

SUMMARY
Technologies for counting protein molecules are enabling single-cell proteomics at increasing depth and scale. New advances in single-molecule methods by Brinkerhoff and colleagues promise to further increase the sensitivity of protein analysis and motivate questions about scaling up the counting of the human proteome.
The need for single-cell protein analysis
As biological investigations increasingly focus on in vivo systems composed of diverse cell types and states, the necessity for single-cell analysis increases. This necessity has motivated the rapid proliferation of high-throughput methods for analyzing nucleic acids at single-cell resolution, which in turn have increased the appreciation for cellular diversity (Keren-Shaul et al., 2017). The aim to understand the molecular mechanisms underpinning this diversity motivates expanding single-cell analysis beyond nucleic acids to include proteins and their modifications, activities, and localization (Slavov, 2020). This need for single-cell proteomics has propelled the development of methods for quantifying thousands of proteins across single human cells (Vistain and Tay, 2021). A recent study by Brinkerhoff et al. (2021) takes a step toward single-molecule peptide fingerprinting, which promises to expand the single-cell proteomic toolkit.
Proteomics methods often simplify protein analysis by first digesting proteins to peptides and then counting the peptide molecules. They can count either one peptide copy at a time (single-molecule methods) or multiple copies of a peptide sequence, as performed by mass spectrometry (MS). In both cases, the counting needs to be sensitive, specific, and scalable. Scalability to the proteome scale is particularly challenging since a representative human cell con tains about 6×109 protein molecules compared to about 3×105 mRNA molecules (Bekker-Jensen et al., 2017; Milo et al., 2010). Thus, counting enough protein molecules for a representative ensemble is challenging even for a single cell, and the challenge scales with the number of single cells. Nonetheless, the possibility of sequencing single peptides has inspired hopes that single-molecule methods can accomplish for protein analysis what single-molecule DNA sequencing has accomplished for the analysis of nucleic acids (Alfaro et al., 2021).
Indeed, new single-molecule and MS technologies for sensitive protein quantification are surging and receiving much attention and financial support (Alfaro et al., 2021; Marx, 2019). Both technology types are in early development stages and have huge potentials for growth. While MS has a long history of analyzing complex proteomes, its methods for quantifying thousands of proteins in single mammalian cells were developed only recently and point to outstanding opportunities that can increase the sensitivity and depth of proteome coverage by 10-fold (Slavov, 2021). Single-molecule methods are emerging and making rapid progress as illustrated by the recent work of Brinkerhoff et al. (2021).
Significant advances in single-molecule counting
Approaches for single-molecule peptide detection include Edman degradation, tunneling currents, and nanopores (Alfaro et al., 2021). The nanopore approaches use nanometer-sized pores in a membrane separating two electrolyte-filled compartments (Figure 1A). A voltage applied across the membrane creates an ionic current flow through the nanopore, which is modulated by the translocation of molecules. These current modulations can be used to sequence polymers transversing the nanopores. Such nanopore sequencing has been successfully applied to DNA and RNA polymers, but its application to proteins faces many challenges. Unlike the uniformly charged nucleotides, amino acids vary substantially in charge, which complicates unidirectional trans-location of proteins by electrophoresis. Furthermore, protein sequencing requires disrupting secondary and tertiary protein structures and distinguishing 20 amino acids plus many more post-translational modifications.
Figure 1. Prospects for single-cell proteomics by single-molecule counting.
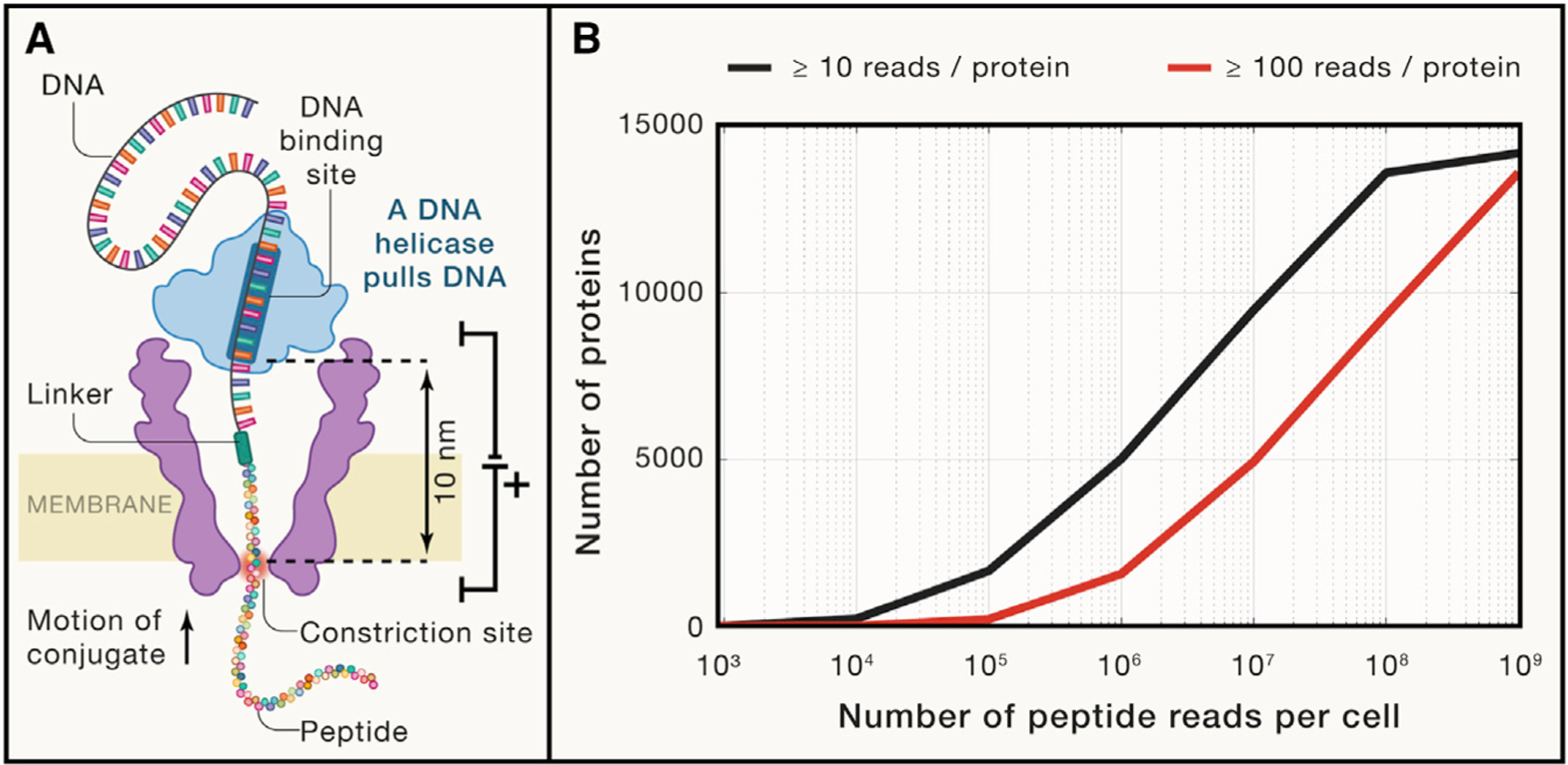
(A) A diagram of a nuclear pore used by Brinkerhoff et al. (2021) to fingerprint peptide variants. The pore is made of the mutant porin A, a channel-forming protein originally derived from Mycobacterium smegmatis. Its channel is about 1nm in diameter and is flanked by regions of larger diameter. The peptides are pulled through the pore by the attached DNA molecule pulled by a DNA helicase. As the peptide passes through the constriction zone (marked in red), the current blockades provide information for the amino acid sequence.
(B) An estimated number of proteins detected per single HeLa cell as a function of the number of peptide reads. The estimation sampled proteins with probability proportional to their abundance in HeLa cells (Bekker-Jensen et al., 2017). Peptide-specific biases and unmappable peptides were not considered by this estimation; such effects will increase the required number of reads.
These challenges have motivated creative solutions for enabling peptide finger-printing by nanopores. For example, DNA molecules conjugated to peptides can drive the translocation of the peptides through the pores independent of the highly non-uniform charges on amino acid residues. Using such DNA-peptide conjugate, Brinkerhoff et al. (2021) were able to pull its 80-nucleotide DNA strand by a DNA helicase and to fingerprint the conjugated 26 amino acid synthetic peptide (Figure 1A). To increase the fidelity of identifying amino acid variants, the authors read the same peptide molecule multiple times by rewinding it. This process generated consensus current traces that enabled low error in distinguishing peptide variants.
The significance of this progress for single-cell proteomics should be evaluated in the context of its potential to achieve high sensitivity, specificity, and scalability. Such an evaluation is preliminary since Brinkerhoff et al. (2021) analyzed a few synthetic peptides, not a complex proteome. Nonetheless, assuming efficient sample preparation and DNA-peptide conjugation, nanopore peptide fingerprinting has the potential to detect proteins present at only a few copies per sample, which can result very high sensitivity. Brinkerhoff et al. demonstrated excellent specificity in distinguishing a few synthetic peptide variants, but it is challenging to extrapolate this specificity to the analysis of a complex mammalian proteome. The ability to perform multiple reads of the same peptide molecule is likely to be very helpful as nanopore methods are applied to biological protein mixtures. Scaling nanopore sequencing to count enough molecules for quantifying a mammalian proteome is likely to be the most formidable challenge in achieving single-cell proteomics by single-molecule protein sequencing.
Scaling up
When single-molecule methods deliver on their promise of sequencing proteins from complex mixtures, the next frontier will be to scale them up to sequence the billions of protein molecules in a single human cell (Milo et al., 2010). Single-cell RNA sequencing methods detect thousands of RNA molecules per cell, which corresponds to about a tenth of the transcripts present in a cell. To sample a tenth of the protein molecules, single-molecule proteomics will need to sequence hundreds of millions of protein molecules. Unbiased sampling of protein molecules from a HeLa cell (Figure 1B) indicates the single-molecule methods need to read hundreds of thousands of peptides per single cell to achieve the proteome coverage of current single-cell MS proteomics (Specht et al., 2021). Reading hundreds of millions of peptides can result in nearly comprehensive proteome coverage of a single human cell (Figure 1B). Thus, practical single-cell proteomics by single-molecule methods would require scaling their throughput beyond the scale currently achieved by DNA sequencing methods. Alternatively, single-molecule methods may be combined with separation and fractionation methods that obviate the need to count the most abundant proteins and thus lower the number of peptide counts required for characterizing the less abundant proteins of the proteome. A third option is for single-molecule methods to find applications in counting low copy number proteins in simpler samples. These options highlight both exciting opportunities and the necessity for massively parallel single-molecule methods that can scale to the tremendous complexity of eukaryotic proteomes.
The work of Brinkerhoff et al. marks a milestone toward single-molecule protein sequencing. The multiple reads per peptide enhance the specificity of peptide detection, which brings single-molecule protein methods closer to analyzing complex proteomes. If scaled to counting millions of protein molecules per cell, the technology will hold an enticing promise for single-cell proteomics.
ACKNOWLEDGMENTS
The author is funded by an Allen Distinguished Investigator award through The Paul G. Allen Frontiers Group, a Seed Networks Award from CZI (CZF2019-002424), and an NIH award 1UG3CA268117.
Footnotes
DECLARATION OF INTERESTS
The author has applied for patents on single-cell proteomics technologies.
REFERENCES
- Alfaro JA, Bohländer P, Dai M, Filius M, Howard CJ, van Kooten XF, Ohayon S, Pomorski A, Schmid S, Aksimentiev A, et al. (2021). The emerging landscape of single-molecule protein sequencing technologies. Nat. Methods 18, 604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen DB, Kelstrup CD, Batth TS, Larsen SC, Haldrup C, Bramsen JB, Sørensen KD, Høyer S, Ørntoft TF, Andersen CL, et al. (2017). An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 4, 587–599.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkerhoff H, Kang ASW, Liu J, Aksimentiev A, and Dekker C (2021). Multiple rereads of single proteins at single-amino acid resolution using nanopores. Science 374, 1509–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Marx V (2019). A dream of single-cell proteomics. Nat. Methods 16, 809–812. [DOI] [PubMed] [Google Scholar]
- Milo R, Jorgensen P, Moran U, Weber G, and Springer M (2010). BioNumbers–the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N (2020). Unpicking the proteome in single cells. Science 367, 512–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N (2021). Driving Single Cell Proteomics Forward with Innovation. J. Proteome Res 20, 4915–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, Kharchenko P, Koller A, and Slavov N (2021). Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 22, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistain LF, and Tay S (2021). Single-Cell Proteomics. Trends Biochem. Sci 46, 661–672. [DOI] [PubMed] [Google Scholar]