

Abstract
Background
Nursing homes have represented important hotspots of viral spread during the initial wave of COVID-19 pandemics. The proximity of patients inside nursing homes allows investigate the dynamics of viral transmission, which may help understand SARS-Cov2 biology and spread.
Methods
SARS-CoV-2 viral genomes obtained from 46 patients infected in an outbreak inside a nursing home in Calabria region (South Italy) were analyzed by Next Generation Sequencing. We also investigated the evolution of viral genomes in 8 patients for which multiple swabs were available. Phylogenetic analysis and haplotype reconstruction were carried out with IQ-TREE software and RegressHaplo tool, respectively.
Results
All viral strains isolated from patients infected in the nursing home were classified as B.1 lineage, clade G. Overall, 14 major single nucleotide variations (SNVs) (frequency > 80%) and 12 minor SNVs (frequency comprised between 20% and 80%) were identified with reference to the Wuhan-H-1 sequence (NC_045512.2).
All patients presented the same 6 major SNVs: D614G in the S gene; P4715L, ntC3037T (F924F) and S5398P in Orf1ab gene; ntC26681T (F53F) in the M gene; and ntC241T in the non-coding UTR region. However, haplotype reconstruction identified a founder haplotype (Hap A) in 36 patients carrying only the 6 common SNVs indicated above, and 10 other haplotypes (Hap B—K) derived from Hap A in the remaining 10 patients. Notably, no significant association between a specific viral haplotype and clinical parameters was found.
Conclusion
The predominant viral strain responsible for the infection in a nursing home in Calabria was the B.1 lineage (clade G). Viral genomes were classified into 11 haplotypes (Hap A in 36 patients, Hap B—K in the remaining patients).
Keywords: SARS-CoV-2, COVID-19, NGS, Haplotypes
1. Introduction
The rapid spread of a novel Severe Acute Respiratory Syndrome CoronaVirus-2 (SARS-CoV-2) infection was declared as a pandemic on March 11th 2020 by the World Health Organization (WHO) (Cucinotta and Vanelli, 2020). Since the beginning of the pandemic, the phylogenetic analysis of viral sequences was used to monitor the pandemic spread (Rambaut et al., 2020; van Dorp et al., 2020). So, the development of international databases (Fang et al., 2021; Massacci et al., 2020; McBroome et al., 2021; O'Toole et al., 2021), sharing viral genomic sequences together with epidemiological and clinical data, has contributed to the design of public health countermeasures. The virus has evolved within an infected host as quasispecies, showing complex and dynamic distributions of intra-host viral populations (Capobianchi et al., 2020; Lythgoe et al., 2021; Z. Shen et al., 2020b; van Dorp et al., 2020; Wertheim, 2020). Specific patterns of substitutions carried on single isolates have been classified as haplotypes, which can be used to determine viral lineage and to understand community spread (L. Shen et al., 2020a).
At the beginning of the first pandemic wave, the two dominant viral lineages were A and B. These two lineages spread worldwide and evolved under different selective pressures, leading to the continuous rise of novel variants in different countries. In Europe, the dominant viral lineages from February to April 2020 were A.2 and A.5, while from January to May 2020 were B.1 and B.2 (Rambaut et al., 2020).
Elderly people in nursing homes are particularly vulnerable to COVID-19 because of their age, the presence of chronic medical conditions, congregate living quarters and routine contact with staff members and outside visitors (M. K. Chen et al., 2021; Strausbaugh et al., 2003; Trecarichi et al., 2020).
On March 26th 2020, the first outbreak in a nursing home in the Calabria Region involved 46 patients, providing a good model to investigate viral spread at the beginning of the pandemic. This outbreak was described in a clinical paper (Trecarichi et al., 2020).
Herein, we report on the results of Next Generation Sequencing (NGS) analysis of SARS-CoV-2 genomes isolated from nasopharyngeal swabs of positive patients residing in the nursing home. Multiple swabs were available for 8 patients so that a longitudinal follow-up was performed. The objective of the study was to investigate the evolution of viral variants both using phylogenetic analysis and haplotype determination. We found that the predominant viral strain responsible for the infection in a nursing home in Calabria was the B.1 lineage (clade G). However, sequenced viral genomes were classified into 11 haplotypes (Hap A in 36 patients, Hap B—K in the remaining 10 patients). This analysis can help track virus transmission and identify relevant mutations having an impact on the biological characteristics of the virus.
2. Materials and methods
2.1. Patients
The study was approved by the Ethical Committee (February 18th, 2021) of the Calabria Region, Italy. Sixty nasopharyngeal swabs samples, collected between March 26th and May 11th, 2020, from 46 subjects infected by SARS-CoV-2 were included in the analysis. For each patient a sample was obtained at admission (baseline) and 14 swabs were obtained at sequential time points from 8 patients of the same cohort. Clinical data were treated in accordance with the Helsinki Declaration (59th World Medical Association General Assembly, Seoul, Korea, October 2008) and the principles of good clinical practice. See Table 1 and Supplementary File S1 for details.
Table 1.
Patient's clinical characteristics (N = 36).
| Patients (N) | |
|---|---|
| Asymptomatic at diagnosis | 5 |
| Symptomatic at diagnosis | 31 |
| Fever | 7 |
| Cough | 1 |
| Respiratory Failure | 2 |
| Fever + Respiratory Failure | 11 |
| Fever + Cough | 5 |
| Cough + Respiratory Failure | 1 |
| Fever + Cough + Respiratory Failure | 4 |
| Comorbidities | |
| Hypertension | 16 |
| Diabetes | 2 |
| Cardiovascular diseases | 3 |
| Hypertension + Cardiovascular diseases | 4 |
| Hypertension + Diabetes | 2 |
| Diabetes + Cardiovascular diseases | 3 |
| Clinical outcome | |
| Hospitalization | 36 |
| Intensive Care Unit admission | 1 |
| Death (exitus) | 11 |
2.2. Diagnostic procedures and samples selection
Viral RNA was extracted using the NUCLISENS® easyMAG® (bioMérieux, Florence, Italy). SARS-CoV-2 RNA detection was performed by GeneFinder COVID-19 Plus RealAmp Kit (Elitech, Turin, Italy) designed to detect envelope (E), RNA-dependent-RNA polymerase (RdRp), and nucleocapsid (N) genes and was subjected to NGS with the SARS-Cov-2 Respiratory panel (Illumina). According to the manufacturer's instructions, a Ct value <40 was considered positive. A total of 60 RNA samples, showing a broad range of representative SARS-CoV-2 Ct values (from 12 to 37) and interpreted as positive, was selected for sequencing. Samples were divided into 3 groups: i) low Ct group (12 ≤ Ct <20), ii) medium Ct group (21 ≤ Ct ≤25) and iii) high Ct group (Ct >25).
2.3. SARS-CoV-2 genome sequencing
RNA extracted from nasopharyngeal swabs were treated with Invitrogen DNase I, Amplification Grade (ThermoFisher Scientific, Waltham, MA, USA). The concentration and the quality of isolated RNA samples were measured with the Qubit 2.0 (ThermoFisher Scientific) and Agilent 2200 Tape Station Instrument, respectively. Total RNA was reverse transcribed into cDNA with Maxima H Minus Double-Stranded cDNA Synthesis Kit (ThermoFisher Scientific, Waltham, MA, USA). The resulting dsDNA was purified using 1.8× Agencourt AMPure XP Beads (Beckman Coulter, Woerden, Netherlands) and quantified with Qubit 2.0 (ThermoFisher Scientific).
Sequencing-ready libraries were prepared using the SARS-Cov-2 Respiratory panel with the DNA Prep with Enrichment and IDT for Nextera DNA UD Indexes from Illumina (San Diego, CA, USA) according to the manufacturer's instructions. Viral enriched libraries were sequenced on the MiSeq System at 2 × 75 bp read length using MiSeq v3 reagents (Illumina).
2.4. Full-length analysis of genome variation
Raw sequencing reads (Fastq files) from each sample were aligned to the Wuhan-H-1 reference genome (accession number NC_045512.2) using the bwa aligner (Li and Durbin, 2009). Reads with quality or alignment scores of less than 30 were discarded. For each sample, a consensus sequence formed by the assembly of the most representative nucleotide at each position was generated. At positions with coverage of less than 50 reads the reference nucleotide was assigned. The Lofreq software was used for variant calling (Wilm et al., 2012). Single nucleotide variations (SNVs) were defined as variants exceeding 25% frequency within a sample.
2.5. Haplotype determination
Haplotype reconstruction was performed using sequence variation identified in all samples through different tools (Shorah2, Haploclique, RegressHaplo). However, linkage analysis executed by all three algorithms provided solutions with poor fits while haplotype reconstruction using RegressHaplo provided good fits (Leviyang et al., 2017). In all cases, we chose the haplotype reconstruction that contained the consensus and had the minimum number of haplotypes with analysis restricted to variants showing frequency ≥ 25%.
2.6. Phylogenetic analysis of SARS-CoV-2 isolates
Phylogenetic analysis was carried out using a total of 5095 full-length SARS-CoV-2 sequences, collected in Italy by March 2021 and downloaded from the GISAID database on June 4th 2021 (Elbe and Buckland-Merrett, 2017). We pairwise aligned each sequence to the reference and selected, for subsequent analysis, a subset of 913 sequences that had been uploaded in the period March 2020—March 2021. The selected 913 sequences included sequences from every PANGO lineage and every region of Italy. We also included 80 sequences from PANGO lineages A and B as outliers. Finally, two additional sequences were included: the founder sequence, which was the most common consensus sequence among the samples analyzed in this study and the reference sequence (NC_045512.2). We used MAFTT to align the 913 sequences and IQ-TREE (parameters -m GTR + G) to compute a maximum likelihood phylogeny (Katoh and Standley, 2013). We used the R packages ggtree (Nguyen et al., 2015; Yu et al., 2018) and ggnetwork (F. Briatte, R package, CRAN) to generate figures.
2.7. Public availability of sequencing data
The newly whole-genome sequences were submitted to GenBank® database (Benson et al., 2015). All sequences can be retrieved from GISAID under accession numbers listed in Supplementary File S2.
3. Results
3.1. NGS sequencing of COVID-19 patients
During the early pandemic phase in March–May 2020, we collected 60 nasopharyngeal swabs from 46 subjects residing in a nursing home in Calabria (Southern Italy). Of these, 46 samples were obtained at admission (baseline), 14 samples were obtained at sequential time points from 8 different patients. RNA from nasopharyngeal swabs was subjected to NGS with the SARS-Cov-2 Respiratory panel (Illumina). The median depth of sequenced SARS–CoV-2 genome was 3693 (range, 36–19,461), with >90% of the genome covered more than 50-fold in 58 samples. A median number of 111,747,841 reads (range 1,126,001- 574,097,776) was generated for each sample (Supplementary file S3). To analyze NGS results, we constructed a viral consensus sequence for each COVID-19 patient within the nursing home. The consensus sequence was generated by assembling viral sequences relative to the reference genome of Wuhan-H-1 (NC_045512.2), using the most frequent nucleotide obtained by NGS at each position of the viral genome. The 60 consensus sequences obtained showed more than 99% sequence identity to the SARS-CoV-2 reference Wuhan-H-1 genome.
3.2. Viral classification
All the consensus sequences generated from the 60 isolates were submitted to the PANGO lineage tool to reconstruct viral phylogeny and identify viral lineages. On average, samples analyzed in this work contained 4–8 nucleotide changes relative to the reference sequence. The molecular analysis made use of viral sequences from March 2020 to May 2021 downloaded from the public database GSAID (May 2021). The 60 viral isolates from analyzed patients were classified as B.1 lineage, clade G (Fig. 1 ). The cluster of the B.1 lineage in the viral isolates of this study was characterized by the presence of three SNVs: ntC241T in the 5′ untranslated region, D614G in the S gene and P4715L in the Orf1ab (NSP12) gene, in agreement with epidemiological data available in March and early April 2020 in Europe (Rambaut et al., 2020).
Fig. 1.

Phylogenetic analysis of SARS-CoV-2 genomic sequences collected in the Chiaravalle patients. Multilineage phylogenetic tree reconstructed using 913 SARS-CoV-2 sequences downloaded from GISAID database. The SARS-CoV-2 consensus genomes of the 60 Chiaravalle patients are indicated with a red dot. Inclusion of all 60 consensus genomes led to a cluster of genomes around the founder genome. The reliability of the phylogenetic clustering was evaluated using bootstrap analysis with 1000 replicates. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Major and minor SNVs
Overall 26 single nucleotide variants (SNVs) passing quality control and frequency filters were identified in the study, including 65.5% transitions and 24.1% transversions. Transitions were distributed as follows: C > T (44.8%), T > C (6.8%), A > G (10.3%) and, G > A (3.5%) whereas transversions were: G > C (3.5%), A > C (6.9%), C > A (6.9%), G > T (6.9%) (Fig. S1Supplementary Fig. S1). The most frequent substitution was the transversion C > T (44.8%) whereas the least frequent was G > A (3.5%). In agreement with previous studies (Graudenzi et al., 2021; Tonkin-Hill et al., 2021), our data show that the ratio C > T /G > A (values 44.8 and 3.5, respectively) was consistent with an excess of substitutions in the plus strand.
SNVs in the viral genome were classified as major when identified in >80% of virus population or minor when identified in a percentage of virus population included between 20% and 80%. In total, of the 26 SNVs identified in this study, 14 were regarded as major SNVs and 12 were regarded as minor SNVs. Major SNVs were distributed across 4 out of the 10 protein-coding genes of the viral genome (ORF1ab, ORF3a, S and M).
Among major SNVs, 7 were in the Orf1ab gene, 2 were in the S gene, 1 was in the Orf3a gene, 3 were in the M gene and, 1 was in 5’-UTR. See Supplementary file S4 (sheet 1) for more details.
The SNVs in the coding regions of Orf1ab gene were both synonymous (3037C > T:A964A; 3157C > T: A964A) and non-synonymous (11,083 G > T: L3606F; 11,103C > T: P3613L; 14,408C > T: P4715L; 15,328C > T: L5022F; 16,456 T > C: S5398P); the 2 SNVs in the coding regions of S gene were non-synonymous (21,575C > T: L5F; 23,403 A > G: D614G); the small indel 26,155 G > d (V255-) in the Orf3a gene was a 1-bp deletion; the 3 SNVs in the coding regions of M gene were both synonymous (26,555 A > G: E11E; 26,681C > T: F53F) and non-synonymous (26,774 G > T: M84I) while the remaining SNV located in the 5’-UTR was non-coding (ntC241T). See Fig. 2 for a graphic representation.
Fig. 2.

Distribution of major and minor SNVs along the SARS-CoV-2 genome. The figure shows major SNVs on the top and minor SNVs under the 0-orizzontal axis. SNVs are indicated according to their position in the SARS-CoV-2 genome. SNVs are depicted as follows: synonymous (green), non-synonymous (red), non-coding (blue), small indel (black). Frequencies of |SNVs are reported on the left. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Six major SNVs were common to all samples at baseline, except for patient #26 (viral isolate 39): 241C > T in the 5’-UTR region, 3037C > T (F924F) in the Orf1ab gene, 14,408C > T (P4715L) in the Orf1ab gene, 16,456 T > C (S5398P) in the Orf1ab gene, 23,403 A > G (D614G) in the S gene and 26,681C > T (F53F) in the N gene.
Four major SNVs were identified in single patients: 3157C > T (A964A) in the ORF1ab, 21,575C > T (L5F) in the S gene, 26555 A > G (E11E) and 26,774 G > T (M84I) in the M gene. They were present in patients #38, #6, #10 and #46, respectively. The remaining 4 major SNVs were found in 2 additional patients: patient #39 carried 2 SNVs in ORF1a gene (11,083 G > T: L3606F and 11,103C > T: P3613L) while patient #34 carried a SNV in ORF1a (15,328C > T: L5022F) and a SNV in del255 in ORF3a gene.
Conversely, minor SNVs were distributed across 5 out of the 10 protein-coding genes of the viral genome: ORF1ab, ORF3a, ORF6, M and N. See Fig. 2 for graphical details. Identified minor SNVs included 6 in the Orf1ab gene, 3 in the Orf3a gene and, 1 in the ORF6, M and N genes, respectively. See Supplementary file S4 (sheet 2) for more details. Minor SNVs in the coding regions of the Orf1ab gene were both synonymous (2) and non-synonymous (4); SNVs in the coding regions of ORF3 and N genes were non-synonymous (3 and 1, respectively) while SNVs in the coding regions of ORF6 and M were synonymous (2). See Fig. 2.
SNVs in the Orf1ab gene were 1684C > T (I473I), 4898C > T (H1545Y), 5765 G > A (G1834T), 7594C > T (G2443G), 8208C > A (T2648N) and 18,211 A > C (M5983L); SNVs in the ORF3a gene were 25,945C > A (Q185K), 25,771C > T (L127F) and 26,057 A > C (D222A); SNVs in the ORF6, M and N were 27,354 A > G (Q51Q), 27,138 T > C (L206L) and 29,067C > T (T265I), respectively. See to Supplementary File S4 for more details. It is of note that 10 out of 12 minor SNVs were specific of a single patient while the remaining 2 (5765 G > A (G1834T) in the ORF1ab gene and 26,057 A > C (D222A) in the ORF3a gene) were shared between patients #7 and #25.
Notably, of all SNVs identified, 13 occurred in the ORF1ab gene. However, when gene length was taken into account, SNVs resulted enriched only in the ORF3a and M genes (binomial test, p-values 1E-4 and 0.04, respectively). The dNdS test for enrichment of non-synonymous versus synonymous SNVs within genes suggested that a significant enrichment of non-synonymous mutations was observed only in the ORF3a gene (dNdS = 3.1, p-value 0.04).
3.4. Analysis of viral haplotypes
The analysis of the 60 consensus viral sequences suggested the presence of different SARS-CoV-2 haplotypes co-circulating in a single nursing home within a short time frame during the first wave of the pandemic outbreak. In particular, the sequences from the 46 viral isolates at baseline collapsed into 11 haplotypes: from Hap A to Hap K (Table 2 ; Fig. 3 ).
Table 2.
Haplotypes A-K.
| Haplotype | SNVs | Patient | Isolate |
|---|---|---|---|
| A | 5′UTR: ntC241T, ORF1ab: F924F, ORF1ab: P4715L, ORF1ab: S5398P, S: D614G, M: F53F | (*) | (*) |
| B | ORF3a: D222A | #7 | 13 |
| C | S: L5F | #6 | 18 |
| D | M: E11E | #10 | 2 |
| E | ORF1ab: T2648N | #4 | 26 |
| F | 5’UTR: ntT241C, ORF1ab: P5398S, S: G614D | #26 | 39 |
| G | ORF1ab: L5022F, ORF3a: ntG26155-, ntG26156-, ntG26157- | #34 | 48 |
| H | ORF1ab: A964A | #38 | 56 |
| I | ORF1ab: L3606F, ORF1ab: P3613L | #39 | 5 |
| J | N: T265I | #41 | 59 |
| K | M: M84I | #46 | 64 |
(*) The complete list of patients/isolates with the Hap A is reported in Supplementary File S1 and S5.
Fig. 3.
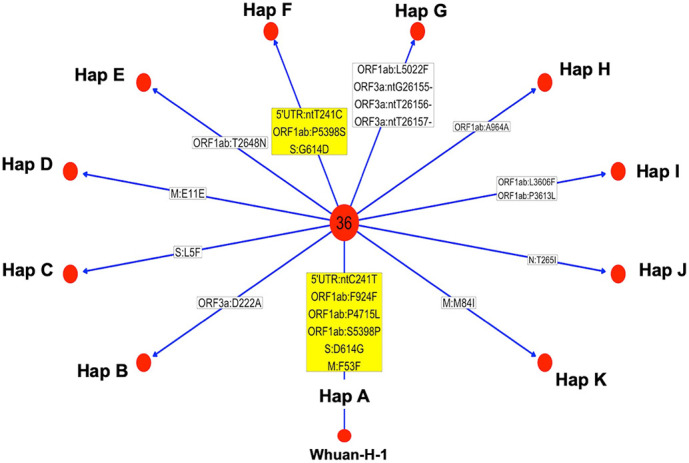
Haplotype identification in the SARS-CoV-2 isolates at baseline. The figure shows SNVs with >50% frequency present in the consensus sequences. Each red dot identifies a specific haplotype (hap A-K). In the center of the figure is shown a hub composes of 36 genomes with the same haplotype. In boxes are shown SNVs (relative to the Wuhan-H-1 sequence) that are characteristic of specific isolates: yellow boxes show major SNVs; white boxes show minor SNVs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The most prevalent haplotype was present in 36 patients and allowed us to identify a founder haplotype (Hap A), which may have initiated the local transmission of infection. Hap A was characterized by 6 major SNVs [241C > T in the 5’-UTR region, 3037C > T (F924F) in the Orf1ab gene, 14,408C > T (P4715L) in the Orf1ab gene, 16,456 T > C (S5398P) in the Orf1ab gene, 23,403 A > G (D614G) in the S gene, and 26,681C > T (F53F) in the N gene].
Taking into account the consensus sequences of viral isolates at baseline, we found that 36 patients were infected by a virus carrying Hap A. Conversely, 10 patients were infected by virus carrying additional substitutions with respect to Hap A, giving rise to 10 novel haplotypes, Hap B to Hap K. See Table 2 for details.
The isolates from 2 patients (#10, and #38) were each characterized by the additional presence of 1 synonymous SNV (Hap D: E11E inM; Hap H: A964A in ORF1ab, respectively); 5 isolates (patients #4, #6, #7, #41 and #46) were characterized by the presence of 1 non-synonymous SNV (Hap E: T2648N in ORF1ab; Hap C: L5F in t S; Hap B: D222A in ORF3a; Hap J: T265I in N; Hap K: M84I in M); the haplotype of patient #34 (Hap G) was characterized by 1 non-synonymous SNV in the ORF1ab gene (L5022F) and by a 3-nt deletion (ntG26155, ntT26156, ntT26157) in the ORF3a gene that results in the deletion of V255; the haplotype of patient #39 (Hap I) presented two non-synonymous SNVs in the ORF1ab gene (L3606F, P3613L). It is of note that patient #26 (isolate 39) carried 3 SNVs that reverted to the sequence corresponding to Whuan-H-1 (ntT241C, P5398S and G614D) (Hap F).
3.5. Dynamic of haplotypes in patients with longitudinal follow-up
Patients for whom multiple swabs at different time points were available are: #1 (swabs taken at days 0 and 15; isolates 65 and 12), #2 (swabs taken at days 0 and 7; isolates 52 and 53), #3 (swabs taken at days 0 and 10; isolates 34, and 35), #4 (swabs taken at days 0, 8 and 16; isolates 26–28), #5 (swabs taken at days 0, 15 and 23; isolates 22–24), #6 (swabs taken at days 0 and 7; isolates 18, and 19), #7 (swabs taken at days 0, 18, 25, 33, and 41; isolates 13–17) and #8 (swabs taken at days 0, 7 and 15; isolates 8–10). Haplotype distribution was analyzed longitudinally in the 8 patients for whom multiple swabs were available.
The analysis revealed that, at baseline, the viral haplotype of patients #1, #2, #3, #5 and, #8 was Hap A while the viral haplotypes of patients #4, #6 and, #7 were Hap E, Hap C and Hap B, respectively. For patients #1, #2, #3, #5 and #8 our data show that the founder haplotype (Hap A) remained unchanged. Conversely, at baseline patient #4 carried two different viral populations (Hap A and Hap E). At baseline, the frequencies of viral haplotypes were 69% for Hap E and 31% for Hap A. Haplotypes' frequencies were almost unchanged at day 8 (75% for Hap E and 25% for Hap A) whereas at day 16 the frequencies of haplotypes inverted significantly (34% for Hap E and 66% for Hap A) (Fig. 4 and Supplementary File S5).
Fig. 4.

Haplotypes across samples at different time points. The plot shows haplotypes (Hap) across samples obtained at different time points from baseline swab in patients 1, #2, #3, 4, #5, 6, #7 and #8. Haplotypes'code is shown in brackets. The numbers on the right represent the percentage of the viral population characterized by the indicated haplotype.
As regard patient #6, a single haplotype (Hap C) was present at baseline while 2 haplotypes (Hap C and Hap C.1, with frequencies of 67% and 33%, respectively) were present on day 7 (Fig. 4 and Supplementary File S5). Haplotype Hap C was characterized by SNV 21575C > T (L5F) in the S gene while haplotype Hap C.1 was characterized by SNVs 21,575C > T (L5F) in the S gene and 1684C > T (I473I) in the ORF1ab gene. As regard patient #7 (isolates 13–17), 3 viral haplotypes were detected in the swab at baseline (Hap A, Hap B, Hap B.1), with frequencies of 46% for Hap A and 27% for Hap B and Hap B.1(Fig. 4). Hap A was the founder haplotype; Hap B was characterized by the SNV 26057 A > C (D222A) in the ORF3a; Hap B1 was characterized by the SNV 26057 A > C (D222A) in the ORF3a gene and 5765 G > A (G1834S) and 5766 G > C (G1834A) in the ORF1a gene. Interestingly, on day 18 haplotypes Hap B and Hap B.1 disappeared and the only viral haplotype detected was the Hap A. At days 25 and 41 a novel haplotype (Hap B.2), characterized by SNVs 18211A > C (M5983L) in the ORF1a gene and 25771C > T (L127F) in the ORF3a gene, was detected with a frequency of 34% (Fig. 4.) See also supplementary File S5.
3.6. Clinical characteristics of patients and correlation with viral haplotypes
Clinical and follow-up information was available for 36 patients. The mean age of the patients was 80 years (range 56–97). Hospitalization was required for 36 patients, while ICU admission was required for only one patient. As to symptoms at the time of diagnosis, 5 were classified as asymptomatic (#11, #14, #26, #34 and #35) and 31 as symptomatic. Reported symptoms were of varying severity, ranging from mild to critical. Of the patients with symptoms, 7 presented with fever, 1 with cough, 2 with respiratory failure, 11 with fever and respiratory failure, 5 with fever and cough, 1 with cough and respiratory failure, and 4 with fever, cough, and respiratory failure. Concomitant diseases (comorbidities) were reported in 31 individuals except for patients #7, #15, #24, #35, #41 and #42. Reported comorbidities included hypertension, diabetes, cardiovascular disease or a combination thereof. By August 2021, 11 of the patients accrued for the study had died while 25 patients were still alive. All deceased patients presented comorbidities except for patients #15, #24 and #42: 8 patients presented cardiovascular disease, 3 patients had also hypertension, 2 had also diabetes. Similarly, also the majority of surviving patients presented comorbidities except for patients #7, #41 and #3. Among surviving patients 16 had only hypertension, 2 only diabetes, 1 diabetes and cardiovascular disease, 1 cardiovascular disease and hypertension, 2 diabetes and hypertension. It is of note that patient #11, asymptomatic at diagnosis, and subsequently admitted in ICU, presented the founder haplotype Hap A. The clinical characteristics of the patients studied are summarized in Table 1. Additional demographic and clinical information can be found in Supplementary file S1.
When patients were stratified for viral haplotype, we found that 9/11 deceased patients carried the haplotype Hap A; while the remaining 2 patients (#6 and #38) presented the Hap C and Hap H haplotypes, respectively. As regard the 25 patients who were still alive by August 2021, 19 presented the founder haplotype Hap A whereas 6 patients presented different haplotypes (#4, Hap E; #7, Hap B; #10, Hap D; #26, Hap F; #34, Hap G; #41, Hap J).
Conversely, of the 5 asymptomatic patients, 3 (patients #11, #14 and #35) showed the founder haplotype (Hap A), while patients #34 presented Hap G and #26 presented Hap F. As for the symptomatic patients, 6 patients who had fever only presented haplotype Hap A (patients #1, #13, #23, #25, #32, #36) and 1 presented haplotype Hap B (patient #7); the patient who had cough only (#41) presented haplotype Hap J; the patients with respiratory failure (#6 and #16) presented Hap A and Hap C; four of the five patients who had fever and cough (#2, #31, #33 and #44) had Hap A; most of the patients with fever and respiratory failure (10/11) had Hap A and the remaining patients presented Hap H (#38); finally, all 4 patients with fever, cough and respiratory failure (#12, #18, #20, #37) presented Hap A. In summary, of the 28 patients for whom clinical information was available who were infected by SARS-Cov2 virus with haplotype Hap A, 3 were symptomatic while 9 had died. Conversely, of the 8 patients infected by the virus with haplotypes different from Hap A, 6 were symptomatic (1 cough, 1 respiratory failure, 1 fever, 1 fever and cough, 2 fever and respiratory failure) and 2 had died (#6 with respiratory failure and #38 with fever and respiratory failure). Notably, both patients #6 and 38 (84 and 97 years old, respectively) presented important comorbidities, including hypertension and cardiovascular disease. In conclusion, the analysis reported here failed to find a significant association between viral haplotypes and any of the clinical parameters analyzed.
4. Discussion
Nursing homes have represented important hotspots of SARS-Cov2 spread during the initial wave of COVID-19 pandemics. The close proximity of patients inside nursing homes allows investigate the dynamics of viral transmission, which may improve our understanding of SARS-Cov2 biology and ability to spread. In this study, we have retrospectively sequenced 60 SARS-CoV-2 genomes isolated from 46 patients hospitalized in a nursing home in the Calabria Region.
Overall, the main findings of this NGS analysis are: 1) the main viral strain responsible for infection in the nursing home belonged to the B.1 lineage, clade G, with 14 major SNVs and 12 minor SNVs with reference to the Wuhan-H-1 sequence; 2) the reconstruction of the haplotypes based on the analysis of major and minor SNVs led to the identification of 11 haplotypes. A founder haplotype (Hap A) characterized by 6 major SNVs [D614G in the S gene; P4715L, ntC3037T (F924F) and S5398P in Orf1ab gene; ntC26681T (F53F) in the M gene; and ntC241T in the non-coding UTR region] was found in 36 patients. Conversely, haplotypes Hap B—K derived from Hap A through acquisition of additional mutations were identified in the 10 remaining patients; 3) the sequencing of viral genomes indicated that mutations in genes other than S (i.e. Orf1ab, Orf3a, and M) may have a pathogenic role; 4) no significant association between a specific viral haplotype and clinical parameters was observed.
The phylogenetic analysis of viral isolates whose genome has been sequenced in this study indicated that the main viral strains responsible for infection in the nursing home belonged to the B.1 lineage, clade G. Accordingly, between January and May 2020, lineages B.1 and B.2 were responsible for the large outbreak of the pandemics in Italy (Rambaut et al., 2020). At that time, the spreading of SARS-CoV-2 was characterized by three main differences compared to the Wuhan H1 strain: ntC241T in the 5′ untranslated region, D614G in the S gene and, P4715L in the ORF1ab (NSP12) gene. In particular, the D614G SNV is known to increase viral infectivity (Yuan et al., 2020). Remarkably, all virus isolates analyzed at baseline presented all three SNVs, which is again consistent with the viral strain predominant in Italy in March 2020. However, 2 patients (#2 and #3) presented the D residue at position 614 in the follow-up samples, suggesting that both strains (with G and D substitutions) were co-circulating or emerged in Italy since March 2020, although strains with D substitutions circulated in a limited fraction of patients (Korber et al., 2020).
The whole-genome analysis performed in this study identified 26 SNVs with respect to the sequence of the Wuhan-H1 isolate. Half of the 26 SNVs are located in the ORF1ab gene, though the ORF3a was the gene in which SNVs were enriched when taking into account the length. In fact, based on odNdS analysis, significant enrichment of nonsynonymous mutations was found only in the ORF3a gene. Interestingly, the accessory protein ORF3a plays a crucial role in virus entry due to its localization on the cell surface (Issa et al., 2020; Liu et al., 2014; Siu et al., 2019). ORF3a is also involved in the production of pro-inflammatory cytokines and chemokines via the JNK and NF-kB pathways (Siu et al., 2019). In addition, ORF3a is involved in the formation of ion channels that play a role in the release of viruses from host cells (Liu et al., 2014). Specifically, we found mutations of the ORF3a protein that fall in the cysteine-rich domain (L127F) and in the C-terminal domain (Q185K, D222A, V255del), which are known to be involved in ORF3a protein homodimerization, intracellular protein sorting and trafficking (Issa et al., 2020; Lu et al., 2006). Notably, the D222A substitution was shared by 2 patients (#7 and #25), suggesting a possible close contact between them, possibly supporting the increased infectivity associated with Orf3a mutations.
The 46 viral isolates collected at baseline were classified into 11 different haplotypes. Thirty-six of the 46 patients shared a founder haplotype, characterized by the six most common SNVs (ntC241T in the UTR noncoding region; F924F, P4715L, and S5398P in ORF1ab; D614G in the spike protein; and F53F in the N protein), which may have initiated local transmission of infection. Of the founder mutations, S5398P in ORf1ab falls in the region encoding the nsp13 helicase enzyme and mutation P4715L falls in the region encoding nsp12 (NiRAN-RdRp). Notably, the S5398P (S74P) substitution is located in the ZBD domain of nsp13, which is expected to have disruptive effects on in vitro helicase activity and viral replication (Hao et al., 2017; Jia et al., 2019). Based on recent studies providing evidence for the cooperation of nsp13 and nsp12 in RNA replication (Biswas and Mudi, 2020; J. Chen et al., 2020; Gurung, 2020; Pachetti et al., 2020) it is conceivable that these mutations could negatively affect the aggressiveness of the virus. Given the central role of helicase in SARS-CoV-2 replication and its potential as an antiviral drug target, we have searched for the presence of S5398P substitution which was not found in the additional isolates analyzed (data not shown). This observation suggests that S5398P substitution may be considered a neutral variant emerged in the patients residents at the nursing home that had not spread like more aggressive mutations.
The remaining ten haplotypes identified were characterized by the presence of additional mutations compared to the founder strain, Hap A (Fig. 3). In particular, one patient (#6) carried the L5F substitution, which has been shown to be responsible for increased infectivity by promoting protein folding, assembly and secretion of the virus (Zhang et al., 2020). Previous reports consider L5F as a kind of positive Darwinian selection in the spike gene of SARS-CoV-2, which clarifies the adaptation mechanism of this virus (Zhan et al., 2021). Notably, our results showed that patient #6, who unfortunately died, carried the L5F in both baseline samples (isolate 18) and follow-up samples (isolate 19) in major and minor haplotypes (67% vs. 33%). One patient (#46) carried the M84I substitution within M protein, located in a region closer to the I82T typical of the delta variant, which currently reaches a frequency of 70%, thus suggesting that this substitution may provide some sort of fitness advantage. It is of note that one of the deceased patients (#38) carried a synonymous SNV (A964A, Hap H) in the non-structural protein nsp3 encoded by ORF1ab. Nsp3 is known to be an essential component of the SARS virus replication complex and has been reported to block the host immune response (Lei et al., 2018).
Lastly, we report on the results of a longitudinal analyses of samples from 8 patients. In five patients (#1, #2, #3, #5 and #8) the haplotype Hap A identified at baseline has remained unchanged while in 3 patients (#4, #6 and #7) a dynamic evolution of the SARS-CoV-2 genome detected at baseline apparently occurred. In particular, patient #4 had two distinct haplotypes, one of which (Hap E, T2648N ORF1ab) was more prevalent at baseline and reduced on day 16. Patient #6 carried the L5F mutation in the S protein in both the major and minor viral populations, while the I473I in ORF1ab was only present in a lower viral population (33%). Patient #7 had three haplotypes at baseline (Hap A, Hap B and Hap B.1), which were completely replaced by a single haplotype (Hap B.2) carrying 2 SNVs (L127F and M5983L in ORF3a and ORF1ab, respectively) that may enhance viral infectivity as discussed above.
In conclusion, in this study we report on NGS analysis performed on patients infected by SARS-CoV2 in a nursing home in Calabria. We have found that the main viral strain responsible for infection in the nursing home belonged to the B.1 lineage, clade G. The analysis of the viral spread in the nursing home is of certain interest because elderly people in nursing homes are particularly vulnerable to COVID-19. On the other hand, infection in the nursing home has spread rapidly and allowed to study the dynamic of infection in a very controlled way. The results reported here highlight the high potential of virus evolution in a limited period of time during a single outbreak from a founder viral haplotype, supporting the importance to monitor the appearance of novel mutations and to classify viral haplotypes in order to determine their biological and clinical significance.
Data availability
SARS-CoV-2 genome sequences are deposited in Gisaid (www.gisaid.org) and the access identifiers are listed in Supplementary File S2.
Funding
This research received specific financial support from Department of Experimental and Clinical Medicine (COVID19-DMSC) and Interdepartmental Center of Services (CIS), Molecular Genomics and Pathology, “Magna Græcia” University of Catanzaro, Italy. The work was also supported by Regione Calabria (noCOVID19@UMG POR Calabria - FESR/FSE 2014-2020 D.D.R.C. n. 4584 del 4/5/2021- Azione 10.5.12).
Aknowledgements
IDTM UMG COVID-19 Group
Enrico Maria Trecarichi, Alessandro Russo, Maria Mazzitelli, Francesca Serapide, Maria Chiara Pelle, Bruno Tassone, Eugenio Arrighi, Graziella Perri, Paolo Fusco, Vincenzo Scaglione, Chiara Davoli, Rosaria Lionello, Valentina La Gamba, Giuseppina Marrazzo, Maria Teresa Busceti, Amerigo Giudice, Marco Ricchio, Anna Cancelliere, Elena Lio, Giada Procopio, Salvatore Rotundo, Helen Morrone, Maria Teresa Tassone, Vincenzo Olivadese, Chiara Costa, Francesco Saverio Costanzo, Daniela Patrizia Foti, Giovanni Matera, Carlo Torti, Federico Longhini, Andrea Bruni, Eugenio Garofalo, Eugenio Biamonte, Domenico Laganà, Maria Petullà, Bernardo Bertucci, Angela Quirino, Giorgio Settimo Barreca, Aida Giancotti, Luigia Gallo, Angelo Lamberti, Maria Carla Liberto, Nadia Marascio, Adele Emanuela De Francesco.
CRediT authorship contribution statement
Carmela De Marco: Investigation, Visualization, Formal analysis, Writing – review & editing. Nadia Marascio: Resources, Investigation, Writing – original draft. Claudia Veneziano: Investigation, Data curation, Visualization. Flavia Biamonte: Data curation, Writing – review & editing. Enrico Maria Trecarichi: Resources, Writing – review & editing. Gianluca Santamaria: Investigation, Methodology, Data curation. Sivan Leviyang: Investigation, Writing – review & editing. Maria Carla Liberto: Resources. Maria Mazzitelli: Resources. Angela Quirino: Resources. Federico Longhini: Resources, Writing – review & editing. Daniele Torella: Writing – review & editing. Aldo Quattrone: Writing – review & editing. Giovanni Matera: Resources, Writing – review & editing. Carlo Torti: Supervision, Funding acquisition, Resources, Writing – review & editing. Francesco Saverio Costanzo: Funding acquisition, Resources. Giuseppe Viglietto: Conceptualization, Methodology, Supervision, Funding acquisition, Writing – review & editing, Project administration.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.meegid.2022.105253.
Appendix A. Supplementary data
The following are the supplementary data related to this article.
Supplementary Fig. S1.
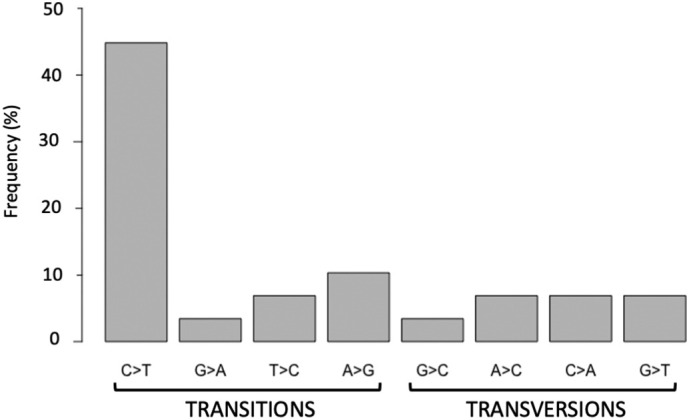
Classification of variants identified in SARS-CoV-2 samples. The bar graph shows the distribution of transitions and transversions.
Supplementary File S1. Patients’ clinical data
Supplementary File S2. Accession numbers of GISAID sequencies
Supplementary file S3. Next Generation Sequencing metrics
Supplementary file S4. Major and minor SNVs
Supplementary file S5. Haplotypes of viral isolates
References
- Benson D.A., et al. GenBank. Nucleic Acids Res. 2015;43(Database issue):D30–D35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.K., Mudi S.R. Spike protein D614G and RdRp P323L: the SARS-CoV-2 mutations associated with severity of COVID-19. Genom. Inform. 2020;18(4) doi: 10.5808/GI.2020.18.4.e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capobianchi M.R., et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020;26(7):954–956. doi: 10.1016/j.cmi.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., et al. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020;182(6):1560–1573. doi: 10.1016/j.cell.2020.07.033. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.K., Chevalier J.A., Long E.F. Nursing home staff networks and COVID-19. Proc. Natl. Acad. Sci. U. S. A. 2021;118(1) doi: 10.1073/pnas.2015455118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucinotta D., Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020;91(1):157–160. doi: 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Global Chall. 2017;1(1):33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S., et al. GESS: a database of global evaluation of SARS-CoV-2/hCoV-19 sequences. Nucleic Acids Res. 2021;49(D1):D706–d14. doi: 10.1093/nar/gkaa808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graudenzi A., et al. Mutational signatures and heterogeneous host response revealed via large-scale characterization of SARS-CoV-2 genomic diversity. iScience. 2021;24(2) doi: 10.1016/j.isci.2021.102116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung A.B. In silico structure modelling of SARS-CoV-2 Nsp13 helicase and Nsp14 and repurposing of FDA approved antiviral drugs as dual inhibitors. Gene Rep. 2020;21 doi: 10.1016/j.genrep.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W., et al. Crystal structure of Middle East respiratory syndrome coronavirus helicase. PLoS Pathog. 2017;13(6) doi: 10.1371/journal.ppat.1006474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa E., et al. SARS-CoV-2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems. 2020;5(3) doi: 10.1128/mSystems.00266-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z., et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47(12):6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182(4):812–827. doi: 10.1016/j.cell.2020.06.043. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Kusov Y., Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antivir. Res. 2018;149:58–74. doi: 10.1016/j.antiviral.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leviyang S., et al. A penalized regression approach to haplotype reconstruction of viral populations arising in early HIV/SIV infection. Bioinformatics. 2017;33(16):2455–2463. doi: 10.1093/bioinformatics/btx187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., et al. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014;109:97–109. doi: 10.1016/j.antiviral.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., et al. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. U. S. A. 2006;103(33):12540–12545. doi: 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lythgoe K.A., et al. SARS-CoV-2 within-host diversity and transmission. Science. 2021;372(6539) doi: 10.1126/science.abg0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massacci A., et al. Design of a companion bioinformatic tool to detect the emergence and geographical distribution of SARS-CoV-2 spike protein genetic variants. J. Transl. Med. 2020;18(1):494. doi: 10.1186/s12967-020-02675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBroome J., et al. A daily-updated database and tools for comprehensive SARS-CoV-2 mutation-annotated trees. Mol. Biol. Evol. 2021;38(12):5819–5824. doi: 10.1093/molbev/msab264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.T., et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32(1):268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole Á., et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7(2):veab064. doi: 10.1093/ve/veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachetti M., et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020;18(1):179. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5(11):1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., et al. Comprehensive genome analysis of 6,000 USA SARS-CoV-2 isolates reveals haplotype signatures and localized transmission patterns by state and by country. Front. Microbiol. 2020;11 doi: 10.3389/fmicb.2020.573430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 2020;71(15):713–720. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.L., et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33(8):8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausbaugh L.J., Sukumar S.R., Joseph C.L. Infectious disease outbreaks in nursing homes: an unappreciated hazard for frail elderly persons. Clin. Infect. Dis. 2003;36(7):870–876. doi: 10.1086/368197. [DOI] [PubMed] [Google Scholar]
- Tonkin-Hill G., et al. Patterns of within-host genetic diversity in SARS-CoV-2. Elife. 2021;10 doi: 10.7554/eLife.66857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trecarichi E.M., et al. Clinical characteristics and predictors of mortality associated with COVID-19 in elderly patients from a long-term care facility. Sci. Rep. 2020;10(1):20834. doi: 10.1038/s41598-020-77641-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dorp L., et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020;83 doi: 10.1016/j.meegid.2020.104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim J.O. A glimpse into the origins of genetic diversity in the severe acute respiratory syndrome coronavirus 2. Clin. Infect. Dis. 2020;71(15):721–722. doi: 10.1093/cid/ciaa213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A., et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40(22):11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., et al. Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol. Biol. Evol. 2018;35(12):3041–3043. doi: 10.1093/molbev/msy194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F., et al. Global SNP analysis of 11,183 SARS-CoV-2 strains reveals high genetic diversity. Transbound. Emerg. Dis. 2020;68(6):3288–3304. doi: 10.1111/tbed.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X.Y., et al. Elderly male with cardiovascular-related comorbidities has a higher rate of fatal outcomes: a retrospective study in 602 patients with coronavirus disease 2019. Front. Cardiovasc. Med. 2021;8:680604. doi: 10.3389/fcvm.2021.680604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File S1. Patients’ clinical data
Supplementary File S2. Accession numbers of GISAID sequencies
Supplementary file S3. Next Generation Sequencing metrics
Supplementary file S4. Major and minor SNVs
Supplementary file S5. Haplotypes of viral isolates
Data Availability Statement
SARS-CoV-2 genome sequences are deposited in Gisaid (www.gisaid.org) and the access identifiers are listed in Supplementary File S2.