

Abstract
Despite the enormous therapeutic potential of immune checkpoint blockade (ICB), it benefits only a small subset of patients. Some chemotherapeutics can switch ‘immune-cold’ tumours to ‘immune-hot’ to synergize with ICB. However, safe and universal therapeutic platforms implementing such immune effects remain scarce. We demonstrate that sphingomyelin-derived camptothecin nanovesicles (camptothesomes) elicit potent granzyme-B- and perforin-mediated cytotoxic T lymphocyte (CTL) responses, potentiating PD-L1/PD-1 co-blockade to eradicate subcutaneous MC38 adenocarcinoma with developed memory immunity. In addition, camptothesomes improve the pharmacokinetics and lactone stability of camptothecin, avoid systemic toxicities, penetrate deeply into the tumour and outperform the antitumour efficacy of Onivyde. Camptothesome co-load the indoleamine 2,3-dioxygenase inhibitor indoximod into its interior using the lipid-bilayer-crossing capability of the immunogenic cell death inducer doxorubicin, eliminating clinically relevant advanced orthotopic CT26-Luc tumours and late-stage B16-F10-Luc2 melanoma, and achieving complete metastasis remission when combined with ICB and folate targeting. The sphingomyelin-derived nanotherapeutic platform and doxorubicin-enabled transmembrane transporting technology are generalizable to various therapeutics, paving the way for transformation of the cancer immunochemotherapy paradigm.
Although ICB therapy (for example, α-CTLA-4, α-PD-L1 or α-PD-1) has transformed the cancer treatment paradigm, only a select subset of patients are responsive1,2. Against colorectal cancer (CRC) specifically, ICB is mostly ineffective—the exception being the ~4% of patients with mismatch repair deficiency or microsatellite instability-high tumours3,4. Extensive efforts have centred on employing therapeutic modalities (for example, chemotherapy, radiation, viral and targeted therapies, and therapeutic vaccines) that can turn ‘immune-cold’ into ‘immune-hot’ tumours to potentiate ICB immunotherapy5–10. Among these, immunogenic chemotherapy has shown remarkable potential to synergize with ICB (for example, increasing tumour-infiltrating CTLs). However, owing to poor pharmacokinetics, limited tumour accumulation and nonspecific toxicities to healthy tissues and immune cells, chemotherapeutic utility in enhancing the efficacy of ICB has been hindered.
Camptothecin (CPT), a potent chemotherapeutic against various cancers, including CRC, has shown the potential to enhance CTL-mediated tumour cell killing11. Nevertheless, its poor water solubility, severe adverse effects and lactone ring instability12 limit the clinical application of CPT and its combination with ICB. We have developed a sphingomyelin (SM)-derived CPT (SM-CPT) liposomal nanotherapeutic platform with different tumour-sensitive linkages (ester, glycine and disulfide bonds) and varied linker lengths. SM, a naturally occurring phospholipid and major component of high-density lipoprotein and animal cell membrane, contains a hydroxyl group, enabling conjugation to functional moieties (for example, hydroxyl and carboxylic groups) of therapeutics. SM-CPTs enhanced lactone stability and self-assembled into camptothesomes in aqueous medium driven by the amphiphilicity of SM. Structure–activity relationship analysis showed that a disulfide-bridged conjugate with a longer linker (SM-CSS-CPT, camptothesome-4) outperformed free CPT and other counterparts, exhibiting a prolonged circulation half-life, enhanced tumour uptake, no overt side effects, deep tumour penetration and efficient intratumoral CPT release. Camptothesome-4 outperformed Onivyde (an FDA-approved liposomal irinotecan, a CPT derivative) by bolstering tumour reduction and prolonging mouse survival. Furthermore, camptothesome-4 significantly induced tumour-infiltrating CD8, granzyme B, perforin, IFN-γ and cleaved caspase-3 (CC3) in CRC tumours, demonstrating CTL-elicited antitumour immunity. The increase in IFN-γ upregulated intratumoral PD-L1/PD-1 expression, which has been associated with an improved response to ICB13. These findings justified combining PD-L1/PD-1 blockade with camptothesome-4, leading to the eradication of MC38 tumours in 83.3% of immunocompetent mice.
Indoleamine 2,3-dioxygenase (IDO1), another independent crucial immune checkpoint, enzymatically degrades tryptophan, causing CTL anergy while activating regulatory T cells (Treg cells), resulting in immunosuppression in various tumours14–16. Consistent with the literature17,18, we found that IDO1 is expressed in CRC tumours, and that this increases in response to INF-γ production (Supplementary Figs. 20 and 21). To reverse IDO1-mediated immunosuppression, we proposed to use camptothesome-4 to co-deliver an IDO1 inhibitor, indoximod (IND) (due to its safety and potency)15,19. However, the limited solubility of IND rendered direct loading challenging. To tackle this problem, we conjugated IND to the immunogenic cell death (ICD) inducer doxorubicin (DOX), using DOX as a membrane-crossing carrier to import IND into the camptothesome. We specifically designed a pH-sensitive hydrazone linkage20 between DOX and IND that would break inside the nanovesicle under the acidic pH produced by a protonating agent, forming drug precipitates that cannot diffuse back across the lipid membrane. IND has been reported to synergize with DOX to elicit tumour regression21. With ICD-eliciting potential, DOX offers additional antitumour immunity benefits22. Strikingly, DOX-IND/camptothesome-4 cured a significant proportion of mice bearing advanced metastatic orthotopic CRC or late-stage subcutaneous CRC and melanoma tumours when functionalized with folate targeting and/or combined with PD-L1/PD-1 inhibitors.
Establishment of the camptothesome nanotherapeutic platform
Four different SM-CPTs were initially synthesized (Fig. 1c–f and Supplementary Schemes 1–4)—one with an ester bond (SM-ester-CPT), one with a disulfide linkage (SM-SS-CPT), one with a glycine bond (SM-glycine-CPT) and one with a disulfide linkage and a longer linker (SM-CSS-CPT). These linkages are sensitive to high hydrolase, cathepsin B and glutathione levels, respectively, in tumour tissues and cells23–26. Their chemical structures were confirmed by 1H NMR, 13C NMR and electrospray ionization mass spectrometry (Supplementary Methods and Supplementary Figs. 1–4). The longer linker was used to link the disulfide bond with SM by introducing succinate to form a noncarbonate ester (Supplementary Scheme 4), preventing spontaneous pyrolysis—a process leading to premature carbonate ester bond breakdown and CO2 release (Supplementary Scheme 2)27. All SM-CPTs greatly enhanced CPT lactone stability and efficiently self-assembled into liposomal nanovesicles (camptothesomes), as visualized by cryo-electron microscopy (cryo-EM); the camptothesomes showed significantly higher CPT drug loading capacity (DLC) and improved formulation stability compared to CPT physically encapsulated in conventional liposomes (Fig. 1h,i,l, and Supplementary Figs. 9d,e and 10). In addition, the self-assembly of SM-CPTs was substantiated using 1H NMR (Supplementary Fig. 5). Typical proton spectra were obtained for SM-CPTs, SM, cholesterol and DSPE-PEG2K in CDCl3 due to their free dispersion in this solvent. Camptothesomes in CDCl3 expressed all of the typical proton signals for each individual constituent. However, when collected in D2O, nearly all of the proton signals from individual components in the camptothesomes were suppressed, which can be attributed to their spontaneous self-assembly into camptothesomes (Supplementary Fig. 5), disrupting their free dispersion. The proton signals from the CPT in SM-CPTs were not seen in camptothesomes in D2O (Supplementary Fig. 5), suggesting successful packaging of the CPT into the lipid bilayer. Additionally, the fluorescence of the SM-CPTs was significantly quenched upon their incorporation into camptothesomes, indicating a strong π–π stacking interaction among SM-CPT molecules (CPT contains several aromatic rings) and further corroborating the self-assembly process (Fig. 1j and Supplementary Fig. 6). The camptothesomes displayed a narrow size distribution as reflected by a low polydispersity index (Fig. 1k, and Supplementary Fig. 10b). Camptothesome-4 had a smaller size and significantly longer formulation stability compared to camptothesome-2 and camptothesome-3 (Fig. 1m,n).
Fig. 1 |. Development of SM-derived CPT liposomal nanovesicles (camptothesomes).
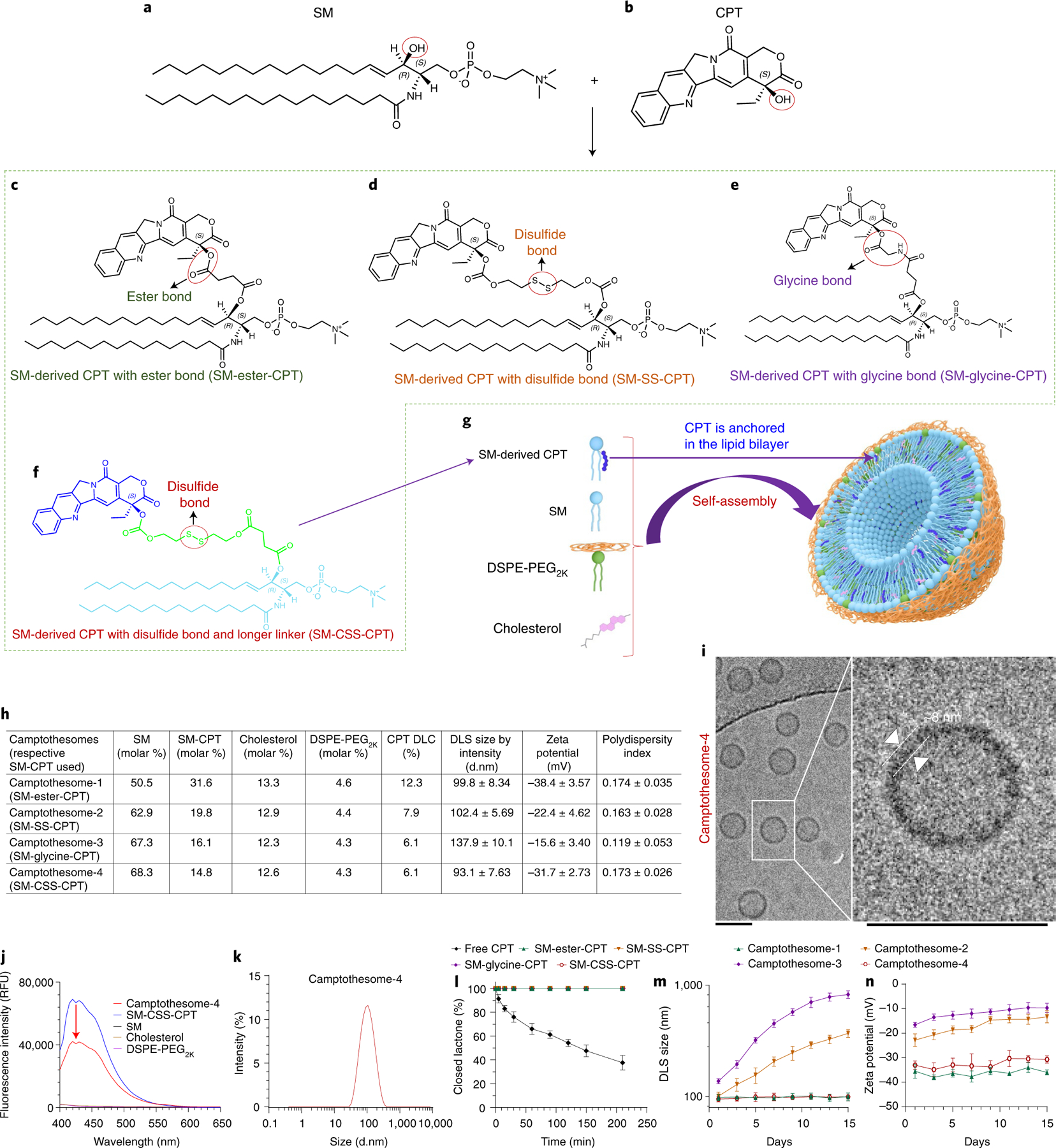
a,b, Chemical structures of SM (a) and CPT (b). c–f, Conjugation of SM and CPT resulted in SM-derived CPTs with either an ester bond (SM-ester-CPT) (c), a disulfide linkage (SM-SS-CPT) (d), a glycine bond (SM-glycine-CPT) (e), or a disulfide linkage and a longer linker (SM-CSS-CPT) (f). g, Schematic depicting the self-assembly of SM-CPT into a camptothesome. h, A table delineating the physicochemical characterizations of the four camptothesomes. DLS, dynamic light scattering; d.nm, diameter values in nanometres. i, cryo-EM for camptothesome-4. Scale bars, 100 nm. j, The fluorescence intensities (relative fluorescence units, RFU) for camptothesome-4, SM-CSS-CPT, SM, cholesterol and DSPE-PEG2K in methanol at equivalent concentrations. The significant fluorescence quenching for SM-CSS-CPT after self-assembly into camptothesome-4 demonstrates that there are strong π–π stacking interactions among SM-CSS-CPT molecules since CPT contains several aromatic rings. k, Representative DLS size distribution by intensity for camptothesome-4. For i–k, three independent experiments were performed to confirm the results. l, Percent closed lactone in PBS (pH 7.4) as a function of time for free CPT and the four different SM-CPT conjugates. SM-conjugated CPTs strongly prevented the lactone form from being converted into the inactive carboxylate form, enhancing CPT stability. m,n, Monitoring of the DLS size by intensity (m) and zeta potential (n) for each of the four different camptothesomes over time in 5% dextrose at 4 °C. Data in h (right portion) and l–n are expressed as mean ± s.d. (n = 3 independent experiments).
The maximum tolerated doses (MTDs) of the four different camptothesomes were evaluated in healthy BALB/c mice in comparison to free CPT. Various doses of camptothesomes and free CPT at a single intravenous (i.v.) injection were tested. The camptothesomes increased the MTD of CPT (5 mg kg−1) by 5- to 23-fold (to 30–120 mg CPT kg−1) without incurring evident adverse effects on healthy tissues, immune cells (leukocytes), erythrocytes and thrombocytes (Fig. 2a–d and Supplementary Fig. 11). Although CPT at MTD did not cause marked mouse weight loss, it induced severe systemic toxicities by significantly deviating the alkaline phosphatase, alanine transaminase, blood urea nitrogen, creatinine, glucose and total protein levels from their normal values, decreasing lymphocytes counts and haemoglobin concentrations, and inducing overt hepatic steatosis and diffuse microvesicular degeneration of hepatocytes in liver and haemorrhage in interstitial tissue in kidney (Fig. 2e,f). These data demonstrate the remarkable in vivo safety profile of camptothesomes and their potential to maximize the therapeutic efficacy of CPT against tumours.
Fig. 2 |. Camptothesomes increased the MTD of CPT without inducing systemic toxicities in healthy mice.
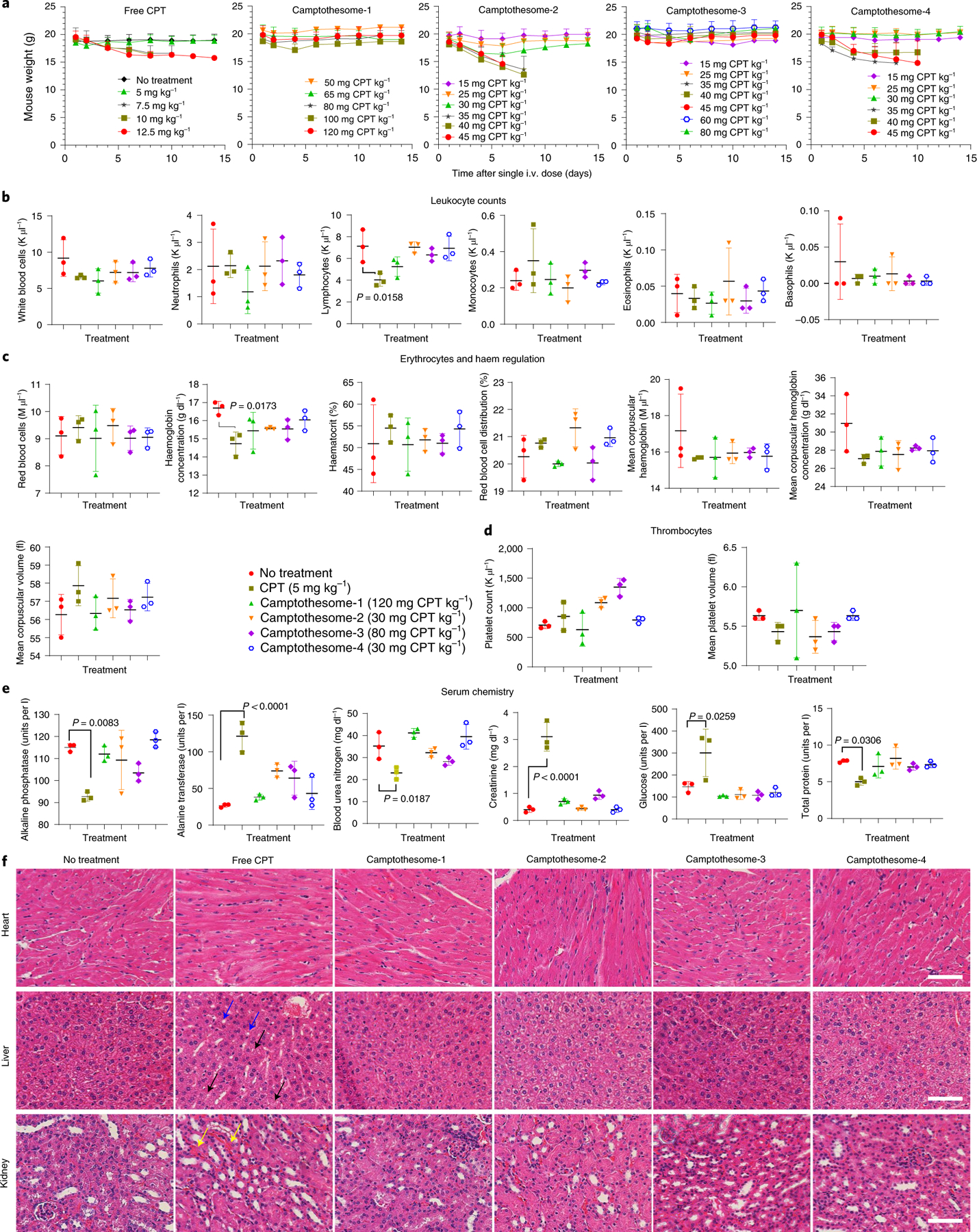
a, Weight changes in healthy BALB/c mice in a MTD study of free CPT and the four camptothesomes administered intravenously once at various doses (as indicated) via a tail vein. Mouse body weight and survival were monitored for 2 weeks. The MTD was defined as the dose that did not cause mouse death or a weight loss of more than 15% during the whole period34,39,40. Weight monitoring was terminated for the mice in the same dose group when there was any mouse death. b–f, On day 14 post i.v. injection, blood was withdrawn for leukocytes (b), erythrocytes (c), thrombocytes (d) and serum chemistry (e) analysis, and the heart, liver (blue arrow, hepatic steatosis; black arrow, diffuse microvesicular degeneration of hepatocytes)41,42 and kidney (yellow arrow, haemorrhage in interstitial tissue)43 were isolated from the mice in the MTD groups (free CPT, 5 mg kg−1; camptothesome-1, 120 mg CPT kg−1; camptothesome-2, 30 mg CPT kg−1; camptothesome-3, 80 mg CPT kg−1; and camptothesome-4, 30 mg CPT kg−1) and the no-treatment group for haematoxylin and eosin (H&E) staining (f). Scale bar, 100 μm. Data in a–e are expressed as mean ± s.d. (n = 3). For H&E staining, representative samples were taken from three independent mice in each group. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test.
Blood kinetics, biodistribution and tumour penetration
The pharmacokinetics and tissue distributions of the camptothesomes in CT26 tumour-bearing mice were evaluated. Free CPT was rapidly eliminated from the blood, while the camptothesomes significantly prolonged circulation half-lives and delivered more drug into tumours with efficient CPT release (Fig. 3a–c), affirming the hypothesis that the tumour-sensitive linkages bridging SM and CPT were readily cleaved in tumours. Camptothesome-4 presented the highest tumour uptake by delivering 23.3-fold more drug into tumours compared to free CPT, 50.2% of which was converted to free CPT (Fig. 3c), and it minimized drug release in the blood (Supplementary Fig. 15a), further corroborating its superior safety profile. The 5.11% of the injected dose in tumour at 24 h achieved with camptothesome-4 is higher than the percentage attained with many FDA-approved nanomedicines (2–3%) in mice28–31; nonetheless, camptothesome-4 had lower distributions to heart, liver, spleen, lung and kidney, thereby minimizing nonspecific systemic toxicities (Fig. 3b). We then evaluated the tumour delivery efficiency of camptothesome-4 in live animals bearing CT26 tumours. We labelled camptothesome-4 with a near-infrared (NIR) dye, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cyanine 5.5) (DSPE-Cy5.5), and tested various amounts to ensure that the dye-doped nanoparticle resembled parental camptothesome-4. At 0.2 % weight of total lipids, Cy5.5/camptothesome-4 had a similar size and zeta potential to camptothesome-4 without dye (Supplementary Fig. 12). Free DSPE-Cy5.5-injected mice exhibited no observable signal in the tumours during the entire monitored period. In stark contrast, Cy5.5/camptothesome-4 peaked in tumours as early as 2.5 h and retained significant fluorescence intensity after 24 h post i.v. administration (Fig. 3d), consistent with ex vivo imaging and tumour uptake levels at 2.5 h and 24 h analysed by high-performance liquid chromatography (HPLC) (Fig. 3b,d,e and Supplementary Fig. 15b). To unravel whether camptothesome-4 can extravasate and penetrate deeply into tumours following tumour delivery, we stained platelet endothelial cell adhesion molecule 1 (PECAM1) to visualize the tumour vasculature (Fig. 3f). The widespread green immunofluorescence revealed an extensive distribution of blood vessels in CT26 tumours. Interestingly, red fluorescence signals from Cy5.5/camptothesome-4 were distributed throughout the tumour section, suggestive of a deep tumour penetration capability (Fig. 3f), which is crucial for antitumour efficacy.
Fig. 3 |. Improved circulation half-life and tumour delivery with efficient intratumoral drug release and deep tumour penetration.
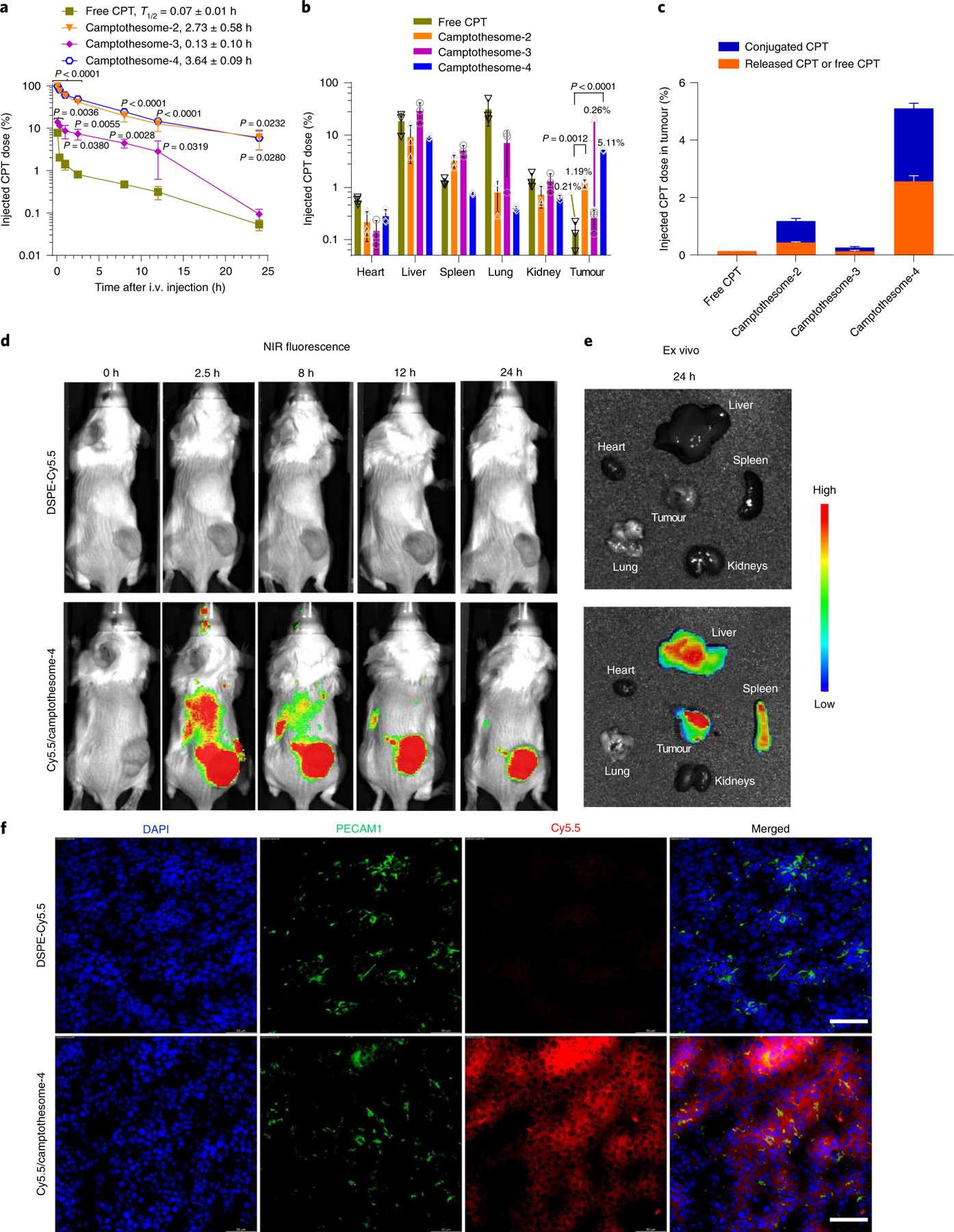
a, Blood kinetics of CPT in subcutaneous CT26 tumour-bearing mice (n = 3 mice; tumours, ~300 mm3) following single i.v. injection of camptothesomes (20 mg CPT kg−1) and free CPT (5 mg kg−1, MTD). b,c, Tissue distribution (b) and CPT intratumoural release (c) at 24 h in mice in (a). The per cent injected doses in camptothesomes represent the released CPT and SM-conjugated CPT. Drug contents in plasma and major tissues were measured by HPLC (Supplementary Fig. 13). d,e, Real-time Lago optical imaging (d) and ex vivo imaging of different organs (e) from a representative mouse after single i.v. administration of free DSPE-Cy5.5 and DSPE-Cy5.5-labelled camptothesome-4 into subcutaneous CT26 tumour-bearing BALB/c mice (n = 3 mice; tumours, ~300 mm3). f, Investigation of the ability of camptothesome-4 to extravasate and penetrate the tumour after i.v. administration into mice with subcutaneous CT26 tumours (n = 3 mice, tumours, ~300 mm3). 24 h after i.v. injection of Cy5.5/camptothesome-4 (red), confocal laser scanning microscopy of sections of CT26 tumours was performed. Blood vessels were marked with PECAM1 antibody followed by Alexa Fluor 488 secondary antibody staining (green). Cell nuclei were stained by DAPI (blue). Scale bars, 50 μm. The results in a–c are expressed as mean ± s.d. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test and compared to free CPT.
Camptothesome potentiates PD-L1/PD-1 blockade therapy
To elucidate whether the camptothesomes retain the anticancer activity of CPT, we compared the effects of treating CT26 tumour-bearing mice with a single dose of camptothesome, free CPT or Onivyde. Tumours in the vehicle control group grew rapidly, demonstrating their aggressive nature, whereas treatment with free CPT, Onivyde, camptothesome-1 or camptothesome-3 reduced the tumour significantly (Fig. 4a,b). The two disulfide-bonded camptothesomes, particularly camptothesome-4, enhanced tumour inhibition, which further prolonged mouse survival (Fig. 4a–c). This is probably attributable to its higher chemical and formulation stability and increased tumour uptake (Supplementary Schemes 2 and 4, and Figs. 1m,n and 3b). As camptothesome-4 outperformed the other camptothesomes in terms of stability, tumour uptake and efficacy, we focused on camptothesome-4 in our subsequent therapeutic and immune investigations. Interestingly, PD-L1, PD-1 and IFN-γ were markedly upregulated in tumours following camptothesome-4 treatment. The PD-L1/PD-1 induction was dictated by IFN-γ, as depleting IFN-γ significantly dampened PD-L1/PD-1 expression (Supplementary Fig. 17f). Thus, these data provide solid support for combining camptothesome-4 with PD-L1/PD-1 blockade to treat CRC. We then intravenously delivered a single dose of camptothesome-4 plus three intraperitoneal (i.p.) injections of anti-PD-L1 monoclonal antibody (α-PD-L1) into CT26 tumour-bearing mice and compared the effects to those of the individual therapies. Consistent with the results shown in Fig. 4a,b and the literature32, camptothesome-4 or α-PD-L1 significantly impeded tumour development, but combination therapy was far more effective and improved mouse survival (Supplementary Fig. 17a–e). These effects were further enhanced when co-blocking PD-L1 and PD-1 along with the use of camptothesome-4, as this combination regimen eradicated the tumour in one out of five mice (Fig. 4d). Camptothesome-4 boosted tumour-infiltrating CD8 and enhanced granzyme B, perforin, IFN-γ and CC-3 production, indicative of CTL-elicited anti-CRC adaptive immunity, which was further bolstered in combination therapies (Fig. 4g and Supplementary Fig. 18). To verify whether similar therapeutic effects can be obtained in other CRC type, we investigated the efficacy of combination therapy in a MC38 tumour model. We found that α-PD-L1 elicited a noticeable tumour reduction, which was more significant when α-PD-L1 was combined with α-PD-1 (Fig. 4h–j). Strikingly, a single i.v. dose of camptothesome-4 shrank tumours in six out of six mice and eliminated the tumour in one out of six mice. Co-blocking PD-L1/PD-1 in combination with camptothesome-4 eradicated tumours in five out of six mice and appreciably extended the mouse survival rate (Fig. 4h–k). Surprisingly, the five surviving mice remained tumour-free after being rechallenged with MC38 cells, signifying the development and activation of the memory immunity that is vital for preventing tumour recurrence (Fig. 4l). To elucidate the mechanism of action for camptothesome-4-induced antitumour immunity, we systemically knocked down IFN-γ in CT26 tumour-bearing mice, which drastically decreased anti-CRC efficacy and immune response in the combination therapies as manifested by significantly attenuated tumour growth suppression and diminished granzyme B, perforin and CC-3), suggesting an IFN-γ-dependent antitumour immunity (Fig. 4g and Supplementary Fig. 18).
Fig. 4 |. Camptothesome synergizes with PD-L1/PD-1 blockade to eradicate CRC tumours.
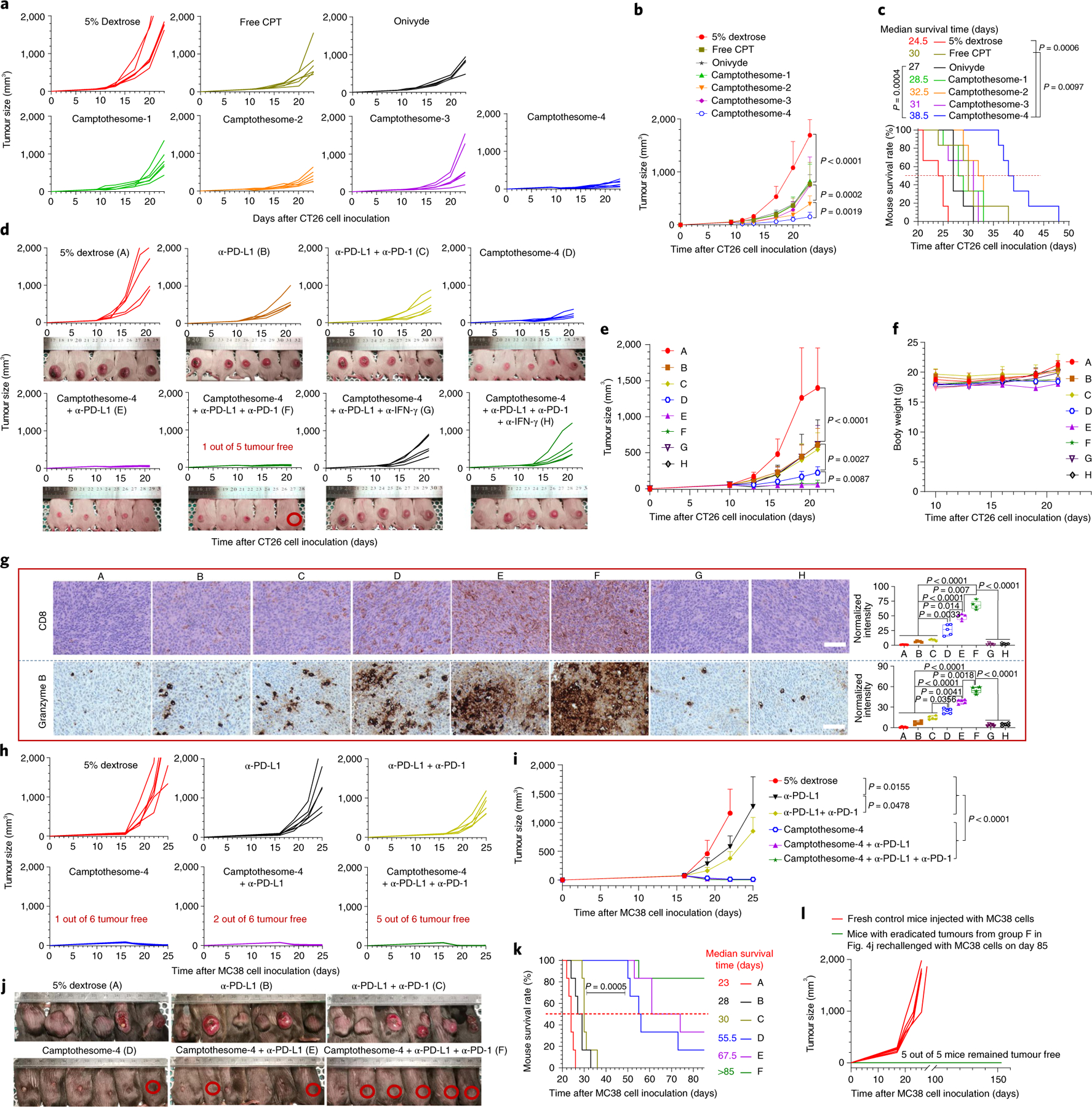
a, Individual tumour growth curves (TGCs) in subcutaneous CT26 tumour-bearing mice (n = 6 mice; tumours, ~50 mm3) receiving a single i.v. injection of free CPT (5 mg kg−1), camptothesome or Onivyde at 20 mg CPT or irinotecan per kg on day 9; 5% dextrose was the vehicle control. b, Average TGCs. c, Kaplan–Meier survival curves. d, Individual TGCs for CT26 tumour-bearing mice (n = 5) receiving a single i.v. dose of camptothesome-4 (20 mg CPT kg−1) alone or combined with i.p. α-PD-L1 or α-PD-L1/α-PD-1 (100 μg per mouse per 3 day on three occasions) with or without α-IFN-γ on or from the tenth day6. α-IFN-γ was intraperitoneally injected (200 μg per mouse per 3 days)44. Mouse images were taken on day 21. Red circle, tumour-free mouse. e, Average TGCs. f, Mouse body weight. g, Immune phenotypic analysis of the tumours in d using immunohistochemistry (IHC). Labels A to H in g correspond to those in d. In the box-and-whisker plots (right panel), boxes delineate lower and upper quartiles, middle lines show median values, dots show individual points, and whiskers show minimal and maximal values for each dataset. Scale bar, 100 μm. h, Individual TGCs for MC38 tumour-bearing mice (n = 6 mice; tumours, ~50 mm3) receiving a single i.v. dose of camptothesome-4 (20 mg CPT kg−1). α-PD-L1 and α-PD-1 were used similarly. i, Average TGCs. j–l, Tumour-bearing mouse images taken on day 26 (j, one mouse death from the vehicle control group on day 23); red circle, tumour-free mouse. Kaplan–Meier survival curves (k); five tumour-free mice from group F in j were rechallenged with MC38 cells on day 85 (l). Labels A to F in k correspond to those in j. Data in b, e, f, i are expressed as mean ± s.d. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test; survival curves were compared using the log-rank Mantel–Cox test.
Loading of DOX-derived IND into camptothesome-4
Consistent with previous findings17,18, we found that IDO1 is expressed in CT26 and MC38 CRC tumours and was further induced by camptothesome-4-elicited INF-γ (Supplementary Figs. 20 and 21). These data prompted us to investigate whether integrating the IDO1 inhibitor IND into camptothesome-4 enhances therapeutic potential. We discovered that the direct loading of IND into camptothesome-4 was limited (<0.5% DLC, Supplementary Fig. 24). To facilitate IND encapsulation, we customized a DOX-conjugated IND (DOX-IND) with a hydrazone bond on DOX and a disulfide linkage on the IND side, based on their unique chemical properties (Fig. 5a). We posited that DOX would serve as a membrane-crossing carrier to bring IND into the interior of camptothesome-4, as DOX can readily cross the lipid bilayer. The pH-sensitive hydrazone bond was meticulously devised to be cleaved in the acidic environment generated by prefilling with a protonating agent, (NH4)2SO4 inside camptothesome-4, leading to the formation of drug precipitates, which can avoid drug leakage. This intraliposomal mild acidic pH is also conducive to stabilizing the CPT lactone ring (Supplementary Fig. 7). We found that DOX-IND was transported into camptothesome-4, where it efficiently broke apart to release DOX and IND intermediate, yielding drug precipitates in the interior (Fig. 5e and Supplementary Fig. 24b). DOX-IND-laden camptothesome-4 exhibited a uniform size distribution and accommodated up to 22% of DOX-IND (5.1% IND), which is markedly higher than that achieved with direct IND loading (Supplementary Fig. 24a). Since folate receptor is overexpressed in many tumour cells, including CRC33, we extrapolated that adding a folate ligand to camptothesome-4 would further enhance the intratumoral uptake and retention of delivered drugs34. Incorporating folate (using DSPE-PEG2K-folate) onto the surface of camptothesome-4 negligibly impacted its size, polydispersity index, morphology and drug loading (Supplementary Fig. 24a,c,d). The antitumour efficacy of DOX-IND/camptothesome-4 with or without folate was tested in large MC38 tumour-bearing mice. Free DOX-IND did not significantly suppress tumour growth compared to the vehicle control, while camptothesome-4 appreciably controlled tumour development. Co-delivering DOX-IND with camptothesome-4 shrank tumours in six out of six mice, while co-administering free DOX-IND and camptothesome-4 only marginally improved efficacy compared to camptothesome-4 alone (Fig. 5f). Remarkably, when coupled with folate targeting, DOX-IND/camptothesome-4 eradicated ~300 mm3 MC38 tumours in two out of six mice, probably due to the improved intratumoral uptake efficiency (Fig. 5i and Supplementary Fig. 30). In addition, DOX-IND/camptothesome-4 dramatically inhibited the IDO1 pathway by boosting P-S6K and reducing IL-6 levels, and it significantly increased calreticulin (the ‘eat-me’ signal for dendritic cell uptake during ICD), CD8, IFN-γ, granzyme B, perforin and CC-3 while simultaneously stunting the Foxp3+ Treg cells in tumours; these effects were more prominent with folate targeting (Fig. 5g,h,j and Supplementary Fig. 27d). Of note, camptothesome-4 elicited better anti-CRC efficacy when co-delivering DOX-IND than the co-injection of Doxil plus IND, which could be due to improved pharmacokinetics and tumour uptake of IND (Fig. 5k–n and Supplementary Fig. 30). Upon depleting CD8α systemically, the therapeutic efficacy of DOX-IND/camptothesome-4 was significantly hindered, with reduced IFN-γ, granzyme B, perforin and CC-3, indicating the pivotal role of CTL-elicited immunity in anti-CRC efficacy (Fig. 5f,i,j and Supplementary Fig. 27d). When combined with PD-L1/PD-1 co-blockade, DOX-IND/camptothesome-4 eliminated ~400 mm3 tumours in three out of five mice, and extended mouse survival. The efficacy of this synergistic combination immunochemotherapy was IFN-γ dependent, as neutralizing IFN-γ significantly diminished its antitumour effects (Fig. 5k–n).
Fig. 5 |. Co-encapsulating DOX-IND in camptothesome-4 using DOX as a transmembrane-enabling carrier.
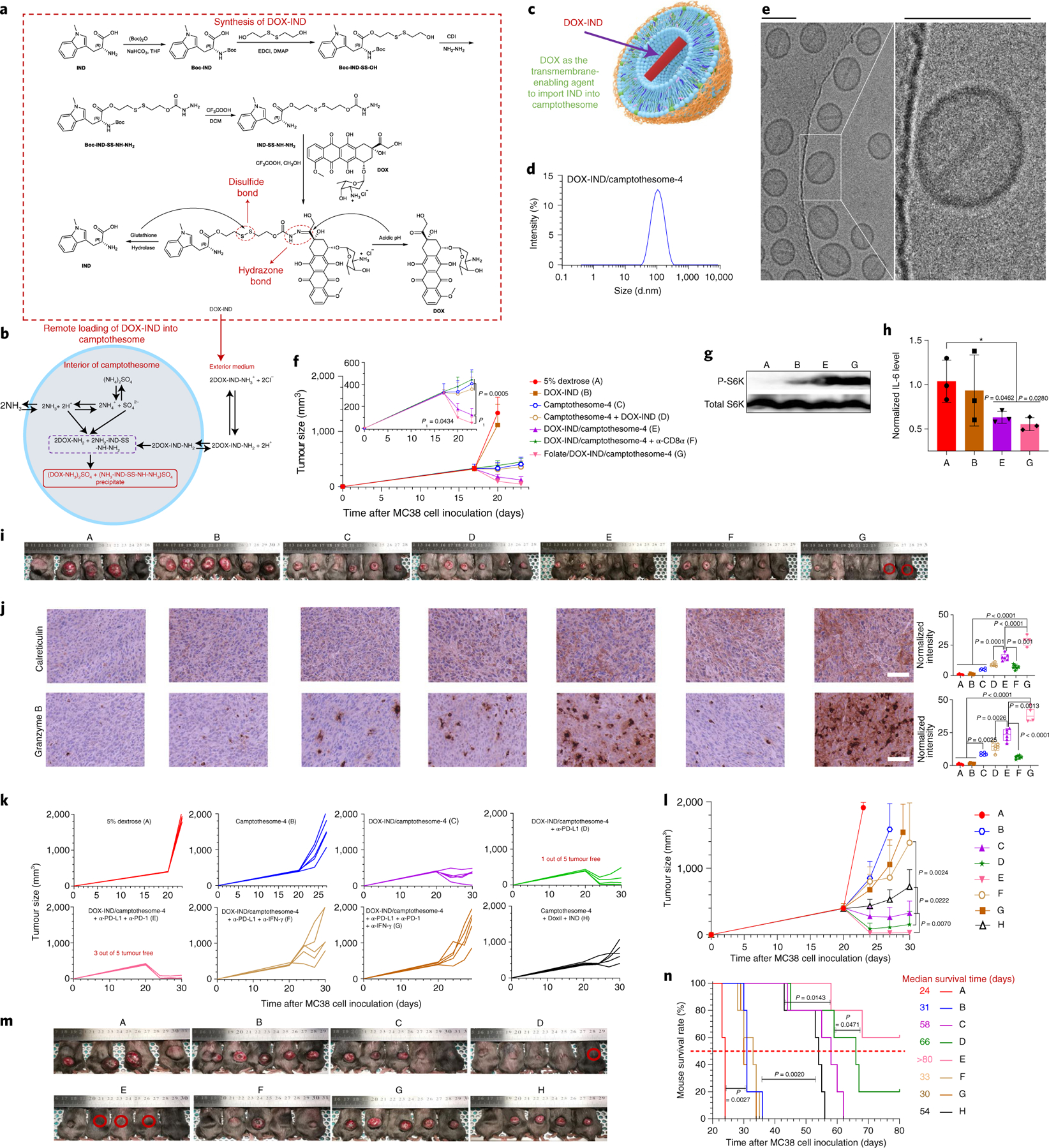
a, Synthesis of DOX-IND. b, Schematic of the remote incorporation of IND into camptothesome utilizing DOX as a lipid membrane-crossing carrier with (NH4)2SO4 as a pH gradient. Once DOX-IND is inside camptothesome-4, the acidic pH produced by (NH4)2SO4 breaks the hydrazone bond, releasing free DOX and IND intermediate. The –NH2 groups from both DOX and IND-SS-NH-NH2 enable the formation of (DOX-NH3)2SO4 and (NH3-IND-SS-NH-NH3)SO4 aggregated salts with dissociated SO42−, avoiding drug leakage or escape. c, Illustration of the co-encapsulation of DOX-IND into camptothesome. d,e, Representative DLS size distribution (d) and cryo-EM (e) of DOX-IND/camptothesome-4 from three independent experiments. Scale bar, 100 nm. f–j, Antitumour efficacy in subcutaneous MC38 tumour-bearing mice (n = 6 mice; tumours, ~300 mm3) receiving a single i.v. injection on day 17 at the equivalent of 20 mg CPT kg−1 and 6.7 mg DOX-IND kg−1. α-CD8α (monoclonal antibody neutralizes mouse CD8α, 200 μg per mouse per 3 days) was given from day 17 via i.p. injection45. Shown are average TGCs (f), representative western blotting for P-S6K (g) and qRT-PCR for IL-6 (h) from three independent tumours. Also shown are mouse images on day 23 (i, one mouse death from the vehicle control group on day 22; red circle, tumour-free mouse), and representative IHC staining and semi-quantitative analysis for calreticulin and granzyme B from independent tumours in i (j). Labels A to G in g and i correspond to those in f. In the box-and-whisker plots (j, right panel), boxes delineate lower and upper quartiles, middle lines show median values, dots show individual data points, and whiskers show minimal and maximal values for each dataset). Scale bar, 100 μm. k–n, Subcutaneous MC38 tumour-bearing mice (n = 5 mice; tumours, ~400 mm3) received a single i.v. injection on day 20 at the equivalent of 15 mg CPT kg−1 and 5 mg DOX-IND kg−1. α-PD-L1, α-PD-1 and α-IFN-γ were administered via i.p. injection as described above. Individual TGCs (k), average TGCs (l), mouse images on day 24 (m, one mouse death from vehicle control group on day 21; red circle, tumour-free mouse) and Kaplan–Meier survival curves (n). Labels A to H in m and n correspond to those in l. Data in f, h, l are presented as mean ± s.d. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test; survival curves were compared using the log-rank Mantel–Cox test.
Elimination of advanced tumours with metastasis remission
To further explore the therapeutic potential of the co-delivery camptothesome-4, we established advanced syngeneic murine models bearing late-stage and metastatic orthotopic CRC or melanoma. In the orthotopic CRC mouse model (~300 mg, Supplementary Fig. 29), tumours in the vehicle control mice grew uncontrollably, and one mouse died on day 18 post inoculation of CT26-Luc cancer cells into the caecum subserosa35; they metastasized to various internal organs, including the liver, spleen, kidney, stomach, intestines, colon and rectum (Fig. 6a–e), demonstrating the remarkable aggressiveness and invasiveness of this malignant tumour. α-PD-L1 plus α-PD-1 had a negligible effect in terms of controlling the primary tumour and preventing metastasis, with one mouse died on day 18, suggesting that this tumour type responds poorly to ICB (Fig. 6a–e). Camptothesome-4 monotherapy inhibited tumour growth significantly and reduced tumour spread; these effects were markedly enhanced by co-delivering DOX-IND (Fig. 6a–e). When used with folate targeting, DOX-IND/camptothesome-4 further detained tumour growth and eradicated tumours in 33.3% of mice. Combined with PD-L1/PD-1 co-blockade, folate/DOX-IND/camptothesome-4 helped 66.7% of mice to survive tumour free with no detectable tumour metastasis (Fig. 6a–e); this treatment boosted innate and adaptive antitumour immune responses by dramatically increasing calreticulin and HMGB1 (ICD hallmarks), LRP1 and TLR4 (receptors on dendritic cells for calreticulin and HMGB-1 uptake, respectively, during ICD)36, CD8, perforin, granzyme B, and proinflammatory cytokines (IL-12 and IFN-γ) while concurrently interfering with Foxp3+ Treg cells development and inhibiting anti-inflammatory IL-10 in tumours (Supplementary Fig. 31c). Similarly, in mice bearing late-stage melanoma (~400 mm3), folate/DOX-IND/camptothesome-4 eliminated large primary melanoma tumours in one out of five mice, enhancing the CTL anticancer immune response. Upon co-blocking PD-L1/PD-1, folate/DOX-IND/camptothesome-4 eradicated primary tumours in 40% of the mice, with complete metastasis remission (Fig. 6f–i and Supplementary Fig. 32). To rule out the possibility that luciferase might contribute to the immunological effects and thus to the observed antitumour efficacy, the same treatments were investigated in a late-stage and metastatic orthotopic CT26 tumour model and in large-sized subcutaneous CT26 and B16-F10 tumour models without the luciferase expression. Similar results were achieved in these aggressive tumour models (Supplementary Figs. 33–35), demonstrating that the observed therapeutic efficacy is not luciferase dependent.
Fig. 6 |. Eradication of advanced and metastatic orthotopic CRC and subcutaneous melanoma tumours.

a–e, Therapeutic efficacy in orthotopic CRC tumour mouse model. Mice were inoculated with 2 × 106 CT26-Luc cells (RPMI-1640/Matrigel, 3/1, v/v) into the caecum subserosa35. On day 8, mice (n = 6 mice; tumours, ~300 mg) were intravenously administered one dose of camptothesome-4, DOX-IND/camptothesome-4 or folate/DOX-IND/camptothesome-4 at the equivalent of 15 mg CPT kg−1 and 5 mg DOX-IND kg−1. α-PD-L1 and α-PD-1 were injected as described above. Lago imaging is shown for live mice (a). Red circles, tumour-free mice (one mouse each from the vehicle control and α-PD-L1 + α-PD-1 groups died on day 18). Quantitative bioluminescence intensity (QBI) for whole mouse tumour burden (b), QBI in various organs (c), a heatmap summarizing the tumour metastatic rate (d) and representative ex vivo photographs (e, upper panel) and bioluminescence imaging (e, lower panel) of various organs on day 18. f–i, Antitumour efficacy in melanoma-bearing C57BL/6 mice. Animals were subcutaneously inoculated with 0.1 × 106 B16-F10-Luc2 cells. On day 14, mice (n = 5 mice; tumours, ~400 mm3) received the same treatments as in a. Lago imaging is shown for live mice (f). Red circle, tumour-free mouse. Two mice from the vehicle control group died on day 20. Normalized tumour size measured by a digital caliper (g), QBI in various organs (h) and a heatmap presenting the tumour metastatic rate (i). Data in b, c, g, h are expressed as mean ± s.d. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test.
Most FDA-approved cancer nanomedicines are liposome based because of the excellent biocompatibility and biodegradability, and favourable in vivo stability. While current liposomal platforms work well for hydrophilic drugs, incorporating hydrophobic therapeutics involuntarily jeopardizes lipid bilayer integrity. Inspired by the clinical success and suitability of liposomes, we invented a novel liposomal platform—camptothesome—by conjugating CPT covalently to SM (a backbone component used in liposomes), which led to the secure anchoring of CPT between lipid bilayers following a spontaneous self-assembly process (Fig. 1). The amide linkage in SM increases intermolecular hydrogen bonding and formulation stability compared to the ester bonds of diacyl chains in other phospholipids under physiological conditions37. This SM-conjugation approach has the potential to rescue CPT and overcomes the limitations (e.g.,, poor solubility and pharmacokinetics) associated with 50–60% of current small-molecule anticancer drugs containing a functional group38.
Although camptothesome significantly improved the anticancer efficacy of CPT, tumour eradication was not achieved due to the intrinsic negative feedback mechanisms of tumours (e.g., upregulation of immune checkpoints PD-L1/PD-1 and IDO1 in response to IFN-γ), which allow them to escape the chemotherapy-induced antitumour immunity, avoiding complete elimination (Supplementary Figs. 17f, 20 and 21). Surprisingly, when combined with PD-L1/PD-1 co-blockade, camptothesome-4 eradicated MC38 tumours (~50 mm3) in 83.3% of mice and activated memory immunity, preventing tumour recurrence after rechallenging tumour-eradicated mice with the same cancer cells (Fig. 4h–l). Taking advantage of the membrane-crossing capability of DOX, camptothesome enables co-delivery of the IDO1 inhibitor IND by remote loading, further enhancing antitumour efficacy and immune responses, and eradicating large MC38 tumours (~400 mm3) in 60% of mice when co-blocking PD-L1/PD-1 (Fig. 5). Remarkably, folate/DOX-IND/camptothesome-4 plus PD-L1/PD-1 co-blockade eliminated advanced metastatic orthotopic CT26-Luc tumours (~300 mg) in 66.7% of mice; and similar effects were observed in a late-stage metastatic melanoma (~400 mm3) murine model (Fig. 6).
This DOX-enabled transmembrane transportation technology opens up a new field of research into the temporally and spatially controlled and synergistic co-delivery of various therapeutics (hydrophobic and hydrophilic) that cannot be co-encapsulated by existing liposomal platforms. The resulting synergistic combination immunochemotherapy is, we believe, attributable to: (1) improved pharmacokinetics, enhanced tumour accumulation and retention, and efficient extravasation and tumour penetration, as well as efficient and sustained intratumoural drug release; (2) generation of the first round of immune responses by camptothesome-4 through augmentation of the CTL killing of tumour cells and PD-L1/PD-1 expression to potentiate PD-L1/PD-1 blockade; and (3) subsequent IND and DOX release from camptothesome-4, which further enhanced and/or sustained the magnitude of antitumour immunity by overcoming IDO1-induced immunosuppression (for example, by stunting the Treg cells) and concurrently eliciting ICD (for example, by stimulating calreticulin and LRP1, and HMGB1 and TLR4) (Figs. 3–5 and Supplementary Figs. 15, 17, 18, 21, 27 and 30–32).
Conclusions
Here we report a nanotherapeutic platform based on a SM-conjugated drug, and a DOX-enabled transmembrane transporting technology, both of which are generalizable to various therapeutics. Given that (1) SM is used as the backbone constituent in the FDA-approved liposomal nanotherapeutic Marqibo, (2) the manufacturing procedure for camptothesome or co-delivery camptothesome is facile, standardized and well established, similar to traditional liposomal nanoformulations, (3) remarkable efficacy was achieved against both early-stage and clinically difficult-to-treat late-stage metastatic tumours, and (4) IDO1 is expressed in diverse cancer cells, the robust and multipronged camptothesome immunochemotherapy framework presented here has promising clinical relevance and the potential to revolutionize cancer treatment paradigms.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41565-021-00950-z.
Methods
Preparation of camptothesome nanovesicles.
The self-assembly of SM-derived CPT conjugates into liposomal nanovesicles was achieved by a standard thin-film hydration method36. In brief, SM, cholesterol, DSPE-PEG2K (Avanti Polar Lipids) and SM-conjugated CPT (SM-ester-CPT, SM-SS-CPT, SM-glycine-CPT or SM-CSS-CPT) in an appropriate ratio (listed in Fig. 1h) were dissolved in ethanol in a 100 ml round-bottom glass flask. The solvent was evaporated under a rotatory evaporator (RV 10 digital, IKA) to generate a thin film, which was further dried under ultra-high vacuum (MaximaDry, Fisherbrand) for 0.5 h. The film was hydrated with 5% dextrose aqueous solution at 50 °C for 30 min and then sonicated for 12 min using a 3/2 s on/off pulse at a power output of 60 W. To remove any unencapsulated SM-CPT, the nanoparticles underwent ultracentrifugation at 100,000×g for 45 min. The dynamic light scattering (DLS) size and the zeta potential, the morphology and the CPT content of the purified camptothesome nanovesicles were determined by a Zetasizer Nano (Zetasizer software (v. 7.13), Nano-ZS, Malvern Panalytical), cryo-EM (Tecnai User Interface software (v. 3.1.5) and EMMenu) and HPLC (ChemStation Rev.A. software (v. 10.01, Agilent Technologies)), respectively. The CPT DLC was calculated as:
| (1) |
Preparation of DOX-IND/camptothesome-4 and folate/DOX-IND/camptothesome-4 nanoformulations.
SM, SM-CSS-CPT, cholesterol and DSPE-PEG2K (addition of 0.5 molar% DSPE-PEG2K-folate (Avanti Polar Lipids) for folate/DOX-IND/camptothesome-4) at the ratio of 68.3/14.8/12.6/4.3 as shown in Fig. 1h were dissolved in ethanol in a 100 ml round-bottom glass flask. The solvent was evaporated under a rotatory evaporator to generate a thin film, which was further dried under ultra-high vacuum for 0.5 h. The film was hydrated with a 80 mM (NH4)2SO4 aqueous solution at 50 °C for 30 min and then sonicated for 12 min using a 3/2 s on/off pulse at a power output of 60 W. The free (NH4)2SO4 was removed by a PD-10 column (Sephadex G-25, GE Healthcare) using PBS as eluent. The remote DOX-IND loading was achieved by incubating (NH4)2SO4/camptothesome-4 with 2–10 mg ml−1 DOX-IND at 65 °C for 1 h. After cooling at 4 °C for 30 min, the free DOX-IND was removed by running through a PD-10 column. The size and zeta potential, morphology and drug content of the DOX-IND/camptothesome-4 and folate/DOX-IND/camptothesome-4 nanovesicles were determined by DLS, cryo-EM and HPLC, respectively. The DOX-IND DLC and drug loading efficiency (DLE) were calculated as:
| (2) |
| (3) |
Fluorescence quenching assay.
Camptothesomes and DOX-IND/camptothesome-4 were prepared as described above. Controls included the respective SM-CPT conjugates, SM, cholesterol, DSPE-PEG2K and/or DOX-IND. Various samples in Fig. 1j and Supplementary Figs. 6, 22 and 23 at the equivalent of 100 μM in 200 μl were placed into a 96-well plate (Bio-One UV-Star, Greiner), and the fluorescence intensity was detected on a SpectraMax M3 reader (SoftMax Pro (v. 7.1.0), Molecular Devices), employing an excitation wavelength of 360 nm and emission wavelengths of 400–650 nm for CPT and SM-CPT conjugates, and an excitation wavelength of 470 nm and emission wavelengths of 520–700 nm for DOX-IND.
Pharmacokinetics and biodistribution studies.
In subcutaneous CT26 tumour-bearing mice.
Following a single i.v. injection of free CPT (5 mg CPT kg−1, MTD) or different camptothesomes (20 mg CPT kg−1) into subcutaneous CT26 tumour-bearing mice (n = 3, ~300 mm3) via a tail vein, blood was withdrawn at 0.083, 0.333, 1, 2.5, 8, 12 and 24 h, and plasma was collected (using a plasma tube, BD Microtainer) and digested in methanol (90 μl methanol per 10 μl serum) for HPLC analysis to measure the released CPT and SM-derived CPT conjugates. 24 h after i.v. administration of the drug, tumour tissues and major organs (heart liver, spleen, lung and kidneys) were collected, weighed, and then homogenized in acidified methanol (0.075 M HCl, 900 μl per 100 mg tissue) before the drug content was determined using an established HPLC method (Supplementary Fig. 13). To unravel the intratumoral CPT release rate after short and long time periods for camptothesome-4, we performed a separate biodistribution experiment in which subcutaneous CT26-bearing mice (n = 3, ~300 mm3) received a single i.v. injection of camptothesome-4 (20 mg CPT kg−1). Mice were euthanized at 2.5 h and 72 h post drug administration, whereupon the drug contents in the collected tumour tissues and organs were processed and analysed as described above.
In orthotopic CT26-Luc tumour-bearing mice.
Various drug formulations (free CPT (5 mg kg−1), IND (1.6 mg kg−1), Doxil (3.9 mg kg−1), DOX-IND/camptothesome-4 (6.7/20 mg DOX-IND/CPT kg−1, 2% DOX-IND) and folate/DOX-IND/camptothesome-4 (6.7/20 mg DOX-IND/CPT kg−1)) were intravenously injected into orthotopic CT26-Luc tumour-bearing mice (n = 3, tumour weight, ~300 mg) via a tail vein, based on the MTD (Supplementary Fig. 25). At 0.083, 0.333, 1, 2.5, 8, 12 and 24 h post drug administration, blood was withdrawn, and plasma was digested in methanol prior to HPLC measurement of the DOX, IND, DOX-IND, CPT and SM-CSS-CPT. At 24 h, tumour tissues and major organs were collected and homogenized in acidified methanol (0.075 M HCl, 900 μl per 100 mg tissue) before HPLC drug content analysis was performed (Supplementary Figs. 13 and 26). The various pharmacokinetic parameters were assessed using the PKSolver software (version 2.0)46.
Visualization of in vivo tumour delivery and deep tumour penetration.
To visualize camptothesome-4 tumour delivery in vivo, camptothesome-4 was labelled with 0.2% w/w DSPE-Cy5.5. subcutaneous CT26 tumour-bearing mice (n = 3, ~300 mm3) were intravenously injected once with Cy5.5/camptothesome-4 (20 mg CPT kg−1). Mice were imaged at 0 h (prior to drug administration) and at 2.5, 8, 12 and 24 h post i.v. injection (excitation at 675 nm; emission at 710 nm). At 24 h, tumours, heart, liver, spleen, lung and kidneys were imaged using a Lago optical imager (Aura 64 bit analysis software (v. 3.2.0)). For the tumour penetration investigation, tumours were frozen in an acetone–dry ice mixture prior to immunofluorescence examination. The blood vessels in the tumour were stained with a primary anti-CD31 (also known as PECAM1) antibody (ab28364, 1:50, Abcam) followed by an Alexa Fluor 488-conjugated secondary antibody (ab150073, 1:400, Abcam). DAPI (4,6-diamidino-2-phenylindole) was used to localize the cellular nuclei. Tumour tissues were cut into 5 μm slide sections and subjected to confocal laser scanning microscopy using a Leica SP5-II confocal microscope (Leica LAS-AF software (v. 2.7.3.9723)) at the University of Arizona Cancer Center (UACC) Tissue Acquisition and Cellular/Molecular Analysis Shared Resource (TACMASR).
Therapeutic efficacy investigation of camptothesome nanovesicles.
In subcutaneous CT26 tumour-bearing mice.
BALB/c mice (n = 5 or 6, Charles Rivers) were subcutaneously injected with 1 × 105 CT26 cells in 100 μl of serum-free medium at the right flank. When tumours grew to ~50 mm3 in size, the mice received one dose of intravenously administered 5% dextrose (vehicle control), free CPT (5 mg CPT kg−1), Onivyde (20 mg irinotecan kg−1, Ipsen Biopharmaceuticals, Inc.) and different camptothesomes (20 mg CPT kg−1) or a combination of camptothesome-4 and intraperitoneally injected α-PD-L1 (clone 10 F.9G2, BioXCell; Supplementary Fig. 17) with or without α-PD-1 (BioXCell, clone RMP1–14) at 100 μg per mouse per 3 days on three occasions and with or without intraperitoneally administered α-IFN-γ (BioXcell, clone R4–6A2, 200 μg per mouse per 3 days). To determine the effect on the survival rate in each group (n = 6), Kaplan–Meier plots were used to express the animal survival rate. For the efficacy study in Fig. 4d–g, mice were euthanized on day 21; tumours were collected and soaked in 4% paraformaldehyde overnight prior to the immune phenotypic (CD8, granzyme B, perforin, CC-3 and IFN-γ) analysis using IHC staining by UACC TACMASR.
In an independent study, CT26 tumour-bearing mice (n = 3, ~200 mm3) received a single i.v. injection of camptothesome-4 (20 mg CPT kg−1) with or without α-IFN-γ (as described above), with 5% dextrose used as the vehicle control. Seven days later, IHC staining for PD-L1, PD-1, IFN-γ and IDO in tumours was performed.
In another independent study, CT26 tumour-bearing mice (n = 6, ~400 mm3) were intravenously administered one dose of camptothesome-4, DOX-IND/camptothesome-4, or folate/DOX-IND/camptothesome-4 at the equivalent of 15 mg CPT kg−1 (5 mg DOX-IND kg−1). α-PD-L1 and α-PD-1 were injected as described above. Tumour growth and mice body weight and survival were closely monitored as indicated.
In subcutaneous MC38 tumour-bearing mice.
C57BL/6 mice (n = 6, Charles Rivers) were subcutaneously injected with 2 × 105 cells per mouse in 100 μl of serum-free medium (Fig. 4h–k). When tumours reached ~50 mm3, the mice were intravenously injected once with camptothesome-4 (20 mg CPT kg−1) alone or in combination with α-PD-L1 or α-PD-L1 + α-PD-1. α-PD-L1 and α-PD-1 were intraperitoneally administered as mentioned above. Tumour growth, mouse body weight and survival were closely monitored as indicated.
Rechallenging mice with eradicated tumours with subcutaneous MC38 cells to demonstrate memory immunity.
Five tumour-free survivors in the camptothesome-4 + α-PD-L1 + α-PD-1 group and five fresh, healthy C57BL/6 mice (control mice) were subcutaneously injected in the contralateral flank with MC38 cells (2 × 105 cells per mouse) on day 85. The five control mice developed tumours uncontrollably, while the five surviving mice from the camptothesome-4 + α-PD-L1 + α-PD-1 group remained tumour free (Fig. 4l).
C57BL/6 mice (n = 6) were subcutaneously injected with 4 × 105 cells per mouse in 100 μl of serum-free medium (Fig. 5f–j). Mice were intravenously injected once with various treatments at the equivalent of 20 mg CPT kg−1 and 6.7 DOX-IND mg kg−1 when tumours grew to ~300 mm3. α-CD8α (BioXCell, clone 53–6.7) was intraperitoneally injected at 200 μg per mouse per 3 days from day 17. On day 23, the mice were euthanized and tumour tissues were isolated and cut into three equal pieces: one part for IHC staining for CD8, Foxp3, calreticulin, IFN-γ, perforin, granzyme B and CC-3) by the UACC TACMASR, and the other two parts for western blotting of P-S6K and qRT-PCR (the reverse transcription polymerase chain reaction (RT-PCR) and quantitative real-time PCR (qPCR) combined technique) for IL-6, respectively. Tumour growth, mouse body weight and survival were closely monitored as indicated.
C57BL/6 mice (n = 5) were subcutaneously injected with 2 × 105 cells per mouse (Fig. 5k–n). When tumours reached ~400 mm3, the mice received a single i.v. injection with various treatments at the equivalent of 15 mg CPT kg−1 and 5 mg DOX-IND kg−1 (IND, 1.2 mg IND kg−1; Doxil, 2.9 mg DOX kg−1) or in combination with α-PD-L1 or α-PD-L1 + α-PD-1, with or without α-IFN-γ. α-PD-L1, α-PD-1 and α-IFN-γ were intraperitoneally administered as depicted above. Tumour growth, mice body weight and survival were closely monitored as indicated.
In a separate study, subcutaneous MC38 tumour-bearing mice (n = 3, ~200 mm3) were intravenously injected once with camptothesome-4 (20 mg CPT kg−1) with or without α-IFN-γ, as described above. 5% Dextrose served as the vehicle control. Seven days later, tumours were collected and subjected to PD-L1, PD-1, IFN-γ and IDO1 analysis using IHC staining.
In orthotopic CT26-Luc tumour-bearing mice.
Six- to eight-week-old BALB/c mice were anaesthetized by isoflurane. The fur in the abdominal area of each mouse was removed by clippers. Then the surgical area underwent three alternating scrubs of betadine/povidone iodine followed by 70% ethanol. A subcutaneous injection of buprenorphine SR (1.0 mg kg−1) was administered prior to surgery. Afterwards, an abdominal incision (~1 cm) was created with a sterile disposable scalpel, and the caecum was exteriorized. 2 × 106 CT26-Luc or CT26 cells in 50 μl of RPMI-1640 medium with Matrigel (Corning, Discovery Labware) (3/1, v/v) were inoculated into the caecum subserosa using a 26-gauge needle (BD PrecisionGlide). The caecum was then placed back into the peritoneal cavity after sterilizing the injection site with 70% ethanol to kill any cancer cells that may have leaked out35. The abdominal wall and skin were closed with size 6–0 absorbable sutures (PDS II, Ethicon) and size 5–0 nonabsorbable sutures (PROLENE, Ethicon), respectively. Surgical glue was applied to ensure good apposition of the skin. Animals were placed on a heating pad during and after surgery and closely monitored until ambulatory, after which they were returned to a clean cage. The whole mouse body tumour burden was determined by bioluminescence radiance intensity using Lago optical imaging 10 min after the mice were injected intraperitoneally with 150 mg kg−1 D-Luciferin (GoldBio). Eight days after cancer cell inoculation (primary tumour weight, ∼300 mg; Supplementary Figs. 29 and 34a), orthotopic CT26-Luc or CT26 tumour-bearing BALB/c mice were randomly allocated to six groups (n = 6). Mice were then intravenously administered a single dose of DOX-IND/camptothesome-4 (5/15 mg DOX-IND/CPT kg−1) with or without folate targeting or combined with i.p. α-PD-L1 + α-PD-1 as described above. Controls included 5% dextrose, α-PD-L1 + α-PD-1, and camptothesome-4. For the CT26-Luc model, the whole-body tumour burden was monitored with a Lago optical imager on days 8, 11, 15 and 18 and quantified as the luminescence radiance intensity (p s−1 cm2 sr−1) using Aura 3.2.0 imaging software. On day 18, following the injection of d-Luciferin, the mice were dissected after 5 min, their gastrointestinal tracts and other major organs (heart, liver, spleen, lung, kidneys, stomach, small and large intestines, caecum and rectum) were quickly obtained, and then photography and ex vivo Lago imaging were used to investigate tumour metastasis. Afterwards, tumours were isolated and placed in 4% paraformaldehyde overnight prior to IHC analysis of various immune biomarkers (PD-L1, PD-1, IDO1, CD8, Foxp3, perforin, granzyme B, IFN-γ, calreticulin, LRP1, HMGB1, TLR4, IL-10 and IL-12). For the CT26 model, on day 18, mouse images were taken with the abdomen opened to expose the tumours, and various tissues and organs were extracted and photographed ex vivo. Tumours were subjected to IHC analysis for granzyme B, TLR4, LRP1 and IL-12. Mouse survival was investigated in an independent efficacy study using the orthotopic CT26 tumour model.
In subcutaneous melanoma-bearing mice.
C57BL/6 mice (n = 5 or 6) were subcutaneously injected with 1 × 105 of B16-F10-Luc2 or B16-F10 cells in 100 μl of serum-free medium. When tumours reached ~400 mm3, the mice received a single i.v. dose of DOX-IND/camptothesome-4 (5/15 mg DOX-IND/CPT kg−1) with or without folate targeting or combined with i.p. α-PD-L1 + α-PD-1 as described for the orthotopic CRC tumour model with the same control groups. For the B16-F10-Luc2 model, the whole mouse body tumour burden was evaluated by Lago optical imaging on days 14, 17 and 20. Primary tumours were also measured by a digital caliper as described above. On day 20, the tumour metastasis in the digestive tract and other major organs was deciphered, and immune phenotypic analysis (PD-L1, PD-1, IDO1, CD8, Foxp3, calreticulin, LRP1, IFN-γ, granzyme B and perforin) in orthotopic B16-F10-Luc2 tumours was conducted as described above. For the B16-F10 model, tumour-bearing mice were photographed on day 19, and tumours were collected and used for granzyme B, perforin, calreticulin and LRP1 staining. Mouse survival was evaluated in an independent efficacy study using the B16-F10 tumour model.
IDO1 pathway inhibition in tumours.
IDO1-mediated immunosuppression entails a series of downstream signalling, such as suppressing mTOR (mammalian target of rapamycin) and enhancing the GCN2 (general control nonderepressible 2) and AHR (aryl hydrocarbon receptor) pathways (Supplementary Fig. 20), where phosphorylation of S6K (P-6SK) and IL-6 are critically involved36,47. To elucidate the impact of camptothesome-4-delivered DOX-IND on IDO1 inhibition, western blotting for P-6SK and qRT-PCR for IL-6 were carried out as previously reported36 with a slight modification. Specifically, total S6K was used as the control when determining P-6SK levels. Tumours shown in Fig. 5i were cut into small pieces with scissors and homogenized in RIPA buffer containing a mixture of proteinase and phosphatase (250 μl per 50 mg tissue) within 15 min. The lysates were then centrifuged at 12,000 r.p.m. for 10 min, after which equal amounts of proteins in supernatants were loaded onto a 12% Tris-glycine gel (Novex gel, Invitrogen), which was subsequently transferred to a PDVF (polyvinylidene difluoride) membrane. The membrane was blocked by 5% BSA inTBST. This was followed by incubation with the primary antibody (phospho-p70 S6 kinase (Thr389) no. 9205, dilution 1/1000; p70 S6 kinase no. 9202, dilution 1/1000; Cell Signaling) and horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit IgG, HRP-linked antibody no. 7074, dilution 1/3000; Cell Signaling) that target P-S6K and total S6K. Finally, the blots were developed by soaking the membrane in ECL substrate (Thermo Scientific). Western blot images were acquired using Azure Biosystems software (v. 1.5.0.0518).
For RT-PCR analysis of IL-6 messenger RNA expression, total RNA was extracted from tumours with an miRNeasy Mini Kit (cat. no. 217004, Qiagen) and then treated with an RNase-free Dnase Set (cat. no. 79254, Qiagen) and reverse transcribed using a SuperScript III First-Strand Synthesis System (cat. no. 18080–051, Invitrogen). Quantitative real-time PCR (qPCR) was conducted with QuantStudio 3 (Design & Analysis Software (v. 1.5.1, QuantStudio), Thermo Scientific) and PrimeTime probe-based qPCR assays with Gene Expression Master Mix (Integrated DNA Technologies). For IL-6, assay ID: Mm.PT.58.10005566, forward primer 5′-AGC CAG AGT CCT TCA GAG A-3′, reverse primer 5′-TCC TTA GCC ACT CCT TCT GT-3′, probe 5′-/56-FAM/CCTACCCCA/ZEN/ATTTCCAATGCTCTCCT/3IABkFQ/−3′; for GAPDH, assay ID: Mm.PT.39a.1, forward primer 5′-AAT GGT GAA GGT CGG TGT G-3′, reverse primer 5′-GTG GAG TCA TAC TGG AAC ATG TAG-3′, probe 5′-/56-FAM/TGCAAATGG/ZEN/CAGCCCTGGTG/3IABkFQ/−3′. PCR procedures involved a 3 min step at 95 °C followed by 40 cycles at 95 °C for 5 s, 60 °C for 30 s.
Statistical analysis.
The level of significance in all statistical analyses was set at P < 0.05. Data are presented as mean ± s.d. and were analysed using the two-tailed, unpaired Student’s t-test for two groups or one-way analysis of variance (ANOVA) for three or more groups followed by Tukey’s multiple comparisons test using Prism 8.0 (GraphPad Software). Kaplan–Meier survival curves were compared with the log-rank Mantel–Cox test.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the article and the Supplementary Information. All relevant data are available from the authors upon reasonable request.
Supplementary Material
Acknowledgements
This work was supported in part by a Startup Fund from the College of Pharmacy at the University of Arizona and two Seed Grants from the University of Arizona BIO5 Institute and the State of Arizona’s Technology and Research Initiative Fund (TRIF), and by National Institutes of Health (NIH) grants (NIEHS P30 ES006694, NCI P30 CA023074 and NCI R01 CA092596). We acknowledge Statistical Consulting at the University of Arizona Information Technology for their assistance with the statistical analysis; the use of mass spectrometry equipment in the Analytical and Biological Mass Spectrometry Core Facility at the University of Arizona BIO5 Institute; the University of Arizona Translational Bioimaging Resource Core for the Lago live animal imaging; the University of Arizona University Animal Care Pathology Services for the serum chemistry and haematological counts; the TACMASR of the UACC for the IHC and H&E staining, immunofluorescence and confocal laser scanning microscopy; and Arizona State University’s John Cowley Center for High Resolution Electron Microscopy (the specific instrumentation used was supported by the National Science Foundation (NSF), MRI grant NSF1531991) for the cryo-EM.
Footnotes
Competing interests
J.L. has applied for patents related to this study. The other authors have no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41565-021-00950-z.
Peer review information Nature Nanotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Haslam A & Prasad V Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2, e192535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catenacci DVT, Hochster H & Klempner SJ Keeping checkpoint inhibitors in check. JAMA Netw. Open 2, e192546 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Overman MJ, Ernstoff MS & Morse MA Where we stand with immunotherapy in colorectal cancer: deficient mismatch repair, proficient mismatch repair, and toxicity management. Am. Soc. Clin. Oncol. Educ. Book 38, 239–247 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Huyghe N, Baldin P & Van den Eynde M Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol. Rep. 8, 11–24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galon J & Bruni D Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Song W et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 9, 2237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paz-Ares L et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 379, 2040–2051 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Whiteside TL, Demaria S, Rodriguez-Ruiz ME, Zarour HM & Melero I Emerging opportunities and challenges in cancer immunotherapy. Clin. Cancer Res. 22, 1845–1855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebert PJR et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ribas A et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 170, 1109–1119.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YF et al. Host immune response to anti-cancer camptothecin conjugated cyclodextrin-based polymers. J. Biomed. Sci. 26, 85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J et al. The self-assembling camptothecin-tocopherol prodrug: an effective approach for formulating camptothecin. Biomaterials 62, 176–187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dosset M et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 7, e1433981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prendergast GC et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 63, 721–735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou DY et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 67, 792–801 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Lob S, Konigsrainer A, Rammensee HG, Opelz G & Terness P Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat. Rev. Cancer 9, 445–452 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Phan T et al. Salmonella-mediated therapy targeting indoleamine 2,3-dioxygenase 1 (IDO) activates innate immunity and mitigates colorectal cancer growth. Cancer Gene. Ther. 27, 235–245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes B et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-(L)1 blockade therapy. Mol. Cancer Ther. 17, 2530–2542 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Metz R et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 1, 1460–1468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du JZ, Du XJ, Mao CQ & Wang J Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 133, 17560–17563 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E & Prendergast GC Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 11, 312–319 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Kroemer G, Galluzzi L, Kepp O & Zitvogel L Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Gamcsik MP, Kasibhatla MS, Teeter SD & Colvin OM Glutathione levels in human tumors. Biomarkers 17, 671–691 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong YJ, Shao LH & Li Y Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 42, 373–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw JP & Chou IN Elevation of intracellular glutathione content associated with mitogenic stimulation of quiescent fibroblasts. J. Cell. Physiol. 129, 193–198 (1986). [DOI] [PubMed] [Google Scholar]
- 26.Jessani N, Liu Y, Humphrey M & Cravatt BF Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc. Natl Acad. Sci. USA 99, 10335–10340 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bucher G Ester pyrolysis of carbonates: bis(benzene hydrate) carbonate as potential precursor for monomeric carbonic acid. Eur. J. Org. Chem. 2010, 1070–1075 (2010). [Google Scholar]
- 28.Bhattacharyya J et al. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat. Commun. 6, 7939 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabizon A, Tzemach D, Mak L, Bronstein M & Horowitz AT Dose dependency of pharmacokinetics and therapeutic efficacy of pegylated liposomal doxorubicin (DOXIL) in murine models. J. Drug Target 10, 539–548 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Soundararajan A, Bao A, Phillips WT, Perez R 3rd & Goins BA [(186) Re]liposomal doxorubicin (Doxil): in vitro stability, pharmacokinetics, imaging and biodistribution in a head and neck squamous cell carcinoma xenograft model. Nucl. Med. Biol. 36, 515–524 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalra A et al. Abstract 2065: Magnetic resonance imaging with an iron oxide nanoparticle demonstrates the preclinical feasibility of predicting intratumoral uptake and activity of MM-398, a nanoliposomal irinotecan (nal-IRI). Cancer Res. 74, 2065–2065 (2014). [Google Scholar]
- 32.Song W et al. Trapping of lipopolysaccharide to promote immunotherapy against colorectal cancer and attenuate liver metastasis. Adv. Mater. 30, e1805007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Low PS, Henne WA & Doorneweerd DD Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc. Chem. Res. 41, 120–129 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Lu J et al. Targeted delivery of doxorubicin by folic acid-decorated dual functional nanocarrier. Mol. Pharmaceutics 11, 4164–4178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Davis C, Ryan J, Janney C & Pena MM Development and characterization of a reliable mouse model of colorectal cancer metastasis to the liver. Clin. Exp. Metastasis 30, 903–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu J et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 8, 1811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slotte JP The importance of hydrogen bonding in sphingomyelin’s membrane interactions with co-lipids. Biochim. Biophys. Acta 1858, 304–310 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Narvekar M, Xue HY, Eoh JY & Wong HL Nanocarrier for poorly water-soluble anticancer drugs—barriers of translation and solutions. AAPS PharmSciTech 15, 822–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J et al. PEG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials 34, 1591–1600 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J et al. An improved d-alpha-tocopherol-based nanocarrier for targeted delivery of doxorubicin with reversal of multidrug resistance. J. Control. Release 196, 272–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao Y et al. Bouchardatine analogue alleviates non-alcoholic hepatic fatty liver disease/non-alcoholic steatohepatitis in high-fat fed mice by inhibiting ATP synthase activity. Br. J. Pharmacol. 176, 2877–2893 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konstantopoulos P et al. Metabolic effects of Crocus sativus and protective action against non-alcoholic fatty liver disease in diabetic rats. Biomed. Rep. 6, 513–518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdel-Salam OME, Nada SA, Salem NA, El-Shamarka ME-S & Omara E Effect of Cannabis sativa on oxidative stress and organ damage after systemic endotoxin administration in mice. Comp. Clin. Pathol. 23, 1069–1085 (2013). [Google Scholar]
- 44.Clemente-Casares X et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 530, 434–440 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Wang W et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 46.Zhang Y, Huo M, Zhou J & Xie S PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Meth. Prog. Bio. 99, 306–314 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Litzenburger UM et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 5, 1038–1051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and the Supplementary Information. All relevant data are available from the authors upon reasonable request.