, Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol 2021, 141, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bulawa CE; Connelly S; Devit M; Wang L; Weigel C; Fleming JA, . . . Labaudinière R, Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tycko R, Amyloid polymorphism: structural basis and neurobiological relevance. Neuron 2015, 86, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Broersen K; Jonckheere W; Rozenski J; Vandersteen A; Pauwels K; Pastore A, . . . Schymkowitz J, A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng. Des. Sel 2011, 24, 743–750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NMR chemical shifts have been deposited in the Biological Magnetic Resonance Bank (BMRB) with ID numbers 51008 and 51009.