

Abstract
Parathyroid hormone (PTH) signaling downstream of the PTH 1 receptor (Pth1r) results in both bone anabolic and catabolic actions by mechanisms not yet fully understood. In this study, we show that Pth1r signaling upregulates the expression of several components of the Notch pathway and that Notch signals contribute to the catabolic actions of PTH in bone. We found that constitutive genetic activation of PTH receptor signaling in osteocytes (caPth1rOt) or treatment with PTH daily increased the expression of several Notch ligands/receptors in bone. In contrast, sustained elevation of endogenous PTH did not change Notch components expression. Deletion of the PTH receptor or Sclerostin overexpression in osteocytes abolished Notch increases by PTH. Further, deleting the canonical Notch transcription factor Rbpjk in osteocytes decreased bone mass and increased resorption and Rankl expression in caPth1rOt mice. Moreover, pharmacological bone-targeted Notch inhibition potentiated the bone mass gain induced by intermittent PTH by reducing bone resorption and preserving bone formation. Thus, Notch activation lies downstream of anabolic signaling driven by PTH actions in osteocytes, and Notch pharmacological inhibition maximizes the bone anabolic effects of PTH.
Keywords: Notch, osteocytes, PTH, osteoclasts, resorption, bone, osteoblasts
Introduction
Osteoporosis is a common skeletal disorder characterized by compromised bone strength and increased risk of fractures. Osteoporosis typically occurs due to a negative balance in which bone resorption exceeds new bone formation (1, 2). Anti-resorptive agents are frequently used to treat osteoporosis due to their ability to decrease bone resorption and increase/maintain bone mass (3, 4). However, anti-resorptives do not stimulate bone formation and thus have limited capacity to restore bone architecture (3, 4). Teriparatide (parathyroid hormone (PTH) 1-34) is a PTH receptor 1 (Pth1r) agonist approved by the FDA for the treatment of osteoporosis (5). PTH1-34 robustly stimulates osteoblast differentiation and new bone formation (6, 7). However, PTH 1-34 also increases Receptor activator of nuclear factor kappa-B ligand (Rankl) expression and stimulates osteoclast-mediated bone resorption, progressively diminishing the gain in bone mass (6, 7). Understanding the molecular mechanisms responsible for the anabolic and catabolic effects of PTH on bone has become imperative to identify new therapeutic targets to potentiate or prolong the bone gain induced by anabolic regimens of this hormone.
The Notch signaling pathway regulates several aspects of bone remodeling by mediating cell-to-cell communication between adjacent bone cells (8). Notch signaling is activated through cell-surface receptors (Notch 1-4) that transduce signals from Notch ligands that belong to the Delta-like and Jagged (Jag) families. PTH increases the expression of the Notch ligand Jag1 in osteoblastic cells, and its deletion from osteoprogenitors accelerates the bone gain induced by daily PTH injections (9, 10). These findings suggest that Jag1 inhibits bone anabolism induced by PTH by controlling the transition of osteoprogenitors to mature osteoblasts. Yet, whether PTH regulates other Notch signaling components (ligands, receptors, target genes) and whether Notch signals in osteocytes contribute to the skeletal actions of PTH is unknown.
Herein, we report that daily administration of PTH upregulates the expression of several Notch components and activates Notch signaling in bone. Notch activation by PTH is mediated by direct actions on osteocytes downstream of the Pth1r and requires downregulation of the gene Sost, which encodes the Wnt antagonist and inhibitor of bone formation Sclerostin. Further, PTH activation of Notch signaling in osteocytes restrains bone resorption enabling full bone gain, as demonstrated by genetic approaches. Moreover, pharmacologic inhibition of Notch signaling with a bone-targeted Notch inhibitor decreases bone resorption and preserves bone formation induced by daily PTH, thus maximizing the bone gain caused by the hormone. Together, these findings provide novel evidence supporting the role of Notch signals as mediators of the pro-resorptive skeletal effects of PTH through actions on osteocytes and identify the Notch pathway as a new target to potentiate the bone gain induced by anabolic regimens of PTH.
Methods
Animals.
Wild type (WT) and heterozygous Dmp1-8kb-caPth1r mice (caPth1rOt) littermate female mice (RRID: MGI:5657312) were generated as shown before (11). WT and caPth1rOt mice with conditional deletion of the Rbpjk in osteocytes (RbpjkΔOt) were generated by a 2-step breeding strategy using caPth1rOt, Rbpjkfl/fl (12), and Dmp1-8kb-Cre mice (13). caPth1rOt mice were crossed with Rbpjkfl/fl, and the resulting caPth1rOt; Rbpjkfl/wt were subsequently crossed with Rbpjkfl/wt;Dmp1-8kb-Cre−/+ mice. Littermates of the following genotypes were used for the studies: Rbpjkf/fl, Rbpjkfl/fl;Dmp1-8kb-Cre−/+, Rbpjkf/fl;caPth1rOt, and Rbpjkfl/fl;Dmp1-8kb-Cre−/+;caPth1rOt (caPth1rOt;RbpjkΔOt). Mice with conditional deletion of Pth1r in osteocytes (Pth1rΔOt) were generated by crossing Pth1rfl/fl mice with DMP1-8kb-Cre mice, as reported before (14). The generation of transgenic mice overexpressing the human SOST cDNA (Dmp1-SOST) and littermate controls was published previously (14). Four-month-old WT, Pth1rΔOt, and Dmp1-SOST mice were injected daily with PTH 1-34 (100ng/g, Bachem, Torrance, CA, USA) or vehicle for 28 days, as previously reported (14). Four-month-old C57BL/6 female mice (Harlan, Indianapolis, IN, USA) received daily injections of PTH 1-34 (100 ng/g/day) or vehicle and i.p injections of BT-GSI (5 mg/kg, 3x/wk) or saline for 14 days. Mice were fed a regular diet (Harlan), received water ad libitum, and maintained a 12-h light/dark cycle. To test the effect of continuous elevation of endogenous PTH, 4-month-old C57BL/6 female mice (Harlan) were fed a diet containing 0.01% calcium and 0.4% phosphorous (low calcium diet, TD.95027, Harlan), and control mice were fed a diet containing 0.6% calcium and 0.4% phosphorous (normal calcium diet, TD.97191, Harlan). For all the studies, mice were randomized to the experimental groups based on total BMD before treatment.
Justification of the use of Dmp1 promoter and interpretation of the data.
For all these studies, we used the Dmp1-8kb promoter to direct our genetic interventions to osteocytes. Although Dmp1 promoters remain the most widely used genetic approach to target osteocytes, we acknowledge they can also target other cell populations in bone, including mature osteoblasts potentially destined to become osteocytes (15). Yet, distinct skeletal phenotypes are found when the same gene(s) are deleted using DMP1-Cre drivers vs. Cre promoters targeting osteoblasts and their progeny (i.e., osteocalcin and collagen) (11, 16–23). Thus, we interpret that osteocytes are the main targets of the DMP1-8kb promoter and, therefore, major contributors to the skeletal phenotypes described in this manuscript.
Generation of a bone-targeted notch inhibitor (BT-GSI).
BT-GSI was generated following methods as described before (24). Briefly, the bone-targeted GSI (BT-GSI) is a conjugate formed by 1) a modified, less active bisphosphonate moiety with high bone affinity designed to direct the conjugate to the skeleton, 2) a pH-sensitive labile linker that binds the bone targeting moiety (BT) to the cargo, and 3) the cargo, the small molecule GSI-XII that inhibits Notch signaling by preventing the cleavage of the Notch intracellular domain of Notch receptors. BT-GSI was dissolved in DMSO and diluted in PBS for in vivo administration. We reported before that at equimolar doses, BT-GSI decreased Notch signaling in the bone, whereas no effects were seen with BT (24). Further, we showed that the BT alone exhibits some residual anti-resorptive activity, but its ability to inhibit local bone resorption and reduce osteoclast is lower than BT-GSI (24). Because our main goal was to examine the contribution of the Notch signaling to PTH’s skeletal effects, we selected BT-GSI for these experiments.
Analysis of the skeletal phenotype.
BMD measurements and micro-CT analysis were performed as previously described (22, 25). For micro-computed tomography (μCT) analysis, bones were scanned at 6-μm resolution (Skyscan 1172; SkyScan, Aartselaar, Belgium), and measurements were done 60 μm away from the growth plate. Static and dynamic bone histomorphometric analyses were performed using the OsteoMeasure High-Resolution Digital Video System (OsteoMetrics, Decatur, GA, USA) as previously described (22). Analyses were performed in the cancellous bone of an 800-μm region of the vertebrae, starting 200 μm below the growth plate.
Serum biochemistry.
Procollagen type 1 N-terminal propeptide (P1NP), osteocalcin (OCN), and C-telopeptide fragments of type I collagen (CTX) were measured using enzyme-linked immunosorbent assays (Biomedical Technologies, Stoughton, MA, USA, and Immunodiagnostic Systems and Immunodiagnostic Systems, Fountain Hill, AZ, USA), as previously published (22, 25).
Quantitative PCR.
Bones were harvested, the periosteum was removed, and total RNA from whole bones (including cortical, cancellous, and bone marrow) was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. qPCR was performed using primer-probe sets from Applied Biosystems or Roche Applied Science (Indianapolis, IN, USA) as previously reported (22, 25). For experiments using intermittent PTH, mRNA samples were collected 3h after treatment administration. Relative mRNA expression levels were normalized to the housekeeping gene ribosomal protein S2 using the ΔCt method (26). Fold changes are shown in all figures and were calculated by dividing the treatment values by the control/vehicle values.
Statistical analysis.
Data were analyzed using SigmaStat (SPSS Science, Chicago, IL, USA). All values are reported as mean ± standard deviation (SD). Differences between group means were evaluated using t-Test or two-way ANOVA (with genotype and treatment as independent variables), followed by multiple pairwise comparisons using the Tukey method. p=0.05 values were considered statistically significant. Samples sizes were estimated based on previous studies and results obtained with the in vivo models employed in this manuscript. Two independent investigators performed all these analyses in a blinded manner.
Institutional approval.
All animal procedures were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine, and animal care was carried out following institutional guidelines.
Results
Activation of anabolic Pth1r signaling in osteocytes increases Notch signaling in bone.
Bones from female mice with genetic constitutive active Pth1r in osteocytes (caPth1rOt) displayed elevated mRNA expression of components of the Notch pathway, including Notch2 and 4, ligands Jag1, Delta-like (Dll) 1 and 4, and the Notch target genes Hairy and enhancer of split-1 (Hes1) and Hairy/enhancer-of-split related with YRPW motif-like protein (HeyL) compared to control littermates (WT) (Fig. 1). The mRNA expression of Notch-related genes was also elevated in bones from control Pth1rfl/fl mice receiving intermittent injections of PTH (iPTH) (Fig. 2) but absent in mice with conditional deletion of Pth1r in osteocytes (Pth1rΔOt). In contrast to these observations made in models of hormonal or genetic Pth1r activation leading to bone gain, the catabolic skeletal actions of chronic elevation of endogenous PTH induced by a calcium-deficient diet (27) did not change the expression of Notch genes in bone (Suppl. Fig. 1). Together, these results support the notion that PTH signals leading to bone gain increase the expression of Notch components and activate Notch signaling in bone through mechanisms downstream of the Pth1r in osteocytes.
Figure 1. Osteocytic Pth1r signaling increases Notch signaling in bone.
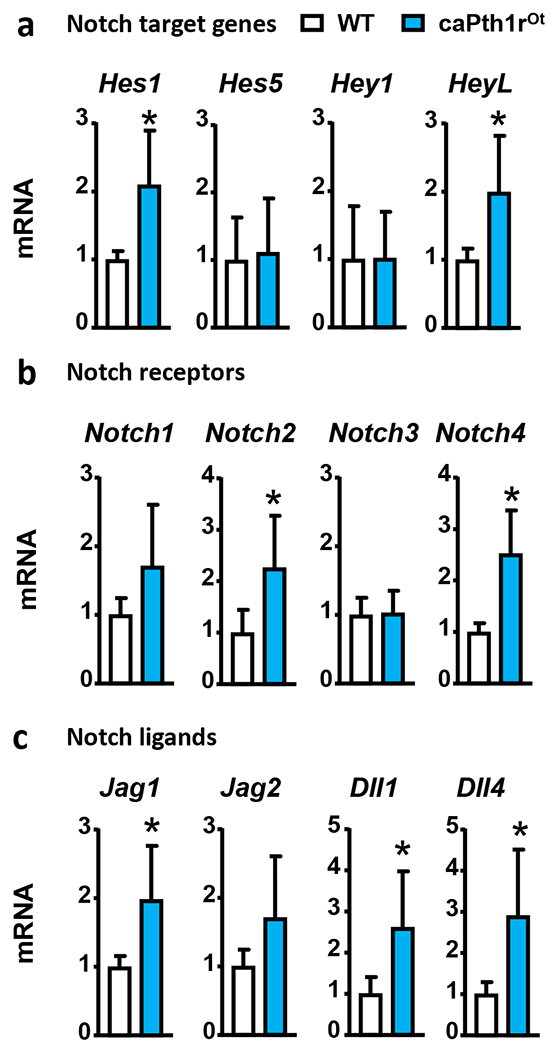
mRNA gene expression of Notch target genes (a), Notch receptors (b), and Notch ligands (c) in bones (whole tibia) from WT and caPth1rOt littermate female mice. n=10-11/group; *p≤0.05 vs. WT mice by t-Test. Bars represent means ± SD.
Figure 2. Osteocytic Pthr1r signaling mediates the increases in Notch components induced by intermittent PTH.
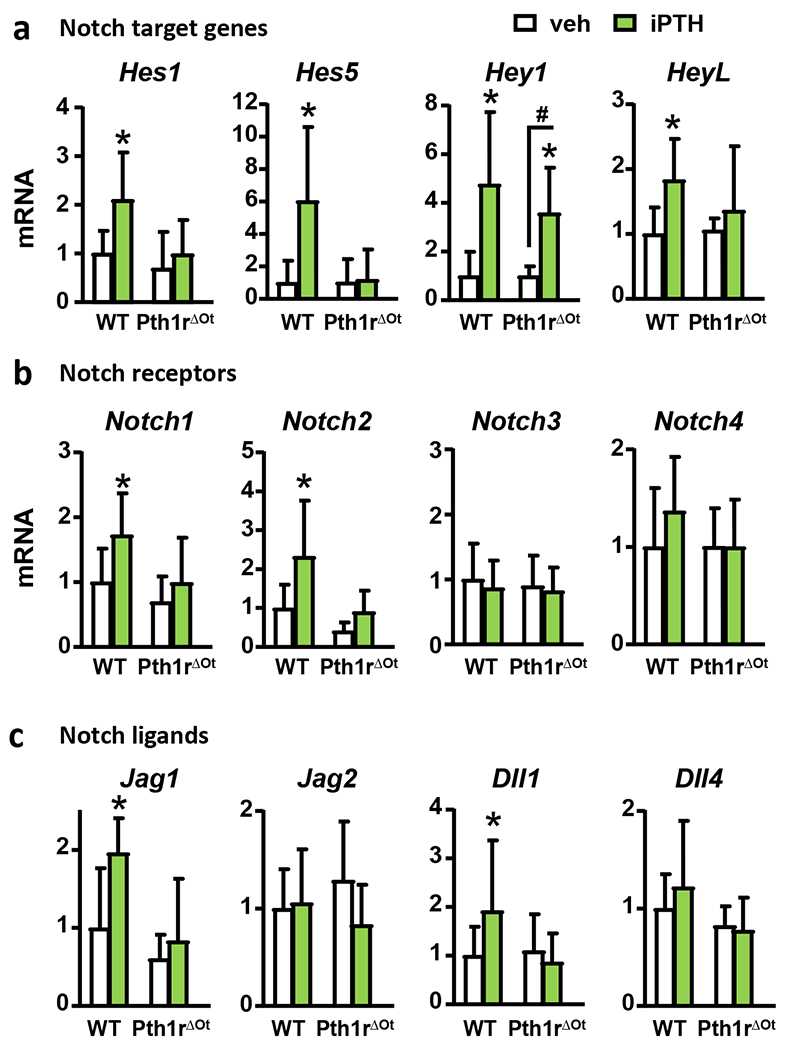
mRNA gene expression of Notch target genes (a), Notch receptors (b), and Notch ligands (c) in bones (whole tibia) from wt and Pth1rΔOt littermate female mice receiving vehicle (veh) or iPTH daily injections. n=8-10/group; *p≤0.05 vs. WT (veh) mice and #p≤0.05 vs. other groups as indicated by bars by two-way ANOVA. Bars represent means ± SD.
Sclerostin downregulation is required for PTH-induced activation of Notch signaling.
PTH downregulates Sost/Sclerostin expression and activates Wnt/β-catenin signaling in bone (28, 29), a pathway that can influence Notch signaling (21, 22). Thus, we sought to determine whether the Notch activation induced by Pth1r signaling in osteocytes is mediated by Sclerostin downregulation. Consistent with the data displayed in Fig. 2, control mice receiving iPTH injections exhibited increased mRNA expression of Notch components (Fig. 3). In contrast, the elevation of Notch components was not detected in mice overexpressing Sclerostin in osteocytes. These results suggest that PTH signaling in osteocytes activates Notch through a mechanism downstream of Sost/Sclerostin downregulation.
Figure 3. Sclerostin mediates the increase in Notch signaling induced by intermittent PTH.
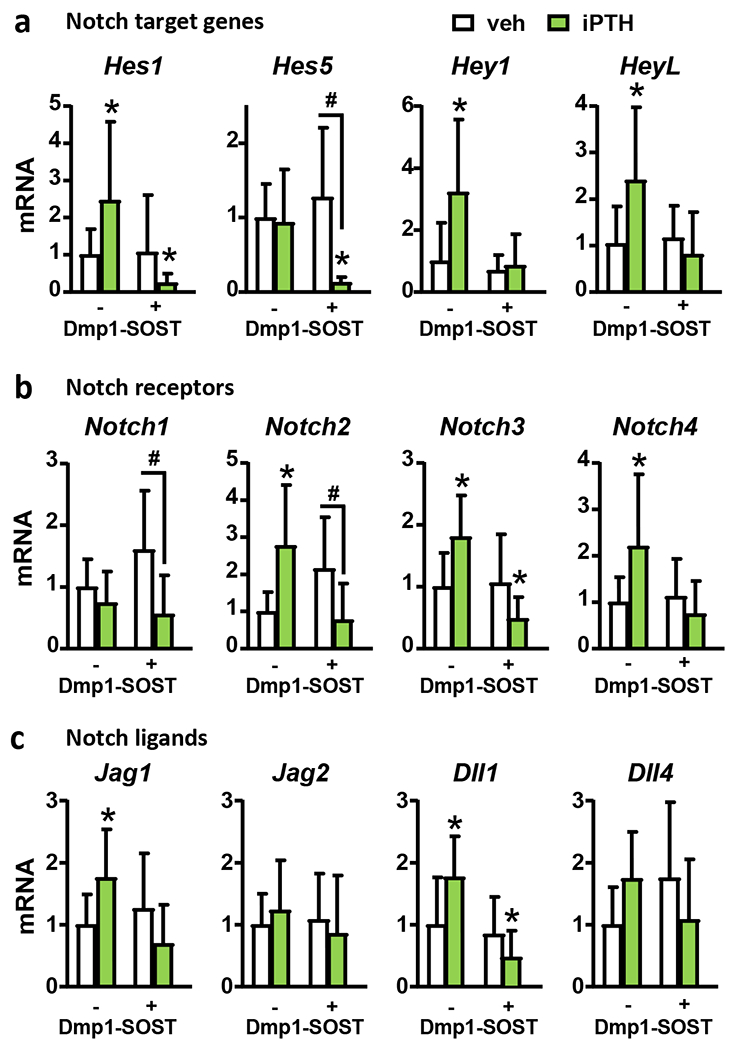
mRNA gene expression of Notch target genes (a), Notch receptors (b), and Notch ligands (c) in bones (vertebrae, L4) from wt and Dmp1-SOST littermate female mice receiving vehicle (veh) or iPTH daily injections. n=9-10/group; *p≤0.05 vs. WT (veh) mice and #p≤0.05 vs. other groups as indicated by bars by two-way ANOVA. Bars represent means ± SD.
Genetic inhibition of canonical Notch signaling in osteocytes increases bone resorption and decreases the bone gain induced by PTH.
Next, we dissected the contribution of canonical Notch signaling in osteocytes to the bone gain seen in caPth1rOt mice (11, 30). We genetically deleted the canonical Notch transcription factor recombination signal binding protein for immunoglobulin kappa J region (Rbpjk) in osteocytes in both wt and caPth1rOt mice. RbpjkΔOt and caPth1rOt;RbpjkΔOt mice exhibited a ~50% reduction in Rbpjk mRNA expression in bone compared to bones from their respective control littermates and Rbpjk deletion blunted in caPth1rOt mice the increased Hes1 and Jag1 seen in bone tissue (Suppl. Fig. 2). At 6 months of age, no differences in BMD, cortical or cancellous bone volume, or bone formation or resorption markers were found between wt and RbpjkΔOt mice (Fig. 4), suggesting that osteocytic canonical Notch signaling is dispensable for physiological bone homeostasis. In contrast, caPth1rOt mice with genetic deletion of Rbpjk (caPth1rOt;RbpjkΔOt) exhibited 5% lower total BMD (Fig. 4A), decreased femoral cortical bone area (−9%), and reduced L5 cancellous BV/TV (−16%), and trabecular thickness (−30%) compared to control caPth1rOt mice (Fig. 4B–C). Moreover, caPth1rOt;RbpjkΔOt mice displayed increased serum CTX (40%) and higher Rankl/Opg (osteoprotegerin) ratio in bone than caPth1rOt mice (Fig. 4D–E). In contrast, the increase in circulating P1NP levels and Sost downregulation exhibited by caPth1rOt mice remained unchanged in caPth1rOt;RbpjkΔOt mice (Fig. 4 D–F). Altogether, these findings support the notion that osteocytic Pth1r signaling activates canonical Notch in bone to restrain bone resorption and facilitate bone gain.
Figure 4. Genetic deletion of Rbpjk in osteocytes decreases PTH-induced bone gain by increasing resorption.
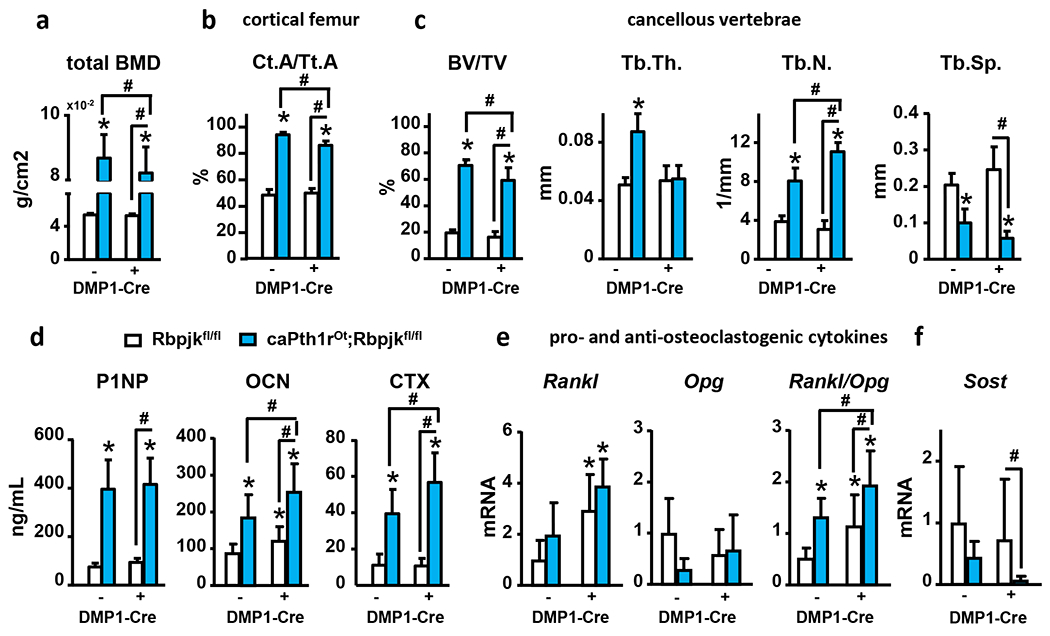
Analysis of (a) total bone mineral density (tBMD), (b) femoral cortical area over tissue area (Ct.A/Tt.A), (c) vertebral cancellous bone volume over tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb. Sp), (d) serum levels of P1NP, osteocalcin (OCN) and CTX, and (e-f) mRNA gene expression (calvaria) of pro- and anti-osteoclastogenic genes and Sost in bones from WT and caPth1rOt littermate female mice with/without genetic deletion of Rbpjk in osteocytes. n=8-6/group; *p≤0.05 vs. WT (Rbpjkfl/fl) mice and #p≤0.05 vs. other groups as indicated by bars by two-way ANOVA. Bars represent means ± SD.
Pharmacologic bone-targeted inhibition of Notch signaling inhibits resorption and potentiates the bone gain induced by iPTH.
We recently developed a bone-targeted Notch inhibitor (BT-GSI) and showed it inhibits Notch signaling in the bone/bone marrow environment and exerts potent anti-resorptive effects in bone (24). Here, we examined the impact of BT-GSI on the skeletal effects of iPTH. iPTH increased Hes1/5/7 and Hey1/L mRNA expression in bone, and co-administration of BT-GSI decreased it to control levels (Fig. 5A). iPTH increased total (7%) and femoral BMD (13%), and preserved spinal BMD (0%). Co-administration of BT-GSI potentiated BMD increases induced by iPTH at all bone sites (10, 17, and 7%, respectively) (Fig. 5B). Further, co-administration of BT-GSI increased by 25% the gain in cancellous BV/TV (L4 and distal femur) and trabecular thickness induced by iPTH, but it did not alter iPTH-induced increases in cortical bone area (8%) (Fig. 5C–E). Co-administration of BT-GSI decreased serum P1NP (−30%); however, it preserved the increased bone formation rate (20%) and osteoblast surface (20%) induced by iPTH in cancellous bone (Fig. 6A–C). Moreover, co-treatment with BT-GSI did not change the iPTH-induced increases in alkaline phosphatase (Alpl), Runt-related transcription factor 2 (Runx2), osteocalcin (Bglap), and Wnt target genes mRNA expression or Sost downregulation in bone (Fig. 6D–E). iPTH increased serum CTX (30%) and osteoclast surface (25%), whereas co-treatment with BT-GSI reduced these indexes to values below those seen in control mice receiving vehicle (Fig. 7A–B). The increased Rankl/Opg expression ratio (1.5-fold) in bone induced by iPTH remained unchanged by BT-GSI, and no changes were found in Colony-stimulating factor 1 (M-Csf) expression. (Fig. 7C) These results demonstrate that bone-targeted inhibition of Notch in the frame of anabolic PTH signaling inhibits bone resorption and induces a superior bone gain compared to iPTH alone.
Figure 5. BT-GSI prevents PTH-induced Notch signaling activation, increases bone mass, and potentiates PTH-induced bone gain.
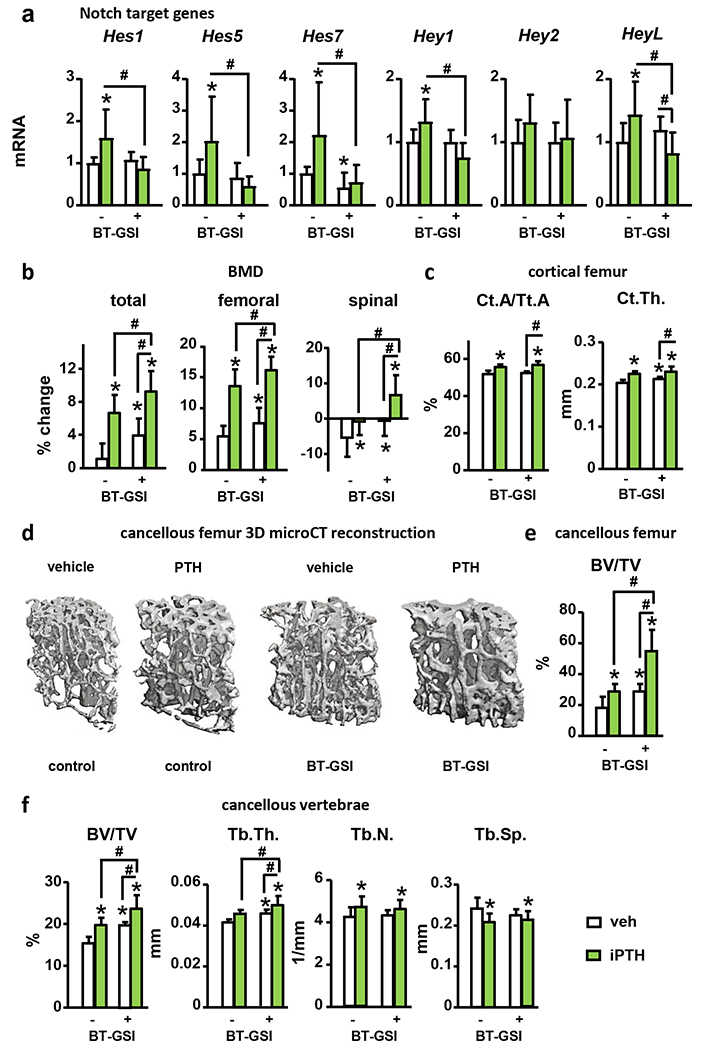
Analysis of (a) mRNA gene expression (vertebrae, L4) of Notch target genes in bone, (b) total, femoral and spinal bone mineral density (BMD), (c) femoral cortical area over tissue area (Ct.A/Tt.A.) and cortical thickness (Ct. Th.), (d) representative microCT 3D images of femoral cancellous bone, (e) femoral cancellous bone volume over tissue volume (BV/TV), and (f) vertebral cancellous BV/TV, trabecular thickness (Tb. Th.), trabecular number (Tb.N.), and trabecular separation (Tb. Sp.), in WT female mice receiving vehicle (veh) or iPTH daily injections with/without BT-GSI. n=10/group; *p≤0.05 vs. WT (veh) mice and #p≤0.05 vs. other groups as indicated by bars by two-way ANOVA. Bars represent means ± SD.
Figure 6. Bone-targeted Notch inhibition preserves the increase in osteoblasts number and bone formation induced by iPTH.
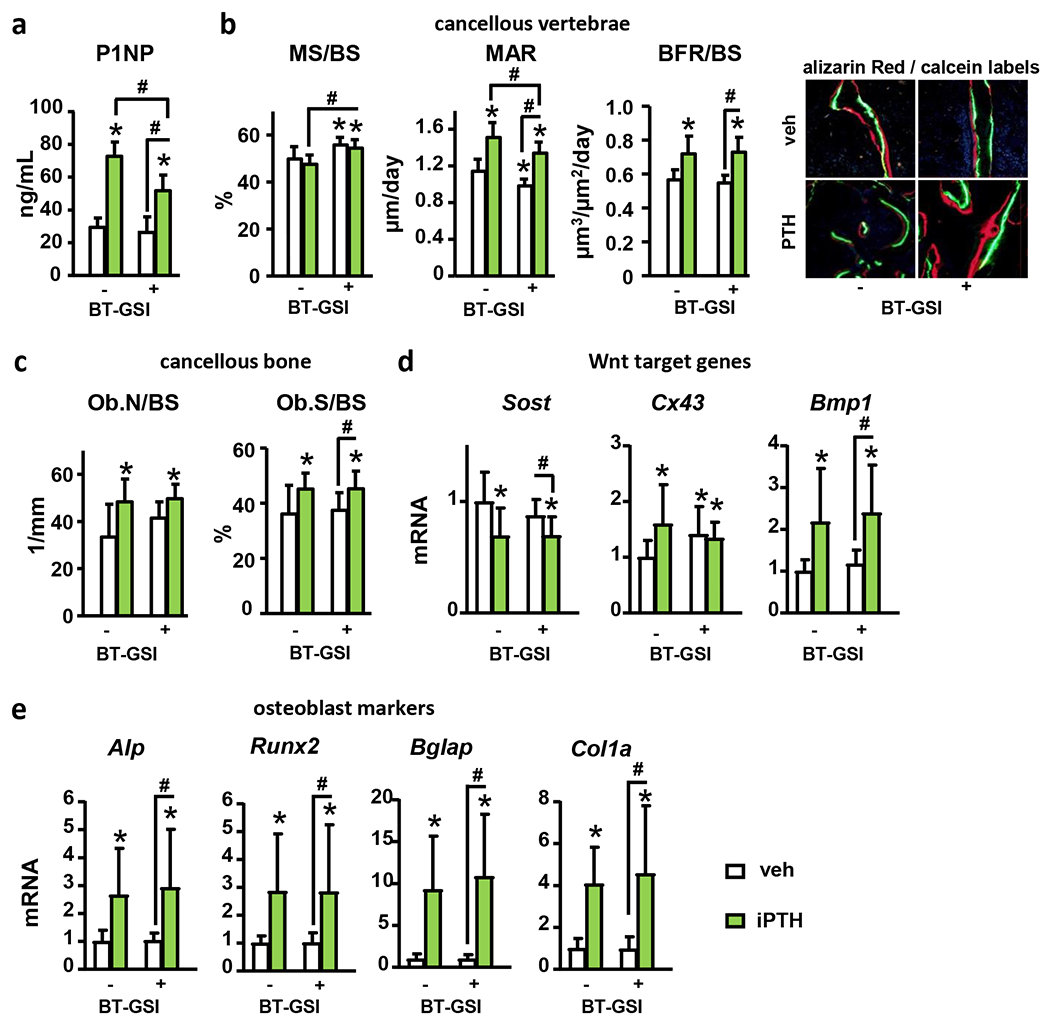
Analysis of (a) serum P1NP, (b) mineralizing surfaces over bone surface (MS/BS), mineral apposition rate (MAR), and bone formation rate (BFR/BS), (c) osteoblast number (Ob.N/BS) and surface (Ob.S/BS) per bone surface, and (d-e) mRNA gene expression (vertebrae, L4) of Wnt target genes and osteoblast markers in bones from WT female mice receiving vehicle (veh) or iPTH daily injections with/without BT-GSI. n=10/group; *p≤0.05 vs. WT (veh) mice and #p≤0.05 vs. other groups as indicated by bars by two-way ANOVA. Bars represent means ± SD.
Figure 7. BT-GSI-XII decreases iPTH induced bone resorption.
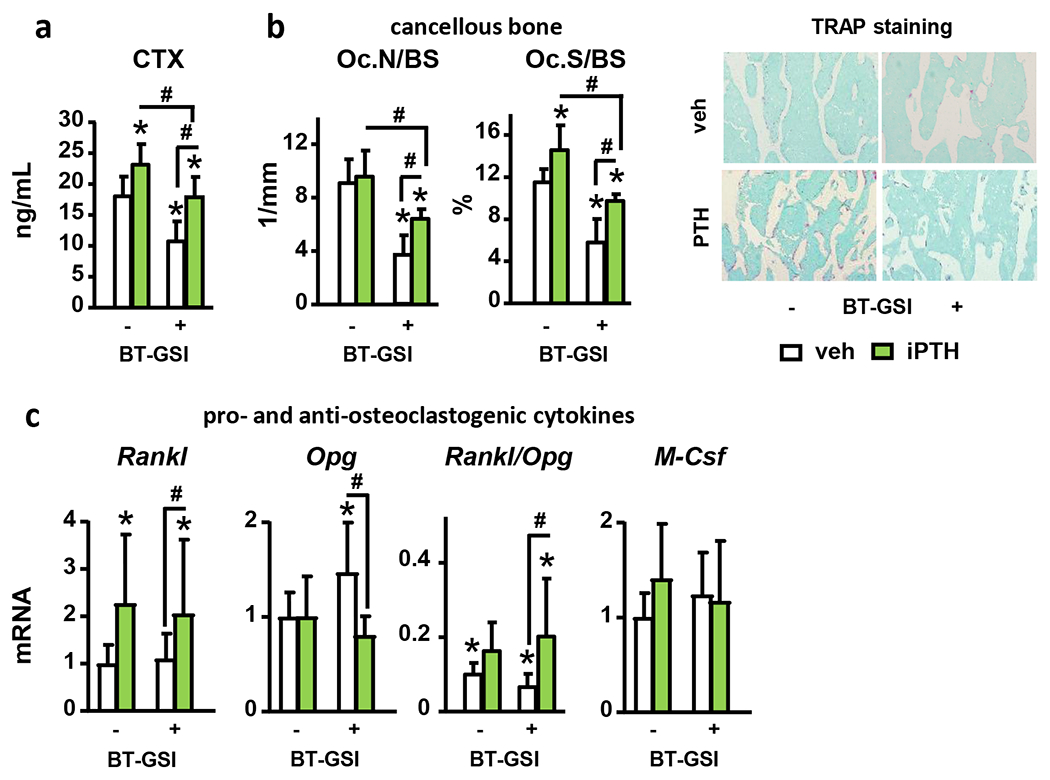
Analysis of (a) serum CTX, (b) osteoclast number (Oc.N/BS) and surface (Oc.S/BS) per bone surface, and (c) mRNA gene expression (vertebrae, L4) of pro- and anti-osteoclastogenic cytokines in bones from WT female mice receiving vehicle (veh) or iPTH daily injections with/without BT-GSI. Representative images of TRAP stained histological sections are shown. n=10/group; *p≤0.05 vs. WT (veh) mice and #p≤0.05 vs. other groups as indicated by bars by two-way ANOVA. Bars represent means ± SD.
Discussion
Activation of PTH receptor signaling by binding of the traditional ligand PTH or new PTH analogs is a major pathway leading to bone anabolism. Yet, the cellular and molecular mechanisms responsible for the skeletal effects of this pathway are not fully understood. Using a combination of genetic and pharmacologic approaches, we demonstrated in this study that anabolic PTH signals activate Notch signaling in bone and that Notch activation modulates the pro-resorptive effects of PTH in the skeleton. First, we found that genetic or pharmacologic activation of PTH receptor signaling in osteocytes, which leads to bone gain, upregulates the expression of several Notch components and Notch target genes in bone. Second, we showed that PTH requires the expression of Pthr1 in osteocytes and downregulation of the Wnt antagonist Sclerostin to activate Notch signaling in bone. Third, using a genetic approach, we found that osteocytic canonical Notch signals downstream of the transcription factor Rbpjk decrease bone resorption to favor bone gain in a mouse model of constitutive genetic activation of Pth1r signaling in osteocytes. Fourth, using a novel bone-targeted pharmacological Notch inhibitor, we demonstrated that Notch signaling can be regulated to favor the bone gain induced by daily injections of PTH. In concert, our findings identify Notch signals as mediators of the pro-resorptive actions triggered by anabolic PTH administration on the skeleton and show the potential of targeting the Notch pathway to maximize and preserve the bone gain induced by intermittent PTH.
Earlier work revealed that PTH upregulates the expression of Jag1 in osteoblasts (10). Our current results demonstrate that in addition to Jag1, PTH upregulates the expression of other Notch components in bone, particularly Notch2 and Dll1, which were consistently upregulated by Pth1r signaling in all our studies. Canalis and collaborators have shown that genetic activation of Notch2 signaling stimulates osteoclast diffrentation and resorption (31, 32). Notch4, which was increased in some of our experiments, is transiently increased during osteogenic differentiation of humans osteoblastic cells (33, 34).Further studies are needed to determine the specific contribution of Notch2 vs. other Notch receptors, or that of Dll1 vs. Jag1, to the skeletal actions of PTH. In addition, we show that the increases in Notch ligands and receptors lead to Notch activation, as PTH also elevated the expression of several Notch target genes in bone, particularly Hes1 that was elevated across all the experiments. It is important to note that the comparison of Notch components’ expression among experiments in our studies is limited by the use of different bones (calvaria vs. tibia vs. vertebrae), which could explain the differences among samples receiving similar treatments. The increases in Notch components found in our studies contrast with a recent report showing short treatment (6h) with PTH appears to downregulate the expression of Notch target genes in osteocytes (35). The length of the treatment (30 days vs. hours) and the model used (mice vs. bone organ cultures) may account for the differences between these two studies. Lastly, we found that chronic PTH elevation did not affect Notch-related genes in bone. These findings are consistent with the demonstration that expression of the Pthr1 in osteocytes is dispensable to reach bone loss with chronic PTH elevation, which instead requires the expression of interleukin-17 receptor in osteocytes (36, 37). Nevertheless, these pieces of evidence imply that intermittent PTH prompts different cellular/molecular actions leading to Notch activation compared to chronic elevation and demand further investigation.
We previously reported that osteocytes are essential for the skeletal responses to anabolic PTH (14, 38). This study shows that the genetic activation of Pth1r signaling in osteocytes is sufficient to activate Notch, and deletion of Pth1r in osteocytes prevents Notch activation by intermittent PTH. Taken together, these findings further support the notion that osteocytes are the bone cells responsible for integrating anabolic PTH signals in bone, leading to the activation of the Notch pathway. In addition to anabolic Pth1r signaling in osteocytes, we also demonstrate that downregulation of Sclerostin is required for PTH to activate Notch. We and others showed that activation of Wnt signaling in osteoblasts or osteocytes increases Notch signaling in bone, suggesting a crosstalk between these two pathways exists (21, 22). We also showed that activation of anabolic Pth1r signaling, either by genetic means or daily PTH injections, upregulates Wnt signaling in control mice (14, 27). Thus, PTH’s downregulation of Sclerostin may contribute to Notch activation by facilitating Wnt signaling upregulation. Future studies are needed to dissect the specific molecular mechanisms through which Sclerostin regulates Notch activation by PTH.
The Notch signaling pathway exerts critical roles in skeletal development, homeostasis, and disease, which are cell and context-dependent (8). We show here that canonical Notch signaling in osteocytes is dispensable for physiological bone homeostasis. Consistent with our findings, Canalis and colleagues reported the lack of a skeletal phenotype in mice with conditional deletion of Rbpjk in osteocytes (39). In contrast, we found that canonical Notch signaling in osteocytes contributes to the regulation of bone resorption and bone gain in response to Pth1r signaling activation. Our results show that canonical Notch in osteocytes limits the increases in Rankl/Opg ratio and bone resorption induced by PTH, suggesting that canonical Notch signaling is required for the full anabolic effects caused by constitutively active Pth1r signaling in osteocytes.
The study of the role of Notch in adult bone is challenging because the skeletal phenotypes result from combined developmental and postnatal effects. We bypassed Notch’s developmental effects and inhibited endoproteolysis downstream of all Notch receptors using a novel bone-targeted Notch inhibitor (24). We found that the main effect of Notch inhibition in adult bone is suppression of bone resorption, which increases bone mass. This observation is consistent with previous literature showing that Notch signals determine the differentiation and function of osteoclasts (40), and our in vitro studies showing that osteoclast differentiation is suppressed by Notch inhibitors (24). Although Notch also regulates the differentiation and function of osteoblast precursors and osteoblasts (8), pharmacological Notch inhibition with BT-GSI did not alter physiological bone formation or osteoblast number. Recently, Zaidi and colleagues employed a genetic approach to activate Notch in adult bone, but only in osteoblasts and osteocytes. Notch activation via Notch1 overexpression in mature osteoblastic cells resulted in high bone mass, but this effect was due to higher osteoblasts and bone formation (41). Yet, similar to our observation, Notch1 activation in adult bone also increased osteoclasts and bone resorption, supporting a direct link between Notch and bone resorption in adult bone. Collectively, these observations highlight the complexity of Notch signaling in adult bone and show that the bone phenotypes depend on the Notch components, the cell lineage, and the differentiation stage being targeted.
Co-administration of BT-GSI blunted Notch activation, potentiated the increases in bone mass, and preserved the elevated bone formation caused by intermittent PTH. Our results are consistent with clinical studies in humans showing that PTH exerts bone anabolic actions even when co-administered with anti-resorptives (3, 42, 43). These findings are also similar to our previous report showing co-administration of a KD014, a pharmacological inhibitor of matrix metalloproteinase 14/soluble Rankl, potentiates the PTH bone gain by inhibiting bone resorption (30). Together, these studies further support the notion that bone resorption limits the gain in cancellous bone induced by intermittent PTH in the early stages of the therapy. Yet, antiresorptive drugs can reduce the remodeling space and impair bone gain caused by PTH during the second year of treatment. It remains to be determined if long-term administration of BT-GSI causes a sustained inhibition of bone resorption without interfering with the anabolic effects of PTH.
Our genetic and pharmacologic interventions result in apparent disparate outcomes regarding the effects of Notch in PTH-induced bone resorption. Upon stimulation of Pth1r signaling, inhibition of canonical Notch in osteocytes by genetic means increases bone resorption, whereas suppression of Notch with BT-GSI decreases it. One possible explanation is that our genetic approach specifically inhibits canonical, Rbpjk-dependent Notch signaling. In contrast, BT-GSI can interfere with both canonical and non-canonical Notch, with the skeletal effects of the latter being poorly understood. Another potential difference is that our genetic approach targets only mature osteoblastic cells, while BT-GSI can suppress Notch in any cell present in the bone microenvironment, including cells of the osteoclastic lineage. Thus, the anti-resorptive effects and additional bone gain seen with co-administration of BT-GSI may be due to direct inhibition of Notch in osteoclasts. Although we reported that osteoclasts are more sensitive to GSI than to the modified bisphosphonate moiety (BT) (24), we cannot exclude the possibility that BT also contributed to the reduced bone resorption. Lastly, Pth1r signaling was activated by different means, genetic constitutive Pth1r overexpression vs. intermittent PTH administration, which both lead to net bone gain but could trigger different cellular/molecular events. Nevertheless, our findings reveal a previously unknown link between Notch and bone resorption in the context of anabolic Pth1r activation and show the potential of interfering with Notch signals to modulate the bone gain induced by PTH.
In summary, our in vivo studies and genetic and pharmacologic interventions revealed that Notch components are direct targets of anabolic PTH signaling in osteocytes and that the Notch pathway can be targeted to modulate the pro-resorptive effects of PTH and potentiate bone gain. These findings provide the basis to investigate combined therapeutic strategies using our novel bone-targeted Notch inhibitor to reduce bone resorption while simultaneously preserving bone anabolism. Given the cell and context-dependent nature of Notch signaling (44) and complexity of some bone disorders, we anticipate that the efficacy of BT-GSI in bone will have to be determined on a disease-specific basis (i.e., neuroendocrine disorders, primary and secondary osteoporosis).
Supplementary Material
Acknowledgments
We thank Matthew Olson, Meloney Cregor, and Dr. Amy Y. Sato for their assistance in tissue collection. This work was supported by the National Institutes of Health (R01-AR059357 to TB and R37-CA251763 to J.D.C.), the U.S. Department of Veteran Affairs (I01 BX002104 and IK6BX004596 to TB), and the Arkansas COBRE program (NIGMS P20GM125503 to J.D.C.).
List of abbreviations
- PTH
parathyroid hormone
- Pth1r
PTH 1 receptor
- caPth1rOt
mice with constitutive genetic activation of PTH receptor signaling in osteocytes
- Dmp1-SOST
mice overexpressing the human SOST cDNA
- RbpjkΔOt
mice with conditional deletion of the Rbpjk in osteocytes
- Pth1rΔOt
mice with conditional deletion of Pth1r in osteocytes
- GSI
gamma-secretase inhibitor
- BT
bone targeting moiety
- BT-GSI
bone-targeted GSI
- μCT
micro-computed tomography
- P1NP
procollagen type 1 N-terminal propeptide
- OCN
osteocalcin
- CTX
c-telopeptide fragments of type I collagen
- iPTH
intermittent injections of PTH
- BMD
bone mineral density
- BV/TV
bone volume over tissue volume
Footnotes
Conflict of interest statement
H.E. is an employee of BioVinc LLC. The authors have declared that no conflict of interest exists.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Manolagas SC (2018) The Quest for Osteoporosis Mechanisms and Rational Therapies: How Far We’ve Come, How Much Further We Need to Go. Journal of Bone and Mineral Research 33, 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armas LA, and Recker RR (2012) Pathophysiology of osteoporosis: new mechanistic insights. Endocrinol Metab Clin North Am 41, 475–486 [DOI] [PubMed] [Google Scholar]
- 3.Cosman F, Nieves JW, and Dempster DW (2017) Treatment Sequence Matters: Anabolic and Antiresorptive Therapy for Osteoporosis. J Bone Miner Res 32, 198–202 [DOI] [PubMed] [Google Scholar]
- 4.Reid IR (2015) Short-term and long-term effects of osteoporosis therapies. Nat Rev Endocrinol 11, 418–428 [DOI] [PubMed] [Google Scholar]
- 5.Martin TJ, Sims NA, and Seeman E (2021) Physiological and Pharmacological Roles of PTH and PTHrP in Bone Using Their Shared Receptor, PTH1R. Endocr Rev 42, 383–406 [DOI] [PubMed] [Google Scholar]
- 6.Cosman F (2005) Anabolic therapy for osteoporosis: parathyroid hormone. Curr Osteoporos Rep 3, 143–149 [DOI] [PubMed] [Google Scholar]
- 7.Jilka RL (2007) Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40, 1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canalis E (2018) Notch in skeletal physiology and disease. Osteoporos Int 29, 2611–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber JM, Forsythe SR, Christianson CA, Frisch BJ, Gigliotti BJ, Jordan CT, Milner LA, Guzman ML, and Calvi LM (2006) Parathyroid hormone stimulates expression of the Notch ligand Jagged1 in osteoblastic cells. Bone 39, 485–493 [DOI] [PubMed] [Google Scholar]
- 10.Lawal RA, Zhou X, Batey K, Hoffman CM, Georger MA, Radtke F, Hilton MJ, Xing L, Frisch BJ, and Calvi LM (2017) The Notch Ligand Jagged1 Regulates the Osteoblastic Lineage by Maintaining the Osteoprogenitor Pool. J Bone Miner Res 32, 1320–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, and Bellido T (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One 3, e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, Ikuta K, and Honjo T (2002) Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol 14, 637–645 [DOI] [PubMed] [Google Scholar]
- 13.Bivi N, Nelson MT, Faillace ME, Li J, Miller LM, and Plotkin LI (2012) Deletion of Cx43 from osteocytes results in defective bone material properties but does not decrease extrinsic strength in cortical bone. Calcif. Tissue Int 91, 215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgado-Calle J, Tu X, Pacheco-Costa R, McAndrews K, Edwards R, Pellegrini GG, Kuhlenschmidt K, Olivos N, Robling A, Peacock M, Plotkin LI, and Bellido T (2017) Control of Bone Anabolism in Response to Mechanical Loading and PTH by Distinct Mechanisms Downstream of the PTH Receptor. J Bone Miner Res 32, 522–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, Bonewald L, Manolagas SC, and O’Brien CA (2015) Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS. ONE 10, e0138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, Dedic C, Maeda A, Lotinun S, Baron R, and Pajevic PD (2013) Parathyroid Hormone (PTH)/PTH-related Peptide Type 1 Receptor (PPR) Signaling in Osteocytes Regulates Anabolic and Catabolic Skeletal Responses to PTH. Journal of Biological Chemistry 288, 20122–20134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu T, Xian L, Crane J, Wen C, Hilton M, Lu W, Newman P, and Cao X (2015) PTH receptor signaling in osteoblasts regulates endochondral vascularization in maintenance of postnatal growth plate. J Bone Miner Res 30, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, and Schipani E (2001) Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest 107, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, and Karsenty G (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8, 751–764 [DOI] [PubMed] [Google Scholar]
- 20.Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, and Williams BO (2005) Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem 280, 21162–21168 [DOI] [PubMed] [Google Scholar]
- 21.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, Brum A, Park D, Galili N, Mukherjee S, Teruya-Feldstein J, Raza A, Rabadan R, Berman E, and Kousteni S (2014) Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, Taketo MM, Burr DB, Plotkin LI, and Bellido T (2015) Osteocytes mediate the anabolic actions of canonical Wnt/b-catenin signaling in bone. Proc. Natl. Acad. Sci U. S. A 112, E478–E486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Feng J, Bao Q, Li A, Zhang B, Shen Y, Zhao Y, Guo Q, Jing J, Lin S, and Zong Z (2015) Adverse Effects of Osteocytic Constitutive Activation of ss-Catenin on Bone Strength and Bone Growth. J Bone Miner Res 30, 1184–1194 [DOI] [PubMed] [Google Scholar]
- 24.Sabol HM, Ferrari AJ, Adhikari M, Amorim T, McAndrews K, Anderson J, Vigolo M, Lehal R, Cregor M, Khan S, Cuevas PL, Helms JA, Kurihara N, Srinivasan V, Ebetino FH, Boeckman RK, Roodman GD, Bellido T, and Delgado-Calle J (2021) Targeting Notch inhibitors to the myeloma bone marrow niche decreases tumor growth and bone destruction without gut toxicity. Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-Awadh A, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI, and Bellido T (2014) Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 155, 2797–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, and Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 27.Delgado-Calle J, Hancock B, Likine EF, Sato AY, McAndrews K, Sanudo C, Bruzzaniti A, Riancho JA, Tonra JR, and Bellido T (2018) MMP14 is a novel target of PTH signaling in osteocytes that controls resorption by regulating soluble RANKL production. Faseb Journal 32, 2878–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, and Jilka RL (2005) Chronic elevation of PTH in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146, 4577–4583 [DOI] [PubMed] [Google Scholar]
- 29.Keller H, and Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37, 148–158 [DOI] [PubMed] [Google Scholar]
- 30.Delgado-Calle J, Hancock B, Likine EF, Sato AY, McAndrews K, Sanudo C, Bruzzaniti A, Riancho JA, Tonra JR, and Bellido T (2018) MMP14 is a novel target of PTH signaling in osteocytes that controls resorption by regulating soluble RANKL production. FASEB J 32, 2878–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu J, Schilling L, Eller T, and Canalis E (2021) Hairy and enhancer of split 1 is a primary effector of NOTCH2 signaling and induces osteoclast differentiation and function. J Biol Chem 297, 101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canalis E, Schilling L, Yee SP, Lee SK, and Zanotti S (2016) Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis, and Bone Resorption. J Biol Chem 291, 1538–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagheri L, Pellati A, Rizzo P, Aquila G, Massari L, De Mattei M, and Ongaro A (2018) Notch pathway is active during osteogenic differentiation of human bone marrow mesenchymal stem cells induced by pulsed electromagnetic fields. J Tissue Eng Regen Med 12, 304–315 [DOI] [PubMed] [Google Scholar]
- 34.Chakravorty N, Hamlet S, Jaiprakash A, Crawford R, Oloyede A, Alfarsi M, Xiao Y, and Ivanovski S (2014) Pro-osteogenic topographical cues promote early activation of osteoprogenitor differentiation via enhanced TGFβ, Wnt, and Notch signaling. Clin Oral Implants Res 25, 475–486 [DOI] [PubMed] [Google Scholar]
- 35.Zanotti S, and Canalis E (2017) Parathyroid hormone inhibits Notch signaling in osteoblasts and osteocytes. Bone 103, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delgado-Calle J, Tu X, Pacheco-Costa R, McAndrews K, Edwards R, Pellegrini G, Kuhlenschmidt K, Olivos N, Robling A, Peacock M, Plotkin LI, and Bellido T (2017) Control of bone anabolism in response to mechanical loading and PTH by distinct mechanisms downstream of the PTH receptor. J. Bone Miner. Res 32, 522–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li JY, Yu M, Tyagi AM, Vaccaro C, Hsu E, Adams J, Bellido T, Weitzmann MN, and Pacifici R (2018) IL-17 Receptor Signaling in Osteoblasts/Osteocytes Mediates PTH-Induced Bone Loss and Enhances Osteocytic RANKL Production. J. Bone Miner. Res [DOI] [PubMed] [Google Scholar]
- 38.Delgado-Calle JM, and Bellido T (2021) The Osteocyte as a Signaling Cell. Physiol Rev [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canalis E, Bridgewater D, Schilling L, and Zanotti S (2016) Canonical Notch activation in osteocytes causes osteopetrosis. Am J Physiol Endocrinol Metab 310, E171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu J, and Canalis E (2020) Notch and the regulation of osteoclast differentiation and function. Bone 138, 115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu P, Ping Y, Ma M, Zhang D, Liu C, Zaidi S, Gao S, Ji Y, Lou F, Yu F, Lu P, Stachnik A, Bai M, Wei C, Zhang L, Wang K, Chen R, New MI, Rowe DW, Yuen T, Sun L, and Zaidi M (2016) Anabolic actions of Notch on mature bone. Proc Natl Acad Sci U S A 113, E2152–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eriksen EF, and Brown JP (2016) Commentary: Concurrent administration of PTH and antiresorptives: Additive effects or DXA cosmetics. Bone 86, 139–142 [DOI] [PubMed] [Google Scholar]
- 43.Leder BZ, Tsai JN, Burnett-Bowie SA, Bouxsein ML, and Neer RM (2016) Letter to the editor in response to the commentary, “Concurrent administration of PTH and antiresorptives: Additive effects or DXA cosmetics. Bone 89, 73–74 [DOI] [PubMed] [Google Scholar]
- 44.Bray SJ (2006) Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol 7, 678–689 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.