

ABSTRACT
T cell engagers represent a novel promising class of cancer-immunotherapies redirecting T cells to tumor cells and have some promising outcomes in the clinic. These molecules can be associated with a mode-of-action related risk of cytokine release syndrome (CRS) in patients. CRS is characterized by the rapid release of pro-inflammatory cytokines such as TNF-α, IFN-γ, IL-6 and IL-1β and immune cell activation eliciting clinical symptoms of fever, hypoxia and hypotension. In this work, we investigated the biological mechanisms triggering and amplifying cytokine release after treatment with T cell bispecific antibodies (TCBs) employing an in vitro co-culture assay of human PBMCs or total leukocytes (PBMCs + neutrophils) and corresponding target antigen-expressing cells with four different TCBs. We identified T cells as the triggers of the TCB-mediated cytokine cascade and monocytes and neutrophils as downstream amplifier cells. Furthermore, we assessed the chronology of events by neutralization of T-cell derived cytokines. For the first time, we demonstrate the contribution of neutrophils to TCB-mediated cytokine release and confirm these findings by single-cell RNA sequencing of human whole blood incubated with a B-cell depleting TCB. This work could contribute to the construction of mechanistic models of cytokine release and definition of more specific molecular and cellular biomarkers of CRS in the context of treatment with T-cell engagers. In addition, it provides insight for the elaboration of prophylactic mitigation strategies that can reduce the occurrence of CRS and increase the therapeutic index of TCBs.
KEYWORDS: Cancer immunotherapy, T cell engagers, T cell bispecific antibodies, cytokine release syndrome, neutrophil, PBMC
Introduction
T cell bispecific antibodies (TCBs) or T cell engagers are bispecific antibodies capable of simultaneously binding a tumor-associated antigen (TAA) and the T cell receptor, which trigger T cell activation, proliferation, cytokine secretion and cytotoxicity toward tumor cells.1 We have previously described cibisatamab (CEA-TCB) and glofitamab (CD20-TCB) which harbor a 2+1 format with one binder for the CD3ε chain of the T cell receptor and two binders for the CEA or CD20 antigen.2–4 Their Fc region has been engineered with a P329G LALA mutation, preventing FcγR signaling without affecting functional binding to FcRn maintaining an IgG-like half-life.5–7 TCBs represent an accessible “off the shelf” alternative to Chimeric Antigen Receptor (CAR) T cells to eliminate tumors.8–13
One of the major on-target safety liabilities associated with the use of TCBs is cytokine release, which can be excessive and initiate a cytokine release syndrome (CRS) early after treatment.10,14,15 CRS is characterized by the clinical symptoms such as fever, hypotension and respiratory insufficiency, which are associated with a release of pro-inflammatory cytokines such as IL-6, IL-1β and TNF-α.10,11 Clinicians have agreed on an ASTCT (American Society for Transplantation and Cellular Therapy) consensus to diagnose CRS severity at the patient's bedside and recommend treatment for syndrome management.16,17 Current approaches to manage CRS rely on glucocorticoids but also inhibition of IL-6R with tocilizumab, and blocking IL-6 with siltuximab are applied.16–22 If the symptoms do not resolve, patients receive supportive care to stabilize blood pressure and oxygen saturation (e.g. administration of vasopressors or oxygen). Dose-escalating regimens are also used to prevent the risk of high grade CRS for T cell engagers entering the clinic.23 Nevertheless, CRS still remains the dose-limiting toxicity associated with T-cell engaging therapies. This highlights the importance of better understanding the biological mechanisms and biomarkers involved in CRS in order to develop prophylactic treatments or build models that guide step-up dosing schedules.24 Therefore, we investigated the key cellular and molecular “triggers” and “amplifiers” involved in the cascade of TCB-mediated cytokine release.
To dissect in further detail the sequence of events in this cytokine release cascade, we used an in vitro T-cell dependent cellular cytotoxicity model (“TDCC”). In this system, peripheral blood mononuclear cells (PBMCs), monocyte-depleted PBMCs or total blood leukocytes (PBMCs and neutrophils) as effector cells were co-cultured with target cells expressing tumor-associated antigens (TAAs) in the presence of a TCB. We used four different 2+1 format TCBs directed to solid tumor surface antigens (CEA, FolR1, Tyrp1) or an hematological tumor antigen, CD20. To clearly assess the role of neutrophils in cytokine release, we used a transcriptomic analysis by single-cell RNA sequencing of human whole blood treated with CD20-TCB.
In line with the findings of Li et al. and Godbersen-Palmer et al., we identified T cells as the trigger of the cytokine release induced by T cell engagers and monocytes as amplifier cells producing TNFα, IL-6 and IL-1β.25,26 Furthermore, our study highlights for the first time the contribution of neutrophils to TCB-mediated cytokine release. Finally, our results also provide additional evidence that TCB-mediated cytokine release can be counteracted by treatment intervention targeted against T cell-derived cytokines, to mitigate CRS.
Material and methods
Antibodies
The 2 + 1 T cell bispecific antibodies are IgG1-based with bivalent binding entities to a target antigen and monovalent binding to the CD3ε chain of the T cell receptor. They have a silent Fc region engineered with a P329G LALA mutation, which prevents binding to the FcγR. DP47-TCB used as a control has the same IgG1-based format but bears two non-binding active binders in place of the target antigen binders. DP47-TCB, Tyrp1-TCB, CEA-TCB, FolR-TCB and CD20-TCB were produced internally. The commercial compounds adalimumab and tocilizumab were used to block TNF-α and IL-6, respectively. The anti-IFN-γ blocking antibody was purchased from BioXcell (BE0245).
Cell lines
CHO cells were engineered to stably express the Tyrp1, FolR or CEA antigen on the cell surface under Puromycin selection. CHO cells were cultured as adherent cells in T flasks (TTP), harvested with Trypsin (Gibco) and passaged twice per week at a density of 20 000 cells/cm2 in DMEM/F12 medium (Gibco) containing 10% FBS (Gibco) and supplemented with 6 µg/mL Puromycin.
MKN45- and NucLight Red (NLR)-labeled MKN45 cells are human gastric cancer cell lines used as target cells in TDCC assays with CEA-TCB (DMSZ). MKN45 cells are adherent cells, harvested with Trypsin (Gibco) and passaged twice per week at a density of 60 000 cell/cm2 in RPMI Glutamax (Gibco) containing 10% FBS (Gibco).
PBMCs isolation
Human fresh blood was collected from anonymous healthy volunteers through the Roche internal employee donation program. Buffy coats were collected from anonymous healthy volunteers through the Zürich blood donation center, in accordance with the declaration of Helsinki. PBMCs were isolated from fresh whole blood or buffy coat by density gradient centrifugation. A 20–25 mL of fresh blood or diluted buffy coat (1:2, PBS) was layered on 15 mL of Ficoll (Stemcell) in 50 mL tubes. Tubes were centrifuged at 2000 rpm for 30 min at RT without braking. The PBMC layer was removed, washed in PBS and centrifuged 3 times for 5 min at 1700 rpm, 1400 rpm and 1100 rpm (RT) to remove the remaining Ficoll. The PBMCs were washed again and centrifuged at 800 rpm (RT) for 10 min to remove remaining platelets. The PBMCs were counted using a Beckmann Coulter cell counter and diluted in medium to the targeted cell concentration.
Monocyte depletion
Monocyte-depleted PBMCs were isolated from fresh whole blood using RosetteSepTM human monocyte depletion cocktail (Stemcell). Fresh blood was first incubated with an antibody cocktail (50 μL/mL of blood sample, 20 min, RT) which targets monocytic surface markers and crosslinks them together with red blood cells (RBC). Monocytes sediment together with RBCs at the bottom of the tube after density gradient centrifugation. The monocyte-depleted PBMC fraction was collected and the cells were washed 4 times as described above to remove any remaining Ficoll and platelets.
Isolation of total leukocytes
Total leukocytes were isolated from fresh whole blood by magnetic removal of RBCs using the EasySepTM RBC depletion kit (Stemcell). Fresh whole blood was diluted 1:2 in PBS with 6 mM EDTA. 10 mL diluted fresh whole blood was incubated together with beads targeting RBCs (50 μL/mL of undiluted blood volume) in a 14 mL polystyrene tube (BD) using a magnetic tube holder (EasyEightsTM, Stemcell) for 5 min at RT. The cell suspension was carefully pipetted out and poured into a new 14 mL polystyrene tube. RBC magnetic isolation was then repeated three times (5 min, RT). The clear yellowish cell suspension was then collected and placed for 5 min in the magnetic tube holder to remove the remaining magnetic beads, washed with PBS twice by centrifugation at 800 rpm, for 10 min (RT) to remove the platelets. The total leukocytes were counted using a Beckmann Coulter cell counter and diluted in medium to reach the same lymphocyte concentration as in the PBMC preparation.
In vitro T cell dependent cellular cytotoxicity assays
One day before the assay, the target cells (MKN45 or transfected CHO cells) were plated in flat-bottomed 96-well plates (TPP) at a concentration of 25000 cells/well in 100 μL of culture medium. On day 0, it was replaced with 100 μL of fresh medium and 50 μL of a stock solution containing 6.0 × 106 lymphocytes/mL was placed in each well (300 000 lymphocytes/well, effector cells: target cells (E:T) = 10:1). For each antibody, a series of eight dilutions (1:10) was prepared and 50 µL was transferred to each well. At the 24 hrs and/or 48 hrs endpoints, the plates were centrifuged and supernatants were collected to measure LDH release (75 μL, RT) and cytokines (75 μL, stored at −80°C). The effector cells were washed twice in 100 μL PBS (1500 rpm, 5 min, RT) and stained for flow cytometry analysis.
In vitro TDCC assays in IncuCyte
For TDCC assays with 5000 adherent NLR-labeled MKN45 cells/well, the assay medium was replaced with fresh medium (100 μL/well) and 50 000 effector cells/well (50 μL) were transferred to obtain a final E:T ratio of approximatively 10:1. The antibody solutions (50 μL) were then added to initiate killing. The assay plates were covered with lids, and placed in the incubator or IncuCyte at 37°C, 5% CO2 (1 scan every 3 hrs, zoom 10x, phase and red, 400 ms acquisition time).
Cytokine analysis
Cytokines were analyzed in the TDCC assay supernatants (stored at −80°C) using the Luminex technology with a human8Plex Assay kit (Biorad) and additional IL-1β and MCP-1 beads. Pre-diluted supernatants were incubated with beads for 1 hr and then centrifuged at 800 rpm. The plate was washed with wash buffer and detection antibodies were added for 1 hr before centrifugation at 800 rpm. The plate was washed again and streptavidin was added for 1 hr before centrifugation at 800 rpm. After another washing, samples were re-suspended in assay buffer before being measured by fluorescence reading using a Luminex plate reader from Biorad. Data were analyzed using the Biorad Bio-Plex Software.
Intracellular cytokine staining
After 4–6 hrs of TCB stimulation, 20 μL of medium containing 1/150 (final dilution: 1/1500) Brefeldin A (Golgiplug, BD) and 1/100 (final dilution: 1/1000) Monensin A (Golgistop, BD) were added in each well to block cytokine secretion for 12–14 hrs. The PBMCs were collected, and washed twice in PBS by centrifugation at 1500 rpm for 5 min (RT). 1/50 Fc-blocker solution (True Monocyte Staining, Biolegend) was added and incubated for 20 min at RT in the dark to prevent nonspecific staining. The PBMCs were washed in PBS and centrifuged for 5 min at 1500 rpm (RT) and stained for 30 min at 4°C in the dark for the following surface markers: CD19 (pacific blue, Biolegend), CD4 (FITC, Biolegend), CD8 (BV605, Biolegend), CD14 (BV711, Biolegend), CD16 (BUV735, BD) and live dead (aqua zombie, Biolegend) in PBS. The PBMCs were fixed with BD Cytofix/Cytoperm (BD) (80 μL/well, 30 min at 4 C). The PBMCs were washed with PBS, centrifuged and washed again with Perm/Wash buffer (BD) and incubated in Perm/Wash buffer (30 min, 4°C, no light). Perm/Wash buffer containing antibodies to IL-6 (PE, Biolegend), IL-1β (Alexa 647, Biolegend), TNF-α (APC-Cy7, Biolegend) and IFN-γ (BUV737, BD) was added (50 μL/well) and incubated for 30 min at 4°C in the dark. The PBMCs were washed with Perm/Wash buffer and with FACS buffer and re-suspended in 100 μL FACS buffer. Data acquisition was performed using a BD Fortessa flow cytometer and the DIVA software.
Target cell killing – LDH release
CytoTox-GloTM (Promega) cytotoxicity assay was used to measure LDH release as a reporter of target cell killing. A 75 μL of supernatants was collected in a 96-well white plate and 25 μL of CytoTox-Glo reagents was added in each well. The plate was agitated for 15 minutes, 600 rpm, RT and luminescence was measured using a Perkin Elmer plate reader. Relative Luminescence Units (RLU) were then reported against the TCB concentration.
Flow cytometry analysis
After stimulation, PBMCs, monocyte-depleted PBMCs or total leukocytes were washed twice in PBS before centrifugation at 1500 rpm for 5 min (RT) and stained for the following surface markers: CD45 (Alexa fluor 700, Biolegend) CD4 (FITC, Biolegend), CD8 (BV605, Biolegend), CD25 (BUV395, BD), CD69 (APC-Cy7, Biolegend) and live dead (aqua zombie, Biolegend) for 30 min at 4°C in FACS buffer. The staining of TNFR1 and TNFR2 on T cells and monocytes was conducted using the following antibodies: CD4 (FITC, Biolegend), CD8 (BV605, Biolegend), CD14 (BV711, Biolegend), CD16 (BUV735, BD), CD120a (APC, Biolegend), CD120b (PE, Biolegend). Antibodies to CD16 (BUV395, BD), CD11b (Pe-Cy7, Biolegend) and CD62L (APC-Cy7, Biolegend) were used to analyze the phenotype of neutrophils. After staining, the cells were washed twice in FACS buffer and re-suspended in 100 μL/well FACS buffer for analysis. Acquisition was performed using a BD Fortessa coupled to an HTS platform.
Data analysis
Flow cytometry data were analyzed using Flowjo V10. Cytokine data were analyzed using the Bio-Plex software from Biorad. GraphPad Prism 8 was used to generate the graphs and for statistical analysis. EC50 values were determined using nonlinear regression curves, variable slope fit (four parameters) and least-square fit. For dose-titration curves, areas under the curves were calculated and used for statistical comparison. Data are shown as means with SD or SEM or as individual curves. The statistical tests used are indicated in the figure legends for each experiment.
Single cell RNA sequencing of whole blood
Whole blood from four donors was treated with 0.2 μg/mL CD20-TCB, or incubated in the absence of CD20-TCB. At baseline (before addition of TCB) and assay endpoints (2, 4, 6, and 20 hrs), blood was collected for total leukocyte isolation using EasySepTM red blood cell depletion reagent (Stemcell). Briefly, cells were counted and processed for single-cell RNA sequencing using the BD Rhapsody platform. To load several samples on a single BD Rhapsody cartridge, sample cells were labeled with sample tags (BD Human Single-Cell Multiplexing Kit) following the manufacturer’s protocol prior to pooling. Briefly, 1 × 106 cells from each sample were re-suspended in 180 μL FBS Stain Buffer (BD, PharMingen) and sample tags were added to the respective samples and incubated for 20 min at RT. After incubation, two successive washes were performed by addition of 2 mL stain buffer and centrifugation for 5 min at 300 g. Cells were then re-suspended in 620 μL cold BD Sample Buffer, stained with 3.1 μL of both 2 mM Calcein AM (Thermo Fisher Scientific) and 0.3 mM Draq7 (BD Biosciences) and finally counted on the BD Rhapsody scanner. Samples were then diluted and/or pooled equally in 650 μL cold BD Sample Buffer. The BD Rhapsody cartridges were then loaded with up to 40 000–50 000 cells. Single cells were isolated using Single-Cell Capture and cDNA Synthesis with the BD Rhapsody Express Single-Cell Analysis System according to the manufacturer’s recommendations (BD Biosciences). cDNA libraries were prepared using the Whole Transcriptome Analysis Amplification Kit following the BD Rhapsody System mRNA Whole Transcriptome Analysis (WTA) and Sample Tag Library Preparation Protocol (BD Biosciences).
Indexed WTA and sample tag libraries were quantified and quality controlled on the Qubit Fluorometer using the Qubit dsDNA HS Assay, and on the Agilent 2100 Bioanalyzer system using the Agilent High Sensitivity DNA Kit. Sequencing was performed on a Novaseq 6000 (Illumina) in paired-end mode (64-8-58) with Novaseq6000 S2 v1 or Novaseq6000 SP v1.5 reagent kits (100 cycles).
Single cell RNA sequencing data analysis
Sequencing data was processed using the BD Rhapsody Analysis pipeline (v 1.0 https://www.bd.com/documents/guides/user-guides/GMX_BD-Rhapsody-genomics-informatics_UG_EN.pdf) on the Seven Bridges Genomics platform. Briefly, read pairs with low sequencing quality are first removed and the cell label and UMI identified for further quality check and filtering. Valid reads are then mapped to the human reference genome (GRCh38-PhiX-gencodev29) using the aligner Bowtie2 v2.2.9, and reads with the same cell label, same UMI sequence and same gene are collapsed into a single raw molecule while undergoing further error correction and quality checks. Cell labels are filtered with a multi-step algorithm to distinguish those associated with putative cells from those associated with noise. After determining the putative cells, each cell is assigned to the sample of origin through the sample tag (only for cartridges with multiplex loading). Finally, the single-cell gene expression matrices are generated and a metrics summary is provided.
After pre-processing with BD’s pipeline, the count matrices and metadata of each sample were aggregated into a single data object and loaded into the besca v2.3 pipeline for the single-cell RNA sequencing analysis.27 First, we filtered low-quality cells with less than 200 genes, less than 500 counts or more than 30% of mitochondrial reads. This permissive filtering was used in order to preserve the neutrophils. We further excluded potential multiplets (cells with more than 5,000 genes or 20,000 counts), and genes expressed in less than 30 cells. Normalization, log-transformed UMI counts per 10,000 reads [log(CP10K+1)], was applied before downstream analysis. After normalization, technical variance was removed by regressing out the effects of total UMI counts and percentage of mitochondrial reads, and gene expression was scaled. The 2,507 most variable genes (having a minimum mean expression of 0.0125, a maximum mean expression of 3 and a minimum dispersion of 0.5) were used for principal component analysis (PCA). Finally, the first 50 PCs were used as input for calculating the 10 nearest neighbors and the neighborhood graph was then embedded into the two-dimensional space using the UMAP algorithm.28 Cell clustering was performed using the Leiden algorithm at a resolution of 2.29
Cell-type annotation was performed using the Sig-annot semi-automated besca module, which is a signature-based hierarchical cell annotation method.30 To identify neutrophils, this signature was used: ELANE, MPO, PRTN3, CTSG, AZU1 and FCGR3B. FCGR3B was the most expressed gene and highly specific, together with negative expression of other lineage markers such as CD3D or CD79A. Finally, neutrophils were selected in order to generate further visualizations, such as the expression level of selected cytokines across conditions, by using a custom script with mainly besca and scanpy functions.
Results
T cell activation and target cell killing are associated with cytokine release upon first TCB stimulation
In an in vitro TDCC assay using PBMCs as effector cells, treatment with CEA-TCB resulted in a rapid dose-dependent T cell activation, as indicated by CD69 and CD25 upregulation on CD4+ and CD8+ T cells after 24 hrs, concomitant with the killing of MKN45 tumor cells [Figure 1(a,b)]. The negative control DP47-TCB bearing two non-binding active domains in place of the CEA binder did not produce these effects, revealing dependence on target engagement. Subsequently, IL-2, IFN-γ, TNF-α, IL-6, IL-1β and IL-8 were detected in the supernatants of the CEA-TCB TDCC assays [Figure 1(c)].
Figure 1.
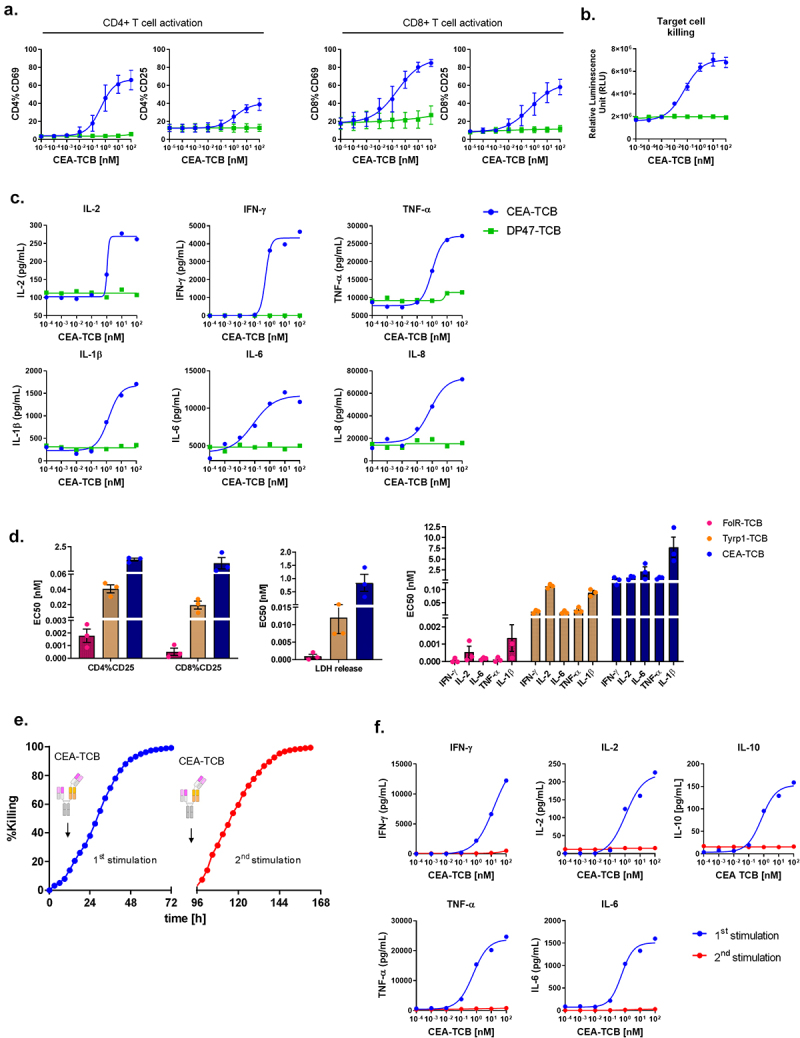
Cytokine release is associated with TCB-induced T cell activation and cytotoxicity upon first stimulation. PBMCs were stimulated with CEA-TCB in a TDCC assay using MKN45 target cells (E:T = 10:1) for 48 hrs. DP47-TCB was used as a negative control. (a) CD69 and CD25 expression on CD4+ and CD8+ T cells was measured by flow cytometry. Cells from technical duplicates were pooled for analysis. Means of n = 3 donors ±SD. (b) Killing of MKN45 target cells was measured by LDH release. Means of n = 3 donors ± SD. (c) The supernatants from technical duplicates were pooled and cytokine levels were measured by Luminex for 1 donor. (d) EC50 values for CD25 induction on CD4+ and CD8+ T cells, LDH release and cytokine release in TDCC assays with FolR-TCB (CHOK1SV target cells), Tyrp1-TCB (CHOK1SV-Tyrp1 target cells), CEA-TCB (CHOK1SV-CEACAM5 target cells), individual data and means of n = 3 donors ± SD (24 hrs). (e) Real time killing of NLR-labeled MKN45 tumor cells by 10 nM CEA-TCB during first and second stimulation (Incucyte). (f) Comparison of cytokine release, 96 hrs after first and second stimulation with 10 nM CEA-TCB. Supernatants from technical replicates were pooled and cytokine levels were analyzed by Luminex. (e,f). Data are shown for 1 donor out of 3 tested.
As these cytokines are elevated in the serum of patients treated with TCBs and developing CRS, the TDCC assay system appears as a relevant assay system for mechanistic studies of TCB-mediated cytokine release.10 EC50 values for T cell activation measured by expression of CD25 on CD4+ and CD8+ T cells, for target cell killing and for cytokine release were calculated for CEA-TCB as well as for two other TCBs, Tyrp1- and FolR-TCB [Figure 1(d)]. FolR-TCB, which is the most potent of these TCBs, had the lowest EC50 for these three parameters followed by Tyrp1-TCB and finally CEA-TCB [Figure 1(d)]. For all three TCBs, the EC50 values indicate that target cell killing is a more sensitive parameter than T cell activation and cytokine release, suggesting that there are dose windows at which TCBs induce killing while not triggering cytokine release [Figure 1(d)]. This observation is also used for a MABEL starting dose selection.31
To model a repeated TCB treatment, PBMCs harvested 4 d after a first stimulation were re-stimulated with CEA-TCB in the presence of fresh NLR-labeled MKN45 tumor cells. The real-time tumor cell killing was followed by Incucyte. After the second stimulation, the kinetics of tumor cell killing remained the same as after the first stimulation [Figure 1(e)]. However, IFN-γ, IL-2, IL-10, TNF-α, and IL-6 levels detected 4 d after the second stimulation were significantly lower than 4 d after the first one, reflecting clinical experience with step-up fractionated dosing32 [Figure 1(f)]. This data shows that CEA-TCB-induced cytokine release is decoupled from T cell cytotoxicity upon re-stimulation, as previously reported for other CD3 bispecific antibodies.25
The in vitro TDCC assay system allows modeling TCB-mediated cytokine release early after T cell stimulation, upon initiation of target cell killing and is an appropriate tool to recapitulate the sequence of events and to identify key cellular and molecular players involved in TCB-mediated cytokine release.
T cells contribute to TNF-α and IFN-γ but not to IL-6 release
Since T cells are directly targeted by TCBs through CD3 engagement and TNF-α, IFN-γ and IL-6 are detected after TCB stimulation, we used intracellular cytokine staining and flow cytometry to determine whether these cytokines were produced by CD4+ and CD8+ T cells. When PBMCs were treated with 100 nM CEA-TCB, 24.9% of CD4+ cells and 18.9% of CD8+ cells were TNF-α positive after 20 hrs of incubation. Only 0.4% and 0.7% of CD4+ and CD8+ T cells, respectively, were TNF-α positive with the DP47-TCB negative control [Figure 2(a)]. With CEA-, FolR- and Tyrp1-TCB, TNF-α production by CD4+ and CD8+ T cells was dose-dependent [Figure 2(c), Supp. Fig 1A, B, C, G].
Figure 2.

CD4+ and CD8+ T cells produce TNF-α and IFN-γ but not IL-6 upon TCB treatment. (a,b) PBMCs were stimulated with CEA-TCB or DP47-TCB (negative control) in a TDCC assay using MKN45 target cells (E:T = 10:1). Golgistop and Golgiplug were added to block cytokine secretion after 6 hrs and intra-cellular staining was performed 20 hrs after stimulation with TCB. Flow cytometry plots of TNF-α (a) and IFN-γ (b) positive cells gated among CD4+ and CD8+ T cells after stimulation with 100 nM CEA- or DP47-TCB for 1 donor. (c,d) PBMCs were stimulated with escalating concentrations of CEA-TCB, Tyrp1-TCB, FolR-TCB or DP47-TCB (negative control) in TDCC assays using MKN45, CHOK1SV-Tyrp1 or CHOK1SV-FolR target cells respectively (E:T = 10:1). Intracellular staining of TNF-α (c) and IFN-γ (d) in CD4+ and CD8+ T cells, means of 2 technical replicates ± SEM. Data shown are from 1 individual donor and from independent experiment For CEA-TCB, PBMCs from n = 3 donors were tested.
Interferon-γ production by T cells was also detectable after stimulation with CEA-TCB [Figure 2(b)]. Among CD4+ and CD8+ T cells, 3.8% and 8.8%, respectively, were IFN-γ-positive 20 hrs after stimulation with 100 nM CEA-TCB. With DP47-TCB, respectively, only 0.5% and 0.5% scored positive. The production of IFN-γ by CD8+ and CD4+ T cells was also TCB dose-dependent for all three TCBs tested [Figure 2(d), supp. Fig 1D, E, G].
The elevated levels of IL-6 in the supernatants of TDCC assays after TCB treatment suggested that T cells might contribute to its release. However, as found using intracellular immunostaining and flow cytometry, CD4+ and CD8+ T cells failed to produce IL-6 after stimulation with 100 nM CEA-TCB [supp. Fig 1F].
Last, we also looked whether NK cells might contribute to cytokine release in the TDCC assay. Using intra-cellular cytokine staining, we could not detect any increase in IFN-γ, TNF-α, or IL-6 production following stimulation with 100 nM Tyrp1-TCB [supp. Fig 2A-B].
Altogether, these data demonstrate that on-target TCB activity triggers the dose-dependent release of TNF-α and IFN-γ but not of IL-6 by activated CD4+ and CD8+ T-cells.
Monocytes contribute to TNF-α production together with T cells and are the main mediators of IL-6 and IL-1β release
In the same assay, we set the gating on CD14+CD16− classical monocytes to evaluate their contribution to cytokine production. When PBMCs were treated for 20 hrs with 100 nM CEA-TCB, 6.0% of CD14+CD16- monocytes were double positive for IL-6 and TNF-α, 3.9% for IL-1β and TNF-α and 26.5% for IL-1β and IL-6 [Figure 3(a)]. In comparison, these percentages were considerably lower (2.4%, 1.5% and 10.9%, respectively) when the cells were treated with 100 nM of the negative control DP47-TCB [Figure 3(a)]. This indicates that CD14+CD16− classical monocytes can simultaneously produce at least two of the three pro-inflammatory cytokines (IL-1β, TNF-α and IL-6) in response to TCB stimulation. For CEA-, Tyrp1- and FolR-TCB, this concomitant production of IL-1β, IL-6 and/or TNF-α by monocytes was TCB dose dependent [Figure 3(b), supp. Fig 3A-C]. For CEA-TCB, a higher background production of IL-1β, IL-6 and TNF-α by monocytes was observed in control conditions (in the absence of TCB or with DP47-TCB). We found that the interaction of monocytes with the MKN45 cell line triggered a baseline production of IL-1β, IL-6 and to a lower extent of TNF-α [Figure 3(b,f)].
Figure 3.
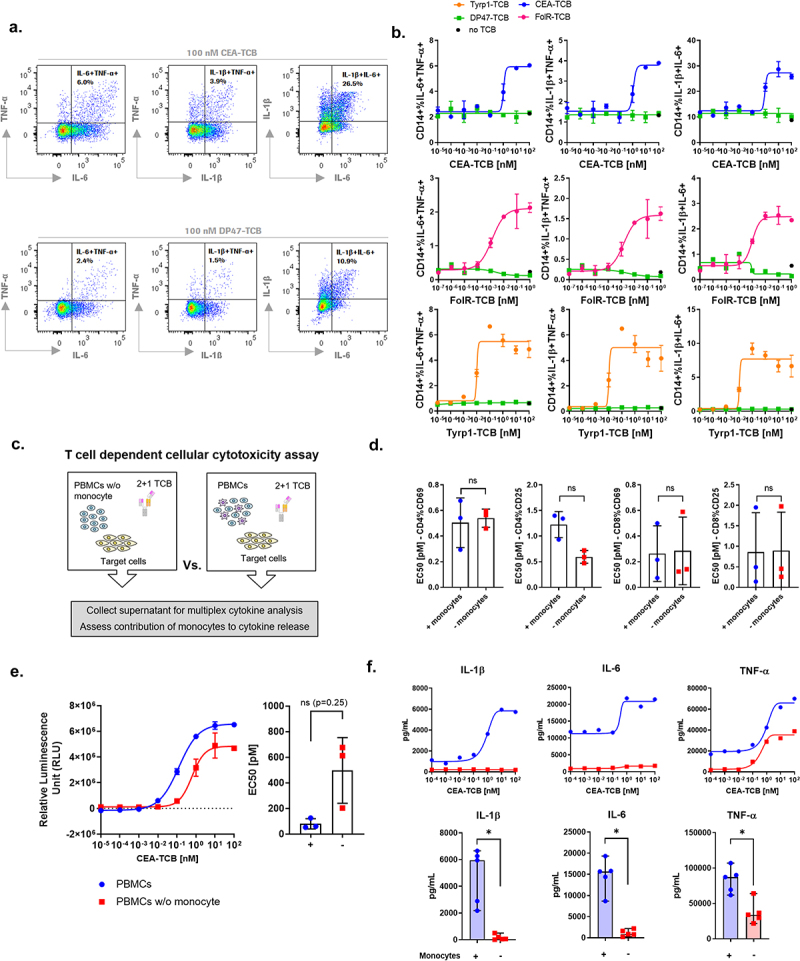
Monocytes produce IL-1β and IL-6 and contribute to TNF-α release upon TCB treatment. PBMCs were stimulated with CEA-TCB, Tyrp1-TCB, FolR-TCB or DP47-TCB (negative control) in a TDCC assay using MKN45, CHOK1SV-Tyrp1 or CHOK1SV-FolR target cells respectively (E:T = 10:1). Golgistop and Golgiplug were added at 6 hrs to block cytokine secretion and intracellular staining was performed 20 hrs after stimulation with TCB. (a). Representative flow cytometry plots of IL-6+ TNF-α+, IL-1β+TNF-α+, IL-1β+IL-6+ populations among CD14+ monocytes after treatment with 100 nM CEA- or DP47-TCB. (b) Intracellular staining of IL-1β, TNF-α and IL-6 in CD14+ monocytes after treatment with escalating concentrations of CEA-TCB, FolR-TCB, Tyrp1-TCB and DP47-TCB. The black dots represent the cytokine levels in absence of TCB (no TCB). Means of 2 technical replicates ± SEM. Data shown are from 1 individual donor and independent experiments. For each TCB, PBMCs from n = 3 donors were tested. (c) PBMCs or monocyte-depleted PBMCs from the same donor were stimulated with CEA-TCB or DP47-TCB (negative control) in a TDCC assay using MKN45 tumor cells (E:T = 10:1). (d) The expression of CD69 and CD25 expression on CD4+ and CD8+ T cells upon treatment with CEA-TCB was measured by flow cytometry after 24 hrs. EC50 values, means of n = 3 donors ± SD with ns = non-significant by Wilcoxon t test. (e) The killing of MKN45 tumor cells was measured by LDH release. The dose-response plots show data for 1 donor representative of 3 and the bar plots show the EC50 values for n = 3 donors, mean ± SD with ns = non-significant by Wilcoxon t test. (f) Cytokine levels were measured in the supernatants by Luminex (24 hrs). The dose-response plots show data for 1 donor representative of 5 and the bar plots show the means of n = 5 donors ± SD for 10 nM CEA-TCB with *p < 0.05 by Wilcoxon t test.
To verify to which extent monocytes contribute to the release of IL-6, IL-1β and TNF-α, we set-up a TDCC assay comparing PBMCs with monocyte-depleted PBMCs from the same donor (effector cells: target cell = 10:1) [Figure 3(c)]. Following stimulation with CEA-TCB, T cell activation profiles and tumor cell killing did not significantly differ neither in the presence or absence of monocytes in effector cells [Figure 3(d,e)]. In contrast, no IL-1β and drastically reduced IL-6 levels were released when the PBMCs were depleted of monocytes [Figure 3(f), supp. Fig 4A]. This result indicates that monocytes are the main producers of IL-1β and IL-6.
Figure 4.
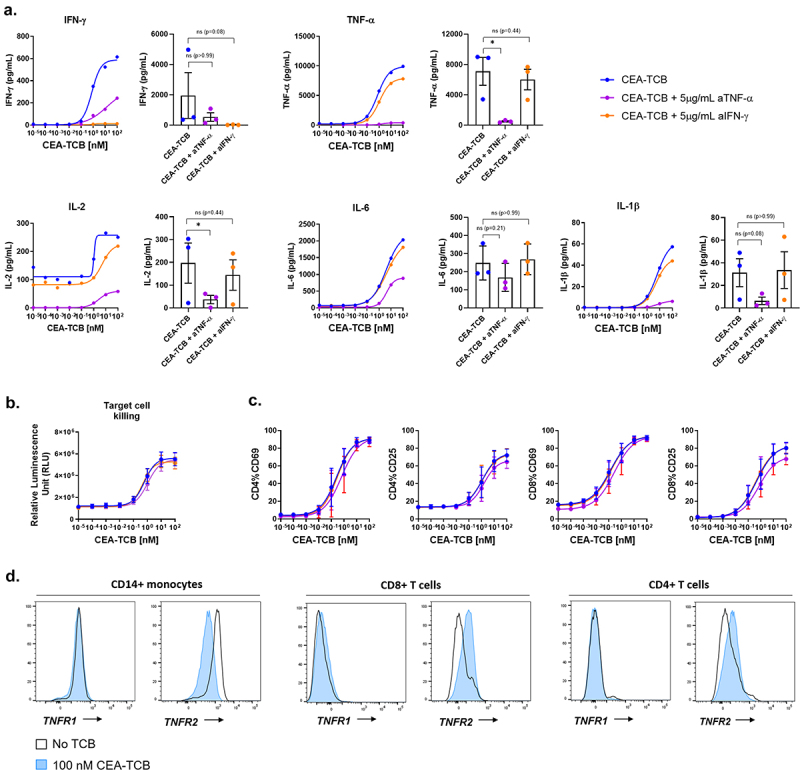
The release of IL-6 and IL-1β is dependent on TNF-α but not IFN-γ release. PBMCs were stimulated with CEA-TCB in a TDCC assay using CHOK1SV-CEACAM5 target cells with CEA-TCB in the presence or absence of 5 μg/mL anti-TNF-α (Adalimumab) or anti-IFN-γ. (a) After 24 hrs, the culture supernatant from technical duplicates were pooled and the levels of IFN-γ, IL-2, TNF-α, IL-1β and IL-6 were measured by Luminex. The dose-response plots show data for 1 donor representative of 3 and the bar plots show values for n = 3 donors treated with 10 nM CEA-TCB with * p < 0.05 by Friedman test. (b). The killing of CHOK1SV-CEACAM5 target cells was measured by LDH release (t = 24 hrs). Means of n = 3 donors ±SEM. (c) The expression of CD69 and CD25 on CD4+ and CD8+ T cells 24 hrs after treatment with CEA-TCB was measured by flow cytometry. Means of n = 3 donors ±SEM. (d) Flow cytometry histogram plots showing the modulation of TNFR1 (CD120a) and TNFR2 (CD120b) (TNF-α receptors) on CD14+ monocytes, CD4+ and CD8+ T cells after 24 hrs in TDCC assays with 100 nM CEA-TCB or in the absence TCB.
TNF-α release was still detected when monocyte-depleted PBMCs were used as effector cells stimulated with CEA-TCB, but its levels remained lower than when PBMCs were used [Figure 3(f)]. This shows that monocytes contributed to TNF-α release, together with CD4+ and CD8+ T cells [Figure 2(a,b)] as previously found by Li et al. and Godbersen-Palmer et al.25,26 In contrast, the levels of IFN-γ and IL-2 were not significantly impacted by the absence of monocytes in effector cells [Supp. Fig 4B].
Altogether, these results highlight that monocytes get indirectly activated after on-target activation of T cells and subsequently release IL-1β and IL-6, in addition to contributing to TNF-α production together with CD4+ and CD8+ T cells.
The release of IL-6 and IL-1β is dependent on TNF-α but not IFN-γ release
To verify if the production of monocyte-derived cytokines was dependent on TNF-α and IFN-γ produced by T cells after stimulation with TCB, we blocked each of the cytokines using 5 μg/mL anti-IFN-γ or anti-TNF-α (adalimumab) in TDCC assays with CEA-, Tyrp1- and FolR-TCB. The blockade of TNF-α but not of IFN-γ resulted in a decrease of IFN-γ, IL-2, IL-6 and IL-1β indicating that these T-cell or monocyte-derived cytokines are dependent on TNF-α release [Figure 4(a), supp. Fig 5A, B, C]. The blockade of IFN-γ or TNF-α did not impair TCB-mediated T cell activation and tumor cell killing, as shown by the expression of CD25 and CD69 on CD4+ and CD8+ T cells as well as the LDH release measured in the supernatants of these TDCC assays, respectively [Figure 4(b,c), supp. Fig 5D]. This indicates that these cytokines do not influence TCB-mediated killing. Interestingly, the expression of the TNF-α receptor CD120b (TNFR2) but not CD120a (TNFR1) was upregulated on CD4+ and CD8+ T cells and downregulated on CD14+ monocytes upon treatment with CEA-TCB [Figure 4(d)]. These results show that monocytes are activated by T cell-derived TNF-α, amplifying the release of TNF-α and mediating the production IL-6 and IL-1β. To note, the release of IFN-γ, TNF-α, IL-6 and IL-1β was not affected by IL-6R blockade, as indicated by the cytokine levels measured in a TDCC assay using Tyrp1-TCB in combination with anti-IL-6R (actemra) [supp. Fig 6A, B].
Figure 5.
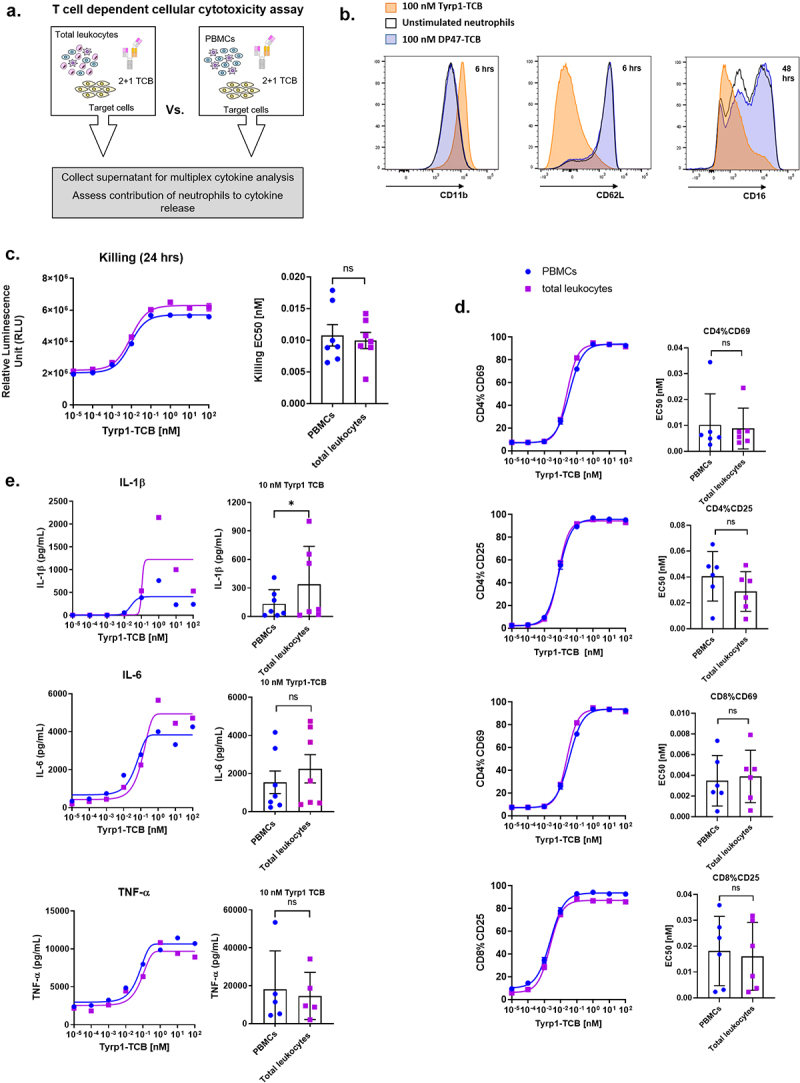
Neutrophils are activated following TCB treatment and contribute to IL-1β release. (a) PBMCs and total leukocytes (PBMCs + neutrophils) isolated from whole blood from one donor were stimulated with Tyrp1-TCB in a TDCC assay using CHOK1SV-Tyrp1 target cells (E:T = 10:1). (b) Flow cytometry histogram plots representing the expression of CD11b (6 hrs), CD62L (6 hrs) and CD16 (48 hrs) on neutrophils before or after incubation with Tyrp1- or DP47-TCB for 1 donor. (c) The killing of CHOK1SV-Tyrp1 target cells was measured by LDH release in the presence or absence of neutrophils (24 hrs). (d) The expression of CD69 and CD25 on CD4+ and CD8+ T cells upon treatment with CEA-TCB was measured by flow cytometry in the presence or absence of neutrophils (24 hrs). (c,d) The dose-response plots show data for 1 donor representative of 6 or 7 and the bar plots show the means of EC50 values ± SD for n = 6 or 7 donors.* p < 0.05 by Wilcoxon t test. (e) The levels of IL-1β, IL-6 and TNF-α were measured by Luminex at assay endpoint in the presence and absence of neutrophils (24 hrs) in pooled technical duplicates. The dose-response plots show data for 1 donor representative of 7 and the bar plots show the means of n = 7 donors treated with 10 nM Tyrp1-TCB with * p < 0.05 by Wilcoxon t test.
Figure 6.
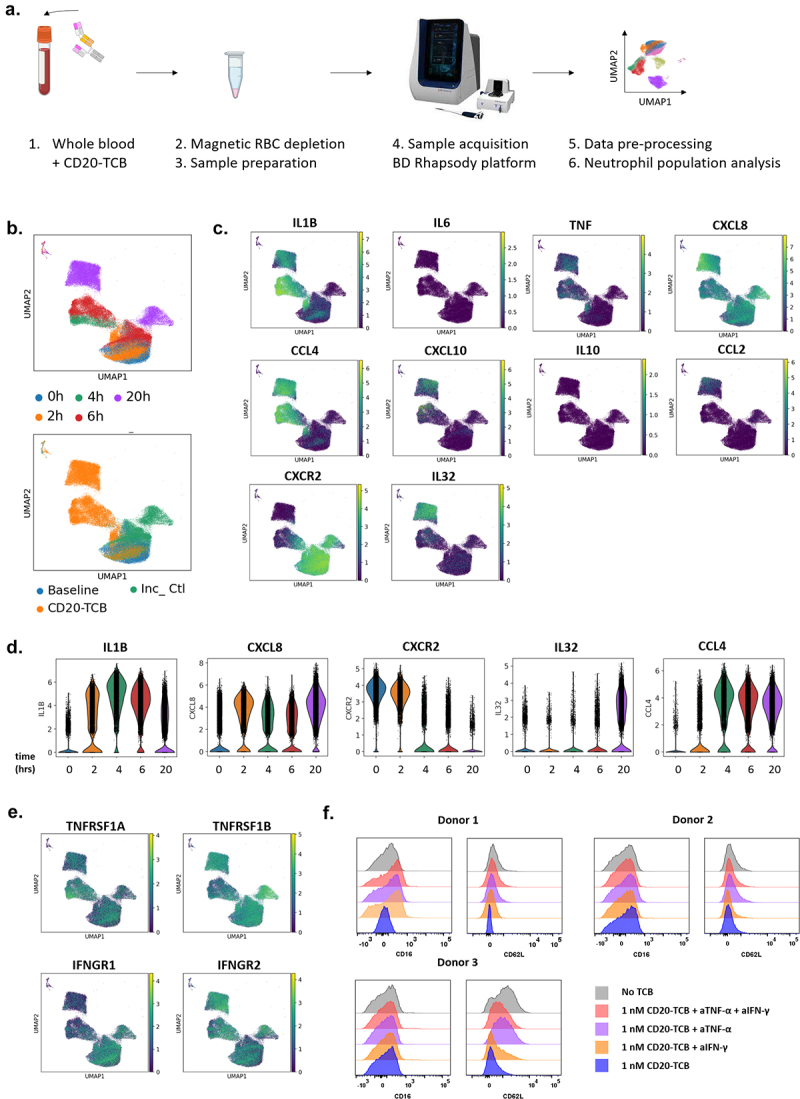
IL-1β and other cytokine and cytokine receptor genes are differentially upregulated in neutrophils from whole blood treated with CD20-TCB. (a) Single cell RNA sequencing of whole blood treated with 0.2 μg/mL CD20-TCB was performed using the BD Rhapsody platform. (b) UMAP plots of neutrophils colored by timepoint (upper panel, pooled data from 2 to 4 donors) or by treatment (lower panel, pooled data from 4 donors), Inc_Ctl stands for incubation control. (c) UMAP plots showing IL-1β (IL1B), IL-6 (IL6), TNF-α (TNF), IL-8 (CXCL8), MIP-1β (CCL4), IP-10 (CXCL10), IL-10 (IL10), MCP-1 (CCL2), IL-8R (CXCR2), IL-32 (IL32) gene expression within the neutrophils clusters identified above. The color code on the right of UMAP plots represents the level of gene expression. (d) Violin plots showing the expression distribution of IL-1β (IL-1B), IL-8 (CXCL8), IL-8R (CXCR2), IL-32 (IL32), MIP-1β (CCL4) genes within neutrophils at each treatment timepoint. (e) UMAP plots showing TNFR1a (TNFRSF1A), TNFR1b (TNFRSF1B), IFNR1 (IFNGR1) and IFNR2 (IFNGR2) gene expression within the neutrophils clusters identified above. (a-e) Each dot represents one cell. N = 4 donors (baseline, and 24 hrs) and n = 2 donors (2 hrs, 4 hrs and 6 hrs). Neutrophils were identified by scRNA sequencing of whole blood based on the ELANE, MPO, PRTN3 CTSG, AZU1 and FCGR3B gene signature. (f) Effects of anti-TNF-α and/or anti-IFN-γ blocking antibodies on CD62L and CD16 expression by neutrophils after 24 hrs incubation of 1 nM CD20-TCB in whole blood, measured by flow cytometry. The histogram plots show data from 3 different blood donors.
The use of neutralizing anti-cytokine antibodies confirmed the chronology of the events triggering TCB-mediated cytokine release as well as the dependence of IFN-γ, IL-2, IL-6 and IL-1β on the early release of TNF-α by T cells.
Neutrophils are activated following TCB treatment and contribute to the release of IL-1β
Since neutrophils are the most abundant leukocyte population in blood, we aimed to assess their potential contribution to TCB-mediated cytokine release. For this purpose, we first conducted in vitro TDCC assays using total leukocytes (PBMCs and neutrophils) as effector cells co-cultured with CHOK1-Tyrp1 or CHOK1-FolR target cells and Tyrp1-TCB or FolR-TCB, respectively (effector cells: target cell = 10 lymphocytes: 1 target cell) [Figure 5(a)]. The expression of surface markers CD11b, CD62L and CD16 was measured by flow cytometry on neutrophils gated based on their FSC/SSC profile to assess the effect of TCB treatment on their phenotype. Both Tyrp1-TCB and FolR-TCB induced an upregulation of CD11b, which was associated with a downregulation of CD62L and CD16. There was no change in phenotype observed with the negative control DP47-TCB or in the absence of TCB [Figure 5(b), supp. Fig 7A]. CD11b upregulation as well as CD62L and CD16 downregulation was also observed on the surface of neutrophils in whole blood treated with CD20-TCB [supp. Fig 8A]. This change in phenotype shows that neutrophils are activated following TCB stimulation.
In TDCC assays comparing PBMCs and total leukocytes as effector cells in the same experimental conditions, T cell activation and target cell killing remained unchanged [Figure 5(c,d) and supp. Fig 7B, C]. However, based on results from seven blood donors, IL-1β but not IL-6 or TNF-α levels were significantly higher when neutrophils were present in the effector cells, suggesting that they contribute to IL-1β release together with monocytes [Figure 5(e), supp. Fig 7D, 8B].
ScRNAseq of whole blood treated with CD20-TCB reveals the contribution of neutrophils to TCB-mediated cytokine release
To confirm these findings, we conducted single-cell RNA sequencing of whole blood using the BD Rhapsody platform at baseline and 2 hrs, 4 hrs, 6 hrs and 20 hrs after treatment with 0.2 μg/mL CD20-TCB [Figure 6(a), supp. Fig 8]. Neutrophil populations were identified based on the ELANE, MPO, PRTN3 CTSG, AZU1 and FCGR3B gene signature together with negative expression of lineage markers such as CD3D or CD79A. Of note, FCGR3B was the main gene expressed among those identified in the neutrophil gene signature and had high specificity. Additionally, the proportions of neutrophils were matching those measured by flow cytometry in the corresponding samples [supp. Fig 9A-C]. As shown in the UMAP plot, they clustered differently after 4 hrs, 6 hrs or 20 hrs stimulation with CD20-TCB, indicating a change in their phenotype [Figure 6(b)]. As a control, neutrophil populations from untreated samples incubated for 2 hrs, 6 hrs and 20 hrs overlapped with baseline samples, confirming that the observed change in phenotype was due to treatment with CD20-TCB [Figure 6(b)].
In line with our above findings, the IL-1β (IL1B) but not the IL-6 (IL6) gene was expressed in neutrophil populations after stimulation with CD20-TCB, as indicated by the UMAP plots [Figure 6(c)]. Only sparse expression of the TNF-α (TNF) gene was observed. Interestingly, the gene encoding the IL-8 receptor (CXCR2) was expressed at baseline and rapidly downregulated upon CD20-TCB treatment [Figure 6(d)]. The IL-8 gene (CXCL8) was induced early after TCB treatment, suggesting that neutrophil-derived IL-8 might act in an autocrine fashion. Additionally, we verified whether transcripts for IL-10 (IL10), MCP-1 (CCL2), MIP-1β (CCL4), IP-10 (CXCL10), described as molecular players in the biology of TCB-mediated CRS, were induced in neutrophils. Among them, IL-8 (CXCL8) and MIP-1β (CCL4) were upregulated upon treatment with CD20-TCB, indicating that neutrophils may broadly contribute to TCB-mediated cytokine release [Figure 6(c,d)]. The IL-32 (IL32R) gene was also identified among the top 40 genes induced in neutrophils after CD20-TCB stimulation. In the kinetics of events, IL-1β (IL1B) and MCP-1 (CCL2) transcripts were upregulated already 2 hrs after treatment with CD20-TCB and peaked at 4 hrs, while IL-32 (IL32), IL-8 (CXCL8) and MIP-1β (CCL4) were mainly upregulated at later timepoints [Figure 6(d), supp. Fig 10A-B]. These data demonstrate the rapid onset of cytokine release by neutrophils.
The TNF receptor genes (TNFRSF1A and TNFRSF1B) and IFN receptor genes (IFNGR1 and IFNGR2) were also found expressed on neutrophils at baseline and after treatment, suggesting that IFN-γ or TNF-a released by T cells and monocytes may signal in neutrophils [Figure 6(e)]. To verify this hypothesis, we blocked either or both of these cytokines using 5 μg/mL anti-IFN-γ or anti-TNF-α (adalimumab) in a whole blood assay with CD20-TCB. The blockade of TNF-α, and to a lower extent of IFN-γ, inhibited the downregulation of CD16 in one of three donors and of CD62L in all three donors tested, suggesting that both cytokines may contribute to neutrophil activation [Figure 6(f)].
Overall, these results confirm the early contribution of neutrophils to IL-1β release at the transcriptional level and show that activated neutrophils broadly contribute to the cytokine release cascade following TCB treatment, notably through the additional release of CCL2, MIP-1β, IL-8 and IL-32.
Discussion
On-target activation of T cells is associated with a release of cytokines that can potentially result in a cytokine release syndrome, one of the major safety liabilities intrinsic to the mode-of-action of T cell engagers. The in vitro TDCC assay system allows to recapitulate TCB-induced cytokine release and to identify the contribution of the immune cell populations present among the effector cells. Early after activation with a TCB, CD4+ and CD8+ T cells release IFN-γ and TNF-α, but not IL-6. T cell-derived cytokines further activate T cells and other immune cells present in PBMCs. Among them, monocytes were identified as the main producers of IL-1β and IL-6 and found to contribute to TNF-α release together with CD4+ and CD8+ T cells. This was clearly shown by intracellular cytokine immunostaining and the comparison of cytokine levels in supernatants from TDCC assays using either monocyte-depleted PBMCs or total PBMCs as effector cells. Our data with TCBs confirm the findings of Li J. et al. and Godbersen-Palmer C. et al., who highlighted the contribution of myeloid cells to IL-6, IL-1β and TNF-α production in vitro and in vivo early after treatment with T cell engagers.25,26
Subsequently, we focused on the role of neutrophils in the cytokine release induced by T cell engagers, which had not yet been reported. Neutrophils show an activated phenotype in TDCC assays using total leukocytes as effector cells, or in whole blood treated with CD20-TCB. This suggested that they might well contribute to TCB-induced cytokine release. We first showed that neutrophils released IL-1β, by comparing IL-1β levels in culture supernatants from TDCC assays using either total leukocytes or PBMCs as effector cells. This result was confirmed by single-cell RNA sequencing of whole blood treated with CD20-TCB. Additionally, genes of other cytokines such as IL-8 (CXCL8), IL-32 (IL32), MIP-1β (CCL4) were found rapidly upregulated in neutrophils upon treatment with CD20-TCB, indicating a broader contribution of neutrophils to TCB-mediated cytokine release. Along with the upregulation of these cytokine genes, we also observed that the IL-8R genes were downregulated upon treatment with CD20-TCB, denoting that IL-8 might act there in an autocrine fashion. T-cell derived TNF-α and IFN-γ may as well stimulate neutrophils, as the gene coding for their receptors was expressed in neutrophil clusters identified in whole blood treated with CD20-TCB.
Although they represent a large proportion of white blood cells, the contribution of neutrophils to TCB-mediated cytokine release may be underestimated both in in vitro functional assays using PBMCs as effector cells and in vivo mouse models where the proportion of neutrophils does not translate to the human amounts. Our comparison of cytokine levels in TDCC using either PBMCs or total leukocytes shows significantly higher levels of IL-1β production when total leukocytes are used. Therefore, neutrophils are likely to contribute to the release of IL-1β and other cytokines induced in the serum of patients treated with T cell engagers. However, it may be difficult to assess the relative contribution of neutrophils versus monocytes to the levels found in the serum of patients, as monocytes also produce some of these cytokines. Interestingly, neutrophils were found infiltrating tissues expressing the targeted tumor-associated antigens in cynomolgus monkeys and in mice (A.M. Giusti, unpublished results) following TCB treatment. This suggests that neutrophils also directly contribute to tissue inflammation.
In the clinic, not every patient treated with a T-cell engager experienced CRS.33,34 In a phase I trial with glofitamab, CRS occurred in 86 of 171 (50.3%) B-cell lymphoma patients (grade 3 or 4: 3.5%).33 In our in vitro TDCC assay, while cytokine release was consistent across donors, there was a strong variability in the absolute levels of cytokines observed. This observation goes in line with the recent work from Ye et al.,35 describing donor variability with respect to cytokine release in a humanized mouse model after stimulation with compounds known to be associated with CRS in the clinic. In addition to patient’s genetic polymorphism, factors independent of cytokine release by immune cells including tumor burden and endothelial cell activation have been reported to correlate with the risk of CRS after treatment with T cell engaging therapies.36–38
Presently, CRS is managed in the clinic with the use of glucocorticoids and anti-IL-6R (tocilizumab) or anti-IL-6 (siltuximab) antibodies.16 As demonstrated, IL-6 is released by myeloid cells downstream of the TCB-induced cytokine cascade, stimulated by TNF-α. Current therapeutic antibodies blocking the IL-6 pathway may mitigate IL-6-derived toxicities such as endothelial cell activation, which eventually can lead to vascular leakage, but may not affect the levels of other cytokines implicated in CRS.39 The use of anti-IL-1Ra (anakinra) antibody has been proposed for the mitigation of CAR-T cell-induced CRS and neurotoxicity.19,40 With IL-1β produced by myeloid cells and neutrophils, downstream of TCB-induced T cell activation, the specific neutralization of IL-1Ra would likely mitigate IL-1β-driven toxicities but not strongly reduce the overall cytokine release.
It is confirmed that TCB-mediated cytokine release is first initiated by T cell-derived cytokines, as a result of on-target TCB activity. This highlights that prophylactic intervention at the T cell level could more broadly reduce the cytokine cascade. As shown by Li J. et al., and by our in vitro results, TNF-α blockade could reduce both IL-6 and IL-1β release by monocytes without impacting TCB-induced T cell cytotoxicity.26 Interestingly, etanercept and adalimumab were shown to counteract CRS in patients failing to respond to IL-6/IL-6R blockade.41
Our results indicate that IFN-γ is mainly released by T cells upon treatment with TCB. They also suggest that IFN-γ blockade may not efficiently reduce the release of other cytokines implicated in CRS. However, our model systems may not fully capture its potential effects since macrophages are lacking. As IFN-γ is known to activate macrophages, its blockade could reduce the release of macrophage-derived IL-6 and IL-1β s upon treatment with T cell engaging therapies.40,42 The neutralization of IFN-γ with emapalumab is indicated for the management of hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS) symptoms, which are closely related to CRS in patients treated with CAR-T cells.11,43 Nevertheless, IFN-γ blockade was also shown to alter the recruitment of circulating T cells to the tumor site, suggesting that it might have a deleterious effect on anti-tumor efficacy, thus making it a less favorable prophylactic approach for the mitigation of CRS for T cell engagers targeting solid tumors.44
Step-up dosing of T cell engagers is also widely used to prevent the occurrence of CRS. As shown by others and in the present study, upon re-administration, peaks of inflammatory cytokines are attenuated while tumor cell killing remains effective.26,45 However, despite the use of IL-6/IL-6R neutralizing antibodies, glucocorticoids and step-up dosing, the risk of CRS still remains a major dose-limiting toxicity associated with the treatment of T cell engagers, highlighting that CRS mitigation by pre-treatment may open the way for clinical schedules that require less intense monitoring and get faster to the efficacious dose. Beside TNF-α blockade, promising T-cell targeted approaches for the mitigation of CRS include the use of kinase inhibitors targeting signaling pathways downstream of TCR stimulation46–48
Our data on the mechanisms of TCB-induced cytokine release also provide insight to develop re-stimulation TDCC in vitro models that may help guide escalation factor, and time intervals for step-up dosing schedules. Further investigations are required with respect to understanding biological mechanisms that counter balance cytokine release. Additionally, our work highlights the key cytokines upregulated intrinsically by the mode-of-action of TCBs, and may contribute to the selection of more specific clinical biomarker panels reflecting the cytokine release by monocytes and neutrophils, which are the major source of cytokines such as IL-1β, IP-10 or MIP-1β. Finally, exploring the phenotypes of neutrophils and monocytes and their propensities to produce high levels of pro-inflammatory cytokines could be beneficial for the identification of potential personalized biomarkers, which may predict whether a patient is at risk for CRS after treatment with T cell engaging therapies.
Supplementary Material
Acknowledgments
The authors thank all their colleagues from Cancer Immunotherapy, Oncology, Large Molecule Research and Pharmaceutical Sciences at Roche Pharmaceutical Research and Early Development (pRED) who contributed to this work.
Funding Statement
All funding for the studies were provided by Roche. The authors do not declare a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Abbreviations
- CAR:
chimeric antigen receptor
- CRS:
cytokine release syndrome
- NLR:
NucLight Red
- TAA:
tumor-associated antigen
- PBMCs:
peripheral blood mononuclear cells
- RBC:
red blood cell
- TCB:
T cell bispecific antibody
- TCR:
T cell receptor
- TDCC:
T-cell dependent cellular cytotoxicity
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Disclosure statement
All authors, except A. Odermatt, are employees of Roche or were employed by Roche at the time of this study and declare patent. All the authors, except A. Odermatt, G. Leclercq, N. Steinhoff, declare ownership of Roche stock.
Authors’ contributions
Concept & Experimental design G. Leclercq, H. Haegel, A. Schneider, C. Bossen, S. Danilin, N. Steinhoff, M. Bacac.
Acquisition of data: G. Leclercq, S. Danilin.
Data analysis and interpretation: G. Leclercq, L. Alberti-Servera, H. Haegel, A. Schneider, A. Giusti, M. Bacac, C. Klein.
Writing, review and/or revision of the manuscript: G. Leclercq, H. Haegel, A. Odermatt, L. Alberti-Servera, S. Danilin, C. Klein, M. Bacac.
Administrative, technical or material support: J. Challier, S. Danilin.
Study supervision: H. Haegel, A. Schneider, C. Klein, M. Bacac, P. Umana.
Availability of data and material
All data relevant to the study are included in the article or uploaded as online supplementary information.
References
- 1.Zhou S, Liu M, Ren F, Meng X, Yu J.. The landscape of bispecific T cell engager in cancer treatment. Biomarker Res. 2021;9(1):38. doi: 10.1186/s40364-021-00294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacac M, Colombetti S, Herter S, Sam J, Perro M, Chen S, Bianchi R, Richard M, Schoenle A, Nicolini V, et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin Cancer Res. 2018;24(19):4785–15. doi: 10.1158/1078-0432.CCR-18-0455. [DOI] [PubMed] [Google Scholar]
- 3.Bacac M, Klein C, Umana P. CEA TCB: a novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5(8):e1203498. doi: 10.1080/2162402X.2016.1203498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22(13):3286–3297. doi: 10.1158/1078-0432.CCR-15-1696. [DOI] [PubMed] [Google Scholar]
- 5.Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. mAbs. 2016;8(6):1010–1020. doi: 10.1080/19420862.2016.1197457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Engineering Design and Selection. 2016;29(10):457–466. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 7.Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–175. doi: 10.1016/j.pharmthera.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 9.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17:197–223. [DOI] [PubMed] [Google Scholar]
- 10.Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, Kochanek M, Böll B, Von Bergwelt-baildon MS. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. doi: 10.1186/s40425-018-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subklewe M. BiTEs better than CAR T cells. Blood Adv. 2021;5(2):607–612. doi: 10.1182/bloodadvances.2020001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishiguro T, Sano Y, Komatsu SI, Kamata-Sakurai M, Kaneko A, Kinoshita Y, Shiraiwa H, Azuma Y, Tsunenari T, Kayukawa Y, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med. 2017;9(410). doi: 10.1126/scitranslmed.aal4291. [DOI] [PubMed] [Google Scholar]
- 14.Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022;22(2):85–96. doi: 10.1038/s41577-021-00547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karki R, Kanneganti TD. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42(8):681–705. doi: 10.1016/j.it.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, Maus MV, Park JH, Mead E, Pavletic S, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riegler LL, Jones GP, Lee DW. Current approaches in the grading and management of cytokine release syndrome after chimeric antigen receptor T-cell therapy. Ther Clin Risk Manag. 2019;15:323–335. doi: 10.2147/TCRM.S150524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. 2016;2016(1):567–572. doi: 10.1182/asheducation-2016.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, Zhao J. Cytokine release syndrome: grading, modeling, and new therapy. J Hematol Oncol. 2018;11(1):121. doi: 10.1186/s13045-018-0653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, Wu T, Chen D, Chang AH, Gao Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):15. doi: 10.1038/s41408-020-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khadka RH, Sakemura R, Kenderian SS, Johnson AJ. Management of cytokine release syndrome: an update on emerging antigen-specific T cell engaging immunotherapies. Immunotherapy. 2019;11(10):851–857. doi: 10.2217/imt-2019-0074. [DOI] [PubMed] [Google Scholar]
- 23.Pishvaian M, Morse MA, McDevitt J, Norton JD, Ren S, Robbie GJ, Ryan PC, Soukharev S, Bao H, Denlinger CS, et al. Phase 1 dose escalation study of MEDI-565, a bispecific T-cell engager that targets human carcinoembryonic antigen, in patients with advanced gastrointestinal adenocarcinomas. Clin Colorectal Cancer. 2016;15(4):345–351. doi: 10.1016/j.clcc.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Hosseini I, Gadkar K, Stefanich E, Li -C-C, Sun LL, Chu Y-W, Ramanujan S. Mitigating the risk of cytokine release syndrome in a Phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. NPJ Syst Biol Appl. 2020;6(1):28. doi: 10.1038/s41540-020-00145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godbersen-Palmer C, Coupet TA, Grada Z, Zhang SC, Sentman CL. Toxicity induced by a bispecific T cell-redirecting protein is mediated by both T cells and myeloid cells in immunocompetent mice. J Immunol. 2020;204(11):2973–2983. doi: 10.4049/jimmunol.1901401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Piskol R, Ybarra R, Chen YJ, Li J, Slaga D, Hristopoulos M, Clark R, Modrusan Z, Totpal K, et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med. 2019;11(508). doi: 10.1126/scitranslmed.aax8861. [DOI] [PubMed] [Google Scholar]
- 27.Mädler SC, Julien-Laferriere A, Wyss L, Phan M, Sonrel A, Kang ASW, Ulrich E, Schmucki R, Zhang JD, Ebeling M, et al. Besca, a single-cell transcriptomics analysis toolkit to accelerate translational research. NAR Genom Bioinform. 2021;3(4). doi: 10.1093/nargab/lqab102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McInnes L, Healy J. UMAP: uniform manifold approximation and projection for dimension reduction. ArXiv. 2018. [Google Scholar]
- 29.Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep. 2019;9(1):5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mädler SC, Julien-Laferriere A, Wyss L, Phan M, Kang ASW, Ulrich E, et al. Besca, a single-cell transcriptomics analysis toolkit to accelerate translational research. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudal S, Hinton H, Giusti AM, Bacac M, Muller M, Fauti T, Colombetti S, Heckel T, Giroud N, Klein C, et al. Application of a MABEL approach for a T-cell-bispecific monoclonal antibody: CEA TCB. J Immunother. 2016;39(7):279–289. doi: 10.1097/CJI.0000000000000132. [DOI] [PubMed] [Google Scholar]
- 32.Dickinson MJ, Morschhauser F, Iacoboni G, Carlo-Stella C, Offner FC, Sureda A, Salles G, Martinez J, Crump M, Thomas DN, et al. CD20-TCB (RG6026), a novel “2:1” format T-cell-engaging bispecific antibody, induces complete remissions in relapsed/refractory B-cell non-Hodgkin’s lymphoma. Hematol Oncol. 2019;37(S2):92–93. doi: 10.1002/hon.59_2629.31187519 [DOI] [Google Scholar]
- 33.Hutchings M, Morschhauser F, Iacoboni G, Carlo-Stella C, Offner FC, Sureda A, Salles G, Martínez-Lopez J, Crump M, Thomas DN, et al. Glofitamab, a novel, bivalent CD20-Targeting T-cell-engaging bispecific antibody, induces durable complete remissions in relapsed or refractory B-cell lymphoma: a phase I trial. J Clin Oncol. 2021;39(18):1959–1970. doi: 10.1200/JCO.20.03175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimabukuro-Vornhagen A, Subklewe M, Von Bergwelt-baildon M. Challenges in immuno-oncology improve deficits, exploit the potentials. Deut Med Wochenschr. 2018;143:1022–1029. [DOI] [PubMed] [Google Scholar]
- 35.Ye C, Yang H, Cheng M, Shultz LD, Greiner DL, Brehm MA, Keck JG. A rapid, sensitive, and reproducible in vivo PBMC humanized murine model for determining therapeutic-related cytokine release syndrome. FASEB J. 2020;34(9):12963–12975. doi: 10.1096/fj.202001203R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tedesco V, Mohan C. Biomarkers for predicting cytokine release syndrome following CD19-targeted CAR T cell therapy. J Immunol. 2021;206(7):1561–1568. doi: 10.4049/jimmunol.2001249. [DOI] [PubMed] [Google Scholar]
- 37.Hong F, Shi M, Cao J, Wang Y, Gong Y, Gao H, Li Z, Zheng J, Zeng L, He A, et al. Predictive role of endothelial cell activation in cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukaemia. J Cell Mol Med. 2021;25(24):11063–11074. doi: 10.1111/jcmm.17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimabukuro-Vornhagen A, Garcia-Marquez M, Fischer RN, Iltgen-Breburda J, Fiedler A, Wennhold K, Rappl G, Abken H, Lehmann C, Herling M, et al. Antigen-presenting human B cells are expanded in inflammatory conditions. J Leukocyte Biol. 2017;101(2):577–587. doi: 10.1189/jlb.5A0416-182R. [DOI] [PubMed] [Google Scholar]
- 39.Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, López JA, Chen J, Chung D, Harju-Baker S, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Wang S, Xu J, Zhang R, Zhu H, Wu Y, Zhu L, Li J, Chen L. Etanercept as a new therapeutic option for cytokine release syndrome following chimeric antigen receptor T cell therapy. Exp Hematol Oncol. 2021;10(1):16. doi: 10.1186/s40164-021-00209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol. 2020;9(1):15. doi: 10.1186/s40164-020-00171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, Degar B, Garrington TP, Sevilla J, Putti M-C, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. 2020;382(19):1811–1822. doi: 10.1056/NEJMoa1911326. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Ybarra R, Mak J, Herault A, De Almeida P, Arrazate A, Ziai J, Totpal K, Junttila MR, Walsh KB, et al. IFNγ-induced chemokines are required for CXCR3-mediated T-cell recruitment and antitumor efficacy of anti-HER2/CD3 bispecific antibody. Clin Cancer Res. 2018;24(24):6447–6458. doi: 10.1158/1078-0432.CCR-18-1139. [DOI] [PubMed] [Google Scholar]
- 45.Hernandez G, Huw L-Y, Belousov A, Wilson D, Koeppen H, McCord R, Peng K, Bartlett NL, Budde LE, Assouline S, et al. Pharmacodynamic effects and immune correlates of response to the CD20/CD3 bispecific antibody mosunetuzumab in relapsed or refractory non-Hodgkin lymphoma. Blood. 2019;134(Supplement_1):1585. doi: 10.1182/blood-2019-123933.31558469 [DOI] [Google Scholar]
- 46.Leclercq G, Haegel H, Schneider A, Berger EM, Walz A, Boetsch C, Pulko V, Ferlini C, Klein C. 653 Dasatinib as a rapid pharmacological ON/OFF switch for T cell bispecific antibody-induced T cell activation and cytokine release. J Immunother Cancer. 2020;8(Suppl 3):A690–A. doi: 10.1136/jitc-2020-SITC2020.0653. [DOI] [Google Scholar]
- 47.Leclercq G, Haegel H, Schneider A, Giusti AM, Marrer-Berger E, Boetsch C, Walz A-C, Pulko V, Sam J, Challier J, et al. Src/lck inhibitor dasatinib reversibly switches off cytokine release and T cell cytotoxicity following stimulation with T cell bispecific antibodies. J Immunother Cancer. 2021;9(7):e002582. doi: 10.1136/jitc-2021-002582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leclercq G, Haegel H, and Toso A, et al. JAK and mTOR inhibitors prevent cytokine release while retaining T cell bispecific antibody in vivo efficacy. J Immunother Cancer. 2022;10:e003766. doi: 10.1136/jitc-2021-003766 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as online supplementary information.