

Abstract
COVID-19 is a new infectious disease caused by SARS-COV-2 virus of the coronavirus Family. The identification of drugs against this serious infection is a significant requirement due to the rapid rise in the positive cases and deaths around the world. With this concept, a molecular docking analysis for vitamins and their derivatives (28 molecules) with the active site of SARS-CoV-2 main protease was carried out.
The results of molecular docking indicate that the structures with best binding energy in the binding site of the studied enzyme (lowest energy level) are observed for the compounds; Folacin, Riboflavin, and Phylloquinone oxide (Vitamin K1 oxide). A Molecular Dynamic simulation was carried out to study the binding stability for the selected vitamins with the active site of SARS-CoV-2 main protease enzyme. Molecular Dynamic shows that Phylloquinone oxide and Folacin are quite unstable in binding to SARS-CoV-2 main protease, while the Riboflavin is comparatively rigid. The higher fluctuations in Phylloquinone oxide and Folacin indicate that they may not fit very well into the binding site. As expected, the Phylloquinone oxide exhibits small number of H-bonds with protein and Folacin does not form a good interaction with protein. Riboflavin exhibits the highest number of Hydrogen bonds and forms consistent interactions with protein. Additionally, this molecule respect the conditions mentioned in Lipinski's rule and have acceptable ADMET proprieties which indicates that Riboflavin (Vitamin B2) could be interesting for the antiviral treatment of COVID-19.
Keywords: Coronavirus, COVID-19, Vitamins, SARS-CoV-2 main protease, Molecular docking, Molecular dynamic
Graphical abstract
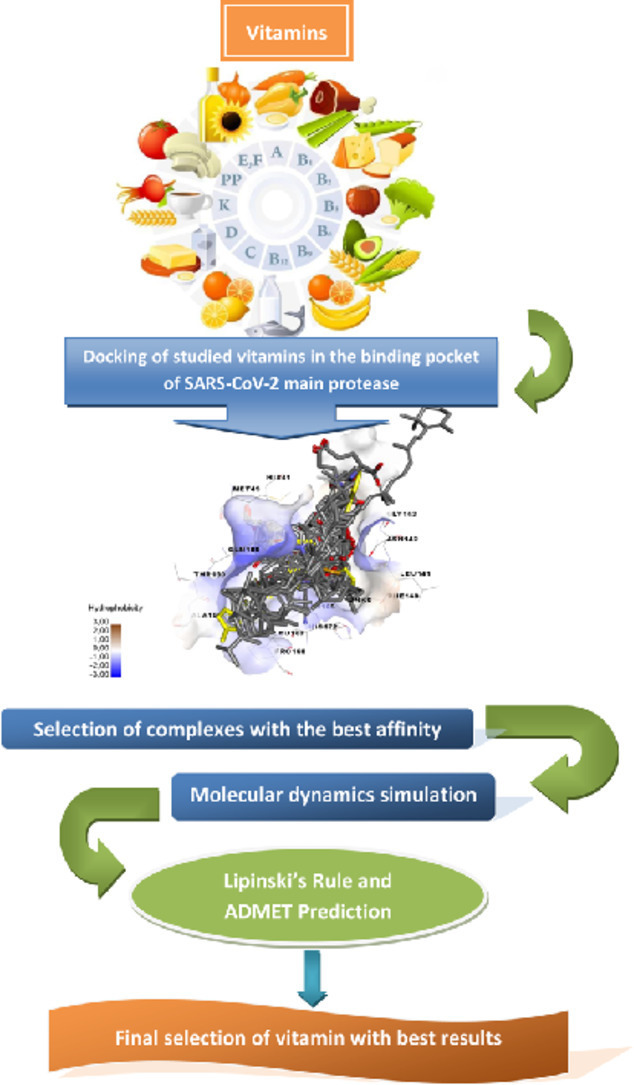
1. Introduction
Coronavirus (Covid-19) was reported for the first time in December 2019 in Wuhan (China). Then it spread all over the world causing more than 374,547,999 confirmed cases and more than 5,678,981 deaths until early January 2022, according to the world health organization [1].
COVID-19 virus infects cells by binding the spike glycoprotein (S) to the Angiotensin-converting enzyme 2 (ACE2) which is attached to the cell membranes of cells located in the lungs, arteries, heart, kidney, and intestines. In order to complete the entry into the cell, the TMPRSS2 protease enzyme must prime the Spike glycoprotein. In fact, the activation by TMPRSS2 as a protease is needed to attach virus Spike protein to human's cellular ligand. The viral genome is transcribed and then translated after the virus enters the host cell and uncoats. Therefore, targeting and disturbing the operation of one or many of those enzymes involved in the viral replication, transcription or translation can be effective to stop the emergency of this pandemic [2].
The crystal structure of main protease (also named 3CLpro or 3-chymotrypsin-like protease [2]) has many PDB structures (pdb code: 6LU7, 6M03, 6W4B, 6Y84, 6YB7, 5R7Y, 5R7Z, 5R80, 5R81, 6W63, 6M3M…) [3]. This study focuses on the crystal structure of 3CLpro (pdb code: 6LU7).The protease activity that is responsible for the cleavage of the polyprotein is presented in this enzyme (nsp5 protein) [4], this crystallographic structure is encoded within the viral genome, in combined with N3 inhibitor [5]. The potential treatment for coronaviruses can be applied in two different ways depending on the target, one acts on the human immune system and the other acts on the virus itself [2].
There is currently no specific treatment approved for coronavirus infection [6,7], although some treatments including anti-inflammatory, bronchodilator and anticoagulant drugs are used. Furthermore, some antiparasitic and antiviral drugs including Ivermectin and remdesivir have also been used [8,9]. The two antimalarial drugs; chloroquine and hydroxychloroquine were discontinued as COVID-19 treatment by the WHO [10], [11], [12], [13]. Certainly, there are many ways in which these drugs could exert their anti-SARS-CoV-2 effects. One of the most interesting SARS-CoV-2 targets in the main protease (Mpro) which is essential in the virus life cycle including its replication [14].
Treatment with vitamins or using them to reduce COVID -19 side effects was one of the treatment protocols approved by WHO. Vitamins are organic chemical compounds classified as essential nutrients needed in small quantities (less than 100 mg/day). They do not provide energy but are essential for the proper functioning of the body, especially for the metabolism of living organisms [15]. Vitamins are thirteen in number and fall into two categories:
-
•
The fat-soluble vitamins which are absorbed at the same time as the fats and stored. They are soluble in organic solvents (vitamins A, D, E and K).
-
•
Water-soluble vitamins which are not stored for a prolonged period and are excreted in the urine when their intake is excessive. They are soluble in water (vitamins C, B1, B2, PP, B5, B6, B8, B9, B12).
Vitamin A reduces morbidity and mortality in various infectious diseases, such as measles, diarrheal diseases, measles-related pneumonia, infection with the human immunodeficiency virus (HIV) and malaria [16]. Vitamin B2 and UV light effectively reduce the titer of MERS-CoV in human plasma products [17]. Vitamin C increases the resistance of chick embryo tracheal organ cultures to avian coronavirus infection [18]. Vitamin D has an effect on coronavirus infections, indeed, the decreased vitamin D status in calves had been reported to cause the infection of Bovine coronavirus [19]. In addition, Vitamin E deficiency had been reported to intensify the myocardial injury of coxsackievirus B3 (a kind of RNA viruses) infection in mice [20].
Repurposing medications may be the only response to the epidemic of unexpected infectious diseases, due to the longtime of producing new medicines. Among the drugs proposed as inhibitors against COVID-19, vitamin A, vitamin D, vitamin C, vitamin B12 and nicotinamide are suggested against SARS-CoV-2 in many research papers [21], [22], [23], [24], [25], [26]. Based on this effect, the study of interactions between vitamins and their derivatives compounds against SARS-CoV-2 main protease recently crystallized are recommended. These molecules would be much cheaper and simpler than the other drugs as a therapeutic option to be tested.
With this concept, a molecular docking analysis for vitamins and their derivatives (28 molecules) with the active site of SARS-CoV-2 main protease was carried out to predict the mode of binding between these molecules and their potential target (SARS-CoV-2 main protein), then determine the stability of these molecules in the binding site of the same enzyme using Molecular Dynamic simulation following by evaluation of their Lipinski's rule violations and their ADMET proprieties prediction to select the structures with acceptable proprieties which could be interesting for the antiviral treatment of COVID-19 [27].
2. Materials and methods
2.1. Data set
The studied compounds were evaluated against novel Coronavirus (SARS-CoV-2 main protease) as shown in Table 1 .
Table 1.
Chemical structures of vitamins and their derivatives compounds.
| N° | Vitamin | Chemical name | Molecular structure | ChemSpider ID |
|---|---|---|---|---|
| 1 | Vitamin A1 | All-trans-Retinol | 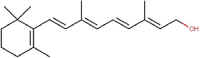 |
393012 |
| 2 | Vitamin A aldehyde | All-trans-retinal | 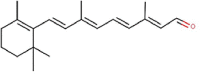 |
553582 |
| 3 | Vitamin A derivatives | 9-cis-Retinol | 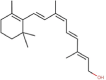 |
8123435 |
| 4 | Retinol stearate | 4942878 | ||
| 5 | Retinyl acetate | 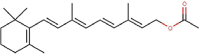 |
553599 | |
| 6 | Retinyl palmitate | 4444162 | ||
| 7 | Tretinoin | 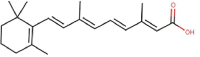 |
392618 | |
| 8 | Vitamin A myristate | 8227106 | ||
| 9 | Vitamin A propionate | 4908583 | ||
| 10 | β-Carotene | 4444129 | ||
| 11 | Vitamin B2 | Riboflavin | 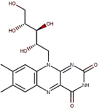 |
431981 |
| 12 | Vitamin B3 | Niacin | 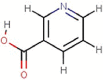 |
913 |
| 13 | Vitamin B3 | Niacinamide | 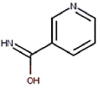 |
911 |
| 14 | Vitamin B3 | Nicotinamide riboside | 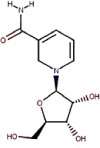 |
10136936 |
| 15 | Vitamin B5 | Pantothenic acid | 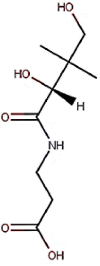 |
6361 |
| 16 | Vitamin B6 | Pyridoxine | 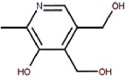 |
1025 |
| 17 | Vitamin B6 | Pyridoxamine | 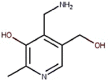 |
1023 |
| 18 | Vitamin B6 | Pyridoxal | 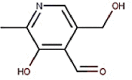 |
1021 |
| 19 | Vitamin B7 | Biotin | 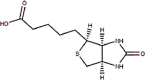 |
149962 |
| 20 | Vitamin B9 | Folacin or folic acid | 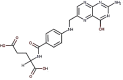 |
5815 |
| 21 | Vitamin C | Ascorbic acid or ascorbate | 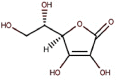 |
10189562 |
| 22 | Vitamin D2 | Ergocalciferol | 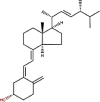 |
4444351 |
| 23 | Vitamin D3 | Cholecalciferol or colecalciferol | 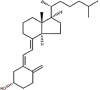 |
4444353 |
| 24 | Vitamin E | Tocopherol | 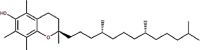 |
14265 |
| 25 | Vitamin E | Tocotrienol | 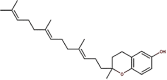 |
8105532 |
| 26 | Vitamin K1 | Phylloquinone | 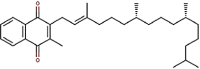 |
4447652 |
| 27 | Vitamin K2 | Menaquinone | 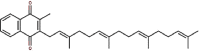 |
4445530 |
| 28 | Vitamin K1 oxide | Phylloquinone oxide | 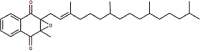 |
4444391 |
2.2. Minimization of studied molecules
All the studied compounds were obtained from chemical structure databases ChemSpider (An Online Chemical Information Resource) [28].
The minimization of the studied compounds was performed using SYBYL-X2.0 molecular modeling software [29]. Each structure of vitamins and their derivatives compounds was optimized with SYBYL program to ensure that the lowest-energy structure is used for the docking study, using the standard tripos molecular mechanics force field and energy gradient convergence criterion of 0.01 kcal/(mol.Å), the partial atomic charges required for calculation of the electrostatic potential were assigned using the Gasteiger_Huckel method [30], [31], [32], [33].
2.3. Molecular docking
A docking of studied compounds in the binding pocket of SARS-CoV-2 main protease (pdb code 6LU7) was performed [34], to determine binding energy and to study the intermolecular interactions of the studied molecules in the specific target.
Autodock vina [35] and Autodock tools 1.5.6 [36] were used to carry out the molecular docking study; Discovery Studio 2016 program [37] was used to obtain the binding site of crystallographic structure of SARS-CoV-2 main protease (pdb code 6LU7) [38,39]. The center of the active site (the coordinates: x= -10.782, y = 15.787 and z=71.277) has been determined on the basis of the co-crystallized ligand N3 [40]. 20 × 34 × 20 xyz points with a grid spacing of 1 Å is the grid size to cover the folic acid binding site in the enzyme (generated using the co-crystallized ligand (N3) as the center for docking) [40]. For ligand and enzyme preparations, an extended PDB format, termed PDBQT was used to coordinate the files, which includes atomic partial charges and atom types using Autodock tools 1.5.6. Torsion angles were calculated to assign the flexible and non-bonded rotation of molecules. All results were consequently analyzed using Discovery studio program [27].
2.4. Molecular dynamics
GROMACS simulation package (GROMACS 2020.4) was used to perform molecular dynamics simulations. MD simulations of the complexes (Phylloquinone oxide, Riboflavin and Folacin protein complexes) with the best affinities (after docking calculation) were carried out for 100 ns in water using CHARMM36 force field; trajectory and energy files were written every 10 ps.
The system was solvated in a truncated octahedral box, containing TIP3P water molecules. The protein was centered in the simulation box within minimum distance to the box edge of 1 nm to efficiently satisfy the minimum image convention. 4 Potassium ions were added to Phylloquinone oxide, Riboflavin and folacin protein complexes to neutralize the overall system, each containing 66884, 66887, and 66873 atoms, respectively.
Minimization was carried out for 5000 steps using Steepest Descent Method and the convergence was achieved within the maximum force < 1000 (KJ mol−1 nm−1), to remove any steric clashes. All three systems were equilibrated at NVT and NPT ensembles for 100ps (50,000 steps) and 1000ps (1,000,000 steps), respectively, using time steps 0.2 and 0.1 fs, respectively, at 300K to ensure a fully converged system for production run.
Production run for simulation was carried out at a constant temperature of 300 K and a pressure of 1 atm or bar (NPT) using weak coupling velocity-rescaling (modified Berendsen thermostat) and Parrinello-Rahman algorithms, respectively. Relaxation times were set to τ T = 0.1 ps and τ P = 2.0 ps. All bond lengths involving hydrogen atom were kept rigid at ideal bond lengths using the Linear Constraint Solver (lincs) algorithm, allowing for a time step of 2 fs. Verlet scheme was used for the calculation of non-bonded interactions. Periodic Boundary Conditions (PBC) were used in all x, y, z directions. Interactions within a short-range cutoff of 1.2 nm were calculated in each time step. Particle Mesh Ewald (PME) was used to calculate the electrostatic interactions and forces to account for a homogeneous medium outside the long-range cutoff. The production was run for 100ns for all three complexes.
2.5. Lipinski's rule and ADMET prediction
Lipinski's rule and ADMET [41] parameters of studied molecules were calculated using Swissadmet [42] and preADMET [43] web servers, respectively. Lipinski's rule including; molecular weight, number of rotatable bonds, number of hydrogen bonds acceptor, number hydrogen bonds donor and logP were determinate. Molecules violating more than one of these parameters may have problems with bioavailability and a high probability of failure to display drug-likeness [44].
We used preADMET server to predict the Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties of the studied molecules. We predicted BBB: in vivo blood-brain barrier penetration (C.brain/C.blood), Buffer_solubility: Water solubility in buffer system (SK atomic types, mg/L), HIA: Human intestinal absorption (HIA, %); Pgp_inhibition: in vitro P-glecoprotein inhibition, SK logD in pH 7.4 (SK atomic types), SK logP (SK atomic types). We also predicted the metabolism of these molecules by certain CYP such as CYP450_2C19, CYP450_2C9, CYP_2D6, CYP450_3A4. The toxicity profiling includes testing of acute algae, daphnia and fish toxicity, Ames test for mutagenicity testing of several Salmonella typhimurium strains. Carcinogenicity testing is also included through 2 years carcinogenicity bioassay in rats and mice in addition to in vitro human ether-a-go-go related gene channel (hERG) inhibition testing.
3. Results
3.1. Molecular docking
Molecular docking was performed to find the binding energy of the studied vitamins in the studied enzyme. 28 different molecules have been evaluated for their binding energy against SARS-CoV-2 main protease (pdb code 6LU7), the results of molecular docking study are presented in Table 2 Fig. 1. shows the position of the best conformation of all studied vitamins and derivatives in the active site of SARS-CoV-2 main protease [45]. As indicated in Fig. 1, the binding site has four pockets S1, S1’, S2 and S4. The S1 pocket has the residues Phe140, Asn142, Glu166, His163, Leu141 and His172, while the adjacent S1’ pocket which holds hydrophobic benzyl group of inhibitors is surrounded by hydrophobic residues Tyr24, Thr25, Thr26 and Leu27. The S2 pocket is surrounded by residues His41, Met49, Tyr54, Met165 and Asp187, while the S4 pocket is surrounded by residues Leu167, Phe185 and Gln192 [46].
Table 2.
Docking results: Binding energy of the best conformation in the binding pocket of SARS-CoV-2 main protease.
| Chemical name | Binding energy (Kcal/mol) | Binding constant | Ki(µM) | Chemical name | Binding energy (Kcal/mol) | Binding constant | Ki(µM) |
|---|---|---|---|---|---|---|---|
| Folacin | -7.9 | -4.20 | 833.35 | Biotin | -6.1 | -4.33 | 666.48 |
| Riboflavin | -7.7 | -6.77 | 10.87 | Vitamin A propionate | -6.0 | -5.94 | 44.49 |
| Phylloquinone oxide | -7.1 | -4.04 | 1.09 | All-trans-Retinol | -5.8 | -6.11 | 13.13 |
| Ergocalciferol | -7.1 | -6.33 | 10.11 | Tocopherol | -5.7 | -4.90 | 256.17 |
| Cholecalciferol | -6.7 | -7.04 | 6.95 | Retinol stearate | -5.5 | -1.56 | 72.14 |
| Nicotinamide riboside | -6.7 | -4.36 | 640.0 | Retinyl palmitate | -5.4 | -1.54 | 74.81 |
| Retinyl acetate | -6.5 | -5.46 | 99.71 | Pantothenic acid | -5.2 | -4.16 | 890.28 |
| β-Carotene | -6.5 | -4.74 | 333.44 | Ascorbic acid | -5.2 | -1.23 | 125.82 |
| Phylloquinone | -6.4 | -3.45 | 2.95 | Vitamin A myristate | -4.9 | -1.23 | 125.82 |
| 9-cis-Retinol | -6.4 | -7.23 | 5.03 | Pyridoxine | -4.8 | -4.86 | 272.8 |
| All-trans-retinal | -6.3 | -6.65 | 13.26 | Pyridoxal | -4.8 | -5.06 | 194.19 |
| Tretinoin | -6.3 | -5.67 | 69.56 | Pyridoxamine | -4.6 | -4.92 | 249.13 |
| Menaquinones | -6.3 | -4.92 | 248.74 | Niacinamide | -4.2 | -4.45 | 543.13 |
| Tocotrienol | -6.3 | -4.95 | 233.73 | Niacin | -4.2 | -4.31 | 695.0 |
Fig. 1.

Position of the best conformation of all studied compounds in the binding pocket of SARS-CoV-2 main protease (the co-crystallized ligand N3 is presented by yellow color).
The results of docking study show that the best energies of interaction with SARS-CoV-2 main protease are observed for vitamins; Folacin (Vitamin B9), Riboflavin (Vitamin B2) and Phylloquinone oxide (Vitamin K1 oxide).
The interaction results of Folacin, Riboflavin and Phylloquinone oxide in the SARS-CoV-2 main protease are presented in Fig. 2, Fig. 3, Fig. 4 , and Table 3 .
Fig. 2.

2D and 3D presentations of interactions between Folacin and SARS-CoV-2 main protease.
Fig. 3.

2D and 3D presentations of interactions between Riboflavin and SARS-CoV-2 main protease.
Fig. 4.

2D and 3D presentations of interactions between Phylloquinone oxide and SARS-CoV-2 main protease.
Table 3.
Detailed information of chemistry of the selected vitamins and their mode interaction with SARS-CoV-2 main protease.
| Name | Distance | Category | Types | From | Chemical Role | To | Chemical Role |
|---|---|---|---|---|---|---|---|
| Folacin protein complex | |||||||
| A:THR24:OG1 - :Folacin:O24 | 2.98648 | Hydrogen Bond | Conventional Hydrogen Bond | A:THR24:OG1 | H-Donor | :Folacin:O24 | H-Acceptor |
| A:THR25:OG1 - :Folacin:O24 | 3.03082 | A:THR25:OG1 | :Folacin:O24 | ||||
| A:THR26:N - :Folacin:O16 | 3.01864 | A:THR26:N | :Folacin:O16 | ||||
| A:THR45:OG1 - :Folacin:O23 | 2.78028 | A:THR45:OG1 | :Folacin:O23 | ||||
| A:SER46:N - :Folacin:O23 | 3.03409 | A:SER46:N | :Folacin:O23 | ||||
| A:SER46:OG - :Folacin:O23 | 2.84247 | A:SER46:OG | :Folacin:O23 | ||||
| A:SER144:N - :Folacin:O5 | 3.27256 | A:SER144:N | :Folacin:O5 | ||||
| A:SER144:OG - :Folacin:O5 | 2.9634 | A:SER144:OG | :Folacin:O5 | ||||
| A:SER144:OG - :Folacin:N3 | 3.23905 | A:SER144:OG | :Folacin:N3 | ||||
| A:CYS145:N - :Folacin:O5 | 3.21622 | A:CYS145:N | :Folacin:O5 | ||||
| A:CYS145:SG - :Folacin:N7 | 3.35245 | A:CYS145:SG | :Folacin:N7 | ||||
| A:CYS145:SG - :Folacin:O5 | 3.33679 | A:CYS145:SG | :Folacin:O5 | ||||
| A:GLU166:N - :Folacin:N33 | 3.30538 | A:GLU166:N | :Folacin:N33 | ||||
| :Folacin:O5 - A:LEU141:O | 2.99553 | :Folacin:O5 | A:LEU141:O | ||||
| :Folacin:N1 - A:PHE140:O | 3.18167 | :Folacin:N1 | A:PHE140:O | ||||
| :Folacin:N1 - A:GLU166:OE2 | 3.17136 | :Folacin:N1 | A:GLU166:OE2 | ||||
| :Folacin - A:CYS145 | 5.07158 | Hydrophobic | Pi-Alkyl | :Folacin | Pi-Orbitals | A:CYS145 | Alkyl |
| :Folacin - A:CYS145 | 5.27906 | :Folacin | A:CYS145 | ||||
| Riboflavin protein complex | |||||||
| A:SER144:OG - :Riboflavin:O6 | 2.98399 | Hydrogen Bond | Conventional Hydrogen Bond | A:SER144:OG | H-Donor | :Riboflavin:O6 | H-Acceptor |
| :Riboflavin:O3 - A:GLU166:O | 3.26207 | :Riboflavin:O3 | A:GLU166:O | ||||
| :Riboflavin:O5 - A:HIS163:NE2 | 2.98949 | :Riboflavin:O5 | A:HIS163:NE2 | ||||
| :Riboflavin:O6 - A:LEU141:O | 3.18437 | :Riboflavin:O6 | A:LEU141:O | ||||
| A:MET165:SD - :Riboflavin:O2 | 3.31579 | Other | Sulfur-X | A:MET165:SD | Sulfur | :Riboflavin:O2 | O,N,S |
| A:HIS41 - :Riboflavin | 4.70797 | Hydrophobic | Pi-Pi T-shaped | A:HIS41 | Pi-Orbitals | :Riboflavin | Pi-Orbitals |
| A:HIS41 - :Riboflavin | 5.25721 | A:HIS41 | :Riboflavin | Pi-Orbitals | |||
| :Riboflavin - A:MET49 | 5.01167 | Pi-Alkyl | :Riboflavin | A:MET49 | Alkyl | ||
| :Riboflavin - A:MET165 | 4.90586 | :Riboflavin | A:MET165 | ||||
| Phylloquinone oxide protein complex | |||||||
| A:GLY143:N - :Phylloquinone oxide:O2 | 2.94657 | Hydrogen Bond | Conventional Hydrogen Bond | A:GLY143:N | H-Donor | :Phylloquinone oxide:O2 | H-Acceptor |
| A:SER144:N - :Phylloquinone oxide:O2 | 3.04349 | A:SER144:N | :Phylloquinone oxide:O2 | ||||
| A:CYS145:N - :Phylloquinone oxide:O2 | 3.08497 | A:CYS145:N | :Phylloquinone oxide:O2 | ||||
| A:MET165 - :Phylloquinone oxide | 4.77648 | Hydrophobic | Alkyl | A:MET165 | Alkyl | :Phylloquinone oxide | Alkyl |
| :Phylloquinone oxide - A:LEU27 | 5.4606 | Pi-Alkyl | :Phylloquinone oxide | Pi-Orbitals | A:LEU27 | ||
| :Phylloquinone oxide - A:CYS145 | 4.63063 | A:CYS145 | |||||
The interaction results of Folacin, Riboflavin and Phylloquinone oxide in the SARS-CoV-2 main protease (Fig. 2, Fig. 3, Fig. 4, and Table 3) show Hydrogen bond interaction, π-alkyl, Sulfure-X, π-π T-shaped and Alkyl interactions.
3.2. Molecular dynamic
The molecular dynamics simulations of (Phylloquinone oxide, Riboflavin, and Folacin) – protein complexes with SARS-CoV-2 main protease, were performed for 100 ns. The trajectory was performed in terms of root mean square deviation (RMSD), root mean square fluctuations (RMSF), the radius of gyration (Rog), the center of mass distance (COM) between the protein and (Phylloquinone oxide, Riboflavin and Folacin) templates and the number of H-bonds, dynamic cross correlation matrix analysis (DCCM), and MMPBSA [47] ligand-protein binding energy.
Simply root-mean-square deviation (RMSD) calculated for (Phylloquinone oxide, Riboflavin, and Folacin) – protein complexes based on ‘C-alpha’ atoms using gromacs program as shown in Fig. 5 . The mean RMSD values are: (a) Phylloquinone oxide-protein complex = 0.26 ± 0.04 nm; (b) Riboflavin-protein complex = 0.28 ± 0.04 nm; and (c) Folacin-protein complex = 0.30 ± 0.05 nm. RMSD shows the structure remained stable throughout simulation time. However, fluctuations are observed throughout the 100ns of simulation which indicate local conformational changes. The Folacin-protein complex shows the highest RMSD values with higher standard deviation as compared to the other two complexes. Also, (RMSD) calculated for all three ligands based on respective ligand's atoms and the mean RMSD values were: (a)* Phylloquinone oxide = 0.22 ± 0.04 nm; (b)* Riboflavin = 0.09 ± 0.01 nm; and (c)* Folacin = 0.14 ± 0.03 nm as shown in Fig. 5. RMSD shows that Phylloquinone oxide and Folacin are quite flexible in protein binding site while the Riboflavin is comparatively rigid. The higher fluctuations in Phylloquinone oxide and Folacin indicate that they may not fit very well in to the binding site.
Fig. 5.

RMSD for (a) Phylloquinone oxide-protein complex; (b) Riboflavin-protein complex; and (c) folacin-protein complex based on ‘C-alpha’ atoms. RMSD for (a)* Phylloquinone oxide-protein complex; (b)* Riboflavin-protein complex; and (c)* Folacin-protein complex based on respective ligand's atoms.
Root Mean Square Fluctuations (RMSF) calculated for all three complexes as shown in Fig. 6 . (a) Phylloquinone oxide; (b) Riboflavin and (c) Folacin based on ‘C-alpha’ atoms using gromacs program. Overall, the fluctuation pattern is same for all complexes. However, the C terminals are comparatively more flexible in all complexes; being the highest in Folacin (c). The higher N-terminal fluctuation also support fluctuations in RMSD values.
Fig. 6.

RMSF calculated for (a) Phylloquinone oxide; (b) Riboflavin and (c) Folacin based on ‘C-alpha’ atoms.
Radius of gyration (Rog) calculated for all three complexes based on ‘C-alpha’ atoms using gromacs program as shown in Fig. 7 . The mean Rog values for (a) Phylloquinone oxide; (b) Riboflavin and (c) Folacin protein complexes are 2.23 ± 0.01 nm, 2.24 ± 0.01 nm and 2.23 ± 0.02 nm, respectively. The fluctuation within 0.05 nm Rog value during the MD simulation time indicates a slight change in the compactness of protein. These fluctuations in Rog are more likely due to the freely moving N-terminal of the proteins.
Fig. 7.

Rog calculated (a) Phylloquinone oxide = 2.23 ± 0.01 nm; (b) Riboflavin = 2.24 ± 0.01 nm and (c) Folacin 2.23 ± 0.02 nm.
3.3. Lipinski's rule and ADMET prediction
The Lipinski's rule including logP, number of hydrogen bonds acceptor, number hydrogen bonds donor, number of rotatable bonds, and molecular weight were shown in Table 4.
Table 4.
Lipinski's role of studied compounds.
| Compound |
Property |
Lipinski Violations |
|||||
|---|---|---|---|---|---|---|---|
| Log P | H-bond Acceptor | H-bond Donor | Rotatable bonds | Molecular weight g/mol | |||
| Rule | <4.15 | ≤10 | <5 | <10 | ≤500 | ≤1 | |
| N° | Name | ||||||
| 1 | Folacin | -0.69 | 10 | 6 | 10 | 441.40 | 2 |
| 2 | Riboflavin | -0.54 | 8 | 5 | 5 | 376.36 | 0 |
| 3 | Phylloquinone oxide | 2.42 | 13 | 6 | 27 | 906.08 | 3 |
The ADMET prediction was used in this study to calculate the pharmacokinetics parameters of three compounds (Tables 5 and 6).
Table 5.
In silico ADME properties of selected compounds.
| Folacin | Riboflavin | Phylloquinone oxide | |
|---|---|---|---|
| AlogP98_value | 0.189600 | -0,4209 | 9,1128 |
| AMolRef | 110,9088 | 94,2542 | 141,6875 |
| BBB | 0.0490799 | 0.0459537 | 10,9441 |
| Buffer_solubility_mg_L | 953,596 | 6812,13 | 0.000887422 |
| Caco2 | 17,003 | 17,8008 | 54,9688 |
| CYP_2C19_inhibition | No | No | Inhibitor |
| CYP_2C9_inhibition | Inhibitor | No | Inhibitor |
| CYP_2D6_inhibition | No | No | No |
| CYP_2D6_substrate | No | No | No |
| CYP_3A4_inhibition | Inhibitor | Inhibitor | Inhibitor |
| CYP_3A4_substrate | Weakly | Weakly | Substrate |
| HIA | 23,337322 | 43,932172 | 97,568445 |
| MDCK | 4,83621 | 4,65534 | 67.2502* |
| Pgp_inhibition | Non | Non | Inhibitor |
| Plasma_Protein_Binding | 48,514267 | 38,924672 | 100 |
| Pure_water_solubility_mg_L | 5,53254 | 760,174 | 1.73297e-005 |
| Skin_Permeability | -4,69446 | -5,03441 | -0.682794* |
| Solvation_Free_Energy | -46.120000** | -32.990000** | -5,4 |
BBB: in vivo blood-brain barrier penetration (C.brain/C.blood), Buffer_solubility: Water solubility in buffer system (SK atomic types, mg/L), HIA: Human intestinal absorption (HIA, %); Pgp_inhibition: in vitro P-glecoprotein inhibition, SK logD in pH 7.4 (SK atomic types), SK logP (SK atomic types).
Table 6.
In silico Toxicity of selected compounds.
| Folacin | Riboflavin | Phylloquinone oxide | |
|---|---|---|---|
| algae_at | 0.0299479 | 0.0791428 | 0.000499828 |
| Ames_test | mutagen | mutagen | non-mutagen |
| Carcino_Mouse | negative | negative | positive |
| Carcino_Rat | negative | negative | positive |
| daphnia_at | 0.372889 | 0.880183 | 0.000999859 |
| hERG_inhibition | high_risk | low_risk | medium_risk |
| medaka_at | 0.296093 | 1,25728 | 2.3438e-006 |
| minnow_at | 0.291013 | 2,6361 | 3.93629e-007 |
| TA100_10RLI | negative | positive | negative |
| TA100_NA | negative | negative | negative |
| TA1535_10RLI | negative | negative | negative |
| TA1535_NA | positive | negative | negative |
4. Discussions
4.1. Docking study
The results of docking study are shown in Table 2; the best energies of interaction with SARS-CoV-2 main protease (pdb code 6LU7) (lowest energy level) are observed for compounds; Folacin (Vitamin B9), Riboflavin (Vitamin B2), Phylloquinone oxide (Vitamin K1 oxide) and Ergocalciferol (Vitamin D2), which indicates that these molecules could be interesting for the antiviral treatment of COVID-19 [48].
These result are supported by other results, Vitamin B, Vitamin K, and Vitamin D are suggested against SARS-CoV-2 in many research papers [21], [22], [23], [24], [25], [26], in addition, these vitamins indicate a large variety of therapeutic mechanism ofaction [49], according to their properties in boosting immunity and maintaining the neurological system such as vitamin B group against HIV disease [50]. In cardiovascular diseases vitamin E was proved effective [51]. Vitamin K was reported to have an inhibitory effect on HIV [52]. Several studies support the immunomodulatory properties of vitamin D and its important role in the maintenance of immune homeostasis [53]. Additionally, vitamin D was reported to be proper candidates in improving body immunity towards COVID-19 [54], as low levels of these supplements increase viral infections with bovine coronavirus in cattle [19]. So, the evaluation of in vitro activity against COVID-19 of these molecules could be interesting, and it would be much cheaper and simpler than the other drugs as a therapeutic option to be tested.
4.2. Docking validation protocol
To validate the accuracy of the docking procedure; re-docking of the Co-Crystallized ligand was applied. The superimposed view between the docked ligand conformation and the co-crystallized ligand (inhibitor N3) is clarified in Fig. 8 .
Fig. 8.

Overlay of default conformation (green colored) on docked conformation (purple colored) of the co-crystallized ligand validating docking protocol.
As observed in Fig. 8, the inhibitor N3 (co-crystallized ligand) within the active cavity of SARS-CoV-2 in a similar position and interacted with similar residues, compared to that observed for the docked ligand conformation (RMSD = 0.88 Å, binding energy −7.5 kcal/mol). Thus, the docking process in this study was successfully validated.
It could be seen from Fig. 9 that the bonds are presented by Alkyl and/or π-Alkyl interactions with Met 167, Leu 167, Ala 191 and Pro 168, Met 49 and His 41 residues, Conventional Hydrogen Bonds interactions with Thr 190, Gln 189, His 163 Glu 166, His 164, Gly 143 and Phe 140 residues, amide-π Staked interactions with Leu 141 residue and Carbon Hydrogen Bond with Met 165 and His 172 residues.
Fig. 9.

Interactions between inhibitor N3 (co-crystallized ligand) and SARS-CoV-2 main protease.
The selected molecules with the best binding energy present the interactions with the same residues. Thus, these vitamins could have a potent inhibition of SARS-CoV-2 main protease (Fig. 2, Fig. 3, Fig. 4, and Table 3 show Hydrogen bond interaction, π-alkyl, Sulfure-X, π-π T-shaped and Alkyl interactions).
Folacin (binding energy − 7.9 kcal mol−1) seems to accommodate in a polar binding pocket forming a key hydrogen bond interactions with Thr24, Thr25 and Thr26 residue from S1’ pocket and Phe140, Leu141 and Glu166 residues from S1 pocket (Fig. 2 and Table 3). One of the hydroxyl substituent on core flavone moiety forms a hydrogen bond with Ser144, a residue present adjacent to S1 pocket.
Riboflavin (binding energy − 7.7 kcal mol−1) accommodates in S2 pocket where the pi–pi T-shaped and pi-alkyl interactions were observed with His41, Met 49 and Met165 residues respectively. Riboflavin also showed the hydrogen bonds with Ser144, Glu166, His163 and Leu141 residues (S1 pocket), and Sulfur-X interaction with Met165 (Fig. 3 and Table 3).
Phylloquinone oxide (binding energy − 7.1 kcal mol−1) showed the hydrogen bond with Ser144 residue (S1 pocket), pi-Alkyl interactions with Leu27 and Cys145 residues and Alkyl interaction with Met165 residue (S2 pocket) as seen in Fig. 4 and Table 3.
4.3. Molecular dynamic study
Figs. (S1–S3) show the total number of hydrogen bonds formed between ligand and protein during 100ns of simulation time. Fig. S4 shows the mean numbers of H-bonds are: (a) Phylloquinone oxide-protein = 0.24 ± 0.45; (b) Riboflavin = 3.87 ± 1.22; and (c) Folacin = 0.38 ± 0.98. As expected, the Phylloquinone oxide exhibits small number of H-bonds with protein. Folacin does not form a good interaction with protein as indicated in (c) graphs. Riboflavin exhibits the highest number of Hydrogen bonds and forms consistent interactions with protein. Also, the values of average Center-of-Mass Distance (COM) between ligand and protein during 100ns of simulation timeprove both Phylloquinone oxide and riboflavin exhibit close interaction with protein while Folacin does not bind with protein as indicated in (c) graph Fig. S5. The mean distances values are: (a) Phylloquinone oxide-protein = 2.18 ± 0.11; (b) Riboflavin-protein = 2.02 ± 0.15; and (c) Folacin-protein = 3.90 ± 1.01.
Principal component analysis of molecular dynamics simulations is a popular method for accounting for essential system dynamics over a low dimensional free energy landscape. Using Cartesian coordinates, the translation and global rotation must first be removed from the trajectory. As the rotation depends via the moment of inertia on the structure of the molecule, this separation is only simple for relatively rigid systems [55]. Principal component analysis of all 3 complexes (a) Phylloquinone oxide; (b) Riboflavin and (c) Folacin) calculated from Bio3D program of R. All three pictures capture 53% (a), 56.6% (b), and 68.8% (c) of structural variance in protein as shown in Fig. S6. And the dynamic cross correlation matrix analysis (DCCM) for protein Residue dynamic cross correlated motions obtained for all 3 complexes calculated from Bio3D program of R. Colors varying from red to white to Blue indicate intensity of correlated motion, where blue colors indicate positive correlation, white shows no correlation and pink color indicates negative correlated motions between residues as shown in Fig. S7.
Kinetic energy, Potential energy, Total energy, Total pressure, and Temperature for (a) Phylloquinone oxide; (b) Riboflavin and (c) Folacin system during 100 ns of Molecular Dynamic simulation as obtained from Gromacs 2020.4 energy file are shown in Figs. S8–S12, and the data of MM-PBSA calculations of binding free energy of selected complexes are summarized in Table 7.
Table 7.
MM-PBSA calculations of binding free energy of selected complexes.
| Average | Std. Dev | Std. Err. of Mean | ||
|---|---|---|---|---|
| Riboflavin protein complex | VDWAALS | -36.772 | 6.116 | 0.612 |
| Electrostatic energy | -2.327 | 3.700 | 0.370 | |
| Electrostatic solvation energy | 24.896 | 7.300 | 0.730 | |
| Enpolar | -4.056 | 0.478 | 0.048 | |
| ΔGgas | -39.099 | 7.178 | 0.718 | |
| ΔGsolv | 20.841 | 6.977 | 0.698 | |
| ΔGtotal | -18.258 | 4.525 | 0.453 | |
| Folacin protein complex | VDWAALS | -35.99 | 5.69 | 0.57 |
| Electrostatic energy | -9.71 | 9.48 | 0.95 | |
| Electrostatic solvation energy | 47.70 | 8.14 | 0.81 | |
| Enpolar | -3.58 | 0.28 | 0.03 | |
| ΔGgas | -45.71 | 11.62 | 1.16 | |
| ΔGsolv | 44.13 | 8.05 | 0.81 | |
| ΔGtotal | -1.58 | 8.53 | 0.85 | |
| Phylloquinone_oxide protein complex | VDWAALS | -5.52 | 9.75 | 0.98 |
| Electrostatic energy | -3.49 | 9.76 | 0.98 | |
| Electrostatic solvation energy | 7.91 | 16.02 | 1.60 | |
| Enpolar | -0.71 | 1.18 | 0.12 | |
| ΔGgas | -9.02 | 16.93 | 1.69 | |
| ΔGsolv | 7.20 | 14.96 | 1.50 | |
| ΔGtotal | -1.82 | 4.64 | 0.46 |
Among the selected complexes, Mpro- Riboflavin complex showed the lowest binding free energy followed by Mpro- Folacin, and Mpro- Phylloquinone_oxide complex.
All results indicated that Phylloquinone oxide and Folacin not fit very well in to the binding site of SARS-CoV-2 main protease because Phylloquinone oxide forms small number of H-bonds and Folacin does not form a good interaction with SARS-CoV-2 main protease. Whereas Riboflavin (Vitamin B2) exhibits the highest number of Hydrogen bonds and forms consistent interactions with protein and this consistent with some recent studies [56,57].
4.4. Lipinski's rule and ADMET prediction
As observed in Table 4 Riboflavin respect all the conditions mentioned in Lipinski's rule, Folacin has two Lipinski violations, and Phylloquinone oxide has three Lipinski violations.
The selected molecules have low BBB, except Phylloquinone oxide. Riboflavin and Phylloquinone oxide molecules have different and acceptable absorption percentages (44% and 98%, respectively). For metabolism, all these molecules could be inhibitors for the cytochromes CYP450_3A4 (Table 5).
Examination of the preADMET toxicity screening results for the selected compounds are shown in Table 6. The results revealed that Phylloquinone oxide showed negative AMES mutagenicity to all salmonella strains. Riboflavin and Folacin showed positive AMES mutagenicity to only one of salmonella strains (TA100_10RLI and TA1535_NA, respectively). Moreover, Riboflavin and Folacin showed the highest toxicity against algae, daphnia, and fish. Additionally, Riboflavin and Folacin showed negative carcinogenicity for either rat, and negative carcinogenicity for mice. Also, Riboflavin have a low risk for hERG_ inhibition, but Folacin and Phylloquinone oxide have high and medium risk, respectively (Table 6).
From these results, we can conclude that Riboflavin is the structure with best binding energy in the binding site of the studied enzyme, this molecule respect the conditions mentioned in Lipinski's rule and have acceptable ADMET proprieties; so, this compounds could have more potent antiviral treatment of COVID-19 than the studied compounds.
A molecule classified as a medicinal drug or helps treat a specific disease must reach the protein or enzyme that causes that disease in sufficient concentration and give a high biological binding and response. Drug discovery and development are highly dependent on assessing absorption, distribution, metabolism, and excretion (ADME) characteristics. Discovering coronavirus protease inhibitors targeting Mpro posed a significant challenge due to poor pharmacokinetic properties of peptidomimetic/macromolecular compounds and poor inhibitory potency of non-peptidomimetic low molecular weight compounds [58], [59], [60]. Since the beginning of the COVID-19 pandemic, WHO endorsed treatment with vitamins or used them to reduce the side effects of COVID-19 among the treatment protocols. Because of their superior inhibitory potency and selectivity, vitamins inhibit SARS-CoV-2 Mpro more effectively than other compounds [21], [22], [23], [24], [25], [26].
In conclusion, for COVID-19 patients, vitamins could be an exciting treatment option because they can help alleviate pain and strengthen the immune system. Clinical evaluation results (published in other literature) proved it after following the approved treatment protocol by WHO. In addition, it is cheaper than other drugs as a treatment option to be tested.
5. Conclusion
In this study, a docking study of 28 vitamins and their derivatives with the active site of SARS-Cov-2 main protease was carried out. The result indicates that the structures with the best binding energy in the binding site of the studied enzyme are observed for compounds; Folacin (Vitamin B9), Riboflavin (Vitamin B2), and Phylloquinone oxide (Vitamin K1 oxide), which indicates that these molecules could be interesting for the antiviral treatment of COVID-19. These three structures with the best binding energy in the binding site of the studied enzyme are test by MD simulation; to explain their stability in the studied enzyme. The Folacin-protein complex shows the highest RMSD values with higher standard deviation as compared to the other two. RMSD shows that Phylloquinone oxide and Folacin are quite flexible in protein binding site while the Riboflavin is comparatively rigid. The higher fluctuations in Phylloquinone oxide and Folacin indicate that they may not fit very well in to the binding site. The Phylloquinone oxide exhibits small number of H-bonds with protein and Folacin does not form a good interaction with SARS-CoV-2 main protease. Riboflavin (Vitamin B2) exhibits the highest number of Hydrogen bonds and forms consistent interactions with SARS-CoV-2 main protease. Additionally, this molecule respect the conditions mentioned in Lipinski's rule and have acceptable ADMET proprieties; In conclude Vitamin B2 can play another therapeutic and nutritional role, which can help COVID-19 patients to overcome pain and strengthen the immune system, the clinical evaluation against COVID-19 of this vitamin could be interesting, and it would be much cheaper and simpler than the other drugs as a therapeutic option to be tested.
Credit author statement
Assia Belhassan and Samir Chtita designed the structure and carried out the molecular docking calculations. Hanane Zaki carried out ADMET properties calculations. Marwa Alaqarbeh, Firas Almohtaseb and Nada Alsakhen carried out the theoretical calculations (MD). Mohammed Bouachrine and Tahar Lakhlifi wrote up the article.
Supporting information
Reponse to Reviewers.
Supplementary material.
Declaration of Competing Interest
Conflict of interest declared none.
All authors read and approved the final version of manuscript.
Acknowledgements
We are grateful to the “Association Marocaine des ChimistesThéoriciens” (AMCT) for its their pertinent help concerning the programs.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2022.132652.
Appendix. Supplementary materials
References
- 1.https://www.worldometers.info/coronavirus/access on (2/11/ 2021), (n.d.).
- 2.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robson B. Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput. Biol. Med. 2020;119 doi: 10.1016/j.compbiomed.2020.103670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pooladanda V., Thatikonda S., Godugu C. The current understanding and potential therapeutic options to combat COVID-19. Life Sci. 2020 doi: 10.1016/j.lfs.2020.117765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.R. Hatada, K. Okuwaki, Y. Mochizuki, K. Fukuzawa, Y. Komeiji, Y. Okiyama, S. Tanaka, Fragment molecular orbital based interaction analyses on COVID-19 main protease - inhibitor N3 complex (PDB ID:6LU7), (2020). 10.26434/chemrxiv.11988120.v1. [DOI] [PubMed]
- 6.Hui D.S., Zumla A. Severe acute respiratory syndrome: historical, epidemiologic, and clinical features. Infect. Dis. Clin. 2019;33:869–889. doi: 10.1016/j.idc.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., Qin C. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11:59. doi: 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caly L., Druce J.D., Catton M.G., Jans D.A., Wagstaff K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020;178 doi: 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Tawfiq J.A., Al-Homoud A.H., Memish Z.A. Remdesivir as a possible therapeutic option for the COVID-19. Travel Med. Infect. Dis. 2020;34 doi: 10.1016/j.tmaid.2020.101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radke J.B., Kingery J.M., Maakestad J., Krasowski M.D. Diagnostic pitfalls and laboratory test interference after hydroxychloroquine intoxication: a case report. Toxicol. Rep. 2019;6:1040–1046. doi: 10.1016/j.toxrep.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsang A.C., Ahmadi S., Hamilton J., Gao J., Virgili G., Coupland S.G., Gottlieb C.C. The diagnostic utility of multifocal electroretinography in detecting chloroquine and hydroxychloroquine retinal toxicity. Am. J. Ophthalmol. 2019;206:132–139. doi: 10.1016/j.ajo.2019.04.025. [DOI] [PubMed] [Google Scholar]
- 12.Sahraei Z., Shabani M., Shokouhi S., Saffaei A. Aminoquinolines against coronavirus disease 2019 (COVID-19): chloroquine or hydroxychloroquine. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh A.K., Singh A., Shaikh A., Singh R., Misra A. Chloroquine and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: a systematic search and a narrative review with a special reference to India and other developing countries. Diabetes Metab. Syndr. Clin. Res. Rev. 2020;14:241–246. doi: 10.1016/j.dsx.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tahir ul Qamar M., Alqahtani S.M., Alamri M.A., Chen L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020;10:313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnet M., Lagier J.C., Raoult D., Khelaifia S. Bacterial culture through selective and non-selective conditions: the evolution of culture media in clinical microbiology. New Microbes New Infect. 2020;34 doi: 10.1016/j.nmni.2019.100622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semba R.D. Vitamin A and immunity to viral, bacterial and protozoan infections. Proc. Nutr. Soc. 1999;58:719–727. doi: 10.1017/s0029665199000944. [DOI] [PubMed] [Google Scholar]
- 17.Keil S.D., Bowen R., Marschner S. Inactivation of M iddle E ast respiratory syndrome coronavirus (MERS-C o V) in plasma products using a riboflavin-based and ultraviolet light-based photochemical treatment. Transfusion. 2016;56:2948–2952. doi: 10.1111/trf.13860. (Paris) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atherton J.G., Kratzing C.C., Fisher A. The effect of ascorbic acid on infection of chick-embryo ciliated tracheal organ cultures by coronavirus. Arch. Virol. 1978;56:195–199. doi: 10.1007/BF01317848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nonnecke B.J., McGill J.L., Ridpath J.F., Sacco R.E., Lippolis J.D., Reinhardt T.A. Acute phase response elicited by experimental bovine diarrhea virus (BVDV) infection is associated with decreased vitamin D and E status of vitamin-replete preruminant calves. J. Dairy Sci. 2014;97:5566–5579. doi: 10.3168/jds.2014-8293. [DOI] [PubMed] [Google Scholar]
- 20.Beck M.A., Kolbeck P.C., Rohr L.H., Shi Q., Morris V.C., Levander O.A. Vitamin E deficiency intensifies the myocardial injury of coxsackievirus B3 infection of mice. J. Nutr. 1994;124:345–358. doi: 10.1093/jn/124.3.345. [DOI] [PubMed] [Google Scholar]
- 21.Kandeel M., Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251 doi: 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pawar A.Y. Combating devastating COVID -19 by drug repurposing. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., Xie B., Hashimoto K. Current status of potential therapeutic candidates for the COVID-19 crisis. Brain Behav. Immun. 2020 doi: 10.1016/j.bbi.2020.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruta H., He H. PAK1-blockers: potential therapeutics against COVID-19. Med. Drug Discov. 2020;6 doi: 10.1016/j.medidd.2020.100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silberstein M. Vitamin D: a simpler alternative to tocilizumab for trial in COVID-19? Med. Hypotheses. 2020;140 doi: 10.1016/j.mehy.2020.109767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khodadadi E., Maroufi P., Khodadadi E., Esposito I., Ganbarov K., Espsoito S., Yousefi M., Zeinalzadeh E., Kafil H.S. Study of combining virtual screening and antiviral treatments of the Sars-CoV-2 (Covid-19) Microb. Pathog. 2020 doi: 10.1016/j.micpath.2020.104241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aanouz I., Belhassan A., El Khatabi K., Lakhlifi T., El Idrissi M., Bouachrine M. Moroccan medicinal plants as inhibitors of COVID-19: computational investigations. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1758790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pence H.E., Williams A. ChemSpider: an online chemical information resource. J. Chem. Educ. 2010;87:1123–1124. doi: 10.1021/ed100697w. [DOI] [Google Scholar]
- 29.Tripos Inc., St. Louis, MO, USA, SYBYL-X 2.0, (n.d.). http://www.tripois.com.
- 30.Clark M., Cramer R.D., Van Opdenbosch N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989;10:982–1012. [Google Scholar]
- 31.Purcell W.P., Singer J.A. A brief review and table of semiempirical parameters used in the Hueckel molecular orbital method. J. Chem. Eng. Data. 1967;12:235–246. [Google Scholar]
- 32.A. Belhassan, S. Chtita, T. Lakhlifi, M. Bouachrine, 2D-and 3D-QSRR Studies of Linear Retention Indices for Volatile Alkylated Phenols, in: Dysfunct. Olfactory Syst., IntechOpen, 2019.
- 33.Zaki H., Belhassan A., Benlyas M., Lakhlifi T., Bouachrine M. New Dehydroabietic Acid (DHA) derivatives with anticancer activity against HepG2 cancer cell lines as a potential Drug targeting EGFR kinase Domain. CoMFA study and virtual ligand-based screening. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1759452. [DOI] [PubMed] [Google Scholar]
- 34.Protein Data Bank PDB, http://www.rcsb.org. (n.d.).
- 35.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunter C.A., Lawson K.R., Perkins J., Urch C.J. Aromatic interactions. J. Chem. Soc. Perkin Trans. 2001:651–669. doi: 10.1039/B008495F. 2. 0. [DOI] [Google Scholar]
- 37.Dassault systèmes BIOVIA discovery studio modeling environment, release 2017 dassault systèmes, (2016). http://accelrys.com/products/collaborative-science/biovia-discovery-studio/.
- 38.A. Belhassan, H. Zaki, A. Aouidate, M. Benlyas, T. Lakhlifi, M. Bouachrine, Interactions between (4Z)-hex-4-en-1-ol and 2-methylbutyl 2-methylbutanoate with olfactory receptors using computational methods, Moroc. J. Chem. 7 (n.d.) 7–1.
- 39.Belhassan A., Zaki H., Chtita S., Benlyas M., Lakhlifi T., Bouachrine M. Study of interactions between odorant molecules and the hOR1G1 olfactory receptor by molecular modeling. Egypt. J. Ear Nose Throat Allied Sci. 2017;18:257–265. [Google Scholar]
- 40.Hakmi M., Bouricha E.M., Kandoussi I., El Harti J., Ibrahimi A. Repurposing of known anti-virals as potential inhibitors for SARS-CoV-2 main protease using molecular docking analysis. Bioinformation. 2020;16:301. doi: 10.6026/97320630016301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.C.B. Jalkute, S.H. Barage, Identification of angiotensin converting enzyme inhibitor : an in silico perspective, (2015) 107–115. 10.1007/s10989-014-9434-8.
- 42.Daina A., Michielin O., Zoete V. SwissADME : a free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Nat. Publ. Group. 2017:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.PreADMET | Prediction of ADME/Tox, PreADMET Predict. ADMETox. (n.d.). https://preadmet.bmdrc.kr/ (accessed June 14, 2020).
- 44.Y.H. Zhao, M.H. Abraham, J. Le, A. Hersey, C.N. Luscombe, G. Beck, B. Sherborne, I. Cooper, Rate-limited steps of human oral absorption and QSAR studies, 19 (2002) 1446–1457. [DOI] [PubMed]
- 45.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 46.Sawant S., Patil R., Khawate M., Zambre V., Shilimkar V., Jagtap S. Computational assessment of select antiviral phytochemicals as potential SARS-Cov-2 main protease inhibitors: molecular dynamics guided ensemble docking and extended molecular dynamics. Silico Pharmacol. 2021;9:44. doi: 10.1007/s40203-021-00107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller B.R., McGee T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA. py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 48.Belhassan A., et al. Molecular docking analysis of N-substituted oseltamivir derivatives with the SARS-Cov-2 main protease. Bioinformation. 2020;16:404–410. doi: 10.6026/97320630016404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.A. Linani, K. Benarous, M. Yousfi, Novel structural mechanism of glutathione as a potential peptide inhibitor to the main protease (Mpro): CoviD-19 treatment, Molecular Docking and SAR Study, (2020). 10.26434/chemrxiv.12153021.v1. [DOI] [PMC free article] [PubMed]
- 50.L.T. Miller, Vitamin B group and the immune system, (1998).
- 51.Han J., Zhao C., Cai J., Liang Y. Comparative efficacy of vitamin supplements on prevention of major cardiovascular disease: Systematic review with network meta-analysis. Complement. Ther. Clin. Pract. 2020 doi: 10.1016/j.ctcp.2020.101142. [DOI] [PubMed] [Google Scholar]
- 52.Min B.S., Miyashiro H., Hattori M. Inhibitory effects of quinones on RNase H activity associated with HIV-1 reverse transcriptase. Phytother. Res. 2002;16:57–62. doi: 10.1002/ptr.808. [DOI] [PubMed] [Google Scholar]
- 53.Fabbri A., Infante M., Ricordi C. Editorial–Vitamin D status: a key modulator of innate immunity and natural defense from acute viral respiratory infections. Eur. Rev. Med. Pharmacol. Sci. 2020;24:4048–4052. doi: 10.26355/eurrev_202004_20876. [DOI] [PubMed] [Google Scholar]
- 54.Wang L., Wang Y., Ye D., Liu Q. A review of the 2019 novel coronavirus (COVID-19) based on current evidence. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sittel F., Jain A., Stock G. Principal component analysis of molecular dynamics: on the use of Cartesian vs. internal coordinates. J. Chem. Phys. 2014;141 doi: 10.1063/1.4885338. [DOI] [PubMed] [Google Scholar]
- 56.Jovic T.H., Ali S.R., Ibrahim N., Jessop Z.M., Tarassoli S.P., Dobbs T.D., Holford P., Thornton C.A., Whitaker I.S. Could vitamins help in the fight against COVID-19? Nutrients. 2020;12:2550. doi: 10.3390/nu12092550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shakoor H., Feehan J., Mikkelsen K., Al Dhaheri A.S., Ali H.I., Platat C., Ismail L.C., Stojanovska L., Apostolopoulos V. Be well: a potential role for vitamin B in COVID-19. Maturitas. 2021;144:108–111. doi: 10.1016/j.maturitas.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 59.Drag M., Salvesen G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amin Sk.A., Banerjee S., Ghosh K., Gayen S., Jha T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021;29 doi: 10.1016/j.bmc.2020.115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.