

ABSTRACT
Immune checkpoint inhibitors (ICI) targeting the PD-1/PD-L1 axis have changed the treatment paradigm for advanced solid tumors; however, many patients experience treatment resistance. In preclinical models 4-1BB co-stimulation synergizes with ICI by activating cytotoxic T- and NK-cell-mediated anti-tumor immunity. Here we characterize the mechanism of action of a mouse-reactive Fc-inert PD-L1×4-1BB bispecific antibody (mbsAb-PD-L1×4-1BB) and provide proof-of-concept for enhanced anti-tumor activity. In reporter assays mbsAb-PD-L1×4-1BB exhibited conditional 4-1BB agonist activity that was dependent on simultaneous binding to PD-L1. mbsAb-PD-L1×4-1BB further blocked the PD-L1/PD-1 interaction independently of 4-1BB binding. By combining both mechanisms, mbsAb-PD-L1×4-1BB strongly enhanced T-cell proliferation, cytokine production and antigen-specific cytotoxicity using primary mouse cells in vitro. Furthermore, mbsAb-PD-L1×4-1BB exhibited potent anti-tumor activity in the CT26 and MC38 models in vivo, leading to the rejection of CT26 tumors that were unresponsive to PD-L1 blockade alone. Anti-tumor activity was associated with increased tumor-specific CD8+ T cells and reduced regulatory T cells within the tumor microenvironment and tumor-draining lymph nodes. In immunocompetent tumor-free mice, mbsAb-PD-L1×4-1BB treatment neither induced T-cell infiltration into the liver nor elevated liver enzymes in the blood. Dual targeting of PD-L1 and 4-1BB with a bispecific antibody may therefore address key limitations of first generation 4-1BB-agonistic antibodies, and may provide a novel approach to improve PD-1/PD-L1 checkpoint blockade.
KEYWORDS: PD-L1, 4-1BB, bispecific antibody, checkpoint immunotherapy, T cells
Introduction
While PD-1/PD-L1 immune checkpoint inhibitors (ICI) have changed the treatment paradigm and prognosis for patients with advanced solid tumors, durable clinical responses are observed only in a limited number of patients.1,2 Many patients experience innate or acquired treatment resistance, and overcoming resistance remains a major challenge. Therefore, combination therapies designed to enhance immune responses by targeting complementary pathways may be necessary to address the unmet need in treating the patient population with ICI resistant cancer.
ICI treatment resistance may be overcome by simultaneously targeting immunostimulatory receptors such as 4-1BB, which is a co-stimulatory member of the TNF receptor superfamily and is predominantly expressed by activated T cells and NK cells.3 Binding to its natural ligand 4-1BBL induces clustering of 4-1BB and intracellular signaling, resulting in cellular activation, proliferation and cytokine secretion. 4-1BB represents an attractive immunotherapy target, as its engagement activates T-cell and NK-cell-mediated anti-tumor immunity.4 Various therapeutic 4-1BB monoclonal antibodies (mAbs) have been developed, which induce 4-1BB clustering directly by high-avidity binding and/or through Fcγ receptor (FcγR)-mediated antibody crosslinking.5 Promising initial efficacy signals were observed with urelumab, a fully human IgG4 mAb with strong 4-1BB agonist activity. However, further development of this mAb was hampered by dose-limiting hepatotoxicity, which was characterized by elevations in serum transaminase levels.6–8 By contrast, utomilumab, a fully human IgG2 mAb with weak 4-1BB agonist activity, was well tolerated, but showed limited efficacy as monotherapy.6–8 Preclinical studies reported that immune checkpoint blockade combined with 4-1BB co-stimulation enhanced activation of CD8+ T cells and induced potent anti-tumor activity in murine models,9–13 leading to the evaluation of 4-1BB and PD-1/PD-L1 mAb combinations in patients with advanced solid tumors.14,15
An alternative approach is to develop therapies that activate 4-1BB in a conditional manner, i.e. dependent on crosslinking to a tumor-associated antigen using an Fc-inert bispecific antibody (bsAb). Targeting 4-1BB agonist activity to the tumor microenvironment (TME) may provide for an effective anti-tumor immune response.16–18 Bispecific next-generation checkpoint immunotherapies that provide dual targeting of PD-L1 and 4-1BB have the potential to activate T cells through PD-L1 blockade and simultaneous conditional 4-1BB co-stimulation. This PD-L1 × 4-1BB bispecific approach offers the advantage of acting on two complementary immune-oncology targets within one compound and may also be safer than a combination of PD-(L)1 and 4-1BB-specific mAbs, due to the conditional nature of 4-1BB co-stimulation.
Here, we characterized an Fc-inert bsAb that monovalently binds to mouse PD-L1 and mouse 4-1BB (mbsAb-PD-L1×4-1BB). In reporter assays we demonstrated that mbsAb-PD-L1×4-1BB activated the 4-1BB receptor in a conditional manner, dependent upon simultaneous binding to PD-L1. mbsAb-PD-L1×4-1BB blocked the PD-1/PD-L1 interaction independently of 4-1BB binding. By combining these mechanisms, mbsAb-PD-L1×4-1BB enhanced T-cell activation and effector functions in vitro. In mice bearing subcutaneous tumors, mbsAb-PD-L1×4-1BB induced potent antitumor activity that was superior to PD-L1 blockade alone. Anti-tumor activity was dose-dependent and required simultaneous engagement of both targets. Anti-tumor activity was further associated with an increased prevalence of tumor antigen-specific T cells as well as reduced immunosuppression in the TME and tumor-draining lymph nodes (tdLNs). Furthermore, changes in liver immune cell composition or transaminase activity in the peripheral blood were not detected in mice treated with mbsAb-PD-L1×4-1BB. While some of our findings are in line with data previously published on PD-L1×4-1BB bispecific antibodies,18–21 we now provide further insights into the mode-of-action in vivo, thereby highlighting specific advantages of targeting both pathways simultaneously with a bispecific agent compared to PD-1/PD-L1 blockade alone or a combination of mAbs.
Materials and methods
Antibodies and cell lines
Commercially available antibodies used in the experiments are listed in Table S1. Monoclonal antibodies (mAbs) that are listed in Table S2 (amino acid sequences in Table S3) were recombinantly produced in a mouse IgG2a backbone, using previously described methods.22 Double matched point mutations in the CH3 domain (F405L/R411T in one and T370K/K409R in the other) were introduced to facilitate controlled Fab-arm exchange (cFAE). In addition, L234A and L235A Fc-silencing mutations were introduced that were previously described to reduce binding to FcγR and C1q.22,23 BsAbs were generated by cFAE as described previously,22 a process in which the matched CH3 mutations drive heterodimerization of the Fab arms and formation of bispecific molecules (Figure 1a). The HIV-1 gp120-specific mAb-ctrl that comprises CDR sequences based on IgG1-b1224 were used to generate bsAbs with one non-binding Fab arm. Antibodies were biochemically characterized using hydrophobic interaction chromatography (HIC), high-performance size-exclusion chromatography (HP-SEC), and capillary electrophoresis-sodium dodecyl sulfate (CE-SDS) (Table S2).
Figure 1.
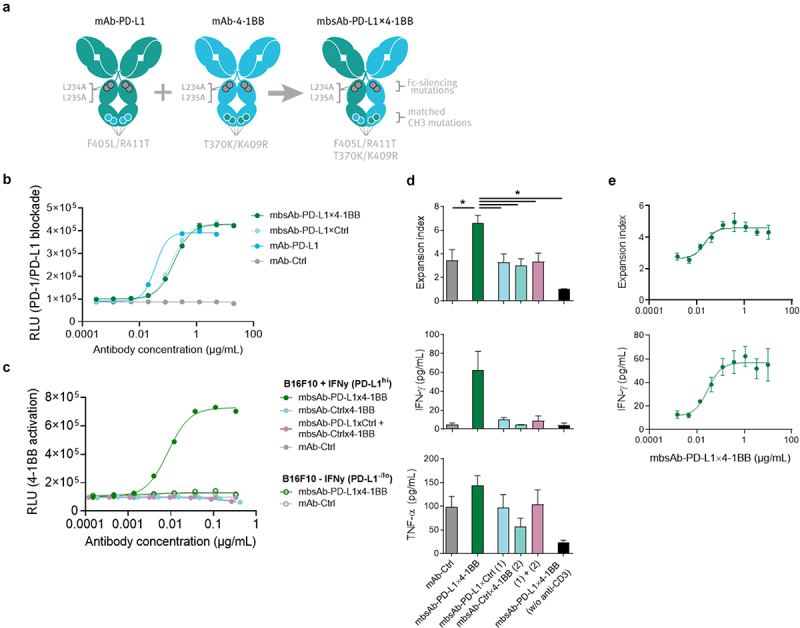
Generation of mouse-reactive Fc-inert mbsAb-PD-L1×4-1BB and characterization in vitro.
(a) Generation of mbsAb-PD-L1x4-1BB by controlled Fab arm exchange (cFAE) of Fc-silenced analogues of PD-L1 clone MPDL3280A (mAb-PD-L1) and 4-1BB clone 3H3 (mAb-4-1BB). The parental antibodies contain double matched point mutations in the CH3 domain (F405L/R411T in one and T370K/K409R in the other [EU numbering]) that drive heterodimerization of the Fab arms and formation of bispecific molecules during cFAE, as well as L234A and L235A Fc-silencing mutations that abrogate binding to FcγR and C1q. (b) Blockade of PD-1/PD-L1 interaction by mbsAb-PD-L1×4-1BB or the indicated control antibodies was assessed using a mouse PD-1/PD-L1 blockade bioassay. RLU, relative light units. (c) Induction of 4-1BB signaling by mbsAb-PD-L1×4-1BB or the indicated control antibodies was assessed using a mouse 4-1BB reporter assay. (d) CFSE-labeledCD8+ T cells from C57BL/6 mice were co-cultured with autologous BMDCs (T-cell:DC ratio 15:1) and stimulated with 5 ng/mL anti-CD3, or left unstimulated (w/o anti-CD3). mbsAb-PD-L1×4-1BB or the indicated control antibodies were added at 1 µg/mL each. Proliferation was measured by CFSE dilution after four days (pooled data are shown in upper panel) and expressed as expansion index (a measure of T-cell expansion at population level). IFN-γ and TNF-α concentrations in the supernatants were measured 48 hours after initiation of the culture (one representative experiment is shown in the middle and lower panels). (e) CFSE-labeledCD8+ T cells were co-cultured with autologous BMDCs and stimulated with anti-CD3 in the presence of increasing concentrations of mbsAb-PD-L1×4-1BB. Proliferation and IFN-γ concentration were measured as in (d). Data shown are mean (± SD) of triplicate wells of one representative experiment. *, P < .05; One-way ANOVA with Sidak’s multiple comparisons test.
The MC38 colon carcinoma cell line (Kerafast, cat. no. ENH204-FP) and the CT26 colon carcinoma cell line (ATCC, cat. no. CRL-2638) were expanded in MC38 or CT26 culture medium, respectively (Table S4). The cell lines were passaged at least three times after culture initiation and harvested when growing in log-phase for in vivo experiments. The B16_OVA-MO4 cell line (a kind gift of Kris Thielemans, University College London) and the B16-F10 cell line (ATCC, cat. No. CRL-6475) were cultured in B16 culture medium (Table S4).
Mice and tumor models
C57BL/6 were initially purchased from Charles River or Envigo. BALB/c and OT-I mice were initially purchased from Janvier and Charles River, respectively. Throughout all experiments, age-matched (8–12 weeks) female animals were used. Procedures and experimental group sizes were approved by the animal welfare committee appointed by local regulatory authorities of Rhineland-Palatinate (ID: 00009690-1-X). All mice were kept in accordance with federal and state policies on animal research.
C57BL/6 mice were injected subcutaneously (SC) in the upper flank with 5 × 105 MC38 cells in 100 μL DPBS (Life Technologies GmbH, cat. no. 14190250). BALB/c mice were injected SC in the upper flank with 5 × 105 CT26 cells in 100 μL Dulbecco’s phosphate-buffered saline (DPBS). Treatment was commenced when tumors had reached median volumes indicated in the figure legends. Mice were randomized with the ANOVA-P method using Daniel’s XL Toolbox, an open-source add-in for Microsoft Excel for data analysis and visualization. Tumor-bearing mice were treated by intraperitoneal (IP) injection of mbsAb-PD-L1×4-1BB or control antibodies (Table S2) in 200 µL PBS.
Tumor growth was evaluated at least twice per week using a caliper and tumor volumes (mm3) were calculated from caliper measurements as ([length] × [width]2)/2, where the length is the longest tumor dimension and the width is the longest tumor dimension perpendicular to the length.
For liver immune profiling experiments, tumor-free C57BL/6 mice were treated with 250 µg mbsAb-PD-L1×4-1BB or control antibodies (Table S2) in 200 µL PBS by IP injection.
Reporter assays
The effect of mbsAb-PD-L1×4-1BB on the functional interaction of PD-1 with PD-L1 was determined using the mouse PD-1/PD-L1 Blockade Bioassay (Promega, cat.no. CS303201), according to manufacturer’s instructions. Briefly, a PD-L1-expressing CHO-K1 cell line functioning as artificial APC was co-cultured with a PD-1-expressing effector cell line in the presence of mbsAb-PD-L1×4-1BB or control antibodies for 6 h. Luciferase reporter activity was determined by luminescence measurement using an Infinite F200 Pro plate reader (Tecan Life Sciences). 4-1BB agonist activity was evaluated using mouse 4-1BB effector cells (Promega, early access) and the BioGloTM Luciferase Assay system (Promega, cat. no. G7940). 4-1BB effector cells were co-cultured with B16F10 cells, which had been stimulated overnight with 5 ng/mL IFN-γ (Biolegend, Cat. No. 714006) to achieve high levels of PD-L1 expression, or left unstimulated (PD-L1lo/-). Luminescence was recorded using an Infinite F200 Pro plate reader.
Cell isolation and enrichment
Spleens were mechanically dissociated to a single-cell suspension and washed. Erythrocytes were lysed with erythrocyte lysis buffer (Table S4) for 5 min at RT. Draining (inguinal) as well as non-draining (axillary, brachial, mandibular) lymph nodes were mechanically dissociated and incubated with 1 mg/mL Collagenase D (Sigma-Aldrich, cat. No. 11088866001) for 10 min at 37°C. Blood was collected by puncture of the retrobulbar venous plexus with heparinized microhematocrit capillary tubes (Hirschmann, cat. No. 9030208). Tumor single-cell suspensions were prepared by enzymatic digestion using the mouse tumor dissociation kit (Miltenyi, cat. no. 130–096-730) and the gentleMACS dissociator (Miltenyi, cat. no. 130–093-235) following the manufacturer’s manual.
CD8+ T cells were enriched by negative selection from mouse splenocytes by magnetic activated cell sorting (MACS), using the mouse CD8a+ T cell isolation kit (Miltenyi, cat. no. 130–104-075) and LS columns (Miltenyi, cat. no. 130–042-401), according to the manufacturer’s instructions. In some experiments, CD8+ T cells were labeled with CFSE using the Vybrant CFDA SE Cell tracer kit (Life Technologies GmbH, cat. no. V12883).
Bone marrow was flushed from resected femurs and tibias and collected into a culture dish using mature dendritic cell (mDC) medium (Table S4). Bone marrow cells were resuspended, filtered through a 70 μm cell strainer, centrifuged and incubated with 5 mL erythrocyte lysis buffer for 3 min at RT, filtered through a 70 μm cell strainer and washed.
Differentiation of bone marrow-derived dendritic cells (BMDCs)
Bone marrow cells were incubated in a T75 culture flask (Greiner Bio-One, cat. no. 658175) in mDC medium supplemented with 1000 U/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; Peprotech, cat. no. 315–03). After two days, 5 mL fresh mDC medium with 1000 U/mL GM-CSF was added. Four days after culture initiation, non-adherent cells were pelleted by centrifugation (300 × g, 6 min), resuspended in 20 mL mDC medium supplemented with 1000 U/mL GM-CSF and transferred to a new T75 culture flask. On day 7, non-adherent cells (designated BMDCs) were collected, washed, frozen and stored in liquid nitrogen until further use.
T-cell proliferation assay
CFSE-labeled CD8+ T cells and BMDCs (T-cell:DC ratio 15:1 or 30:1) were plated in mDC medium in round-bottom 96-well plates (Costar, cat. no. 734–1797). Cells were treated simultaneously with anti-CD3 antibody and mbsAb-PD-L1×4-1BB or control antibodies (Tables S1-S2) for 72 h. Supernatants were harvested for cytokine analysis. Cells were analyzed for T-cell proliferation by flow cytometry.
Cytotoxicity assay
Splenocytes from OT-I or C57BL/6 mice were stimulated with 50 ng/mL OVA257-264 and 50 U/mL rhIL-2 overnight (O/N). CD8+ T cells were enriched and co-cultured with B16_OVA-MO4 cells that had been seeded in an xCELLigence E-plate at 7 × 103 cells per well on the previous day (1.05 × 105 T cells per well, effector:target cell ratio 15:1). mbsAb-PD-L1×4-1BB or control antibodies (Table S2) were added at 1 µg/mL and the cells incubated for 120 h. The impedance was measured in 30-minute intervals in an xCELLigence Real-Time Cell Analysis Instrument (Acea Biosciences). Data from impedance measurements were first normalized to the time-point of co-culture start for each treatment condition individually. Subsequently, data were expressed relative to the isotype control-treated co-cultures (mAb-Ctrl set to 100%).
After 48 h of incubation supernatants were removed for cytokine analysis from replicate wells that were not subjected to impedance measurement. Fresh media containing Brefeldin A (BD Biosciences, cat. No. 555029) was added to the cells, followed by incubation for 4 h and flow cytometry analysis. Proliferation analysis based on CFSE dilution was performed using the proliferation modeling tool from FlowJo, the generation peaks were automatically fit and expansion index values were calculated using the integrated formula.
Flow cytometry
Cells were isolated from spleens, lymph nodes, and tumors as described under ‘cell isolation and enrichment’. Cultured cells, splenocytes, lymph node cells, peripheral blood samples or tumor digests were stained for cell surface antigens, and in some experiments an MHC class I-gp70 tetramer (Table S1), for 30 min at 4°C. For intracellular staining, cells were fixed and permeabilized followed by intracellular staining for 30 min at 4°C. Blood samples were incubated with erythrocyte lysis buffer (BD Biosciences, cat. No. 349202) after completion of staining. Data were acquired on a FACSCanto™ or a FACSCelesta™ flow cytometer (BD Biosciences). Data was analyzed using FlowJo™ software V10.3.
Cytokine analysis
Cell culture supernatants were analyzed using the V-PLEX Proinflammatory Panel 1 Mouse Kit (MSD LLC, cat. no. K15048D) on a MESO QuickPlex SQ 120 instrument (MSD LLC., cat. no. R31QQ-3), according to the manufacturer’s instructions.
ELISpot
A 96-well ELISpot plate (Millipore, cat. no. MSIPS4510) was coated O/N at 2–8°C with an anti-IFN-γ antibody (Table S1). 2 × 105 freshly isolated lymph node cells per well were added and restimulated with 2 µg/mL H2-Ld restricted gp70 peptide (SPSYAYHQF),25 5 × 104 CT26 cells or media only. After O/N incubation at 37°C, cells and supernatant were removed and ELISpot plates developed using a biotinylated anti-IFN-γ antibody followed by biotin-coupled enzyme (ExtrAvidin-Alkaline Phosphatase; Sigma-Aldrich, cat. no. E2636) and BCIP/NBT substrate (Sigma-Aldrich, cat. no. B1911). IFN-γ spots were counted using the ImmunoSpot® Series S6CORE analyzer (CTL Europe) and the ImmuniCaptureTM Image Acquisition as well as the ImmunoSpot® Professional Software.
Immunohistochemistry
From the liver immune profiling experiments, livers were dissected, fixed in formalin and paraffin embedded and sectioned (4 µm). For histologic assessment, the liver sections were deparaffinized and stained with Mayer’s hematoxylin (Carl Roth, cat. no. T865.2) and Eosin-G 0.5% (Carl Roth, cat. no. X883.2) using the Leica ST5020 multistainer. For evaluation of CD8+ T cells or F4/80+ cells within the liver, liver sections were deparaffinized, antigens were retrieved using antigen retrieval buffer, followed by quenching of endogenous peroxidase and blocking of unspecific binding sites with blocking buffer (Table S4). Subsequently, sections were incubated with primary and HRP-labeled secondary Abs (Table S1), and HRP was visualized using the VECTOR®NovaRED® Peroxidase (HRP) Substrate kit (Vector Laboratories, cat. no. SK-4800), according to manufacturer’s instructions. Nuclei were counterstained by incubation with Mayer’s hematoxylin. Stained slides were subjected to whole-slide imaging (Zeiss; AxioScan Z1), and whole slide images were uploaded to and analyzed with Halo software (Indica Labs, Albuquerque) using pre-programmed software analysis tools to determine CD8a and F4/80 cells (Immune Cell v 1.3), lymphocytes (Multiplex IHC v 1.2), and tissue slice area (Area Quantification v 1.0). For the number of lymphocytes per area, the number of lymphocytes was determined and related to the respective area of the cut.
Clinical chemistry
Alanine aminotransferase (ALT), aspartate aminotransferase (AST) and glutamate dehydrogenase (GLDH) activity levels in serum samples were analyzed using the INDIKO clinical chemistry system (Thermo Fisher Scientific, cat. no. 98631000), according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism. Statistical analysis including multiple conditions were conducted by One-Way ANOVA with either Dunnett’s (comparison to mbsAb-PD-L1×4-1BB) or Tukey’s (comparison between all groups, or paired test) multiple comparisons test. Survival analysis was conducted by Log-Rank (Mantel-Cox) test. Tumor growth curves were analyzed by Two-Way ANOVA with Tukey’s multiple comparisons test.
Results
Generation of mbsAb-PD-L1×4-1BB
To evaluate the immunostimulatory and anti-tumor potential of simultaneously targeting PD-L1 and 4-1BB with a bsAb, we generated mbsAb-PD-L1×4-1BB by cFAE (Figure 1a). The bsAb was designed to induce crosslinking of 4-1BB exclusively upon simultaneous binding to PD-L1, by introducing Fc-silencing mutations that abrogate binding to FcγR and C1q.22 The binding affinities of the PD-L1 specific and 4-1BB specific Fab arms in mbsAb-PD-L1×4-1BB to mouse PD-L1 and mouse 4-1BB were in the nanomolar range (KD PD-L1: 5.9 nM; KD 4-1BB: 5.0 nM; Figure S1, Table S5).
mbsAb-PD-L1×4-1BB exhibited conditional T-cell stimulatory activity
We performed studies using reporter assays and primary T cells to assess the functional activity of mbsAb-PD-L1×4-1BB and dissect its mechanism-of-action (MoA). To evaluate whether mbsAb-PD-L1×4-1BB exhibited blocking or agonist activity by binding to a single target, or whether simultaneous engagement of both targets was required, we included monovalent control bsAbs comprised of a PD-L1 or 4-1BB binding Fab arm and a non-binding Fab arm (mbsAb-PD-L1×Ctrl and mbsAb-ctrl×4-1BB) individually and/or in combination. First, the effect of mbsAb-PD-L1×4-1BB on PD-1/PD-L1 inhibitory signaling was assessed using a cell-based reporter assay. mbsAb-PD-L1×4-1BB and mbsAb-PD-L1×Ctrl efficiently blocked PD-1/PD-L1 inhibitory signaling, as maximal blocking activity was comparable to bivalent mAb-PD-L1 (Figure 1b). As mbsAb-PD-L1×4-1BB blocked the PD-1/PD-L1 axis also in the absence of 4-1BB binding, the PD-L1-specific Fab arm of mbsAb-PD-L1×4-1BB functioned as a classical immune checkpoint inhibitor. Furthermore, mbsAb-PD-L1×4-1BB activated 4-1BB signaling in reporter cells upon co-culture with IFN-γ pre-stimulated B16F10 cells, which expressed high levels of PD-L1 (Figure 1c and Figure S2). Importantly, 4-1BB activation was not achieved by a combination of PD-L1-specific and 4-1BB specific monovalent control bsAbs. In addition, mbsAb-PD-L1×4-1BB did not activate 4-1BB in co-cultures with B16F10 cells that had not been pre-stimulated with IFN-γ. Therefore, 4-1BB agonist activity of mbsAb-PD-L1×4-1BB was strictly conditional, i.e. dependent upon crosslinking to PD-L1-expressing cells.
To further explore the immunostimulatory effect of mbsAb-PD-L1×4-1BB on primary cells, T-cell proliferation and cytokine production was analyzed in co-cultures of C57BL/6-derived anti-CD3 stimulated CD8+ T cells and autologous BMDCs. mbsAb-PD-L1×4-1BB enhanced proliferation of CD8+ T cells (Figure 1d; Figure S3A), which was associated with increased production of IFN-γ and TNF-α (Figure 1d). A combination of mbsAb-PD-L1×Ctrl and mbsAb-Ctrl×4-1BB was unable to enhance proliferation or cytokine secretion, confirming that the effect of mbsAb-PD-L1×4-1BB was strictly dependent on simultaneous binding to both targets. In addition, mbsAb-PD-L1×4-1BB-induced T-cell proliferation and cytokine production were strictly dependent on prior T-cell stimulation, as no effect was observed in the absence of anti-CD3. Similar to what was previously described in the literature,26–29 bivalent 4-1BB antibodies based on clone 3H3-induced T-cell proliferation and cytokine production both in the presence or absence of Fc-mediated crosslinking, to an extent comparable to mbsAb-PD-L1×4-1BB (Figure S3B). The induction of T-cell proliferation and IFN-γ production by mbsAb-PD-L1×4-1BB was dose-dependent (Figure 1e), with EC50 values of 0.05 ± 0.05 µg/mL (proliferation) and 0.03 µg/mL (IFN-γ), i.e. in the subnanomolar range (0.32 ± 0.34 nM [proliferation] and 0.18 nM [IFN-γ]). Comparable dose-dependent induction of T-cell proliferation and cytokines by mbsAb-PD-L1×4-1BB was observed using T cells derived from BALB/c mice (Figure S3C).
The capacity of mbsAb-PD-L1×4-1BB to enhance T-cell mediated cytotoxicity was tested using co-cultures of ovalbumin (OVA257-264) peptide-stimulated CD8+ OT-I T cells and ovalbumin-expressing B16 (B16_OVA-MO4) tumor cells. mbsAb-PD-L1×4-1BB enhanced antigen-specific T cell-mediated cytotoxicity, which became apparent at 20 h after start of the bsAb treatment and was maintained over time until the termination of the experiment after 5 days (Figure 2a). mbsAb-PD-L1×4-1BB-induced T-cell-mediated cytotoxicity was similar to mAb-PD-L1 or the combination mbsAb-PD-L1×ctrl and mbsAb-ctrl×4-1BB, suggesting that cytotoxicity was enhanced predominantly by PD-L1 checkpoint blockade activity under these experimental conditions. Co-culture with B16-OVA cells alone was sufficient to induce high levels of granzyme B in essentially all OT-I T cells across experimental conditions (Figure S4), indicating an overall strong stimulation in co-culture. However, expression levels of granzyme B (P < .0001) and the degranulation marker CD107a (P < .01) on a per-cell level were significantly increased in mbsAb-PD-L1×4-1BB-treated cells compared to controls that only blocked PD-L1 (Figure 2b). In addition, mbsAb-PD-L1×4-1BB was able to enhance cytokine production in the B16-OVA co-culture setting, as shown by significantly increased intracellular IFN-γ in CD8+ T cells (P < .05, Figure 2c and Figure S4) and increased secretion of IL-2 and TNF-α (Figure 2d) compared to all control groups. Therefore, additional 4-1BB co-stimulation was able to elicit functional changes that may confer enhanced cytotoxicity in the presence of weaker TCR stimuli or a lower frequency of cytotoxic T cells.
Figure 2.
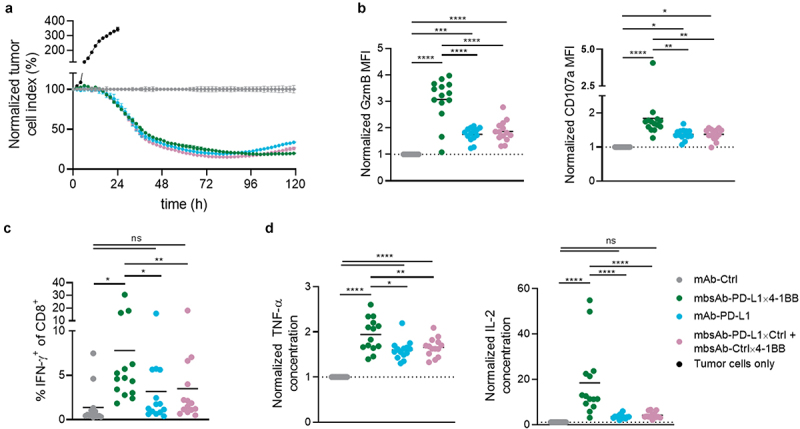
mbsAb-PD-L1×4-1BB enhances antigen-specific T-cell effector functions in vitro.
CD8+ T cells were isolated from OT-I splenocytes by magnetic separation following overnight activation with OVA257-264. The isolated CD8+ T cells were co-cultured with OVA-expressing B16 tumor cells in the presence of mbsAb-PD-L1×4-1BB or the indicated control antibodies for 2–5 days. (a) Cytotoxicity of the CD8+ T cells toward B16-OVA cells was monitored over time using the xCELLigence real-time cell analysis system. Representative data from co-cultures derived from one individual mouse are shown (n = 14). Data were normalized to the time-point of co-culture start, and expressed as relative to the isotype control-treated co-cultures (mAb-Ctrl set to 100%). (b and c) The expression of cytotoxic mediator granzyme B, degranulation marker CD107a and IFN-γ by the CD8+ T cells was assessed by intracellular staining. Pooled data from three experiments (n = 14) are shown. (d) Cytokines measured in supernatant after 48 h with multiplexed ECLIA. Pooled data from three experiments (n = 14) are shown. Data are expressed as relative to mAb-Ctrl. ****, P < .0001; ***, P < .001; **, P < .01; **, P < .05; ns, not significant; One-Way ANOVA with Tukey’s multiple comparisons test.
Taken together, mbsAb-PD-L1×4-1BB dose-dependently enhanced T-cell proliferation and effector functions in vitro. The 4-1BB agonist activity was conditional as it required TCR stimulation and 4-1BB crosslinking by simultaneous binding to PD-L1, whereas PD-L1 blocking activity of mbsAb-PD-L1×4-1BB was also retained in the absence of 4-1BB. Furthermore, mbsAb-PD-L1×4-1BB demonstrated superior immunostimulatory effects compared to PD-L1 blocking agents alone.
mbsAb-PD-L1×4-1BB-treatment did not alter the prevalence of CD8+ T cell or F4/80+ cells in the liver, or liver enzyme levels
Treatment of mice with 4-1BB agonistic mAbs, including clone 3H3 that provided the 4-1BB-specific Fab arm of mbsAb-PD-L1×4-1BB, has previously been associated with liver inflammation, which manifested as elevated transaminase activity and liver immune cell infiltration.26–28,30 Since 4-1BB agonist activity of mbsAb-PD-L1×4-1BB was conditional, we assessed whether liver inflammation was reduced compared to the bivalent agonist 4-1BB antibody. Therefore, immune cells in the liver and liver enzyme activity in the blood were evaluated in healthy C57BL/6 mice after treatment with 250 μg/mouse (approximately 12.5 mg/kg) mbsAb-PD-L1×4-1BB, mAb-PD-L1 or agonistic, bivalent 4-1BB mAbs (three IP injections on days 0, 3, and 6). Compared to isotype control and mAb-PD-L1, mbsAb-PD-L1×4-1BB did not induce any significant increase in lymphocyte, CD8+ T cell or F4/80+ cell density (Figure 3a–c and Figure S5A), liver weight (Figure 3d) or liver enzyme activity in the blood (Figure 3e and Figure S5B) on day 16. By contrast, bivalent agonistic 4-1BB mAbs induced a significant increase in CD8+ T cell density (P < .0001), liver weight (P < .05) and liver enzyme activity (P < .05) compared to mbsAb-PD-L1×4-1BB. The increase in these parameters was further aggravated after combined administration of mAb-PD-L1 together with the bivalent Fc-active 4-1BB mAb (P < .0001 for CD8+ T cell density and liver enzyme activity, P < .01 for liver weight). Transaminase activity levels were similarly increased in agonistic 4-1BB mAb-treated, but not mbsAb-PD-L1×4-1BB-treated mice 8 days after administration, with slightly lower overall ALT and AST levels compared to day 16 (data not shown).
Figure 3.
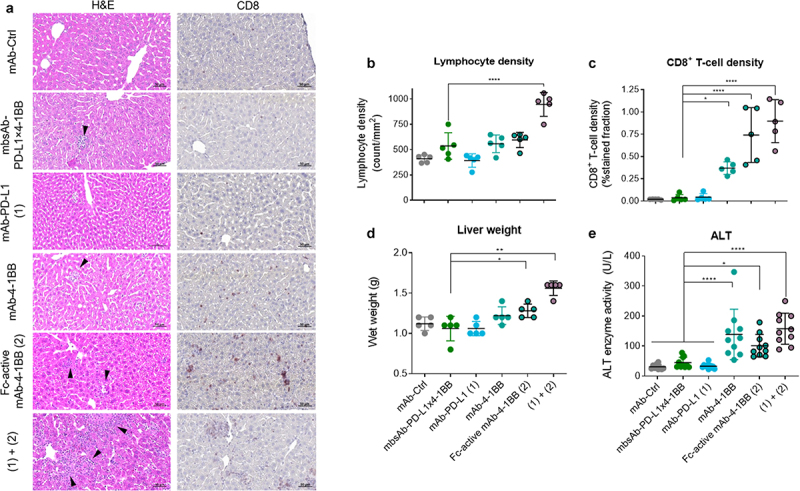
mbsAb-PD-L1×4-1BB does not induce infiltration of CD8+ T cells in the liver, increase liver wet weight or liver enzyme activity in peripheral blood in healthy C57BL/6 mice.
C57BL/6 mice received three IP injections with mbsAb-PD-L1×4-1BB or the indicated control antibodies (250 µg each) on days 0, 3, and 6. (a) Livers were resected on day 16, and sections were stained with H&E (left panels) or using CD8 antibodies (right panels). Representative images are shown. Arrowheads indicate lymphocytic infiltrates. (b) Quantification of lymphocytes in the liver sections (n = 5) (c) Quantification of CD8+ cells in the liver sections. (d) Wet weights of livers 16 days after treatment (n = 5). (e) ALT enzyme activity was measured in sera collected 16 days after the first treatment before the mice were sacrificed. Data from individual mice are shown, as well as group mean ± SD (n = 10). Statistical significance was determined by Ordinary one-way ANOVA with Dunnetts’s multiple comparisons test (treatment groups vs. isotype control). *P < .05; **, P < .01; ***, P < .001; ****, P < .0001.
Given that signs of immune-mediated liver inflammation were not detected in mbsAb-PD-L1×4-1BB-treated mice, we aimed to confirm that mbsAb-PD-L1×4-1BB is pharmacologically active in vivo. To this end, the capacity of mbsAb-PD-L1×4-1BB to expand antigen-specific T cells was determined in C57BL/6 mice that received an adoptive OT-I CD8+ T-cell transfer and were subsequently vaccinated with ovalbumin (OVA). Both 20 and 100 μg/mouse mbsAb-PD-L1×4-1BB significantly enhanced expansion of OVA-specific OT-I CD8+ T cells, compared to monovalent control antibodies targeting either PD-L1 or 4-1BB (P < .001, Figure S6). Importantly, mbsAb-PD-L1×4-1BB-mediated T-cell expansion was similar to that of the agonist mAb-4-1BB with an active Fc region.
Taken together, these findings have shown that while mbsAb-PD-L1×4-1BB treatment did not alter liver immune cell homeostasis or liver enzyme activity in the blood, it promoted the expansion of vaccine epitope-specific CD8+ T cells in vivo.
mbsAb-PD-L1×4-1BB exhibited potent anti-tumor activity in vivo
Given the strong immunostimulatory properties of mbsAb-PD-L1×4-1BB, we next evaluated anti-tumor activity of mbsAb-PD-L1×4-1BB in vivo using murine syngeneic tumor models. We first treated C57BL/6 mice bearing subcutaneous MC38 tumors with mbsAb-PD-L1×4-1BB at 50 or 250 µg/mouse (approximately 2.5 or 12.5 mg/kg) or with mAb-PD-L1 at 250 µg/mouse (twice weekly for two weeks, IP, starting at day 8 after tumor inoculation). mbsAb-PD-L1×4-1BB elicited strong antitumor activity at both dose levels, so that 7/8 mice remained tumor-free until the end of the observation period (Figure 4a and data not shown). mAb-PD-L1 also inhibited tumor growth, with slightly higher mean tumor volumes compared to mbsAb-PD-L1×4-1BB, resulting in 4/8 tumor-free mice. In the isotype control group rapid tumor outgrowth was observed in all animals. Both mbsAb-PD-L1×4-1BB and mAb-PD-L1 significantly increased survival compared to the isotype control (P < .001, Figure 4b). To demonstrate anti-tumor activity against larger tumors, we conducted an additional study where MC38 tumors were allowed to grow until they had reached a median size of 46 mm3 before treatment with mbsAb-PD-L1×4-1BB (50 µg/mouse, twice weekly for 2 weeks, IP) was commenced at day 14 after tumor inoculation. In this setting, mbsAb-PD-L1×4-1BB treatment also significantly inhibited tumor growth (P < .05) and conferred significant survival benefit (P < .01) compared to isotype control treatment (Figure S7).
Figure 4.
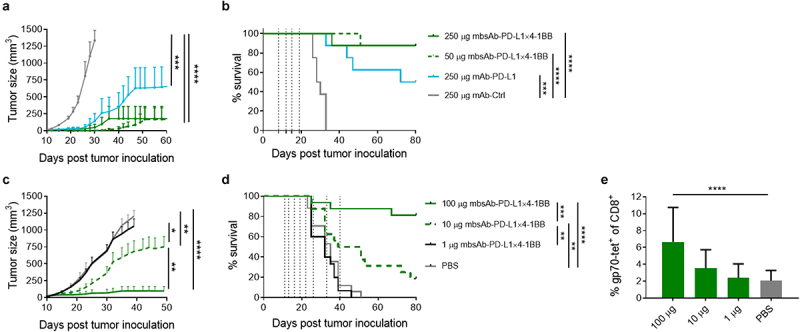
mbsAb-PD-L1×4-1BB exhibits anti-tumor activity.
(a and b) C57BL/6 mice were inoculated SC with 5 × 105 MC38 cells. Groups of mice were administered IP with the indicated doses of mbsAb-PD-L1×4-1BB, mAb-PD-L1 or isotype control antibody on days 8, 12, 15 and 19 after tumor inoculation (vertical dotted lines; median tumor volume at the start of treatment 7.8 mm3). Tumor growth curves shown in (a) depict group mean ± SEM tumor volume over time. Growth curves were discontinued when less than 3 mice remained alive within groups (last observation carried forward [LOCF] ≥ 3). ***, P < .001; ****, P < .0001; Two-way ANOVA with Tukey’s multiple comparisons test. Percent survival is shown in (b). **, P < .01; ***, P < .001; ****, P < .0001; log-rank (Mantel-Cox) test (n = 8). (c and d) BALB/c mice were inoculated SC with 5 × 105 CT26 cells. Groups of mice were administered IP with the indicated doses of mbsAb-PD-L1×4-1BB or PBS only, on days 11, 13, 16, 19, 22, 26, 33 and 40 after tumor inoculation (vertical dotted lines; median tumor volume at the start of treatment 18 mm3). Tumor growth curves shown in (c) depict group mean ± SEM tumor volume over time (LOCF ≥ 3). *, P < .05; **, P < .01; ****, P < .0001. Percent survival is shown in (d). Statistical significance was determined as in (b). (e) The percentage of CD8+ T cells recognizing the tumor-associated gp70 epitope in peripheral blood samples was determined by tetramer staining 18 days after tumor inoculation. ****, P < .0001; One-way ANOVA with Dunnett‘s multiple comparisons test (treatment groups vs. PBS).
In BALB/c mice bearing subcutaneous CT26 tumors, mbsAb-PD-L1×4-1BB significantly inhibited tumor growth (P < .01) at 10 and 100 µg/mouse (approximately 0.5 or 5 mg/kg; twice weekly for 3 weeks followed by weekly for 2 weeks, IP; Figure 4c). Accordingly, a dose-dependent improvement of survival was observed upon treatment with mbsAb-PD-L1×4-1BB (Figure 4d). Anti-tumor activity was associated with a dose-dependent increase in the expansion of CD8+ T cells specific for a gp70 peptide, an immunodominant CT26 tumor antigen, in the peripheral blood. This increase was statistically significant at 100 μg/mouse mbsAb-PD-L1×4-1BB compared to vehicle control (P < .0001, Figure 4e).
To determine whether the anti-tumor activity of mbsAb-PD-L1×4-1BB was conditional, its activity was compared to monospecific controls mbsAb-PD-L1×Ctrl and mbsAb-Ctrl×4-1BB in the CT26 model, either alone or in combination (twice weekly for 3.5 weeks, IP; 100 µg/mouse [approximately 5 mg/kg]). mbsAb-PD-L1×4-1BB significantly inhibited tumor growth compared to the combination of mbsAb-PD-L1×Ctrl and mbsAb-Ctrl×4-1BB (P < .001, Figure 5a). Furthermore, mbsAb-PD-L1×4-1BB treatment significantly improved survival in comparison to treatment with isotype control mAb (P < .05), whereas the combination of monovalent control antibodies did not (Figure 5b).
Figure 5.
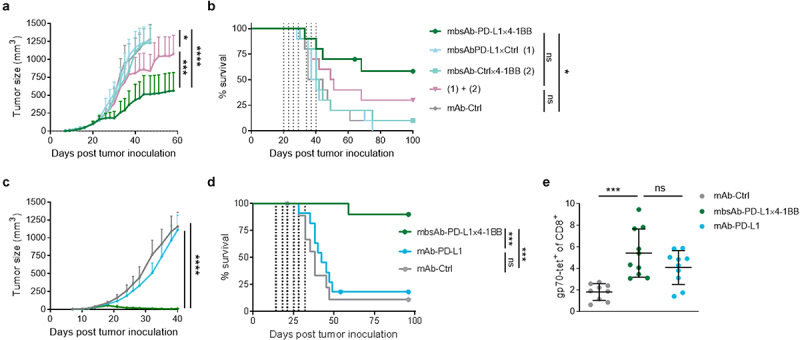
Anti-tumor effect of mbsAb-PD-L1×4-1BB is dependent on conditional agonist activity.
(a and b). BALB/c mice were inoculated SC with 5 × 105 CT26 cells. Groups of mice were administered IP with 100 µg/dose mbsAb-PD-L1×4-1BB, the monovalent control antibodies mbsAb-PD-L1×Ctrl and mbsAb-Ctrl×4-1BB alone or in combination, or isotype control antibody (mAb-Ctrl) on days 20, 23, 26, 29, 34, 37 and 40 after tumor inoculation (vertical dotted lines; median tumor volume at the start of treatment 76.1 mm3). Tumor growth curves shown in (a) depict group mean ± SEM tumor volume over time (LOCF ≥ 3). *, P < .05; ***, P < .001; ****, P < .0001; Two-way ANOVA with Tukey’s multiple comparisons test. Percent survival is shown in (b). *, P < .05; ns, not significant; log-rank (Mantel-Cox) test. (c and d) Groups of CT26 tumor-bearing BALB/c mice (n = 10) were administered IP with 100 µg/dose mbsAb-PD-L1×4-1BB, mAb-PD-L1 or isotype control antibody (mAb-Ctrl), twice weekly for three weeks on days 14, 18, 21, 25, 28, 32 and 40 days after tumor inoculation (vertical dotted lines; median tumor volumes at the start of treatment 28.7 mm3). Tumor growth curves shown in (c) depict group mean ± SEM tumor volume over time (LOCF≥3). ****, P < .0001. Percent survival is shown in (d). ***, P < .001. (e) The percentage of CD8+ T cells recognizing the tumor-associated gp70 epitope in peripheral blood samples of mice treated as in (c and d) was determined by tetramer staining 27 days after tumor inoculation. ***, P < .001; One-way ANOVA with Dunnett’s multiple comparisons test.
Next, the anti-tumor activity of mbsAb-PD-L1×4-1BB was compared to bivalent mAb-PD-L1 in the CT26 model (twice weekly for 3.5 weeks, IP; 100 µg/mouse). Similar to previous experiments in the CT26 model, mbsAb-PD-L1×4-1BB strongly and significantly inhibited tumor growth compared to both mAb-PD-L1 and the isotype control (P < .0001), and led to tumor rejection in the majority of mice (Figure 5c). Furthermore, mbsAb-PD-L1×4-1BB induced a significant increase in survival compared to both the mAb-PD-L1 and isotype control groups (P < .001, Figure 5d). Both mbsAb-PD-L1×4-1BB and mAb-PD-L1 augmented expansion of CD8+ T cells recognizing the immunodominant tumor antigen gp70 in the peripheral blood (Figure 5e). Notably, comparable expansion was observed in both groups, despite the great difference between the two treatments in controlling tumor growth.
Taken together, mbsAb-PD-L1×4-1BB demonstrated strong anti-tumor activity in two murine tumor models. Anti-tumor activity was dose-dependent and required the simultaneous engagement of both targets in vivo. Importantly, mbsAb-PD-L1×4-1BB treatment greatly improved survival even in tumor models where PD-L1 blockade alone was completely ineffective. Furthermore, anti-tumor activity was observed at dose levels that did not alter liver immune cell homeostasis or liver enzyme activity in tumor-free mice.
mbsAb-PD-L1×4-1BB enhanced anti-tumor immunity by modulating immune responses in the tdLNs and the TME
To further characterize the mbsAb-PD-L1×4-1BB-dependent anti-tumor immune responses in vivo, we performed immunophenotyping of the tdLNs and TME in the CT26 model. Within the TME, the proportion of CD8+ T cells was significantly increased in mbsAb-PD-L1×4-1BB-treated mice both compared to groups treated with mAb-PD-L1 and isotype control (P < .001), whereas the proportion of NK cells was reduced in mbsAb-PD-L1×4-1BB-treated mice (Figure 6a and Figure S8A) Intratumoral CD8+ T cells were mostly gp70-specific and expressed PD-1 across treatment groups. As a result, gp70-specific CD8+ T cells were significantly more frequent among hematopoietic cells and among total cells in mbsAb-PD-L1×4-1BB-treated mice compared to the other groups (P < .01, Figure 6a and Figure S8B). Intratumoral gp70-specific CD8+ T cells contained a subset that expressed the transcription factor Tcf-1, which is endowed with a stem cell-like capacity of self-renewal and may mediate extended tumor control.31 The frequency of the Tcf-1+ gp70-specific cells was significantly higher in mbsAb-PD-L1×4-1BB-treated mice compared to the other groups (P < .01, Figure 6a). Together these findings indicated that mbsAb-PD-L1×4-1BB treatment enhanced tumor-specific CD8+ T cell responses. In contrast, a substantially increased frequency of FoxP3+CD25+CD4+ regulatory T cells (Treg) was observed in the TME of mice treated with mAb-PD-L1. As a result, the tumor-specific CD8+ T cell-to-Treg ratio was significantly increased in mbsAb-PD-L1x4-1BB-treated mice compared to other groups (P < .001, Figure 6b and Figure S8C).
Figure 6.
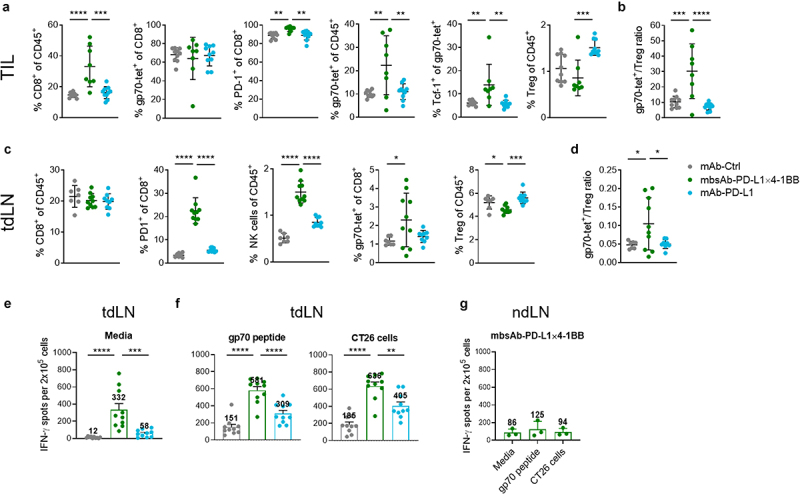
mbsAb-PD-L1×4-1BB modulates the tdLNs and TME in favor of anti-tumor immunity.
BALB/c mice were inoculated SC with 5 × 105 CT26 cells. Groups of mice were administered IP with 100 µg of the indicated antibodies on days 12 and 17 after tumor inoculation (median tumor volume at the start of treatment 37.7 mm3). Mice were sacrificed on day 18 after inoculation for analysis. (a and b) Flow cytometry analysis of enzymatically dissociated tumors. (c and d) Flow cytometry analysis of tdLNs. (e and f) tdLN cell suspensions were either left unstimulated (medium control; e) or restimulated with a gp70 peptide or CT26 cells (f), and IFN-γ secretion was assessed by ELISpot. (g) IFN-γ ELISpot using distant ndLN cell suspensions of mbsAb-PD-L1×4-1BB treated mice. Data from individual mice are shown, as well as group mean (± SD). *, P < .05; **, P < .01; ***, P < .001; ****, P < .0001; One-way ANOVA with Dunnett‘s multiple comparisons test, (treatment groups vs. isotype control group, n = 8–10 [Two mbsAb-PD-L1×4-1BB-treated mice were excluded from analyses shown in (a and b), because their tumors were too small for flow cytometry analysis]).
While the frequency of total CD8+ T cells in the tdLNs was comparable across all treatment groups, proportions of PD-1+ activated CD8+ T cells and NK cells were significantly increased in mbsAb-PD-L1×4-1BB-treated mice compared to both isotype control and mAb-PD-L1 (P < .0001, Figure 6c). Importantly, the frequency of gp70-specific CD8+ T cells was also significantly increased compared to the isotype control group (P < .05). In contrast, Treg proportions were reduced in mbsAb-PD-L1×4-1BB-treated mice, resulting in a significantly increased ratio of gp70-specific CD8+ T cells to Treg (P < .05, Figure 6c and d). Increased proportions of tumor-specific CD8+ T cells and PD-1+ T cells were also detected in the peripheral blood (Figure S8D). IFN-γ production was increased in ex vivo tdLN cell suspensions of mbsAb-PD-L1x4-1BB-treated mice compared to mAb-PD-L1 or isotype control-treated mice (Figure 6e). IFN-γ production was further augmented upon restimulation with the tumor-derived gp70 peptide or CT26 tumor cells, which resulted in the highest proportions of IFN-γ secreting cells in the mbsAb-PD-L1×4-1BB-treated mice (Figure 6f). The spontaneous IFN-γ production is hypothesized to be a consequence of residual tumor antigen presented in the tdLN, as IFN-γ production in distant non-draining lymph nodes (ndLN) was much lower (Figure 6g). Taken together, the mbsAb-PD-L1x4-1BB treatment-induced modulation of lymphocyte populations within the TME was consistent with enhanced tumor-specific CD8+ T-cell responses and a reduction of Treg abundance in the tdLN. These findings suggested that mbsAb-PD-L1×4-1BB treatment led to a pronounced shift within the balance of tumor-reactive CD8+ T cells and immunosuppressive Tregs in favor of anti-tumor immunity.
Discussion
Here we demonstrated PD-L1 blocking and conditional 4-1BB agonist activity of the bispecific antibody mbsAb-PD-L1×4-1BB in multiple in vitro systems and tumor models. mbsAb-PD-L1×4-1BB elicited potent anti-tumor activity, as it was recently described for another Fc-inert mouse PD-L1×4-1BB bsAb.18 We further provide evidence for in vivo activation and expansion of vaccine epitope-specific or tumor antigen-specific T cells as well as a qualitatively improved immune response within the TME and the tdLNs compared to PD-L1 blockade. In addition, mbsAb-PD-L1×4-1BB did not affect liver function as assessed by liver enzyme levels or liver immune cell composition in treated tumor-free mice at an efficacious dose level. Collectively, these findings provided proof-of-concept for dual targeting of PD-L1 and 4-1BB with an Fc-inert bsAb as an efficacious cancer immunotherapy approach that may further present with reduced liver pharmacology, and provided novel insights into the mode-of-action in vivo.
Agents designed to focus 4-1BB agonist activity to the TME typically combine a component with 4-1BB agonist activity and a component that specifically targets tumor-associated antigens.16–18,32 Targeting inhibitory pathways by blocking PD-L1 offers the advantage of conferring dual immunomodulatory activity to the bispecific agent in addition to localizing its activity to the tumor and tdLNs. As such, mbsAb-PD-L1×4-1BB functioned as a checkpoint inhibitor independently of 4-1BB binding. On the other hand, 4-1BB co-stimulation by mbsAb-PD-L1×4-1BB was strictly conditional upon simultaneous binding to PD-L1. By combining these mechanisms, mbsAb-PD-L1×4-1BB enhanced several T-cell functions, including proliferation, cytokine production and cytotoxicity compared to PD-L1 blockade, consistent with the acquisition of a potent and highly activated T-cell effector phenotype.
Combinations of PD-1/PD-L1 and 4-1BB mAbs mediate potent anti-tumor activity in mouse models, including aggressively growing and poorly immunogenic tumors.9–11,13,33 Importantly, the combination treatment conferred significantly and substantially improved survival in models where both treatments as monotherapy are ineffective in previous studies.13,34 These underlying synergies could also be addressed by next-generation bispecific agents. Indeed, here we showed that treatment with mbsAb-PD-L1×4-1BB resulted in the rejection of CT26 tumors, which were insensitive to PD-L1 blockade alone. While this was in line with previous observations,35,36 we also provided novel mechanistic insights that allow for further dissection of the immune responses triggered by simultaneous targeting of both pathways compared to PD-L1 blockade alone. Hence, this bispecific approach is a promising strategy to guide the immune system to attack the tumor, and may have potential in treating cancers resistant to PD-1/PD-L1 blockade.
In agreement with previous studies showing that 4-1BB+ intratumoral CD8+ T cells are enriched for tumor antigen-specific T cells,11,37 mbsAb-PD-L1×4-1BB induced a significant expansion of CD8+ T cells within the TME, the majority of which was tumor-antigen specific. Since PD-L1 blockade alone did not result in a similar increase, this effect required conditional 4-1BB agonist activity and led to a profound shift in the CD8+ T cell-to-Treg ratio, which may have contributed to the potency of mbsAb-PD-L1×4-1BB in the PD-L1 blockade-insensitive CT26 model. While the precise mechanism causing this shift remains unclear, it may be attributable to a direct effect of mbsAb-PD-L1×4-1BB on Tregs, as Tregs were reported to express both PD-1 and 4-1BB.38–40 Alternatively, reduced Treg numbers may be an indirect effect of a pro-inflammatory TME, as high local IFN-γ concentrations may impair Treg stability.41 In addition, both PD-1/PD-L1 blockade and 4-1BB co-stimulation were shown to inhibit differentiation of conventional T cells into adaptive Tregs,42,43 which could therefore be effectively achieved by simultaneous modulation of both pathways in vivo.
Our immunophenotyping data further showed that mbsAb-PD-L1×4-1BB treatment exhibited effects beyond the TME, since modulation of CD8+ T cell and Treg-prevalence in tdLNs was consistent with the changes in TME immune composition. It is conceivable that mbsAb-PD-L1×4-1BB supported the expansion of recently primed T cells in the tdLNs, which were specific for tumor antigens released as a consequence of mbsAb-PD-L1×4-1BB-induced TME inflammation and tumor cell lysis. PD-L1 upregulation by DCs in tdLNs has been demonstrated in several murine tumor models,44 suggesting that crosslinking with recently primed 4-1BB+ T cells could occur in the tdLN. Our observation that mbsAb-PD-L1×4-1BB boosted the expansion of vaccine epitope-specific T cells in tumor-free mice further supported the idea that PD-L1 expressing cells in lymphoid tissue are able to support target crosslinking and thereby augment antigen-specific T-cell responses. Importantly, the expansion of antigen-specific T cells required conditional 4-1BB agonist activity in both settings, since PD-L1 blocking antibodies only had a modest effect.
While compelling evidence has validated 4-1BB as a therapeutic target, clinical development of 4-1BB mAbs was hampered by the occurrence of severe hepatotoxicity at efficacious doses.32 Murine studies have demonstrated that toxicity in 4-1BB mAb-treated animals is dependent on T cells and 4-1BB expression, and that activation of liver-resident 4-1BB+ T cells may critically contribute to the inflammatory cascade that leads to liver damage.45,46 In line with previous studies,26–28,30 the parental anti-4-1BB mAb clone 3H3 in a bivalent format induced elevated transaminase levels and increased intrahepatic CD8+ T-cell numbers, which was further aggravated by combination treatment with mAb-PD-L1. Notably, comparable liver inflammation was observed in mice treated with Fc-inert or Fc-active bivalent 3H3, indicating that agents with strong intrinsic 4-1BB agonist activity may induce liver pathology through high target-binding avidity, without the requirement for Fc receptor-mediated crosslinking.28 Although the 4-1BB-specific Fab arm of mbsAb-PD-L1×4-1BB was derived from clone 3H3, the 4-1BB agonist activity is directed to the TME and tdLNs by its dependence on crosslinking to PD-L1. As a consequence, mbsAb-PD-L1×4-1BB treatment did not evoke manifestations of hepatic inflammation such as elevated serum transaminase activity levels or CD8+ T-cell infiltration of the liver, at dose levels that provided anti-tumor activity in vivo.
In conclusion, through its unique MoA, mbsAb-PD-L1×4-1BB combined PD-1/PD-L1 immune checkpoint blockade and conditional 4-1BB co-stimulation to boost CD8+ T-cell activation and effector functions in vitro. Furthermore, we provided proof-of-concept that dual targeting of PD-L1 and 4-1BB using Fc-inert bispecific antibodies can enhance tumor antigen-specific CD8+ T-cell responses while reducing the prevalence of immunosuppressive cells, resulting in potent antitumor activity in murine tumor models, including those unresponsive to PD-L1 blockade. Our combined targeted approach of a bispecific antibody may have the potential to improve the clinical benefit observed with anti-PD-1/PD-L1 therapy. These findings supported the development of a clinical candidate, GEN1046 (BNT311/DuoBody-PD-L1x4-1BB), which is currently being evaluated in the clinical setting for the treatment of advanced solid tumors (NCT03917381).
Supplementary Material
Acknowledgments
The authors would like to thank Paul Parren for his contributions to conceptualizing the PD-L1x4-1BB bsAb format; Nele Brüne, Yvonne Feuchter, Bernadette Jesionek, Julia Mühl, and Ann-Kathrin Wallisch for technical expertise and support on animal studies and in vitro experiments; Gilbert van den Tillaart and René Vӧlker-van der Meijden for technical support on the BLI experiments.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
U.S. and Ö.T. are management board members and employees at BioNTech SE (Mainz, Germany); A.M., F.G., K.S., A.T., D.E., L.M.K., M.V., M.D. and S.K. are employees at BioNTech SE. Some of the authors have securities from BioNTech SE. I.A., S.M.B., T.W.S., D.V., D.P.E.S., K.S., T.A., E.C.W.B., and M.J. are employees at Genmab and own stock and/or stock options. I.A., D.P.E.S., U.S., A.M., F.G. and C.G. are inventors on patents and patent applications related to PD-L1×4-1BB bispecific antibodies.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Gong J, Chehrazi-Raffle A, Reddi S, Salgia R.. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8. doi: 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Betof Warner A, Palmer JS, Shoushtari AN, Goldman DA, Panageas KS, Hayes SA, Bajwa R, Momtaz P, Callahan MK, Wolchok JD, et al. Long-term outcomes and responses to retreatment in patients with melanoma treated with PD-1 blockade. J Clin Oncol. 2020;38(15):1655–13. doi: 10.1200/JCO.19.01464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snell LM, Lin GH, McPherson AJ, Moraes TJ, Watts TH. T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy. Immunol Rev. 2011;244(1):197–217. doi: 10.1111/j.1600-065X.2011.01063.x. [DOI] [PubMed] [Google Scholar]
- 4.Bartkowiak T, Curran MA. 4-1BB agonists: multi-potent potentiators of tumor immunity. Front Oncol. 2015;5:117. doi: 10.3389/fonc.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, Sen D, Nager AR, Oakdale NS, Brown C, et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun. 2018;9(1):4679. doi: 10.1038/s41467-018-07136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. 2018;131(1):49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 7.Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, Schmidt H, Robert C, Chiarion-Sileni V, Ascierto PA, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res. 2017;23(8):1929–1936. doi: 10.1158/1078-0432.CCR-16-1272. [DOI] [PubMed] [Google Scholar]
- 8.Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010;37(5):508–516. doi: 10.1053/j.seminoncol.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Azpilikueta A, Agorreta J, Labiano S, Perez-Gracia JL, Sanchez-Paulete AR, Aznar MA, Ajona D, Gil-Bazo I, Larrayoz M, Teijeira A, et al. Successful immunotherapy against a transplantable mouse squamous lung carcinoma with anti-PD-1 and anti-CD137 monoclonal antibodies. J Thorac Oncol. 2016;11(4):524–536. doi: 10.1016/j.jtho.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Lee L-F, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3(2):149–160. doi: 10.1158/2326-6066.CIR-14-0118. [DOI] [PubMed] [Google Scholar]
- 11.Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8(+) T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol Res. 2018;6(1):14–24. doi: 10.1158/2326-6066.CIR-17-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morales-Kastresana A, Sanmamed MF, Rodriguez I, Palazon A, Martinez-Forero I, Labiano S, Hervas-Stubbs S, Sangro B, Ochoa C, Rouzaut A, et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin Cancer Res. 2013;19(22):6151–6162. doi: 10.1158/1078-0432.CCR-13-1189. [DOI] [PubMed] [Google Scholar]
- 13.Qu QX, Zhu XY, Du WW, Wang HB, Shen Y, Zhu YB, Chen C. 4-1BB agonism combined with PD-L1 blockade increases the number of tissue-resident CD8+ T cells and facilitates tumor abrogation. Front Immunol. 2020;11:577. doi: 10.3389/fimmu.2020.00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atkins PW, Thompson DM. Combination avelumab and utomilumab immunotherapy can induce diabetic ketoacidosis. Diabetes Metab. 2018;44(6):514–515. doi: 10.1016/j.diabet.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, Di Gravio D, Huang B, Gambhire D, Chen Y. Phase Ib study of utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res. 2017;23(18):5349–5357. doi: 10.1158/1078-0432.CCR-17-1243. [DOI] [PubMed] [Google Scholar]
- 16.Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, Albrecht R, Herter S, Schlenker R, Hüsser T, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. 2019;11(496). doi: 10.1126/scitranslmed.aav5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinner MJ, Aiba RSB, Jaquin TJ, Berger S, Durr MC, Schlosser C, Allersdorfer A, Wiedenmann A, Matschiner G, Schüler J, et al. Tumor-localized costimulatory T-cell engagement by the 4-1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin Cancer Res. 2019;25(19):5878–5889. doi: 10.1158/1078-0432.CCR-18-3654. [DOI] [PubMed] [Google Scholar]
- 18.Lakins MA, Koers A, Giambalvo R, Munoz-Olaya J, Hughes R, Goodman E, Marshall S, Wollerton F, Batey S, Gliddon D, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res. 2020;26(15):4154–4167. doi: 10.1158/1078-0432.CCR-19-2958. [DOI] [PubMed] [Google Scholar]
- 19.Jeong S, Park E, Kim HD, Sung E, Kim H, Jeon J, Kim Y, Jung U-J, Son Y-G, Hong Y, et al. Novel anti-4-1BBxPD-L1 bispecific antibody augments anti-tumor immunity through tumor-directed T-cell activation and checkpoint blockade. J Immunother Cancer. 2021;9(7):e002428. doi: 10.1136/jitc-2021-002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai T, Wang C, Xu Y, Huang W, Yuan Z, Wang T, Dai S, Peng S, Pang T, Jiang W, et al. Generation of a safe and efficacious llama single-domain antibody fragment (vHH) targeting the membrane-proximal region of 4-1BB for engineering therapeutic bispecific antibodies for cancer. J Immunother Cancer. 2021;9(6):e002131. doi: 10.1136/jitc-2020-002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geuijen C, Tacken P, Wang LC, Klooster R, van Loo PF, Zhou J, Mondal A, Liu Y-B, Kramer A, Condamine T, et al. A human CD137xPD-L1 bispecific antibody promotes anti-tumor immunity via context-dependent T cell costimulation and checkpoint blockade. Nat Commun. 2021;12(1):4445. doi: 10.1038/s41467-021-24767-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labrijn AF, Meesters JI, Bunce M, Armstrong AA, Somani S, Nesspor TC, Chiu ML, Altintaş I, Verploegen S, Schuurman J, et al. Efficient generation of bispecific murine antibodies for pre-clinical investigations in syngeneic Rodent models. Sci Rep. 2017;7(1):2476. doi: 10.1038/s41598-017-02823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arduin E, Arora S, Bamert PR, Kuiper T, Popp S, Geisse S, Grau R, Calzascia T, Zenke G, Kovarik J, et al. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol Immunol. 2015;63(2):456–463. doi: 10.1016/j.molimm.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Barbas CF 3rd, Collet TA, Amberg W, Roben P, Binley JM, Hoekstra D, Cababa D, Jones TM, Williamson RA, Pilkington GR, et al. Molecular profile of an antibody response to HIV-1 as probed by combinatorial libraries. J Mol Biol. 1993;230(3):812–823. doi: 10.1006/jmbi.1993.1203. [DOI] [PubMed] [Google Scholar]
- 25.Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13(4):529–538. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 26.Eskiocak U, Guzman W, Wolf B, Cummings C, Milling L, Wu H-J, Ophir M, Lambden C, Bakhru P, Gilmore DC. Differentiated agonistic antibody targeting CD137 eradicates large tumors without hepatotoxicity. JCI Insight. 2020;5(5). doi: 10.1172/jci.insight.133647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho SK, Xu Z, Thakur A, Fox M, Tan SS, DiGiammarino E, Zhou L, Sho M, Cairns B, Zhao V, et al. Epitope and Fc-mediated cross-linking, but not high affinity, are critical for antitumor activity of CD137 agonist antibody with reduced liver toxicity. Mol Cancer Ther. 2020;19(4):1040–1051. doi: 10.1158/1535-7163.MCT-19-0608. [DOI] [PubMed] [Google Scholar]
- 28.Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, Yang X. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity. Nat Commun. 2019;10(1):2141. doi: 10.1038/s41467-019-10088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, Brown TJ, Emswiler J, Raecho H, Larsen CP. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186(1):47–55. doi: 10.1084/jem.186.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartkowiak T, Jaiswal AR, Ager CR, Chin R, Chen CH, Budhani P, Ai M, Reilley MJ, Sebastian MM, Hong DS, et al. Activation of 4-1BB on liver myeloid cells triggers hepatitis via an interleukin-27-dependent pathway. Clin Cancer Res. 2018;24(5):1138–1151. doi: 10.1158/1078-0432.CCR-17-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.PMID: 30635237 ; doi: 10.1016/j.immuni.2018.12.021. [DOI]
- 32.Etxeberria I, Glez-Vaz J, Teijeira A, Melero I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open. 2020;4(Suppl3). doi: 10.1136/esmoopen-2020-000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, Delgoffe GM. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215(4):1091–10100. doi: 10.1084/jem.20171068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei H, Zhao L, Li W, Fan K, Qian W, Hou S, Wang H, Dai M, Hellstrom I, Hellstrom KE. Combinatorial PD-1 blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin. PLoS One. 2013;8(12):e84927. doi: 10.1371/journal.pone.0084927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis KE, Selby MJ, Masters G, Valle J, Dito G, Curtis WR, Garcia R, Mink KA, Waggie KS, Holdren MS, et al. Interleukin-21 combined with PD-1 or CTLA-4 blockade enhances antitumor immunity in mouse tumor models. Oncoimmunology. 2017;7(1):e1377873. doi: 10.1080/2162402X.2017.1377873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, Coffman RL, Guiducci C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A. 2016;113(46):E7240–E9. doi: 10.1073/pnas.1608555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME. PD-1 identifies the patient-specific CD8+tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, Kochel CM, Lan RY, Zhai J, Ghasemzadeh A, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest. 2020;130(3):1405–1416. doi: 10.1172/JCI128672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov D, Rudensky A. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122–1134. doi: 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toker A, Nguyen LT, Stone SC, Yang SYC, Katz SR, Shaw PA, Clarke BA, Ghazarian D, Al-Habeeb A, Easson A. Regulatory T cells in ovarian cancer are characterized by a highly activated phenotype distinct from that in melanoma. Clin Cancer Res. 2018;24(22):5685–5696. doi: 10.1158/1078-0432.CCR-18-0554. [DOI] [PubMed] [Google Scholar]
- 41.Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. Interferon-gamma drives treg fragility to promote anti-tumor immunity. Cell. 2017;169(6):1130–41 e11. doi: 10.1016/j.cell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105(27):9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madireddi S, Schabowsky RH, Srivastava AK, Sharma RK, Yolcu ES, Shirwan H. SA-4-1BBL costimulation inhibits conversion of conventional CD4+ T cells into CD4+ FoxP3+ T regulatory cells by production of IFN-gamma. PLoS One. 2012;7(8):e42459. doi: 10.1371/journal.pone.0042459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. 2018;128(2):805–815. doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubrot J, Milheiro F, Alfaro C, Palazon A, Martinez-Forero I, Perez-Gracia JL, Morales-Kastresana A, Romero-Trevejo JL, Ochoa MC, Hervás-Stubbs S, et al. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol Immunother. 2010;59(8):1223–1233. doi: 10.1007/s00262-010-0846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin GH, Snell LM, Wortzman ME, Clouthier DL, Watts TH. GITR-dependent regulation of 4-1BB expression: implications for T cell memory and anti-4-1BB-induced pathology. J Immunol. 2013;190(9):4627–4639. doi: 10.4049/jimmunol.1201854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.