

Abstract
The rising prevalence of multi-drug resistant bacteria is an urgent health crisis that can only be countered through renewed investment in the discovery and development of antibiotics. There is no panacea for the antibacterial resistance crisis; instead, a multi-faceted approach is called for. In this perspective we make the case that, in the face of evolving clinical needs and enabling technologies, numerous validated antibacterial targets and associated lead molecules deserve a second look. At the same time, many worthy targets lack good leads despite harboring druggable active sites. Creative and inspired techniques buoy discovery efforts; while soil screening efforts frequently lead to antibiotic rediscovery, researchers have found success searching for new antibiotic leads by studying underexplored ecological niches or by leveraging the abundance of available data from genome mining efforts. The judicious use of “polypharmacology” (i.e., the ability of a drug to alter the activities of multiple targets) can also provide new opportunities, as can the continued search for inhibitors of resistance enzymes with the capacity to breathe new life into old antibiotics. We conclude by highlighting available pharmacoeconomic models for antibacterial discovery and development while making the case for new ones.
Keywords: Antibiotics, Natural Product Discovery, Antibiotic Crisis, Compound Optimization, Pharmacoeconomic Models
Graphical Abstract
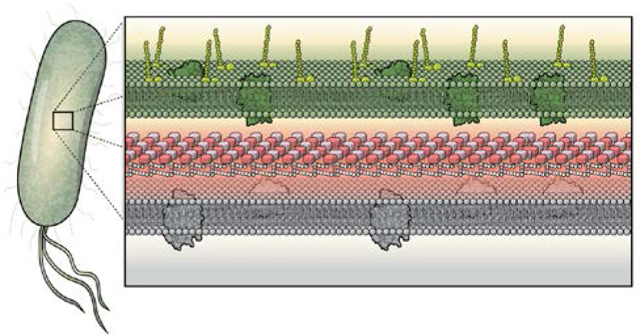
The rise of resistance to clinically approved antibacterial agents poses a major threat to the use of these antibiotics in the future. Multidrug-resistant (MDR) organisms are becoming increasingly common and limit our ability to treat infections in the clinic.1,2 Particularly problematic Gram-positive MDR organisms include Staphylococcus aureus, Enterococcus faecalis, and Streptococcus pneumoniae, whereas Gram-negative pathogens of special concern include Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa (Figure 1).1 Meanwhile, with MDR tuberculosis accounting for approximately half of the globally diagnosed cases of this infectious disease, Mycobacterium tuberculosis remains a perpetual threat. All of these organisms show high rates of not only MDR phenotypes, but also extensive-drug resistance (XDR) and even pan-drug resistance (PDR), leaving few or no available options for treatment.3 This issue is most pressing for Gram-negative strains, with XDR or PDR strains of all of the bacteria listed above being consistently reported.3,4,5
Figure 1.

(a) The cell wall of a Gram-negative bacterium is composed of an outer membrane, the peptidoglycan, and the periplasm, all of which localize many antibiotic targets. (b) Gram-positive bacterium have a thick peptidoglycan layer surrounding its plasma membrane and cytoplasm. Illustrations courtesy of Eric Smith.
However, the risk in Gram-positive organisms cannot be overlooked. Methicillin-resistant S. aureus (MRSA) now commonly carries resistance to multiple other antibiotics as well, as does S. pneumoniae.6,7 While agents such as vancomycin and daptomycin typically retain effectiveness against MRSA, isolated cases of resistance to all of these have been reported, and it seems only a matter of time before many or all of these phenotypes are found in a single strain.3,6,7 The reduced ability to treat bacterial pathogens leads to worse outcomes for patients with community-derived as well as hospital-acquired infections.8
Rising antibiotic resistance is not a new problem, and has plagued medicine since antibiotics were first discovered.9 Within five years of penicillin (1, Scheme 1) entering into the clinic, 50% of S. aureus isolates showed resistance to it.10 It is assumed that resistance is inevitable once an antibiotic enters widespread use, and that any given antibiotic has a “shelf life” before resistance to it becomes common enough to render the drug ineffective.11 During the “Golden age” of antibiotic discovery from the 1930s to the 1960s, this problem was mitigated by the ongoing development of new antibiotics.9,11 Resistance to 1 was answered by the development of methicillin (2), and then other agents once resistance to 2 developed.9,11 However, the rate of antibiotic discovery has dropped significantly, with rediscovery of existing antibiotics or classes of antibiotics being a particular issue with traditional natural product-based approaches.12,13 Notwithstanding this drought, we argue in this Perspective that there are in fact many excellent protein targets and small molecule leads capable of serving as springboards to reinvigorate the antibiotic drug discovery pipeline.
Scheme 1.

Penicillins
While scientific advances afford fundamentally new opportunities,14 perhaps the most significant change in mindset is prompted by mankind’s recent experiences with SARS-CoV-2, an unrelated but clearly problematic human pathogen. Despite not being bacterial in nature, the COVID-19 pandemic has taught us two vitally important lessons with profound implications for fighting old and new bacterial pathogens. From a technical standpoint, the value of partially validated targets and leads has never been more compelling. Indeed, the most successful targets (e.g., RNA-dependent RNA polymerase, Spike protein, MPro protease) and leads (e.g., remdesivir, anti-Spike monoclonal antibodies, conformationally stabilized Spike muteins, PF-07321332) for COVID-19 therapy and prophylaxis emerged from relatively mature R&D programs on related RNA viruses. From an economic perspective, public-private partnerships proved essential to de-risking expensive clinical development campaigns necessary for new drug and vaccine approval. Here we draw upon both these lessons to motivate a renewed focus on antibacterial drug discovery.
Many validated target/lead combinations deserve a second look
The search for a novel antibacterial is most efficiently launched from a foundation of a well-studied biological target with a good chemical lead, neither of which have been subjected to the evolutionary pressure of clinical drug resistance. As it turns out, there is a surprisingly long list of such targets and leads. Classic examples can be found among cell membrane inhibitors (Scheme 2) such as the polymyxins including 3, which were discovered in the 1940s and enjoyed a few decades of clinical use before their usage dropped due to nephrotoxicity issues.15 However, as resistance to other classes of antibiotics has spiked in the past few decades, 3 has been re-enlisted to treat particularly vexing pathogens such as drug-resistant A. baumannii and P. aeruginosa.15,16 More recently, another membrane-targeting antibiotic, daptomycin (4), was approved for use against complicated skin and skin structure infections in 2003.17
Scheme 2.

Cell membrane targeting antibiotics
Beyond cell membranes, another example of an under-exploited target with attractive leads is the essential translation factor EF-Tü, which delivers aminoacyl-tRNAs to the ribosome during translation. Unlike its partner elongation factor EF-G, which is targeted by multiple antibiotics that have been approved for human or veterinary use (e.g., fusidic acid (5), thiostrepton (6)), EF-Tü represents a validated but untapped antibacterial target. At least four families of potent and structurally diverse leads have been identified (Scheme 3), including kirromycin (7), enacyloxin IIa (8), pulvomycin (9), and GE2270A (10).18 These antibiotics are active against Gram-negative and Gram-positive bacteria.19,20 Crystal structures of prototypical inhibitors complexed with EF-Tü have been solved; not only do 7 and 9 have distinct binding sites but they also exploit radically different inhibitory mechanisms – while 9 is a competitive inhibitor of aminoacyl-tRNA binding to the ribosome, 7 locks the EF-Tü•GDP complex to the ribosome.18,19 In light of the fact that bacteria contain fewer than 10,000 ribosomes whereas EF-Tü is one of the most abundant cytoplasmic proteins in the cell, the latter mechanism may be less prone to the evolution of resistance via mere target overexpression.
Scheme 3.

EF-G and EF-Tü inhibitors
Clearly neither any of the above-mentioned leads nor their target is devoid of medicinal challenges. For example, development of 10 encountered difficulties associated with compound solubility and pharmacokinetics.19 One might also presume that, because human mitochondria contain a homolog of eubacterial EF-Tü, safety considerations must be carefully vetted via comparative structure-activity relationship analysis.19,21 In this context it is noteworthy that kirromycin is a considerably weaker inhibitor of mitochondrial translation than eubacterial protein translation.22 As one encouraging example, LFF571 (11), a semi-synthetic analog of 10, showed efficacy in Phase II clinical trials against C. difficile infections.21
Two more examples of underexploited targets with promising leads are illustrative of this point (Scheme 4). First, the Sec transport system in bacteria exports proteins across the cell membrane and harbors at least one medicinally viable active site in its signal peptidase I (SPase I, also known as the leader peptidase LepB).23,24 This serine protease utilizes a Ser-Lys catalytic dyad mechanism to cleave the signal sequence of the nascent protein during export, which differentiates it from most eukaryotic serine proteases (including its mammalian ortholog in the endoplasmic reticulum) that have a Ser-His-Asp catalytic triad in their active sites.25 Arylomycin (12) is a potent SPase I inhibitor with narrow-spectrum activity;26 however, a widespread, naturally occurring mutation was found to confer resistance to 12 in pathogens such as S. aureus, E. coli, and P. aeruginosa.26 This hurdle was overcome through elegant medicinal chemistry efforts, leading to the development of actinocarbasin (13)27 and G0775 (14),28 both with broad-spectrum activity against MRSA, K. pneumoniae, A. baumannii, P. aeruginosa, and other pathogens. While 12 is a noncovalent inhibitor,29 14 covalently binds the catalytic Lys of SPaseI.28
Scheme 4.

Signal peptidase and aminoacyl-tRNA synthetase inhibitors
Aminoacyl-tRNA synthetases (AaRSs) are essential for activating amino acids during ribosomal protein translation.30 They remain a heavily underexploited class of targets with numerous medicinal interesting leads. While safety concerns are legitimate due to the presence of AaRS homologs in humans, many leads do in fact show strong selectivity for bacterial AaRSs.31,32 Nonetheless, to date the only clinically approved AaRS inhibitor is mupirocin (15), a topically used selective inhibitor of bacterial isoleucyl-tRNA synthetase.30
The synthetically accessible phenyl-thiazolylurea-sulfonamide (16) class of competitivePhe-tRNA synthetases (PheRS) teach a valuable lesson, as they have shown good in vitro potency, selectivity, and in vivo activity.31 Enthusiasm for clinical development, however, has been lukewarm because in animal models of infection their activity decreases with increased dietary phenylalanine. While this limitation is understandable for a competitive enzyme inhibitor, it further underscores the promise of exploring other overlooked classes of AaRS inhibitors in the hope of identifying leads that are less amenable to this deactivation mechanism either through higher affinity binding or allosteric mechanisms.
In addition to those mentioned above, there are many other targets that could be options for further development. We have compiled a list of previously reported targets with at least one known antibacterial lead, as well as a partial list of such leads (see Tables S1 and S2). It is important to note that while many of these antibiotics, especially those produced by nature, possess promising mechanisms of action, or have excellent activity profiles, their structural complexity will necessitate substantial development efforts. There will likely be a need for advances in material availability (such as through metabolic pathway engineering) and significant medicinal chemistry campaigns to modify them for SAR improvements, which could prove challenging due to the limited medicinal chemistry accessibility of many large, complex natural products.
Many worthy targets lack good leads despite harboring druggable active sites
By now there appears to be widespread acceptance of the cliché that good targets fall into two classes – druggable and undruggable – with the existence of an evolutionarily well-sculpted active site being the key differentiator. However, the field of antibacterial drug discovery continues to challenge this dogma with its long and growing list of seemingly druggable targets that lack medicinally useful leads. The Mur ligases involved in peptidoglycan biosynthesis are a classic but often overlooked example. The druggability of the Mur pathway (Scheme 5) is incontrovertible; not only is its first step (MurA) blocked by fosfomycin (17),33 but downstream reactions in peptidoglycan formation are the targets of celebrated antibiotics such as β-lactams34 and glycopeptides.35 However, between these clinically validated bookends lie four homologous ATP-dependent amide synthetases (MurC, MurD, MurE and MurF)36 with a troublesome lack of leads against seemingly simple active sites.37 While inhibitors against these enzymes have undoubtedly been reported,33,37,38 none have yielded medicinally viable leads. This paucity of drug-like leads cannot be blamed on a lack of structural insight38 or assay capabilities.39,40,41 In fact, a compelling case can be made for MurC-F being universal benchmarks for innovative in silico or in vitro library technologies that purport to spawn medicinal leads, with pathway assays as a possible tool for facilitating the discovery of such leads. In advance of a future bacterial pandemic, imagine how drug hunters would be empowered by a panel of selective inhibitors of these amide synthetases with excellent in vitro and in vivo potency, good safety and ADME characteristics, and compatibility with automated synthesis methods!
Scheme 5.

Mur ligase pathway
More recently, several prokaryote-specific macromolecular machines have emerged as attractive drug targets that nonetheless lack medicinally appropriate leads. The Clp proteases are a vivid example; they are proteasome-like, ATP-dependent enzymatic machines in bacteria that degrade misfolded or aggregated proteins.42 A multimeric ATPase subunit (ClpC) unfolds a protein substrate, which is then funneled to another multimeric proteolytic subunit (ClpP), where it is degraded. While ClpP homologs are found in both humans and bacteria, the ATPase unit is more variable.43,44 The chemistry and biology of Clp proteases has been extensively studied in recent years.45 While Clp function is only essential in mycobacteria,46 activators of Clp protease action (Scheme 6) have broad-spectrum activity. For example, cyclomarin A (18) activates ATPase activity, leading to cell death by uncontrolled protein degradation,47 while acyl-depsipeptides such as 19 (ADEPs) achieve the same goal through allosteric activation of ClpP.48,49 While neither is a viable class of lead molecules unto themselves (18 depends upon reactivity of an electrophilic epoxide, whereas naturally occurring 19 has poor aqueous solubility and high clearance), these molecules set the stage for a more expansive approach toward medicinal lead identification against this target family.50,51 A particularly intriguing mode of action might involve borrowing a page from proteolysis-targeting chimeras (PROTACS), bifunctional small molecules that facilitate the degradation of specific proteins in eukaryotes.52 The Clp protease system could be analogously hijacked through the engineering of chimeric molecules that target proteolysis of specific proteins in bacteria.53 One example of a target protein is FtsZ, discussed below.
Scheme 6.

Clp protease activators and FtsZ inhibitors
FtsZ, an analog of eukaryotic tubulin that is essential for cell division in bacteria,54 is also an attractive antibiotic target in search of new leads. Polymerization of FtsZ forms the cytokinetic “Z-ring” at the division site, which then recruits accessory factors required for peptidoglycan remodeling and other aspects of cell division (collectively known as the divisome).55 While analogous to tubulin in function, it differs considerably in sequence and structure.55 Inhibitors of FtsZ are active against S. aureus, S. pneumoniae, M. tuberculosis, and P. aeruginosa, among others.56,57,58 For example, SRI-3072 (20) kills M. tuberculosis inside mouse macrophages,55 while the Zantrins58 (21–25) are synthetic noncompetitive inhibitors with broader spectrum activity.59 Whereas the FtsZ inhibitor TXA709 (26), formulated as a prodrug, has shown good safety in humans,60 no antibiotic targeting the divisome has been approved to date notwithstanding the identification of at least three druggable sites within this assembly.61 Undoubtedly, as classical enablers of drug discovery such as improved binding assays62 and additional X-ray crystal structures63,64,65 become available, the opportunities for superior clinical candidates will continue to grow. At the same time, the tight geometric constraints for FtsZ function at the cell division plate suggest that an alternative PROTACS-like approach for FtsZ degradation may be particularly promising.
Non-traditional strategies for antibacterial discovery must be encouraged
Prior to the COVID-19 pandemic, a vast majority of our repertoire of clinically used antivirals had been engineered against essential targets encoded within the genome of the pathogenic virus. The stunning capacity of SARS-CoV-2 (as well as earlier coronaviruses such as SARS and MERS) to exacerbate from a seemingly innocuous upper respiratory tract infection into a systemic, life-threatening illness has prompted a shift in focus towards the identification of host-acting antiviral mechanisms that could function as broader-spectrum antivirals that might even be less prone to resistance. While the implications of this new approach to antiviral therapy remain to be seen, analogous paradigmatic shifts are starting to gain momentum in antibacterial drug discovery and must be nurtured (Scheme 7).
Scheme 7.

Antibiotics discovered from non-traditional approaches
During the Golden age of antibiotic discovery, new antibiotics were rapidly discovered through systematic screening of secondary metabolites produced by soil-dwelling bacteria, predominantly the actinomycetes.66 Notwithstanding its immense success, this strategy ultimately led to diminishing returns.67 With the sequencing of the Haemophilus influenzae genome, the prospect of identifying new bacterial targets led to early investment by many pharmaceutical companies.68,69 Unfortunately, genomics-driven antibacterial discovery failed to quickly spawn a second “golden” era of antibiotic discovery (as discussed in many retrospective analyses67,68,69,70) as targets determined to be essential often failed to yield useful leads. This highlights how targets with even a weak lead may be preferable to those without any, regardless of other properties of the targets themselves. Genome mining approaches to new antibacterial lead discovery assist in the discovery of new leads based on the recognition that biosynthetic genes for secondary metabolite production in bacteria and fungi are clustered, which allows educated guesses of both the natural product’s broad structural features as well as its antibacterial mode of action.71 The former guess derives from the identification of putative enzymes involved in polyketide, peptide, or isoprenoid biosynthesis, whereas the latter stems from co-localization of a putative self-resistance enzyme that protects the host from its own natural product.72 Examples of self-resistance enzymes colocalized with biosynthetic pathways include ones involved in protein biosynthesis,73 DNA replication,74 and lipid metabolism.75 An illustrative case of this kind of genome mining is highlighted by the discovery of thiotetronic acid (27), which blocks fatty acid synthesis in bacteria.76
Another promising trend in antibacterial drug discovery involves harnessing natural products from under-explored genera and ecological niches. As a simple example, while actinomyces have been extensively screened for new antibiotics, filamentous fungi have been not as thoroughly mined notwithstanding their track record of yielding immensely valuable β-lactams (including of course penicillin).77
Similarly, the nematode-inhabiting bacteria Xenorhabdus and Photorhabdus have gained attention as promising sources of new antibiotics based on the discovery of two promising new antibiotics, odilorhabdin (28)78,79 and darobactin (29),80 respectively. 28 inhibits a new site in the ribosomes of both Gram-positive and Gram-negative bacteria (thus showing no cross-resistance to other clinically used ribosome inhibitors),78 whereas 29 targets a hitherto unrecognized target BamA, an outer membrane protein in Gram-negative bacteria that catalyzes the folding and insertion of essential outer membrane proteins.81 The attractiveness of BamA as an antibiotic target has been further underscored by the more-or-less concomitant identification of three other classes of leads, including a murepavadin-polymyxin B chimera,82 MRL-494 (30),83 and even an antibody.84 Collectively, these antibiotics have demonstrated potency against many particularly problematic Gram-negative pathogens such as A. baumannii, P. aeruginosa, and K. pneumoniae.82 It goes without saying that these antibiotics have the added benefit of acting at the cell surface, thereby circumventing cell permeability issues. While attractive leads are known for these targets, additional medicinal chemistry will be necessary to bring any of them to the clinic.
One final paradigm-shifting approach to antibacterial discovery deserves mention here. While research on M. tuberculosis has extensively documented the phenomenon of a “persister” phenotype in this pathogen (one that achieves antibiotic resistance by persisting in a non-replicating, metabolically inert stage within the human host for long durations), there is growing recognition that persisters represent a major challenge to antibiotic-driven elimination of chronic infections caused by a variety of human pathogens. A better understanding of the bacterial defense system in the persister state holds the key to conquering this phenotype.85 For example, in response to oxidative stress or antibiotics, bacteria increase the production of NO and H2S as signaling molecules.86,87 H2S is produced en zymatically by orthologues of mammalian cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), or 3-mercaptopyruvate sulfurtransferase.85 Recently, inhibitors with an indole moiety such as NL1 (31) were found to inhibit CSE from S. aureus and P. aeruginosa and demonstrated promising potentiation effects when dosed in combination with major bactericidal antibiotic classes such as the fluoroquinolones, β-lactams, and aminoglycosides, but not with bacteriostatic antibiotics such as tetracycline or chloramphenicol.85 While much remains to be learned about the remarkable metabolic networks used by persister bacteria to evade antibiotics, it is likely that these fundamental investigations will shine light on promising targets for future antibacterial drug discovery.
Polypharmacology is under-exploited in antibacterial drug development
A particularly effective strategy to hinder the emergence of drug resistance involves engineering a drug that synergistically blocks more than one target. By now polypharmacology is well established in oncology especially through the emergence of multiple pan-kinase inhibitors as frontline cancer chemotherapeutics.88,89 In contrast, although combination therapy could be envisioned as a rudimentary flavor of bacterial polypharmacology (e.g., trimethoprim (32) and sulfamethoxazole (33) in co-trimoxazole90), it has received scant attention in anti-infective drug discovery.
In fact, there are many examples of promising antibiotic leads selective for multiple bacterial targets (Scheme 8). For example, kibdelomycin (34), a polyketide/non-ribosomal peptide, targets the ATPase activities of type II topoisomerases including GyrB and ParE, both legitimate antibacterial targets in their own right.91 Structural studies of 34 bound to S. aureus GyrB showed that it binds the ATP-binding site pocket with a unique “dual-arm” binding mode with multiple contact points, perhaps explaining the low resistance frequency to this antibiotic observed in S. aureus.92 Kibdelomycin has potent activity in a hamster model of C. difficile colitis, conferring 100% protection at a 6.25 mg/kg oral dose with low systemic clearance.93 Notably, it also has strong activity against a panel of clinical isolates of A. baumannii.94 Another example is teixobactin (35), a depsipeptide that binds to lipid II and lipid III and thus blocks peptidoglycan and cell wall teichoic acid synthesis, respectively.95 This leads to a disorganized, damaged bacterial cell envelope and delocalized autolysins, culminating in cell death.96,97 Due to its promising mechanism of action and moderate complexity, it has been the subject of multiple SAR studies.98 Last but not least, corbomycin (36) and complestatin (37) inhibit multiple autolysins,99 a class of peptidoglycan hydrolases required for cell wall turnover and cell separation during replication.100
Scheme 8.

Antibacterial agents exhibiting polypharmacology
While these examples of multitarget agents act on related targets in the same pathway, multifunctional antibiotics acting via unrelated mechanisms are also known. For example, Irresistin-16 (38) permeabilizes the bacterial cell membrane while also targeting dihydrofolate reductase (DHFR).101 DHFR catalyzes the NADPH-dependent production of the tetrahydrofolate, an essential compound used for the de novo synthesis of dTMP involved in DNA building block synthesis.102 It demonstrates efficacy in Gram-negative and Gram-positive bacteria such as MRSA, N. gonorrhoeae and A. baumannii with no sign of resistance.101 Similarly, while the use of rifampin (39) is limited to combination therapies owing to the emergence of resistance against this antibiotic at appreciable frequency, TNP-2092 (40), combining the pharmacophores of RNA polymerase-targeting 39 with the DNA gyrase-targeting quinolizinone, maintains the potent activity of rifamycin against persistent pathogens and at the same time minimizes the development of rifamycin resistance.103 Recently, a “tribrid” siderophore-cephalosporin-oxazolidinone conjugate (41) was engineered; after internalization via siderophore-mediated uptake, the oxazolidinone is released by the bacteria’s inherent β-lactamase activity.104
The quest for resistance enzyme inhibitors that breathe new life into old antibiotics must continue
The potency of a failing antibiotic can be preserved by combining it with an agent that inhibits a clinically relevant resistance mechanism. For example, β-lactamase inhibitors (Scheme 9) have found widespread utility. β-lactamases fall into four distinct classes, with Class A and Class C being the most important from a clinical standpoint.105,106 Classes A, C, and D rely on a serine protease type hydrolytic mechanism, whereas class Bs are zinc metallo-hydrolases.105,106 Clavulanic acid (42) is a prototypical example of a clinically successful β-lactamase inhibitor. Itself a weak β-lactam antibiotic produced by Streptomyces clavuligerus, 42 is a potent irreversible inhibitor of common β-lactamase enzymes.107,108,109 It found initial use in combination with amoxicillin in the early 1980s.110,111 However, much like with β-lactam antibiotics themselves, 42 has become susceptible to the evolution of resistance.110,111 In response, newer β-lactamase inhibitors with broader ranges of activities were developed, with one of the most successful being avibactam (43).112 43 lacks a β-lactam core, but still covalently inhibits β-lactamases, including many of the most important Class A, C, and D enzymes.112,113,114 It is typically paired with the third-generation cephalosporin ceftazidime (44) a combination that retains activity against many key Gram-negative organisms. However, 43 has poor oral bioavailability, and is therefore only used as an injectable. A prodrug variant of 43, ARX-1796 (45), is entering clinical trials with the companion β-lactam antibiotic ceftibuten (46).115 Notably, while leads against Class B β-lactamases such as QPX7728 (47)116 and aspergillomarasmine A (48)117,118,119 have been discovered, a clinically useful inhibitor of this class of metalloenzymes has not yet emerged.
Scheme 9.

β-lactamase inhibitors and relateds molecules
Whereas β-lactamase inhibitors represent a clear success in targeting resistance mechanisms to restore activity of existing molecules, similar approaches have not been successful against aminoglycoside antibiotic modifying enzymes (AMEs). Aminoglycosides (Scheme 10) are a diverse class of molecules that typically inhibit protein synthesis through direct binding to the ribosome.120 Most aminoglycosides bind inside or very close to the A-site of the ribosome and interfere with the translocation step of protein synthesis,120 although some like streptomycin are known to bind elsewhere in the ribosome.121 They were in common use until the late 1970s, when they began to be phased out due to discovery of newer antibiotics with fewer toxicity issues,120 but have undergone somewhat of a renaissance due to increasing resistance to other antibiotics.120,122 Three major types of aminoglycoside modifying enzymes are classified by the modification they catalyze: aminoglycoside N-acetyltransferases (AAC), aminoglycoside O-adenyltransferases (ANT), and aminoglycoside O-phosphotransferases (APH).122,123,124 They are further sub-classified by the actual position on the aminoglycoside, such as kanamycin (49), they modify. For instance, an AAC(6’) enzyme acetylates the 6’ nitrogen of susceptible aminoglycosides. Because a given aminoglycoside modifying enzyme is usually most effective against only a small subset of aminoglycosides,122 inhibitors need not be broadly active against this superfamily of resistance enzyme in order to be useful. For example, AAC(6’) leads to resistance against the second-generation aminoglycoside amikacin (50);122 therefore a medicinally effective inhibitor of this N-acetyltransferase would have considerable clinical impact. Aranorosin (51) is an inhibitor of the bifunctional AAC(6’)-APH(2”) enzyme from methicillin-resistant S. aureus that potentiates amikacin activity in vitro.125 More recently, a small-scale screen identified additional inhibitors of AAC(6’)-APH(2”) enzymes.126 As a target class though, aminoglycoside modifying enzymes are not as economically attractive as β-lactamases due to their inability to breathe new life into multiple broad-spectrum antibiotics. An alternative approach to rejuvenating the effectiveness of aminoglycosides has already been taken with plazomicin (52), a semisynthetic aminoglycoside that is not a substrate for most AMEs.127 Unfortunately, despite receiving FDA approval, 52 was a major financial failure and partially responsible for the bankruptcy of its developer Achaogen.128 When attempting to address the problems of antibiotic resistance, economic challenges must be considered as well.
Scheme 10.

Aminoglycosides and aminoglycoside modifying enzyme inhibitors
The future of antibacterial discovery will remain bleak absent new pharmacoeconomic models
The growing list of promising antibacterial agents that either failed to complete clinical trials due to insufficient resources or worse became economically non-viable products after gaining new drug approval has prompted calls from experts and laymen for alternative business models to promote innovative antibiotic discovery and development.
At the legislative level, Congress passed the Generating Antibiotic Incentives Now (GAIN) Act of 2012, which provided for an incremental five-year exclusivity period from generic competition (over and beyond the minimum five-year exclusive period for new small molecule drugs as well as any applicable orphan drug or pediatric exclusivity) plus Fast Track and Priority Review incentive programs by the FDA. Many experimental antibiotics have qualified for GAIN Act designation, and a few have even been approved (dalbavancin (53, Scheme 11) being a notable example).129 However, nearly a decade later, most objective assessments would conclude that the impact of the GAIN Act alone on the antibiotic pipeline has been modest at best. The unfortunate example of 52, which collapsed as a product (along with its developer, Achaogen) notwithstanding FDA approval following GAIN Act qualification, is a vivid testimonial.
Scheme 11.

Dalbavancin
New government-philanthropy partnerships have also been explored over the past decade to fill the void in antibacterial discovery and development created by negligible pharma or venture capital investment. A particularly notable example is the Combating Antibiotic-Resistant Bacteria Accelerator (CARB-X). Over the past five years, CARB-X has curated an impressive pipeline of ca. 50 anti-infective programs including small molecules, proteins, non-traditional agents (e.g., bacteriophages, microbiome therapies), and vaccines.130 While the roadmap for these agents through pivotal clinical studies and beyond is anything but straightforward, one hopes that judicious stewardship until clinical proof-of-concept is at hand will give these programs their best shot at long-term success.
Looking forward, at least two recently precedented pharmacoeconomic strategies have the potential to rejuvenate our pipeline of innovative antibacterials. One approach would involve expanding the scope of the Priority Review Voucher program created by the United States Congress in 2007 to accelerate drug development for rare pediatric disorders as well as neglected diseases that predominantly burden the developing world. These vouchers, which reduce the time to new drug approval by approximately six months, have been issued for the successful development of more than 50 drugs thus far (including a few anti-infective agents), and have an established value of~$100M (https://sites.fuqua.duke.edu/priorityreviewvoucher/). If applied more broadly to antibacterial drug development, such an economic bonanza can incentivize both investors and the pharmaceutical industry to invest more heavily in the most promising programs that emerge from the academic sector. An alternative (and complementary) strategy has been successfully test-piloted during the COVID-19 pandemic with the U.S. government substantially de-risking SARS-CoV-2 drug and vaccine development through a three-pronged approach comprising co-investments in clinical trials, an invigorated Emergency Use Authorization (EUA) effort by the FDA, and post-approval purchase guarantees from multiple national governments. The breakneck speeds at which innovative products such as mRNA vaccines and anti-infective monoclonal antibodies have been launched is a vivid testimonial to the power of such an integrated strategy. While scientific advances in microbiology and chemistry will remain the fountainhead for new tools that ward off the persistent threat of new and old bacterial pathogens, the will to translate these discoveries into clinical practice must ultimately come from society. Absent such a collective commitment from all of us, future local or global pandemics will continue to reverse our hard-won gains in life expectancy and quality of life afforded by the Golden Era of Antibiotics in the past century.
Supplementary Material
ACKNOWLEDGMENT
We thank E. Smith for figure preparation and graphics support.
Funding Sources
C.K. is supported by the National Institutes of Health (R35 GM141799). G.D.W is supported by funded by a Canadian Institutes of Health Research grant (FRN-148463), and by a Canada Research Chair. A.M.S acknowledges support from the National Science Foundation Graduate Research Fellowship under Grant DGE-1656518.
Footnotes
ASSOCIATED CONTENT
This material is available free of charge via the Internet at http://pubs.acs.org.
Supporting Information: Lists of selected antibiotic targets and leads.
J.W. is consulting for Soltego. C.T.W. is a scientific advisor to Hua Biomedicines, T Cell Therapeutics, and the Vividion division of Bayer. G.D.W. is an advisor to Prokaryotics Inc. N.S.G. is a founder, scientific advisory board member (SAB) and equity holder in Syros, C4, Allorion, Jengu, B2S, Inception, EoCys, Larkspur (board member) and Soltego (board member). C.K. is on the Board of Directors of ImmunogenX.
REFERENCES
- (1).Vivas R; Barbosa AAT; Dolabela SS; Jain S Multidrug-Resistant Bacteria and Alternative Methods to Control Them: An Overview. Microb. Drug Resist. 2019, 25 (6), 890–908. [DOI] [PubMed] [Google Scholar]
- (2).Aslam B; Wang W; Arshad MI Khurshid M, Muzammil S; Rasool MH; Nisar MA; Alvi RF; Aslam MA; Qamar MU; Salamat MKF; Baloch Z Antibiotic Resistance: A Rundown of a Global Crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Magiorakos AP; Srinivasan A; Carey RB; Carmeli Y; Falagas ME; Giske CG; Harbarth S; Hindler JF; Kahlmeter G; Olsson-Liljequist B; Paterson DL; Rice LB; Stelling J; Struelens MJ; Vatopoulos A; Weber JT; Monnet DL Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18 (3), 268–281. [DOI] [PubMed] [Google Scholar]
- (4).Livermore DM Current Epidemiology and Growing Resistance of Gram-Negative Pathogens. Korean J. Intern. Med. 2012, 27 (2), 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rossolini GM; Arena F; Pecile P; Pollini S Update on the Antibiotic Resistance Crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. [DOI] [PubMed] [Google Scholar]
- (6).Almanaa TN; Alyahya SA; Khaled JM; Shehu MR; Alharbi NS; Kadaikunnan S; Alobaidi AS; Khalid Alzahrani A The Extreme Drug Resistance (XDR) Staphylococcus Aureus Strains among Patients: A Retrospective Study. Saudi J. Biol. Sci. 2020, 27 (8), 1985–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Adeiza SS; Ademola Onaolapo J; Olalekan Olayinka B Prevalence, Risk Factors and Antimicrobial Susceptibility Profile of Methicillin-Resistant Staphylococcus Aureus (MRSA) Obtained From Nares of Patients and Staff of Sokoto State-Owned Hospitals in Nigeria. GMS Hyg. Infect. Control. 2020, 15, Doc25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).van Duin D; Paterson DL Multidrug-Resistant Bacteria in the Community: Trends and Lessons Learned. Infect. Dis. Clin. North. Am. 2016, 30 (2), 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Podolsky SH The Evolving Response to Antibiotic Resistance (1945–2018). Palgrave Commun. 2018, 4 (1). 10.1057/s41599-018-0181-x. (Accessed 2021-08-16) [DOI] [Google Scholar]
- (10).Wenzel RP The Antibiotic Pipeline — Challenges, Costs, and Values. N. Engl. J. Med. 2004, 351 (6), 523–526. [DOI] [PubMed] [Google Scholar]
- (11).Walsh C Where Will New Antibiotics Come From? Nat. Rev. Microbiol. 2003, 1 (1), 65–70. [DOI] [PubMed] [Google Scholar]
- (12).Nathan C Antibiotics at the Crossroads. Natur. 2004, 431 (7011), 899–902. [DOI] [PubMed] [Google Scholar]
- (13).Baltz RH Marcel Faber Roundtable: Is Our Antibiotic Pipeline Unproductive Because of Starvation, Constipation or Lack of Inspiration? J. Ind. Microbiol. Biotechnol. 2006, 33 (7), 507–513. [DOI] [PubMed] [Google Scholar]
- (14).Fischbach MA; Walsh CT Antibiotics for Emerging Pathogens. Science. 2009, 325 (5944), 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yahav D; Farbman L; Leibovici L; Paul M Colistin: New Lessons on an Old Antibiotic. Clin. Microbiol. Infect. 2012, 18 (1), 18–29. [DOI] [PubMed] [Google Scholar]
- (16).Boucher HW; Talbot GH; Bradley JS; Edwards JE; Gilbert D; Rice LB; Scheld M; Spellberg B; Bartlett J Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- (17).Heidary M; Khosravi AD; Khoshnood S; Nasiri MJ; Soleimani S; Goudarzi M Daptomycin. J. Antimicrob. Chemother. 2018, 73 (1), 1–11. [DOI] [PubMed] [Google Scholar]
- (18).Parmeggiani A; Nissen P Elongation Factor Tu-Targeted Antibiotics: Four Different Structures, Two Mechanisms of Action. FEBS Lett. 2006, 580 (19), 4576–4581. [DOI] [PubMed] [Google Scholar]
- (19).Prezioso SM; Brown NE; Goldberg JB Elfamycins: Inhibitors of Elongation Factor-Tu. Mol. Microbiol. 2017, 106 (1), 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Leeds JA; LaMarche MJ; Brewer JT; Bushell SM; Deng G; Dewhurst JM; Dzink-Fox J; Gangl E; Jain A; Lee L; Lilly M; Manni K; Mullin S; Neckermann G; Osborne C; Palestrant D; Patane MA; Raimondi A; Ranjitkar S; Rann EM; Sachdeva M; Shao J; Tiamfook S; Whitehead L; Yu D In Vitro and in Vivo Activities of Novel, Semisynthetic Thiopeptide Inhibitors of Bacterial Elongation Factor Tu. Antimicrob. Agents Chemother. 2011, 55 (11), 5277–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mullane K; Lee C; Bressler A; Buitrago M; Weiss K; Dabovic K; Praestgaard J; Leeds JA; Blais J; Pertel P Multicenter, Randomized Clinical Trial to Compare the Safety and Efficacy of LFF571 and Vancomycin for Clostridium Difficile Infections. Antimicrob. Agents Chemother. 2015, 59 (3), 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang L;Ging NC;Komoda T;Hanada T;Suzuki T; Watanabe K Antibiotic Susceptibility of Mammalian Mitochondrial Translation. FEBS Lett. 2005, 579 (28), 6423–6427. [DOI] [PubMed] [Google Scholar]
- (23).Paetzel M Structure and Mechanism of Escherichia Coli Type I Signal Peptidase. Biochim. Biophys. Acta. 2014, 1843 (8), 1497–1508. [DOI] [PubMed] [Google Scholar]
- (24).Paetzel M; Dalbey RE; Strynadka NCJ The Structure and Mechanism of Bacterial Type I Signal Peptidases: A Novel Antibiotic Target. Pharmacol. Ther. 2000, 87 (1), 27–49. [DOI] [PubMed] [Google Scholar]
- (25).Auclair SM; Bhanu MK; Kendall DA Signal Peptidase I: Cleaving the Way to Mature Proteins. Protein Sci. 2012, 21 (1), 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Craney A; Romesberg FE The Inhibition of Type I Bacterial Signal Peptidase: Biological Consequences and Therapeutic Potential. Bioorg. Med. Chem. Lett. 2015, 25 (21), 4761–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Therien AG; Huber JL; Wilson KE; Beaulieu P; Caron A; Claveau D; Deschamps K; Donald RGK; Galgoci AM; Gallant M; Gu X; Kevin NJ; Lafleur J; Leavitt PS; Lebeau-Jacob C; Lee SS; Lin MM; Michels AA; Ogawa AM; Painter RE; Parish CA; Park YW; Benton-Perdomo L; Petcu M; Phillips JW; Powles MA; Skorey KI; Tam J; Tan CM; Young K; Wong S; Waddell ST; Miesel L Broadening the Spectrum of β-Lactam Antibiotics through Inhibition of Signal Peptidase Type I. Antimicrob. Agents Chemother. 2012, 56 (9), 4662–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Smith PA; Koehler MFT;G irgis HS; Yan D; Chen Y; Chen Y; Crawford JJ; Durk MR; Higuchi RI; Kang J; Murray J; Paraselli P; Park S; Phung W; Quinn JG; Roberts TC; Rougé L; Schwarz JB; Skippington E; Wai J; Xu M; Yu Z; Zhang H; Tan MW; Heise CE Optimized Arylomycins Are a New Class of Gram-Negative Antibiotics. Nature. 2018, 561 (7722), 189–194. [DOI] [PubMed] [Google Scholar]
- (29).Luo C; Roussel P; Dreier J; Page MGP; Paetzel M Crystallographic Analysis of Bacterial Signal Peptidase in Ternary Complex with Arylomycin A2 and a β-Sultam Inhibitor. Biochemistry. 2009, 48 (38), 8976–8984. [DOI] [PubMed] [Google Scholar]
- (30).Hurdle JG; O’Neill AJ; Chopra I Prospects for Aminoacyl-tRNA Synthetase Inhibitors as New Antimicrobial Agents. Antimicrob. Agents Chemother. 2005, 49 (12), 4821–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Beyer D; Kroll HP; Endermann R; Schiffer G; Siegel S; Bauser M; Pohlmann J; Brands M; Ziegelbauer K; Haebich D; Eymann C; Brötz-Oesterhelt H New Class of Bacterial Phenylalanyl-tRNA Synthetase Inhibitors with High Potency and Broad-Spectrum Activity. Antimicrob. Agents Chemother. 2004, 48 (2), 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Purnapatre KP; Rao M; Pandya M; Khanna A; Chaira T; Bambal R; Upadhyay DJ; Masuda N In Vitro and in Vivo Activities of DS86760016, a Novel Leucyl-tRNA Synthetase Inhibitor for Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2018, 62 (4), e01987–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Silver LL Fosfomycin: Mechanism and Resistance. Cold Spring Harb. Perspect. Med. 2017, 7 (2), a025262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wivagg CN; Bhattacharyya RP; Hung DT Mechanisms of β-Lactam Killing and Resistance in the Context of Mycobacterium Tuberculosis. J. Antibiot. (Tokyo). 2014, 67 (9), 645–654. [DOI] [PubMed] [Google Scholar]
- (35).Reynolds PE Structure, Biochemistry and Mechanism of Action of Glycopeptide Antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8 (11), 943–950. [DOI] [PubMed] [Google Scholar]
- (36).Barreteau H; Kovac A; Boniface A; Sova M; Gobec S; Blanot D Cytoplasmic Steps of Peptidoglycan Biosynthesis. FEMS Microbiol. Rev. 2008, 32 (2), 168–207. [DOI] [PubMed] [Google Scholar]
- (37).ElZoeiby A; Sanschagrin F; Levesque RC Structure and Function of the Mur Enzymes: Development of Novel Inhibitors. Mol. Microbiol. 2003, 47 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- (38).Kouidmi I; Levesque RC; Paradis-Bleau C The Biology of Mur Ligases as an Antibacterial Target. Mol. Microbiol. 2014, 94 (2), 242–253. [DOI] [PubMed] [Google Scholar]
- (39).Munshi T; Gupta A; Evangelopoulos D; Guzman JD; Gibbons S; Keep NH; Bhakta S Characterisation of ATP-Dependent Mur Ligases Involved in the Biogenesis of Cell Wall Peptidoglycan in Mycobacterium Tuberculosis. PLoS One. 2013, 8 (3), e60143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Eniyan K; Rani J; Ramachandran S; Bhat R; Khan IA; Bajpai U Screening of Antitubercular Compound Library Identifies Inhibitors of Mur Enzymes in Mycobacterium Tuberculosis. SLAS Discov. 2020, 25 (1), 70–78. [DOI] [PubMed] [Google Scholar]
- (41).Eniyan K; Kumar A; Rayasam GV; Perdih A; Bajpai U Development of a One-Pot Assay for Screening and Identification of Mur Pathway Inhibitors in Mycobacterium Tuberculosis. Sci. Rep. 2016, 6 (35134). 10.1038/srep35134. (Accessed 2021-08-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kirstein J; Molière N; Dougan DA; Turgay K Adapting the Machine: Adaptor Proteins for Hsp100/Clp and AAA+ Proteases. Nat. Rev. Microbiol. 2009, 7 (8), 589–599. [DOI] [PubMed] [Google Scholar]
- (43).Kang SG; Dimitrova MN; Ortega J; Ginsburg A; Maurizi MR Human Mitochondrial ClpP Is a Stable Heptamer That Assembles into a Tetradecamer in the Presence of ClpX. J. Biol. Chem. 2005, 280 (42), 35424–35432. [DOI] [PubMed] [Google Scholar]
- (44).Culp E; Wright GD Bacterial Proteases, Untapped Antimicrobial Drug Targets. J. Antibiot. (Tokyo). 2017, 70, 366–377. [DOI] [PubMed] [Google Scholar]
- (45).Yu AYH; Houry WA ClpP: A Distinctive Family of Cylindrical Energy-Dependent Serine Proteases. FEBS Lett. 2007, 581 (19), 3749–3757. [DOI] [PubMed] [Google Scholar]
- (46).Gavrish E; Sit CS; Cao S; Kandror O; Spoering A; Peoples A; Ling L; Fetterman A; Hughes D; Bissell A; Torrey H; Akopian T; Mueller A; Epstein S; Goldberg A; Clardy J; Lewis K Lassomycin, a Ribosomally Synthesized Cyclic Peptide, Kills Mycobacterium Tuberculosis by Targeting the ATP-Dependent Protease ClpC1P1P2. Chem. Biol. 2014, 21 (4), 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Maurer M; Linder D; Franke KB; Jäger J; Taylor G; Gloge F; Gremer S; Le Breton L; Mayer MP; Weber-Ban E; Carroni M; Bukau B; Mogk A Toxic Activation of an AAA+ Protease by the Antibacterial Drug Cyclomarin A. Cell Chem. Biol. 2019, 26 (8), 1169–1179.e4. [DOI] [PubMed] [Google Scholar]
- (48).Sass P; Josten M; Famulla K; Schiffer G; Sahl H-G; Hamoen L; Brötz-Oesterhelt H Antibiotic Acyldepsipeptides Activate ClpP Peptidase to Degrade the Cell Division Protein FtsZ. Proc. Natl. Acad. Sci. 2011, 108 (42), 17474–17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kirstein J; Hoffmann A; Lilie H; Schmidt R; Rübsamen-Waigmann H; Brötz-Oesterhelt H; Mogk A; Turgay K The Antibiotic ADEP Reprogrammes ClpP, Switching It from a Regulated to an Uncontrolled Protease. EMBO Mol. Med. 2009, 1 (1), 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hinzen B; Raddatz S; Paulsen H; Lampe T; Schumacher A; Häbich D; Hellwig V; Benet-Buchholz J; Endermann R; Labischinski H; Brötz-Oesterhelt H Medicinal Chemistry Optimization of Acyldepsipeptides of the Enopeptin Class Antibiotics. ChemMedChem. 2006, 1 (7), 689–693. [DOI] [PubMed] [Google Scholar]
- (51).Arvanitis M; Li G; Li DD; Cotnoir D; Ganley-Leal L; Carney DW; Sello JK; Mylonakis E A Conformationally Constrained Cyclic Acyldepsipeptide Is Highly Effective in Mice Infected with Methicillin-Susceptible and -Resistant Staphylococcus Aureus. PLoS One. 2016, 11 (4), e0153912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Nalawansha DA; Crews CM PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27 (8), 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Morreale FE; Kleine S; Leodolter J; Ovchinnikov S; Kley J; Kurzbauer R; Hoi DM; Meinhart A; Hartl M; Haselbach D; Kaiser M; Clausen T BacPROTACs Mediate Targeted Protein Degradation in Bacteria. bioRxiv. 2021, 10.1101/2021.06.09.447781. (Accessed 2021-06-11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bi E; Lutkenhaus J FtsZ Ring Structure Associated with Division in Escherichia Coli. Nature. 1991, 354 (6349), 161–164. [DOI] [PubMed] [Google Scholar]
- (55).White EL; Suling WJ; Ross LJ; Seitz LE; Reynolds RC 2-Alkoxycarbonylaminopyridines: Inhibitors of Mycobacterium Tuberculosis FtsZ. J. Antimicrob. Chemother. 2002, 50 (1), 111–114. [DOI] [PubMed] [Google Scholar]
- (56).Schaffner-Barbero C; Martín-Fontecha M; Chacón P; Andreu JM Targeting the Assembly of Bacterial Cell Division Protein FtsZ with Small Molecules. ACS Chem. Biol. 2012, 7 (2), 269–277. [DOI] [PubMed] [Google Scholar]
- (57).Wang J; Galgoci A; Kodali S; Herath KB; Jayasuriya H; Dorso K; Vicente F; González A; Cully D; Bramhill D; Singh S Discovery of a Small Molecule That Inhibits Cell Division by Blocking FtsZ, a Novel Therapeutic Target of Antibiotics. J. Biol. Chem. 2003, 278 (45), 44424–44428. [DOI] [PubMed] [Google Scholar]
- (58).Margalit DN; Romberg L; Mets RB; Hebert AM; Mitchison TJ; Kirschner MW; RayChaudhuri D Targeting Cell Division: Small-Molecule Inhibitors of FtsZ GTPase Perturb Cytokinetic Ring Assembly and Induce Bacterial Lethality. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (32), 11821–11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Anderson DE; Kim MB; Moore JT; O’Brien TE; Sorto NA; Grove CI; Lackner LL; Ames JB; Shaw JT Comparison of Small Molecule Inhibitors of the Bacterial Cell Division Protein Ftsz and Identification of a Reliable Cross-Species Inhibitor. ACS Chem. Biol. 2012, 7 (11), 1918–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kaul M; Mark L; Zhang Y; Parhi AK; Lyu YL; Pawlak J; Saravolatz S; Saravolatz LD; Weinstein MP; LaVoie EJ; Pilcha DS TXA709, an FtsZ-Targeting Benzamide Prodrug with Improved Pharmacokinetics and Enhanced in Vivo Efficacy against Methicillin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 2015, 59 (8), 4845–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Tripathy S; Sahu SK FtsZ Inhibitors as a New Genera of Antibacterial Agents. Bioorg. Chem. 2019, 91 (103169). 10.1016/j.bioorg.2019.103169. (Accessed 2021-08-28) [DOI] [PubMed] [Google Scholar]
- (62).Kusuma KD; Payne M; Ung AT; Bottomley AL; Harry EJ FtsZ as an Antibacterial Target: Status and Guidelines for Progressing This Avenue. ACS Infect. Dis. 2019, 5 (8), 1279–1294. [DOI] [PubMed] [Google Scholar]
- (63).Matsui T; Yamane J; Mogi N; Yamaguchi H; Takemoto H; Yao M; Tanaka I Structural Reorganization of the Bacterial Cell-Division Protein FtsZ from Staphylococcus Aureus. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 (9), 1175–1188. [DOI] [PubMed] [Google Scholar]
- (64).Fujita J; Maeda Y; Mizohata E; Inoue T; Kaul M; Parhi AK; Lavoie EJ; Pilch DS; Matsumura H Structural Flexibility of an Inhibitor Overcomes Drug Resistance Mutations in Staphylococcus Aureus FtsZ. ACS Chem. Biol. 2017, 12 (7), 1947–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Alnami A; Norton RS;Pena HP; Haider S; Kozielski F Conformational Flexibility of A Highly Conserved Helix Controls Cryptic Pocket Formation in FtsZ. J. Mol. Biol. 2021, 433 (15), 167061. [DOI] [PubMed] [Google Scholar]
- (66).Hutchings MI; Truman AW; Wilkinson B Antibiotics: Past, Present and Future. Curr. Opin. Microbiol. 2019, 51, 72–80. [DOI] [PubMed] [Google Scholar]
- (67).Lewis K The Science of Antibiotic Discovery. Cell. 2020, 181 (1), 29–45. [DOI] [PubMed] [Google Scholar]
- (68).Livermore DM Discovery Research: The Scientific Challenge of Finding New Antibiotics. J. Antimicrob. Chemother. 2011, 66 (9), 1941–1944. [DOI] [PubMed] [Google Scholar]
- (69).Payne DJ; Gwynn MN; Holmes DJ; Pompliano DL Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discov. 2007, 6 (1), 29–40. [DOI] [PubMed] [Google Scholar]
- (70).Atanasov AG; Zotchev SB; Dirsch VM; International Natural Product Sciences Taskforce; Supuran, C. T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20 (3), 200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Foulston L Genome Mining and Prospects for Antibiotic Discovery. Curr. Opin. Microbiol. 2019, 51, 1–8. [DOI] [PubMed] [Google Scholar]
- (72).Yan Y; Liu N; Tang Y Recent Developments in Self-Resistance Gene Directed Natural Product Discovery. Nat. Prod. Rep. 2020, 37 (7), 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).El-Sayed AK; Hothersall J; Cooper SM; Stephens E; Simpson TJ; Thomas CM Characterization of the Mupirocin Biosynthesis Gene Cluster from Pseudomonas Fluorescens NCIMB 10586. Chem. Biol. 2003, 10 (5), 419–430. [DOI] [PubMed] [Google Scholar]
- (74).Kling A; Lukat P; Almeida DV; Bauer A; Fontaine E; Sordello S; Zaburannyi N; Herrmann J; Wenzel SC; König C; Ammerman NC; Barrio MB; Borchers K; Bordon-Pallier F; Brönstrup M; Courtemanche G; Gerlitz M; Geslin M; Hammann P; Heinz DW; Hoffmann H; Klieber S; Kohlmann M; Kurz M; Lair C; Matter H; Nuermberger E; Tyagi S; Fraisse L; Grosset JH; Lagrange S; Müller R Targeting DnaN for Tuberculosis Therapy Using Novel Griselimycins. Science. 2015, 348 (6239), 1106–1112. [DOI] [PubMed] [Google Scholar]
- (75).Liu X; Fortin PD; Walsh CT Andrimid Producers Encode an Acetyl-CoA Carboxyltransferase Subunit Resistant to the Action of the Antibiotic. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (36), 13321–13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Tang X; Li J; Millán-Aguiñaga N; Zhang JJ; O’Neill EC; Ugalde JA; Jensen PR; Mantovani SM; Moore BS Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-Directed Genome Mining. ACS Chem. Biol. 2015, 10 (12), 2841–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Nielsen JC; Grijseels S; Prigent S; Ji B; Dainat J; Nielsen KF; Frisvad JC; Workman M; Nielsen J Global Analysis of Biosynthetic Gene Clusters Reveals Vast Potential of Secondary Metabolite Production in Penicillium Species. Nat. Microbiol. 2017, 2 (17044). 10.1038/nmicrobiol.2017.44. (Accessed 2021-09-19) [DOI] [PubMed] [Google Scholar]
- (78).Pantel L; Florin T; Dobosz-Bartoszek M; Racine E; Sarciaux M; Serri M; Houard J; Campagne JM; de Figueiredo RM; Midrier C; Gaudriault S; Givaudan A; Lanois A; Forst S; Aumelas A; Cotteaux-Lautard C; Bolla JM; Vingsbo, Lundberg C; Huseby DL; Hughes D; Villain-Guillot P; Mankin AS; Polikanov YS; Gualtieri M Odilorhabdins, Antibacterial Agents That Cause Miscoding by Binding at a New Ribosomal Site. Mol. Cell. 2018, 70 (1), 83–94. [DOI] [PubMed] [Google Scholar]
- (79).Racine E; Nordmann P; Pantel L; Sarciaux M; Serri M; Houard J; Villain-Guillot P; Demords A; Lundberg CV; Gualtieri M In Vitro and In Vivo Characterization of NOSO-502, a Novel Inhibitor of Bacterial Translation. Antimicrob. Agents Chemother. 2018, 62 (9), e01016–e01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Imai Y; Meyer KJ; Iinishi A; Favre-Godal Q; Green R; Manuse S; Caboni M; Mori M; Niles S; Ghiglieri M; Honrao C; Ma X; Guo JJ; Makriyannis A; Linares-Otoya L; Böhringer N; Wuisan ZG; Kaur H; Wu R; Mateus A; Typas A; Savitski MM; Espinoza JL; O’Rourke A; Nelson KE; Hiller S; Noinaj N; Schäberle TF; D’Onofrio A; Lewis K A New Antibiotic Selectively Kills Gram-Negative Pathogens. Nature. 2019, 576 (7787), 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Kim KH; Aulakh S; Paetzel M The Bacterial Outer Membrane β-Barrel Assembly Machinery. Protein Sci. 2012, 21 (6), 751–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Luther A; Urfer M; Zahn M; Müller M; Wang SY; Mondal M; Vitale A; Hartmann JB; Sharpe T; Lo Monte F; Kocherla H; Cline E; Pessi G; Rath P; Modaresi SM; Chiquet P; Stiegeler S; Verbree C; Remus T; Schmitt M; Kolopp C; Westwood MA; Desjonquères N; Brabet E; Hell S; LePoupon K; Vermeulen A; Jaisson R; Rithié V; Upert G; Lederer A; Zbinden P; Wach A; Moehle K; Zerbe K; Locher HH; Bernardini F; Dale GE; Eberl L; Wollscheid B; Hiller S; Robinson JA; Obrecht D Chimeric Peptidomimetic Antibiotics against Gram-Negative Bacteria. Nature. 2019, 576 (7787), 452–458. [DOI] [PubMed] [Google Scholar]
- (83).Hart EM; Mitchell AM; Konovalova A;Grabowicz M; Sheng J; Han X; Rodriguez-Rivera FP; Schwaid AG; Malinverni JC; Balibar CJ; Bodea S; Si Q; Wang H; Homsher MF; Painter RE; Ogawa AK; Sutterlin H; Roemer T; Black TA; Rothman DM; Walker SS; Silhavy TJ A Small-Molecule Inhibitor of BamA Impervious to Efflux and the Outer Membrane Permeability Barrier. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (43), 21748–21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Storek KM; Auerbach MR; Shi H; Garcia NK; Sun D; Nickerson NN; Vij R; Lin Z; Chiang N; Schneider K; Wecksler AT; Skippington E; Nakamura G; Seshasayee D; Koerber JT; Payandeh J; Smith PA; Rutherford ST Monoclonal Antibody Targeting the β-Barrel Assembly Machine of Escherichia Coli Is Bactericidal. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (14), 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Shatalin K;Nuthanakanti A; Kaushik A;Shishov D; Peselis A; Shamovsky I; Pani B; Lechpammer M; Vasilyev N; Shatalina E; Rebatchouk D; Mironov A; Fedichev P; Serganov A; Nudler E Inhibitors of Bacterial H2S Biogenesis Targeting Antibiotic Resistance and Tolerance. Science. 2021, 372 (6547), 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Luhachack L; Nudler E Bacterial Gasotransmitters: An Innate Defense against Antibiotics. Curr. Opin. Microbiol. 2014, 21, 13–17. [DOI] [PubMed] [Google Scholar]
- (87).Shatalin K;Shatalina E; Mironov A;Nudler E H2S: A Universal Defense against Antibiotics in Bacteria. Science. 2011, 334 (6058), 986–990. [DOI] [PubMed] [Google Scholar]
- (88).Reddy AS; Zhang S Polypharmacology: Drug Discovery for the Future. Expert Rev. Clin. Pharmacol. 2013, 6 (1), 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Apsel B; Blair JA; Gonzalez B; Nazif TM; Feldman ME; Aizenstein B; Hoffman R; Williams RL; Shokat KM; Knight ZA Targeted Polypharmacology: Discovery of Dual Inhibitors of Tyrosine and Phosphoinositide Kinases. Nat. Chem. Biol. 2008, 4 (11), 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Manyando C; Njunju EM; D’Alessandro U; Van Geertruyden JP Safety and Efficacy of Co-Trimoxazole for Treatment and Prevention of Plasmodium Falciparum Malaria: A Systematic Review. PLoS One. 2013, 8 (2), e56916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Phillips JW; Goetz MA; Smith SK; Zink DL; Polishook J; Onishi R; Salowe S; Wiltsie J; Allocco J; Sigmund J; Dorso K; Lee S; Skwish S; De La Cruz M; Martín J; Vicente F; Genilloud O; Lu J; Painter RE; Young K; Overbye K; Donald RGK; Singh SB Discovery of Kibdelomycin, A Potent New Class of Bacterial Type II Topoisomerase Inhibitor by Chemical-Genetic Profiling in Staphylococcus Aureus. Chem. Biol. 2011, 18 (8), 955–965. [DOI] [PubMed] [Google Scholar]
- (92).Lu J; Patel S; Sharma N; Soisson SM; Kishii R; Takei M; Fukuda Y; Lumb KJ; Singh SB Structures of Kibdelomycin Bound to Staphylococcus Aureus GyrB and ParE Showed a Novel U-Shaped Binding Mode. ACS Chem. Biol. 2014, 9 (9), 2023–2031. [DOI] [PubMed] [Google Scholar]
- (93).Miesel L; Hecht DW; Osmolski JR; Gerding D; Flattery A; Li F; Lan J; Lipari P; Polishook JD; Liang L; Liu J; Olsen DB; Singh SB Kibdelomycin Is a Potent and Selective Agent against Toxigenic Clostridium Difficile. Antimicrob. Agents Chemother. 2014, 58 (4), 2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Singh SB; Dayananth P; Balibar CJ; Garlisi CG; Lu J; Kishii R; Takei M; Fukuda Y; Ha S; Young K Kibdelomycin Is a Bactericidal Broad-Spectrum Aerobic Antibacterial Agent. Antimicrob. Agents Chemother. 2015, 59 (6), 3474–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Ling LL; Schneider T; Peoples AJ; Spoering AL; Engels I; Conlon BP; Mueller A; Schäberle TF; Hughes DE; Epstein S; Jones M; Lazarides L; Steadman VA; Cohen DR; Felix CR; Fetterman KA; Millett WP; Nitti AG; Zullo AM; Chen C; Lewis K A New Antibiotic Kills Pathogens without Detectable Resistance. Nature. 2015, 517 (7535), 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Hussein M; Karas JA; Schneider-Futschik EK; Chen F; Swarbrick J; Paulin OKA; Hoyer D; Baker M; Zhu Y; Li J; Velkov T The Killing Mechanism of Teixobactin against Methicillin-Resistant Staphylococcus Aureus: An Untargeted Metabolomics Study. mSystems. 2020, 5 (3), e00077–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Homma T; Nuxoll A; Gandt AB; Ebner P; Engels I; Schneider T; Götz F; Lewis K; Conlon BP Dual Targeting of Cell Wall Precursors by Teixobactin Leads to Cell Lysis. Antimicrob. Agents Chemother. 2016, 60 (11), 6510–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Gunjal VB; Thakare R; Chopra S; Reddy DS Teixobactin: A Paving Stone toward a New Class of Antibiotics? J. Med. Chem. 2020, 63 (21), 12171–12195. [DOI] [PubMed] [Google Scholar]
- (99).Culp EJ; Waglechner N; Wang W; Fiebig-Comyn AA; Hsu Y-P; Koteva K; Sychantha D; Coombes BK; Van Nieuwenhze MS; Brun YV; Wright GD Evolution-Guided Discovery of Antibiotics That Inhibit Peptidoglycan Remodelling. Nature. 2020, 578 (7796), 582–587. [DOI] [PubMed] [Google Scholar]
- (100).Blackman SA; Smith TJ; Foster SJ The Role of Autolysins during Vegetative Growth of Bacillus Subtilis 168. Microbiology. 1998, 144 (1), 73–82. [DOI] [PubMed] [Google Scholar]
- (101).Martin JK; Sheehan JP; Bratton BP; Moore GM; Mateus A; Li SHJ; Kim H; Rabinowitz JD; Typas A; Savitski MM; Wilson MZ; Gitai Z A Dual-Mechanism Antibiotic Kills Gram-Negative Bacteria and Avoids Drug Resistance. Cell. 2020, 181 (7), 1518–1532.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Raimondi MV; Randazzo O; La Franca M; Barone G; Vignoni E; Rossi D; Collina S DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules. 2019, 24 (6), 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Ma Z; Lynch AS Development of a Dual-Acting Antibacterial Agent (TNP-2092) for the Treatment of Persistent Bacterial Infections. J. Med. Chem. 2016, 59 (14), 6645–6657. [DOI] [PubMed] [Google Scholar]
- (104).Liu R; Miller PA; Vakulenko SB; Stewart NK; Boggess WC; Miller MJ A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria to Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem. 2018, 61 (9), 3845–3854. [DOI] [PubMed] [Google Scholar]
- (105).Shahid M; Sobia F; Singh A; Malik A; Khan HM; Jonas D; Hawkey PM Beta-Lactams and Beta-Lactamase-Inhibitors in Current-or Potential-Clinical Practice: A Comprehensive Update. Crit. Rev. Microbiol. 2009, 35 (2), 81–108. [DOI] [PubMed] [Google Scholar]
- (106).Livermore DM Beta-Lactamase-Mediated Resistance and Opportunities for Its Control. J. Antimicrob. Chemother. 1998, 41 (Suppl. D), 25–41. [DOI] [PubMed] [Google Scholar]
- (107).Brown AG; Butterworth D; Cole M; Hanscomb G; Hood JD; Reading C; Rolinson GN Naturally-Occurring β-Lactamase Inhibitors with Antibacterial Activity. J. Antibiot. (Tokyo). 1976, 29 (6), 668–669. [DOI] [PubMed] [Google Scholar]
- (108).Neu HC; Fu KP Clavulanic Acid, a Novel Inhibitor of β-Lactamases. Antimicrob. Agents Chemother. 1978, 14 (5), 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Saudagar PS; Survase SA; Singhal RS Clavulanic Acid: A Review. Biotechnol. Adv. 2008, 26 (4), 335–351. [DOI] [PubMed] [Google Scholar]
- (110).Mancabelli L; Mancino W; Lugli GA; Argentini C; Longhi G; Milani C; Viappiani A; Anzalone R; Bernasconi S; van Sinderen D; Ventura M; Turroni F Amoxicillin-Clavulanic Acid Resistance in the Genus Bifidobacterium. Appl. Environ. Microbiol. 2021, 87 (7), e03137–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Hoban DJ; Bouchillon SK; Johnson JL; Zhanel GG; Butler DL; Saunders KA; Miller LA; Poupard JA; Surveillance Study Research Group. Comparative in Vitro Potency of Amoxycillin-Clavulanic Acid and Four Oral Agents against Recent North American Clinical Isolates from a Global Surveillance Study. Int. J. Antimicrob. Agents. 2003, 21 (5), 425–433. [DOI] [PubMed] [Google Scholar]
- (112).Zhanel GG; Lawson CD; Adam H; Schweizer F; Zelenitsky S; Lagacé-Wiens PRS; Denisuik A; Rubinstein E; Gin AS; Hoban DJ; Lynch JP; Karlowsky JA Ceftazidime-Avibactam: A Novel Cephalosporin/β-Lactamase Inhibitor Combination. Drugs. 2013, 73 (2), 159–177. [DOI] [PubMed] [Google Scholar]
- (113).Ehmann DE; Jahic H; Ross PL; Gu RF; Hu J; Kern G; Walkup GK; Fisher SL Avibactam Is a Covalent, Reversible, Non-β-Lactam β-Lactamase Inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (29), 11663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Ehmann DE; Jahic H; Ross PL; Gu RF; Hu J; Durand-Réville TF; Lahiri S; Thresher J; Livchak S; Gao N; Palmer T; Walkup GK; Fisher SL Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases. J. Biol. Chem. 2013, 288 (39), 27960–27971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Veeraraghavan B; Bakthavatchalam YD; Sahni RD Oral Antibiotics in Clinical Development for Community-Acquired Urinary Tract Infections. Infect. Dis. Ther. 2021, 10, 1815–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Sabet M; Tarazi Z; Griffith DC In Vivo Activity of QPX7728, an Ultrabroad-Spectrum Beta-Lactamase Inhibitor, in Combination with Beta-Lactams against Carbapenem-Resistant Klebsiella Pneumoniae. Antimicrob. Agents Chemother. 2020, 64 (11), e01267–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).King AM; Reid-Yu SA; Wang W; King DT; De Pascale G; Strynadka NC; Walsh TR; Coombes BK; Wright GD Aspergillomarasmine A Overcomes Metallo-β-Lactamase Antibiotic Resistance. Nature. 2014, 510 (7506), 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Zhang J; Wang S; Wei Q; Guo Q; Bai Y; Yang S; Song F; Zhang L; Lei X Synthesis and Biological Evaluation of Aspergillomarasmine A Derivatives as Novel NDM-1 Inhibitor to Overcome Antibiotics Resistance. Bioorg. Med. Chem. 2017, 25 (19), 5133–5141. [DOI] [PubMed] [Google Scholar]
- (119).Bergstrom A; Katko A; Adkins Z; Hill J; Cheng Z; Burnett M; Yang H; Aitha M; Mehaffey MR; Brodbelt JS; Tehrani KHME; Martin NI; Bonomo RA; Page RC; Tierney DL; Fast W; Wright GD; Crowder MW Probing the Interaction of Aspergillomarasmine A with Metallo-β-Lactamases NDM-1, VIM-2, and IMP-7. ACS Infect. Dis. 2018, 4 (2), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Becker B; Cooper MA Aminoglycoside Antibiotics in the 21st Century. ACS Chem. Biol. 2013, 8 (1), 105–115. [DOI] [PubMed] [Google Scholar]
- (121).Carter AP; Clemons WM; Brodersen DE; Morgan-Warren RJ; Wimberly BT; Ramakrishnan V Functional Insights from the Structure of the 30S Ribosomal Subunit and Its Interactions with Antibiotics. Nature. 2000, 407 (6802), 340–348. [DOI] [PubMed] [Google Scholar]
- (122).Ramirez MS; Tolmasky ME Aminoglycoside Modifying Enzymes. Drug Resist. Updat. 2010, 13 (6), 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Wright GD Aminoglycoside-Modifying Enzymes. Curr. Opin. Microbiol. 1999, 2 (5), 499–503. [DOI] [PubMed] [Google Scholar]
- (124).Shaw KJ; Rather PN; Hare RS; Miller GH Molecular Genetics of Aminoglycoside Resistance Genes and Familial Relationships of the Aminoglycoside-Modifying Enzymes. Microbiol. Rev. 1993, 57 (1), 138–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Suga T; Ishii T; Iwatsuki M; Yamamoto T; Nonaka K; Masuma R; Matsui H; Hanaki H; Omura S; Shiomi K Aranorosin Circumvents Arbekacin-Resistance in MRSA by Inhibiting the Bifunctional Enzyme AAC(6')/APH(2). J. Antibiot. (Tokyo). 2012, 65 (10), 527–529. [DOI] [PubMed] [Google Scholar]
- (126).Zhu L; Liu R; Liu T; Zou X; Xu Z; Guan H A Novel Strategy to Screen Inhibitors of Multiple Aminoglycoside-Modifying Enzymes with Ultra-High Performance Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. J. Pharm. Biomed. Anal. 2019, 164, 520–527. [DOI] [PubMed] [Google Scholar]
- (127).Cox G; Ejim L; Stogios PJ; Koteva K; Bordeleau E; Evdokimova E; Sieron AO; Savchenko A; Serio AW; Krause KM; Wright GD Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes. ACS Infect. Dis. 2018, 4 (6), 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Clancy CJ; Nguyen MH Estimating the Size of the U.S. Market for New Antibiotics with Activity against Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2019, 63 (12), e01733–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Chen AY; Zervos MJ; Vazquez JA Dalbavancin: A Novel Antimicrobial. Int. J. Clin. Pract. 2007, 61 (5), 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Duffy EM; Buurman ET; Chiang SL; Cohen NR; Uria-Nickelsen M; Alm RA The CARB-X Portfolio of Nontraditional Antibacterial Products. ACS Infect. Dis. 2021, 7 (8), 2043–2049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.