

Abstract
Optimized NMR experiments are developed for isolating magnetization belonging to the I=1/2 manifolds of 13CH3 methyl groups in proteins, enabling the manipulation of the magnetization of a 13CH3 moiety as if it were an AX (1H-13C) spin-system. These experiments result in the same ‘simplification’ of a 13CH3 spin-system that would be obtained from the production of {13CHD2}-methyl-labeled protein samples. The sensitivity of I=1/2 manifold-selection experiments is a factor of approximately 2 less than that of the corresponding experiments acquired on {13CHD2}-labeled methyl groups. The methodology described here is primarily intended for small-to-medium sized proteins, where the losses in sensitivity associated with the isolation of I=1/2 manifold transitions can be tolerated. Several NMR applications that benefit from simplification of the 13CH3 (AX3) spin-systems are described, with an emphasis on the measurements of methyl 1H-13C residual dipolar couplings in a {13CH3}-methyl-labeled deletion mutant of the human chaperone DNAJB6b, where modulation of NMR signal intensities due to evolution of methyl 1H-13C scalar and dipolar couplings follows a simple cosine function characteristic of an AX (1H-13C) spin-system, significantly simplifying data analysis.
Keywords: magnetization, methyl groups, nuclear magnetic resonance, proteins, residual dipolar couplings
The design of multinuclear, multi-pulse experiments has been critical to the development of NMR spectroscopy as a powerful tool for studying protein structure and dynamics. NMR experiments targeting methyl groups as unique probes of molecular structure and motion, play a special role in these developments yielding considerable insights into numerous biochemical processes.[1] The degree of sophistication of NMR experiments is to a large extent predicated on the complexity of targeted spin-systems. The eigenstates of complex spin systems containing magnetically equivalent nuclei such as AX3 or AX2 groups in protein side-chains, are usually divided onto several classes (manifolds) using the standard rules of angular momentum theory.[2] For example, in the case of an AX3 (13CH3) methyl moiety, the energy level diagram contains one manifold with spin I=3/2 and two with I=1/2. The magnetization of each of the two I=1/2 manifolds, as well as their sum, behave in a manner analogous to that of a simple AX spin system. Thus, the ability to manipulate the magnetization in a manner that enables separation of I=3/2 and I=1/2 manifolds would reduce the complexity of the 13CH3 spin-system to the simple AX case. The problem of separation of 13CH3 methyl transitions belonging to I=3/2 and I=1/2 manifolds was tackled previously,[3] where NMR experiments were designed that allow the 1H coherences of the two manifolds to be separated using extensive phase-cycling of radio-frequency (RF) pulses. This earlier work was primarily geared towards the measurement of Carr-Purcell-Meiboom-Gill (CPMG)[4,5] relaxation dispersion profiles, targeting the isolated 1H magnetization of the I=1/2 manifolds of 13CH3 methyls in protein systems undergoing chemical exchange.
Here, we address the problem of simplification of the 13CH3 spin-system in a more comprehensive manner. Making use of a simpler and more sensitive method for isolation of the 1H magnetization of the I=1/2 manifolds, we design optimized NMR experiments aimed at the sequestration of both 1H and 13C single quantum coherences of the I=1/2 manifolds, allowing one to manipulate the magnetization of a 13CH3 methyl as if it were an AX spin-system, not unlike that available via {13CHD2}-labeling of methyl groups. The methodology is tested on two {13CH3}-methyl-labeled proteins: {U-[2H]; Ileδ1-[13CH3]; Leu,Val-[13CH3,12CD3]}-labeled human ubiquitin (8.5 kDa; 5 and 25°C) and {U-[2H]; Alaβ-[13CH3]}-labeled malate synthase G (MSG; 82 kDa; 37°C). We show that the I=1/2 manifold-selection experiments are only a factor of ~2 less sensitive than the (same) NMR experiments acquired on {13CHD2}-methyl-labeled proteins, {U-[2H]; Ileδ1-[13CHD2]; Leu,Val-[13CHD2,12CD3]}-ubiquitin and {U-[2H]; Alaβ-[13CHD2]}-MSG. Finally, the application of I=1/2 manifold selection to the measurement of methyl 1H-13C residual dipolar couplings (RDCs) in a {U-[2H]; Ileδ1-[13CH3]; Leu,Val-[13CH3,12CD3]}-labeled deletion mutant of the human DNAJB6b chaperone, ΔST-DNAJB6b (25 kDa; 25°C), is described. The resulting modulation of NMR signal intensities due to evolution of methyl 1H-13C scalar and dipolar couplings follows that of an AX (1H-13C) spin-system, significantly simplifying data analysis.
An energy level diagram of a spin system consisting of three magnetically equivalent nuclei in a methyl group (H3) is shown in Figure 1. Although a 13CH3 methyl group is considered in practice, the transitions of 13C nuclei have been omitted from the figure for simplicity, as the state of the density matrix of a 13CH3 group can be represented as a tensorial product of the 13C spin operator and the density matrix containing 1H states. The I=3/2 and two I=1/2 manifolds are independent of each other under the effect of RF pulses or any coherent NMR interactions but can be coupled through spin relaxation. In the macro-molecular limit, transverse spin relaxation of the inner 1H transitions of the I=3/2 manifold and the 1H transitions of the I=1/2 manifold occurs with slower rates (red arrows), while the outer transitions of the I=3/2 manifold are characterized by faster rates of decay (blue arrows).[6]
Figure 1.
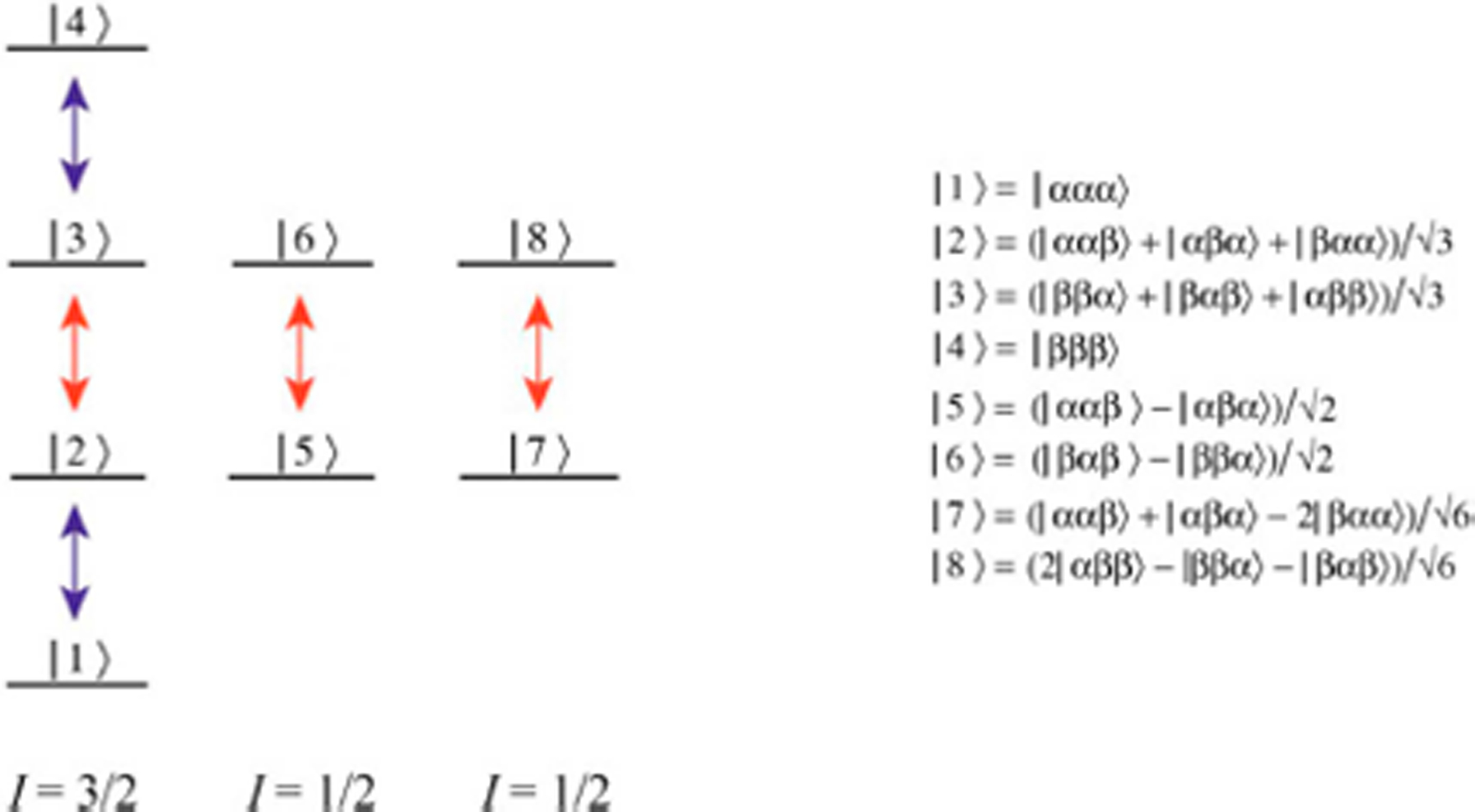
Energy level diagram for the X3 spin-system of a methyl group. Slow and fast relaxing single-quantum 1H transitions are shown by the red and blue arrows, respectively. The eight 1H eigenstates are depicted by |i,j,k>(i,j,k ∈ {α,β}). The spin quantum numbers, I, of the three manifolds are specified below the diagram. For simplicity, only the 1H part of the 13CH3 spin-system is shown.
The optimized pulse schemes that have been developed to sequester the coherences belonging to the I=1/2 manifolds are shown in Figure 2. A detailed description of these schemes ‘tracking’ how each of the transitions illustrated in Figure 1 evolves during the course of the experiment using single transition spin operators is provided in the SI. Briefly, the 13CH3 methyl magnetization is prepared first in a state in which only the slowly relaxing coherences (shown with red arrows in Figure 1) are present. Subsequent application of a 1H pulse of flip-angle α =sin−1(2/3)=41.81° (shown in green in Figure 2) is central to the approach developed here. This pulse eliminates the slow-relaxing 1H transitions of the I=3/2 manifold, ‘recreates’ some fast-relaxing 1H coherences (shown with blue arrows in Figure 1), and produces a mixture of methyl 1H coherences: double quantum, ‘DQ’, triple-quantum, ‘TQ’, and zero-quantum, ‘ZQ’ (see SI for details). The cycling of the phase of the 1H pulse of flip-angle α with concomitant retention of the receiver phase eliminates all the 1H coherences of even order (ZQ and DQ), while the fast-relaxing 1H coherences of the I=3/2 manifold are eliminated by the second application of the element of duration 2τb and the associated phase-cycling of the subsequent 13C 90° pulse, so that after the elements of the scheme enclosed in dashed boxes in Figure 2, the state of the density matrix of methyl 1H magnetization, σH can, to within a multiplication factor, be described by,
| (1) |
Figure 2.
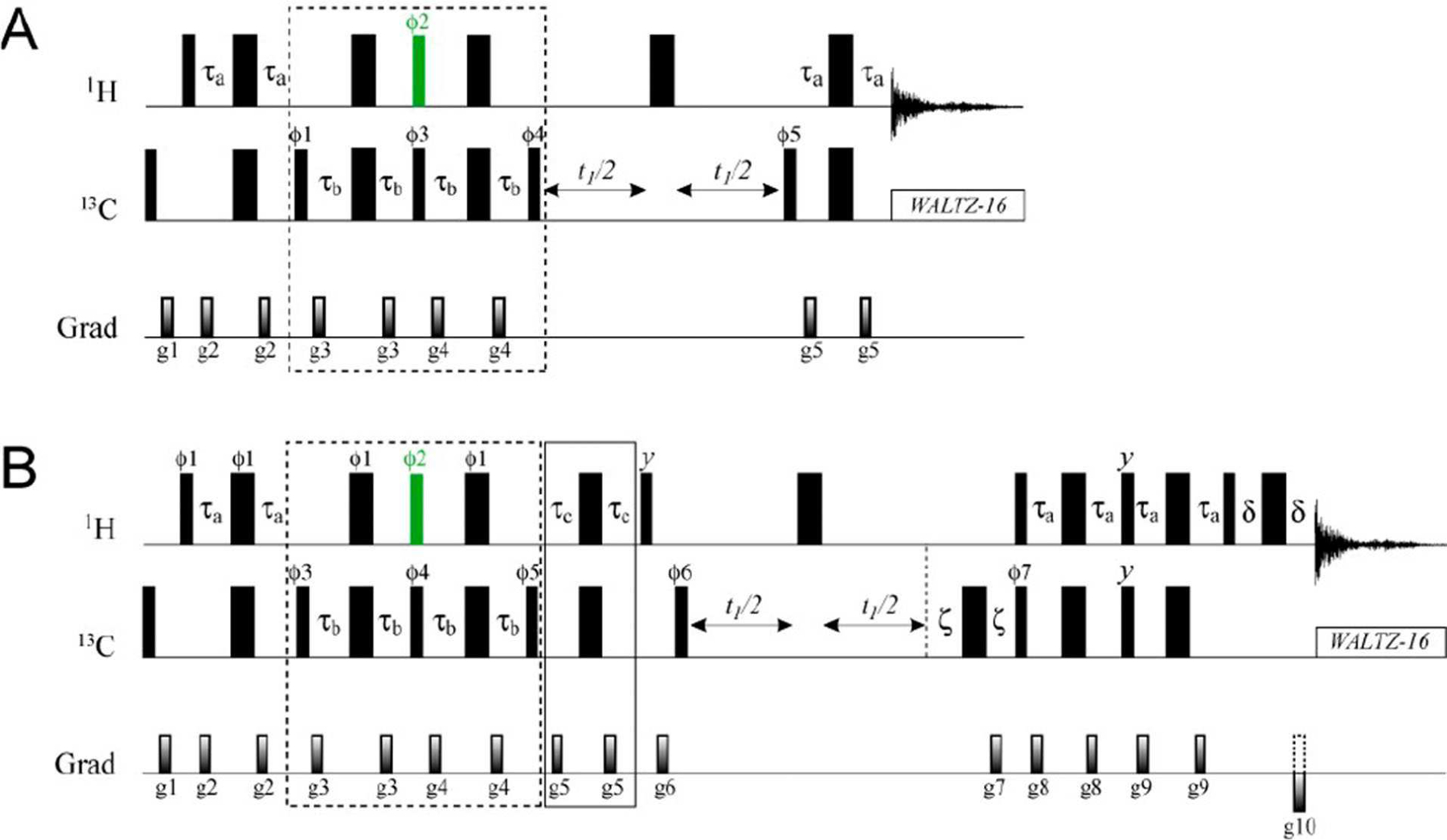
Pulse sequences for separation of I=1/2 manifold transitions in 13CH3 methyl groups with (A) a HMQC-’read-out’, and (B) active elimination of 1H TQ coherences and a gradient-selected sensitivity enhanced HSQC ‘read-out’ scheme.[7,8] Details of experimental parameters are provided in the Supplementary Information (Figure S1).
If the order of 1H coherences is not ‘perturbed’ in the rest of the experiment as is the case, for example, in HMQC-type ‘read-out’ schemes,[9,10] the selection of the I=1/2 manifold transitions (the first two terms in Eq. (1)) occurs ‘naturally’, as the third term containing 1H TQ coherences will not lead to observable magnetization at the end of the experiment. An example of such a scheme is presented in Figure 2A. The ratio of sensitivity of the I=1/2 selection in Figure 2A, where, according to Eq. (1), √5/3 of the I=1/2 magnetization is recovered in a single scan, to the previously published scheme[3] (where 3/5 of the I=1/2 magnetization is recovered per scan) is 5√5/9=1.24. We have been able to verify this gain in sensitivity on a sample of {13CH3}-labelled ubiquitin: the average ratio of signal intensities was 1.26±0.02 for the two experiments. In addition, using a straightforward extension of the scheme in Figure 2A to measure methyl 1H transverse spin relaxation rates, R2, we verified that the I=1/2 1H coherences in ubiquitin relax slower (6.3±1.7 s−1 at 25°C) than the slow relaxing 1H coherences of the I=3/2 manifold (7.6±1.7 s−1; selected as described earlier[3]) by ~20% on average - in agreement with expectations based on earlier predictions that the I=1/2 1H coherences relax 2.2-times slower than the inner transitions of the I=3/2 manifold due to interactions with external proton spins.[3] Note the contribution of external protons to methyl 1H transverse relaxation rates is relatively small in perdeuterated proteins, where methyl 1H-13C dipolar interactions constitute the main relaxation mechanism.
Many NMR applications require that the selected I=1/2 magnetization be manipulated as a ‘standalone’ AX spin-system. In particular, if isolation of single-quantum (SQ) 13C coherences of the I=1/2 manifolds is desired and/or gradient-selected sensitivity enhancement schemes[7,8] are employed, the third term of Eq. (1), containing 1H TQ coherences, will unavoidably lead to generation of SQ 13C coherences of the I= 3/2 manifold. Depending on the details of the NMR pulse-scheme employed, these coherences will be either directly observable during the indirect and direct acquisition periods of a 2D experiment or will generate detectable magnetization due to large differences in transverse relaxation properties of individual 13C transitions. It is important, therefore, to achieve ‘clean’ selection of I=1/2 magnetization by active elimination of any terms belonging to the I=3/2 manifold. Such a scheme is shown in Figure 2B, where elimination of 1H TQ coherences is achieved via their selective refocusing with respect to 13C spins (the element of duration 2τc =1/(6JCH) enclosed in the solid box in Figure 2B; see SI for details). Of note, only when 1H SQ magnetization deriving from the I=1/2 manifold is completely isolated and not ‘contaminated’ by 1H TQ coherences, sensitivity enhancement by preservation of equivalent pathways (PEP),[11] employed in the scheme of Figure 2B, provides the expected gain of √2 in signal-to-noise (39±2 % on average measured for {13CH3}-ubiquitin). The theoretical efficiency E of the elements that ensure the proper isolation of the I=1/2 manifold, which we define as the fraction of the I=1/2 magnetization that is recovered at the end of the experiment in the absence of losses due to relaxation and pulse imperfections, is equal to (√5/3) cos(π/6) = 0.645. This loss in signal is largely compensated for by the sensitivity enhancement used in the scheme of Figure 2B. The measured average sensitivity ratios of 0.165±0.015 and 0.283±0.022 obtained for ubiquitin (25°C) in the experiment of Figure 2B compared to [1H-13C]-HMQC spectra without and with selection of slow-relaxing transitions,[6] respectively, are therefore in good agreement with expectations considering that the total amount of detectable 1H magnetization contained in the I=1/2 manifolds is 1/6 of that in the whole of the (13C)H3 group and 1/3 if only slow-relaxing transitions are selected.
Since the magnetization from I=1/2 manifolds isolated by the scheme in Figure 2B is equivalent to that of an AX (1H-13C) spin-system with respect to any coherent NMR interactions, it is of interest to compare the sensitivity attainable in this experiment with that of its counterparts targeting {13CHD2}-methyl-labeled samples. In the absence of relaxation, the total amount of 1H magnetization contained in I=1/2 manifolds of a 13CH3 group is one half that available in a 13CHD2 spin-system. As the selection for the I=1/2 manifolds can only be performed with a given efficiency E (defined as above), any deviations from a factor of ~E/2=0.32 in sensitivity ratios between the experiment of Figure 2B recorded on the {13CH3}-labeled samples and the sensitivity-enhanced HSQC on the {13CHD2}-labeled samples (the scheme in Figure 2B excluding the elements enclosed in boxes), under otherwise identical experimental conditions, must arise from relaxation and pulse imperfections. Indeed, the recovery of methyl 1H magnetization to equilibrium between the scans of NMR experiments is much slower in {13CHD2}-methyls[12] (Figure 3A), and the results of sensitivity comparisons will therefore depend on the scan repetition rate. Figure 3B shows average ratios of 2D cross-peak intensities obtained in the experiment of Figure 2B recorded on a sample of {13CH3}-ubiquitin and the sensitivity-enhanced HSQC acquired on the {13CHD2}-labeled sample of the same protein concentration (1.3 mM) as a function of inter-scan delay. From almost 50% of {13CHD2} intensity at an inter-scan delay of 1 s, the ratio slowly approaches the value of E/2 for slower repetition rates. The same comparison performed between Alaβ-{13CH3}- and Alaβ-{13CHD2}-labeled samples of MSG (0.6 mM; 37°C; 33 peaks) yields somewhat higher ratios (0.62±0.12 for an inter-scan delay of 1 s) as the 1H longitudinal relaxation rate (R1) in both methyl isotopomers is slower on average in alanines of MSG. We note that alternative methyl isotope labeling schemes of Val and Leu residues can be expected to provide somewhat different results in sensitivity comparisons between the experiments targeting {13CHD2}-labeled samples and those performed on 13CH3 methyls with I=1/2 manifold selection. For example, if both methyl groups of isopropyl moieties of Val and Leu are labeled with {13CHD2} methyl isotopomers, one would expect a substantial increase in 1H R1 rates due to spatial proximity of the two prochiral methyl groups.
Figure 3.
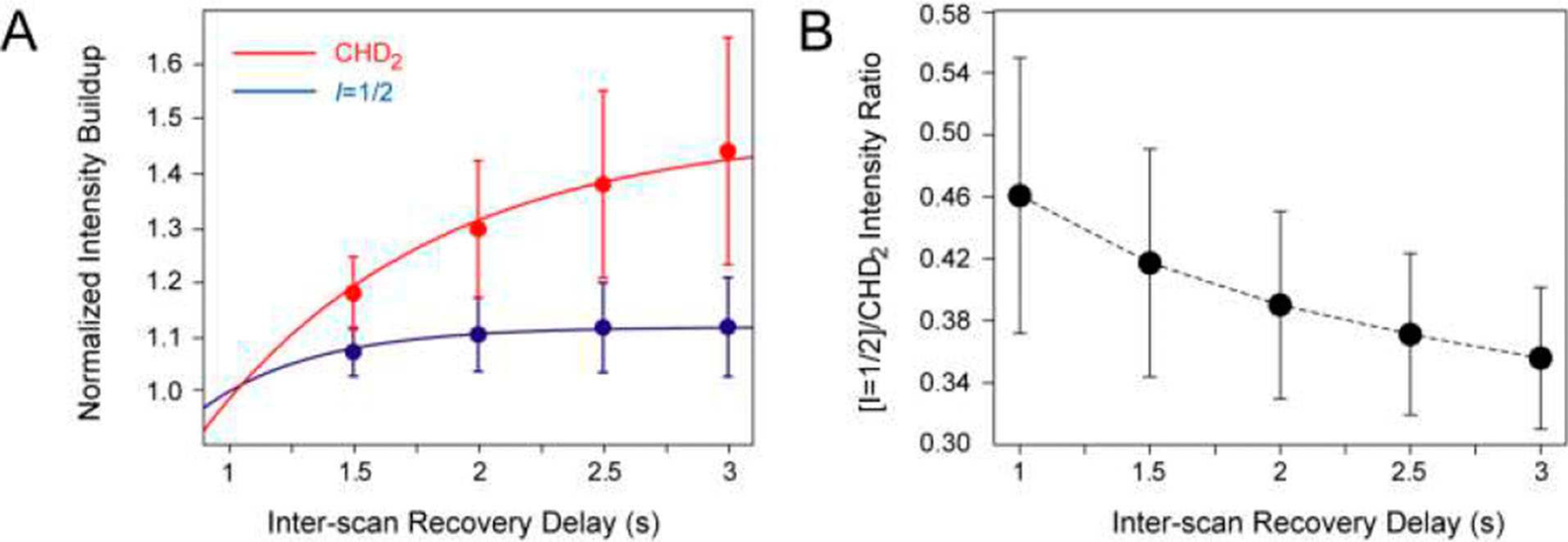
(A) Build-up of an average 2D cross-peak intensity normalized to the intensity at an inter-scan delay equal to 1 s (y-axis) versus the inter-scan delay duration (x-axis) in the gradient-selected sensitivity enhanced HSQC acquired on 1.3 mM {U-[2H]; Ileδ1-[13CHD2]; Leu,Val-[13CHD2,12CD3]}-ubiquitin (red filled circles) and the I=1/2 manifold selection experiment in Figure 2B acquired on 1.3 mM {U-[2H]; Ileδ1-[13CH3]; Leu,Val-[13CH3,12CD3]}-ubiquitin (blue filled circles). The error bars derive from the r.m.s.d. of the intensities of 29 methyl cross-peaks. Least-squares best-fits of the data to the function I0[1 - exp(−R1d1)], where I0 is the intensity at equilibrium, R1 is the average longitudinal methyl 1H spin relaxation rate and d1 is the duration of the inter-scan delay (shown with red and blue curves), provide R1 rates of 1.09±0.20 s−1 and 2.23±0.25 s−1 for the {13CHD2}- and {13CH3}-labeled samples, respectively. (B) Average ratios of 2D cross-peak intensities obtained in the experiment of Figure 2B recorded on {13CH3}-labeled ubiquitin and the gradient-selected sensitivity-enhanced HSQC acquired on {13CHD2}-labeled ubiquitin (y-axis) as a function of the inter-scan delay duration (x-axis). The error bars derive from the r.m.s.d. of the ratios of intensities over 29 cross-peaks. The dashed curve connecting the experimental ratios is drawn to guide the eye. All the spectra were recorded at 500 MHz and 5°C. Very similar values are obtained at 25°C.
Methyl-{13CHD2}-labeled samples can be used to estimate the experimental losses of magnetization at each stage of I=1/2 selection in the scheme of Figure 2B. Comparisons of 2D signal intensities obtained using this experiment with and without each of the elements enclosed in boxes in Figure 2B using {13CHD2}-labeled ubiquitin (25°C) provided the following average efficiencies associated with each successive element: 0.73 for the first element (enclosed in the dashed box; Figure 2B) compared with the theoretical factor (√5/3) = 0745, and 0.84 for the second element (enclosed in the solid box) compared with the theoretical factor of (√3/2) = 0.866. Each of the two experimental values and their product (0.61; to be compared with the value of I=1/2 selection efficiency E=0.64) are slightly lower than the corresponding theoretical estimates reflecting minor losses of magnetization due to relaxation and pulse imperfections.
A number of NMR applications can be envisaged that would benefit from the simplification of the 13CH3 spin-system achieved using the approach described above. For example, using the I=1/2 manifold selecting elements of the scheme in Figure 2B, we developed a SQ 1H CPMG relaxation dispersion experiment (SI, Figure S2) that was tested on {13CH3}-labeled ubiquitin providing flat dispersion profiles for all methyl sites, as well as on a sample of {13CH3}-labeled ΔST-DNAJB6b, where relaxation dispersions were observed for the methyl sites that participate in chemical exchange with sparsely-populated, oligomeric states of the protein. The experiment in Figure S2 is on average ~2.5-fold more sensitive than that developed previously with I=1/2 manifold selection,[3] due to the use of gradients to eliminate 1H TQ coherences in the latter, and the use of more efficient I=1/2 manifold selection and sensitivity enhancement in the former. However, a much more sensitive methyl-TROSY-based SQ methyl-1H CPMG experiment, developed recently by Kay and co-workers[13] for 13CH3 methyls, largely eliminates the need for the simplification of the 13CH3 spin-system in NMR studies that use methyl protons as probes of chemical exchange on the micro-to-millisecond timescale. Likewise, more sensitive SQ 13C CPMG experiments exist that target 13CH3 methyls directly.[14] With these considerations in mind, we choose to focus in more detail on applications that benefit from a reduction in the complexity of a 13CH3 spin-system that is not achievable without isotopic labeling of methyl moieties in proteins with 13CHD2 groups that (if deuterons are treated as ‘silent’ nuclei) effectively represent fast-rotating AX spin-systems.[15–17]
One such application is the measurement of methyl 1H-13C scalar (JCH) and residual dipolar couplings (RDCs; DCH) in the indirect (13C) dimension of 2D methyl 1H-13C correlation spectra of proteins. Figure 4A shows the pulse-scheme that makes use of the I=1/2 selecting elements of the experiment in Figure 2B to effectively reduce the 13CH3 spin-system to a 1H-13C one with respect to the evolution of magnetization due to JCH and DCH couplings. Aiming at determination of methyl-containing side-chain rotamer distributions and refinement of side-chain conformations, we measured methyl 1H-13C RDCs in a {13CH3}-labeled ΔST-DNAJB6b first using a simplified version of the constant-time HSQC-based experiment of Ottiger et al.[18] In the absence of relaxation, the modulation of the methyl signal intensity I occurring during the evolution delay t2, is given by, I(t2)=I0{3 cos(3π[JCH+DCH]t2) + cos(π[JCH+DCH]t2)}, where I0 is initial intensity, the first term accounts for evolution of the two outer (fast-relaxing) lines of the HSQC 13C quadruplet, and the second term describes that of the two inner (slow-relaxing) transitions (shown with the dashed red curves in the upper row of Figure 4B). Due to very different transverse relaxation rates of the inner and outer 13C transitions, the experimental modulation patterns (black filled circles in the upper row of Figure 4B) show considerable deviations from ‘relaxation-free’ predictions - especially so for well-ordered methyl sites located deep in the hydrophobic core of the protein structure as is the case, for example, for Val11γ1 (see Figure 4C for location of the corresponding methyl groups in the structure of ΔST-DNAJB6b). Spin relaxation cannot be accounted for in a rigorous manner in these measurements, and approximate approaches have to be devised. However, in the measurements performed with the scheme in Figure 4A, the signal modulation patterns are reduced to a simple cosine function I(t2)=I0cos(π[JCH+DCH]t2) as shown in the lower row of panels in Figure 4B, where all the effects of relaxation during the constant-time period are contained in I0. Although the experiment in Figure 4A is on average a factor of ~4.5 less sensitive than that targeting the whole of 13CH3 moiety in ΔST-DNAJB6b, the analysis of the I=1/2 manifold selected data is significantly simplified without compromising the accuracy of the derived RDC values. The robustness of the experiment in Figure 4A with respect to deviations of the couplings (JCH+DCH) from the nominal value of 125 Hz is estimated in the SI (Figure S4).
Figure 4.
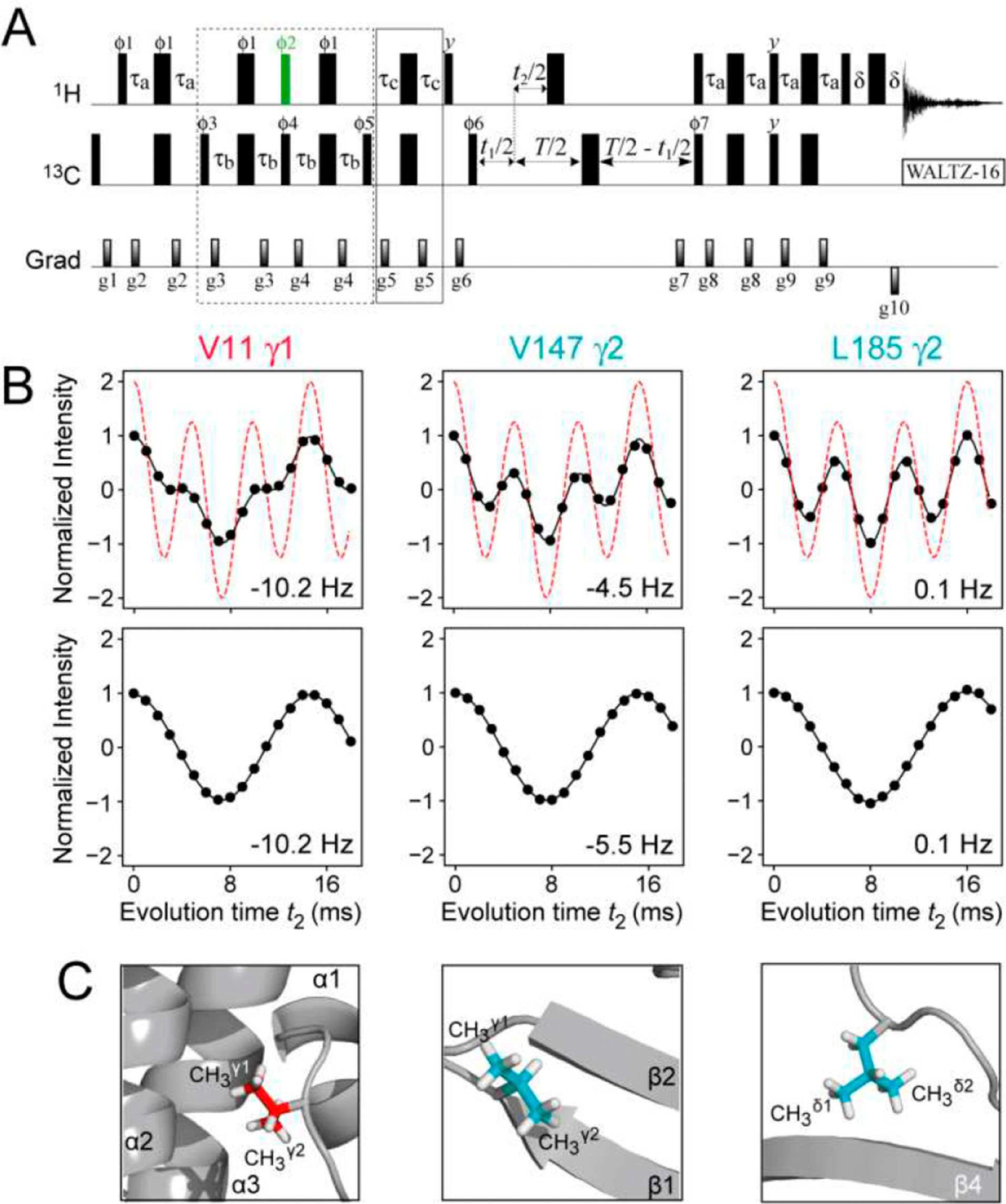
(A) Pulse scheme for the measurement of 1H-13C scalar and residual dipolar couplings in methyl groups of proteins with I=1/2 manifold selection in 13CH3 methyl groups. See SI, Figure S1B, for experimental parameters. A constant-time (CT) delay T=28 ms was used, although sensitivity permitting, any value of T can be used in selectively {13CH3}-labeled protein samples. (B) Plots showing the modulation of the intensity of the 13CH3 correlations in the spectra of {U-[2H]; Ileδ1-[13CH3]; Leu,Val-[13CH3,12CD3]}-labeled ΔST-DNAJB6b (600 MHz; 25°C) recorded using the scheme in panel (A) as a function of the coupling evolution period (t2) without (top row) and with (bottom row) I=1/2 manifold selection. The plot of intensity modulation in the absence of relaxation and without I=1/2 manifold selection is shown with red dashed lines (top row). The data in upper and lower rows are best-fit to the functions, I(t2)=I0{3cos(3πJCHt2)exp(−7RST)+cos(πJCHt2) exp(−RST)} and I(t2)=I0cos(πJCHt2), respectively, where the initial intensity I0, the coupling constant JCH, and the transverse relaxation rate of slow-relaxing carbon coherences RS, are optimized parameters. This function approximately accounts for relaxation of 13C magnetization during the period T. The values of DCH derived from the fits are indicated in each panel. The sample was aligned in 10 mg/mL phage pf1 (see SI for experimental conditions). (C) The locations of the corresponding methyl groups in the structure of ΔST-DNAJB6b are shown in ‘ball-and-stick’ representations.
Using the approach for I=1/2 manifold selection described here, experiments have been designed for the measurement of methyl 1H-13C RDCs directly from the 1H-13C splitting in the indirect (13C) dimension of 2D spectra. As the I=1/2 manifold selection scheme in Figures 2B and 4A effectively converts the 13C quadruplet in HSQC-type correlation maps of 13CH3 groups to a 13C-1H doublet, conventional in-phase/anti-phase (IPAP)[19,20] acquisition scheme developed for AX spin systems, can be applied in these experiments circumventing the need for more complicated signal manipulation schemes,[21] or the use of {13CHD2} methyl isotopomers.[22] We note, however, that for large proteins, the IP/AP scheme for methyl 1H-13C RDC measurements in the acquisition (1H) dimension of 2D spectra developed by Sprangers and Kay,[23] is expected to provide much higher sensitivity since no losses associated with the (unnecessary) simplification of the 13CH3 spin-system would be incurred in this case.
In conclusion, we present optimized NMR experiments for complete isolation of the magnetization belonging to the I=1/2 manifolds of 13CH3 methyl groups, enabling the manipulation of the magnetization of a 13CH3 moiety as if it were an AX (1H-13C) spin-system, thereby avoiding the need for production of {13CHD2}-methyl-labeled samples of small-to-medium sized proteins, where the losses in sensitivity associated with the isolation of the I=1/2 manifold can be tolerated. The sequestration of the I=1/2 manifold coherences described here can be used in practically any NMR applications that benefit from simplification of the 13CH3 (AX3) spin-system, commonly achieved by labeling of methyl sites with 13CHD2 isotopomers. In addition to the examples of such applications provided above, NMR experiments can also be developed for the measurement of 13C R2(R1ρ) and R1 relaxation rates of SQ 13C coherences of the I=1/2 manifold. It is worth noting that these relaxation rates will be equal to those of analogous 13C SQ coherences in 13CHD2 methyl groups only in the limit where the local motions of a methyl group (including rotation around the three-fold symmetry axis) are infinitely fast compared to the rate of global molecular tumbling[24] (i.e. when the auto-relaxation spectral density function is equal to that of cross-correlated relaxation). In methyl groups of small-to-medium sized proteins for which the methodology described here is mainly primarily intended, this condition is likely to be violated resulting in significant differences between the measured 13C relaxation rates. These differences are currently a subject of further investigation.
Supplementary Material
Acknowledgements
The authors thank Ad Bax for useful suggestions and James Baber for technical support. This work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (DK029023 to G.M.C.).
Footnotes
Supporting Information
Description of the evolution of magnetization during the pulse-schemes for I=1/2 manifold selection using single transition spin operators (Figure S1). Figure S2 showing the pulse scheme for a single-quantum (SQ) methyl 1H CPMG relaxation dispersion experiment with I=1/2 manifold selection. Figure S3 showing examples of SQ methyl 1H CPMG relaxation dispersion profiles obtained for selected residues of {13CH3}-labeled ΔST-DNAJB6b. Estimation of the robustness of the experiment for RDC measurements with respect to deviations of (JCH+DCH) couplings from their nominal value in the absence of alignment (125 Hz; Figure S4). ‘Materials and Methods’ section describing the procedures of NMR sample preparation and NMR experimental details.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is available on the WWW under https://doi.org/10.1002/cphc.201900959
References
- [1].Rosenzweig R, Kay LE, Annu. Rev. Biochem 2014, 83, 291–315. [DOI] [PubMed] [Google Scholar]
- [2].Corio PL, Structure of High-Resolution NMR Spectra; Academic Press: New York, 1966. [Google Scholar]
- [3].Tugarinov V, Kay LE, J. Am. Chem. Soc 2007, 129, 9514–9521. [DOI] [PubMed] [Google Scholar]
- [4].Carr HY, Purcell EM, Phys. Rev 1954, 4, 630–638. [Google Scholar]
- [5].Meiboom S, Gill D, Rev. Sci. Instrum 1958, 29, 688–691. [Google Scholar]
- [6].Tugarinov V, Kay LE, J. Am. Chem. Soc 2006, 128, 7299–7308. [DOI] [PubMed] [Google Scholar]
- [7].Kay LE, Keifer P, Saarinen T, J. Am. Chem. Soc 1992, 114, 10663–10665. [Google Scholar]
- [8].Schleucher J, Sattler M, Griesinger C, Angew. Chem. Int. Ed. Engl 1993, 32, 1489–1491. [Google Scholar]
- [9].Bax A, Griffey RH, Hawkings BL, J. Magn. Reson 1983, 55, 301–315. [Google Scholar]
- [10].Mueller L, J. Am. Chem. Soc 1979, 101, 4481–4484. [Google Scholar]
- [11].Palmer AG, Cavanagh J, Wright PE, Rance M, J. Magn. Reson 1991, 93, 151–170. [Google Scholar]
- [12].Ollerenshaw JE, Tugarinov V, Skrynnikov NR, Kay LE, Biomol J. NMR 2005, 33, 25–41. [DOI] [PubMed] [Google Scholar]
- [13].Yuwen T, Huang R, Vallurupalli P, Kay LE, Angew. Chem. Int. Ed. Engl 2019, 58, 6250–6254. [DOI] [PubMed] [Google Scholar]
- [14].Lundstrom P, Vallurupalli P, Religa TL, Dahlquist FW, Kay LE, Biomol J. NMR 2007, 38, 79–88. [DOI] [PubMed] [Google Scholar]
- [15].Ishima R, Louis JM, Torchia DA, J. Am. Chem. Soc 1999, 121, 11589–11590. [Google Scholar]
- [16].Ishima R, Petkova AP, Louis JM, Torchia DA, J. Am. Chem. Soc 2001, 123, 6164–6171. [DOI] [PubMed] [Google Scholar]
- [17].Tugarinov V, Kay LE, Biochemistry 2005, 44, 15970–15977. [DOI] [PubMed] [Google Scholar]
- [18].Ottiger M, Delaglio F, Marquardt JL, Tjandra N, Bax A, J. Magn. Reson 1998, 134, 365–369. [DOI] [PubMed] [Google Scholar]
- [19].Ottiger M, Delaglio F, Bax A, J. Magn. Reson 1998, 131, 373–378. [DOI] [PubMed] [Google Scholar]
- [20].Yang D, Nagayama K, J. Magn. Reson. Ser. A 1996, 118, 117–121. [Google Scholar]
- [21].Kontaxis G, Bax A, Biomol J. NMR 2001, 20, 77–82. [DOI] [PubMed] [Google Scholar]
- [22].Godoy-Ruiz R, Guo C, Tugarinov V, J. Am. Chem. Soc 2010, 132, 18340–18350. [DOI] [PubMed] [Google Scholar]
- [23].Sprangers R, Kay LE, J. Am. Chem. Soc 2007, 129, 12668–12669. [DOI] [PubMed] [Google Scholar]
- [24].Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE, J. Am. Chem. Soc 2003, 125, 10420–10428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.