

Abstract
The next generation anti-androgen drugs, XTANDI® (Enzalutamide), ZYTIGA® (Abiraterone acetate), ERLEADA™ (Apalutamide) and NUBEQA (Darolutamide) extend survival times and improve quality of life in advanced prostate cancer patients. Despite these advances, resistance occurs frequently and there is currently no definitive cure for Castration-Resistant Prostate Cancer (CRPC). Our previous studies identified that similar mechanisms of resistance to enzalutamide or abiraterone occur following treatment and cross-resistance exists between these therapies in advanced prostate cancer. Here we show that enzalutamide and abiraterone resistant prostate cancer cells are further cross-resistant to apalutamide and darolutamide. Mechanistically, we have determined that the AKR1C3/AR-V7 axis confers this cross-resistance. Knockdown of AR-V7 in enzalutamide resistant cells re-sensitize cells to apalutamide and darolutamide treatment. Furthermore, targeting AKR1C3 re-sensitizes resistant cells to apalutamide and darolutamide treatment through AR-V7 inhibition. Chronic apalutamide treatment in C4-2B cells activates the steroid hormone biosynthesis pathway and increases AKR1C3 expression which confers resistance to enzalutamide, abiraterone and darolutamide. In conclusion, our results suggest that apalutamide and darolutamide share similar resistant mechanisms with enzalutamide and abiraterone. The AKR1C3/AR-V7 complex confers cross-resistance to second generation AR-targeted therapies in advanced prostate cancer.
Keywords: Prostate cancer, anti-androgens, AKR1C3, AR-V7, cross-resistance
Introduction
Treatment strategies for castration-resistant prostate cancer (CRPC) have evolved over the past decade. Before 2010, docetaxel was the only treatment option for CRPC patients (1). Following the discovery that CRPC patients remain dependent on androgen receptor (AR) signaling (2–5), targeting AR became the major strategy for the treatment of CRPC. The anti-androgen therapies, including abiraterone, enzalutamide, apalutamide and darolutamide, significantly improved patient overall survival rate and are approved by the FDA (6–9). However, the length of clinical response to these treatments is limited and all patients eventually develop resistance. Thus, it is urgent to understand the underlying mechanisms of the resistance.
Apalutamide is approved for the treatment of non-metastatic CRPC and metastatic castration-sensitive prostate cancer (mCSPC) (6). A missense AR mutant, F877L, is linked to apalutamide resistance in preclinical models and has been detected in plasma DNA from apalutamide treated patients (10). However, recent clinical data suggests the F877L and the T878A mutations might not cause either de novo or acquired apalutamide resistance (11). Data from phase I/II/III clinical trials show that darolutamide has strong activity and minimal toxicity (12,13). In contrast to enzalutamide and apalutamide, darolutamide does not penetrate the blood-brain barrier which lowers the toxicity of this drug. Nevertheless, both apalutamide and darolutamide still target the full-length AR and mechanisms of resistance and cross-resistance with enzalutamide/abiraterone are largely unknown and need to be further investigated.
Constitutively active AR variants such as AR-V7 can activate distinct transcriptional programs and confer enzalutamide and abiraterone resistance (14–17). In addition to AR-V7, AKR1C3 activation has been shown to be a critical mechanism to promote resistance to both enzalutamide and abiraterone (18–20). The primary function of AKR1C3 is to catalyze androgen synthesis (19,20). Our previous study demonstrated a new function of AKR1C3: it can bind with AR-V7 and promote its stabilization (18), suggesting that the AKR1C3/AR-V7 axis plays critical roles in conferring resistance to both enzalutamide and abiraterone. However, the role of the AKR1C3/AR-V7 axis in apalutamide and darolutamide is unknown.
Here we demonstrate cross-resistance between both apalutamide and darolutamide therapies with enzalutamide and abiraterone treatment. Apalutamide and darolutamide target full length AR while having no effect on AR-V7 activity. Chronic apalutamide treatment significantly elevated AKR1C3 and AR-V7 expression in prostate cancer cells. Knockdown of AKR1C3 decreased AR-V7 expression and significantly reversed the cross-resistance to enzalutamide, abiraterone, apalutamide and darolutamide in resistant cells, suggesting that the AKR1C3/AR-V7 axis plays a critical role in cross-resistance among the next generation anti-androgen drugs.
Materials and Methods
Cells lines and tissue culture
C4-2B and CWR22Rv1 were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin and 0.1 mg/ml streptomycin. 293 cells were maintained in DMEM supplemented with 10% FBS, 100 units/ml penicillin and 0.1 mg/ml streptomycin. All experiments with cell lines were performed within 6 months of receipt from the ATCC or resuscitation from cryopreservation. C4-2B cells were kindly provided and authenticated by Dr. Leland Chung, Cedars-Sinai Medical Center (Los Angeles, CA). C4-2B neo and C4-2B AKR1C3 cells were generated by stable transfection of C4-2B cells with either empty vector pcDNA3.1 or pcDNA3.1 encoding AKR1C3 and were maintained in RPMI1640 medium containing 300 μg/mL G418. C4-2B cells were chronically exposed to increasing concentrations of apalutamide (5 μM ~ 40 μM) by passage in media containing apalutamide for >12 months in complete FBS and stored for further analysis. Cells resistant to apalutamide were referred to as C4-2B APALR (C4-2B apalutamide resistant). C4-2B MDVR (C4-2B enzalutamide resistant) and C4-2B AbiR (C4-2B abiraterone resistant) are described previously (15). C4-2B MDVR, C4-2B AbiR and C4-2B APALR cells were maintained in 20 μM enzalutamide containing medium, 10 μM abiraterone acetate containing medium and 20 μM apalutamide respectively. Parental C4-2B cells were passaged alongside the resistant cells as an appropriate control. All cells were maintained at 37°C in a humidified incubator with 5% carbon dioxide. Enzalutamide, abiraterone acetate, apalutamide, darolutamide, and indomethacin were purchased from Selleck Chemicals.
Plasmids and cell transfection
For transfection of small interfering RNA (siRNA), cells were seeded at a density of 0.5×105 cells per well in 12-well plates or 2×105 cells per well in 6-well plates and transfected with 20 nM of siRNA targeting the AR-V7 sequence (GUAGUUGUGAGUAUCAUGA) (Dharmacon), or control siRNA (Catalog# 12935300, Invitrogen) using Lipofectamine-iMAX (Invitrogen). Briefly, cells were plated one day before the transfection. 20 nM siRNA in serum-free Opti-MEM medium was mixed with same volume of Opti-MEM medium containing Lipofectamine-iMAX and incubated for 5 min at room temperature, prior to be added to cells. pcDNA3.1-AR-V7 or AKR1C3 were constructed by PCR amplification followed by sub-cloning into the pcDNA3.1 vector. Cells were transfected by expressing plasmids for pcDNA3.1 vector, AR-V7 and AKR1C3 using Lipofectamine 2000 (Invitrogen). Briefly, cells were plated one day before the transfection. 0.5–1µg plasmids in serum-free Opti-MEM medium was mixed with same volume of Opti-MEM medium containing Lipofectamine2000 and incubated for 20 min at room temperature, prior to be added to cells. The effect of siRNA-mediated gene silencing and ectopic expression plasmids were examined using qRT-PCR or western blot 2–3 days after transfection. Lentiviral plasmid encoding shRNA targeting AKR1C3 (TRCN0000026561) was purchased from Sigma-Aldrich. pLenti-GFP Lentiviral control vector were used as control. Lentiviral particles were produced in 293T cells after co-transfection of the lentivirus vectors, psPAX2 and pMD2.G in 10-cm dishes. The lentiviral plasmids were kindly provided by Dr. Hong-Wu Chen at UC Davis. The lentivirus containing medium was collected and cells were infected.
Protein extraction and western blotting
Whole cell protein extracts were resolved on SDS-PAGE and proteins were transferred to nitrocellulose membranes. After blocking for 1 hour at room temperature in 5% milk in PBS/0.1% Tween-20, membranes were incubated overnight at 4℃ with the indicated primary antibodies: AR (441, 1:1000 dilution, Santa Cruz biotechnology, Inc.), AR (N-20, 1:1000 dilution, Santa Cruz biotechnology, Inc.), AR-V7 (AG10008, Mouse monoclonal antibody, 1:1000 dilution, precision antibody); AKR1C3 (A6229, 1:1000 dilution, Sigma-Aldrich, St. Louis, MO); c-Myc (N262, 1:1000 dilution, Santa Cruz biotechnology, Inc.); Tubulin (T5168, Monoclonal Anti-α-Tubulin antibody, 1:5000 dilution, Sigma-Aldrich, St. Louis, MO). Tubulin was used as loading control. Following secondary antibody incubation, immunoreactive proteins were visualized with an enhanced chemiluminescence detection system (Millipore, Billerica, MA).
Luciferase reporter assay
C4-2B cells were transfected with pGL3-PSA6.0-Luc reporters along with AR-V7 in charcoal stripped FBS (CS-FBS) conditions. Cell lysates (35 μL per well) were used for measurement of luciferase activity in a luminometer by first mixing the cell lysates (25 μL) with 20 μL luciferase assay reagent for measuring firefly luciferase activity and subsequently adding 20 μL Stop-Glo reagent for measuring Renilla luciferase activity. Data were normalized to Renilla luciferase activity.
Cell growth and survival assay
C4-2B parental, C4-2B AbiR, C4-2B MDVR, C4-2B APALR or CWR22Rv1 cells were seeded on 12-well plates at a density of 0.3×105 cells/well in RPMI 1640 media containing 10% FBS and treated with enzalutamide, apalutamide or darolutamide. Total cell number was determined after 0, 3 and 5 days. C4-2B MDVR cells were transiently transfected with siRNA targeting AR-V7 or a control siRNA, and then treated with apalutamide or darolutamide for 3 days. C4-2B MDVR or C4-2B APALR cells were seeded on 12-well plates at a density of 0.5×105 cells/well in RPMI 1640 media containing 10% FBS and co-treated with indomethacin and apalutamide/darolutamide in media containing FBS. The coefficient of drug interaction (CDI) (21) is calculated as follows: CDI = AB/(A×B). A is growth inhibition effect of indomethacin treatment. B is growth inhibition effect of apalutamide/darolutamide treatment. AB is the growth inhibition effect of combination treatment. The CDI were analyzed to determine the synergism of two drugs combination treatment (CDI value <1, =1 or >1 indicates that the drugs are synergistic, additive or antagonistic). C4-2B neo and C4-2B AKR1C3 cells were seeded on 12-well plates at a density of 0.3×105 cells/well in RPMI 1640 media containing 10% FBS and treated with different doses of apalutamide or darolutamide for 3 days. Total cell number was counted and the cell survival rate (%) was calculated. Cell survival rate (%) = (Treatment group cell number / Control group cell number) ×100%.
Clonogenic Assay
C4-2B parental, C4-2B MDVR, C4-2B AbiR or CWR22Rv1 cells were treated with DMSO, 20 μM enzalutamide, 20 μM apalutamide or 5 μM darolutamide in media containing 10% complete FBS. C4-2B APALR cells were treated with 0μM, 20μM or 40μM indomethacin alone or in combination with 20 μM apalutamide. All cell lines were plated at equal densities (600 cells/well) and all treatments lasted for 14 days. The colonies were rinsed with PBS before being stained with 0.5% crystal violet/4% formaldehyde for 30 min and the number of colonies was counted as described previously (22).
Real-Time quantitative RT-PCR
Total RNA was extracted using TriZOL reagent (Invitrogen). cDNA was prepared after digestion with RNase-free RQ1 DNase (Promega) and then subjected to real-time reverse transcription-PCR (RT-PCR) using Sso Fast Eva Green Supermix (Bio-Rad) according to the manufacturer’s instructions and as described previously (23). Each reaction was normalized by co-amplification of actin. Triplicates of samples were run on default settings of a Bio-Rad CFX-96 real-time cycler. Primers used for RT-PCR were: AKR1C3, 5′-gagaagtaaagctttggaggtcaca-3′(forward) and 5′-caacctgctcctcattattgtataaatga-3′(reverse); AKR1C1/2, 5′-ggtcacttcatgcctgtcct-3′ (forward) and 5′-actctggtcgatgggaattg-3′ (reverse); HSD3B, 5′-cgggcccaactcctacaag-3′ (forward) and 5′-ttttccagaggctcttcttcgt-3′(reverse); SRD5A1, 5′-acgggcatcggtgcttaat-3′ (forward) and 5′-ccaacagtggcataggctttc-3′ (reverse) HSD17B2, 5′-tttgccggagttttgaatgaa-3′ (forward) and 5′-gcaggttcttcgcaattcct-3′ (reverse) and actin, 5′-agaactggcccttcttggagg-3′(forward) and 5′-gtttttatgttcctctatggg-3′ (reverse).
Statistical Analysis
The data are presented as means ± standard deviation of the mean (SD) from three independent experiments. Statistical analyses were performed with SPSS16.0. Differences between individual groups were analyzed by two tailed student’s t test or one-way analysis of variance (ANOVA) followed by the Scheffé procedure for comparison of means. p<0.05 was considered statistically significant.
Results
Enzalutamide/abiraterone resistant prostate cancer cells are cross-resistant to apalutamide and darolutamide
Apalutamide and darolutamide are new generation anti-androgens that bind the AR with high affinity and inhibit AR transcriptional activity and nuclear translocation (12,24). To determine the response of enzalutamide/abiraterone resistant cells to apalutamide and darolutamide, C4-2B parental, C4-2B AbiR, C4-2B MDVR and CWR22Rv1 cells were treated with different doses of enzalutamide, apalutamide or darolutamide. We found that C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells were resistant to all the anti-androgen treatments. As shown in Fig. 1A, 20 µM enzalutamide inhibited 41%, 6.3%, 5.5% and 21%; 40 µM apalutamide inhibited 24%, 5.8%, 5.7% and 15%; 10 µM darolutamide inhibited 54%, 26%, 28% and 31% of cell growth in C4-2B, C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells, respectively. In C4-2B parental cells, the rank of cell growth inhibition is darolutamide > enzalutamide > apalutamide. Apalutamide is least potent compared to enzalutamide and darolutamide in the inhibition of cellular growth of C4-2B parental cells. We then determined growth inhibition at different time points by these anti-androgens in different cell lines. As shown in Fig. 1B, C4-2B parental cells respond to all the anti-androgen treatments in a time dependent manner; however, C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells were resistant to all anti-androgen treatments at all time points. At 5 days treatment, 20 µM enzalutamide inhibited 44%, 7.3%, 11% and 19%; 40 µM apalutamide inhibited 23%, 3.0%, 2.9% and 14%; 5 µM darolutamide inhibited 46%, 13.5%, 11% and 13.3% of cell growth in C4-2B, C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells respectively. These data were confirmed using colony formation assays. We found that C4-2B parental cells formed fewer colonies when treated with enzalutamide, apalutamide or darolutamide, whereas C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells were far less affected by anti-androgen treatment (Fig. 1C). Taken together, these data suggest enzalutamide/abiraterone resistant prostate cancer cells are cross-resistant to both apalutamide and darolutamide.
Figure 1. Enzalutamide/abiraterone resistant prostate cancer cells are cross-resistant to apalutamide and darolutamide.
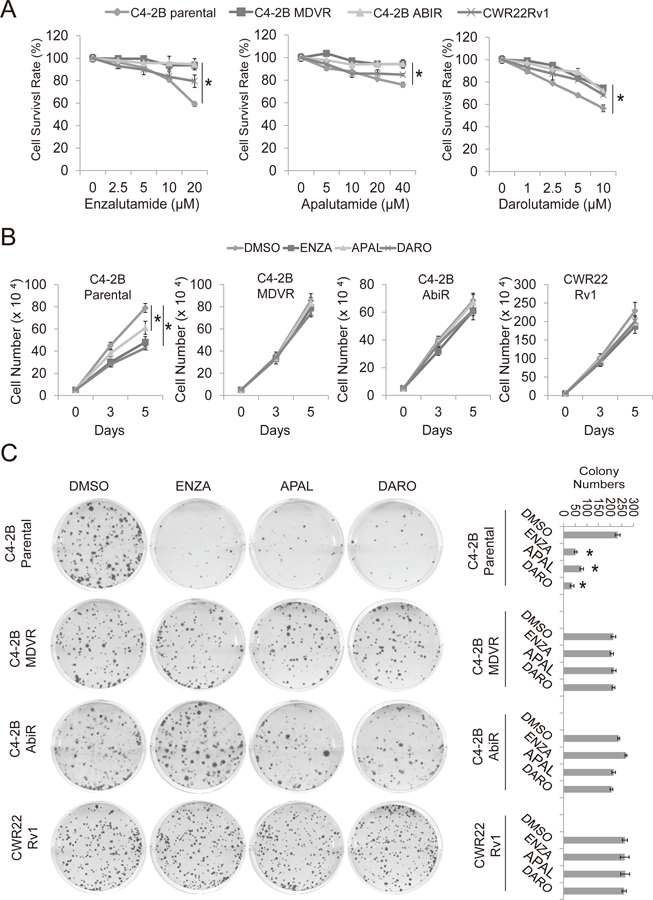
A. C4-2B parental, C4-2B AbiR, C4-2B MDVR and CWR22Rv1 cells were treated with different concentrations of enzalutamide, apalutamide or darolutamide for 3 days. Total cell numbers were counted and cell survival rate was calculated. B. C4-2B parental, C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells were treated with 20 μM enzalutamide, 20μM apalutamide or 5 μM darolutamide for 0, 3 and 5 days and total cell numbers were determined. C. The clonogenic ability of C4-2B parental, C4-2B MDVR, C4-2B AbiR and CWR22Rv1 cells treated with 20 μM enzalutamide, 20 μM apalutamide or 5 μM darolutamide was analyzed. Enza: enzalutamide, APAL: apalutamide, Abi: abiraterone acetate, DARO: darolutamide. * p<0.05.
AR-V7 confers apalutamide and darolutamide resistance
Previous studies showed that AR-V7, a truncated variant of the AR, confers enzalutamide resistance (14,15). To investigate the potential mechanisms of cross-resistance between enzalutamide, apalutamide and darolutamide, we determined levels of full length AR, AR-V7 and AKR1C3 following different anti-androgen treatments in C4-2B parental, C4-2B MDVR and CWR22Rv1 cells. As shown in Fig. 2A, enzalutamide, apalutamide and darolutamide suppressed both full length AR and AR-V7 protein expression in C4-2B parental cells. Darolutamide was the most effective drug at decreasing full length AR in these cells compared to enzalutamide and apalutamide. C4-2B MDVR cells expressed higher level of full length AR and AR variants than C4-2B parental cells. Enzalutamide, apalutamide and darolutamide had less effect on full length AR yet no effect on AR-V7 expression in C4-2B MDVR cells. However, total AR variants expression was inhibited by enzalutamide and darolutamide when detected by pan-AR antibody in C4-2B MDVR cells. None of the anti-androgens had effect on full length AR and AR-V7 expression in CWR22Rv1 cells. Promoter activity assays revealed that enzalutamide, apalutamide and darolutamide significantly suppressed DHT induced but not AR-V7 induced AR transcriptional activity (Fig. 2B). Niclosamide, a previously identified AR-V7 inhibitor (15), significantly suppressed both DHT and AR-V7 induced PSA promoter luciferase activity (Fig. 2B). To further test whether AR-V7 overexpression is one of the mechanisms contributing to apalutamide and darolutamide cross-resistance, AR-V7 was knocked down in C4-2B MDVR cells followed by treatment with apalutamide and darolutamide. As shown in Fig. 2C, AR-V7 knockdown reduced cell growth in C4-2B MDVR cells, and apalutamide or darolutamide further inhibited cell growth in AR-V7 knockdown groups. We also determined the effects of AR-V7 knockdown by western blot. As shown in Fig. 2D, AR-V7 was successfully knocked down by AR-V7 siRNA. AR-V7 knockdown significantly suppressed c-Myc expression but had no effect on AKR1C3 expression. Similar results were observed in CWR22Rv1 cells showing that knockdown of AR-V7 significantly re-sensitized CWR22Rv1 cells to apalutamide and darolutamide treatment (Fig. 2E). Additionally, it reduced c-Myc but no effect on AKR1C3 expression (Fig. 2F). Taken together, these results suggest that AR-V7 confers apalutamide and darolutamide resistance.
Figure 2. AR-V7 confers apalutamide and darolutamide resistance.
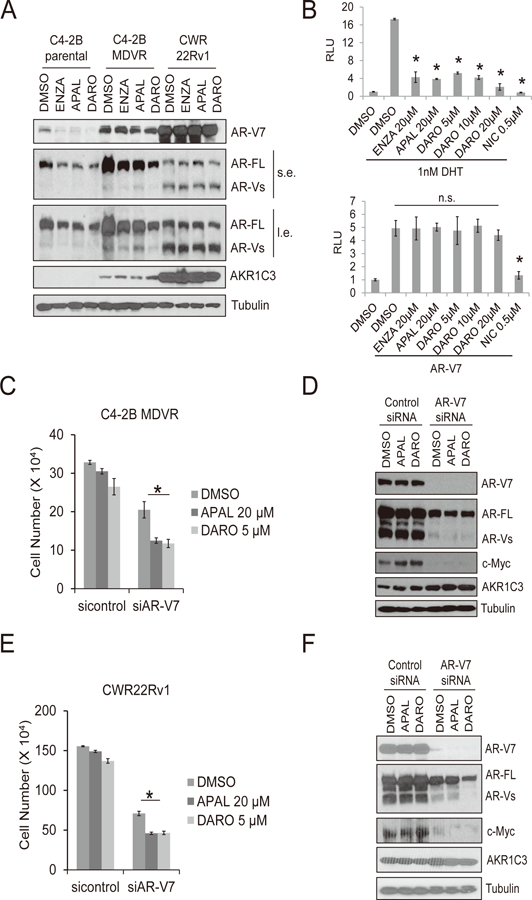
A. C4-2B parental, C4-2B MDVR and CWR22Rv1 cells were treated with 20 μM enzalutamide, 20 μM apalutamide or 5 μM darolutamide for 3 days. Whole cell lysates were collected and subjected to western blot. B. C4-2B cells were transfected with PSA luciferase promoter in CS-FBS condition for 24 hours, followed by treatment with 1 nM DHT with or without 20µM enzalutamide, 20 μM apalutamide, 5, 10 or 20 μM darolutamide or 0.5 μM niclosamide overnight. Whole cell lysates were collected and subjected to luciferase assay (top). C4-2B cells were co-transfected with PSA luciferase promoter and AR-V7 in CS-FBS conditions for 24 hours, followed by treatment with 20µM enzalutamide, 20 μM apalutamide, 5, 10 or 20 μM darolutamide or 0.5 μM niclosamide overnight. Whole cell lysates were collected and subjected to luciferase assay (bottom). C. C4-2B MDVR cells were transiently transfected with control siRNA or AR-V7 siRNA. Following transfection, cells were treated with 20 µM apalutamide or 5 μM darolutamide and cell numbers were determined after 5 days. D. Whole cell lysates were collected and subjected to western blot. E. CWR22Rv1 cells were transiently transfected with control siRNA or AR-V7 siRNA. Following transfection, cells were treated with 20 µM apalutamide or 5 μM darolutamide and cell numbers were determined after 5 days. F. Whole cell lysates were collected and subjected to western blot. Enza: enzalutamide, APAL: apalutamide, Abi: abiraterone acetate, DARO: darolutamide. NIC: niclosamide. n.s.: not-significant. * p<0.05
Targeting AKR1C3 re-sensitizes resistant cells to apalutamide and darolutamide through AR-V7 inhibition
AKR1C3 mediated intracrine androgen biosynthesis is one of the critical mechanisms of enzalutamide resistance (19). Our recent publication established that AKR1C3 binds to AR-V7 and stabilizes the protein level in resistant prostate cancer cells (18). To test if the AKR1C3/AR-V7 axis confers cross-resistance to apalutamide and darolutamide, we determined if knockdown of AKR1C3 re-sensitizes the resistant cells to apalutamide and darolutamide. As shown in Fig. 3A, knockdown of AKR1C3 by lenti-AKR1C3 shRNA inhibited cell growth. AKR1C3 knockdown combined with apalutamide/darolutamide further reduced cell number compared to the control group. We further confirmed that knockdown of AKR1C3 significantly down-regulated AR, AR-V7, and c-Myc expression in C4-2B MDVR cells (Fig. 3B). Similar results were found in CWR22Rv1 cells. Knockdown of AKR1C3 re-sensitized CWR22Rv1 cells to apalutamide and darolutamide treatment (Fig. 3C), and significantly hampered AR, AR-V7 and c-Myc expression (Fig. 3D). To further confirm AKR1C3 is involved in apalutamide and darolutamide resistance, we used C4-2B-AKR1C3 cells to test whether exogenous expression of AKR1C3 induces apalutamide or darolutamide resistance. As shown in Fig. 3E, C4-2B-AKR1C3 cells exhibited greater resistance to apalutamide and darolutamide than C4-2B-neo cells. Western blot showed that both AR and AR-V7 levels were increased in C4-2B-AKR1C3 cells compared to those in the C4-2B-neo cells (Fig. 3F). Finally, AKR1C3 inhibitor indomethacin significantly enhanced both apalutamide and darolutamide treatment in C4-2B MDVR and CWR22Rv1 cells in a time dependent manner (Fig. 3G). At 5 days treatment, the coefficient of drug interaction (CDI) of 20 µM indomethacin with 5 µM apalutamide or 20 µM darolutamide combination treatment were 0.56 and 0.64 respectively in C4-2B MDVR cells, suggesting synergistic interactions (CDI<1). In light of these results, our data indicated that the AKR1C3/AR-V7 axis is one of the major mechanisms conferring enzalutamide/apalutamide/darolutamide cross-resistance in advanced prostate cancer.
Figure 3. AKR1C3 confers apalutamide and darolutamide resistance through AR-V7 regulation.
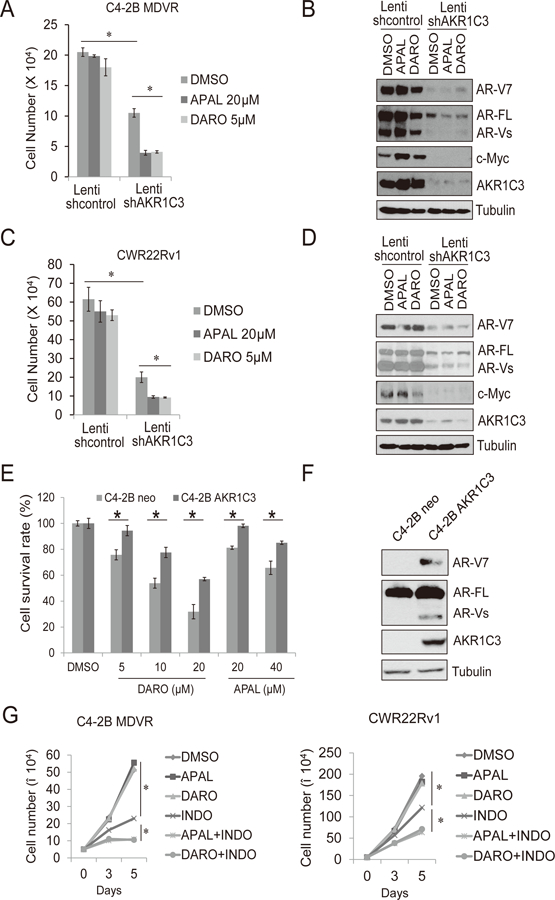
A-B. C4-2B MDVR cells were infected with lenti-control shRNA or lenti-AKR1C3 shRNA and then treated with 20 µM apalutamide or 5 μM darolutamide and cell numbers were determined after 5 days. Whole cell lysates were collected and subjected to western blot. C-D. CWR22Rv1 cells were infected with lenti-control shRNA or lenti-AKR1C3 shRNA and then treated with 20 µM apalutamide or 5 μM darolutamide and cell numbers were determined after 5 days. Whole cell lysates were collected and subjected to western blot. E-F. C4-2B neo or C4-2B AKR1C3 cells were treated with different concentrations of apalutamide or darolutamide for 3 days, total cell numbers were counted and cell survival rate (%) was calculated. Whole cell lysates were collected and subjected to western blot. G. C4-2B MDVR and CWR22Rv1 cells were treated with 20 µM apalutamide or 5 μM darolutamide or 20 µM indomethacin, alone or in combination, total cell numbers were determined on 3 and 5 days. APAL: apalutamide, DARO: darolutamide. INDO: indomethacin * p<0.05.
Chronic apalutamide treatment in C4-2B cells upregulates the steroid biosynthesis pathway
Apalutamide is a new next-generation anti-androgen with a molecular structure very similar to enzalutamide (6). To investigate apalutamide resistance, we have successfully generated a C4-2B apalutamide resistant cell line (C4-2B APALR). As shown in Fig. 4A, C4-2B parental cells were sensitive to apalutamide treatment in a dose dependent manner; however, C4-2B APALR cells were resistant to the treatment. To test if cross-resistance exists between apalutamide and other androgen signal-targeting agents in this cell model, C4-2B APALR cells were treated with apalutamide, darolutamide, enzalutamide and abiraterone. Both cell growth (Fig. 4B) and colony formation assays (Fig. 4C–D) confirmed that C4-2B APALR cells exhibited robust resistance not only to apalutamide but also to darolutamide, enzalutamide and abiraterone. We then examine the transcriptional alterations of steroid hormone biosynthesis related genes in C4-2B APALR cells compared to the parental cells. The steroid hormone biosynthesis related genes were significantly upregulated in C4-2B APALR cells compared to C4-2B parental cells (Fig. 4E). Results from western blot indicated that the protein levels of both AKR1C3 and AR-V7 are overexpressed in C4-2B APALR cells. However, these levels were lower than those in C4-2B MDVR cells (Fig. 4F). These results strongly imply that the AKR1C3/AR-V7 axis is critical in conferring cross-resistance between apalutamide and other anti-androgens.
Figure 4. Chronic apalutamide treatment in C4-2B cells activates the steroid biosynthesis pathway through AKR1C3 upregulation.
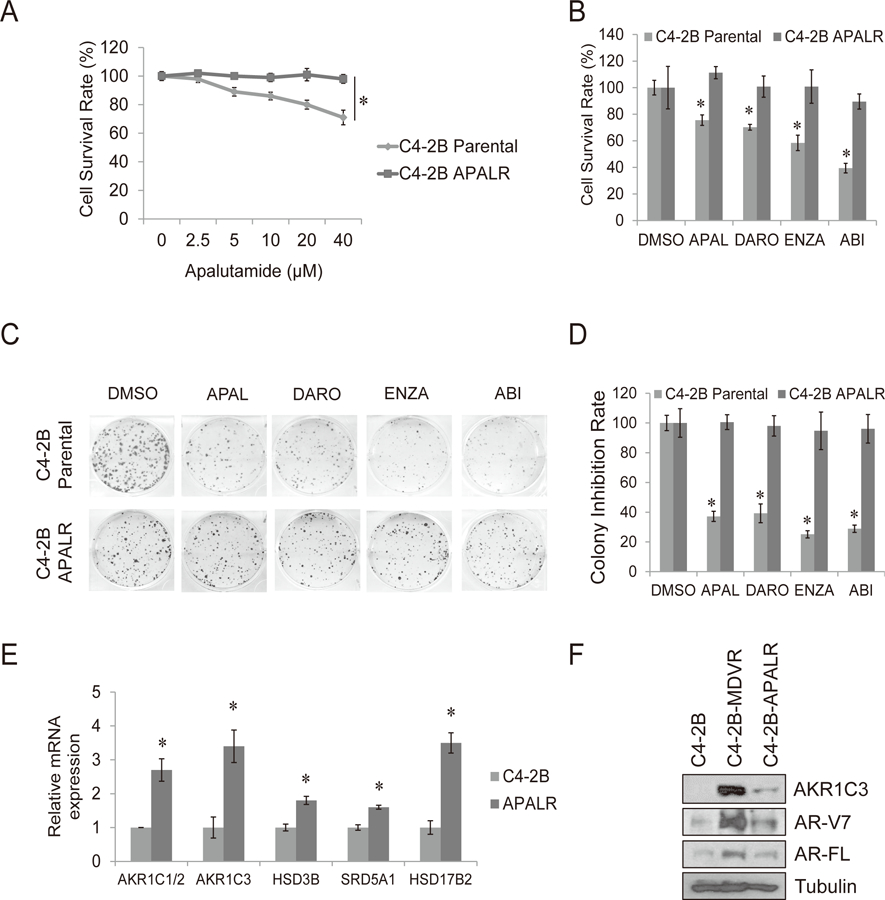
A. C4-2B parental and C4-2B APALR cells were treated with different concentration of apalutamide for 3 days, total cell numbers were determined and cell survival rate was calculated. B. C4-2B parental and C4-2B APALR cells were treated with 20 μM apalutamide, 5 μM darolutamide, 20 μM enzalutamide or 10 μM abiraterone and cell numbers were determined after 3 days. C-D. The clonogenic ability of C4-2B parental and C4-2B APALR cells treated with 20 μM apalutamide, 5 μM darolutamide, 20 μM enzalutamide or 10 μM abiraterone was analyzed. E. The genes involved in steroid hormone biosynthesis pathway were determined by qRT-PCR. F. Whole cell lysates were collected from C4-2B parental, C4-2B MDVR and C4-2B ARNR cells and subjected to western blot. Enza: enzalutamide, APAL: apalutamide, Abi: abiraterone acetate, DARO: darolutamide. * p<0.05
Targeting AKR1C3 resensitize apalutamide resistant cells to apalutamide and enzalutamide
To confirm the contribution of the AKR1C3/AR-V7 axis in cross-resistance between enzalutamide and apalutamide, C4-2B APALR cells were treated with enzalutamide or apalutamide with or without the knockdown of AKR1C3 by lenti-AKR1C3 shRNA (Fig. 5A). Growth of C4-2B APALR cells was largely inhibited by AKR1C3 knockdown. AKR1C3 knockdown in combination with enzalutamide/apalutamide treatment further reduced cell growth. The knockdown of AKR1C3 was accompanied with reduced expression levels of AR-FL, AR-V7 and c-Myc (Fig. 5B), which is consistent with what is observed in C4-2B MDVR cells (18). Finally, C4-2B APALR cells were treated with the AKR1C3 inhibitor indomethacin or apalutamide, alone and in combination. Indomethacin effectively resensitized C4-2B APALR cells to apalutamide treatment (Fig. 5C), the CDI of 20 μM apalutamide with 20 μM indomethacin or 40 μM indomethacin combination treatment were 0.61 and 0.63 respectively, suggesting drug synergism (CDI<1). The results were confirmed by clonogenic assay. 20 μM apalutamide has no effects on colony formation in C4-2B APALR cells, single indomethacin treatment reduced the colony formation and combination treatment significantly suppressed colony size and numbers (Fig. 5D–5E). Collectively, our data suggest that targeting AKR1C3 in apalutamide resistant cells could efficiently reverse cross-resistance between apalutamide and enzalutamide.
Figure 5. Targeting AKR1C3 resensitizes apalutamide resistant cells to apalutamide and enzalutamide.
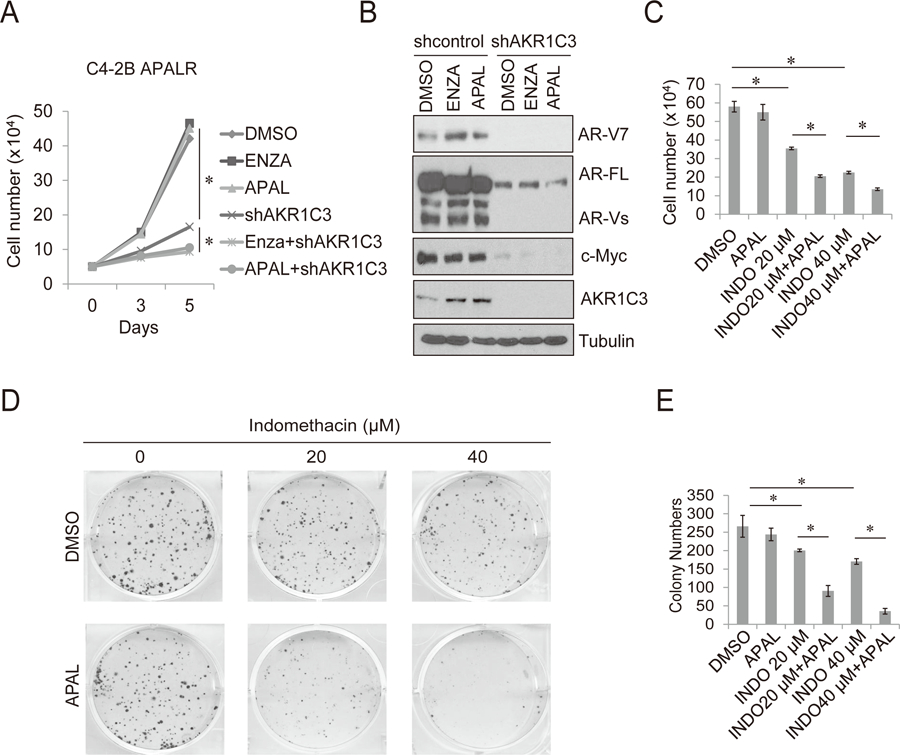
A. C4-2B APALR cells were treated with 20 µM enzalutamide, 20 µM apalutamide or infected with lenti-AKR1C3 shRNA, alone or in combination. Total cell numbers were determined on 3 and 5 days. B. C4-2B APALR cells were infected with lenti-control shRNA or lenti-AKR1C3 shRNA and then treated with 20 µM enzalutamide or 20 µM apalutamide. After 5 days, whole cell lysates were collected and subjected to western blot. C. C4-2B APALR cells treated with 0, 20, 40 µM indomethacin with or without 20 μM apalutamide and cell numbers were determined after 5 days. D-E. The clonogenic ability of C4-2B ARNR cells treated with 0, 20, 40 µM indomethacin with or without 20 μM apalutamide was analyzed. APAL: apalutamide, ENZA: enzalutamide. INDO: indomethacin * p<0.05.
Discussion
Our study provides the first experimental evidence to show that cross-resistance exists between enzalutamide/abiraterone and the newly FDA approved apalutamide and darolutamide. We demonstrated that AKR1C3/AR-V7 axis plays a critical role in cross-resistance among the next generation anti-androgen drugs. Results from this study and our previous publications (25) complete a blueprint regarding the cross-resistance. These findings strengthened the concept that since AR-targeting drugs share similar action of mechanisms, there is non-negligible cross-resistance among them.
The past decade has witnessed the remarkable development of the next generation androgen-targeting agents in the treatment of advanced prostate cancer, which vastly improved the survival outcomes of prostate cancer patients (6–9). However, challenges remain that after a period of efficient treatment, tumor cells inevitably become resistant. Thus, unveiling the mechanism underlying drug-resistance is of great importance to optimize treatment decisions and overcome resistance. In addition, more therapeutic options become available with additional possibilities of diverse sequential treatments. Hence, identifying the key factors responsible for the cross-resistance among different therapies is essential for determining the optimal long-term sequential treatment schemes for mCRPC patients.
The mainstay treatments for advanced prostate cancer consist of two major parts, i.e.: AR-targeting therapy and cytotoxic chemotherapy. Clinical data has implied the presence of cross-resistance between enzalutamide and abiraterone, while docetaxel-based chemotherapy is still effective for mCRPC patients after the failure of enzalutamide or abiraterone and vice versa (26–29). These phenomena were validated in vitro by our previous work using the drug-resistant cell line models we have developed (25). Apalutamide and darolutamide were recently approved for the treatment of non-metastatic (M0) CRPC in clinical settings and significantly extended the metastasis-free survival (MFS) and overall survival (OS) in CRPC patients over placebo (6,13). Recently, apalutamide was also approved by the FDA to treat mCSPC patient due to the fact that the drug was able to prolong the radiographic progression-free survival (rPFS) and OS in the setting of this patient population (30). Darolutamide expresses better in vitro activity than other anti-androgens which is consistent with our present findings. Darolutamide shows higher binding affinity to the AR than enzalutamide and apalutamide in vitro and can target AR mutants, such as F877L and W742L, which drives other anti-androgens resistance (31). Nevertheless, a fact that should be noticed is that darolutamide has worse pharmacokinetic parameters in vivo compared to enzalutamide and apalutamide (32,33). In the recently published phase III clinical trial, the darolutamide oral dose is 1,200 mg per day which is 7.5 fold and 5 fold higher dose than the enzalutamide or apalutamide oral doses, respectively (NCT02799602). Although darolutamide was approved in the treatment of prostate cancer patients, to address its bioavailability is still a necessary topic in future research.
Apalutamide has a very similar chemical structure to enzalutamide and RD162. Compared to RD162, apalutamide only differs by a carbon atom replaced with a nitrogen atom in one of the phenyl rings (4,24). Based on the similar structures of apalutamide to enzalutamide, we postulated that both drugs might share the same resistant mechanisms. Intriguingly, darolutamide has the potential to overcome enzalutamide resistance by targeting AR mutants, such as F877L, which uses enzalutamide or apalutamide as an AR agonist instead of antagonist (34,35). In our study, we found that darolutamide is more effective than enzalutamide and apalutamide at the same dose in C4-2B parental cells in vitro. However, both enzalutamide and abiraterone resistant cell models are still cross-resistant to apalutamide and darolutamide treatment; and importantly, acquired apalutamide resistant prostate cancer cells are cross-resistant to all other anti-androgens. Because apalutamide currently is approved to treat mCSPC patient with ADT, the fact we should not overlook is after these patients become resistant to apalutamide, they are expected to be resistant to other anti-androgens as well, such as enzalutamide, darolutamide and abiraterone based on our pre-clinical data.
Both AR-V7 and AKR1C3 play important roles in leading disease progression and conferring drug-resistance prostate cancer cells (14–19,23,36–38). AR-V7 is a truncated AR variant that lacks the ligand-binding domain and is able to directly translocate into the nucleus without activation by androgen (39). Thus, current anti-androgen treatment cannot target AR-V7, and the tumor cells harboring AR-V7 may escape full length AR targeting therapies and survive. Moreover, detection of AR-V7 in circulating tumor cells is strongly correlated to enzalutamide- and abiraterone-resistance clinically (14). In our previous studies, we have shown that niclosamide, an AR-V7 inhibitor, significantly reverses drug-resistance in enzalutamide and abiraterone therapy (15,23,36). A clinical trial using niclosamide in combination with abiraterone to treat enzalutamide resistant prostate cancer patient is currently ongoing (NCT02807805). However, opposition to AR-V7’s role in the development of anti-androgen resistant CRPC is surging, and the exact position of AR-variants in contributing therapeutic resistance is in debates (40). By using reagents that can bind to AR but not AR-V7 and induce AR degradation, Steven et al. showed that AR instead of AR-V7 is the prominent driver of enzalutamide-resistance (41), which challenged the role of AR-V7 in enzalutamide resistance. Additionally, previous study indicated AR-V7 remains dependent of full length AR in gain of function (42). Intriguingly, AR-V7 can form a heterodimer with full length AR in controlling gene regulation. Targeting AR might block the downstream effector genes of the AR/AR-V7 dimers. Above all, the complex role of AR-V7 in the development of drug-resistant CRPC remains to be revealed in future research.
On the other hand, AKR1C3, a multifunctional enzyme, induces enzalutamide- and abiraterone-resistance by converting the weak androgens androstenedione and 5α-androstenedione to the more active androgens testosterone (T) and dihydrotestosterone (DHT) (19,23). Various inhibitors have been developed to target AKR1C3 activation in xenograft tumor models (43), including indomethacin (44), GTx-560 (45) and ASP9521 (46), however, most untested in the clinical settings so far. Thus, there is a great unmet need to engage AKR1C3 inhibitors for clinical trial initiation. Previously, we have reported that indomethacin, a commonly used nonsteroidal anti-inflammatory drug (NSAID), could efficiently inhibit AKR1C3 activity both in vivo and in vitro (23). Given the promising effect of indomethacin, a phase I/II trial (NCT02935205) aiming at exploring its effect in treating mCRPC patients in combination with enzalutamide is ongoing. Moreover, we recently found that overexpression of AKR1C3 increases AR-V7 expression (18). This finding was supported clinically by a study where positive AKR1C3 expression from prostate biopsy samples was associated with higher positivity of AR-V7 (7/38 [18.4%] vs. 4/76 [5.3%], p=0.025) (47). Consistent with our previous findings in C4-2B MDVR cells, here we showed that inhibition of AKR1C3 decreases AR and AR-V7 levels in C4-2B APALR cells. AKR1C3 has been shown to bind to AR-FL as a co-activator in a previous study (45). Our subsequent study found that AKR1C3 binds with AR-V7 and promotes AR-V7 protein stabilization via the ubiquitin-mediated proteasome pathway. Notably, we found that overexpression of AKR1C3 significantly stabilizes both AR-FL and AR-V7 proteins. Inhibition of AKR1C3 by indomethacin reduced both AR-FL and AR-V7 protein half-lives which could be reversed by proteasome inhibitor MG132 (18). Thus, AKR1C3 may stabilize AR similar to AR-V7 via the same mechanism through ubiquitin-proteasome system regulation. Additionally, we previously showed that knockdown of AR-V7 in C4-2B MDVR cells decreases the level of c-Myc. In the current study, this phenomenon was confirmed in C4-2B APALR cells. c-Myc is an important proto-oncogene in prostate cancer that can deregulate the AR and AR variant signaling pathway. c-Myc activates AR target genes and enhances AR and AR variant protein stability (48). Here our data showed that c-Myc is also a downstream target of AR-V7 which suggests a possible reciprocal regulation of AR-V7 and c-Myc. Knockdown of AKR1C3 suppresses both AR-V7 and c-Myc expression in resistant cells. However, the interplay among AKR1C3, AR-V7 and c-Myc needs to be further investigated. Overall, our findings indicate that AKR1C3/AR-V7 axis plays an important role in controlling anti-androgen resistance.
According to the National Comprehensive Cancer Network (NCCN) guidelines (49), among the four next generation AR-targeting agents, enzalutamide has the widest scope of application and could be used in mCSPC, M0 CRPC or mCRPC stage. Abiraterone is approved in mCSPC and mCRPC patients whereas apalutamide is approved in mCSPC and M0 CRPC patients. So far, darolutamide is only approved in M0 CRPC stage. However, several phase III trials (NCT02799602, NCT02489318) are now ongoing and the application spectrum of these drugs will certainly enlarge in the near future. Thus, our study provides important preclinical clues about the values of AKR1C3 and AR-V7 in distinguishing the optimal candidates of these AR-targeting treatments from the potential non-responders. In addition, we showed that the combination of anti-androgens and the AKR1C3 inhibitor indomethacin effectively reverses the drug resistance, which may be a new treatment strategy to maximize the therapeutic efficacy.
Collectively, our study provides the information for clinicians to have a better understanding about the mechanisms underlying drug-resistance of different next generation AR-targeting drugs. The results demonstrated that enzalutamide and abiraterone resistant cells are cross-resistant to apalutamide and darolutamide, and that AKR1C3/AR-V7 plays a critical role in conferring the cross-resistance among these next generation anti-androgen drugs.
Acknowledgments
This work was supported in part by grants NIH/NCI CA168601, CA179970, DOD PC150229, and the U.S. Department of Veterans Affairs, Office of Research & Development BL&D grant number I01BX0002653 (A.C. G), a Research Career Scientist Award (A.C. G). A.C.G is also a Research Career Scientist at VA Northern California Health Care System, Mather, California.
Financial Support: This work is part by grants NIH/NCI CA168601, CA179970, DOD PC150229, and the U.S. Department of Veterans Affairs, Office of Research & Development BL&D grant number I01BX0002653 (A.C.G)
Footnotes
Conflicts of Interest: All authors have no conflicts of interest.
References
- 1.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine 2004;351(15):1502–12 doi 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nature medicine 2004;10(1):33–9 doi 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 3.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. The American journal of pathology 2004;164(1):217–27 doi 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324(5928):787–90 doi 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005;23(32):8253–61 doi 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 6.Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. The New England journal of medicine 2018;378(15):1408–18 doi 10.1056/NEJMoa1715546. [DOI] [PubMed] [Google Scholar]
- 7.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine 2011;364(21):1995–2005 doi 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine 2012;367(13):1187–97 doi 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 9.Shore N, Zurth C, Fricke R, Gieschen H, Graudenz K, Koskinen M, et al. Evaluation of Clinically Relevant Drug-Drug Interactions and Population Pharmacokinetics of Darolutamide in Patients with Nonmetastatic Castration-Resistant Prostate Cancer: Results of Pre-Specified and Post Hoc Analyses of the Phase III ARAMIS Trial. Targeted oncology 2019;14(5):527–39 doi 10.1007/s11523-019-00674-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer discovery 2013;3(9):1020–9 doi 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 11.Rathkopf DE, Smith MR, Ryan CJ, Berry WR, Shore ND, Liu G, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Annals of oncology : official journal of the European Society for Medical Oncology 2017;28(9):2264–71 doi 10.1093/annonc/mdx283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. The Lancet Oncology 2014;15(9):975–85 doi 10.1016/S1470-2045(14)70240-2. [DOI] [PubMed] [Google Scholar]
- 13.Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. The New England journal of medicine 2019;380(13):1235–46 doi 10.1056/NEJMoa1815671. [DOI] [PubMed] [Google Scholar]
- 14.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine 2014;371(11):1028–38 doi 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(12):3198–210 doi 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer research 2012;72(14):3457–62 doi 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer research 2013;73(2):483–9 doi 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu C, Yang JC, Armstrong CM, Lou W, Liu L, Qiu X, et al. AKR1C3 Promotes AR-V7 Protein Stabilization and Confers Resistance to AR-Targeted Therapies in Advanced Prostate Cancer. Molecular cancer therapeutics 2019;18(10):1875–86 doi 10.1158/1535-7163.MCT-18-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, et al. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer research 2015;75(7):1413–22 doi 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Molecular cancer therapeutics 2017;16(1):35–44 doi 10.1158/1535-7163.MCT-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Gao JL, Ji JW, Gao M, Yin QS, Qiu QL, et al. Cytotoxicity enhancement in MDA-MB-231 cells by the combination treatment of tetrahydropalmatine and berberine derived from Corydalis yanhusuo W. T. Wang. Journal of intercultural ethnopharmacology 2014;3(2):68–72 doi 10.5455/jice.20140123040224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Zhu Y, Lou W, Nadiminty N, Chen X, Zhou Q, et al. Functional p53 determines docetaxel sensitivity in prostate cancer cells. The Prostate 2013;73(4):418–27 doi 10.1002/pros.22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C, Armstrong CM, Lou W, Lombard AP, Cucchiara V, Gu X, et al. Niclosamide and Bicalutamide Combination Treatment Overcomes Enzalutamide- and Bicalutamide-Resistant Prostate Cancer. Molecular cancer therapeutics 2017;16(8):1521–30 doi 10.1158/1535-7163.MCT-16-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer research 2012;72(6):1494–503 doi 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lombard AP, Liu L, Cucchiara V, Liu C, Armstrong CM, Zhao R, et al. Intra versus Inter Cross-resistance Determines Treatment Sequence between Taxane and AR-Targeting Therapies in Advanced Prostate Cancer. Molecular cancer therapeutics 2018;17(10):2197–205 doi 10.1158/1535-7163.Mct-17-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bianchini D, Lorente D, Rodriguez-Vida A, Omlin A, Pezaro C, Ferraldeschi R, et al. Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration-resistant prostate cancer (CRPC) pre-treated with docetaxel and abiraterone. European journal of cancer 2014;50(1):78–84 doi 10.1016/j.ejca.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 27.Loriot Y, Bianchini D, Ileana E, Sandhu S, Patrikidou A, Pezaro C, et al. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Annals of oncology : official journal of the European Society for Medical Oncology 2013;24(7):1807–12 doi 10.1093/annonc/mdt136. [DOI] [PubMed] [Google Scholar]
- 28.Noonan KL, North S, Bitting RL, Armstrong AJ, Ellard SL, Chi KN. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Annals of oncology : official journal of the European Society for Medical Oncology 2013;24(7):1802–7 doi 10.1093/annonc/mdt138. [DOI] [PubMed] [Google Scholar]
- 29.Schrader AJ, Boegemann M, Ohlmann CH, Schnoeller TJ, Krabbe LM, Hajili T, et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. European urology 2014;65(1):30–6 doi 10.1016/j.eururo.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 30.Chi KN, Agarwal N, Bjartell A, Chung BH, Pereira de Santana Gomes AJ, Given R, et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. The New England journal of medicine 2019;381(1):13–24 doi 10.1056/NEJMoa1903307. [DOI] [PubMed] [Google Scholar]
- 31.Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Scientific reports 2015;5:12007 doi 10.1038/srep12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sulochana SP, Saini NK, Daram P, Polina SB, Mullangi R. Validation of an LC-MS/MS method for simultaneous quantitation of enzalutamide, N-desmethylenzalutamide, apalutamide, darolutamide and ORM-15341 in mice plasma and its application to a mice pharmacokinetic study. Journal of pharmaceutical and biomedical analysis 2018;156:170–80 doi 10.1016/j.jpba.2018.04.038. [DOI] [PubMed] [Google Scholar]
- 33.Fizazi K, Smith MR, Tombal B. Clinical Development of Darolutamide: A Novel Androgen Receptor Antagonist for the Treatment of Prostate Cancer. Clinical genitourinary cancer 2018;16(5):332–40 doi 10.1016/j.clgc.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 34.Prekovic S, van Royen ME, Voet AR, Geverts B, Houtman R, Melchers D, et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Molecular cancer therapeutics 2016;15(7):1702–12 doi 10.1158/1535-7163.MCT-15-0892. [DOI] [PubMed] [Google Scholar]
- 35.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer discovery 2013;3(9):1030–43 doi 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 36.Liu C, Armstrong C, Zhu Y, Lou W, Gao AC. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget 2016;7(22):32210–20 doi 10.18632/oncotarget.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17(18):5913–25 doi 1078-0432.CCR-11-0728 [pii] 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrader AJ, Schrader MG, Cronauer MV. Re: Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-resistant Prostate Cancer Cell Lines. European urology 2013;64(1):169–70 doi S0302-2838(13)00387-4 [pii] 10.1016/j.eururo.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 39.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. The Journal of biological chemistry 2012;287(23):19736–49 doi 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo J, Attard G, Balk SP, Bevan C, Burnstein K, Cato L, et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. European urology 2018;73(5):715–23 doi 10.1016/j.eururo.2017.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia (New York, NY) 2020;22(2):111–9 doi 10.1016/j.neo.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences of the United States of America 2010;107(39):16759–65 doi 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penning TM. Aldo-Keto Reductase (AKR) 1C3 inhibitors: a patent review. Expert opinion on therapeutic patents 2017;27(12):1329–40 doi 10.1080/13543776.2017.1379503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flanagan JU, Yosaatmadja Y, Teague RM, Chai MZ, Turnbull AP, Squire CJ. Crystal structures of three classes of non-steroidal anti-inflammatory drugs in complex with aldo-keto reductase 1C3. PloS one 2012;7(8):e43965 doi 10.1371/journal.pone.0043965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19(20):5613–25 doi 10.1158/1078-0432.CCR-13-1151. [DOI] [PubMed] [Google Scholar]
- 46.Kikuchi A, Furutani T, Azami H, Watanabe K, Niimi T, Kamiyama Y, et al. In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17beta-hydroxysteroid dehydrogenase type 5 (17betaHSD5; AKR1C3). Investigational new drugs 2014;32(5):860–70 doi 10.1007/s10637-014-0130-5. [DOI] [PubMed] [Google Scholar]
- 47.Zhao J, Zhang M, Liu J, Liu Z, Shen P, Nie L, et al. AKR1C3 expression in primary lesion rebiopsy at the time of metastatic castration-resistant prostate cancer is strongly associated with poor efficacy of abiraterone as a first-line therapy. The Prostate 2019;79(13):1553–62 doi 10.1002/pros.23875. [DOI] [PubMed] [Google Scholar]
- 48.Bai S, Cao S, Jin L, Kobelski M, Schouest B, Wang X, et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019;38(25):4977–89 doi 10.1038/s41388-019-0768-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarwar M, Semenas J, Miftakhova R, Simoulis A, Robinson B, Gjorloff Wingren A, et al. Targeted suppression of AR-V7 using PIP5K1alpha inhibitor overcomes enzalutamide resistance in prostate cancer cells. Oncotarget 2016;7(39):63065–81 doi 10.18632/oncotarget.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]