

Abstract
We report herein the discovery of exceptionally potent and orally bioavailable PROTAC AR degraders with ARD-2585 being the most promising compound. ARD-2585 achieves DC50 values of ≤0.1 nM in the VCaP cell line with AR gene amplification and in the LNCaP cell line carrying an AR mutation. It potently inhibits cell growth with IC50 values of 1.5 and 16.2 nM in the VCaP and LNCaP cell lines, respectively, and achieves excellent pharmacokinetics and 51% of oral bioavailability in mice. It is more efficacious than enzalutamide in inhibition of VCaP tumor growth and does not cause any sign of toxicity in mice. ARD-2585 is a promising AR degrader for extensive investigations for the treatment of advanced prostate cancer.
Graphical Abstract

INTRODUCTION
Metastatic castration-resistant prostate cancer (mCRPC) remains incurable and lethal. Androgen receptor (AR) antagonists, such as enzalutamide, apalutamide, and darolutamide, are effective for the treatment of mCRPC.1‘2 Unfortunately, patients treated with these AR antagonists ultimately develop drug resistance. In the majority of tumors resistant to AR antagonists, the AR signaling continues to be functional and drives tumor growth and progression.3 Some of the major resistance mechanisms to AR antagonists include AR gene amplification and mutation and expression of AR variants.4,5 Therefore, new therapeutic strategies to effectively target the AR signaling in tumors resistant to AR antagonists are urgently needed.
It has been proposed that induced degradation of AR protein could be potentially more effective in targeting the AR signaling than traditional AR antagonists.6 Similar to classical selective estrogen receptor degraders, selective androgen receptor degraders (SARDs) were discovered.7 SARDs bind to the ligand-binding domain in AR and disrupt AR-coregulator interactions, leading to proteasome-dependent AR degradation.8 Another new and promising strategy to achieve induced AR degradation is based on the proteolysis-targeting chimera (PROTAC) technology platform.9
A PROTAC-based AR degrader is a bifunctional small molecule, consisting of an AR ligand that binds to AR protein, and a ligand that binds to and recruits an E3 ligase complex, tethered together through a linker.10 In 2008, the Crews laboratory reported the first PROTAC AR degrader (1), which was designed using a ligand for the MDM2 E3 ligase.11 While compound 1 degraded AR protein in cells only at micromolar concentrations, it provided an important proof-of-concept. Subsequently, other PROTAC AR degraders were reported using different E3 ligase degradation systems, such as inhibitors of apoptosis protein (IAPs), the von Hippel–Lindau (VHL)/cullin 2, and cereblon/cullin 4A. Scientists from Takeda reported an IAP-based PROTAC AR degrader (2), which was capable of inducing AR degradation at 1 μM concentration.12 Subsequently, a number of highly potent PROTAC-based AR degraders designed using ligands for VHL have been reported. In 2018, Crews et al. reported ARCC-4 (3) as a potent AR degrader designed using a VHL ligand, which achieves AR degradation in multiple cell lines at low nanomolar concentrations.6 Our laboratory reported ARD-69 (4) and ARD-266 (5), which were also designed using VHL ligands.13,14 ARD-69 potently induces AR degradation in cells, effectively reduces AR protein in tumor tissues, and inhibits tumor growth in vivo.13,14 PROTAC AR degraders such as 615 and TD-802 (7)16 were designed using a cereblon ligand. While compound 6 is a weak AR degrader, TD-802 potently degrades AR protein and inhibits cancer cell growth in AR+ cancer cells.15,16 In addition, TD-802 has good microsomal stability and in vivo pharmacokinetic (PK) properties and is capable of retarding tumor growth in the VCaP xenograft model in mice.16
ARV-110 (8) discovered by scientists at Arvinas Inc. was the first-in-class potent and orally active AR degrader advanced into clinical development and its chemical structure was recently disclosed.17 ARV-110 degrades AR protein and inhibits cell growth at low nM concentrations in the LNCaP and VCaP cell lines.18 Importantly, when orally administered to mice, ARV-110 is more efficacious than enzalutamide in inhibition of tumor growth in AR+ prostate cancer xenograft models. Initial clinical data showed that ARV-110 is well tolerated, effectively reduces AR protein in tumor tissue in patients, and achieves clinical objective responses upon oral administration.19 The data for ARV-110 suggest that PROTAC AR degraders are promising new therapies for the treatment of AR+ mCRPC.
Herein, we describe our design, synthesis, and extensive evaluation of a series of PROTAC AR degraders, which led to the discovery of ARD-2585 as an exceptionally potent and orally active AR degrader.
RESULTS AND DISCUSSION
Design of Potent and Orally Bioavailable PROTAC AR Degraders.
A PROTAC degrader consists of a ligand for the target protein of interest, a ligand to bind to and recruit an E3 ligase complex, and a linker tethering these two ligands together. As can be seen from those PROTAC AR degraders in Figure 1, both VHL and cereblon have been used for the design of potent PROTAC AR degraders. However, previously reported potent VHL ligands have MW > 400 and are peptidomimetics. In comparison, cereblon ligands such as thalidomide and lenalidomide have MW of ~250 and possess excellent physiochemical and PK properties. Hence, for the design of orally bioavailable PROTAC AR degraders, we decided to employ cereblon ligands.
Figure 1.

Chemical structures of previously reported representative PROCTAC AR degraders and enzalutamide.
A number of AR ligands have been used in designed PROTAC AR degraders. Compound 9 was previously reported by Pfizer scientists as a potent AR ligand,20 and compound 10, which is a more soluble analogue of 9, has been used in the design of PROTAC AR degraders, including; ARV-110, which was developed by scientists at Arvinas.21,22 We chose to employ compounds 9 and 10 as the AR ligands in our initial exploration in the design and synthesis of PROTAC AR degraders in the present study (Figure 2).
Figure 2.

AR ligands used in our initial design of PROTAC AR degraders.
In a PROTAC degrader, the structure of the linker plays a key role in inducing degradation of the target protein. We thus first determined the optimal linker lengths needed in our AR PROTAC molecules for potent and effective AR degradation. We tethered compound 9 through the para-position of its phenyl group to the 5-position of the isoindoline-1,3-dione moiety in thalidomide through an amine group on both ends with linear alkyl groups of different lengths, which resulted in compounds 11–19 (Table 1). These compounds were evaluated by Western blotting with 24 h treatment time for their ability to reduce the AR protein level in the AR-positive (AR+) VCaP cell line. Their DC50 and maximum degradation (Dmax) values are shown in Table 1 and Figure S1. ARV-110 was also evaluated under the same assay conditions for comparison.
Table 1.
Identification of Optimal Linker Lengths in PROTAC AR Degraders

| |||
|---|---|---|---|
| Compound No | Linker | AR degradation in VCaP cell line |
|
| DC50 (nM) | Dmax (%) | ||
| ARV-110 | 1.6 | 98 | |
| 11 | –NH(CH2)3NH– | 1.0 | 85 |
| 12 | –NH(ch2)4NH– | 1.1 | 89 |
| 13 | –NH(ch2)5NH– | 0.2 | 95 |
| 14 | –NH(ch2)6NH– | 0.6 | 99 |
| IS | –NH(ch2)7NH– | 0.7 | 99 |
| 16 | –NH(CH2)8NH– | 1.S | 97 |
| 17 | –NH(ch2)9NH– | 3.2 | 96 |
| 18 | –NH(ch2)10NH– | >1000 | 25 |
| 19 | –NH(CH2)11NH– | >1000 | 42 |
Consistent with the reported data, we found ARV-110 to be a potent AR degrader, which achieves a DC50 of 1.6 nM and a maximum degradation (Dmax) of 98% in the VCaP cell line.
Compound 11 with a linker containing five nonhydrogen atoms is a potent AR degrader and shows a DC50 value of 1.0 nM. However, the Dmax of compound 11 is 85%, less than that of ARV-110. Increasing the linker length by one methylene group in 11 generated 12, which has a DC50 value of 1.1 nM and a Dmax of 89%. Further increasing the linker length in 12 resulted in 13, 14, and 15, which achieve DC50 values of 0.2, 0.6, and 0.7 nM and Dmax values of 95, 99, and 99%, respectively. Further increasing the linker length in 15 by one or two methylenes resulted in 16 and 17, which have DC50 values of 1.5 and 3.2 nM, and Dmax values of 97 and 96%, respectively. Compound 18 containing a –NH(CH2)10NH–linker and compound 19 containing a –NH(CH2)11NH–linker only have modest effect in reduction of AR protein with Dmax of 25 and 42% respectively, at concentrations up to 1000 nM. The degradation data for compounds 11–19 established optimal linker lengths to achieve the most potent and effective AR degradation, and the linker lengths in compounds 13, 14, and 15 are optimal for this series of compounds. In fact, compounds 13–15 are more potent than ARV-110 in inducing AR degradation based upon their DC50 values and they all have a Dmax value of >95%.
Among compounds 11–19, compound 11 with the shortest linker is a fairly potent and effective AR degrader and compound 13 is a highly potent and very effective AR degrader. We assessed their oral exposures in mice with the data summarized in Table S1. To our disappointment, both compounds 11 and 13 have very low oral exposures in mice (Table S1).
Compounds 11–19 all employ a linear and hydrophobic linker. Conformational restriction has been often used as a strategy to improve oral bioavailability of small-molecule drugs.23 Accordingly, we next designed and synthesized a series of AR degraders containing a conformationally restricted linker with a positively charged amine group with the objective of achieving improved physiochemical properties and oral bioavailability. These compounds were evaluated for their ability to reduce AR protein in the VCaP cell line and the data obtained are summarized in Table 2 and Figure S2.
Table 2.
Restriction of the Linkers of AR PROTACs
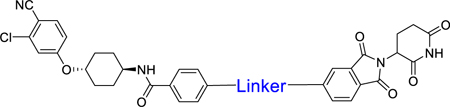
| |||
|---|---|---|---|
| Compound | Linker | AR degradation in VCaP cell line |
|
| No | DC50 (nM) | Dmax (%) | |
|
| |||
| 20 |
|
0.8 | 77 |
| 21 |

|
2.7 | 82 |
| 22 |
|
16.2 | 67 |
| 23 |
|
0.9 | 85 |
| 24 |
|
0.2 | 97 |
| 25 |
|
0.3 | 88 |
| 26 |
|
0.2 | 95 |
| 27 |
|
0.3 | 92 |
| 28 |
|
0.1 | 76 |
Compounds 20–23 have a semi-rigid linker with a positively charged piperazine group. Compound 20 has a DC50 value of 0.8 nM and a Dmax of 77%. Compound 21, which has one more methylene in its linker than 20, has a DC50 value of 2.1 nM and a Dmax of 82%. Compound 22, which has one more methylene in its linker than 21, has a DC50 value of 16.2 nM and a Dmax of 67%. Compound 23, which has one more methylene than 22 in its linker, is more potent than compound 22, with a DC50 value of 0.9 nM and a Dmax of 85%. These data showed that for all of these compounds containing a semi-rigid linker, while they are potent in inducing AR degradation based upon their DC50 values, they are not very effective degraders, failing to achieve a Dmax value of >90%.
In order to induce AR degradation most effectively, a PROTAC AR degrader needs to form a productive complex with AR protein and the cereblon/cullin 4A E3 ligase. We hypothesized that increased conformational restriction of the linker in an AR PROTAC degrader may lead to a more stable and productive ternary complex. Accordingly, we designed and synthesized compounds 24–28 in which the linker conformation is further restricted compared to compounds 20–22 (Table 2). Gratifyingly, compounds 24 and 26 are highly potent and effective AR degraders, achieving DC50 values from 0.2 to 0.3 nM and Dmax of 95–97%. Of interest, although compound 28 with an DC50 value of 0.1 nM is very potent in inducing AR degradation, its Dmax is only 76%, indicating that the linker in 28 is too short for formation of a highly productive ternary complex for efficient AR degradation.
Compound 26, with a highly rigid linker is a very potent and effective AR degrader. We evaluated the pharmacokinetics in mice obtaining the data summarized in Table 3. With intravenous administration at 1 mg/kg, compound 26 has a high exposure (AUC = 2425 h*ng/mL), an excellent volume of distribution (Vss = 3.0 L/kg), a low clearance (Cl = 0.4 L/h/kg), and a long half-life (T1/2 = 6.1 h). At 3 mg/kg oral administration, compound 26 shows a half-life of 5.6 h, a Cmax of 207 ng/mL, an AUC of 2154 h*ng/mL, and an oral bioavailability of 30%. It, therefore, has an excellent PK profile in mice and is a promising lead compound for further optimization.
Table 3.
PKs of Compounds 26, 35, and 40–45 in Mice
| male mice | IV (mg/kg) | T1/2 (h) | AUC(0–t) (h*ng/mL) | Vss (L/kg) | Cl (L/h/kg) | PO (mg/kg) | Tmax (h) | T1/2 (h) | Cmax (ng/mL) | AUC(0–t) (h*ng/mL) | F (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 26 | 1 | 6.1 | 2425 | 3.0 | 0.4 | 3 | 4.0 | 5.6 | 207 | 2154 | 30 |
| 35 | 1 | 6.6 | 2857 | 2.7 | 0.3 | 3 | 5.3 | 10.6 | 251 | 3811 | 44 |
| 40 | 1 | 12.5 | 693 | 17.6 | 1.1 | 3 | 6.0 | 27.0 | 45 | 738 | 35 |
| 41 | 1 | 7.6 | 3366 | 2.6 | 0.3 | 3 | 4.7 | 7.0 | 256 | 3001 | 29 |
| 42 | 1 | 3.8 | 1226 | 3.9 | 0.8 | 3 | 5.3 | 4.3 | 127 | 1111 | 30 |
| 43 | 2 | 5.5 | 6481 | 1.8 | 0.3 | 5 | 2.0 | 4.6 | 1140 | 8254 | 51 |
| 44 | 1 | 8.8 | 1118 | 8.3 | 0.8 | 3 | 4.0 | 8.1 | 92.2 | 1134 | 34 |
| 45 | 1 | 7.5 | 4234 | 2.1 | 0.2 | 3 | 4.7 | 26.8 | 484 | 8637 | 67 |
In a PROTAC AR degrader, the AR antagonist portion plays a critical role in binding to AR and recruiting AR to the cereblon/cullin 4A E3 ligase complex. Using compound 26 as a promising lead compound, we further modified its AR antagonist portion to investigate the structure–activity relationship of the resulting AR degraders on AR degradation. The results are summarized in Table 4 and Figure S3.
Table 4.
Structure–Activity Relationships of the AR Antagonist Portion in Our Potent AR Degrader 26
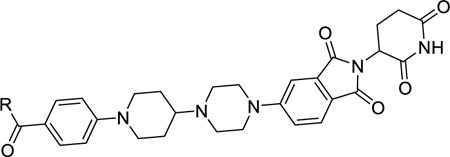
| |||
|---|---|---|---|
| Compound | R Group | AR degradation in VCaP cell line |
|
| No | DC50 (nM) | Dmax (%) | |
|
| |||
| 29 |
|
>1000 | 20 |
| 30 |
|
>1000 | 45 |
| 31 |
|
>1000 | 54 |
| 32 |
|
>1000 | 19 |
| 33 |
|
>1000 | 35 |
| 34 |
|
0.3 | 78 |
| 35 |
|
0.1 | 99 |
| 36 |
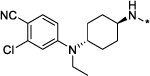
|
0.1 | 99 |
| 37 |
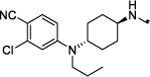
|
1.4 | 88 |
Compound 26 contains a cyclohexane ring in its AR antagonist portion. We synthesized compounds 29, 30, and 31 to investigate the effect of the ring size on AR degradation. Compounds 29 containing a cyclopentane, 30 with a cycloheptane, and 31 containing a cyclooctane are all much less potent and effective in reducing AR protein than compound 26. Compound 32 containing a piperidine and compound 33 containing an azapane group have little or no effect in reducing AR protein.
To assist with our further modifications of the AR ligand portion in compound 26, we performed computational modeling to predict the binding model of compound 9 in complex with human AR.24 Our predicted binding model suggested that compound 9 binds to AR in a manner very similar to an AR ligand S1 (Figure S4). The nitrile group in compound 9 forms hydrogen bonds with side chains of Gln 711 and Arg752 and the rest of the molecule binds in a highly hydrophobic environment and has extensive contacts with side chains of hydrophobic residues. The predicted binding model suggested that the oxygen atom connecting the chlorobenzonitrile and the cyclohexyl moiety in compound 9 plays a key role in controlling the relative conformations of the chlorobenzonitrile and the cyclohexyl groups but forms no specific hydrogen bonding interaction with AR. Interestingly, there is sufficient room around this oxygen atom to accommodate a slightly larger group. Based upon the predicted binding model for compound 9, we proposed several new AR ligands in which the oxygen atom in compound 9 is replaced with an amino, N-methyl, or N-ethyl and predicted their binding models in complex with AR (Figure S4). The predicted binding models suggested that those new AR ligands bind with AR very similarly as compared to compound 9 and S1 (Figure S4).
Encouraged by the modeling predictions, we synthesized compounds 34–37 in which the oxygen atom in the AR ligand portion in compound 26 was replaced by an amine or a substituted N-alkyl group as potential AR degraders. These compounds were evaluated for their AR degradation in the VCaP cell line with the data summarized in Table 4.
Compound 34, in which the oxygen atom in 26 has been replaced by an amine, is a very potent AR degrader with a DC50 value of 0.3 nM but it achieves a Dmax of only 78%. Substituting the amine group in compound 34 with either a methyl or an ethyl group led to compounds 35 and 36, respectively, both of which achieve a DC50 value of 0.1 nM and a Dmax of 99%. Replacing the amine in compound 34 with a n-propyl group yielded compound 37, which has a DC50 value of 1.4 nM and a Dmax of 88%. In a direct comparison, compound 37 is 14 times less potent than compounds 35 and 36 and has a lower Dmax than compounds 35 and 36.
Compound 35 is an exceptionally potent and effective AR degrader with a DC50 value of 0.1 nM and a Dmax of 99% in the VCaP cell line. We evaluated the PKs in mice of compound 35 and the data obtained are summarized in Table 3. Compound 35 has an excellent overall PK profile with improved PK parameters compared to compound 26 in both intravenous and oral administration. In particular, compound 35 achieves an oral bioavailability of 44% in mice, compared to 30% for compound 26.
Encouraged by the exceptional AR degradation potency and an excellent PK profile of compound 35, we next performed further modifications on the linker in compound 35 by employing other rigid linkers of similar lengths and physiochemical properties. This yielded compounds 39–45 (Table 5, Figure S4). These compounds were similarly evaluated in the VCaP cell line for their AR degradation.
Table 5.
Futrther Exploration of the Linker in Compound 35
| |||
|---|---|---|---|
| Compound No |
Linker | AR degradation in VCaP cell line |
|
| DC50 (nM) | Dmax (%) | ||
|
| |||
| 35 |
|
0.1 | 99 |
| 39 |
|
1.4 | 93 |
| 40 |
|
0.5 | 96 |
| 41 |
|
0.1 | 96 |
| 42 |

|
0.01 | 99 |
| 43 |
|
0.04 | 99 |
| 44 |
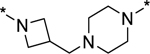
|
0.1 | 100 |
| 45 |
|
0.2 | 96 |
In general, all the compounds in Table 5 are very potent AR degraders with DC50 values of 0.01–1.4 nM and Dmax values of 93–100%. While compound 39 is the least potent with a DC50 value of 1.4 nM and a Dmax of 93%, compound 42 is the most potent with a DC50 value of 0.01 nM and a Dmax of 99%.
Compounds 40–45 are very potent and effective AR degraders. We evaluated the pharmacokinetics in mice of these six compounds and obtained the data summarized in Table 3. Overall, compounds 40, 42, and 44 have inferior PK parameters compared to those of compound 35, as evidenced by their higher clearance following intravenous administration and lower AUC values after either intravenous or oral administration. While compound 41 has very similar PK parameters to those of compound 35, compounds 43 and 45 have a PK profile, which is improved over that of compound 35. Specifically, both 43 and 45 have an excellent volume of distribution (Vss = 1.8–2.1 L/kg), a long half-life (t1/2 = 5.5–7.5 h), and a slow clearance (Cl = 0.2–0.3 L/h/kg) with intravenous administration. With 5 mg/kg of oral administration, compound 43 achieves a Cmax of 1140 ng/mL and an AUC of 8254 h*ng/mL. With 3 mg/kg of oral administration, compound 43 achieves a Cmax of 484 ng/mL and 8637 h*ng/mL. Compounds 43 and 45 have an oral bioavailability of 51 and 67%, respectively.
Bicalutamide, enzalutamide, and apalutamide are AR antagonists that have been approved by the FDA for the treatment of prostate cancer. We investigated if the AR antagonist portion in 41 could be simply replaced by one of these three FDA-approved AR antagonists to obtain potent AR degraders. This led to synthesis and evaluation of compounds 46–48 (Table 6 and Figure S5). Interestingly, compounds 46, 47, and 48 in which the AR antagonist portion in 41 was replaced by bicalutamide, enzalutamide, or apalutamide, respectively, are all ineffective or minimally effective in reducing the AR protein in the VCaP cell line, with Dmax <32%. These results suggest that for the design of potent and effective AR degraders, the linker should be optimized for different AR antagonists.
Table 6.
AR Degraders by Employing Three FDA-Approved AR Antagonists
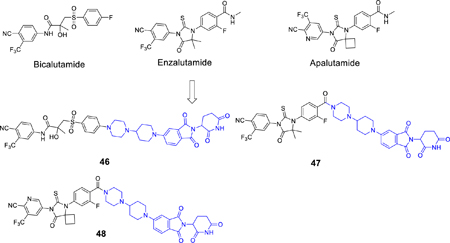
| ||
|---|---|---|
| AR degradation in the VCaP cell line |
||
| Compound No | DC50 (nM) | Dmax (%) |
| 46 | >1000 | 32 |
| 47 | >1000 | 14 |
| 48 | >1000 | 27 |
Evaluation of Cell Growth Inhibition of Potent AR Degraders in the VCaP Cell Line.
Because AR plays a key role in cell proliferation of AR+ prostate cancer cells, reduction of the AR protein level should effectively inhibit cell growth in AR+ prostate cancer cell lines, as has been shown previously.13 We evaluated several highly potent AR degraders for their ability to inhibit cell growth in the VCaP cell line, with ARV-110 included as a control. We also evaluated a number of weak AR degraders as control compounds for their cell growth inhibition in the VCaP cell line. We included enzalutamide as an additional control in the cell growth inhibition assay and the data are summarized in Table 7.
Table 7.
Cell Growth Inbibition of AR Degraders and Control Compounds in the VCaP Cell Linea
| Compound No | DC50 (nM) | Dmax (%) | IC50 (nM) |
|---|---|---|---|
| enzalutamide | NA | NA | 393 ± 0.5 |
| 38 | NA | NA | 61.0 ± 2.3 |
| ARV-110 | 1.6 | 98 | 30.4 ± 3.5 |
| 24 | 0.2 | 97 | 2.7 ± 0.7 |
| 25 | 0.3 | 88 | 7.9 ± 2.2 |
| 26 | 0.2 | 95 | 9.7 ± 3.2 |
| 27 | 0.3 | 92 | 6.7 ± 0.9 |
| 34 | 0.3 | 78 | 16.6 ± 1.3 |
| 35 | 0.1 | 99 | 1.8 ± 0.5 |
| 36 | 0.1 | 99 | 2.1 ± 0.7 |
| 37 | 1.4 | 88 | 83.5 ± 30.5 |
| 41 | 0.1 | 96 | 0.8 ± 0.1 |
| 42 | 0.01 | 99 | 1.1 ± 0.2 |
| 43 | 0.04 | 99 | 1.5 ± 0.3 |
| 44 | 0.1 | 100 | 4.3 ± 0.6 |
| 45 | 0.2 | 96 | 5.1 ± 0.8 |
| 29 | >1000 | 20 | >1000 |
| 32 | >1000 | 19 | >1000 |
| 33 | >1000 | 22 | >1000 |
| 46 | >1000 | 32 | >1000 |
| 47 | >1000 | 14 | >1000 |
| 48 | >1000 | 27 | >1000 |
NA: not available. IC50 value was determined by three independent experiments for each compound.
ARV-110 has an IC50 value of 30.4 nM in inhibition of the VCaP cell growth and enzalutamide has an IC50 value of 393 nM in the same assay. Compounds 24–27 with DC50 values of 0.2–0.3 nM and Dmax of 88–97% in the degradation assay have IC50 values of 2.7–9.7 nM in the cell growth inhibition assay. In direct comparison, compounds 24–27 are >30 times more potent than enzalutamide and are also 3–10 times more potent than ARV-110 in the VCaP cell line.
Compounds 35 and 36 with DC50 values of 0.1 nM and Dmax of 99% are highly potent in inhibition of VCaP cell growth and achieve IC50 values of 1.8 and 2.1 nM, respectively. In comparison, compound 34 with a DC50 value of 0.3 nM and Dmax of 78% is nearly 10 times weaker than compounds 35 and 36 in inhibition of VCaP cell growth, indicating that both DC50 and Dmax affect cell growth inhibition activity for an AR degrader. Consistent with its weaker AR degradation (DC50 =1.4 nM and Dmax = 88%), compound 37 is >40 times less potent than compounds 35 and 36.
Compounds 41–44 with DC50 values of 0.2–0.01 nM and Dmax of 96–100% are all highly potent in inhibition of VCaP cell growth and achieve IC50 values of 0.8–5.1 nM. Among them, compounds 41–43 are the most potent compounds and are >100 times more potent than enzalutamide. Compounds 41–43 are also >10 times more potent than ARV-110 in cell growth inhibition in the VCaP cell line.
Consistent with their minimal AR degradation activity (DC50 > 1000 nM and Dmax = 14–32%), compounds 29, 32–33, and 46–47 are all ineffective in inhibition of VCaP cell growth with IC50 values >1000 nM.
Further Evaluation of Representative AR Degraders in the LNCaP Cell Line.
Mutations in AR are a major mechanism of resistance of castration-resistant prostate cancer to AR antagonists. The LNCaP human prostate cancer cell line carries a T878A mutation and has been extensively used in preclinical studies as a castration-resistance prostate cancer model. We evaluated a number of our potent AR degraders for their ability to reduce the AR T878A mutant protein in the LNCaP cell line, with ARV-110 included as the control (Table 8 and Figure S6).
Table 8.
DC50, Dmax and IC50 Values of Selected Compounds in the LNCaP Cell Linea
| AR degradation in LNCaP cell line |
|||
|---|---|---|---|
| Compound No | DC50 (nM) | Dmax (%) | IC50 in LNCaP cell line (nM) |
| Enzalutamide | NA | NA | 133 ± 5 |
| ARV-110 | 1.5 | 99 | 33.1 ± 3.7 |
| 35 | 0.3 | 95 | 15.3 ± 3.7 |
| 41 | 0.4 | 95 | 22.3 ± 2.9 |
| 42 | 0.9 | 99 | 11.4 ± 2.2 |
| 43 | 0.1 | 98 | 16.2 ± 1.8 |
| 44 | 1.9 | 99 | 11.6 ± 2.0 |
| 45 | 1.3 | 64 | 21.7 ± 5.9 |
NA: not available. IC50 value was determined by three independent experiments for each compound.
ARV-110 effectively reduces degradation of AR T878A mutant protein in the LNCaP cell line, achieving a DC50 value of 1.5 nM and a Dmax of 99%. Compounds 35 and 41–44 are all highly potent and effective in reducing the AR T878A mutant protein level in the LNCaP cell line with DC50 values of 0.1–1.9 nM and Dmax values of 95–99%. Although compound 45 reduces the AR T878A mutant protein, it only achieves a Dmax of 64% and is thus not a very effective AR degrader. Interestingly, compound 45 is a very potent and effective AR degrader in the VCaP cell line, which carries a wild-type AR protein (DC50 = 0.2 nM and Dmax = 96%). The less-effective AR degradation by compound 45 in the LNCaP cell line suggested that a highly potent and effective AR degrader against wild-type AR protein may not be a very effective degrader of a mutated AR protein.
We evaluated the potent AR degraders for their cell growth inhibition activity in the LNCaP cell line with ARV-110 and enzalutamide used as controls.
In the LNCaP cell line, enzalutamide has an IC50 value of 133 nM, whereas ARV-110 has an IC50 value of 33.1 nM. Compounds 35 and 41–45 achieve IC50 values of 11.4–22.3 nM in the same cell line and are therefore 5–10 times more potent than enzalutamide and slightly more potent than ARV-110.
Evaluation of AR Degradation Kinetics in the VCaP and LNCaP Cell Lines.
We investigated the degradation kinetics of compounds 26, 27, 35, 40, 41, 42, 43, 44, and 45 in both the VCaP and LNCaP cell lines and obtained the data shown in Figure S7. Compounds 26, 27, 35, 40, 41, 42, 43, 44, and 45 effectively reduce the AR protein level in both VCaP and LNCaP at 1 h and by >90% at 6 h and 24 h. The kinetics data showed that these AR degraders induce AR degradation with fast kinetics.
Pharmacodynamics and Tissue Distribution Studies.
Based upon their degradation potency, cell growth inhibition, and PK data, compounds 35, 41, 43, and 45 are four promising AR degraders. We next tested their pharmacodynamic (PD) effect in the VCaP xenograft tumor tissue in mice.
We first tested compounds 35, 41, 43, and 45 with a single oral administration at 20 mg/kg in mice bearing the VCaP xenograft tumors. Western blotting analysis of the tumor tissues showed that compounds 41, 43, and 45 are effective in reducing the AR protein level at 6 and 24 h time-points, with a stronger effect at the 24 h time-point (Figure 3). In comparison, compound 35 is less effective than compounds 41, 43, and 45 (Figure 3).
Figure 3.

PD effect of AR degraders on AR protein in VCaP tumors. Mice bearing VCaP tumors were treated with a single oral dose via oral gavage at 20 mg/kg and tumor tissues were collected at 6 and 24 h time-points for Western blotting analysis. GAPDH was used as the loading control.
Compounds 35, 41, 43, and 45 were evaluated for tissue distribution in mice bearing the VCaP xenograft tumor with single oral administration at 20 mg/kg (Table 9). Compounds 35, 41, and 43 have higher concentrations in tumors than in plasma or prostate, while 45 is distributed evenly in plasma and in tumors and prostate. While the drug concentrations of 35 decrease from 6 to 24 h, the drug concentrations for compounds 41, 43 (ARD-2585), and 45 at the 6 and 24 h time-points are similar in plasma, prostate, and tumor. These tissue distribution data are consistent with their PK data in mice.
Table 9.
Tissue Distribution of Compounds 35, 41, 43, and 45 in Mice Bearing VCaP Tumorsa
| concentration ± SD (ng/mL in plasma or ng/ g in tissues) |
|||||
|---|---|---|---|---|---|
| tissue | time-point (h) | 35 | 41 | 43 | 45 |
| Plasma | 6 | 208 ± 80 | 37 ± 6 | 87 ± 25 | 181 ± 31 |
| 24 | 65 ± 41 | 38 ± 14 | 102 ± 57 | 106 ± 112 | |
| Prostate | 6 | 67 ± 57 | 58 ± 6 | 101 ± 28 | 108 ± 28 |
| 24 | 4 ± 6 | 83 ± 14 | 107 ± 35 | 123 ± 29 | |
| Tumor | 6 | 306 ± 138 | 120 ± 15 | 224 ± 57 | 129 ± 26 |
| 24 | 137 ± 33 | 176 ± 49 | 311 ± 122 | 116 ± 54 | |
Three mice were evaluated in each cohort. Tissues were collected at 6 and 24 h after 20 mg/kg PO.
Compounds 41 and 43 effectively reduce the AR protein in the VCaP tumor with a single oral administration in mice (Figure 3). We further tested 41 and 43 with daily oral administration at 10 mg/kg for 3 days in mice bearing the VCaP xenograft tumors (Figure 4). Western blotting analysis of the tumor tissues showed that while compound 41 is effective in reducing the AR protein level at the 3 and 6 h time-points by 74 and 70% (p < 0.05), it has a modest effect at the 24 h time-point, reducing the AR protein level by 20% (p > 0.05). In comparison, 43 (ARD-2585) is effective in reducing the AR protein level at the 3 and 24 h time-points by 78% (p < 0.01) and 60% at the 6 h time-point (p > 0.05). These data show that compound ARD-2585 has a more persistent PD effect in the tumor tissue in reducing AR protein than compound 41.
Figure 4.

PD effect of AR degraders on AR protein in VCaP tumors. Mice bearing VCaP tumors were given a triple oral dose via oral gavage at 10 mg/kg and tumor tissues were collected at 3, 6, and 24 h time-points for Western blotting analysis. GAPDH was used as the loading control.
We further carried out tissue distribution studies of ARD-2585 with a single oral administration at 20 mg/kg in mice bearing VCaP xenograft tumor and the data are summarized in Table 10. These tissue distribution data demonstrate that consistent with its excellent PK profile, ARD-2585 is extensively distributed in tissues.
Table 10.
Tissue Distribution Studies of ARD-2585 in Mice Bearing VCaP Tumorsa
| drug concentration in different tissues at different time-points |
|||
|---|---|---|---|
| tissue | 1 (h) | 6 (h) | 24 (h) |
| plasma (ng/mL) | 70 ± 61 | 134 ± 98 | 214 ± 78 |
| tumor (ng/g) | 47 ± 15 | 201 ± 102 | 301 ± 67 |
| tumor/plasma (ratio) | 0.7 | 1.5 | 1.4 |
| liver (ng/g) | 2638 ± 813 | 3507 ± 1580 | 6873 ± 2470 |
| liver/plasma (ratio) | 37.7 | 26.2 | 32.1 |
| kidney (ng/g) | 437 ± 235 | 661 ± 501 | 366 ± 124 |
| kidney/plasma (ratio) | 6.2 | 4.9 | 1.7 |
| prostate (ng/g) | 25 ± 10 | 76 ± 51 | 109 ± 23 |
| prostate/plasma (ratio) | 0.4 | 0.6 | 0.5 |
| heart (ng/g) | 210 ± 77 | 339 ± 199 | 683 ± 321 |
| heart/plasma (ratio) | 3.0 | 2.5 | 3.2 |
Three mice were evaluated in each cohort. Tissues were collected at 1, 6, and 24 h with a single oral dose at 20 mg/kg.
Investigation of the Mechanism of Action of AR Degradation by ARD-2585.
We investigated the mechanism of action of ARD-2585 in induction of AR degradation in both the VCaP and LNCaP cell lines, and the data are presented in Figure 5. ARD-2585 at 100 nM effectively reduces the AR protein in both VCaP and LNCaP cells with 3 h treatment. AR degradation was effectively blocked by pretreatment of an AR inhibitor, a cereblon ligand thalidomide, a proteasome inhibitor MG-132, and an E1 neddylation inhibitor MLN4924. These mechanistic data provide clear evidence that ARD-2585 induces AR degradation through a cereblon-, proteasome-, and neddylation-dependent mechanism and is therefore a bona fide PROTAC AR degrader.
Figure 5.

Evaluation of the mechanism of action of ARD-2585. VCaP and LNCaP cells were pretreated for 2 h with DMSO, AR inhibitor 38 (10 μM), cereblon ligand thalidomide (10 μM), proteasome inhibitor MG-132 (3 μM), and E1 neddylation inhibitor MLN4924 (0.5 μM). Cells were then treated for 3 h with ARD-2585 at 100 nM. (a) Western blotting. Loading control GAPDH: glyceraldehyde 3-phosphate dehydrogenase, (b) Quantitative bar graph of VCaP of Western blotting. (c) Quantitative bar graph of LNCaP of Western blotting.
Evaluation of the Efficacy of ARD-2585 in the VCaP Xenograft Tumor Model.
Our PD data showed that ARD-2585 effectively reduces AR protein in the VCaP tumor tissue. We next tested the antitumor activity of ARD-2585 in the VCaP xenograft tumor model in mice with enzalutamide included as a control, and the data are summarized in Figure 6.
Figure 6.

Efficacy study of ARD-2585 in the VCaP xenograft tumor model in SCID mice with enzalutamide included as the control. Seven mice were evaluated in each group. Animals were dosed daily via oral garage for 3 weeks. (a) Average tumor volume for each dosing group. (b) Percentage of mouse body weight change in each group.
The efficacy data showed that ARD-2585 effectively inhibits tumor growth at all the three doses tested (Figure 6a). At the end of the treatment (day 37), ARD-2585 inhibits tumor growth by 54.9, 74.3, and 65.9% over the vehicle control group (p < 0.0001 for all three dosing groups). In comparison, enzalutamide at 40 mg/kg inhibits tumor growth by 45.0% (p = 0.0058). Statistically, ARD-2585 at both 20 and 40 mg/kg is more efficacious than enzalutamide at 40 mg/kg in inhibition of tumor growth (p = 0.022 and 0.0058, respectively).
Both ARD-2585 and enzalutamide were well tolerated in this efficacy experiment and did not cause animal weight loss or other signs of toxicity during the entire experiment (Figure 6b).
Further Assessment of AR Degradation of ARD-2585 in AR+ Prostate Cancer Cell Lines.
Our data demonstrate that ARD-2585 is a potent, orally bioavailable, and efficacious AR degrader. We directly compared ARD-2585 with ARV-110, a first-in-class AR degrader in clinical development, for its ability to induce AR degradation in the VCaP cell line with an AR amplification, in the LNCaP cell line carrying a T878A AR mutation, the 22Rvl cell line with F876L mutation, and the MDA-PCa-2b cell line with AR double mutations (L702H and T878A). The data are shown in Figure 7.
Figure 7.

AR degradation of ARD-2585 and ARV-110 in the VCaP, LNCaP, 22Rv1, and MDA-PCa-2b cell lines. Cells were treated with different concentrations of these two AR degraders for 24 h. AR protein was probed by western blotting and GAPDH was used as the loading control.
ARD-2585 is very effective, reducing the AR protein level by >80% at concentrations as low as 0.1 nM in the VCaP cell line, 0.3 nM in the LNCaP cell line, 0.1 nM in the 22Rvl cell line, and 1 nM in the MDA-PCa-2b cell line, respectively. ARD-2585 achieves a Dmax of >95% in each of these four AR+ cell lines. In comparison, ARV-110 reduces the AR protein level by 80% at concentrations as low as 3 nM in both the VCaP and LNCaP cell lines and has a Dmax of >95%. ARV-110 reduces the AR protein level by 80% at 30 nM in the 22Rv1 cell line and has a Dmax of 92%. ARV-110 is not very potent or effective in reducing the AR protein level in the MDA-PCa-2b cell line with AR double mutations and reduces the AR mutated protein by 57% at a concentration of 1000 nM. Thus, ARD-2585 is 30, 10, 300, and 1000 times more potent than ARV-110 in reducing the AR protein level in the VCaP, LNCaP, 22Rvl, and MDA-PCa-2b cell lines, respectively. Collectively, ARD-2585 is an exceptionally potent AR degrader.
Investigation of Microsomal and Plasma Stability and hERG Channel Inhibition for ARD-2585.
We evaluated ARD-2585 for its liver microsomal stability in five different species (human, mouse, rat, dog, and monkey). ARD-2585 has excellent stability in liver microsomes in all the five species with T1/2 > 120 min. The excellent mouse microsomal stability data are consistent with the slow clearance of ARD-2585 based upon the PK data in mice.
We tested ARD-2585 for its plasma stability in five different species (human, mouse, rat, dog, and monkey). ARD-2585 has excellent plasma stability in all 5 species with T1/2 > 120 min.
In vitro inhibition of the human ERG (the human ether-à-go-go-related gene) channel has been used as an important assay to assess potential cardiotoxicities of a drug molecule. We evaluated ARD–2585 for its inhibition of the hERG channel and found that ARD-2585 has no hERG inhibition up to 30 μM, the highest concentration tested.
Chemistry.
The synthesis of compounds 11–19 is shown in Scheme 1. Nucleophilic substitution of 49 with 50 affords the cyclohexyloxy phenyl ether (51), and is followed by TFA deprotection of Boc to provide the free amine (52).24 Buchwald coupling of 53 and 54a–54i gives 55a–i, which upon deprotection gives the amino acid (56a–56i).25 Nucleophilic displacement of fluorine in 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione affords acids 57a–57i. Amide coupling of acids in 57a–57i with amines in 52 produced the degraders (11–19).
Scheme 1. Synthesis of AR Degraders Containing a Linear Linkera.

aReaction conditions: (a) THF, NaH, 0 °C, 4 h; (b) dichloromethane, TFA; (c) dioxane, Pd2(dba)3, Xphos, Cs2CO3, 90 °C, 12 h; (d) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h; and (e) dichloromethane, 52, HATU, DIPEA.
The synthesis of the degraders (20–23) is shown in Scheme 2. Amide coupling of 52 and 58 to afford 59 is followed by Boc deprotection, giving 60. Substitution or reductive animation of 60 and Boc deprotection affords the amines (62a–62d). The amino moiety in 62a–62d displaces F in 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione providing the degraders (20–23).
Scheme 2. Synthesis of Degraders 20–23 with a Semirigid Linkera.

aReaction conditions: (a) dichloromethane, HATU, DIPEA, rt, 0.5 h; (b) dichloromethane, TFA; (c) ACN, N-Boc bromoalkylamine, DIPEA, rt; and (d) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h.
The synthesis of the degraders 24–28 is shown in Scheme 3. Buchwald coupling of 63 with bicyclic secondary amines (64a–64e), and subsequent hydrolysis of the methyl ester provides the acids (66a–66e). Amide coupling followed by deprotection of Boc in 66a–66e provides 68a–68e. Finally, a nucleophilic displacement reaction affords the degraders (24–28).26,27
Scheme 3. Synthesis of 24–28a.

aReaction conditions: (a) dioxane, Pd2(dba)3, Xphos, Cs2CO3, 110 °C, 12 h; (b) (1) MeOH, THF, NaOH (1 N); (2) HCl (1 N); (c) dichloromethane, HATU, DIPEA, rt, 0.5 h; (d) dichloromethane, TFA; and (e) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h.
Intermediates 70a–70b were obtained from substitution or reductive animation reactions (Scheme 4). The F in 49 was displaced by hydroxyl in 71a–71c and 74a–74b or by amine in 71d–71g and 70a–70b, and this, followed by deprotection of Boc afforded the amines (73a–73g) and (76a–76b). Amide coupling and Boc deprotection of these compounds afforded 79a–79b and 81a–81g. The degraders (29–37) were synthesized from 79a–79b and 81a–81g by nucleophilic displacement of 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione.
Scheme 4. Synthesis of Table 3 Compounds 29–37a.

aReaction conditions: (a) DMF, DIPEA, bromoalkane, 70 °C, 12 h; (b) 1,2-dichloroethane, akyl aldehyde, NaB(OAc)3H, AcOH, rt; (c) THF, NaH, 0 °C; (d) DMF, CS2CO3, 90 °C, 12 h; (e) dichloromethane, TFA; and (f) dichloromethane, HATU, DIPEA, rt, 0.5 h; and (g) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h.
The synthesis of the degraders 39–45 is shown in Scheme 5, in a three step sequential amide coupling, deprotection of Boc, and nucleophilic displacement reactions.
Scheme 5. Synthesis of Degraders 39–45a.

aReaction conditions: (a) dichloromethane, HATU, DIPEA, rt, 0.5 h; (b) dichloromethane, TFA; and (c) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h.
The synthesis of the bicalutamide-based degrader (46) is shown in Scheme 6, as a three step sequence of nucleophilic substitution,13 deprotection of Boc and F displacement.
Scheme 6. Synthesis of Bicalutamide-Based Degrader 46a.

aReaction conditions: (a) DMF, DIPEA, 110 °C, 12 h; (b) dichloromethane, TFA; and (c) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h.
The synthesis of enzalutamide- and apalutamide-based degrader 47 and 48 is shown in Scheme 7. Hydrolysis of the amides in enzalutamide and apalutamide affords the acids (89a–89b).13 Amide coupling, Boc deprotection, and F- nucleophilic substitution afford the degraders (47, 48).
Scheme 7. Synthesis of Enzalutamide- and Apalutamide-Based Degraders 47 and 48a.

aReaction conditions: (a) HCl (con.), MeOH, 90 °C, 4 h; (b) dichloromethane, HATU, DIPEA, rt, 0.5 h; (c) dichloromethane, TFA; and (d) 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, DMF, DIPEA, 90 °C, 12 h.
SUMMARY AND DISCUSSION
In this study, we have described the design, synthesis, and evaluation of PROTAC AR degraders using the cereblon ligand, thalidomide, and different classes of AR antagonists. Through extensive optimization of the linker and modifications of the AR antagonist portion of the compounds, we have discovered a set of exceptionally potent and orally bioavailable AR degraders, exemplified by ARD-2585. ARD-2585 achieves picomolar DC50 values and >98% of Dmax in the VCaP cell line with a wild-type AR and in the LNCaP cell line carrying a T878A-mutated AR mutant. In addition, ARD-2585 reduces AR protein by >80% at 0.1 nM in the 22Rvl cell line carrying a AR-V7 variant and at 1 nM in the MDA-PCa-2b cell line carrying a double AR mutation. ARD-2585 potently inhibits cell growth with IC50 values of 1.5 and 16.2 nM in the VCaP and LNCaP AR+ prostate cancer cell lines, respectively. ARD-2585 is very stable in liver microsomes and plasma and has no hERG inhibition liability. It displays excellent PK parameters with both intravenous and oral routes of administration in mice and achieves extensive tissue distribution. Oral administration of ARD-2585 effectively reduces AR protein the VCaP xenograft tumor tissue in mice and inhibits VCaP tumor growth. In direct comparison, ARD-2585 is more efficacious than enzalutamide in inhibition of the VCaP tumor growth. Our data demonstrate that ARD-2585 is a promising AR degrader in further extensive evaluations for the treatment of AR+ prostate cancer and other human diseases in which AR plays a key role.
ARV-110 is the first PROTAC AR degrader advanced into human clinical trials by Arvinas and has demonstrated encouraging clinical activity and safety profile.17 Arvinas has published a number of patents for PROTAC AR degraders using several classes of AR antagonists, including compounds 9 and 10 and cereblon ligands.17,22 However, with the exception of ARV-110,22 no detailed biological and pharmacological characterizations have been published for other PROTAC AR degraders disclosed in the patents. Recently, the chemical structure of ARV-110 was disclosed, and allows us to directly compare the AR degradation potencies between ARV-110 and ARD-2585. Our direct comparative data revealed that ARD-2585 is 30, 10, 300, and 1000 times more potent than ARV-110 in reducing the AR protein level in the VCaP, LNCaP, 22Rvl, and MDA-PCa-2b cell lines, respectively.
This current study has demonstrated that conformational restriction of the linker in PROTAC AR degraders, coupled with modifications of AR antagonist portion are critical in the successful discovery of ARD-2585 as an exceptionally potent and orally active AR degrader.
EXPERIMENTAL SECTION
Chemistry.
Unless otherwise noted, all purchased reagents were used as received without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE 400 MHz spectrometer. 1H NMR spectra are reported in parts per million (ppm) downfield from tetramethylsilane. All 13C NMR spectra are reported in ppm and were obtained with 1H decoupling. In reported spectral data, the format (δ) chemical shift (multiplicity, J values in Hz, integration) is used with the fallowing abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. Mass spectral (MS) analysis was carried out with a Waters ultraperformance liquid chromatography (UPLC) mass spectrometer. The prepared compounds were all purified by a C18 reverse-phase preparative high-performance liquid chromatography (HPLC) column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in CH3CN) as eluents. The purity of all of the final compounds was confirmed to be >95% by UPLC–MS and UPLC.
N-((1R,4R)-4-(3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)propyl)-amino)benzamide (11).
Compound 50 (2.15 g, 10 mmol) was dissolved in anhydrous THF (30 mL) in an ice bath. NaH (60% in mineral oil, 0.9 g, 22.5 mmol) was added in portions, and after 0.5 h the reaction mixture was warmed up to room temperature (rt) for 0.5 h. The reaction was placed in an ice bath and compound 49 (2.30 g, 15 mmol) was added and warmed up to rt for 4 h. After UPLC–MS showing complete conversion of 50, the reaction mixture was quenched with H2O at 0 °C, extracted with EtOAc, dried, and concentrated. Compound 51 (2.89 g 83%) was obtained after purification on a silica gel column (60% EtOAc in hexane). 1H NMR (MeCN-d3): δ 7.69 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 2.4 Hz, 1H), 6.98 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 5.25 (br, 1H), 4.43 (m, 1H), 3.43 (m, 1H), 2.12 (m, 4H), 1.55 (m, 2H), 1.42 (s, 9H), 1.38 (m, 2H).
Compound 51 (0.35 g, 1 mmol) was dissolved in DCM (4 mL) and TFA (0.44 g, 4 mmol) was added at rt. After 0.5 h, all volatile materials were removed in a rotary evaporator to afford 52 in 100% yield.24 1H NMR (MeCN-d3): δ 7.71 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 2.4 Hz, 1H), 7.02 (dd, J = 8.0 Hz, 2.4 Hz, 1H), 6.79 (br, 3H, NH2∙TFA), 4.43 (m, 1H), 3.27 (m, 1H), 2.22 (m, 2H), 2.14 (m, 2H), 1.62 (m, 2H), 1.52 (m, 2H).
To a dry round-bottomed flask, compound 53 (0.304 g, 1 mmol), N-Boc aminopropylamine (54a, 0.174 g, 1 mmol), Pd2(dba)3 (0.092 g, 0.1 mmol), Xphos (0.048 g, 0.1 mmol), and Cs2C03 (0.975 g, 3 mmol) were added in dioxane (10 mL). The reaction mixture was degassed and stirred at 90 °C for 12 h. The reaction was cooled down, partitioned between EtOAc and H2O, and the organic layer was concentrated and purified by CombiFlash chromatography (40% EtOAc in hexane) to afford 55a (0.24 g 75%).25 1H NMR (MeCN-d3): δ 7.79 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 8.4 Hz, 2H), 5.35 (br, 1H), 4.43 (m, 1H), 5.24 (br, H), 3.18 (m, 4H), 1.97 (m, 2H), 1.42 (s, 9H), 1.43 (s, 9H).
Compound 55a (0.261 g, 0.745 mmol) was dissolved in DCM (3 mL) and TFA (0.328 g, 2.98 mmol) was added at rt. After 0.5 h, all volatile compounds were removed using a rotary evaporator to afford 56a in 100% yield. 1H NMR (MeCN-d3): δ 7.81 (d, J = 2.8 Hz, 2H), 7.49 (br, 4H), 6.95 (m, 2H), 3.28 (t, J = 8.0 Hz, 2H), 3.09 (d, J = 8.0 Hz, 2H), 1.97 (quin, J = 8.0 Hz, 2H).
Compound 56a (0.308 g, 1 mmol TFA salt) was dissolved in DMF (3 mL) and basified with DIPEA (0.516 g, 4 mmol). 2-(2,6-Dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (0.414 g, 1.5 mmol) was added to the solution, which was stirred at 90 °C for 12 h. All solvents were removed under vacuum and purified by CombiFlash chromatography (10% MeOH in DCM) to afford 57a in 30%. UPLC–MS: 3.0 min, MS: [M + H]+ found, 450.99 calcd 460.16. Compound 57a (0.113 g, 0.25 mmol) was dissolved in DMF (l mL) and basified with DIPEA (0.097 g, 0.75 mmol), then HATU (0.114 g, 0.3 mmol) was added and the mixture was stirred for 10 min. In a separate vial, compound 52 (0.91 g, 0.25 g TFA salt) was dissolved in DMF (0.8 mL) and basified with DIPEA (0.065 g, 0.5 mmol). A solution of compound 52 was added to the reaction mixture and stirred for 0.5 h. The resulted reaction mixture was acidified with TFA and purified by preparative HPLC to afford 11 in 76% yield. UPLC–MS: 5.0 min, purity >95%, MS: [M + H]+ found, 683.12 calcd 683.23. Prep. HPLC 53% MeCN in water. 1H NMR (MeCN-d3): δ 8.98 (s, 1H, (CO)2NH), 7.71 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.0 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.99 (d, J = 2.2 Hz, 1H), 6.87 (dd, J = 2.2 Hz, 8.4 Hz, 1H), 6.64 (d, J = 2.0 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 6.60 (s, 1H, CONH), 4.95 (m, 1H), 4.46 (quint, J = 3.8 Hz, 1H), 3.91 (m, 1H), 3.70 (m, 1H), 3.33 (m, 2H), 3.27 (m, 2H), 3.16 (m, 3H), 2.65 (m, 4H), 2.18 (m, 2H), 2.08 (m, 2H), 1.59 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)butyl)-amino)benzamide (12).
Compound 12 was synthesized following the procedure used to prepare 11. UPLC–MS: 5.3 min, purity >95%, MS: [M + H]+ found, 697.05 cald 697.25. Prep. HPLC 53% ACN in water. 1H NMR (MeCN-d3): δ 8.95 (s, 1H, (CO)2NH), 7.69 (d, J = 8.8 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 2.2 Hz, 1H), 7.03 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 6.95 (d, J = 2.0 Hz, 1H), 6.87 (m, 1H), 6.66 (d, J = 2.4 Hz, 1H), 6.62 (d, J = 2.0 Hz, 1H), 6.60 (s, 1H, CONH), 4.96 (m, 1H), 4.48 (quint, J = 3.8 Hz, 1H), 3.89 (m, 1H), 3.72 (m, 1H), 3.34 (m, 2H), 3.31 (m, 3H), 3.17 (m, 2H), 2.66 (m, 4H), 2.20 (m, 2H), 2.10 (m, 4H), 1.59 (m, 4H).
N-((1R,4R)-4-(3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)pentyl)-amino)benzamide (13).
Compound 13 was synthesized following the procedure used for 11. UPLC–MS: 5.5 min, purity >95%, MS: [M + H]+ found, 711.10 cald 711.26. Prep. HPLC 56% ACN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H, (CO)2NH), 7.70 (d, J = 8.8 Hz, 1H), 7.66 (dd, J = 8.8 Hz, 2.0 Hz, 2H), 7.55 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 6.96 (d, J = 2.0 Hz, 1H), 6.83 (dd, J = 2.2 HZ, 8.4 Hz, 1H), 6.79 (d, J = 2.2 Hz, 1H, CONH), 6.73 (dd, J = 2.0 Hz, 7.9 Hz, 2H), 4.96 (dd, J = 7.2 Hz, 5.3 Hz, 1H), 4.45 (m, 1H), 3.92 (m, 1H), 3.27 (m, 4H), 3.74 (m, 3H), 2.18 (m, 2H), 2.10 (m, 1H), 2.08 (m, 2H), 1.70 (m, 4H), 1.54 (m, 6H), 1.34 (m, 2H).
N-((1R,4R)-4-(3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)hexyl)-amino)benzamide (14).
Compound 14 was synthesized following the procedure used for 11. UPLC–MS: 5.6 min, purity >95%, MS: [M + H]+ found, 725.30 calcd 725.28. Prep. HPLC 59% ACN in water. 1H NMR (MeCN-d3): δ 8.90 (s, 1H, (CO)2NH), 7.71 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.00 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.97 (d, J = 2.2 Hz, 1H), 6.86 (dd, J = 2.2 HZ, 8.4 Hz, 1H), 6.60 (d, J = 6.8 Hz, 2H), 4.95 (m, 1H), 4.45 (m, 1H), 3.91 (m, 1H), 3.23 (t, J = 7.0 Hz, 2H), 3.16 (t, J = 7.2 Hz, 2H), 2.74 (m, 4H), 2.00 (m, 2H), 2.06 (m, 2H), 1.65 (m, 5H), 1.56 (m, 2H), 1.49 (m, 4H), 1.35 (m, 3H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)heptyl)-amino)benzamide (15).
Compound 15 was synthesized following the procedure used for 11. UPLC–MS: 6.0 min, purity >95%, MS: [M + H]+ found, 739.21, calcd 739.29. Prep. HPLC 61% ACN in water. 1H NMR (MeCN-d3): δ 8.92 (s, 1H, (CO)2NH), 7.71 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.97 (d, J = 2.1 Hz, 1H), 6.86 (dd, J = 2.2 HZ, 8.4 Hz, 1H), 6.60 (d, J = 8.8 Hz, 3H), 4.96 (m, 1H), 4.46 (m, 1H), 3.92 (m, 1H), 3.22 (t, J = 7.0 Hz, 2H), 3.15 (t, J = 8.2 Hz, 2H), 2.74 (m, 4H), 2.18 (m, 2H), 2.06 (m, 2H), 1.64 (m, 6H), 1.43 (m, 5H), 1.35 (m, 5H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)octyl)-amino)benzamide (16).
Compound 16 was synthesized following the procedure used for 11. UPLC–MS: 6.4 min, purity >95%, MS: [M + H]+ found, 753.22 cald 753.31. Prep. HPLC 63% ACN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H, (CO)2NH), 7.71 (d, J = 8.8 Hz, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H), 6.86 (dd, J = 2.2 HZ, 8.4 Hz, 1H), 6.60 (d, J = 8.8 Hz, 3H), 4.93 (m, 1H), 4.45 (m, 1H), 3.89 (m, 1H), 3.70 (m, 1H), 3.21 (t, J = 7.2 Hz, 2H), 3.15 (m, 3H), 2.74 (m, 4H), 2.18 (m, 2H), 2.06 (m, 2H), 1.78 (m, 2H), 1.62 (m, 6H), 1.35 (m, 8H).
N-(1R,4R)-4-((3-chloro-4-cyanophenoxy)cyclohexyl)-4-((9-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)nonyl)-amino)benzamide (17).
Compound 17 was synthesized following the procedure used for 11. UPLC–MS: 6.5 min, purity >95%, MS: [M + H]+ found, 767.20 calcd 767.32. Prep. HPLC 68% MeCN in water. 1H NMR (MeCN-d3): δ 8.92 (s, 1H, (CO)2NH), 7.71 (d, J = 8.8 Hz, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.96 (d, J = 2.0 Hz, 1H), 6.85 (dd, J = 2.2 HZ, 8.4 Hz, 1H), 6.63 (s, 1H), 6.60 (d, J = 8.8 Hz, 2H), 4.95 (m, 1H), 4.45 (m, 1H), 3.91 (m, 1H), 3.70 (m, 2H), 3.23 (t, J = 7.2 Hz, 2H), 3.21 (m, 2H), 2.75 (m, 4H), 2.15 (m, 2H), 2.06 (m, 2H), 1.61 (m, 4H), 1.51 (m, 2H), 1.62 (m, 6H), 1.35 (m, 6H).
N-(lR,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((10-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)decyl)-amino)benzamide (18).
Compound 18 was synthesized following the procedure used for 11. UPLC–MS: 6.6 min, purity >95%, MS: [M + H]+ found, 781.22 calcd 781.34. Prep. HPLC 72% ACN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H, (CO)2NH), 7.71 (d, J = 8.8 Hz, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 2.2 Hz, 8.4 Hz, 1H), 6.97 (d, J = 2.2 Hz, 1H), 6.85 (dd, J = 2.0 HZ, 8.4 Hz, 1H), 6.60 (d, J = 8.8 Hz, 3H), 4.96 (m, 1H), 4.43 (m, 1H), 3.91 (m, 1H), 3.22 (m, 2H), 3.21 (m, 2H), 2.91 (m, 2H), 2.76 (m, 2H), 2.15 (m, 4H), 2.07 (m, 2H), 1.92 (m, 2H), 1.61 (m, 12H), 1.35 (m, 6H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-((11-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)undecyl)-amino)benzamide (19).
Compound 19 was synthesized following the procedure used for 11. UPLC–MS: 7.0 min, purity >95%, MS: [M + H]+ found, 795.30 calcd 795.35. Prep. HPLC 72% ACN in water. 1H NMR (MeCN-d3): δ 8.94 (s, 1H, (CO)2NH), 7.70 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 2.2 Hz, 8.4 Hz, 1H), 6.97 (d, J = 2.2 Hz, 1H), 6.84 (dd, J = 2.0 HZ, 8.4 Hz, 1H), 6.58 (d, J = 8.8 Hz, 3H), 4.96 (m, 1H), 4.45 (m, 1H), 3.90 (m, 1H), 3.68 (m, 2H), 3.21 (m, 2H), 3.15 (m, 4H), 2.73 (m, 2H), 2.13 (m, 2H), 2.06 (m, 2H), 1.79 (m, 2H), 1.61 (m, 10H), 1.40 (m, 6H), 1.34 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)ethyl)-piperazin-1-yl)benzamide (20).
Compound 58 (0.306 g, 1 mmol) was dissolved in DCM (2 mL) and basified with DIPEA (0.39 g, 3 mmol), and HATU (0.49 g, 1.3 mmol) was added and the mixture was stirred for 10 min. In another vial, compound 52 (0.364 g, 1 mmol TFA salt) was dissolved in DCM (2 mL) and basified by DIPEA (0.26 g, 2 mmol). The solution of compound 52 was poured into the above reaction mixture and stirred for 0.5 h. All the volatiles were removed and the residue was purified by CombiFlash chromatography (hexane and EtOAc) to afford 59 in 85% yield. 1H NMR (MeCN-d3): δ 7.72 (m, 3H), 7.20 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 6.97 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 2.4 Hz, 1H, CONH), 4.45 (m, 1H), 3.93 (m, 1H), 3.55 (t, J = 5.2 Hz, 4H), 3.26 (t, J = 5.2 Hz, 4H), 2.15 (m, 2H), 2.03 (m, 2H), 1.63 (m, 4H), 1.49 (s, 9H).
Compound 59 (0.27 g, 0.5 mmol) was dissolved in DCM (2 mL) and TFA (0.5 mL) was added at rt. After 0.5 h, all volatile materials were removed in a rotary evaporator to afford 60 in 100% yield. 1H NMR (MeCN-d3): δ 7.77 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.4 Hz, 1H), 7.21 (d, J = 2.4 Hz, 1H), 7.03 (m, 3H), 6.84 (d, J = 8.0 Hz, 1H, CONH), 5.13 (br, 2H, NH-TFA), 4.46 (m, 1H), 3.95 (m, 1H), 3.53 (m, 4H), 3.35 (m, 4H), 2.17 (m, 2H), 2.07 (m, 2H), 1.59 (m, 4H).
Compound 60 (0.28 g, 0.5 mmol TFA salt) was dissolved in DCE (2 mL), with AcOH (0.03 g, 0.5 mL), then N-Boc amino-propylaldehyde (0.105 g, 0.75 mmol) and NaB(OAc)3H (0.318 g, 1.5 mmol) were added. After 6 h, all volatile materials were removed and the residue was purified by CombiFlash chromatography (DCM and MeOH) to afford compound 61a. This was dissolved in DCM (2 mL) and TFA (l mL) was added at rt. After 0.5 h, all volatile materials were removed by a rotary evaporator to afford 62a in 80% yield. 1H NMR (MeCN-d3): δ 7.75 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.01 (m, 3H), 6.83 (d, J = 8.4 Hz, 1H, CONH), 4.66 (br, 3H, NH-TFA), 3.95 (m, 2H), 3.79 (m, 1H), 3.63 (m, 3H), 3.54 (m, 4H), 3.39 (m, 3H), 3.08 (m, 1H), 2.19 (m, 2H), 2.07 (m, 2H), 1.59 (m, 4H).
Compound 62a (0.099 g, 0.2 mmol) was dissolved in DMF (l mL) and basified by DIPEA (0.0103 g, 0.8 mmol). 2-(2,6-Dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (0.082 g, 0.3 mmol) was added to the above solution and the mixture was stirred at 90 °C for 12 h. The reaction mixture was cooled, acidified with TFA, and purified by preparative HPLC to afford compound 20 in 39% yield. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 738.08 calcd 738.27. Prep. HPLC 42% MeCN in water. 1H NMR (MeCN-d3): δ 8.99 (s, 1H, CONH), 8.93 (s, 1H, (CO)2NH), 7.95 (dd, J = 4.5 Hz, 8.2 Hz, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.63 (m, 1H), 7.56 (m, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.01 (m, 3H), 6.77 (d, J = 8.0 Hz, 1H), 5.01 (m, 1H), 4.46 (m, 1H), 3.94 (m, 1H), 3.71 (m, 1H), 3.68 (m, 2H), 3.40 (m, 2H), 3.14 (m, 2H), 2.83 (m, 2H), 2.77 (m, 2H), 2.72 (m, 2H), 2.19 (m, 4H), 2.14 (m, 2H), 1.55 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)propyl)-piperazin-1-yl)benzamide (21).
Compound 21 was synthesized following the procedure used for 20. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 752.25, calcd 752.29. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.95 (s, 1H, (CO)2NH), 7.76 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.01 (m, 4H), 6.90 (dd, J = 2.0 Hz, 8.9 Hz, 1H), 6.77 (d, J = 6.5 Hz, 1H), 4.97 (m, 1H), 4.46 (m, 1H), 4.14 (m, 1H), 3.68 (m, 3H), 3.36 (m, 2H), 3.22 (m, 2H), 3.12 (m, 4H), 2.75 (m, 3H), 2.77 (m, 2H), 2.16 (m, 4H), 2.04 (m, 3H), 1.56 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)butyl)-piperazin-1-yl)benzamide (22).
Compound 22 was synthesized following the procedure used for 20. UPLC–MS: 4.1 min, purity >95%, MS: [M + H]+ found, 766.12 calcd 766.30. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.97 (s, 1H, (CO)2NH), 7.71 (m, 2H), 7.71 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.01 (m, 4H), 6.90 (m, 1H), 6.77 (d, J = 6.5 Hz, 1H), 4.97 (m, 1H), 4.49 (m, 1H), 4.16 (m, 1H), 3.68 (m, 3H), 3.36 (m, 2H), 3.26 (m, 2H), 3.12 (m, 4H), 2.76 (m, 3H), 2.77 (m, 4H), 2.16 (m, 4H), 2.04 (m, 3H), 1.58 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)pentyl)-piperazin-1-yl)benzamide (23).
Compound 23 was synthesized following the procedure used for 20. UPLC–MS: 4.2 min, purity >95%, MS: [M + H]+ found, 780.20 calcd 780.32. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.90 (s, 1H, (CO)2NH), 7.74 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.12 (m, 1H), 7.01 (m, 2H), 6.85 (dd, J = 2.0 Hz, 8.9 Hz, 1H), 6.82 (d, J = 6.5 Hz, 1H), 6.72 (d, J = 8.4 Hz, 1H), 4.97 (m, 1H), 4.46 (m, 1H), 4.05 (m, 1H), 3.65 (m, 3H), 3.34 (m, 2H), 3.20 (m, 2H), 3.06 (m, 4H), 2.78 (m, 3H), 2.71 (m, 2H), 2.19 (m, 4H), 2.04 (m, 5H), 1.56 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-((1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-methyl)piperazin-1-yl)benzamide (24).
In a dry round-bottomed flask, compound 63 (0.13 g, 0.5 mmol), 64a (0.21 g, 0.75 mmol), Pd2(dba)3 (0.092 g, 0.1 mmol), Xphos (0.048 g, 0.1 mol), and Cs2C03 (0.49 g, 1.5 mmol) were added to dioxane (10 mL). The reaction mixture was degassed and stirred at 90 °C for 12 h. The reaction was cooled down, partitioned between EtOAc and H2O, and the organic layer was concentrated, and purified by CombiFlash chromatography (10% MeOH in DCM) to afford 65a (0.145 g 70%). 1H NMR (CDCI3): δ 7.92 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 4.14 (m, 3H), 3.75 (s, 3H), 3.52 (m, 5H), 2.77 (m, 4H), 1.85 (m, 4H), 1.49 (s, 9H), 1.20 (m, 3H).
Compound 65a (0.145 g, 0.34 mmol) was dissolved in MeOH (2 mL), THF (3 mL), and NaOH (6 N, 2 mL). The reaction mixture was stirred at rt overnight. After removing the majority of the MeOH and THF, the residue was diluted with 2 mL of H2O and the pH was adjusted to 2 with 1 N HCL The precipitate was filtered and dried to afford 66a. Compound 66a (0.09 g, 0.22 mmol) was dissolved in DCM (l mL) and DIPEA (0.129 g, 1 mmol), and HATU (0.126 g, 0.33 mmol) was added. After 0.5 h, 52 (0.091 g, 0.25 mmol TFA salt) was added. After 20 min, the reaction mixture was direcdy treated with CombiFlash chromatography (10% MeOH in DCM) to give 67a. 1H NMR (CDC13): δ 7.89 (m, 1H), 7.46 (m, 1H), 7.32 (m, 1H), 7.20 (m, 2H), 6.89 (m, 2H), 6.72 (m, 1H), 4.49 (m, 1H), 4.02 (m, 2H), 3.76 (m, 3H), 3.49 (m, 3H), 3.45 (m, 2H), 3.33 (m, 2H), 3.10 (m, 3H), 2.72 (m, 4H), 2.20 (m, 2H), 2.08 (m, 1H), 1.93 (m, 1H), 1.56 (m, 3H), 1.48 (s, 9H), 1.40 (m, 2H).
Compound 67a (0.063 g, 0.1 mmol) was dissolved in DCM (0.5 mL) and TFA (0.1 mL) was added. The reaction was stirred at rt for 2 h. All the volatile materials were removed and the residue was dried to give 68a, which was used directly in the next step without purification. Compound 68a (0.064 g, 0.1 mmol) was dissolved in DMF (l mL) and basified by DIPEA (0.051 g, 0.4 mmol). 2-(2,6-Dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (0.414 g, 0.15 mmol) was added to the above solution and stirred at 90 °C for 12 h. The reaction mixture was cooled down and acidified with TFA and purified by preparative HPLC to afford compound 24 in 65% yield. UPLC–MS: 4.2 min, purity >95%, MS: [M + H]+ found, 792.22 calcd 792.32. Prep. HPLC 44% MeCN in water. 1H NMR (MeCN-d3): δ 8.88 (s, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 7.21 (m, 2H), 7.04 (m, 3H), 6.74 (d, J = 7.6 Hz, 1H), 4.98 (m, 1H), 4.46 (m, 1H), 4.06 (m, 2H), 3.92 (m, 3H), 3.69 (m, 3H), 3.48 (m, 2H), 3.35 (m, 2H), 3.15 (m, 3H), 3.05 (m, 4H), 2.74 (m, 4H), 2.20 (m, 2H), 2.01 (m, 1H), 1.91 (m, 1H), 1.58 (m, 3H), 1.43 (m, 2H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-piperazin-1-yl)benzamide (25).
Compound 25 was synthesized following the procedure used for 24. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 778.20 calcd 778.30. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.89 (s, 1H), 7.76 (d, J = 8.9 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.38 (d, J = 2.4 Hz, 1H), 7.26 (dd, J = 2.4 Hz, 8.6 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.02 (td, J = 2.4 Hz, 8.6 Hz, 3H), 6.74 (d, J = 7.0 Hz, 1H), 4.96 (m, 2H), 4.46 (m, 1H), 4.21 (m, 2H), 3.93 (m, 2H), 3.64 (m, 2H), 3.46 (m, 2H), 3.34 (m, 2H), 3.16 (m, 2H), 3.03 (m, 3H), 2.75 (m, 4H), 2.08 (m, 2H), 1.91 (m, 2H), 1.79 (m, 1H), 1.59 (m, 5H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-piperidin-1-yl)benzamide (26).
Compound 26 was synthesized following the procedure used for 24. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 778.25 calcd 778.30. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.96 (s, 1H), 7.96 (m, 1H), 7.73 (m, 3H), 7.59 (m, 1H), 7.40 (m, 1H), 7.21 (m, 2H), 7.02 (m, 2H), 6.74 (m, 1H), 5.03 (m, 1H), 4.46 (m, 1H), 4.04 (m, 1H), 3.93 (m, 1H), 3.68 (m, 2H), 3.38 (m, 1H), 3.13 (m, 2H), 2.85 (m, 4H), 2.64 (m, 3H), 2.17 (m, 3H), 2.04 (m, 1H), 1.90 (m, 1H), 1.59 (m, 3H), 1.37 (m, 8H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-((l-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)azetidin-3-yl)-methyl)piperazin-1-yl)benzamide (27).
Compound 27 was synthesized following the procedure used for 24. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 764.32 calcd 764.291. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.98 (s, 1H, (CO)2NH), 7.76 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8.2 Hz, 1H), 7.19 (d, J = 2.4 Hz, 1H), 7.01 (m, 3H), 6.87 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 2.2 HZ, 1H), 6.63 (dd, J = 2.2 Hz, 8.4 Hz, 1H), 4.94 (m, 1H), 4.45 (m, 1H), 4.27 (m, 2H), 3.94 (m, 1H), 3.87 (m, 3H), 3.48 (m, 2H), 3.41 (m, 2H), 2.74 (m, 2H), 2.64 (m, 3H), 2.10 (m, 5H), 1.58 (m, 5H), 1.39 (m, 2H), 1.19 (m, 1H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cyclohexyl)-4-(4-(1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)azetidin-3-yl)-piperazin-1-yl)benzamide (28).
Compound 28 was synthesized following the procedure for 24. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 750.21 calcd 750.27. Prep. HPLC 41% MeCN in water. 1H NMR (MeCN-d3): δ 8.88 (s, 1H), 7.76 (d, J = 8.9 Hz, 2H), 7.71 (d, J = 4.4 Hz, 1H), 7.69 (d, J = 4.0 Hz, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.02 (m, 3H),6.89 (d, J = 2.0 Hz, 1H), 6.74 (d, J = 2.0 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 4.99 (m, 1H), 4.46 (m, 1H), 4.38 (m, 2H), 4.14 (m, 1H), 3.93 (m, 1H), 3.62 (m, 3H), 3.28 (m, 3H), 2.75 (m, 4H), 2.19 (m, 5H), 2.08 (m, 3H), 1.59 (m, 3H), 1.34 (m, 1H).
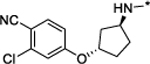
N-(1S,3S)-3-((3-Chloro-4-cyanophenoxy)cyclopentyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-piperidin-1-yl)benzamide (29).
Compound 29 was synthesized following the procedure used for 24. Purity >95%, MS: [M + H]+ found, 763.96 calcd 764.29. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.97 (s, 1H), 8.94 (s, 1H), 7.73 (m, 3H), 7.39 (d, J = 2.2 Hz, 1H), 7.26 (dd, J = 8.5 Hz, 2.3 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 6.99 (m, 3H), 6.87 (d, J = 7.2 Hz, 1H), 5.02 (m, 2H), 4.54 (m, 1H), 4.04 (m, 2H), 3.69 (m, 2H), 3.35 (m, 2H), 3.13 (m, 1H), 2.86 (m, 2H), 2.73 (m, 3H), 2.01 (m, 3H), 1.84 (m, 5H), 1.70 (m, 3H), 1.34 (m, 4H).
N-(1R,4R)-4-((3-Chloro-4-cyanophenoxy)cycloheptyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-piperidin-1-yl)benzamide (30).
Compound 30 was synthesized following the procedure used for 24. Purity >95%, MS: [M + H]+ found, 792.17 calcd 792.32. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H), 7.73 (m, 4H), 7.39 (d, J = 2.2 Hz, 1H), 7.26 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 6.99 (m, 3H), 6.84 (d, J = 7.2 Hz, 1H), 4.97 (m, 1H), 4.63 (m, 2H), 4.11 (m, 2H), 4.03 (m, 2H), 3.37 (m, 3H), 2.81 (m, 5H), 2.74 (m, 6H), 1.90 (m, 6H), 1.83 (m, 6H), 1.39 (m, 1H).
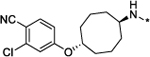
N-(1R,5R)-5-((3-Chloro-4-cyanophenoxy)cyclooctyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-piperidin-1-yl)benzamide (31).
Compound 31 was synthesized following the procedure used for 24. Purity >95%, MS: [M + H]+ found, 806.27 calcd 806.34. Prep. HPLC 46% MeCN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H), 7.74 (m, 4H), 7.39 (d, J = 2.0 Hz, 1H), 7.26 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 6.99 (m, 3H), 6.76 (d, J = 7.2 Hz, 1H), 4.99 (m, 1H), 4.63 (m, 2H), 4.11 (m, 2H), 4.03 (m, 2H), 3.37 (m, 4H), 2.85 (m, 6H), 2.75 (m, 6H), 1.92 (m, 6H), 1.78 (m, 6H), 1.34 (m, 1H).
2-Chloro-4-((1-(4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)benzoyl)piperidin-4-yl)-oxy)benzonitrile (32).
Compound 32 was synthesized following the procedure used for 24. Purity >95%, MS: [M + H]+ found, 764.00 calcd 764.29. Prep. HPLC 42% MeCN in water. 1H NMR (MeCN-d3): δ 9.11 (s, 1H), 8.96 (s, 1H), 7.72 (m, 2H), 7.39 (m, 2H), 7.23 (m, 2H), 7.03 (m, 3H), 5.00 (m, 1H), 4.77 (m, 1H), 4.01 (m, 2H), 3.86 (m, 2H), 3.69 (m, 4H), 3.44 (m, 3H), 3.35 (m, 2H), 3.14 (m, 4H), 2.79 (m, 5H), 2.21 (m, 1H), 2.12 (m, 1H), 2.02 (m, 2H), 1.88 (m, 1H), 1.74 (m, 2H).
2-Chloro-4-((1-(4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yljpiperidin-1-yl)benzoyl)azepan-4-yl)-oxyjbenzonitrile (33).
Compound 33 was synthesized following the procedure used for 24. UPLC–MS: 4.1 min, purity >95%, MS: [M + H]+ found, 778.21 calcd 778.30. Prep. HPLC 41% MeCN in water. 1H NMR (MeCN-d3): δ 8.91 (s, 1H), 7.96 (m, 1H), 7.71 (m, 2H), 7.59 (m, 1H), 7.40 (m, 2H), 7.20 (m, 2H), 6.97 (m, 2H), 4.99 (m, 2H), 4.70 (m, 1H), 4.49 (m, 1H), 4.08 (m, 1H), 3.94 (m, 2H), 3.58 (m, 7H), 3.29 (m, 2H), 3.12 (m, 3H), 2.77 (m, 5H), 1.58 (m, 3H), 1.09 (m, 3H), 0.91 (m, 3H).
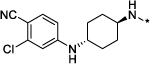
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)amino)cyclohexyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)benzamide (34).
Compound 34 was synthesized following the procedure used for 24. UPLC–MS: 3.8 min, purity >95%, MS: [M + H]+ found, 777.18 calcd 777.32. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.98 (s, 1H), 7.75 (m, 3H), 7.44 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 2.2 Hz, 1H), 7.26 (dd, J = 8.6 Hz, 2.4 Hz, 1H), 6.99 (d, J = 8.2 Hz, 2H), 6.74 (m, 2H), 6.61 (dd, J = 8.8 Hz, 2.2 Hz, 1H), 5.45 (s, 1H), 4.99 (m, 1H), 4.03 (m, 2H), 3.88 (m, 2H), 3.37 (m, 4H), 2.85 (m, 4H), 2.74 (m, 4H), 2.21 (m, 4H), 2.07 (m, 6H), 1.89 (m, 2H), 1.51 (m, 3H).
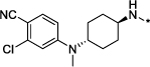
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yljpiperidin-1-yljbenzamide (35).
Trans-(4-methylaminocyclohexyl)carbamate t-butyl ester (2.28 g, 10 mmol) was dissolved in MeCN (30 mL), and compound 49 (2.30 g, 15 mmol) was added. The reaction was stirred at 80 °C for 18 h. After UPLC–MS showed conversion of 50 was complete, the reaction mixture was diluted with H2O, extracted with EtOAc, dried, and concentrated. Compound 72e was obtained in 63% (2.29 g) yield after purification on a silica gel column (70% EtOAc in hexane). 1H NMR (MeCN-d3): δ 7.51 (d, J = 8.4 Hz, 1H), 6.89 (d, J = 2.8 Hz, 1H), 6.78 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 5.21 (br, 1H), 3.73 (m, 1H), 3.38 (m, 1H), 2.84 (s, 3H), 2.16 (m, 4H), 1.74 (m, 4H), 1.42 (s, 9H).
Compound 72e (0.36 g, 1 mmol) was dissolved in DCM (4 mL) and TFA (0.44 g, 4 mmol) was added at rt. After 0.5 h, all volatile compounds were removed using a rotary evaporator to afford 73e in 100% yield. 1H NMR (MeCN-d3): δ 7.52 (d, J = 8.0 Hz, 1H), 7.39 (br, 3H, NH2-TFA), 6.92 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8.0 Hz, 2.4 Hz, 1H), 3.79 (m, 1H), 3.19 (m, 1H), 2.83 (s, 3H), 2.58 (m, 4H), 2.18 (m, 2H), 1.80 (m, 1H), 1.70 (m, 1H).
To a dry round-bottomed flask, compounds 63 (0.13 g, 0.5 mmol) and 64a (0.21 g, 0.75 mmol) were added to dioxane (10 mL). The reaction mixture was degassed and stirred at 90 °C for 12 h. The reaction was cooled down, partitioned between EtOAc and H2O, and the organic layer was concentrated and purified by CombiFlash chromatography (10% MeOH in DCM) to afford 65a (0.145 g, 70%). 1H NMR (CDCl3): δ 7.92 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 4.14 (m, 3H), 3.75 (s, 3H), 3.52 (m, 5H), 2.77 (m, 4H), 1.85 (m, 4H), 1.49 (s, 9H), 1.20 (m, 3H).
Intermediate 77 was synthesized in 70% yield following the procedure used for 66a. 1H NMR (MeCN-d3): δ 7.88 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 4.08 (m, 2H), 3.38 (m, 2H), 2.92 (m, 2H), 2.18 (m, 5H), 1.89 (m, 2H), 1.48 (s, 9H), 1.28 (m, 3H), 0.97 (m, 1H).
Compound 77 (0.389 g, 1 mmol) was dissolved in DCM (2 mL) and basified with DIPEA (0.39 g, 3 mmol) then HATU (0.49 g, 1.3 mmol) was added and stirred for 10 min. In a separate vial, compound 73e (0.378 g, 1 mmol TFA salt) was dissolved in DCM (2 mL) and basified by DIPEA (0.26 g, 2 mmol). The solution of compound 73e was poured into the above reaction mixture and stirred for 0.5 h. All the volatile compounds were removed and the residue was purified by CombiFlash chromatography (hexane and EtOAc) to afford 80e in 80% yield. 1H NMR (MeCN-d3): δ 8.53 (m, 1H), 7.59 (m, 1H), 7.51 (m, 1H), 7.05 (m, 1H), 6.93 (m, 2H), 6.81 (m, 1H), 6.78 (m, 1H), 4.08 (m, 1H), 3.90 (m, 1H), 3.79 (m, 1H), 3.68 (m, 1H), 3.59 (m, 3H), 3.18 (m, 1H), 2.98 (m, 1H), 2.87 (m, 2H), 2.73 (s, 3H), 2.53 (m, 3H), 2.19 (m, 3H), 1.80 (m, 2H), 1.78 (m, 3H), 1.46 (s, 9H), 1.45 (m, 2H), 1.23 (m, 2H), 0.98 (m, 1H).
Compound 80e was dissolved in DCM (2 mL) and TFA (0.5 mL) was added at rt. After 0.5 h, all volatile compounds were removed using a rotary evaporator to afford 81e in 100% yield.
Compound 81e (0.129 g, 0.2 mmol TFA salt) was dissolved in DMF (l mL) and basified by DIPEA (0.0103 g, 0.8 mmol). 2-(2,6-Dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (0.082 g, 0.3 mmol) was added to the above solution and stirred at 90 °C for 12 h. The reaction mixture was cooled down and acidified with TFA and purified by preparative HPLC to afford compound 35 in yield 49%. UPLC–MS: 4.1 min, purity >95%, MS: [M + H]+ found, 791.30 calcd 791.34. Prep. HPLC 40% MeCN in water. 1H NMR (MeCN-d3): δ 8.91 (s, 1H), 7.84 (m, 2H), 7.80 (d, J = 8.8 Hz, 1H), 7.51 (dd, J = 8.8 Hz, 2.4 Hz, 2H), 7.40 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 7.23 (m, 1H), 6.95 (m, 1H), 6.84 (m, 2H), 5.01 (m, 1H), 4.21 (m, 2H), 4.00 (m, 2H), 3.91 (m, 1H), 3.80 (m, 1H), 3.71 (m, 2H), 3.57 (m, 1H), 3.37 (m, 3H), 3.27 (m, 1H), 3.01 (m, 2H), 2.90 (d, 3H), 2.88 (m, 1H), 2.85 (m, 1H), 2.79 (m, 1H), 2.76 (m, 1H), 2.14 (m, 2H), 2.08 (m, 2H), 2.01 (m, 2H), 1.91 (m, 1H), 1.81 (m, 3H), 1.59 (m, 1H), 1.46 (m, 1H). 13C NMR (MeCN-d3): δ 171.97, 169.50, 167.59, 167.13, 165.98, 159.95, 159.60, 137.20, 134.58, 134.25, 128.49, 124.92, 124.85, 120.99, 114.96, 114.59, 112.07, 110.72, 109.09, 109.03, 96.87, 63.37, 56.48, 54.60, 49.21, 48.12, 48.04, 46.80, 44.70, 44.54, 42.96, 42.70, 31.46, 31.31, 31.22, 31.06, 30.82, 27.99, 25.65, 22.27, 17.67, 16.35, 11.80.
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(ethyl)amino)-cyclohexyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yljpiperidin-1-yl)benzamide (36).
Compound 36 was synthesized following the procedure used for 35. UPLC–MS: 4.6 min, purity >95%, MS: [M + H]+ found, 805.05 calcd 805.35. Prep. HPLC 48% MeCN in water. 1H NMR (MeCN-d3): δ 8.95 (m, 2H), 8.14 (m, 1H), 7.73 (m, 2H), 7.51 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 2.2 Hz, 1H), 7.26 (dd, J = 8.6 Hz, 2.4 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.88 (d, J = 2.6 Hz, 1H), 6.78 (d, J = 9.0 Hz, 2.6 Hz, 1H), 6.70 (d, J = 7.8 Hz, 1H), 5.01 (m, 1H), 4.03 (m, 2H), 3.91 (m, 2H), 3.71 (m, 4H), 3.42 (m, 2H), 3.33 (m, 2H), 3.11 (m, 1H), 2.86 (m, 2H), 2.73 (m, 3H), 1.86 (m, 3H), 1.75 (m, 3H), 1.57 (m, 3H), 1.36 (m, 6H), 1.18 (m, 3H).
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(propyl)amino)-cyclohexyl)-4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)benzamide (37).
Compound 37 was synthesized following the procedure used for 35. UPLC–MS: 4.9 min, purity >95%, MS: [M + H]+ found, 819.14 calcd 819.36. Prep. HPLC 51% MeCN in water. 1H NMR (MeCN-d3): δ 8.91 (m, 2H), 8.14 (m, 1H), 7.75 (m, 2H), 7.51 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 2.2 Hz, 1H), 7.26 (dd, J = 8.6 Hz, 2.4 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 2.6 Hz, 1H), 6.76 (d, J = 9.0 Hz, 2.6 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 4.98 (m, 2H), 4.51 (m, 1H), 4.02 (m, 2H), 3.89 (m, 2H), 3.79 (m, 2H), 3.70 (m, 2H), 3.32 (m, 1H), 3.23 (m, 2H), 3.13 (m, 2H), 2.87 (m, 2H), 2.73 (m, 3H), 1.81 (m, 5H), 1.59 (m, 4H), 1.36 (m, 6H), 0.96 (m, 3H).
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(4-((1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)methyl)piperazin-1-yl)benzamide (39).
Compound 39 was synthesized following the procedure used for 35. UPLC–MS: 4.3 min, purity >95%, MS: [M + H]+ found, 805.38 calcd 805.35. Prep. HPLC 45% MeCN in water. 1H NMR (MeCN-d3): δ 8.89 (s, 1H), 7.95 (dd, J = 4.2 Hz, 8.2 Hz, 1H), 7.77 (d, J = 8.9 Hz, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.59 (m, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 2.2 Hz, 1H), 7.19 (dd, J = 2.6 Hz, 8.8 Hz, 1H), 7.02 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 2.6 Hz, 1H), 6.79 (dd, J = 2.6 Hz, 8.8 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 5.01 (m, 2H), 4.06 (m, 2H), 3.90 (m, 2H), 3.79 (m, 2H), 3.68 (m, 2H), 3.34 (m, 2H), 3.13 (m, 2H), 3.6 (m, 2H), 3.00 (m, 1H), 2.88 (s, 3H), 2.82 (m, 2H), 2.75 (m, 4H), 2.15 (m, 5H), 1.80 (m, 3H), 1.60 (m, 1H), 1.43 (m, 1H), 1.36 (m, 1H).
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)benzamide (40).
Compound 40 was synthesized following the procedure used for 35. UPLC–MS: 4.3 min, purity >95%, MS: [M + H]+ found, 805.32 calcd 805.35. Prep. HPLC 45% MeCN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H), 8.07 (s, 1H), 7.84 (m, 1H), 7.76 (m, 1H), 7.46 (m, 1H), 7.27 (m, 1H), 6.98 (m, 1H), 6.74 (m, 1H), 6.67 (m, 1H), 6.62 (m, 1H), 6.00 (m, 2H), 4.99 (m, 1H), 3.88 (m, 2H), 3.76 (m, 3H), 3.66 (m, 1H), 3.29 (m, 4H), 3.14 (m, 1H), 3.06 (m, 1H), 3.01 (m, 2H), 2.89 (m, 1H), 2.79 (m, 3H), 2.06 (s, 3H), 2.02 (m, 3H), 1.75 (m, 2H), 1.72 (m, 2H), 1.70 (m, 2H), 1.69 (m, 2H), 1.64 (m, 2H), 1.38 (m, 1H), 1.34 (m, 1H).
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(4-(1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)piperazin-1-yl)benzamide (41).
Intermediate 66c was synthesized in 65% yield following the procedure used for 66a. 1H NMR (MeCN-d3): δ 7.91 (m, 2H), 6.97 (m, 2H), 4.52 (m, 1H), 4.21 (m, 1H), 4.11 (m, 2H), 3.96 (m, 1H), 3.55 (m, 3H), 3.39 (m, 2H), 2.78 (m, 2H), 2.36 (m, 2H), 2.18 (m, 2H), 1.79 (m, 2H), 1.45 (s, 9H).
Intermediate 82c was synthesized in 75% yield following the procedure used for 80e. 1H NMR (MeCN-d3): δ 8.54 (m, 1H), 8.13 (m, 1H), 7.70 (m, 1H), 7.55 (m, 1H), 7.09 (m, 1H), 6.93 (m, 2H), 6.91 (m, 1H), 4.08 (m, 2H), 3.48 (m, 2H), 3.25 (m, 4H), 2.89 (m, 1H), 2.80 (m, 2H), 2.76 (s, 3H), 2.48 (m, 2H), 2.44 (m, 5H), 2.16 (m, 2H), 1.80 (m, 4H), 1.49 (s, 9H), 1.37 (m, 3H).
Compound 82c was dissolved in DCM (2 mL) and TFA (0.5 mL) was added at rt. After 0.5 h, all volatile materials were removed by rotary evaporator to afford 83c in 100% yield.
Compound 83c (0.129 g, 0.2 mmol TFA salt) was dissolved in DMF (l mL) and basified by DIPEA (0.0103 g, 0.8 mmol). 2-(2,6-Dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (0.082 g, 0.3 mmol) was added to the above solution and stirred at 90 °C for 12 h. The reaction mixture was cooled down and acidified with TFA and purified by preparative HPLC to afford compound 41 in 54% yield. UPLC–MS: 4.2 min, purity >95%, MS: [M + H]+ found, 791.24 calcd 791.34. Prep. HPLC 47% MeCN in water. 1H NMR (MeCN-d3): δ 8.88 (s, 1H), 7.46 (d, J = 8.9 Hz, 2H), 7.72 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 9.0 Hz, 1H), 7.38 (d, J = 2.4 Hz, 1H), 7.25 (dd, J = 2.4 Hz, 8.6 Hz, 1H), 7.00 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 2.6 Hz, 1H), 6.81 (dd, J = 2.5 Hz, 9.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 4.99 (m, 1H), 4.17 (m, 2H), 3.93 (m, 3H), 3.79 (m, 1H), 3.64 (m, 2H), 3.49 (m, 2H), 3.17 (m, 1H), 3.04 (m, 3H), 2.87 (s, 3H), 2.74 (m, 2H), 2.25 (m, 2H), 2.09 (m, 3H), 1.89 (m, 2H), 1.85 (m, 4H), 1.58 (m, 2H), 1.36 (m, 2H). 13C NMR (acetone-d6): δ 171.73, 171.65, 169.30, 169.23, 167.54, 166.96, 165.38, 165.31, 137.06, 134.53, 128.58, 126.41, 124.77, 119.81, 118.52, 117.53, 117.14, 114.72, 112.06, 110.79, 108.46, 97.26, 63.10, 56.67, 56.47, 49.23, 49.21, 48.16, 48.05, 47.94, 46.48, 45.38, 31.57, 31.55, 31.13, 31.10, 30.72, 30.68, 28.13, 27.99, 25.54, 22.53, 16.94.
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(4-((1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)azetidin-3-yl)methyl)piperazin-1-yljbenzamide (42).
Compound 42 was synthesized following the procedure used for 41. UPLC–MS: 4.2 min, purity >95%, MS: [M + H]+ found, 777.16 caled 777.32. Prep. HPLC 45% MeCN in water. 1H NMR (MeCN-d3): δ 8.89 (s, 1H), 7.76 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 2.6 Hz, 1H), 6.81 (dd, J = 2.4 Hz, 8.4 Hz, 2H), 6.75 (d, J = 8.0 Hz, 1H), 6.64 (dd, J = 2.2 Hz, 8.2 Hz, 1H), 4.98 (m, 1H), 4.27 (m 2H), 3.89 (m, 3H), 3.77 (m, 2H), 3.53 (m, 1H), 3.46 (m, 2H), 3.39 (m, 2H), 3.34 (m, 1H), 2.88 (s, 3H), 2.74 (m, 3H), 2.09 (m, 4H), 1.78 (m, 5H), 1.58 (m, 3H), 1.34 (m, 1H).
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(4-(1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)azetidin-3-yl)piperazin-1-yl)benzamide (43).
Intermediate 66e was synthesized in 82% yield following the procedure used fiar 66a. 1H NMR (MeCN-d3): δ 7.87 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 3.92 (m, 1H), 3.78 (m, 1H), 3.36 (m, 2H), 3.11 (m, 1H), 2.49 (m, 2H), 1.67 (m, 2H), 1.45 (s, 9H), 1.29 (m, 2H), 1.21 (m, 1H), 0.95 (m, 1H).
Intermediate 82e was synthesized in 70% yield following the procedure used for 80e. 1H NMR (MeCN-d3): δ 8.57 (m, 1H), 8.13 (m, 1H), 7.69 (m, 1H), 7.57 (m, 1H), 7.09 (m, 1H), 6.90 (m, 2H), 6.77 (m, 1H), 3.92 (m, 3H), 3.78 (m, 2H), 3.51 (m, 2H), 3.31 (m, 3H), 3.19 (m, 1H), 2.86 (m, 1H), 2.76 (s, 3H), 2.52 (m, 5H), 2.18 (m, 1H), 1.78 (m, 2H), 1.61 (m, 1H), 1.46 (s, 9H), 1.40 (m, 1H), 0.89 (m, 1H).
Deprotection of Boc and substitution with 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione following the procedure used for 41 provided compound 43 in 55% yield. UPLC–MS: 4.0 min, purity >95%, MS: [M + H]+ found, 763.15 caled 763.30. Prep. HPLC 43% MeCN in water. 1H NMR (Me2CO-d6): δ 8.89 (s, 1H), 7.83 (d, J = 8.9 Hz, 2H), 7.66 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 2.4Hz, 2H), 6.92 (d, J = 2.6 Hz, 1H), 6.85 (dd, J = 2.4 Hz, 8.6 Hz, 2H), 6.75 (dd, J = 8.2 Hz, 2.2 Hz, 1H), 5.81 (br s, 1H, TFA salt), 5.09 (m, 1H), 4.53 (m, 4H), 3.93 (m, 2H), 3.69 (m, 4H), 3.46 (m, 4H), 2.96 (m, 1H), 2.90 (s, 3H), 2.75 (m, 2H), 2.13 (m, 1H), 2.11 (m, 3H), 1.84 (m, 4H), 1.65 (m, 2H). 13C NMR (acetone-d6): δ 172.58, 170.13, 168.31, 168.07, 166.25, 155.74, 154.25, 152.77, 137.96, 135.43, 135.20, 129.49, 129.46, 127.22, 125.46, 120.21, 118.50, 118.05, 115.82, 115.67, 115.62, 115.57, 115.17, 112.96, 111.69, 105.84, 98.16, 60.87, 57.57, 55.33, 54.28, 50.08, 49.78, 49.62, 48.85, 46.60, 46.29, 32.45, 32.00, 31.63, 23.44. HRMS: [M + H] found, 763.3120 caled 763.3123.
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(3-((4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)azetidin-1-yl)benzamide (44).
Compound 44 was synthesized following the procedure used for 43. UPLC–MS: 4.2 min, purity >95%, MS: [M + H] found, 777.18 caled 777.32. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.93 (s, 1H), 8.07 (s, 1H), 7.77 (m, 2H), 7.47 (m, 2H), 7.38 (m, 1H), 7.24 (m, 1H), 6.86 (m, 2H), 6.51 (m, 1H), 6.14 (m, 1H), 4.97 (m, 2H), 4.52 (m, 1H), 4.14 (m, 1H), 4.03 (m, 1H), 3.91 (m, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.63 (m, 1H), 3.51 (m, 1H), 3.36 (m, 2H), 2.88 (s, 3H), 2.75 (m, 4H), 2.08 (m, 5H), 1.82 (m, 3H), 1.61 (m, 3H), 1.35 (m, 3H).
N-(1R,4R)-4-(((3-Chloro-4-cyanophenyl)(methyl)amino)-cyclohexyl)-4-(3-(4-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)azetidin-1-yl)benzamide (45).
Compound 45 was synthesized following the procedure used for 43. UPLC–MS: 4.1 min, purity >95%, MS: [M + H]+ found, 763.21 ealed 763.30. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 8.89 (s, 1H), 7.76(d, J = 8.9 Hz, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 2.2 Hz, 1H), 7.28 (dd, J = 8.0 Hz, 2.4 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 6.82 (dd, J = 2.4 Hz, 8.6 Hz, 1H), 6.75 (d, J = 8.2 Hz, 1H), 6.54 (d, J = 8.0 Hz, 2H), 5.00 (m, 1H), 4.20 (m, 3H), 4.04 (m, 1H), 3.89 (m, 1H), 3.75 (m, 3H), 3.35 (m, 1H), 3.22 (m, 2H), 2.88 (s, 3H), 2.73 (m, 3H), 2.13 (m, 2H), 2.09 (m, 2H), 2.02 (m, 2H), 1.91 (m, 1H), 1.79 (m, 3H), 1.59 (m, 2H), 1.33 (m, 1H). 13C NMR (MeCN-d3): δ 171.63, 169.18, 166.93, 164.66, 159.19, 153.37, 150.29, 143.98, 141.74, 139.50, 138.30, 136.83, 134.52, 131.25, 128.36, 124.78, 124.09, 120.45, 118.65, 116.84, 112.06, 110.79, 110.37,109.85, 109.04, 98.54, 82.37, 74.02, 56.71, 54.59, 54.10,49.24, 48.61, 47.85, 45.75, 42.83, 31.61, 31.10, 30.72, 22.51, 20.58.
N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-((4-(4-(1-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)piperazin-1-yl)phenyl)sulfonyl)-2-hydroxy-2-methylpropanamide (46).
UPLC–MS: 3.2 min, purity >95%, MS: [M + H]+ found, 836.14 ealed 836.26. Prep. HPLC 38% MeCN in water. 1H NMR (MeCN-d3): δ 9.43 (s, 1H), 8.90 (s, 1H), 8.16 (d, J = 2.0 Hz, 1H), 7.93 (m, 2H), 7.71 (m, 3H), 7.37 (d, J = 2.4 Hz, 1H), 7.25 (dd, J = 8.6 Hz, 2.4 Hz, 1H), 6.90 (d, J = 9.0 Hz, 2H), 4.97 (m, 1H), 4.17 (m, 2H), 4.03 (m, 1H), 3.88 (m, 1H), 3.53 (m, 2H), 3.46 (m, 1H), 3.28 (m, 2H), 3.03 (m, 4H), 2.79 (m, 6H), 2.25 (m, 2H), 2.12 (m, 1H), 1.86 (m, 2H), 1.48 (s, 3H).
4-(3-(4-(4-(1-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)piperazine-1-carbonyl)-3-fluorophenyl)-4,4-dimethyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluoromethyl)-benzonitrile (47).
Compound 89a (0.045 g, 0.1 mmol) was dissolved in DCM (0.5 mL) and basified with DIPEA (0.039 g, 0.3 mmol), then HATU (0.049 g, 0.13 mmol) was added and stirred for 10 min. Boc 4-(piperazin-l-yl)piperidine-l-carboxylate (0.027 g, 0.1 mmol) was added and the reaction mixture was stirred for 0.5 h. All the volatile compounds were removed and the residue was purified by CombiFlash chromatography (10% MeOH in DCM) to afford 90a, which was dissolved in DCM (l mL) and TFA (0.2 mL) and stirred for 0.5 h. All the volatile materials were removed under vacuum to provide 91a as the TFA salt (0.061 g, 85%). 1H NMR (MeCN-d3): δ 8.18 (m, 2H), 7.99 (m, 1H), 7.65 (m, 1H), 7.35 (m, 2H), 5.96 (br, 2H, NH-TFA), 3.72 (m, 5H), 3.59 (m, 2H), 3.43 (m, 1H), 3.18 (m, 3H), 3.04 (m, 2H), 2.65 (m, 1H), 2.36 (m, 2H), 2.09 (m, 2H), 1.60 (s, 6H).
Compound 91a. Displacement of F in 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione following the procedure used for 43 provided compound 47 in 65% yield. UPLC–MS: 4.1 min, purity >95%, MS: [M + H]+ found, 859.20 ealed 859.26. Prep. HPLC 42% MeCN in water. 1H NMR (MeCN-d3): δ 8.89 (s, 1H), 8.15 (m, 2H), 7.99 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.59 (m, 1H), 7.36 (m, 1H), 7.32 (m, 1H), 7.28 (m, 1H), 7.22 (m, 1H), 4.96 (m, 1H), 4.16 (m, 2H), 4.04 (m, 1H), 3.68 (m, 3H), 3.35 (m, 1H), 3.19 (m, 1H), 3.00 (m, 3H), 2.78 (m, 4H), 2.09 (m, 1H), 2.01 (m, 2H), 1.91 (m, 1H), 1.82 (m, 2H), 1.60 (s, 6H).
5-(5-(4-(4-(1-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)piperazine-1-carbonyl)-3-fluorophenyl)-8-oxo-6-thioxo-5,7-diazaspiro[3.4]octan-7-yl)-3-(trifluoromethyl)-picolinonitrile (48).
Compound 48 was synthesized following the procedure used for 47. UPLC–MS: 4.2 min, purity >95%, MS: [M + H]+ found, 872.13 ealed 872.25. Prep. HPLC 43% MeCN in water. 1H NMR (MeCN-d3): δ 9.15 (s, 1H), 8.99 (s, 1H), 8.45 (d, J = 2.6 Hz, 1H), 7.69 (m, 2H), 7.36 (m, 2H), 7.31 (dd, J = 9.0 Hz, 2.0 Hz, 1H), 7.22 (dd, J = 8.6 Hz, 2.2 Hz, 1H), 4.96 (m, 1H), 4.16 (m, 2H), 3.76 (m, 2H), 3.45 (m, 2H), 3.03 (m, 3H), 2.73 (m, 3H), 2.67 (m, 3H), 2.60 (m, 2H), 2.21 (m, 2H), 2.12 (m, 2H), 1.89 (m, 2H), 1.69 (m, 2H), 1.35 (m, 1H), 1.25 (m, 1H).
Cell Lines and Cell Culture.
All cell lines were purchased direedy from American Type Culture Collection (ATCC). LNCaP and 22Rvl cells were grown in RPMI1640 medium (Invitrogen), and VCaP cells were grown in DMEM medium with Glutamax (Invitrogen). MDA-Pca-2b cells were grown with F-12K Medium (Kaighn’s Modification of Ham’s F-12 Medium). All of the cells were supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C in a humidified 5% CO2 incubator. Cell viability was evaluated by a WST-8 assay (Dojindo) following the manufacturer’s instructions. Western blot analysis was performed as previously described.
Western Blotting.
Treated cells were lysed by RIPA buffer supplemented with protease and phosphatase inhibitors. The cell lysates were separated by 4–12% SDS-PAGE gels and blotted into PVDF membranes. ImageJ was used to quantify the percentage of AR degradation.28 The net protein bands and loading controls were calculated by deducting the background from the inverted band value. The final relative quantification values are the ratio of net band to net loading control.
PK, PK/PD, and Efficacy Studies in Mice.
All in vivo studies were performed under animal protocol (PR000009463) approved by the Institutional Animal Care & Use Committee (IACUC) of the University of Michigan, in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
To grow VCaP xenograft tumors, male CB17 SCID mice (Charles River Laboratories) were injected subcutaneously with 5 × 106 VCaP cells in 5 mg/mL Matrigel (Coming).
For determination of oral exposures for AR degraders, each compound was administered in nontumor-bearing male mice via oral gavage using 100% PEG200 as the dosing vehicle. Animals were sacrificed at indicated time-points with three mice for each time-point for each compound and 300 μL of blood was collected from each animal and plasma samples were stored at −80 °C until analysis.
For PK/PD studies in tumor-bearing male SCID mice, each compound was administered in animals via oral gavage using 100% PEG200 as the dosing vehicle when VCaP tumors reached approximately 200 mm3. Animals were sacrificed at indicated timepoints with three mice for each compound at each time-point, and blood (300 μL), prostate and tumor were collected from each animal for analysis. Isolated tumor samples were immediately frozen and ground with a mortar and pestle in liquid nitrogen. All plasma and tumor samples were stored at −80 °C until analysis. For analysis of AR protein levels in tumor samples, resected VCaP xenograft tumor tissues were ground into powder in liquid nitrogen and lysed in CST lysis buffer with halt proteinase inhibitors. Twenty micrograms of whole tumor clarified lysates were separated on 4–20 or 4–12% Novex gels. Western blots were performed as detailed in the previous section.
To prepare tumor samples for LCMS analysis, mixed ultrapure water and acetonitrile solution (4:1) were added to the defrosted tumor tissue samples 5:1, v/w, in order to facilitate homogenization with a Precellys evolution homogenizer at 4 °C. The homogenized tissues solution was denatured using cold acetonitrile (1:3, v/v) with vortex and centrifuged at 13000 rpm 4 °C for 10 min. Following protein precipitation, the final supernatants were collected for LC–MS analysis.
To determine drug concentrations in plasma and tumor samples, a LC–MS/MS method was developed and validated. The LC–MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of a test compound was achieved using a Waters XBridge-C18 column (5 cm X 2.1 mm, 3.5 μm). An AB Sciex QTrap 5500 mass spectrometer equipped with an electrospray ionization source (Applied Biosystems, Toronto, Canada) in the positive-ion multiple reaction monitoring mode was used for detection. For example, the precursor/product ion transitions were monitored at m/z 763.3 for ARD-2585 and internal standard, respectively, in the positive electrospray ionization mode. The mobile phases used on HPLC were 0.1% formic acid in purified water (A) and 0.1% formic acid in acetonitrile (B). The gradient (B) was held at 10% (0–0.3 min), increased to 95% at 0.7 min, then kept at isocratic 95% B for 2.3 min, and then immediately stepped back down to 10% for 2 min re-equilibration. The flow rate was set at 0.4 mL/min. All PK parameters were calculated by noncompartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
For the in vivo efficacy experiments, when VCaP tumors reached an average volume of 150 mm3, mice were tumor size matched and randomly assigned to different experimental groups with seven mice for each group. Drugs or vehicle control were given at the dose schedule as indicated using 100% PEG200 as the dosing vehicle. Tumor sizes and animal weights were measured 2–3 times per week. Tumor volume (mm3) = (length × width2)/2. Tumor growth inhibition was calculated as TGI (%) = (Vc − Vt)/(VC − Vo)*100, where Vc, Vt are the medians of the control and treated groups at the end of the treatment respectively, and Voat the start. Tumor volumes at the end of treatment were statistically analyzed using a two-tailed, impaired t-test (GraphPad Prism 8.0).
Microsomal Metabolic Stability Studies.
An in vitro metabolism study of a test compound was performed in human, mouse, rat dog, and monkey liver microsomes to evaluate its cytochrome P450-mediated metabolism. The metabolic stability was assessed using pooled mouse, rat, dog, monkey, and human liver microsomes, which were purchased from XenoTech (Lenexa, Kansas).
Briefly, 1 μM of the test compound was incubated with 0.75 mg/mL of the respective liver microsome and 1.7 mM cofactor-NADPH in 0.1 M K-phosphate buffer (pH = 7.4) containing 5 mM MgCl2 at 37 °C, with the acetonitrile concentration less than 0.1% in the final incubation solution. After 0, 5, 10, 15, 30, and 45 min of incubation, the reaction was stopped immediately by adding 150 μL cold acetonitrile containing IS to each 45 μL incubation solution in the wells of the corresponding plates, respectively. The incubation without the addition of NADPH was used as the negative control Ketanserin was incubated similarly to the positive control. After quenching, the plate was shaken for 10 min (600 rpm/min) and centrifuged at 6000 rpm for 15 min. 80 μL of the supernatant was then transferred from each well into a 96-well plate containing 140 μL of water for LC–MS/MS analysis, from which the remaining amount of the test compound was determined. The natural log of the remaining amount of the test compound was plotted against time to determine the disappearance rate and the half-life of the test compound. The liver microsomal stability assay was performed by Shanghai Medicilon (Shanghai, China).
Plasma Stability Studies.
The in vitro stability of a test compound was studied in human, mouse, rat, dog, and monkey plasmas. A test compound was dissolved in DMSO to a final concentration of 10 mM and then diluted to 10 μM in 0.1 M K/Mg buffer. 90 μL of prewarmed plasma at 37 °C was added to the wells of a 96-well plate before spiking them with 10 μL of 10 μM test compound to make the final concentration of the test compound to 1 μM. The spiked plasma samples were incubated at 37 °C for 2 h. Reactions were terminated at 0, 5, 15, 30, 60, and 120 min by adding 400 μL of acetonitrile containing IS. After quenching, the plates were shaken for 5 min at 600 rpm and stored at −20 °C if necessary before analysis by LC/MS. Before LC/MS analysis, the samples were thawed at rt and centrifuged at 6000 rpm for 20 min. 100 μL of the supernatant from each well was transferred into a 96-well sample plate containing 100 μL of water for LC/MS analysis. Procaine was used as reference control compound for human, mouse, dog, and monkey plasma stability studies and benfluorex was used as reference control compound for rat plasma stability studies. The in vitro plasma half-life (ti/2) was calculated using the expression t1/2 = 0.693/b, where b is the slope found in the linear fit of the natural logarithm of the fraction remaining of the test compound versus incubation time. The plasma stability assay was performed by Shanghai Medicilon (Shanghai, China).
hERG Channel Inhibition Assay.
ARD-2585 was tested for its in vitro effects on electric current passing through hERG (human ether- a-go-go-related gene) potassium channels stably expressed in a HEK 293 cell line to determine the concentration–response relationship for hERG current inhibition by ARD-2585 using the manual patch- clamp technique. ARD-2585 was tested at 0.3, 1, 3, 10, and 30 μM in duplicate. The hERG assay was performed by Shanghai Medicilon (Shanghai, China).
COMPUTATIONAL MODELING
Computational modeling was performed based on the crystal structure of the AR ligand-binding domain in complex with S-l (PDB ID: 2AXA) and the crystal structure of the AR ligand-binding domain T877A mutant in complex with hydroxyflutamide (PDB ID: 2AX6).24 Both structures were obtained from the RCSB. All of the modeling was conducted using the software package MOE.29 Both structures were imported into MOE and prepared for modeling in a standard fashion.
Briefly, all crystallographic water molecules were removed. The N and C-termini were capped with ACE and NME, respectively, due to unresolved residues. The 2AXA structure had one chain break with residues between ALA843 and CYS852 missing. Those ends were capped with NME and ACE, respectively. The 2AX6 structure also had one chain break with residues between ALA843 and PRO849 missing. These missing residues were built in using MOE utilities. There were no missing side chains except in the break regions. The breaks and termini were distant from the binding site.
The system was parameterized with AMBER 10. Bond orders for the ligands were checked and protonated appropriately. Partial charges for the ligands were then obtained using AM1-BCC. For each complex, all heavy atoms were fixed and the positions of the hydrogen atoms allowed to relax using energy minimization.
Ligands from the 2AXA and 2AX6 were removed from their respective complexes. Their crystallographic poses were randomized before being imported into a MOE database. The other four ligands were built in MOE and imported into the same MOE database. All of the ligands were then charged with AM1-BCC and energy minimized before generating a conformational database for them. The randomization of the 2AXA and 2AX6 ligand coordinates, followed by energy minimization was done to remove conformational bias from the crystallographic poses, which could result in unrealistically good docking results.
The conformational database of the ligands used for the docking was created using the default settings in MOE with the LowModeMD method except that the forcefield partial charges were not recalculated.
Docking was performed on the prepared 2AXA structure using its ligand to define the binding site. As in the generation of the conformational database, default settings in MOE were used for the dockings. The Triangle Matcher algorithm was used to perform the initial placements. The London dG scoring function was used to determine the 30 best scoring placements for each ligand before they were further refined with energy minimization in the presence of the AR The refined poses were then scored with the GBVI/WSA dG function and the five best poses for each ligand were saved to a MOE database.
Of note, MOE was able to reproduce the crystallographic poses for the ligands of the 2AXA and 2AX6 complexes starting from randomized coordinates for those ligands.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by funding from Oncopia Therapeutics, Inc. (now part of Roivant Sciences), the National Cancer Institute, NIH (P50 CA186786), and the University of Michigan Comprehensive Cancer Center Core Grant from the National Cancer Institute, NIH (P30CA046592).
ABBREVIATIONS
- AR
androgen receptor
- mCRPC
metastatic castration-resistant prostate cancer
- PROTAC
proteolysis targeting chimera
- cLAPl
cellular inhibitor of apoptosis protein 1
- VSS
steady-state volume of distribution
- Cmax
maximum drug concentration
- AUC0–24h
area-under-the-curve between 0 and 24 h
- Cl
plasma clearance rate
- T1/2
terminal half-life
- F
oral bio availability
- IV
intravenous administration
- PO
oral administration
- PD
pharmacodynamics
- CYP
cytochrome P450
- ATCC
American Type Culture Collection
- qRT-PCR
quantitative real-time polymerase chain reaction
- SCID
severe combined immunodeficient
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
Footnotes
The authors declare the following competing financial interest(s): The University of Michigan has filed patent applications on these AR degraders, which have been licensed to Oncopia Therapeutics, Inc. W. Xiang, L. Zhao, X. Han, C. Qin, B. Miao and S. Wang are co-inventors on these patent applications and receive royalties from the University of Michigan. S. Wang was a co-founder and served as a paid consultant to Oncopia. S. Wang and the University of Michigan also owned equity in Oncopia, which was acquired by Roivant Sciences. S. Wang is a paid consultant to Roivant Sciences. The University of Michigan has received a research contract from Oncopia (now part of Roivant Sciences) for which S. Wang serves as the principal investigator.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.lc00900.
Western blotting of AR protein for compounds 11–48 in the VCaP cell line; degradation kinetics for 26, 27, 35, and 40–45 in the VCaP and LNCaP cell lines; and 13C NMR spectra for ARD-2585 and HPLC purity spectra for ARD-2585 and other AR degraders tested in vivo; predicted binding models for compound 9 and several other AR ligands; and oral exposure data for several AR degraders in mice(PDF)
Binding model for ligand 9 (PDB)
Binding model for ligand 9a (PDB)
Binding model for ligand 9b (PDB)
Binding model for ligand 9c (PDB)
Molecular formula string for all the final target compounds (CSV)
Contributor Information
Weiguo Xiang, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Lijie Zhao, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Xin Han, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Chong Qin, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Bukeyan Miao, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Donna McEachern, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Yu Wang, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Hoda Metwally, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States.
Paul D. Kirchhoff, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, United States
Lu Wang, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Aleksas Matvekas, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Miao He, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Bo Wen, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Duxin Sun, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Shaomeng Wang, The Rogel Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Internal Medicine, Department of Pharmacology, and Department of Medicinal Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
REFERENCES
- (1).Rice MA; Malhotra SV; Stoyanova T Second-generation antiandrogens: from discovery to standard of care in castration resistant prostate cancer. Front. Oncol. 2019, 9, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mohler ML; Sikdar A; Ponnusamy S; Hwang DJ; He Y; Miller DD; Narayanan R An overview of next-generation androgen receptor-targeted therapeutics in development for the treatment of prostate cancer. Int. J. Mol Sci. 2021, 22, 2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Asangani I; Blair IA; Van Duyne G; Hilser VJ; Moiseenkova-Bell V; Plymate S; Sprenger C; Wand AJ; Penning TM Using biochemistry and biophysics to extinguish androgen receptor signaling in prostate cancer. J. Biol Chem. 2021,296,100240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rathkopf D; Scher HI Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 2013, 19, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ferroni C; Varchi G Non-steroidal androgen receptor antagonists and prostate cancer: a survey on chemical structures binding this fast-mutating target. Curr. Med. Chem. 2019, 26, 6053–6073. [DOI] [PubMed] [Google Scholar]
- (6).Salami J; Alabi S; Willard RR; Vitale NJ; Wang J; Dong H; Jin M; McDonnell DP; Crew AP; Neklesa TK; Crews CM Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ponnusamy S; He Y; Hwang D-J; Thiyagarajan T; Houtman R; Bocharova V; Sumpter BG; Fernandez E; Johnson D; Du Z; Pfeifer LM; Getzenberg RH; McEwan IJ; Miller DD; Narayanan R Orally bioavailable androgen receptor degrader, potential next-generation therapeutic for enzalutamide-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 6764–6780. [DOI] [PubMed] [Google Scholar]
- (8).Hwang D-J; He Y; Ponnusamy S; Mohler ML; Thiyagarajan T; McEwan IJ; Narayanan R; Miller DD New generation of selective androgen receptor degraders: our initial design, synthesis, and biological evaluation of new compounds with enzalutamide-resistant prostate cancer activity. J. Med. Chem. 2019, 62, 491–511. [DOI] [PubMed] [Google Scholar]
- (9).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Burslem GM; Crews CM Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schneekloth AR; Pucheault M; Tae HS; Crews CM Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shibata N; Nagai K; Morita Y; Ujikawa O; Ohoka N; Hattori T; Koyama R; Sano O; Imaeda Y; Nara H; Cho N; Naito M Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J. Med. Chem. 2018, 61, 543–575. [DOI] [PubMed] [Google Scholar]
- (13).Han X; Wang C; Qin C; Xiang W; Femandez-Salas E; Yang C-Y; Wang M; Zhao L; Xu T; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- (14).Han X; Zhao L; Xiang W; Qin C; Miao B; Xu T; Wang M; Yang C-Y; Chinnaswamy K; Stuckey J; Wang S Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. J. Med. Chem. 2019, 62, 11218–11231. [DOI] [PubMed] [Google Scholar]
- (15).Da Y; Liu S; Lin P; Wang F; Yan R; Shu Y; Lin J Design, synthesis, and biological evaluation of small molecule PROTACs for potential anticancer effects. Med. Chem. Res. 2020, 29, 334–340. [Google Scholar]
- (16).Takwale AD; Jo S-H; Jeon YU; Kim HS; Shin CH; Lee HK; Alin S; Lee CO; Du Ha J; Kim J-H; Hwang JY Design and characterization of cereblon-mediated androgen receptor proteolysis-targeting chimeras. Eur. J. Med. Chem. 2020, 208, 112769. [DOI] [PubMed] [Google Scholar]
- (17).Snyder LB Discovery of ARV-110, a first in class androgen receptor degrading PROTAC for the treatment of men with metastatic castration resistant prostate cancer. Cancer Research-, American Association for Cancer Research, 2021; Vol. 81. [Google Scholar]
- (18).Neklesa T; Snyder LB; Willard RR; Vitale N; Pizzano J; Gordon DA; Bookbinder M; Macaluso J; Dong H; Ferraro C; Wang G; Wang J; Crews CM; Houston J; Crew AP; Taylor I AR.V-110: an oral androgen receptor PROTAC degrader for prostate cancer. /. Clin. Oncol. 2019, 37, 259. [Google Scholar]
- (19).Petrylak DP; Gao X; Vogelzang NJ; Garfield MH; Taylor I; Dougan Moore M; Peck RA; Burris H A First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) Sallowing enzalutamide (ENZ) and/or abiraterone (ABI). /. Clin. Oncol 2020, 38, 3500. [Google Scholar]
- (20).Guo C; Linton A; Kephart S; Ornelas M; Pairish M; Gonzalez J; Greasley S; Nagata A; Burke BJ; Edwards M; Hosea N; Kang P; Hu W; Engebretsen J; Briere D; Shi M; Gukasyan H; Richardson P; Dack K; Underwood T; Johnson P; Morell A; Felstead X; Kuruma H; Matsimoto H; Zoubeidi A; Gleave M; Los G; Fanjul AN Discovery of aryloxy tetramethylcyclobutanes as novel androgen receptor antagonists. J. Med. Chem. 2011, 54, 7693–7704. [DOI] [PubMed] [Google Scholar]
- (21).Hu L-Y; Lefker BA; Du DY; Smith YD; Lei H; Harter WG; Downs VL; Boys ML; Iula DM Preparation of 4-cycloalkoxy benzonitriles as androgen modulators. U.S. Patent 20,060,009,427 Al, 2006.
- (22).Crew AP; Homberger KR; Snyder LB; Zimmermann K; Wang J; Berlin M; Crews CM; Dong H Preparation of bifunctional compounds for the targeted degradation of androgen receptor. U.S. Patent 20,180,099,940 Al, 2018.
- (23).DeGoey DA; Chen H-J; Cox PB; Wendt MD Beyond the rule of 5: lessons learned from AbbVie’s drugs and compound collection. J. Med. Chem. 2018, 61, 2636–2651. [DOI] [PubMed] [Google Scholar]
- (24).Bohl CE; Miller DD; Chen J; Bell CE; Dalton JT Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. /. Biol. Chem. 2005, 280, 37747–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Skerratt SE; de Groot MJ; Phillips C Discovery of a novel binding pocket for CYP 2C9 inhibitors: crystallography, pharmacophore modelling and inhibitor SAR. MedChemComm 2016, 7, 813–819. [Google Scholar]
- (26).Kayser-Bricker KJ; Glenn MP; Lee SH; Sebti SM; Cheng J Qy Hamilton, A D. Non-peptidic substrate-mimetic inhibitors of Akt as potential anti-cancer agents. Bioorg. Med. Chem. 2009, 17, 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bai L; Zhou B; Yang CY; Ji J; McEachem D; Przybranowski S; Jiang H; Hu JT; Xu FM; Zhao YJ; Liu L; Femandez-Salas E; Xu J; Dou YL; Wen B; Sun DX; Meagher J; Stuckey J; Hayes DF; Li SQ; Ellis MJ; Wang SM Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017, 77, 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhao L; Han X; Lu J; McEachern D; Wang S A highly potent PROTAC androgen receptor (AR) degrader ARD-61 effectively inhibits AR-positive breast cancer cell growth in vitro and tumor growth in vivo. Neoplasia 2020, 22, 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Molecular Operating Environment (MOE), 2020.09; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.