

Abstract
Cancer therapies have been evolving from conventional chemotherapeutics to targeted agents. This has fulfilled the hope of greater efficacy, but is not accompanied by greater safety. In fact, a broad spectrum of toxicities can be seen, including cardiovascular toxicities. Among these, cardiomyopathy and heart failure have received greatest attention, given their profound implications for continuation of cancer therapies, and cardiovascular morbidity and mortality. Prediction of risk has always been difficult, and even more so with the newer targeted agents. Accurate risk prediction would greatly facilitate decisions on these therapies even before the first dose is given. This is important for agents with a long half-life and high potential to induced life-threatening cardiac complications, such as myocarditis with immune checkpoint inhibitors. An opportunity to address these needs in the field of Cardio-Oncology is provided by the expanding repertoire of “-omics” and other tools in precision medicine. Utilizing a systems biology approach to integrate these personalized tools may facilitate new insights into patho-mechanisms and risk prediction. This would allow for the creation of more precise and cost-effective risk prediction tools, improved therapy decisions, and prevention of cardiovascular complications. Herein, we explore this topic as a future approach to translating the complexity of cardio-oncology to the reality of patient care.
Keywords: cancer, cardiotoxicity, immune checkpoints, precision medicine, systems biology, tyrosine kinases
INTRODUCTION
Since the turn of the millennium, therapeutic strategies have shifted from universal chemotherapies to therapeutic agents that target the molecular footprints of the malignant disease process (signaling pathways, molecules, gene mutations), specific cell clones, or the immune system(1–3). Much against their promise of being safer designer drugs, however, these new targeted therapies are not without cardiovascular complications(4, 5). Toxicities can include cardiomyopathy, heart failure, myocarditis, venous and arterial thrombosis, progression of atherosclerosis, QTc prolongation, and arrhythmias (6). Each therapy is at risk for one or several of these toxicities, with risks being individually different.
One of the eminent demands in this area is optimal risk prediction and management. This, however, requires a thorough understanding of the pathophysiology. The very heterogeneities of the patient populations and the disease spectra have complicated clinical and translation efforts. A systems-based approach may offer a solution to such a broad and heterogeneous field. Integrating data from various angles of view (e.g. genomics, proteomics, kinomics, and metabolomics) can provide mechanistic insight, reveal key risk factors, and may indicate new therapeutic avenues (7, 8). In this review, we will focus on such an approach for cardiomyopathy, considering the response of the cardiovascular system to perturbation by tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs).
Tyrosine Kinase Inhibitors
Since the introduction of imatinib in 2001, there have been over 30 TKIs approved by the US Food and Drug Administration (9). Tyrosine kinases (TK) are ubiquitous phosphorylating enzymes with over 500 being identified in the human genome (1). They operate across a number of signaling pathways of significance for cancer, including proliferation, differentiation, and survival (6, 10–13). Some even take the crucial position of a molecular fingerprint and under such circumstances their inhibition provides a cure. One illustrating example is the expression of the TK oncogene product Bcr-ABL (Abelson murine leukemia gene) in patients with Philadelphia chromosome positive leukemias. Other TKs control such vital processes as angiogenesis, e.g. the VEGF (vascular endothelial growth factor) receptor and other growth factor proliferation pathways, e.g. EGFR (epidermal growth factor receptor) including HER-2. (9). (6, 10–13).
In order to inhibit TK-controlled signaling pathways two main strategies have been pursued: a) antibodies to decrease the activation of receptor TKs (e.g. trastuzumab directed against HER-2) and b) small molecule TK inhibitors (smTKIs) that penetrate into the cell and compete for the ATP (adenosine triphosphate) binding domain of TKs. As this site can be rather similar across several TKs, target specificity is often difficult to accomplish leading to “drug promiscuity” (Table 1). To some degree such multi-targeted approach can be of synergistic action in controlling cancer cell proliferation and survival. However, it may also generate a greater risk of adverse effects with less mechanistic predictability of action.
Table 1.
Small molecule tyrosine kinase inhibitors (modified from Current Oncology Reports, Tyrosine Kinase Inhibitors and Vascular Toxicity: Impetus for a Classification System?, volume 18, issue 6, page 33, year 2016, Joerg Herrmann, with permission of Springer).
| Malignancy targets | Molecular Targets | HTN | Chest pain | MI | Stroke/TIA | PAD | DVT/PE | Additional warnings | |
|---|---|---|---|---|---|---|---|---|---|
| Bcr/Abl inhibitors | |||||||||
| Imatinib | Ph+ CML, Ph+ ALL, GIST, ASM, MDS Off-label melanoma | ABL-1, c-KIT, PDGFR-A, PDGFR-B, FLT-3 | 4% | 7–11% | Edema 11–86% Hypotension 11% | ||||
| Nilotinib | Ph+ CML Off-label GIST | ABL-1, c-KIT, PDGFR-A, PDGFR-B, FLT-3 | 10–11% | 5–9% | 1–3% | 3–4% | Pericardial effusion 2% | ||
| Ponatinib | Ph+ CML, Ph+ ALL | ABL-1, c-KIT, PDGFR-A, PDGFR-B, FLT-1, KDR, FLT-3, FLT-4, FGFR, SRC, TIE2 | 53–71% | 12% | 2% | 8% | 5% | HF6–15% Pericardial effusion 1–3% Atrial fibrillation 4% | |
| Bosutinib | Ph+ CML | ABL-1, FGFR, FLT-1, FLT-2, FLT-3, KDR, PDGFR-A, PDGFR-B, SRC | Edema 14% Pericardial effusion 1–3% | ||||||
| Dasatinib | Ph+ CML, Ph+ ALL Off-label GIST | ABL-1, FGFR2, KIT, PDGFR-A, PDGFR-B, SRC | 4% IHD | Edema 4% Pericardial effusion 4% Cardiac dysfunction 4% QTc prolongation 1% Conduction abnormalities 7% | |||||
| VEGFR inhibitors | |||||||||
| Sorafenib | RCC, Thyroid cancer, HCC, off-label: angiosarcoma and GIST | B-Raf, FLT-1, FLT-3, FLT-4, KDR, KIT, PDGFR-A, PDGFR-B, FGFR, c-fms | 9–41% (grade 3 or 4 10%) | 3% | 1% | HF in 2% | |||
| Sunitinib | RCC, GIST, PNET, off-label: STS, thyroid cancer | ABL-1, c-KIT, PDGFR-A, PDGFR-B, FLT-1, KDR, FLT-3, FLT-4, FGFR, SRC, c-smc | 27–34% (grade 3 or 4 10%) | 13% | 3% each | LVEF drop 16–27% | |||
| Pazopanib | RCC, STS, thyroid cancer | ABL-1, c-KIT, PDGFR-A, PDGFR-B, FLT-1, KDR, FLT-4, FGFR, c-frns | 40% (grade 3 or 4 7%) | 5–10% | 2% | 1% | 1–5% | HF 11% QTc prolongation 2% Bradycardia 2–19% | |
| Axitinib | RCC | c-KIT, PDGFR-A, PDGFR-B, FLT-1, KDR, FLT-4, | 40% (grade 3 or 4 16%) | 2% | 1% | 1–3% | |||
| Regorafenib | GIST, colorectal cancer | PDGFR-B, FLT-1, KDR, FLT-4, TIE2, RET, c-KIT, RAF | 30–59% (grade 3 or 4 8–28%) | 1% | |||||
| Lenvatinib | Thyroid cancer | PDGFR-B, FLT-1, KDR, FLT-4, RET, c-KIT | 73% (grade 3 or 4 44%) | 3% | LVEF drop 2% QTc prolongation 9% | ||||
| EGFR inhibitors | |||||||||
| Erlotinib (+gemcitabine) | NSCLC, pancreatic cancer | EGFR (HER1) | N/a | 12% | (2%) | (3%) | N/a | (4%) | (Cardiac arrhythmia 5%, syncope 5%, thrombosis 11%) |
| Vandetanib | Thyroid cancer | EGFR, KDR, FLT-4, RET | 33% (grade 3 or 4–9%) | 1% | QTc prolongation 14% Heart failure 2% | ||||
| ALK inhibitors | |||||||||
| Crizotinib | NSCLC | ALK,MET, ROS1 | 6% | QTc prolongation 5–6% Bradycardia 5–15% Syncope 1–3% Edema 31–49% | |||||
| Ceritinib | NSCLC | ALK, IGF-1R, ROS1, InsR | QTc prolongation 4% Bradycardia 3% | ||||||
| JAK inhibitors | |||||||||
| Ruxolitinib | Myelofibrosis Polycythemia vera | JAK1, JAK2, TYK2 | 6% |
Adverse effects from TKIs are divided into on- and off-target toxicities (6, 10). “On-target” toxicity results when the intended TK inhibited is responsible for both the anti-neoplastic effect and the unintended toxicity. On the contrary, “off-target” toxicity is the result of inhibition of kinases not intended for the anti-neoplastic effect. An in-depth description of the cardiovascular side effects of TKIs is beyond the scope of this review. However, representative examples of cardiomyopathy induced by antibodies and smTKI will be presented in the following.
Cardiomyopathy of antibody-based receptor tyrosine kinase inhibitors
ERBB (erythroblastic oncogene B)-2, also known as EGFR 2 or human epidermal growth receptor (HER-2), is (over-)expressed in around 15–25% of breast cancer patients (14). Stimulation of the ERBB-2 receptor increases the activity of multiple kinases including mitogen activated protein kinase (MAPK), PI3K, protein kinase C, and signal transducer and activator of transcription (STAT) (14, 15). The subsequent signaling cascade promotes cell replication and inhibits apoptosis. Inhibition of this pathway can be achieved by antibodies (trastuzumab, pertuzumab, trastuzumab emtansine) or by smTKI (lapatinib, neratinib, afatinib), with various combinations purposed. Contrary to the anti-cancer effects, the mechanisms of cardiomyopathy with HER-2 receptor blockade are less well understood.
Several lines of evidence suggest that HER-2 signaling is important in cardiac development as well as for maintenance of cardiac function in adults. HER-2 and -4 receptors are essential co-receptors for developing cardiac myocytes and play a pivotal role in Neuregulin-1 (NRG-1) signaling, which is vital for cardiomyocyte proliferation and regeneration (16). In murine models, mutations in HER-2, HER-4 or NRG-1 result in mid-gestation lethality (17), whereas mice with HER-2 deletion confined to cardiac myocytes are able to survive after birth but develop dilated cardiomyopathy with no compensatory reserve to high afterload conditions (17–19). ERBB-2 and ERBB-4 expression is high during gestation and required for the normal structural development of the heart including ventricular trabeculations, valves, and neuromuscular junctions (16, 20–23). Downregulated shortly after birth, certain stressors such as anthracycline exposure, myocardial ischemia/injury, or high afterload can, however, lead to re-expression of ERBB-2 (20). Blocking the HER-2 pathway under such circumstances decreases the capacity of the myocardium to adapt to such stressors and increases the risk of cardiac dysfunction.
Of note, there are considerable differences in the incidence of cardiomyopathy and heart failure with different HER-2 inhibitors; 2–18% and 0.3–4% with trastuzumab, 3–7% and 0.3–1% with pertuzumab, and 1–2% and 0.1–0.5% with lapatinib, respectively (24, 25). The mechanisms underlying these differences still remain largely unexplained though some studies provide some insight (26). In rodents antibodies against HER-2 (trastuzumab-like) down-regulate BCL-XL, resulting in degradation of mitochondrial membrane potentials and reduction of available ATP (27, 28). This effect has not been appreciated with other types of ERBB kinase inhibitors and would be in keeping with a trastuzumab-specific toxicity profile (27, 28). Further to consider, pertuzumab targets subdomain 2 of HER-2 and not subdomain 4 like trastuzumab (29, 30). The HER-2 subdomain 2 is also known as the “dimerization domain”, and pertuzumab, unlike trastuzumab, blocks the dimerization of HER-2 with HER-1, -3, and -4 (mostly strongly HER-3) (29, 30). This difference in the pharmacokinetics may be a key to the understanding of the lower cardiomyopathy risk of pertuzumab versus trastuzumab. In further distinction from trastuzumab, pertuzumab improves the likelihood of neuregulin-1 (NRG-1) stimulation of HER-4 dimer formation with subsequent activation of P13K/Akt signaling and improvement in cardiac metabolism and repair (18, 31). While HER2:HER4 dimer formation is reduced by pertuzumab, a shift to HER4:HER4 dimerization is conceivable and in keeping with maintained cardiac recovery potential. This theory is supported by work in cultured cardiomyoblasts and fetal cardiac myocytes, which noted different activities of NRG-1 stimulated HER2:HER4 and HER4:HER4 dimerization in response to treatment with different HER2 inhibitors (32). Finally, and interestingly enough, lapatinib has been associated with the lowest rate of cardiotoxicity. This has been attributed to the concomitant increase in AMPK activity and serves as an intriguing example of how off-target effects can be of benefit (33). Different examples of multi-target smTKIs are outlined in the following.
Cardiomyopathy of small molecule (multi-) tyrosine kinase inhibitors
A prime example and one of the most diverse kinase inhibitors is sunitinib, which has been shown to inhibit at least 50 different kinases (34). The main intended targets are the TKs of VEGF receptors 1–3. Multiple RTKs and non-RTKs, however, are affected as well, including platelet derived growth factor receptor (PDGFR), c-KIT, FMS-like tyrosine kinase3 and colony-stimulating factor-1, and RET, for instance, with sunitnib (10, 11). Inhibition of AMPK might be of particular significance for the development of cardiomyopathy/heart failure seen with sunitinib. This off-target effect has been related to ATP depletion and the cascade of oxidative stress, endoplasmic reticulum (ER) stress, mitochondrial damage, and caspase activation (11). Some studies also suggest a multifaceted physiological mechanism to heart failure and cardiomyopathy (35–38). While cellular alteration in myocyte function is suggested, its physiological expression as cardiomyopathy or heart failure may only present in particular settings associated with specific risks. The addition of stressors such as hypertension or or coronary artery disease (ischemia) enhances the risk of cardiomyopathy with or without clinical heart failure (35–38). This might be related to the fact that the VEGF signaling pathways serves a compensatory role under such and other circumstances (39). Up to this point, however, no specific risk prediction models are available.
The risk dimensions reported so far, in terms of incidences of left ventricular dysfunction and clinical heart failure, are in the range of 2 to 33% and 2 to 11%, respectively, with VEGF inhibitors (40–42). Other TKIs not discussed herein are outlined in Table 1. For most of these the exact nature of TKI-induced cardiomyopathy has not been well defined though alterations in cellular metabolism, mitochondrial function, and ATP utilization seem to be common themes (11, 43, 44). In general, TKI-related cardiomyopathies, including those related to VEGF inhibitor use, are thought to be largely reversible and of functional rather than structural nature. This concept, however, is being increasingly challenged.
Immune checkpoint inhibitors
Following the interaction of the major histocompatibility complex (MHC) with the T-cell receptor (TCR); the immune system walks a delicate balance between host defense and autoimmunity, directed by stimulatory and inhibitory signals.(2) Co-stimulation via CD28, CD27/CD70 and CD40/CD40L, promotes effector T-cell activity, whereas interaction with surface molecules like CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), PD-1 (programmed cell death protein 1), and LAG-3 (lymphocyte-activation gene 3) exerts an inhibitory effect (2). Both stimulatory and inhibitory signaling may be referred to as “immune check points”, though most commonly this terminology refers to the inhibitory aspects. Cancer cells can utilize this mechanism to escape the action of the immune system. For instance, melanoma cells are poised to interact with the immune system during lymphatic migration and expression of PD-L1 silences T-cell activity against them. Immune checkpoint inhibitors (ICIs) are designed to remove this “break” and allow for an immune action against the malignant tissue.
First generation ICIs entailed a monoclonal antibody targeted against CTLA-4. Its success led to quick advancement into second generation therapies targeting PD-1 and PD-1L (programmed cell death protein 1 ligand 1) (Table 2). A newer generation of therapies is currently under investigation and has expanded into both inhibitory and stimulatory signaling (Table 2) (2, 45). While extremely successful in terms for tumor response, the flipside of the success has been the induction of immune-related adverse events (irAEs). Among these, myocarditis is clinically infrequent (1%) but the most fatal irAE (up to 40–60% fatality rate). Pericarditis can occur in combination or isolation (with or without clinically relevant pericardial effusion); non-inflammatory cardiomyopathy has also been reported, even more classical Takotsubo-like presentations (46, 47).
Table 2.
Immune checkpoint inhibitors
| IMMUNE CHECK POINT RECEPTOR THERAPIES | |
|---|---|
| Generation of Checkpoint Inhibitor | Location |
| First Generation | |
| CTLA-4 | T-Cell |
| Ipilimumab | |
| Tremelimumab | |
| Second Generation | |
| PD-1 | T-Cell |
| Nivolumab | |
| Pembrolizumab | |
| Cemiplimab | |
| PD-L1 | Tumor, APC |
| Avelumab | |
| Atezolizumab | |
| Durvalumab | |
| Next Generation - Inhibitory pathways | |
| LAG-3 (CD223) | T-Cell |
| IMP321 (Immuntep®) | |
| BMS-986016 | |
| LAG525 | |
| TIM-3 | T-Cell |
| MBG453 | |
| MEDI9447 | |
| TIGIT | T-Cell, NK |
| OMP-31M32 | |
| VISTA | T-Cell |
| JNJ-61610588 | |
| CA-170 | |
| B7-H3 | T-Cell |
| Enoblituzumab (MGA271) | |
| MGD009 | |
| 8H9 | |
| KIR | NK cell |
| Lirilumab | |
| IPH4102 | |
| AlaR | T-Cell |
| CPI-444 | |
| Next Generation - Stimulatory Pathways | |
| OX40 | T-Cell |
| 9B12 | |
| MOXR 0916 | |
| PF-04518600 | |
| MEDI6383 | |
| MEDI0562 | |
| INCAGN01949 | |
| GSK3174998 | |
| GITR | T-Cell |
| TRX-518 | |
| BMS-986156 | |
| AMG 228 | |
| MEDI1873 | |
| MEDI6469 | |
| MK-4166 | |
| INCAGN01876 | |
| GWN323 | |
| ICOS | T-Cell |
| JTX-2011 | |
| GSK3359609 | |
| MEDI-570 | |
| 4–1 BB | NK, T-cell |
| Utomilumab (PF-05082566) | |
| Urelumab | |
| BMS-936561 (MDX-1203) | |
| Varilumab | |
| CD27-CD70 | T-cell/APC, Tumor |
| ARGX-110 | |
| CD 40-CD40L | T-cell/APC, Tumor |
| CP-870893 | |
| APX005M | |
| ADC-1013 | |
| JNJ-64457107 | |
| SEA-CD40 | |
| RO7009789 | |
| IDO | Tumor |
| BMS-986205 | |
| Indoximod | |
| Epacadostat | |
Cardiomyopathy of Immune Checkpoint Inhibitors
The exact mechanisms for myocarditis with ICIs have not been completely elucidated. (4, 46, 47). Of the cases studied, histologic evaluations have demonstrated cardiac infiltration of T-cells and macrophages consistent with lymphocytic myocarditis. There is little evidence of B-cell activation, or a humoral/antibody response (4).
One of the proposed mechanisms of ICI myocarditis is an increased effector T-cell response to an unknown cardiac antigen. This could be facilitated by either a shared epitope or epitope mimicry. While the exact epitope for ICI-induced myocarditis is not known, current theories suspect contractile proteins such as troponin, titin, and myosin (4, 48, 49). This could be supported by findings in melanoma patients where postmortem evaluations, by way of RNA sequencing, noted expression of both troponin and myosin heavy chain (4). Previous works have also investigated the heterogeneity of troponin expression in malignancy. Immunofluorescence studies have shown cardiac troponin protein expression in non-small cell lung cancer, and gene and protein expression have also been shown in cervical and hepatocellular carcinomas and osteosarcoma (50, 51). Thus, it is conceivable that cellular immunity is responding to the same epitope (e.g. troponin), similar epitopes (e.g. “troponin-like”), or two different epitopes (e.g. malignancy antigen and troponin on the same T-cell) (Fig. 1). Moreover, immune cells can be primed to target self in the absence of foreign antigens that are similar to the host. In such cases, the T-cells may have dual or chimeric TCRs (52). In this scenario, T-cells can have one of the dual TCRs reacting to the foreign antigen and the other reacting to self. As most of the potential self-antigen culprits are intra-cardiac proteins, one may speculate that immune exposure to these proteins occurs after cardiomyocyte damage or previous cardiac inflammation (53).
Fig. 1.
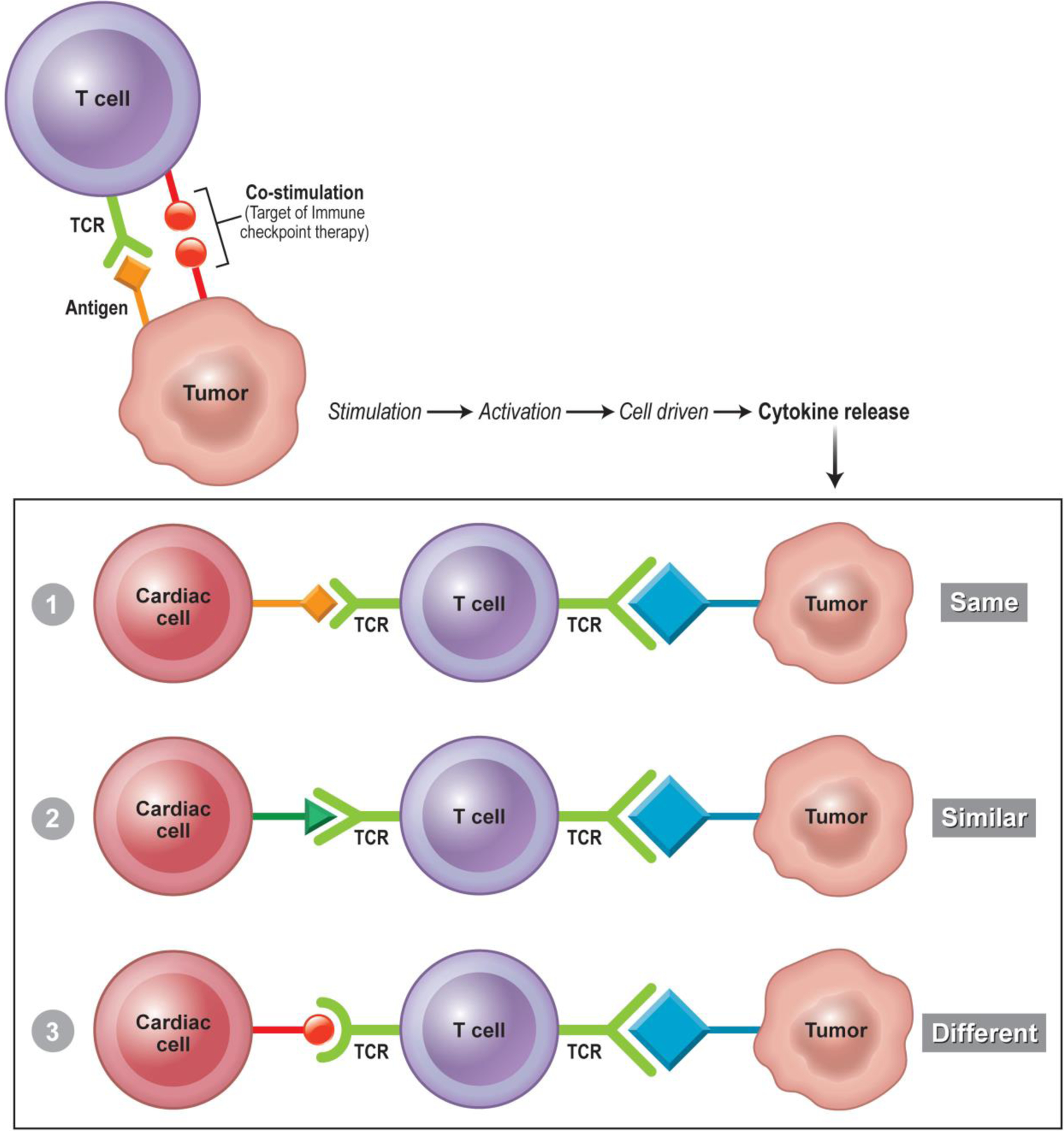
Proposed Mechanisms of Cardiotoxicity from Immune Checkpoint Inhibitors. Top: Tumor cell antigen interaction with TCR. A specific antigen present of the cell surface is recognized by the TCR of the T-cell, co-stimulation is inhibited by immune checkpoint therapy resulting in further T-cell effect. Bottom: Purposed cardiac autoimmunity by one of three mechanisms. 1) “Same antigen” the effector T-cell recognizes the same antigen on both the tumor and self-cardiac myocyte. 2) “Similar antigen” the antigen recognized by the effector T-cell is similar to the antigen recognized but not identical. This similarity is close enough to facilitate T-cell immunity against cardiac myocytes. 3) T-cell chimera where a single T cell has two different TCRs recognizing two different antigens (1 from the tumor, and 1 from the cardiac myocyte) both are recognized leading an effector T-cell response to both tumor and cardiac myocyte. TCR= T-cell receptor.
Another mechanism of cellular autoimmunity may be related to the biological role that immune checkpoint receptors could play in cardiac myocytes. PD-1L expression is found on both human and murine cardiomyocytes, but the functional implications are not fully known (54, 55). Acute ischemia and infarction, in addition to stimulation with various cytokines, have been shown to enhance upregulation of PD-1L (56). Conversely, genetic deficiency of PD-1 and CTLA-4 in mice leads to autoimmune myocarditis (57, 58). It could be purposed that effector T-cells primed for immunity against contractile proteins under traditional circumstances would be inhibited by the expression of PD-1L during cardiac stress; however, in the setting of ICI therapy this protective mechanism would be antagonized.
In addition to myocarditis, recent descriptions of non-inflammatory cardiomyopathies have been recognized and described (59). Whether this is a manifestation of “burned-out” myocarditis, low-level myocarditis, or a separate entity is not completely understood. Importantly, high and low degrees of myocardial inflammation have recently been described with low level myocarditis being clinically subtle or even silent (60). Furthermore, cardiac exposure to adrenergic signals and cytokines released during cancer therapy could result in stress cardiomyopathy, of which several reports have been documented (61, 62). Of interest, an auto-inflammatory etiology has recently been suggested for stress cardiomyopathy (63)While the currently utilized ICI receptors regulate the co-stimulation of T cell-related immune response, receptors such as PD-1 and PD-1L may also play a role in cardiomyocyte viability beyond protection from cellular immunity. Inhibition and downregulation of PD-1 and PD-1L may lead to alterations in transcription and cellular metabolism, which could play a role in non-inflammatory cardiomyopathy.
In summary, several variables likely play a key role in the development of ICI-related cardiomyopathy or myocarditis, including pre-existing cardiovascular disease, immune priming, and the regulation of immune checkpoint receptor expression on cardiac myocytes. It is because of this variability that systems biological and precision medicine approaches could facilitate better understanding of patients’ risks, offering improved monitoring and toxicity prevention.
PRECISION & SYSTEMS CARDIO-ONCOLOGY
Currently, there are no bonafide means of predicting which individuals will develop cardiovascular toxicity from either TKIs or ICIs. Clinical and host variables, as well as drug classification, can help suggest who may be at risk, but these entities by themselves are so far insufficient. Precision and systems medicine approaches may help identify those individuals who are most at risk and personalize their care accordingly. Primarily genomics and transcriptomics related to the pharmacokinetics and pharmacodynamics of VEGF signaling pathways, receptors, and ligands were previously outlined (8). In the following, we extend the precision and systems medicine approaches further to non-receptor signaling genomics, transcriptomics, proteomics, and metabolomics. An important question is whether genomic variants or biomarkers associated with drug efficacy or resistance can also predict cardiovascular toxicity, and if the latter is a reflection of the cancer response to therapy. As such, the opportunities for precision medicine in helping with the understanding, management, and prevention of cardiovascular toxicities from cancer therapies remain yet to be defined in order to advance their use in clinical practice.
Precision Cardio-Oncology in TKI-induced cardiovascular toxicity
Genomics, Transcriptomics, Proteomics, Metabolomics
A genome-wide association study in the fission yeast Schizossaccharomyces pombe identified mitochondrial DNA polymerase gamma 1 (POG1) as a potential mediator of sunitinib-induced cardiovascular toxicity (64). In POG1-deleted heterozygous mutant yeast compared to wild type, more severe cytotoxicity and mitochondrial damage were noted. Knockdown of the human ortholog POLG in HeLA cells by 50–60% using POLG-specific siRNA also associated with significantly increased cytotoxicity in response to sunitinib administration. Interestingly, administration of sunitinib independent of the use of siRNA led to similar downregulation of transcription and translation of POLG by 50%. Prior studies showed that POLG knockdown induces generation of reactive oxygen species and depletion of mitochondrial DNA, resulting in mitochondrial dysfunction and cytotoxicity (65–67). Previous studies also indicate that sunitinib causes abnormal mitochondrial structure and function in human myocardial cells and can associate with cardiomyopathy (35). In mice, POLG mutation causes oxidative stress with consequent loss of mitochondrial DNA and results in abnormal mitochondrial structure and function with subsequent cardiomyopathy (35, 68). Further, human cardiomyocyte expression of POLG has been confirmed by genome-wide integration of transcriptomics and antibody-based proteomics, as a possible mediator of sunitinib-induced cardiomyopathy in humans (69). Taken together, these results suggest that sunitinib downregulates production of POLG mRNA and protein, leading to impaired adaptation to oxidative stress and associated mitochondrial dysfunction, which both mediate cardiovascular toxicity. These studies illustrate the merit of an omics approach in elucidating the potential mechanisms for cardiovascular toxicity as an off-target effect of a TKI, here the VEGF inhibitor sunitinib. Furthermore, studies have indicated a high clinical variability of phenotypic expression in patients carrying POLG mutations (although 3D biophysical modeling would support consistent phenotypes), suggesting the possible influence of other genetic or epigenetic factors (70). In addition, point mutations in mitochondrial DNA can independently associate with cardiomyopathy, which can conceivably be amplified by administration of sunitinib (71). These observations from a systems level may also help explain inter-individual variability in response to sunitinib therapy.
Transcriptomics and proteomics studies in a murine model demonstrated unique differences between sunitinib and sorafenib (72), illustrating the pleomorphic effects of multikinase TKIs and the variability in the genes and mRNA affected by each drug. Therefore, while a class effect may be observed phenotypically regarding hypertension, cardiomyopathy, or other forms of cardiovascular toxicity, at the molecular level, gene and mRNA expression may not be identical. Since both the genome and transcriptome can be influenced by other factors, such as microRNAs or the environment, differential regulation of the genome and transcriptome in various individuals can also help to explain inter-individual variability in cardiovascular toxicity.
In regards to metabolomics, several changes were noted in mice which were treated with sunitinib and developed left ventricular systolic dysfunction within two weeks of drug administration (73). Levels of long chain omega-3 fatty acids docosahexaenoic acid (DHA) and arachidonic acid (AA)/ eicosapentaenoic acid (EPA) were decreased in the heart in addition to the metabolites O-phosphocolamine, 6-hydroxynicotinic acid, and fructose. Cholesterol, sucrose, and similar disaccharides were increased in the liver. Adenosine/inosine and DHA, as well as C11 hydrocarbon and dehydroalanine (possibly from cysteine) were downregulated in skeletal muscle. In distinction, mice treated with erlotinib did not develop left ventricular systolic dysfunction and showed an increase in one metabolite (spermidine) in the heart, downregulation of homoserine and ornithine in the liver as well as C11 hydrocarbon and dehydroalanine (possibly from cysteine) in skeletal muscle. As polyunsaturated fatty acids are important for mitochondrial function and can also act as beneficial inflammatory mediators,(74–76) this study pointed to a metabolic aspect in the pathophysiology of sunitinib-induced cardiotoxicity. Of further interest, treatment with L-carnitine, which transports fatty acids into the mitochondria to facilitate oxidation and energy production, can be protective in the setting of sunitinib therapy (74). Last but not least to mention, one study also noted enhanced glycolysis and reduced oxidative phosphorylation (metabolic switch) in murine myocardium in response to sunitinib (77). Of note, co-administration of an endothelin receptor antagonist protected against these metabolic defects and restored cardiac structure and function, possibly by modulating mitogen-activated protein kinase (MAPK) activity. Collectively these studies illustrate the potential of approaches other than genomics in a systems-based approach in cardio-oncology related to TKIs.
Kinomics in Computational Pathway Analyses
While pathways for cardiotoxicity are multifaceted, they may not necessarily be linearly associated with the number of kinases inhibited. Rather, it seems that it is of greater significance which types of kinases are inhibited. This sentinel kinase concept is also supported by developments in experimental studies on this topic. For example, in one study, TKIs with a known/likely cardiotoxicity profile were contrasted against those with a low cardiotoxicity profile. The comparison indicated that the former group had, in general, a much broader scope of targets (6). While therapies in the “high risk” group were notoriously more kinase-promiscuous, it remains likely that the sheer number of kinases inhibited is less important than the specific kinases inhibited (78, 79). Moreover, not only inhibitory but also the stimulatory effects are of significance. For instance, a very illustrating study revealed that the compared to sunitinib and sorafenib very benign cardiotoxicity profile of erlotinib is not only due to a difference in TKI properties, but also due to the concomitant upregulation of signal transducer and activator of transcription 3 (STAT3) signaling, enabling metabolically adaptive changes (78). Notably, for VEGF inhibitors, activation of the insulin signaling pathway was identified as a compensatory mechanism in an extensive high throughput screen in human inducible pluripotent stem cells. Such recent observations outline the complexity of the matter in both inhibition and upregulation of signaling and lend support for systems biology approaches in assessing the response of the cardiovascular system to TKI-induced perturbation.
Finally, computational systems can be used to address the complexity, mapping pathways and predicting interactions among proteins. Further development, validation, and implementation of such computational predictions could potentially improve clinical and research practice. (80). Integration of various omics may help to optimized computational pathway analyses in their predictive abilities regarding cardiovascular toxicity from cancer therapies.
Precision Cardio-Oncology in ICI-induced cardiovascular toxicity
Gene polymorphisms, Multiple hits, and Autoimmunity
Gene polymorphisms leading to deficiency or dysfunction of CTLA-4, PD-1, or PDL-1 may be associated with myocarditis or cardiomyopathy, or a myriad of other autoimmune or inflammatory conditions, in humans or in mice. Patients carrying genomic variants in CTLA-4 such as the most frequently studied rs231775 (a single nucleotide polymorphism in the 3’ flanking region of the gene) often develop a variety of autoimmune disorders, such as type 1 diabetes, celiac disease, systemic lupus erythematosus, and rheumatoid arthritis (81–86). In one study, patients autoimmune hypothyroidism and with A/G (odds ratio 5.333, p = 0.0004) or G/G (odds ratio 5.779, p = 0.0019) alleles for the CTLA-4 variant rs231775 had lower levels of CTLA-4 mRNA expression than control individuals, with an apparent dose effect on both mRNA levels and susceptibility to autoimmunity (Fig. 2) (87). Other studies found that autoimmune disorders often clustered together in individuals with unfavorable CTLA-4 polymorphisms (88–91). The breadth of autoimmune conditions associated with CTLA-4 polymorphisms is reminiscent of the variety of autoimmune observed in patients treated with inhibitors of CTLA-4, PD-1, PD-L1. It is therefore conceivable that the inter-individual variability in the development of these varied cardiovascular toxicities may in part be mediated by polymorphisms in the genes that encode CTLA-4, PD-1, and PDL-1.
Fig. 2.
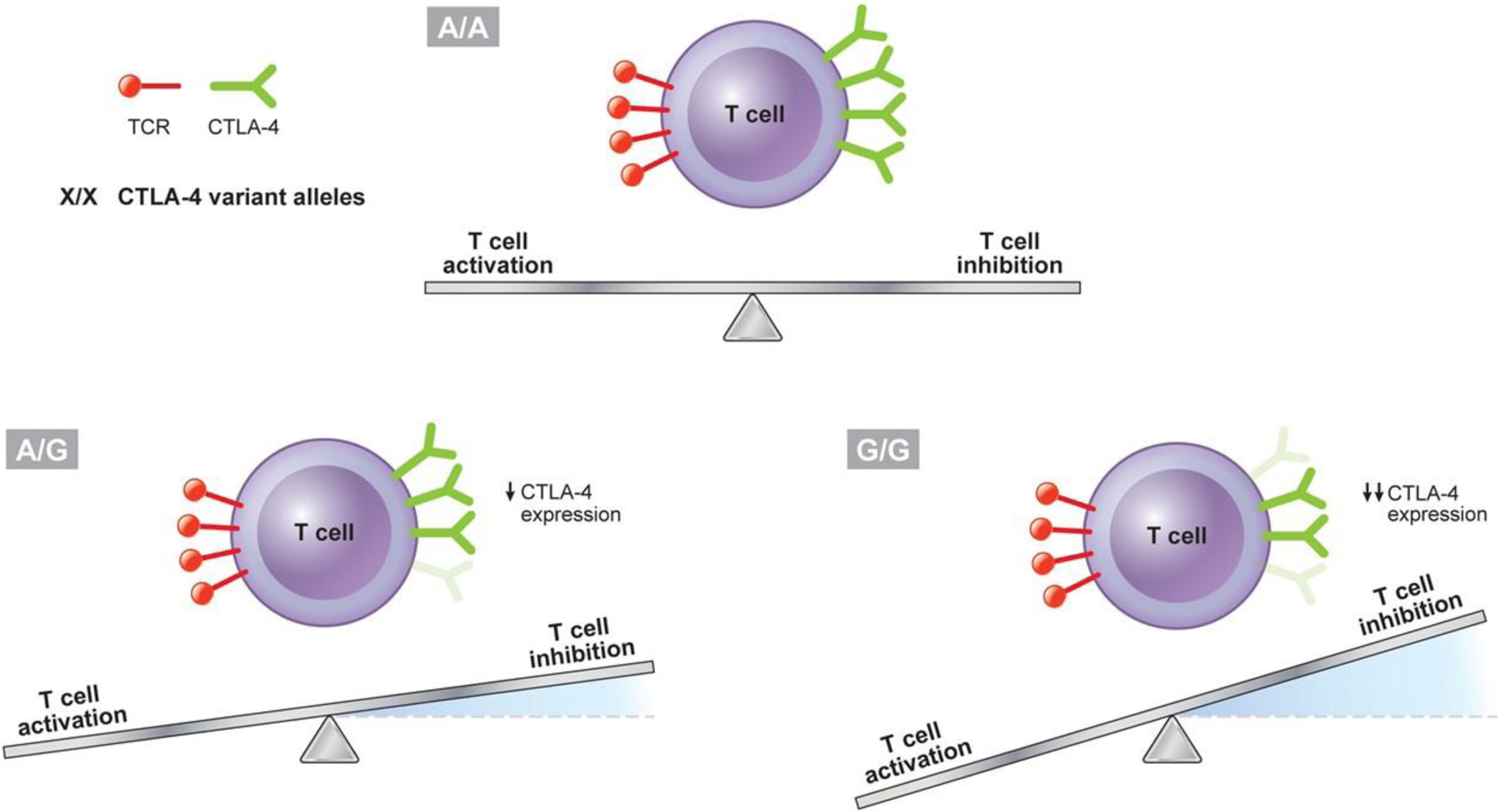
Potential Impact of CTLA-4 Polymorphism on Immune System Activation. An apparent dose effect on both mRNA levels and susceptibility to autoimmunity is noted for the most frequently studied CTLA-4 genomic variant rs231775 (single nucleotide polymorphism in the 3’ flanking region of the gene), with lower levels of CTLA-4 mRNA expression associated with the alleles A/G (odds ratio 5.333, p = 0.0004), and even lower levels of CTLA-4 mRNA expression associated with the alleles G/G (odds ratio 5.779, p = 0.0019), compared to control (A/A) individuals (87); decreased expression of CTLA-4 would be expected to decrease T-cell inhibition, with the equilibrium of T-cell activity shifting towards activation. CTLA-4 = cytotoxic T-lymphocyte-associated protein 4; TCR = T-cell receptor.
It is conceivable that multiple hits may play a role in ICI-induced cardiovascular toxicities. Patients treated with an ICI may have a pre-existing CTLA-4 polymorphism, dual or chimeric TCRs, prior exposure to an infectious agent, leading to prior immune self-priming with cardiac or extra-cardiac proteins, or alterations in stress or sex hormone signaling. Any number of these combinations could potentially be present in patients who present with myocarditis, which is a rare adverse effect in ICI therapy. Further studies in precision cardio-oncology from such a systems perspective may help further elucidate such concurrent mechanisms consistent with multiple hits.
Possible Precision Markers of ICI Efficacy and Toxicity
Several candidate genomic ‘nomograms’ or immunotherapy ‘signatures’ or ‘footprints’ may be useful as precise markers of cardiovascular toxicity or risk. Whole-exome and whole-transcriptome sequencing of surgically resected malignant melanoma have identified an association between mutational load and expression of cytolytic genes and the degree of clinical benefit (92, 93). Whole exome sequencing of non-small-cell lung cancers from patients treated with PD-1 and CTLA-4 inhibitors, for instance, demonstrated that a high tumor mutation burden (or “mutanome”, in “mutanomics”) strongly associated with clinical benefit, including progression-free survival (94, 95). Others have developed genomic and transcriptomic signatures to correlate clinical response to CTLA-4 and PD-1 inhibitor therapy in metastatic melanoma, glioblastoma, and other cancers(96–99). The use of exomics, transcriptomics, and computer simulations has successfully predicted clinical response to PD-1 immunotherapy (100). Perhaps these genomic and transcriptomic signatures could be studied to determine any concurrent association with the likelihood or extent of cardiovascular toxicity from immunotherapy.
Gene polymorphisms alone are not likely to be fully responsible for inter-individual variability in cardiovascular toxicities from ICIs. Methylation of CTLA-4 is related to the clinical response to anti-CTLA-4 and anti-PD-1 immunotherapy (101). DNA regulation, such as histone post-translational modifications and noncoding RNAs, could also play a role (102). Epigenomic changes often occur due to gene-environment interactions and lifestyle habits, such as nutrition, smoking, stress, and pollution, potentially modifying gene expression, and cardiovascular risk (102, 103). Several studies also suggest the utility of microbiomics to predict clinical benefit from ICI therapy (104–107). The endogenous presence or oral administration of bacteria such as Akkermansia muciniphila has been found to improve the response to PD-1 immunotherapy (104, 108, 109), suggesting that the gut microbiome interacts with the host by modulating the function of gut epithelium or directly the local immune system (110). Collectively, these areas within precision medicine could be explored further in cardio-oncology.
Clinical Implementation of Precision Cardio-Oncology for TKI and ICI therapies
Ideally to protect against toxicities, prior to or during cancer therapies, each individual would have a precision nomogram created. This multifaceted nomogram would represent an individual’s risk by way of their genome, exome, mutanome, transcriptome, epigenome, proteome, metabolome, and microbiome, microRNA regulome, exposome, interactome, and so on (Fig. 3). The use of precision Cardio-Oncology with receptor-based therapies could create a fingerprint/signature unique to each patient, thus placing the individual’s nomogram in the context of predicting their specific anti-cancer response and risk for cardiotoxicity. Various tools within precision medicine would be integrated for this purpose, to form a global picture of the patient that could be used to personalize care in Cardio-Oncology via the P*3 pathway (Fig. 4) (7, 8). To preempt and prepare for the possibility of developing cardiovascular toxicity, the data would be integrated into the electronic health record of these patients, along with clinical and other information such as family history, lifestyle, and so on. Imaging data would be incorporated, particularly echocardiography, coronary CT, cardiac MRI and PET scans, as well as more recently developed ‘molecular imaging’ techniques (111). Molecular imaging allows for detection of bimolecular events and trends underlying cardiac physiology and phenotype, using radio-labelled imaging probes. Such advanced testing and nomogram development within the context of precision Cardio-Oncology would require counseling akin to genetic counseling (P1; a comprehensive step 1 shown in the figure). This would help patients understand the probabilistic nature of all their data and also the uncertainty with which we make subsequent predictions (P2). In the end, it it the hope that providing such information will help navigate inter-individual variability and prevent cardiovascular toxicity (P3).
Fig. 3.
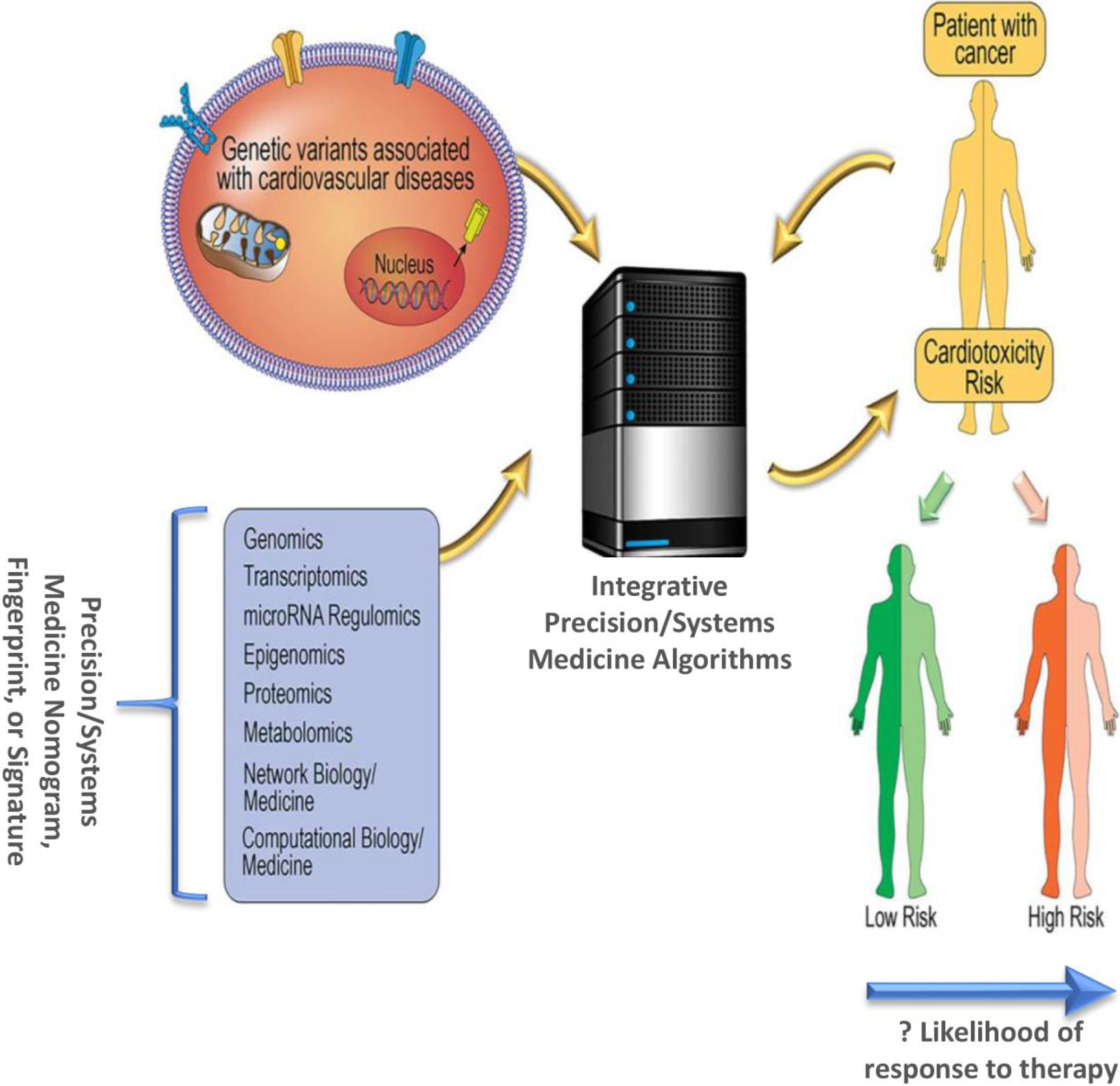
Precision and Systems Medicine Paradigm in Cardio-Oncology. A Precision or Systems Medicine nomogram, fingerprint, or signature consisting of genomic, transcriptomic, microRNA regulomics, epigenomic, proteomic, metabolomic, and other ‘omic’ factors could be determined for each patient, with assessment of their genetic/genomic variants that may associated with cardiotoxicity, potentially utilizing network medicine and computational pathway analyses in integrative algorithms. The output of such algorithms would be prediction of whether a patient with cancer, based on their unique characteristics, might be at low or high risk for developing cardiotoxicity, and whether they may be more or less likely to respond to specific cancer therapies. Adapted from [29], with permission.
Fig. 4.
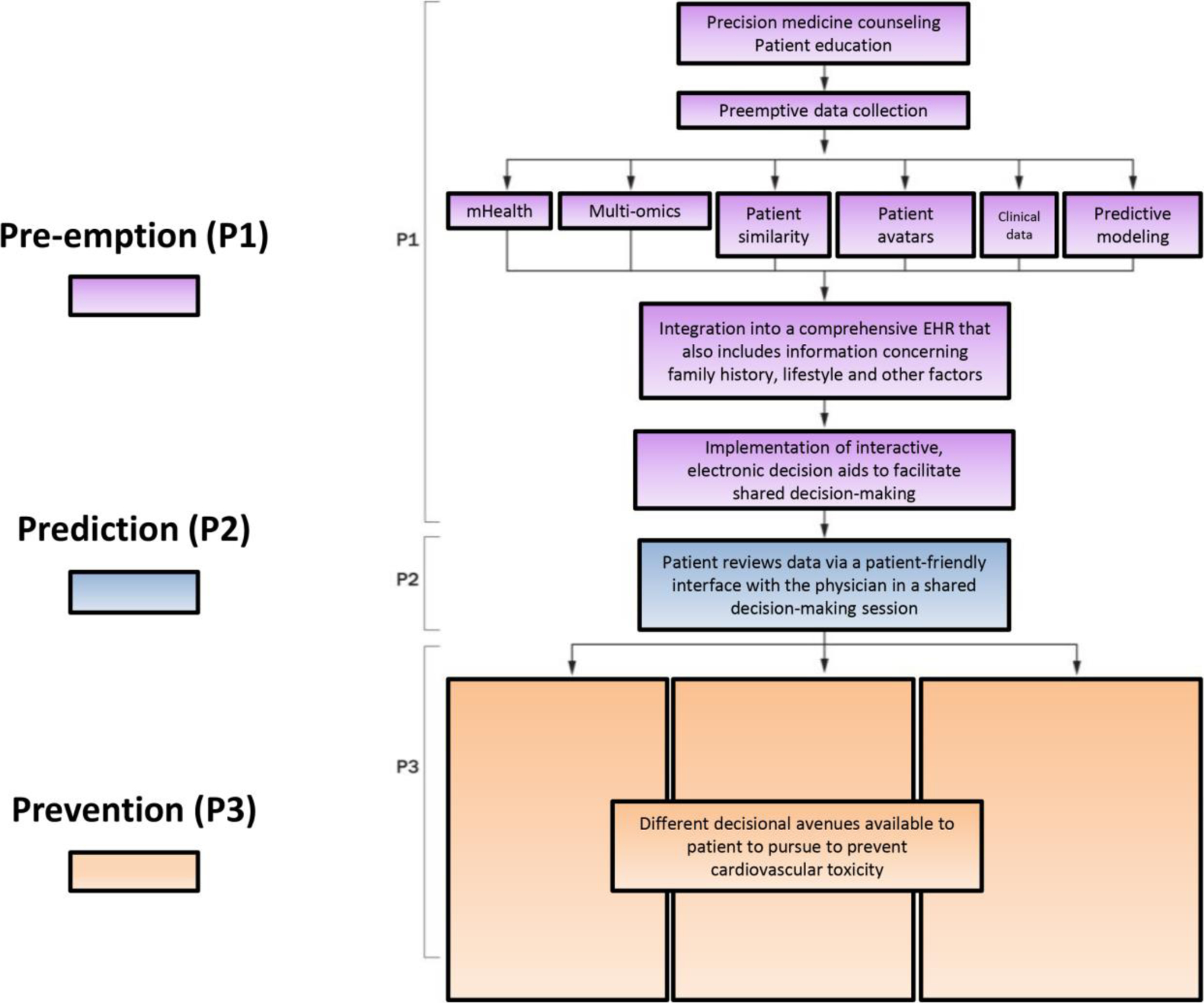
Clinical Implementation of Precision Cardio-Oncology: The P*3 Pathway (7, 8). In the P*3 approach to precision cardio-oncology, pre-emption (P1) is represented in purple: precision medicine data should be integrated into the electronic health record (EHR) along with clinical information and other demographics such as family history and environmental or lifestyle contributors, following precision/systems medicine counseling, and with interactive electronic decision aids to facilitate shared decision-making. Then prediction (P2) is represented in blue, incorporating stratification mechanisms for risk prediction, including both clinical factors and precision medicine data. Individual would be placed into three risk categories: high, intermediate and low. Prevention (P3) is represented in orange, and involves personalized prevention strategies that simultaneously optimize therapeutic efficacy and safety for the individual patient. EHR = electronic health record. Adapted from [93], and used with permission of Springer Nature.
One might consider, for example, the presentation of a patient treated with nivolumab (a PD-1 inhibitor), who subsequently developed ptosis, diplopia, myositis, and myocarditis. This patient’s presenting symptoms are remarkably similar to those seen in patients with multisystem autoimmune conditions associated with CTLA-4 polymorphisms and decreased CTLA-4 receptor levels (Fig. 2) (87). It could be that in the setting of such CTLA-4 polymorphisms, treatment with a PD-1, PD-L1, or CTLA-4 inhibitor creates a multiple hit scenario. Therefore, it would be reasonable to infer that a CTLA-4 agonist such as abatacept in opposition to corticosteroids could rescue individuals with the AG or GG allele combinations, from severe myocarditis, if administered before, during, or after immunotherapy (112). This supports the possibility that CTLA-4 polymorphisms and its associated effect on receptor expression explain the inter-individual variability in the development of myocarditis. As outlined above, it is the hope of developing and implementing precision cardio-oncology in clinical practice to assist with shared decision-making on risk and benefit of ICI therapy. For patients identified to be at high risk for cardiotoxicity, it might be worth testing shorter treatment period or administration of a CTLA-4 agonist (112, 113).
The P*3 pathway for therapy decisions, as well as any other process of incorporating precision medicine to guide imaging or other decisions in cardio-oncology should be useful in the setting of a variety of cancer therapies. In fact, the P*3 pathway was originally developed to guide future decisions for anthracycline therapy in precision cardio-oncology (7). Any drug can be incorporated into such algorithms for either combination or sequential therapies. The overall patient nomogram can be dynamic and updated iteratively over time, e.g., as underlying patient characteristics are updated in the EHR.
Of course, it may not initially be practical or cost-effective to pursue a precision nomogram for every patient in oncology, or for every patient treated with a TKI or an ICI. Initial studies may need to first target patients with factors in their medical history or baseline assessment that may suggest a possibility of high risk of developing cardiovascular toxicity. For example, patients planned to undergo VEGF inhibitor therapy are already known to be at higher risk of cardiovascular toxicity if they have a history of coronary heart disease, arterial thromboembolism, hypertension, or other conditions recently reviewed. (114) (115). Yet, given inter-individual variability in cardiovascular toxicity and poor precise prediction of cardiovascular risk at the level of the individual patient, personalizing their care by use of the P*3 pathway may help tailor treatment plans and improve outcomes. The same holds true for patients undergoing ICI therapy (4, 46). Such patients could also pursue shared decision-making using the P*3 pathway to personalize decisions regarding their treatment and monitoring composition and duration.
However, even more so in this area there is a critical need to systematically catalog and define risk factors for developing ICI-induced cardiovascular toxicity (46, 116). Until precision nomograms become available, it would be reasonable to consider testing patients planned to undergo combination ICI therapy for common genomic variants such as the CTLA-4 rs231775 described above. As additional studies are pursued, more data will become available to guide precision-related decisions and practice guidelines (117).
CONCLUSION
The use of targeted therapies, and especially immune-targeted therapies, continues to grow in cancer care. This is accompanied by the risk of cardiovascular toxicities, which remains extremely difficult to predict by clinical means alone. Integration of information on multiple levels including, but not limited to, genomics, epigenomics, transcriptomics, proteomics, metabolomics, microRNA regulomics, microbiomics, pharmaco-interactomics, and environmentomics might offer solutions in risk prediction and management. Such an approach in precision and systems medicine might also assist with the mechanistic understanding of cardiovascular toxicities seen with relevant drugs. This would allow for a pathophysiology-based approach to the management and prevention of cardiovascular toxicities. With continued preclinical and clinical investigation and innovation, and subsequently clinical implementation, we may perhaps approach an era in which inter-drug and inter-individual differences and variability can be cheaply, reliably, and effectively predicted, to facilitate cancer treatment, remission, and cure without the high burden of cardiovascular toxicity.
Funding:
Funded by the National Institutes of Health/National Cancer Institute at (CA233610).
Conflict of Interest/Funding:
JH is funded by the National Cancer Institute at the National Institutes of Health (CA233610). SAB and JR declare that they have no conflict of interest.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest:
Author SAB declares that she has no conflict of interest.
Author JCR declares that he has no conflict of interest.
Author JH declares that he has not conflict of interest.
REFERENCES
- 1.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature reviews cancer. 2009;9(1):28. [DOI] [PubMed] [Google Scholar]
- 2.Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. Journal of hematology & oncology. 2018;11(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen P Protein kinases—the major drug targets of the twenty-first century? Nature reviews Drug discovery. 2002;1(4):309. [DOI] [PubMed] [Google Scholar]
- 4.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. New England Journal of Medicine. 2016;375(18):1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moslehi JJ, Deininger M. Tyrosine kinase inhibitor–associated cardiovascular toxicity in chronic myeloid leukemia. Journal of Clinical Oncology. 2015;33(35):4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nature Reviews Cancer. 2007;7(5):332. [DOI] [PubMed] [Google Scholar]
- 7.Brown SA, Sandhu N, Herrmann J. Systems biology approaches to adverse drug effects: the example of cardio-oncology. Nat Rev Clin Oncol. 2015;12(12):718–31. [DOI] [PubMed] [Google Scholar]
- 8.Brown SA, Nhola L, Herrmann J. Cardiovascular Toxicities of Small Molecule Tyrosine Kinase Inhibitors: An Opportunity for Systems-Based Approaches. Clin Pharmacol Ther. 2017;101(1):65–80. [DOI] [PubMed] [Google Scholar]
- 9.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends in pharmacological sciences. 2015;36(7):422–39. [DOI] [PubMed] [Google Scholar]
- 10.Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118(1):84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nature reviews Drug discovery. 2011;10(2):111. [DOI] [PubMed] [Google Scholar]
- 12.Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicology and applied pharmacology. 2010;244(2):190–5. [DOI] [PubMed] [Google Scholar]
- 13.Hasinoff BB, Patel D. The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicology and applied pharmacology. 2010;249(2):132–9. [DOI] [PubMed] [Google Scholar]
- 14.Roy V, Perez EA. Beyond trastuzumab: small molecule tyrosine kinase inhibitors in HER-2-positive breast cancer. Oncologist. 2009;14(11):1061–9. [DOI] [PubMed] [Google Scholar]
- 15.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature reviews Molecular cell biology. 2001;2(2):127. [DOI] [PubMed] [Google Scholar]
- 16.D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nature cell biology. 2015;17(5):627. [DOI] [PubMed] [Google Scholar]
- 17.Özcelik C, Erdmann B, Pilz B, Wettschureck N, Britsch S, Hübner N, et al. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proceedings of the National Academy of Sciences. 2002;99(13):8880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuramochi Y, Guo X, Sawyer DB. Neuregulin activates erbB2-dependent src/FAK signaling and cytoskeletal remodeling in isolated adult rat cardiac myocytes. Journal of molecular and cellular cardiology. 2006;41(2):228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crone SA, Zhao Y-Y, Fan L, Gu Y, Minamisawa S, Liu Y, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nature medicine. 2002;8(5):459. [DOI] [PubMed] [Google Scholar]
- 20.D’Uva G, Tzahor E. The key roles of ERBB2 in cardiac regeneration. Cell Cycle. 2015;14(15):2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negro A, Brar BK, Lee KF. Essential roles of Her2/erbB2 in cardiac development and function. Recent progress in hormone research. 2004;59:1–12. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Soria P, Camenisch TD. ErbB signaling in cardiac development and disease. Semin Cell Dev Biol. 2010;21(9):929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azim H, Azim HA, Escudier B. Trastuzumab versus lapatinib: the cardiac side of the story. Cancer Treat Rev. 2009;35(7):633–8. [DOI] [PubMed] [Google Scholar]
- 25.Lenihan D, Suter T, Brammer M, Neate C, Ross G, Baselga J. Pooled analysis of cardiac safety in patients with cancer treated with pertuzumab. Ann Oncol. 2012;23(3):791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenihan D, Suter T, Brammer M, Neate C, Ross G, Baselga J. Pooled analysis of cardiac safety in patients with cancer treated with pertuzumab. Annals of oncology. 2011;23(3):791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grazette LP, Boecker W, Matsui T, Semigran M, Force TL, Hajjar RJ, et al. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. Journal of the American College of Cardiology. 2004;44(11):2231–8. [DOI] [PubMed] [Google Scholar]
- 28.Sendur MA, Aksoy S, Altundag K. Pertuzumab-induced cardiotoxicity: safety compared with trastuzumab. Future Oncology. 2015;11(1):13–5. [DOI] [PubMed] [Google Scholar]
- 29.Adams CW, Allison DE, Flagella K, Presta L, Clarke J, Dybdal N, et al. Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunology, Immunotherapy. 2006;55(6):717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sendur MA, Aksoy S, Altundag K. Pertuzumab in HER2-positive breast cancer. Current medical research and opinion. 2012;28(10):1709–16. [DOI] [PubMed] [Google Scholar]
- 31.Nahta R, Yu D, Hung M-C, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nature Reviews Clinical Oncology. 2006;3(5):269. [DOI] [PubMed] [Google Scholar]
- 32.Fedele C, Riccio G, Malara AE, D’Alessio G, De Lorenzo C. Mechanisms of cardiotoxicity associated with ErbB2 inhibitors. Breast cancer research and treatment. 2012;134(2):595–602. [DOI] [PubMed] [Google Scholar]
- 33.Sendur MA, Aksoy S, Altundag K. Cardiotoxicity of novel HER2-targeted therapies. Current medical research and opinion. 2013;29(8):1015–24. [DOI] [PubMed] [Google Scholar]
- 34.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nature biotechnology. 2008;26(1):127. [DOI] [PubMed] [Google Scholar]
- 35.Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, et al. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci. 2009;2(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thirunavukkarasu M, Juhasz B, Zhan L, Menon VP, Tosaki A, Otani H, et al. VEGFR1 (Flt-1+/−) gene knockout leads to the disruption of VEGF-mediated signaling through the nitric oxide/heme oxygenase pathway in ischemic preconditioned myocardium. Free Radical Biology and Medicine. 2007;42(10):1487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chintalgattu V, Ai D, Langley RR, Zhang J, Bankson JA, Shih TL, et al. Cardiomyocyte PDGFR-β signaling is an essential component of the mouse cardiac response to load-induced stress. The Journal of clinical investigation. 2010;120(2):472–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47(5):887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Touyz RM, Herrmann J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis Oncol. 2018;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. The Lancet. 2007;370(9604):2011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khakoo AY, Kassiotis CM, Tannir N, Plana JC, Halushka M, Bickford C, et al. Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer: Interdisciplinary International Journal of the American Cancer Society. 2008;112(11):2500–8. [DOI] [PubMed] [Google Scholar]
- 42.Telli M, Witteles R, Fisher G, Srinivas S. Cardiotoxicity associated with the cancer therapeutic agent sunitinib malate. Annals of Oncology. 2008;19(9):1613–8. [DOI] [PubMed] [Google Scholar]
- 43.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular cell biology. 2007;8(7):519. [DOI] [PubMed] [Google Scholar]
- 44.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–9. [DOI] [PubMed] [Google Scholar]
- 45.Donini C, D’Ambrosio L, Grignani G, Aglietta M, Sangiolo D. Next generation immune-checkpoints for cancer therapy. Journal of thoracic disease. 2018;10(Suppl 13):S1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. Journal of the American College of Cardiology. 2018;71(16):1755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moslehi JJ, Salem J-E, Sosman JA, Lebrun-Vignes B, Johnson DB. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. The Lancet. 2018;391(10124):933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nature medicine. 2003;9(12):1477. [DOI] [PubMed] [Google Scholar]
- 49.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C, Liu J-B, Bian Z-P, Xu J-D, Wu H-F, Gu C-R, et al. Cardiac troponin I is abnormally expressed in non-small cell lung cancer tissues and human cancer cells. International journal of clinical and experimental pathology. 2014;7(4):1314. [PMC free article] [PubMed] [Google Scholar]
- 51.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 52.Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol. 2012;42(1):102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Root-Bernstein R Rethinking Molecular Mimicry in Rheumatic Heart Disease and Autoimmune Myocarditis: Laminin, Collagen IV, CAR, and B1AR as Initial Targets of Disease. Front Pediatr. 2014;2:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nature medicine. 1999;5(12):1365. [DOI] [PubMed] [Google Scholar]
- 55.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319–22. [DOI] [PubMed] [Google Scholar]
- 56.Baban B, Liu JY, Qin X, Weintraub NL, Mozaffari MS. Upregulation of programmed death-1 and its ligand in cardiac injury models: interaction with GADD153. PloS one. 2015;10(4):e0124059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J, Okazaki I-m, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. International immunology. 2010;22(6):443–52. [DOI] [PubMed] [Google Scholar]
- 58.Love VA, Grabie N, Duramad P, Stavrakis G, Sharpe A, Lichtman A. CTLA-4 Ablation and Interleukin-12–Driven Differentiation Synergistically Augment Cardiac Pathogenicity of Cytotoxic T Lymphocytes. Circulation research. 2007;101(3):248–57. [DOI] [PubMed] [Google Scholar]
- 59.Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, et al. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. Journal for immunotherapy of cancer. 2016;4(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Norwood TG, Westbrook BC, Johnson DB, Litovsky SH, Terry NL, McKee SB, et al. Smoldering myocarditis following immune checkpoint blockade. Journal for immunotherapy of cancer. 2017;5(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geisler BP, Raad RA, Esaian D, Sharon E, Schwartz DR. Apical ballooning and cardiomyopathy in a melanoma patient treated with ipilimumab: a case of takotsubo-like syndrome. Journal for immunotherapy of cancer. 2015;3(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ederhy S, Cautela J, Ancedy Y, Escudier M, Thuny F, Cohen A. Takotsubo-like syndrome in cancer patients treated with immune checkpoint inhibitors. JACC: Cardiovascular Imaging. 2018:2517. [DOI] [PubMed] [Google Scholar]
- 63.Scally C, Abbas H, Ahearn T, Srinivasan J, Mezincescu A, Rudd A, et al. Myocardial and systemic inflammation in acute stress-induced (Takotsubo) cardiomyopathy. Circulation. 2019;139(13):1581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim DM, Kim H, Yeon JH, Lee JH, Park HO. Identification of a Mitochondrial DNA Polymerase Affecting Cardiotoxicity of Sunitinib Using a Genome-Wide Screening on S. pombe Deletion Library. Toxicol Sci. 2016;149(1):4–14. [DOI] [PubMed] [Google Scholar]
- 65.Hudson G, Chinnery PF. Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet. 2006;15 Spec No 2:R244–52. [DOI] [PubMed] [Google Scholar]
- 66.Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17(3):235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Zang QS, Liu Z, Wu Q, Maass D, Dulan G, et al. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am J Physiol Cell Physiol. 2011;301(3):C695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787(5):312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neeve VC, Samuels DC, Bindoff LA, van den Bosch B, Van Goethem G, Smeets H, et al. What is influencing the phenotype of the common homozygous polymerase-γ mutation p.Ala467Thr? Brain. 2012;135(Pt 12):3614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schapira AH. Mitochondrial disease. Lancet. 2006;368(9529):70–82. [DOI] [PubMed] [Google Scholar]
- 72.Stuhlmiller TJ, Zawistowski JS, Chen X, Sciaky N, Angus SP, Hicks ST, et al. Kinome and Transcriptome Profiling Reveal Broad and Distinct Activities of Erlotinib, Sunitinib, and Sorafenib in the Mouse Heart and Suggest Cardiotoxicity From Combined Signal Transducer and Activator of Transcription and Epidermal Growth Factor Receptor Inhibition. J Am Heart Assoc. 2017;6(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jensen BC, Parry TL, Huang W, Ilaiwy A, Bain JR, Muehlbauer MJ, et al. Non-Targeted Metabolomics Analysis of the Effects of Tyrosine Kinase Inhibitors Sunitinib and Erlotinib on Heart, Muscle, Liver and Serum Metabolism In Vivo. Metabolites. 2017;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blanca AJ, Ruiz-Armenta MV, Zambrano S, Miguel-Carrasco JL, Arias JL, Arévalo M, et al. Inflammatory and fibrotic processes are involved in the cardiotoxic effect of sunitinib: Protective role of L-carnitine. Toxicol Lett. 2016;241:9–18. [DOI] [PubMed] [Google Scholar]
- 75.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32. [DOI] [PubMed] [Google Scholar]
- 76.Yang ZH, Emma-Okon B, Remaley AT. Dietary marine-derived long-chain monounsaturated fatty acids and cardiovascular disease risk: a mini review. Lipids Health Dis. 2016;15(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sourdon J, Lager F, Viel T, Balvay D, Moorhouse R, Bennana E, et al. Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism. Theranostics. 2017;7(11):2757–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stuhlmiller TJ, Zawistowski JS, Chen X, Sciaky N, Angus SP, Hicks ST, et al. Kinome and Transcriptome Profiling Reveal Broad and Distinct Activities of Erlotinib, Sunitinib, and Sorafenib in the Mouse Heart and Suggest Cardiotoxicity From Combined Signal Transducer and Activator of Transcription and Epidermal Growth Factor Receptor Inhibition. Journal of the American Heart Association. 2017;6(10):e006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lamore SD, Ahlberg E, Boyer S, Lamb ML, Hortigon-Vinagre MP, Rodriguez V, et al. Deconvoluting kinase inhibitor induced cardiotoxicity. Toxicological Sciences. 2017;158(1):213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suvendu G, Manivannan J, Srinivasan B, Sundaresan L, Gajalakshmib P, Chatterjee S. A proteome-wide systems toxicological approach deciphers the interaction network of chemotherapeutic drugs in the cardiovascular milieu. RSC Advances. 2018;8:20211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Juran BD, Atkinson EJ, Schlicht EM, Fridley BL, Lazaridis KN. Primary biliary cirrhosis is associated with a genetic variant in the 3’ flanking region of the CTLA4 gene. Gastroenterology. 2008;135(4):1200–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang K, Zhu Q, Lu Y, Lu H, Zhang F, Wang X, et al. CTLA-4 +49 G/A Polymorphism Confers Autoimmune Disease Risk: An Updated Meta-Analysis. Genet Test Mol Biomarkers. 2017;21(4):222–7. [DOI] [PubMed] [Google Scholar]
- 83.Nisticò L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet. 1996;5(7):1075–80. [DOI] [PubMed] [Google Scholar]
- 84.Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E, Mougenot JF, Bach JF, Caillat-Zucman S. CTLA-4 gene polymorphism is associated with predisposition to coeliac disease. Gut. 1998;43(2):187–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chang WW, Zhang L, Yao YS, Su H. Association between CTLA-4 exon-1 +49A/G polymorphism and systemic lupus erythematosus: an updated analysis. Mol Biol Rep. 2012;39(9):9159–65. [DOI] [PubMed] [Google Scholar]
- 86.Torres-Carrillo N, Ontiveros-Mercado H, Torres-Carrillo NM, Parra-Rojas I, Rangel-Villalobos H, Ramírez-Dueñas MG, et al. The −319C/+49G/CT60G haplotype of CTLA-4 gene confers susceptibility to rheumatoid arthritis in Mexican population. Cell Biochem Biophys. 2013;67(3):1217–28. [DOI] [PubMed] [Google Scholar]
- 87.Patel H, Mansuri MS, Singh M, Begum R, Shastri M, Misra A. Association of Cytotoxic T-Lymphocyte Antigen 4 (CTLA4) and Thyroglobulin (TG) Genetic Variants with Autoimmune Hypothyroidism. PLoS One. 2016;11(3):e0149441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Birlea SA, Laberge GS, Procopciuc LM, Fain PR, Spritz RA. CTLA4 and generalized vitiligo: two genetic association studies and a meta-analysis of published data. Pigment Cell Melanoma Res. 2009;22(2):230–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huber A, Menconi F, Corathers S, Jacobson EM, Tomer Y. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms. Endocr Rev. 2008;29(6):697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hradsky O, Dusatkova P, Lenicek M, Bronsky J, Nevoral J, Vitek L, et al. The CTLA4 variants may interact with the IL23R- and NOD2-conferred risk in development of Crohn’s disease. BMC Med Genet. 2010;11:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li M, Zheng H, Li T, Gao P, Zhang XL, Liu DW. Cytotoxic T-lymphocyte associated antigen-4 gene polymorphisms and primary biliary cirrhosis: a systematic review. J Gastroenterol Hepatol. 2012;27(7):1159–66. [DOI] [PubMed] [Google Scholar]
- 92.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Subbiah V, Kurzrock R. The Marriage Between Genomics and Immunotherapy: Mismatch Meets Its Match. Oncologist. 2019;24(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell. 2018;33(5):843–52.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun. 2017;8(1):1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362(6411). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brogden KA, Parashar D, Hallier AR, Braun T, Qian F, Rizvi NA, et al. Genomics of NSCLC patients both affirm PD-L1 expression and predict their clinical responses to anti-PD-1 immunotherapy. BMC Cancer. 2018;18(1):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goltz D, Gevensleben H, Vogt TJ, Dietrich J, Golletz C, Bootz F, et al. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight. 2018;3(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zarzour A, Kim HW, Weintraub NL. Epigenetic Regulation of Vascular Diseases. Arterioscler Thromb Vasc Biol. 2019;39(6):984–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ordovás JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. 2010;7(9):510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yi M, Yu S, Qin S, Liu Q, Xu H, Zhao W, et al. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol. 2018;11(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gong J, Chehrazi-Raffle A, Placencio-Hickok V, Guan M, Hendifar A, Salgia R. The gut microbiome and response to immune checkpoint inhibitors: preclinical and clinical strategies. Clin Transl Med. 2019;8(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Silverman GJ, Azzouz DF, Mor A. Immune checkpoint inhibitors and the union of bugs against cancer. Kidney Int. 2018;93(5):1030–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7. [DOI] [PubMed] [Google Scholar]
- 109.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vivarelli S, Salemi R, Candido S, Falzone L, Santagati M, Stefani S, et al. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers (Basel). 2019;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dreyfuss AD, Bravo PE, Koumenis C, Ky B. Precision Cardio-Oncology. J Nucl Med. 2019;60(4):443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Salem J, Allenbach Y, Vozy A, Brechot N, Johnson D, Moslehi J, et al. Abatacept for Severe Immune Checkpoint Inhibitor–Associated Myocarditis. New England Journal of Medicine. 2019;380:2377–9. [DOI] [PubMed] [Google Scholar]
- 113.Shoushtari AN, Friedman CF, Navid-Azarbaijani P, Postow MA, Callahan MK, Momtaz P, et al. Measuring Toxic Effects and Time to Treatment Failure for Nivolumab Plus Ipilimumab in Melanoma. JAMA Oncol. 2018;4(1):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Di Lorenzo G, Autorino R, Bruni G, Cartenì G, Ricevuto E, Tudini M, et al. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann Oncol. 2009;20(9):1535–42. [DOI] [PubMed] [Google Scholar]
- 115.Herrmann J, Yang EH, Iliescu CA, Cilingiroglu M, Charitakis K, Hakeem A, et al. Vascular Toxicities of Cancer Therapies: The Old and the New--An Evolving Avenue. Circulation. 2016;133(13):1272–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl_4):iv119–iv42. [DOI] [PubMed] [Google Scholar]
- 117.Neilan TG, Rothenberg ML, Amiri-Kordestani L, Sullivan RJ, Steingart RM, Gregory W, et al. Myocarditis Associated with Immune Checkpoint Inhibitors: An Expert Consensus on Data Gaps and a Call to Action. Oncologist. 2018;23(8):874–8. [DOI] [PMC free article] [PubMed] [Google Scholar]