

ABSTRACT
Osteoarthritis is a degenerative joint disease and a leading cause of adult disability. Our previous study has reported that mesenchymal stem cell-derived exosomes (MSC-Exo) mediated long non-coding RNA KLF3-AS1 improves osteoarthritis. This study aims to investigate the molecular mechanism of KLF3-AS1 in osteoarthritis. Chondrocytes were treated with IL-1β to induce chondrocyte injury, followed by MSC-Exo treatment. We found that MSC-Exo enhanced KLF3-AS1 expression in IL-1β-treated chondrocytes. IL-1β treatment reduced cell viability and enhanced apoptosis in chondrocytes. MSC-Exo-mediated KLF3-AS1 promoted cell viability and repressed apoptosis of IL-1β-treated chondrocytes. Rapamycin (autophagy activator) promoted cell viability and suppressed apoptosis of chondrocytes by activating autophagy. Moreover, KLF3-AS1 interacted with YBX1 in chondrocytes. MSC-Exo-mediated KLF3-AS1 activated PI3K/Akt/mTOR signaling pathway, which was abrogated by YBX1 silencing. MSC-Exo-mediated KLF3-AS1 repressed autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway. In conclusion, our data demonstrate that MSC-Exo-mediated KLF3-AS1 inhibits autophagy and apoptosis of IL-1β-treated chondrocyte through PI3K/Akt/mTOR signaling pathway. KLF3-AS1 activates PI3K/Akt/mTOR signaling pathway by targeting YBX1 to improve the progression of osteoarthritis. Thus, this work suggests that MSC-Exo-mediated KLF3-AS1 may be a potential therapeutic target for osteoarthritis.
KEYWORDS: Mesenchymal stem cell, exosome, KLF3-AS1, YBX1, autophagy, apoptosis, OA, PI3K/Akt/mTOR
Introduction
Osteoarthritis (OA) is a chronic disabling disease characterized by articular cartilage degeneration, cartilage tissue destruction, and bone hyperplasia around the joints. Articular cartilage is an important part of synovial joints. Once the articular cartilage is damaged, it is difficult to repair, and even cause serious dysfunction and OA [1]. With the rapid development of tissue engineering in recent years, the stem cell tissue engineering to repair the cartilage damage has become a research focus. Mesenchymal stem cells (MSCs) have the potential for self-replication and multi-directional differentiation. Thus, MSCs have become the most studied and widely used seed cells in the construction of cartilage in tissue engineering [2]. The efficacy of autologous or allogeneic MSCs in cartilage repair has been confirmed in animal study and human clinical trial [3,4].
MSCs-derived exosomes (MSC-Exo) are extracellular vesicles secreted by stem cells under conditions such as resting or hypoxic stress, irradiation, and oxidative damage. Exosomes can act as signaling molecules for stem cells and differentiated cells by selectively transporting proteins, liposomes, mRNAs, and microRNAs [5,6]. Previous study has confirmed that the interaction between MSC-Exo and nucleus pulposus cells significantly promotes the repair of degenerated intervertebral disc [7]. In the cartilage defect rat model, MSC-Exo accelerates the filling of new tissues and synthesis of type II collagen, and thus promotes cartilage regeneration [8]. MSC-Exo participates in cartilage tissue repair in OA rats, which attributes to promotes proliferation and invasion, and represses apoptosis of chondrocytes [9,10]. Our previous studies have confirmed that MSC-Exo reduces damage and apoptosis of chondrocytes [11,12]. Long non-coding RNA (lncRNA) KLF3-AS1 in MSC-Exo acts as a competing endogenous RNA (ceRNA) to alleviate OA by regulating miR-206/GIT1 axis [12]. ceRNA is a type of RNA that contains miRNA binding sites, which can competitively bind to miRNA and inhibit the regulation of miRNA on target genes [13]. Thus, KLF3-AS1 may competitively bind to miR-206 and relieve the inhibitory effect of miR-206 on its target gene, GIT1.
Bioinformatics software (http://rbpdb.ccbr.utoronto.ca/) reveals that Y-box binding protein-1 (YBX1) is a target protein of KLF3-AS1 (binding site: UCUGCG). YBX1 is a type of transcription factor that specifically binds to the internal Y-box sequence in the promoter and enhancer of target genes, which is widely present in various species. YBX1, as a MIA/CD-RAP-dependent transcription factor, is closely related to cartilage formation [14]. PI3K/AKT signaling pathway plays a vital role in regulating cell growth, maintaining cell viability and cell lifespan [15]. PI3K/AKT signaling pathway also participates in regulating proliferation, apoptosis and autophagy of chondrocytes [16,17]. YBX1 binds to PI3K promoter to regulate its transcription levels [18]. YBX1 activates PI3K/AKT signaling pathway to participate in regulating tumor progression, such as cutaneous squamous cell carcinomas and nonsmall cell lung cancer [19–21]. Thus, YBX1 may affect proliferation, apoptosis and autophagy of chondrocytes by activating PI3K/AKT signaling pathway.
Autophagy has been reported to be a protective mechanism in normal cartilage to avoid cell death caused by aging and trauma [22]. The compensatory mechanism of chondrocyte autophagy may be one of the pathogenesis of OA. Autophagy is a cell homeostasis mechanism that can eliminate the dysfunctional organelles and macromolecules. The enhancement of autophagy may be a new way to delay joint degeneration and attenuate OA progression. Almontebecerril et al. have confirmed that OA causes autophagy and apoptosis of chondrocytes [23]. Therefore, autophagy is closely associated with articular cartilage repair. As a classic pathway to regulate autophagy, PI3K/Akt/mTOR signaling pathway has a crucial role in the progression of OA [24]. Therefore, we speculate that MSC-Exo-mediated lncRNA KLF3-AS1 inhibits chondrocyte autophagy and reduces chondrocyte apoptosis by activating PI3K/Akt/mTOR signaling pathway. LncRNA KLF3-AS1 activates PI3K/Akt/mTOR signaling pathway by targeting YBX1 to repair cartilage tissue damage.
Materials and Methods
Isolation of chondrocytes
C57BL/6 neonatal mice (5–6 d) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Mice were housed under specific pathogen free condition. C57BL/6 mice were euthanized by cervical dislocation. Cartilage was isolated from caput femoris, femoral condyle and tibial plateau of C57BL/6 mice under sterile condition. Cartilage was cut into pieces (1–2 mm3) with ophthalmic scissors, and then digested with collagenase II (Roche, Basel, Switzerland) at 37°C for overnight. After that, cells were filtered with nylon cell strainer (100 µm porosity) to remove non-digested cartilage. The collected mouse articular chondrocytes were cultured for further use. All protocols were authorized by the Ethics Committee of Luhe People’s Hospital of Nanjing (No. 2,010,034).
Cell culture
Human MSCs were purchased from ATCC (Manassas, VA, USA). Human MSCs and chondrocytes were cultured in Dulbecco’s modified Eagles medium/Nutrient Mixture F-12 (DMEM/F12) (Gibco, Carlsbad, CA, USA) at 37°C and 5% CO2. The medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Procell, Wuhan, China).
Isolation of exosomes
Exosomes were extracted from the culture supernatant of MSCs (MSC-Exo) using Total Exosome Isolation Reagent (from cell culture media) (Invitrogen) as the introduction described. Briefly, MSCs were cultured in exosome-free medium at 37°C and 5% CO2 for 48 h. After that, cell media were collected by centrifugation at 2000 × g for 30 min. Then, the harvested cell media (10 mL) were incubated with the total exosome isolation reagent (5 mL) at 4°C for overnight. The pellets were precipitated by centrifugation at 10,000 × g for 60 min at 4°C. Finally, the pellets were resuspended in PBS and stored at −20°C for further study.
Cell treatment
Chondrocytes were cultured in DMEM/F12 containing 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2. Chondrocytes were treated with 10 mg/mL recombinant IL-1β (Sigma-Aldrich, St. Louis, MO, USA) for 24 h to induce OA cell model as previous reported [25]. IL-1β-induced chondrocytes were cultured in the DMEM/F12 medium containing MSC-Exo (10 μg/mL) or PBS for 24 h. Chondrocytes were cultured in the DMEM/F12 medium containing 100 nM rapamycin (autophagy activator; Sigma-Aldrich) or DMSO (Sigma-Aldrich) for 48 h.
Cell transfection
KLF3-AS1 was subcloned into the vector pcDNA3.1 (pcDNA3.1-KLF3-AS1) (GenePharma, Shanghai, China). The empty vector (pcDNA3.1-NC) served as negative control (NC). Small interference RNA (siRNA) specifically targeting KLF3-AS1 (Si-KLF3-AS1; 5ʹ-GCCUCUUCUUAACUUUGAUTT-3ʹ) or YBX1 (si-YBX1; 5ʹ-GGAGUUUGAUGUUGUUGAAGG-3ʹ), and the corresponding control (Scramble; 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ) were purchased from GenePharma. MSCs or chondrocytes were cultured in a 24-well plate until 50–60% confluence of cell density. Cells were transfected with 0.8 μg plasmids using 2 μL Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) as the protocol described. After 6 to 8 h of incubation, the transfected cells were cultured in complete medium at 37°C and 5% CO2. After 48 h of cultivation, the transfected cells were collected and stored at −20°C for further use.
Quantitative real-time PCR (qRT-PCR)
QRT-PCR was used to measure the gene expression in exosomes and chondrocytes. Total RNA was extracted from chondrocytes using TRIzol reagent (Invitrogen). Total RNA was extracted from exosomes using Total Exosome RNA and Protein Isolation Kit (Invitrogen). The absorbance of RNA at 260 nm/280 nm was examined to assess RNA purity on a NanoDrop 2000 c spectrophotometer (Thermo Scientific, Waltham, MA, USA). 1.5% agarose gel electrophoresis was performed to estimate the integrity of RNA. RNA (500 ng) was reverse transcribed into cDNA using PrimeScript™ RT reagent Kit (Takara, Tokyo, Japan). After that, the cDNA product was amplified by real-time PCR using TB Green® Premix Ex Taq™ II (Takara). The purity and concentrations of cDNA were quantified on a NanoDrop 2000 c spectrophotometer. PCR reaction system (50 μL) contained 25 μL Ex Taq™ II, 2 μL Primer-F, 2 μL Primer-R, 1 μL ROX Reference Dye, 4 μL DNA template and 16 μL sterile water. PCR conditions were shown as follows: initial denaturation at 95°C for 3 min, followed by 40 cycles of 95°C for 5 sec, 60°C for 30 sec and 72°C for 30 sec, followed by a final extension of 72°C for 5 min. PCR reaction was carried out on an ABI 7300 Real-Time PCR System ((Applied Biosystems, Foster City, CA, USA). The primer sequence was shown as follows: KLF3-AS1: F: 5ʹ-AATGAGTGCGTGGAGGAAAT-3ʹ, R: 5ʹ- CCTGGGCAACAGAGTGAGAC-3ʹ; GAPDH (housekeeping gene): F: 5ʹ-AATGGATTTGGACGCATTGGT-3ʹ; R: 5ʹ-TTTGCACTGGTACGTGTTGAT-3ʹ. The relative expression of the target gene was analyzed using 2−∆∆CT method.
Cell viability
CCK-8 assay was carried out to detect cell viability of chondrocytes applying Cell Counting Kit-8 (Beyotime, Shanghai, China). Chondrocytes were seeded into 96-well plate and cultured at 37°C for 48 h. Then, 100 μL chondrocytes were mixed with 10 μL CCK-8 reagent and incubated at 37°C for 1 h. The absorbance of each well was measured at the wavelength of 450 nm.
Cell apoptosis
Apoptosis of chondrocytes was examined using Annexin V-FITC/PI Apoptosis Detection Kit (YEASEN, Shanghai, China) as the instruction described. Chondrocytes were collected by centrifugation and washed with PBS buffer for 2 times. Chondrocytes were mixed with 100 μL 1 × Binding Buffer. The cell suspension was incubated with 5 μL of Annexin V-FITC and 10 μL of PI Staining Solution at darkness for 15 min. Then, the cell suspension was mixed with 400 μL of 1 × Binding Buffer and put on ice. Cell apoptosis was determined by flow cytometry utilizing FACS Calibur instrument (BD Biosciences, San Jose, CA, USA) in an hour.
Western blot (WB)
Total protein was extracted from chondrocytes using Total Protein Extraction Kit (Solarbio, Beijing, China). BCA Protein Assay Kit (Solarbio) was used to assess the concentration of proteins. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred onto the polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA). After that, the membranes were blocked with 5% fat-free milk at room temperature for 2 h. The membranes were then incubated with the primary antibodies, LC3 (1:1000, Proteintech, Wuhan, China), p62 (1:1000, Proteintech), P13 K (1:1000, Abcam), p-AKT (1:2000, Proteintech), Akt (1:1000, Proteintech), p-mTOR (1:1000, Abcam, Cambridge, MA, USA) or mTOR (1:5000, Proteintech) at 4°C for 12 h. Then, the membranes were washed with Tris Buffered saline Tween for several times. Horseradish peroxidase-conjugated second antibody (1:5000, Proteintech) was incubated with the membranes. β-actin antibody (1:5000, Proteintech) was used as a reference protein for normalization. Data were analyzed by Image J software.
Autophagy analysis
Chondrocytes were transfected with 2 µg pcDNA3.1-GFP-LC3 plasmid (HonorGene, Changsha, China) using Lipofectamine 2000. After 48 h of transfection, the autophagy of chondrocytes was observed under an inverted fluorescence microscope (Olympus, Japan, China).
RNA pull-down
The interaction between KLF3-AS1 and YBX1 was verified by RNA pull-down assay using Pierce™ Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific, Waltham, MA, USA). Chondrocytes were lysed by lysis buffer. Then, the target RNA was labeled with Pierce RNA 3ʹ End Desthiobiotinylation Kit. The 3ʹ end desthiobiotinylation-labeled RNA was captured with streptavidin magnetic beads in the chondrocyte lysate. Negative RNA Control [poly(A)25 RNA] served as negative control. Magnetic beads-RNA was incubated with total proteins. After RNA-binding protein complexes was eluted. The cell lysate was used as Input group. Proteins samples were eluted from the RNA-binding protein complexes. Finally, the eluted proteins were analyzed by WB.
RNA binding protein immunoprecipitation (RIP)
The interaction between KLF3-AS1 and YBX1 was verified by RIP assay using Millipore Magna RIP Kit (Merck Millipore). Chondrocytes were lysed by lysis buffer and then incubated with anti-YBX1 (1:1000, Proteintech) or anti-IgG (1:2000, Proteintech) at 4°C for 12 h. Then, a protein-RNA complex was obtained after capturing intracellular specific proteins by the antibody. Proteinase K was used to digest the protein-RNA complex, and the RNAs were collected. Subsequently, the magnetic beads were washed repeatedly with RIP washing buffer to remove the nonspecific adsorption. Finally, qRT-PCR was performed to quantify the immunoprecipitant RNAs (KLF3-AS1).
Statistical analysis
All assays were conducted in triplicate. Measurement data were reported as mean ± standard deviation. SPSS 22.0 statistical software (IBM, Armonk, NY, USA) was used for statistical analysis. Shapiro–Wilk test was used to measure the normality of data. Two-tailed Student’s t test and one-way ANOVA were used to analyze the statistical difference. Least Significance Difference (LSD) test was used for post hoc analysis. P < 0.05 was considered as a significant difference.
Results
MSC-Exo-mediated KLF3-AS1 represses IL-1β-induced chondrocyte injury
In order to determine the biological role of KLF3-AS1 in the progression of OA, we silenced KLF3-AS1 in MSCs by cell transfection. Exosomes were extracted from the KLF3-AS1-silenced MSCs, and the expression of KLF3-AS1 in the exosomes was examined by qRT-PCR. The qRT-PCR data showed that MSCsi-KLF3-AS1-Exo exhibited a down-regulation of KLF3-AS1 (Figure 1(a)). Subsequently, we isolated chondrocytes from C57BL/6 neonatal mice (Supplementary Figure 1), and chondrocytes were treated with IL-1β to induce chondrocyte injury. To explore the effect of MSC-Exo on cartilage injury, the IL-1β-treated chondrocytes were treated with MSC-Exo or MSCsi-KLF3-AS1-Exo. As shown in Figure 1(b), chondrocytes displayed an up-regulation of KLF3-AS1 in the presence of MSC-Exo. Compared with MSCScramble-Exo, MSCsi-KLF3-AS1-Exo treatment led to a decrease of KLF3-AS1 expression in the chondrocytes (Figure 1(b)). Moreover, the results obtained form CCK-8 assay and flow cytometry uncovered that IL-1β treatment repressed cell viability and enhanced apoptosis of chondrocytes. Cell viability of chondrocytes was enhanced, and apoptosis of chondrocytes was reduced by the treatment of MSC-Exo. In contrast, following MSCsi-KLF3-AS1-Exo treatment, chondrocytes exhibited a decrease of cell viability and an increase of apoptosis (Figure 1(c-d)). Thus, these findings suggested that MSC-Exo-mediated KLF3-AS1 repressed IL-1β-induced chondrocyte injury.
Figure 1.
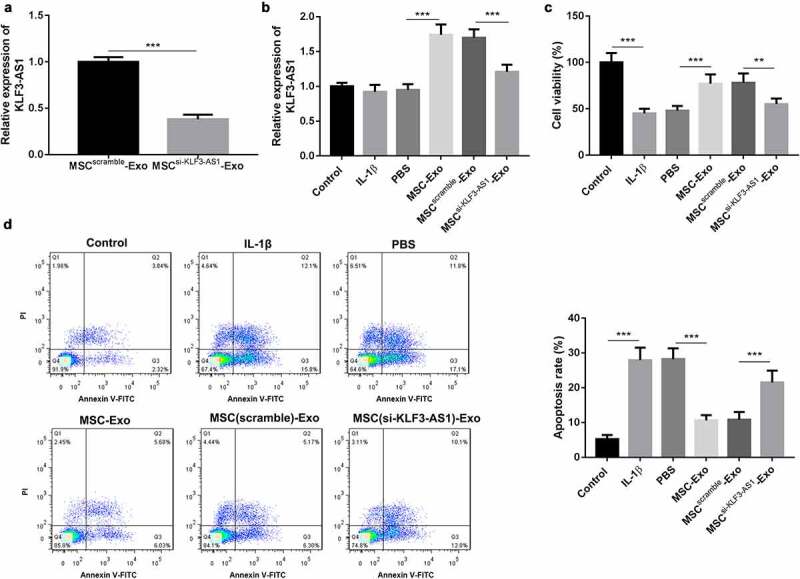
MSC-Exo-mediated KLF3-AS1 represses apoptosis of IL-1β-treated chondrocytes. MSCs were transfected with si-KLF3-AS1 or Scramble. Exosomes were extracted from the modified MSCs. (a) The expression of KLF3-AS1 in the exosomes was assessed by qRT-PCR. MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0001. Chondrocytes were treated with IL-1β, or followed by treatment of PBS, MSC-Exo, MSCScramble-Exo or MSCsi-KLF3-AS1-Exo. Normal chondrocytes served as control. (b) The expression of KLF3-AS1 in the chondrocytes was examined by qRT-PCR. MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P < 0.0001. (c) Cell viability of chondrocytes was detected by performing CCK-8 assay. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P = 0.0008; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0043. (d) Apoptosis of chondrocytes was examined by performing flow cytometry. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0004.
MSC-Exo-mediated KLF3-AS1 inhibits IL-1β-induced chondrocyte injury by repressing autophagy
Next, we further estimated the effect of MSC-Exo-mediated KLF3-AS1 on autophagy of chondrocytes. WB assay was performed to assess the expression of autophagy-related proteins in the chondrocytes. Compared with normal chondrocytes, LC3-II/LC3-I ratio was increased, while p62 expression was decreased in chondrocytes in the presence of IL-1β. MSC-Exo treatment reduced LC3-II/LC3-I expression and enhanced p62 expression in the IL-1β-treated chondrocytes. However, MSCsi-KLF3-AS1-Exo treatment promoted LC3-II/LC3-I expression and reduced p62 expression in the IL-1β-treated chondrocytes (Figure 2(a-c)). Furthermore, we observed the autophagy puncta in the chondrocytes. IL-1β treatment significantly enhanced the number of autophagy puncta in the chondrocytes, which was partly abolished by MSC-Exo treatment. Chondrocytes displayed a boost of autophagy puncta in the presence of MSCsi-KLF3-AS1-Exo (Figure 2(d)). Besides, chondrocytes were treated with rapamycin to activate autophagy. Compared with PBS group, MSC-Exo enhanced cell viability and reduced apoptosis of chondrocytes. However, Rapamycin treatment suppressed cell viability and promoted apoptosis of chondrocytes with respect to DMSO group (Figure 2(e-f)). Overall, these data indicated that MSC-Exo-mediated KLF3-AS1 repressed IL-1β-induced chondrocyte injury by repressing autophagy.
Figure 2.
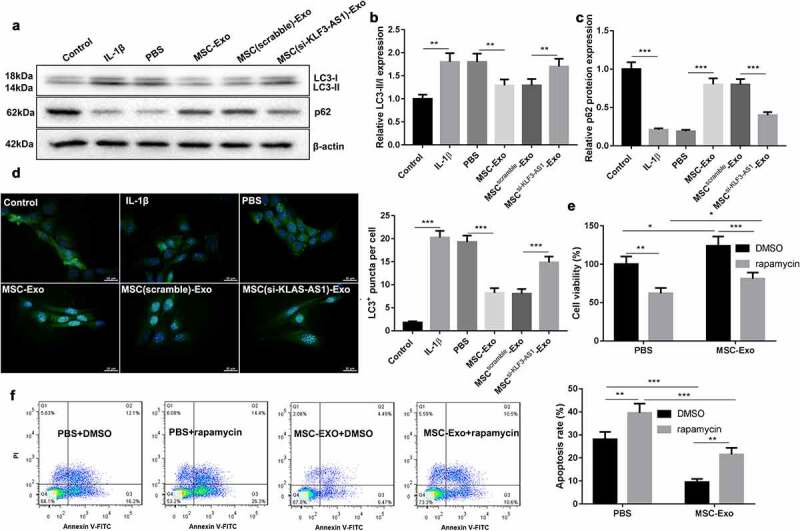
MSC-Exo-mediated KLF3-AS1 represses apoptosis of IL-1β-treated chondrocytes by repressing autophagy. MSCs were transfected with si-KLF3-AS1 or Scramble, and exosomes were extracted from the modified MSCs. Chondrocytes were treated with IL-1β combined with PBS, MSC-Exo, MSCScramble-Exo or MSCsi-KLF3-AS1-Exo. Normal chondrocytes served as control. (a) WB analysis of LC3-I, LC3-II and p62 expressions in the chondrocytes. (b) The ratio of LC3-II/LC3-I in the chondrocytes. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P = 0.0016; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0068. (c) The expression of p62 in the chondrocytes. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P < 0.0001. (d) GFP-LC3 fluorescence was used to observe the autophagy of chondrocytes. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0004. Chondrocytes were treated with IL-1β combined with PBS or MSC-Exo, and then treated with rapamycin or DMSO. (e) Cell viability of chondrocytes was detected by performing CCK-8 assay. Rapamycin + PBS vs. DMSO + PBS: P = 0.0012; Rapamycin + MSC-Exo vs. DMSO + MSC-Exo: P = 0.0005; DMSO + MSC-Exo vs. DMSO + PBS: P = 0.0144; Rapamycin + MSC-Exo vs. Rapamycin + PBS: P = 0.0391. (f) Apoptosis of chondrocytes was examined by performing flow cytometry. Rapamycin + PBS vs. DMSO + PBS: P = 0.0018; Rapamycin + MSC-Exo vs. DMSO + MSC-Exo: P = 0.0013; DMSO + MSC-Exo vs. DMSO + PBS: P < 0.0001; Rapamycin + MSC-Exo vs. Rapamycin + PBS: P < 0.0001.
KLF3-AS1 inhibits IL-1β-induced chondrocyte injury by repressing autophagy
To verify the molecular mechanism of KLF3-AS1 in OA, KLF3-AS1 was overexpressed in chondrocytes. KLF3-AS1 was highly expressed in the KLF3-AS1 following transfection of pcDNA3.1-KLF3-AS1 (Figure 3(a)). WB analysis of autophagy-related proteins showed that IL-1β-treated chondrocytes exhibited an increase of LC3-II/LC3-I and a decrease of p62 expression as compared with normal chondrocytes. KLF3-AS1 overexpression caused a down-regulation of LC3-II/LC3-I and an up-regulation of p62 in the IL-1β-treated chondrocytes (Figure 3(b-d)). Moreover, we observed the levels of autophagy puncta in the chondrocytes, showing that the levels of LC3+ puncta were enhanced in the chondrocytes following treatment of IL-1β. KLF3-AS1 up-regulation reduced levels of LC3+ puncta in the chondrocytes (Figure 3(f-g)). Moreover, KLF3-AS1-transfected chondrocytes were treated with rapamycin to activate autophagy. We estimated cell viability and apoptosis of chondrocytes. Compared with control chondrocytes, KLF3-AS1 overexpression promoted cell viability and repressed apoptosis of chondrocytes. Cell viability of chondrocytes was reduced by the treatment of rapamycin. However, rapamycin promoted apoptosis of chondrocytes (Figure 3e, (h-i)). Taken together, these results showed that KLF3-AS1 inhibited IL-1β-induced chondrocyte injury by repressing autophagy.
Figure 3.
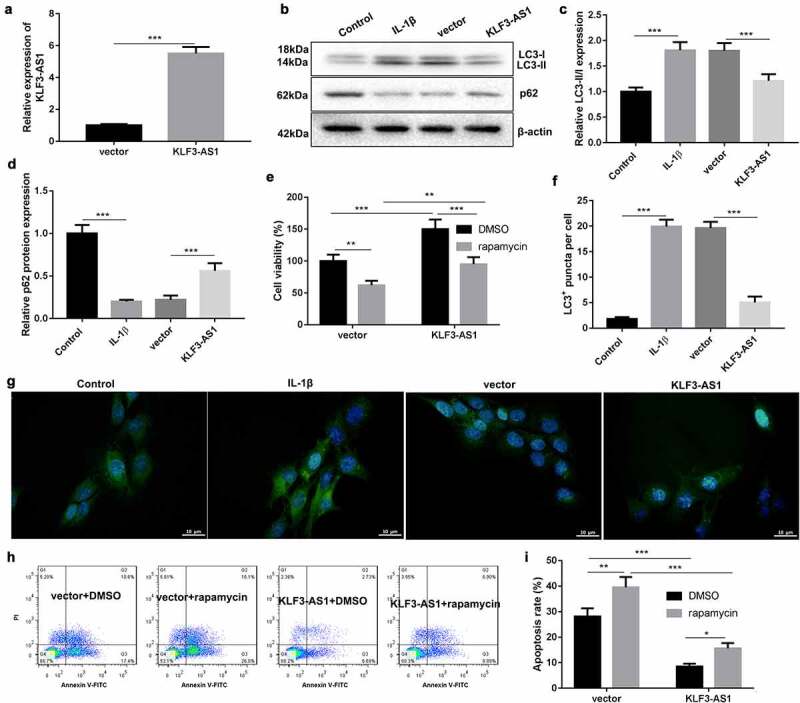
KLF3-AS1 inhibits apoptosis of IL-1β-treated chondrocytes by repressing autophagy. Chondrocytes were transfected with pcDNA3.1-KLF3-AS1 or pcDNA3.1-NC, followed by treatment of IL-1β. Normal chondrocytes served as control. (a) The expression of KLF3-AS1 in chondrocytes was detected by qRT-PCR. (b) WB analysis of LC3-I, LC3-II and p62 expressions in the chondrocytes. (c) The ratio of LC3-II/LC3-I in the chondrocytes. IL-1β vs. Control: P < 0.0001; KLF3-AS1 vs. vector: P = 0.0006. (d) The expression of p62 in the chondrocytes. IL-1β vs. Control: P < 0.0001; KLF3-AS1 vs. vector: P = 0.0004. Chondrocytes were transfected with pcDNA3.1-KLF3-AS1 or pcDNA3.1-NC, and then incubated with rapamycin or DMSO. (e) Cell viability of chondrocytes was detected by performing CCK-8 assay. Rapamycin + vector vs. DMSO + vector: P = 0.0031; Rapamycin + KLF3-AS1 vs. DMSO + KLF3-AS1: P = 0.0003; DMSO + KLF3-AS1 vs. DMSO + vector: P = 0.0006; Rapamycin + KLF3-AS1 vs. Rapamycin + vector: P = 0.0671. (f-g) GFP-LC3 fluorescence was used to observe the autophagy of chondrocytes. IL-1β vs. Control: P < 0.0001; KLF3-AS1 vs. vector: P < 0.0001. (h-i) Apoptosis of chondrocytes was detected by performing flow cytometry. Rapamycin + vector vs. DMSO + vector: P = 0.0012; Rapamycin + KLF3-AS1 vs. DMSO + KLF3-AS1: P = 0.016; DMSO + KLF3-AS1 vs. DMSO + vector: P < 0.0001; Rapamycin + KLF3-AS1 vs. Rapamycin + vector: P < 0.0001.
MSC-Exo-mediated KLF3-AS1 activates PI3K/Akt/mTOR signaling pathway by targeting YBX1 in chondrocytes.
We further demonstrated wehther MSC-Exo-mediated KLF3-AS1 can activate PI3K/Akt/mTOR signaling pathway by targeting YBX1 in chondrocytes. Figure 4(a) showed that the expression of Akt and mTOR had no change in the chondrocytes. IL-1β treatment decreased the levels of PI3K, p-Akt and p-mTOR in the chondrocytes. Compared with PBS, MSC-Exo caused an up-regulation of PI3K, p-Akt and p-mTOR in the chondrocytes. The levels of PI3K, p-Akt and p-mTOR in chondrocytes were severely reduced in the presence of MSCsi-KLF3-AS1-Exo (Figure 4(a-d)). Besides, the interaction between KLF3-AS1 and YBX1 was verified by RNA pull down and RIP assays, showing that KLF3-AS1 interacted with YBX1 in chondrocytes (Figure 4(e-g)). Therefore, these data suggested that MSC-Exo-mediated KLF3-AS1 activated PI3K/Akt/mTOR signaling pathway by targeting YBX1 in chondrocytes.
Figure 4.
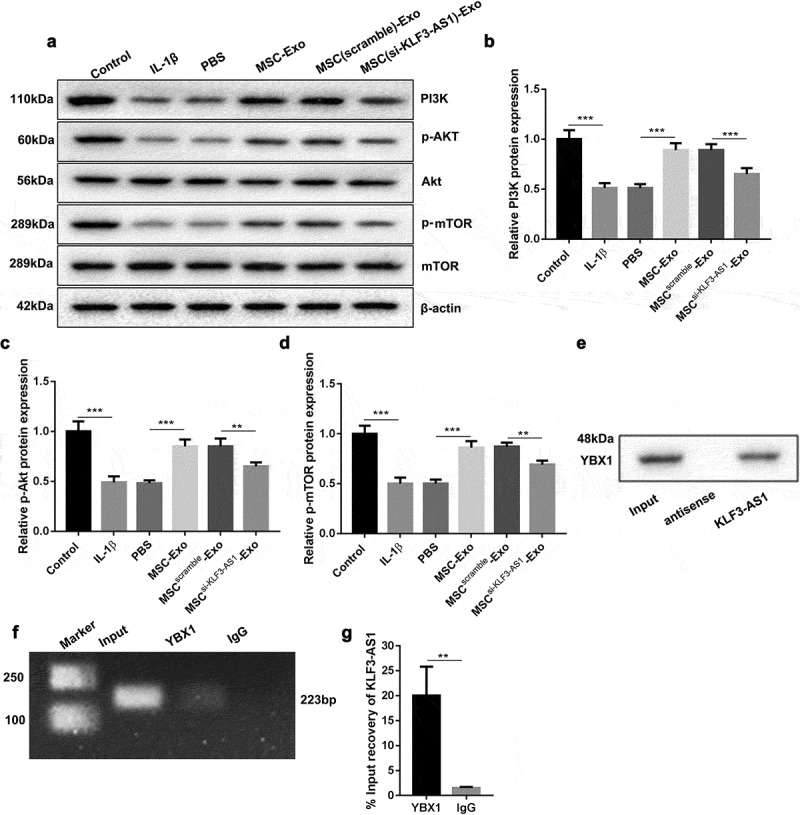
MSC-Exo-mediated KLF3-AS1 interacts with YBX1 to regulate PI3K/Akt/mTOR signaling pathway. MSCs were transfected with si-KLF3-AS1 or Scramble, and exosomes were extracted from the modified MSCs. Chondrocytes were treated with IL-1β combined with PBS, MSC-Exo, MSCScramble-Exo or MSCsi-KLF3-AS1-Exo. Normal chondrocytes served as control. (a) WB analysis of PI3K, Akt, p-AKT, mTOR and p-mTOR expressions in the chondrocytes. (b) The expression of PI3K in the chondrocytes. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0006. (c) The expression of p-AKT in the chondrocytes. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0035. (d) The expression of p-mTOR in the chondrocytes. IL-1β vs. Control: P < 0.0001; MSC-Exo vs. PBS: P < 0.0001; MSCsi-KLF3-AS1-Exo vs. MSCScramble-Exo: P = 0.0021. (e) The interaction between and KLF3-AS1 and YBX1 in chondrocytes was verified by RNA pull-down. (f-g) The interaction between and KLF3-AS1 and YBX1 in chondrocytes was determined by RIP assay. YBX1 vs. IgG: P = 0.0053.
MSC-Exo-mediated KLF3-AS1 represses autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway.
To verify the biologival role of MSC-Exo-mediated KLF3-AS1 and YBX1 in IL-1β-induced chondrocyte injury, chondrocytes were transfected with si-YBX1 to induce YBX1 knockdown. PI3K/Akt/mTOR-related protein expression was examined by WB assay. As shown in Figure 5(a), the expression Akt and mTOR had no change in the chondrocytes. The expression of PI3K, p-Akt and p-mTOR in the chondrocytes was enhanced in the presence of MSC-Exo, which was partly abolished by YBX1 deficiency (Figure 5(a-d)). Furthermore, CCK-8 assay results discovered that MSC-Exo promoted cell viability of chondrocytes. YBX1 silencing abrogated MSC-Exo-mediated promotion of cell viability in chondrocytes (Figure 5(e)). Subsequently, we estimated the expression of autophagy-related proteins in the chondrocytes, showing that MSC-Exo reduced LC3-II/LC3-I expression and enhanced p62 expression in chondrocytes. The influence conferred by MSC-Exo was partly rescued by YBX1 knockdown (Figure 5(f-h)). We also found that the apoptosis of chondrocytes was severely reduced by MSC-Exo treatment. MSC-Exo-mediated decrease of chondrocyte apoptosis was rescued by YBX1 down-regulation (Figure (i-j)). Therefore, these findings suggested that MSC-Exo-mediated KLF3-AS1 repressed autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway.
Figure 5.
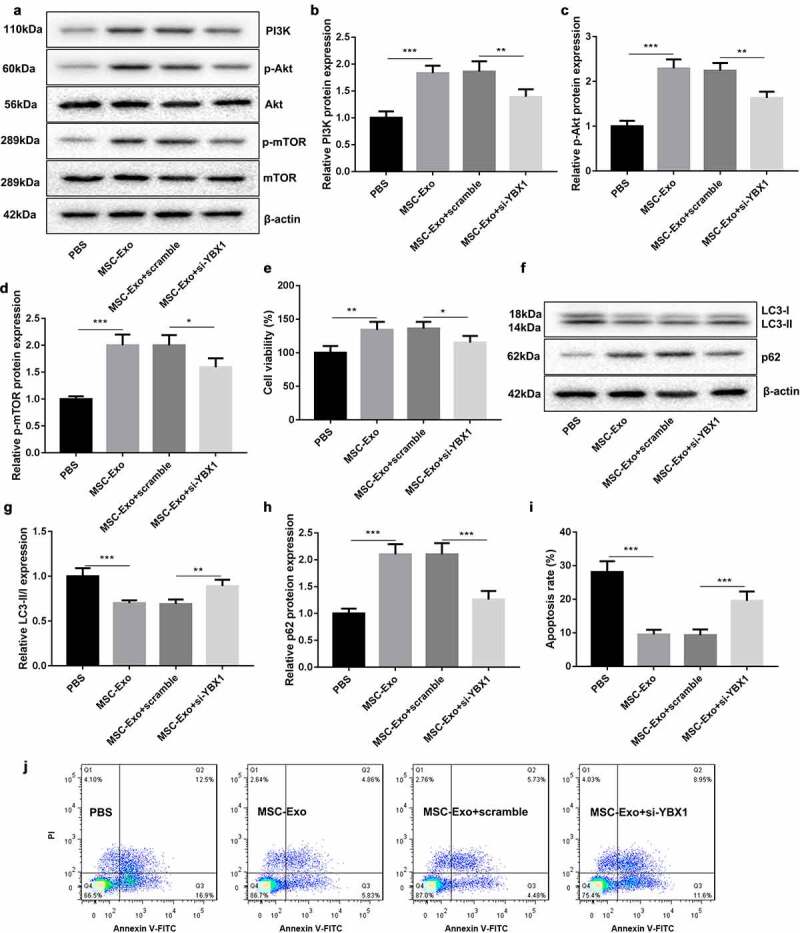
MSC-Exo-mediated KLF3-AS1 represses autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway. Chondrocytes were transfected with si-YBX1 or Scramble, and then treated with IL-1β combined with PBS or MSC-Exo. Normal chondrocytes served as control. (a) WB analysis of PI3K, Akt, p-AKT, mTOR and p-mTOR expressions in the chondrocytes. (b) The expression of PI3K in the chondrocytes. MSC-Exo vs. PBS: P = 0.0001; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0049. (c) The expression of p-AKT in the chondrocytes. MSC-Exo vs. PBS: P < 0.0001; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0016. (d) The expression of p-mTOR in the chondrocytes. MSC-Exo vs. PBS: P < 0.0001; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0155. (e) Cell viability of chondrocytes was detected by performing CCK-8 assay. MSC-Exo vs. PBS: P < 0.0042; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0405. (f) WB analysis of LC3-I, LC3-II and p62 expressions in the chondrocytes. (g) The ratio of LC3-II/LC3-I in the chondrocytes. MSC-Exo vs. PBS: P = 0.0004; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0051. (h) The expression of p62 in the chondrocytes. MSC-Exo vs. PBS: P < 0.0001; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0003. (i-j) Apoptosis of chondrocytes was detected by performing flow cytometry. MSC-Exo vs. PBS: P < 0.0001; MSC-Exo + scramble vs. MSC-Exo + si-YBX1: P = 0.0008.
KLF3-AS1/YBX1 represses autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway.
Finally, we explored the molecular mechanism of KLF3-AS1/YBX1 in regulating PI3K/Akt/mTOR signaling pathway. Chondrocytes were co-transfected with pcDNA3.1-KLF3-AS1 and si-YBX1, and then treated with IL-1β to induce chondrocyte injury. The results of WB revealed that KLF3-AS1 overexpression and YBX1 knockdown had no effect on the expression of Akt and mTOR in the chondrocytes (Figure 6(a)). KLF3-AS1 up-regulation enhanced the expression of PI3K, p-Akt and p-mTOR in the chondrocytes, which was rescued by YBX1 silencing (Figure 6(a-d)). Cell viability obtained from CCK-8 assay showed that KLF3-AS1 overexpression promoted cell viability of chondrocytes. Cell viability of chondrocytes was decreased in the presence of si-YBX1 (Figure 6(e)). Besides, the dada of WB assay revealed that the expression of LC3-II/LC3-I was significantly decreased, and p62 expression was enhanced in the chondrocytes by KLF3-AS1 overexpression. The influence conferred by KLF3-AS1 up-regulation was partly abolished by YBX1 deficiency (Figure 6(f-h)). In addition, KLF3-AS1 overexpression severely reduced apoptosis of chondrocytes. KLF3-AS1 up-regulation induced decrease of apoptosis in chondrocytes was abrogated by YBX1 silencing (Figure 6(i-j)). Thus, these data demonstrated that KLF3-AS1/YBX1 repressed autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway.
Figure 6.
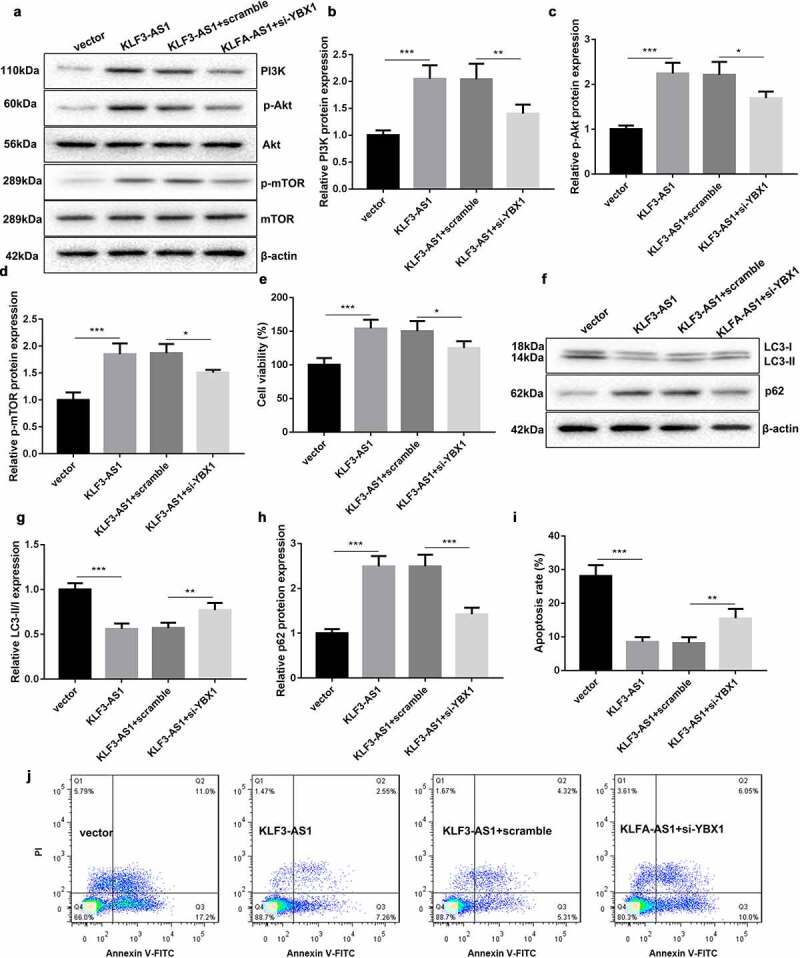
KLF3-AS1/YBX1 represses autophagy and apoptosis of chondrocytes through PI3K/Akt/mTOR signaling pathway. Chondrocytes were transfected with pcDNA3.1-KLF3-AS1 or pcDNA3.1-NC and si-YBX1 or Scramble, and then treated with IL-1β. (a) WB analysis of PI3K, Akt, p-AKT, mTOR and p-mTOR expressions in the chondrocytes. (b) The expression of PI3K in the chondrocytes. KLF3-AS1 vs. vector: P = 0.0003; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.0064. (c) The expression of p-AKT in the chondrocytes. KLF3-AS1 vs. vector: P < 0.0001; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.015. (d) The expression of p-mTOR in the chondrocytes. KLF3-AS1 vs. vector: P = 0.0001; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.0192. (e) Cell viability of chondrocytes was detected by performing CCK-8 assay. KLF3-AS1 vs. vector: P = 0.0006; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.0362. (f) WB analysis of LC3-I, LC3-II and p62 expressions in the chondrocytes. (g) The ratio of LC3-II/LC3-I in the chondrocytes. KLF3-AS1 vs. vector: P < 0.0001; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.007. (h) The expression of p62 in the chondrocytes. KLF3-AS1 vs. vector: P < 0.0001; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.0001. (i-j) Apoptosis of chondrocytes was detected by performing flow cytometry. KLF3-AS1 vs. vector: P < 0.0001; KLF3-AS1 + scramble vs. KLF3-AS1 + si-YBX1: P = 0.0058.
Discussion
The therapeutic role of exosomes in OA has been recognized by scholars [26]. Liang et al. [27] have confirmed that exosomes may become a cell-free therapy for OA. Chondrocyte-affinity peptide-exosomes inhibit cartilage-degrading proteases by delivering miR-140 to the deep cartilage regions, thereby alleviating the progression of OA. Bone MSC-Exo has a promoting effect on proliferation and migration of chondrocytes. MSC-Exo enhances cartilage repair and synthesis of extracellular matrix, thereby alleviating knee pain in OA rats [28]. MSC-Exo can suppress the progression of rheumatoid arthritis by intercellular transferring miR-320a. MiR-320a in MSC-Exo represses activation, migration, and invasion of fibroblast-like synoviocytes, and thus improves arthritis and bone damage in rheumatoid arthritis mice [29]. Our previous study has reported that human MSC-Exo has a promoting effect on cartilage repair and chondrocyte proliferation in OA by delivering KLF3-AS1 [11]. In this work, we conducted further research on the mechanism of action of MSC-Exo-mediated KLF3-AS1 in the progression of OA. We found that MSC-Exo treatment enhanced the expression of KLF3-AS1 in IL-1β-treated chondrocytes. IL-1β treatment repressed cell viability and promoted apoptosis of chondrocytes, which was partly rescued by MSC-Exo treatment. However, MSCsi-KLF3-AS1-Exo treatment reduced cell viability and enhanced apoptosis in IL-1β-treated chondrocytes. Thus, we speculated that MSC-Exo-mediated KLF3-AS1 repressed IL-1β-induced chondrocyte injury.
Autophagy is reported to be associated with the progression of OA. In IL-1β-treated chondrocytes, hsa_circ_0005567 promotes autophagy activation via repressing miR-495 expression, thereby inhibiting apoptosis of chondrocytes [30]. Sirt7 has a protective effect on chondrocyte degeneration in the progression of OA by activating autophagy [31]. In our study, MSC-Exo reduced LC3-II/LC3-I expression and enhanced p62 expression in IL-1β-treated chondrocytes. The levels of autophagy puncta in the chondrocytes were repressed by MSC-Exo, suggesting that MSC-Exo inactivated autophagy in IL-1β-treated chondrocytes. In addition, KLF3-AS1 overexpression repressed apoptosis, and promoted cell viability of IL-1β-treated chondrocytes. However, rapamycin treatment suppressed cell viability and promoted apoptosis of chondrocytes by activating autophagy. Thus, these results revealed that MSC-Exo-mediated KLF3-AS1 repressed apoptosis of IL-1β-treated chondrocytes by repressing autophagy.
KLF3-AS1 participates in the development of many diseases. For example, KLF3-AS1 is down-regulated in esophageal squamous cell carcinoma, and KLF3-AS1 inhibits cell migration and invasion of esophageal squamous cell carcinoma cells via miR-185-5p/KLF3 axis [32]. In myocardial infarction, human MSC-Exo-mediated KLF3-AS1 promotes Sirt1 expression by sponging miR-138-5p. KLF3-AS1/miR-138-5p/Sirt1 signaling pathway inhibits pyroptosis of cardiomyocytes, thereby ameliorating the progression of myocardial infarction [33]. Our data confirmed that KLF3-AS1 interacted with YBX1 in the chondrocytes. Furthermore, MSC-Exo enhanced the expression of PI3K, p-Akt, and p-mTOR in the chondrocytes, while these proteins were down-regulated in the chondrocytes following treatment of MSCsi-KLF3-AS1-Exo. Thus, these data suggested that MSC-Exo-mediated KLF3-AS1 activated PI3K/Akt/mTOR signaling pathway by interacting with YBX1.
PI3K/Akt/mTOR signaling pathway-mediated autophagy is closely associated with the progression of OA. UHRF1 deficiency effectively improves the progression of OA, and UHRF1 knockdown promotes autophagy to protect chondrocytes from apoptosis by regulating PI3K/AKT/mTOR signaling pathway [34]. LncRNA MALAT1 regulates PI3K/AKT/mTOR signaling pathway in OA by repressing miR-146a expression, and MALAT1 plays a vital role in regulating proliferation of LPS treated-articular chondrocytes [35]. MiR-27a acts as a regulator of PI3K-Akt-mTOR axis in OA, and miR-27a enhances autophagy and apoptosis of articular chondrocytes [36]. In our study, we found that MSC-Exo activated PI3K/AKT/mTOR signaling pathway and repressed autophagy and apoptosis of chondrocytes, which was partly abolished by YBX1 knockdown. Moreover, KLF3-AS1 up-regulation promoted activation of PI3K/AKT/mTOR signaling pathway and suppressed autophagy and apoptosis of chondrocytes. The influence conferred by KLF3-AS1 overexpression was partly rescued by YBX1 silencing. Taken together, these data showed that MSC-Exo-mediated KLF3-AS1 repressed autophagy and apoptosis of chondrocytes by activating PI3K/Akt/mTOR signaling pathway.
In conclusion, our data demonstrate that MSC-Exo-mediated lncRNA KLF3-AS1 inhibits autophagy and apoptosis of IL-1β-treated chondrocyte by activating PI3K/Akt/mTOR signaling pathway. LncRNA KLF3-AS1 activates PI3K/Akt/mTOR signaling pathway by targeting YBX1 in the progression of OA. Thus, MSC-Exo-mediated lncRNA KLF3-AS1 may be a potential therapeutic target for OA.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China (81902197); Medical Scientific Research Project of Health Commission of Jiangsu Province (M2020073).
Funding Statement
This work was supported by the National Natural Science Foundation of China [81902197]; Medical Scientific Research Project of Health Commission of Jiangsu Province [M2020073].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Haemer JM, Carter DR, Giori NJ.. The low permeability of healthy meniscus and labrum limit articular cartilage consolidation and maintain fluid load support in the knee and hip. J Biomech. 2012;45(8):1450–1456. [DOI] [PubMed] [Google Scholar]
- [2].Richardson SM, Kalamegam G, Pushparaj PN, et al. Mesenchymal stem cells in regenerative medicine: focus on articular cartilage and intervertebral disc regeneration. Methods. 2016;99:69–80. [DOI] [PubMed] [Google Scholar]
- [3].Wang Z, Han L, Sun T, et al. Extracellular matrix derived from allogenic decellularized bone marrow mesenchymal stem cell sheets for the reconstruction of osteochondral defects in rabbits. Acta Biomater. 2020;118:54–68. [DOI] [PubMed] [Google Scholar]
- [4].Kamei N, Ochi M, Adachi N, et al. The safety and efficacy of magnetic targeting using autologous mesenchymal stem cells for cartilage repair. Knee Surg Sports Traumatol Arthrosc. 2018;26(12):3626–3635. [DOI] [PubMed] [Google Scholar]
- [5].Camussi G, Deregibus MC, Bruno S, et al. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78(9):838–848. [DOI] [PubMed] [Google Scholar]
- [6].Ratajczak J, Wysoczynski M, Hayek F, et al. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20(9):1487–1495. [DOI] [PubMed] [Google Scholar]
- [7].Strassburg S, Hodson NW, Hill PI, et al. Bi-directional exchange of membrane components occurs during co-culture of mesenchymal stem cells and nucleus pulposus cells. PloS One. 2012;7(3):e33739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang S, Chu WC, Lai RC, et al. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthritis Cartilage. 2016;24(12):2135–2140. [DOI] [PubMed] [Google Scholar]
- [9].Toh WS, Lai RC, Hui JHP, et al. MSC exosome as a cell-free MSC therapy for cartilage regeneration: implications for osteoarthritis treatment. Semin Cell Dev Biol. 2017;67:56–64. [DOI] [PubMed] [Google Scholar]
- [10].Zhang S, Chuah SJ, Lai RC, et al. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials. 2018;156:16–27. [DOI] [PubMed] [Google Scholar]
- [11].Liu Y, Zou R, Wang Z, et al. Exosomal KLF3-AS1 from hMSCs promoted cartilage repair and chondrocyte proliferation in osteoarthritis. Biochem J. 2018;475(22):3629–3638. [DOI] [PubMed] [Google Scholar]
- [12].Liu Y, Lin L, Zou R, et al. MSC-derived exosomes promote proliferation and inhibit apoptosis of chondrocytes via lncRNA-KLF3-AS1/miR-206/GIT1 axis in osteoarthritis. Cell Cycle (Georgetown, Tex). 2018;17(21–22):2411–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Miotto M, Marinari E, De Martino A.. Competing endogenous RNA crosstalk at system level. PLoS Comput Biol. 2019;15(11):e1007474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Astanehe A FM, Hojabrpour P, To K, et al. YBX1 is a modulator of MIA/CD-RAP-dependent chondrogenesis. PloS One. 2013;8(12):e82166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu J, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–3060. [DOI] [PubMed] [Google Scholar]
- [16].Feng F, Qiu H. Effects of Artesunate on chondrocyte proliferation, apoptosis and autophagy through the PI3K/AKT/mTOR signaling pathway in rat models with rheumatoid arthritis. Biomed Pharmacothe. 2018;102:1209–1220. [DOI] [PubMed] [Google Scholar]
- [17].Xue J, Shi Z, Zou J, et al. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed Pharmacother. 2017;89:1252–1261. [DOI] [PubMed] [Google Scholar]
- [18].Astanehe A, Finkbeiner MR, Hojabrpour P, et al. The transcriptional induction of PIK3CA in tumor cells is dependent on the oncoprotein Y-box binding protein-1. Oncogene. 2009;28(25):2406–2418. [DOI] [PubMed] [Google Scholar]
- [19].Xu W, Ma Y, Ma H, et al. Co-targeting CK2α and YBX1 suppresses tumor progression by coordinated inhibition of the PI3K/AKT signaling pathway. Cell Cycle (Georgetown, Tex). 2019;18(24):3472–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Troiano A, Lomoriello I, Di Martino O, et al. Y-box Binding Protein-1 Is Part of a Complex Molecular Network Linking ΔNp63α to the PI3K/akt Pathway in Cutaneous Squamous Cell Carcinoma. J Cell Physiol. 2015;230(9):2067–2074. [DOI] [PubMed] [Google Scholar]
- [21].Zheng H, Zhan Y, Zhang Y, et al. Elevated expression of G3BP1 associates with YB1 and p-AKT and predicts poor prognosis in nonsmall cell lung cancer patients after surgical resection. Cancer Med. 2019;8(16):6894–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Caramés B, Taniguchi N, Otsuki S, et al. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheumatism. 2010;62(3):791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, et al. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis. 2010;15(5):631–638. [DOI] [PubMed] [Google Scholar]
- [24].Sun K, Luo J, Guo J, et al. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: a narrative review. Osteoarthritis Cartilage. 2020;28(4):400–409. [DOI] [PubMed] [Google Scholar]
- [25].Wang X, Fan J, Ding X, et al. Tanshinone I Inhibits IL-1β-Induced Apoptosis, Inflammation And Extracellular Matrix Degradation In Chondrocytes CHON-001 Cells And Attenuates Murine Osteoarthritis. Drug Des Devel Ther. 2019;13:3559–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jeyaraman M, Muthu S, Gulati A, et al. Mesenchymal Stem Cell-Derived Exosomes: a Potential Therapeutic Avenue in Knee Osteoarthritis. Cartilage. 2020;1947603520962567. 10.1177/1947603520962567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liang Y, Xu X, Li X, et al. Chondrocyte-Targeted MicroRNA Delivery by Engineered Exosomes toward a Cell-Free Osteoarthritis Therapy. ACS Appl Mater Interfaces. 2020;12(33):36938–36947. [DOI] [PubMed] [Google Scholar]
- [28].He L, He T, Xing J, et al. Bone marrow mesenchymal stem cell-derived exosomes protect cartilage damage and relieve knee osteoarthritis pain in a rat model of osteoarthritis. Stem Cell Res Ther. 2020;11(1):276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Meng Q, Qiu B. Exosomal MicroRNA-320a Derived From Mesenchymal Stem Cells Regulates Rheumatoid Arthritis Fibroblast-Like Synoviocyte Activation by Suppressing CXCL9 Expression. Front Physiol. 2020;11:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang J, Cheng F, Rong G, et al. Hsa_circ_0005567 Activates Autophagy and Suppresses IL-1β-Induced Chondrocyte Apoptosis by Regulating miR-495. Front Mol Biosci. 2020;7:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu SY, Du YC, Yue CF. Sirt7 protects chondrocytes degeneration in osteoarthritis via autophagy activation. Eur Rev Med Pharmacol Sci. 2020;24(18):9246–9255. [DOI] [PubMed] [Google Scholar]
- [32].Liu J-Q, Deng M, Xue -N-N, et al. lncRNA KLF3-AS1 Suppresses Cell Migration and Invasion in ESCC by Impairing miR-185-5p-Targeted KLF3 Inhibition. Mol Ther Nucleic Acids. 2020;20:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mao Q, Liang XL, Zhang CL, et al. LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis. Stem Cell Res Ther. 2019;10:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shi X, Han L, Sun T, et al. Silencing UHRF1 enhances cell autophagy to prevent articular chondrocytes from apoptosis in osteoarthritis through PI3K/AKT/mTOR signaling pathway. Biochem Biophys Res Commun. 2020;529:1018–1024. [DOI] [PubMed] [Google Scholar]
- [35].Li H, Xie S, Li H, et al. LncRNA MALAT1 mediates proliferation of LPS treated-articular chondrocytes by targeting the miR-146a-PI3K/Akt/mTOR axis. Life Sci. 2020;254:116801. [DOI] [PubMed] [Google Scholar]
- [36].Cai C, Min S, Yan B, et al. MiR-27a promotes the autophagy and apoptosis of IL-1β treated-articular chondrocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging (Albany NY). 2019;11(16):6371–6384. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.