

Abstract
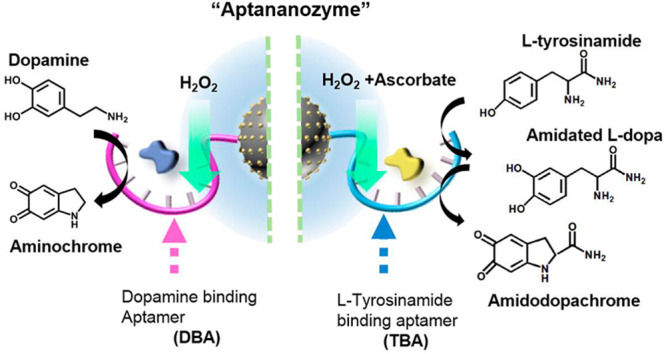
The covalent linkage of aptamer binding sites to nanoparticle nanozymes is introduced as a versatile method to improve the catalytic activity of nanozymes by concentrating the reaction substrates at the catalytic nanozyme core, thereby emulating the binding and catalytic active-site functions of native enzymes. The concept is exemplified with the synthesis of Cu2+ ion-functionalized carbon dots (C-dots), modified with the dopamine binding aptamer (DBA) or the tyrosinamide binding aptamer (TBA), for the catalyzed oxidation of dopamine to aminochrome by H2O2 or the oxygenation of l-tyrosinamide to the catechol product, which is subsequently oxidized to amidodopachrome, in the presence of H2O2/ascorbate mixture. Sets of structurally functionalized DBA-modified Cu2+ ion-functionalized C-dots or sets of structurally functionalized TBA-modified Cu2+ ion-functionalized C-dots are introduced as nanozymes of superior catalytic activities (aptananozymes) toward the oxidation of dopamine or the oxygenation of l-tyrosinamide, respectively. The aptananozymes reveal enhanced catalytic activities as compared to the separated catalyst and respective aptamer constituents. The catalytic functions of the aptananozymes are controlled by the structure of the aptamer units linked to the Cu2+ ion-functionalized C-dots. In addition, the aptananozyme shows chiroselective catalytic functions demonstrated by the chiroselective-catalyzed oxidation of l/d-DOPA to l/d-dopachrome. Binding studies of the substrates to the different aptananozymes and mechanistic studies associated with the catalytic transformations are discussed.
Introduction
Substantial research efforts are directed to the development of inorganic, organic, or metal–organic framework nanoparticles that mimic the functions of native enzymes, “nanozymes”.1 Inorganic nanoparticles, such as Fe3O4,2 V2O5,3 CeO2,4 MoO3,5 Au,6 Ag,7 Pd,8 carbon-based materials, for example, carbon-dots (C-dots)9 or graphene quantum dots,10 Prussian Blue nanoparticles,11 organic nanoparticles, such as melanin nanoparticles,12 and metal–organic framework nanoparticles (NMOFs), such as Zr-based NMOFs13 or MOF-818,14 were reported as nanozymes. Different chemical transformations emulating native enzymes were demonstrated by synthetic nanozymes including oxidase,15 peroxidase,16 laccase,17 catalase,18 superoxide dismutase,19 and hydrolase activities.20 Diverse applications of nanozymes were reported including their use as sensors,21 imaging agents,22 biomedical applications,23 such as cancer therapies,24 and the treatment of other diseases, for example, Alzheimer’s25 or Parkinson’s diseases.26 In addition, nanozymes were applied as antibacterial agents27 and materials for environmental degradation of pollutants.28
In contrast to native enzymes exhibiting active sites composed of recognition binding sites and catalytic units that cooperatively lead to superior catalytic activities, nanozymes lack the substrate binding feature and concentration of the substrate at the catalytic interface (lack of high molarity at the catalytic sites). Thus, the development of a versatile means to concentrate the substrates at the nanozymes catalytic surface could be a major advance in the field of nanozymes. Several approaches to reach these goals were reported including the functionalization of nanozymes with β-cyclodextrin receptors9 or the coating of the nanocatalysts with molecularly imprinted matrices.29
Aptamers are sequence-specific nucleic acids that are selected by the systematic evolution of ligands by exponential enrichment (SELEX) procedure.30 Aptamers revealing high affinity and selective binding properties toward low-molecular-weight ligands and macromolecules were developed.31 These selective binding properties were extensively used for analytical applications, for example, the development of sensors,32 separation matrices,33 and for biomedical applications,34 such as stimuli-responsive drug carriers,35 functional protein inhibitors,36 targeting of cancer cells,37 and imaging38 of cells. Also, the binding and dissociation of aptamer/ligand complexes were used to switch DNA nanostructures and to trigger DNA machines.39 Thus, the conjugation of ligand-binding aptamers to nanozymes could provide a versatile means to mimic native enzymes by concentrating ligand-substrate complexes at the catalytic sites of nanozymes.
In the past few years, we developed the concept of nucleoapzymes, where aptamers were conjugated to catalytic nucleic acids or homogeneous catalysts, as a versatile approach to yield hybrid homogeneous catalytic structures mimicking a native enzyme.40−43 The versatility and diversity of this approach were reflected by the possibility to link the catalytic units to the 5′ or 3′ end of the aptamers and to link the catalyst to the aptamer through spacer bridges or to position the catalyst in middle positions of the aptamers. These catalyst-aptamer structures have established a small library of sets of nucleoapzymes revealing variable catalytic activities originating from different binding affinities of the substrates to the modified aptamers or from steric constraints dictating the spatial positioning of the catalytic sites in respect to the binding sites. Molecular dynamic simulations were applied to elucidate the structure–function relationships within the catalyst-aptamer conjugates to account for the variable catalytic activities of the nucleoapzyme structures. Different nucleoapzymes catalyzing the oxidation of dopamine or N-hydroxy-l-arginine by H2O2 to yield aminochrome or citruline, respectively,40 the hydrolysis of ATP to ADP by mimicking ATPase,42 and the oxygenation of aryl C–H bonds of l-tyrosinamide to yield its catechol derivatives43 have been demonstrated. In principle, this concept can be applied to develop libraries of aptamer-nanozyme hybrids of variable catalytic activities by the conjugation of the 3′ or 5′ end of the aptamer to the nanozyme, the introduction of spacer units that modulate the flexibility of the aptamers in respect to the heterogeneous nanozyme, and by further engineering the DNA bridging modules linking the aptamers to the nanozymes, for example, by introducing stimuli-responsive bridging units. Although substantial advances in developing homogeneous catalytic conjugates consisting of catalyst-aptamer conjugates were demonstrated, the analogous heterogeneous catalyst-aptamer conjugates as functional catalytic units are unprecedented. Thus, realizing the limitations of catalytic nanoparticles (nanozymes) that lack substrate concentration sites, the conjugation of aptamers to the nanozymes could yield a versatile approach to synthesize heterogeneous catalytic modules, thereby bringing homogeneous catalyst-aptamer structures with heterogeneous aptamer hybrid as a unified approach for DNA-based catalytic structures. Realizing the topic of nanozymes is a rapidly developing area in catalysis, we believe that the integration of aptamers into heterogeneous catalysts as selective substrate binding and concentration means yields a new insight into heterogeneous nanozyme catalysis.
In the present study, we describe the conjugation of the dopamine binding aptamer (DBA) or the l-tyrosinamide binding aptamer (TBA) to Cu2+ ion-functionalized C-dots. We demonstrate that the anchoring mode of the aptamer and the spacers separating the aptamer from the catalyst affect the resulting catalytic activities of the aptamer-nanozyme conjugates (aptananozymes). We believe that the present study paves a versatile approach to develop nanozymes of enhanced catalytic performances.
Resulsts and Discussion
Carbon dots, C-dots, were prepared by a microwave treatment of mixture of citric acid and urea, according to the reported procedure.44 The resulting C-dots (ca. 10 nm diameter, Figure S1) include carboxylic acid and amine functionalities on their surface. The C-dots were functionalized with Cu2+ ions through coordination interactions. X-ray photoelectron spectroscopy (XPS) measurements confirmed the association of Cu2+ ions to the C-dots, and Fourier-transform infrared spectroscopy (FTIR) indicated the formation of metal–ligand (Cu–N, 1045 cm–1)45 bonds on the surface of the C-dots particles, Figures S2 and S3. The binding of Cu2+ ions to C-dots support yields a stable complex, Kd = 10.5 ± 1.2 nM. Figure S4 shows the experimental curves corresponding to the isothermal titration calorimetry (ITC) for evaluating the dissociation constants. In addition, inductively coupled plasma mass spectrometry (ICP-MS) measurements analyzing possibly released Cu2+ ions to the bulk solution indicated only trace concentrations (15 ng mL–1) and even higher concentration of Cu2+ ions in the bulk solution that did not reveal any catalytic performance on the substrates described in the study, Figure S9C. The respective amino-functionalized DBA46 or TBA43 were covalently coupled to the free carboxylic acid functionalities associated with the Cu2+ ion-functionalized C-dots using 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) as coupling reagents. The surface coverage of the different aptamers on the Cu2+ ion-functionalized C-dots was evaluated spectroscopically; see Figure S5 and the accompanying discussion. The loading degrees of all aptamers used in the study were very similar and corresponded to four aptamer strands per C-dot. ICP-MS analyses indicated that the loading of Cu2+ ions corresponded to 40 ± 3 μg per mg of C-dots, Table S1. The amino-aptamer-modified Cu2+ ion-functionalized C-dots were then applied to examine the catalyzed oxidation of dopamine to aminochrome by H2O2 in the presence of the DBA-modified Cu2+ ion-functionalized C-dots and the catalyzed oxygenation and oxidation of l-tyrosinamide to amidodopachrome by H2O2 and ascorbate (AA–), in the presence of the TBA-modified Cu2+ ion-functionalized C-dots, as schematically outlined in Figure 1.
Figure 1.
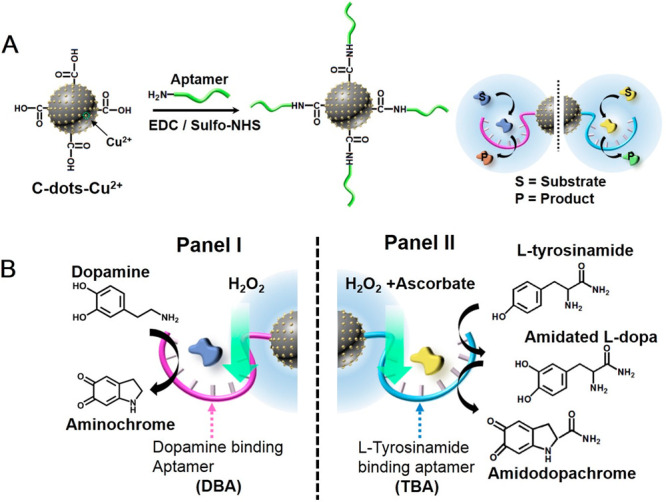
(A) Schematic synthesis of aptamer-modified Cu2+ ion-functionalized C-dots-aptananozyme. (B) Schematic chemical transformations driven by the synthetic aptananozymes. (Panel I) Catalyzing the oxidation of dopamine. (Panel II) Catalyzing oxygenation and oxidation of tyrosinamide.
A small library of DBA-modified Cu2+ ion-functionalized C-dots was prepared, Figure 2A, that included the 5′-end-amino-DBA (1) and 3′-end-amino-DBA (2) as aptamer-modified Cu2+ ion-functionalized C-dots acting as aptananozymes I and II. In addition, aptananozymes consisting of the 5′-end-amino-DBA linked to the Cu2+ ion-functionalized C-dots through spacer units were synthesized and included a (TGTA) spacer, aptananozyme III, (TGTA)2 spacer, aptananozyme IV, or (TGTA)3 spacer, aptananozyme V. The oxidation rates of dopamine to aminochrome by H2O2, in the presence of a constant weight of the respective DBA-modified Cu2+ ion-functionalized C-dots, were examined at variable concentrations of dopamine (in all of the experiments, H2O2, 5 mM, was used). The time-dependent absorbance changes corresponding to aminochrome formation were applied to follow the kinetics of dopamine oxidation by the respective aptananozymes (e.g., the time-dependent absorption spectra generated by aptananozyme (I) upon oxidation of dopamine to aminochrome, Figure S6, and the time-dependent absorbance changes at λ = 480 nm for the entire set of aptananozymes, Figure S7, panels i–vi). These kinetic data were used to derive the rates of oxidation of dopamine to aminochrome as a function of dopamine concentration, as outlined in Figure 2B, curves (a) to (e). For all aptananozymes, Michaelis–Menten-type saturation kinetic curves are observed. For comparison, the rates of oxidation of dopamine by H2O2, in the presence of the separated, Cu2+ ion-functionalized C-dots and the 5′-end-amino-modified DBA (1), at identical concentrations of the constituents associated with the aptananozymes is shown in curve (f). A very inefficient oxidation rate of dopamine is observed. A linear relation between the oxidation rates as a function of the concentrations of dopamine is observed, consistent with a bimolecular reaction process in the homogeneous solution. Also, Figure 2B, curve (g) shows the rate of oxidation of dopamine at different concentrations of the substrate in the presence of Cu2+ ion-functionalized C-dots modified with a scrambled base sequence of the DBA (1a) that consists of the sequence (1). A substantially lower rate of oxidation of dopamine by the (1a)-modified Cu2+ ion-functionalized C-dots, as compared to the set of aptananozymes, is observed, yet is higher than the control system of separated constituents. The enhanced activity of the scrambled strands (1a)-modified C-dots as compared to the separated constituents is attributed to the electrostatic attraction of the protonated dopamine to the negatively charged (1a)-modified C-dots that results in a local concentration of the substrate close to the catalytic interface.
Figure 2.
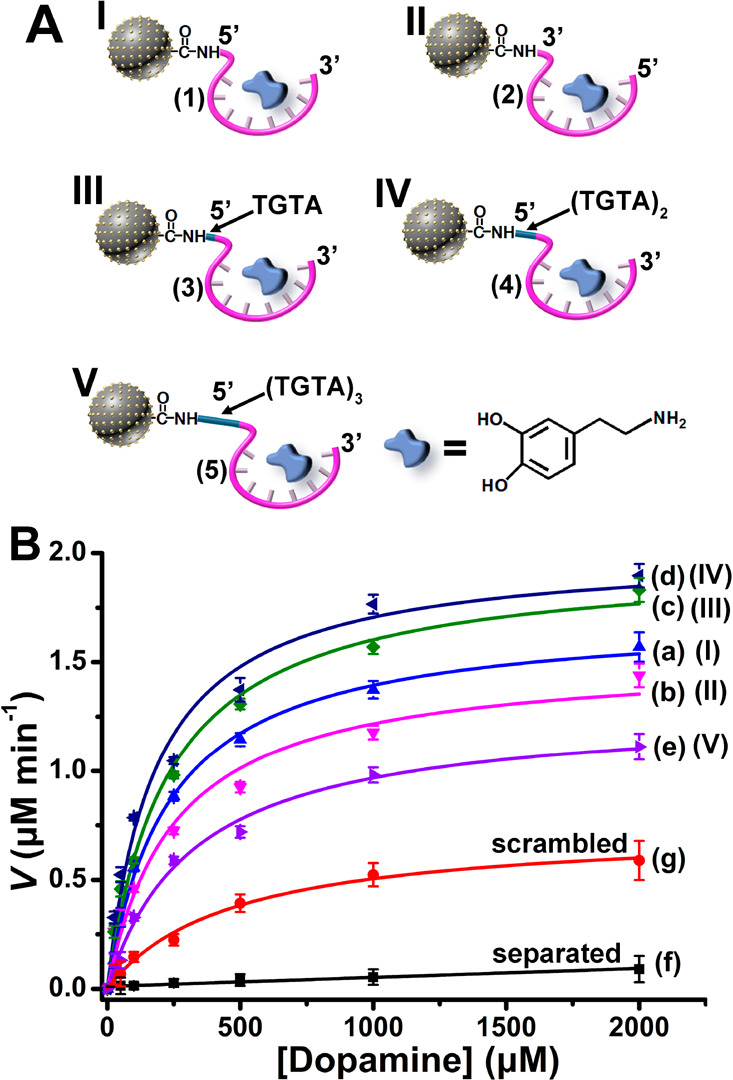
(A) The set of aptananozymes used for the catalyzed oxidation of dopamine to aminochrome (For the detailed sequences of the aptamers conjugated to the Cu2+ ion-functionalized C-dots see the Experimental Section). (B) Rates of oxidation of dopamine to aminochrome by H2O2 using variable concentrations of dopamine in the presence of the set of aptananozymes and the respective control systems. Error bars derived from N = 3 experiments.
The rates of oxidation of dopamine by the set of aptananozymes reveal a dependence on the structure of the aptamer constituents linked to the Cu2+ ion-functionalized C-dots: (i) The aptananozyme composed of the 5′-end-DBA and catalyst, aptananozyme I, reveals a higher activity as compared to the 3′-end-DBA-modified catalyst, aptananozyme II. (ii) The activities of the 5′-end-DBA-modified aptananozymes are affected by the length of the spacer bridging the aptamer to the Cu2+ ion-functionalized C-dots. As the spacer bridging units increase from (TGTA) to (TGTA)2, the catalytic activity of the aptananozymes increases as compared to the aptananozyme composed of the DBA aptamer directly bound to the catalyst, aptananozyme I, following the order IV ≈ III > I. In contrast, the aptananozyme V composed of the longer spacer (TGTA)3 conjugated to the 5′-end-amino DBA aptamer reveals a lower catalytic activity as compared to aptananozyme I, leading to the order of aptananozymes activities IV ≈ III > I > V. The saturation curves corresponding to the rates of oxidation of dopamine at different dopamine concentrations were analyzed in terms of the Michaelis–Menten model, and the kinetic parameters of the set of aptananozymes and the control systems are summarized in Table 1. Specifically, the aptananozyme I reveals a 50-fold enhancement of catalytic activity as compared to the separated Cu2+ ion-functionalized C-dots/DBA constituent. Note that fitting the rates of the catalyzed oxidation of dopamine as a function of the substrate concentration according to the Michaelis–Menten model for the catalytic configurations of aptananozyme I, II, III, IV, and V leads to a best fit using a Hill coefficient of n ≈ 1. Thus, although four aptamer binding units are associated with the core catalytic nanoparticles, no cooperative effect between the different binding sites and the catalytic function is observed. This may be attributed to the low coverage of the Cu2+ ion-functionalized C-dots particles by the aptamers and the spatial separation of the binding aptamers that prohibits cooperative interactions (for a further discussion on the Hill coefficients of all aptananozymes, see the Supporting Information, Pages S6–S7). We find that the aptananozymes reveal a high stability (see Figure S8A and the accompanying discussion). Note that the catalytic activity of the DBA-modified Cu2+ ion-functionalized C-dots toward the oxidation of dopamine is selective for the Cu2+-modified particles, and other DBA-modified metal ion-functionalized C-dots such as Zn2+, Mn2+, Co2+, and Cd2+ ion-modified C-dots did not reveal any catalytic activity. The DBA-modified Fe3+ ion-functionalized C-dots revealed a catalytic activity toward the oxidation of dopamine, yet it was substantially lower as compared to that of the Cu2+ ion-functionalized C-dots, Figure S9A, Supporting Information. Note that the catalytic oxidation of dopamine by H2O2 in the presence of the aptamer-modified Cu2+ ion-functionalized C-dots is controlled by the loading of Cu2+ ions on the C-dots. (for further discussion and experimental results, see Figure S10).
Table 1. Kinetic Parameters Associated with the Aptananozymes I–V and Control Systemsa.
| aptananozyme | Vmax (μM min–1) | KM (μM) | kcat (10–3 s–1) | kcat/KM (s–1 M–1) |
|---|---|---|---|---|
| IV | 2.01 ± 0.1 | 181 ± 32 | 2.02 ± 0.1 | 11.2 |
| III | 1.97 ± 0.08 | 230 ± 31 | 1.98 ± 0.08 | 8.6 |
| I | 1.71 ± 0.04 | 232 ± 17 | 1.72 ± 0.04 | 7.4 |
| II | 1.52 ± 0.1 | 252 ± 57 | 1.53 ± 0.1 | 6.0 |
| V | 1.28 ± 0.05 | 324 ± 43 | 1.29 ± 0.05 | 4.0 |
| scrambled | 0.73 ± 0.04 | 464 ± 70 | 0.74 ± 0.04 | 1.6 |
| separatedb | 0.04 | 0.04 |
All experiments were performed in a 5 mM MES buffer solution, pH 5.5, that included 5 mM MgCl2, 100 mM NaCl, and 0.2 μg·mL–1 of the respective aptananozymes or control system and 5 mM H2O2.
The separated Cu2+ ion-functionalized C-dots/DBA system shows pseudo-first-order kinetics: k = 0.04 × 10–3 s–1.
Realizing that the composition of the Cu2+ ion-functionalized C-dots is identical for all aptananozymes, we searched for a possible origin for the different catalytic activities of the aptananozymes. Toward this goal, we examined the dissociation constants of the dopamine/aptananozyme complexes using ITC, and the derived dissociation constants are summarized in Table S2. (For the experimental binding curves leading to the derived Kd values and the respective control experiments, see Figure S11, panels i–vi, and the accompanying discussion).
From Table S2, we realize that the dissociation constant of the aptananozyme II is significantly higher than the Kd value of aptananozyme I. Thus, the lower binding affinity of the dopamine substrate to the aptamer units of aptananozyme II may account for the lower catalytic activity of aptananozyme II, as compared to that of aptananozyme I. The Kd values of the aptananozymes III and IV, are, however, very similar to the Kd value of aptananozyme I, and thus the binding affinities of dopamine to the aptamer binding sites cannot account for the enhanced catalytic activities of aptananozymes III and IV as compared to that of aptananozyme I. Presumably, the spacer units bridging the aptamer units to the catalytic Cu2+ ion-functionalized C-dots introduce a steric flexibility that allows an enhanced spatial proximity between the dopamine/DBA complex and the catalytic core that facilitates the oxidation of dopamine to aminochrome. The lower catalytic activity of longer-spaced aptananozyme, (TGTA)3, aptananozyme V, as compared to aptananozyme I, is, however, surprising in view of the flexibility anticipated for this aptananozyme. Nonetheless, the binding affinity of the aptananozyme V, Table S2, reveals a significantly higher dissociation constant (Kd = 3.9 ± 0.2 μM) as compared to that of aptananozymes I, III, and IV. The lower binding affinity of dopamine to aptananozyme V may well account for the lower catalytic activity of this aptananozyme. At present, the origin for the high Kd value of aptananozyme V is not known, yet it should be noted that all free, non-C-dots-modified DBA for aptananozymes I, II, III, IV, and V reveal very similar Kd values (Kd ≈ 1.09 μM).
Besides the function of the aptamer units as binding sites for the concentration of the substrate at the catalytic core, the chiral features of the aptamer strands suggest that the aptananozymes could potentially induce chiroselective catalytic transformations. This feature was supported by the chiroselective oxidation of L- or D-DOPA (DOPA = 3,4-dihydroxyphenylalanine) by the aptananozyme I, composed of the Cu2+ ion-functionalized C-dots modified with the 5′-end-amino DBA (1), Figure 3. The diastereoisomeric discrimination between L-DOPA and D-DOPA by the nucleic acid aptamer led to a ca. two-fold enhanced oxidation of L-DOPA to L-dopachrome, Figure 3, curve (a), as compared to the oxidation of D-DOPA to D-dopachrome, curve (b). For comparison, curves (c) and (d) show the rates of oxidation of L-DOPA and D-DOPA in the presence of the separated Cu2+ ion-modified C-dots and the DBA (1), demonstrating the concentration effect of the aptananozyme on the oxidation of the L-/D-DOPA substrates. The catalytic oxidation of L-DOPA and D-DOPA by the aptananozyme I reveals Michaelis–Menten saturation curves, and the kinetic parameters corresponding to the respective oxidation processes are summarized in Table S3, Supporting Information. The enhanced catalytic performance of the aptananozyme I toward L-DOPA is attributed to the diastereoisomeric discrimination of L-DOPA and D-DOPA toward the aptamer that leads to a higher binding affinity of L-DOPA to the aptamer (Kd = 1.7 ± 0.4 μM) as compared to the lower binding affinity of D-DOPA to the aptamer (Kd = 6.6 ± 1 μM), Figure S11A and Table S4, Supporting Information.
Figure 3.
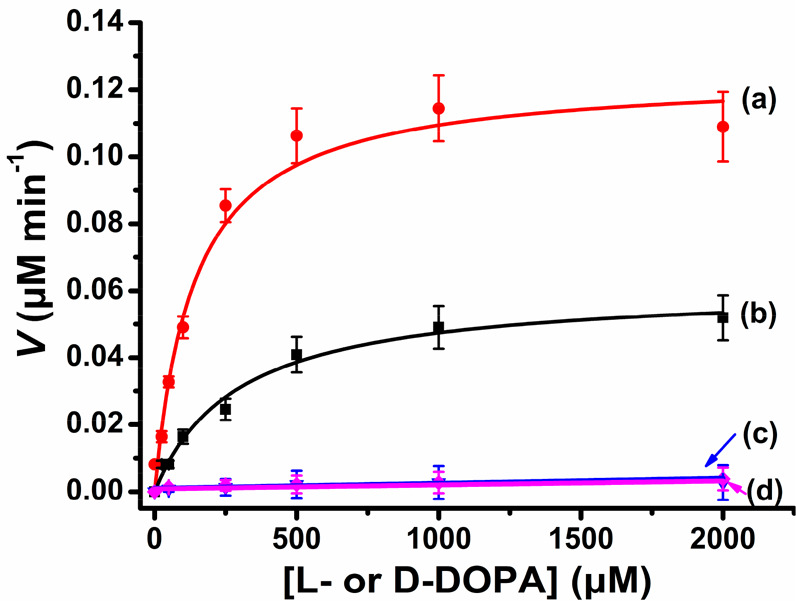
Rates corresponding to (a) the aptananozyme I-catalyzed oxidation of L-DOPA by H2O2 to generate L-dopachrome in the presence of variable concentrations of L-DOPA and (b) the aptananozyme I-catalyzed oxidation of D-DOPA by H2O2 to form D-dopachrome in the presence of variable concentrations of D-DOPA. (c, d) Rates corresponding to the oxidation of L-DOPA and D-DOPA in the presence of variable concentrations of L-/D-DOPA by H2O2, using the separated aptananozyme I and the aptamer (1), respectively. Error bars are derived from N = 3 experiments.
To demonstrate the versatility of the aptananozyme concept, we synthesized a second set of Cu2+ ion-functionalized C-dots modified with the l-tyrosinamide binding aptamer (TBA) for the catalyzed oxygen insertion into the C–H bond of the l-tyrosinamide substrate to yield the catechol product that is further oxidized to amidodopachrome, Figure 1B. The set of aptananozymes is schematically presented in Figure 4A and includes the 5′-end-amino TBA directly linked to the Cu2+ ion-functionalized C-dots, aptananozyme X, the 3′-end-amino TBA directly linked to the Cu2+ ion-functionalized C-dots, aptananozyme XI, the 5′-end-amino TBA conjugated to the Cu2+ ion-functionalized C-dots through a (TGTA) spacer unit, aptananozyme XII, the 5′-end-amino TBA linked to the Cu2+ ion-functionalized C-dots through a (TGTA)2 spacer, aptananozyme XIII, and the 5′-end-amino TBA linked to the Cu2+ ion-functionalized C-dots through a (TGTA)3 spacer bridging unit, aptananozyme XIV. This set of aptananozymes was prepared, as before, by the coupling of the respective amino-modified TBA to the carboxylic acid functionalities associated with the C-dots using EDC/sulfo-NHS as coupling reagents that form the respective amide covalent-bond links. (The sequences of the different TBA strands are detailed in the Experimental Section.) The ICP-MS analyses indicated a loading of Cu2+ ions corresponding to 40 ± 3 μg per mg of C-dots, and spectroscopic measurements indicated an average loading of 4.3 ± 0.2 TBA units per single C-dots (see Table S5, Supporting Information). As before, the catalytic activities of the set of aptananozymes were compared to the activities of separated Cu2+ ion-functionalized C-dots and TBA (6), under the same experimental conditions, and to the Cu2+ ion-functionalized C-dots modified with the scrambled base-sequence (6a) comprising the TBA (6). Note that the scrambled strands associated with the C-dots reveals a residual small catalytic activity as compared to the separated system. This is because l-tyrosinamide is protonated and positively charged at experimental conditions, pH = 7.2, and thus electrostatically attracted and concentrated at the catalytic interface by the negatively charged scrambled strand.
Figure 4.

(A) The set of aptananozymes used for the catalyzed oxidation of l-tyrosinamide to amidodopachrome. (For the detailed sequences of the TBA conjugated to the Cu2+ ion-functionalized C-dots see the Experimental Section.) (B) Rates of oxidation of l-tyrosinamide to amidodopachrome by H2O2 using variable concentrations of l-tyrosinamide in the presence of the set of aptananozymes and the respective control systems. Error bars derived from N = 3 experiments.
The set of aptananozymes was applied as heterogeneous nanozymes for the oxygenation of l-tyrosinamide to the catechol product and subsequent oxidation to amidodopachrome, by the “magic” reaction mixture consisting of H2O2 and ascorbate (AA–). Treatment of l-tyrosinamide in the presence of the set of aptananozyme catalysts and the H2O2/AA– mixture led to the formation of amidodopachrome that was spectroscopically followed at λ = 475 nm, Figure S12. The oxidation product suggests that the l-tyrosinamide substrate was transformed to the catechol product that was further oxidized to amidodopachrome. Control experiments indicated that no oxidation of l-tyrosinamide to amidodopachrome occurred in the presence of only H2O2 or only AA–, indicating that the mixture of the constituents is essential to drive the oxidation of l-tyrosinamide (Figure S13). Figure 4B, curves (a) to (e), depicts the rates of l-tyrosinamide oxidation to amidodopachrome, as a function of the l-tyrosinamide concentrations, using H2O2/AA– as an oxidizing mixture, in the presence of the set of aptananozymes X–XIV, the scrambled and the separated TBA with Cu2+ ion-functionalized C-dots (for the time-dependent oxidation of the l-tyrosinamide substrates in the presence of variable substrate concentrations by the different aptananozymes and control systems, see Figure S14). For comparison, the rate of oxidation of l-tyrosinamide at different concentrations of l-tyrosinamide by the Cu2+ ion-functionalized C-dots modified with a strand (6a) consisting of the scrambled bases of TBA (6) are presented in Figure 4B, curve (f). In addition, the rate of oxidation of l-tyrosinamide at different concentrations of l-tyrosinamide in the presence of the separated Cu2+ ion-functionalized C-dots and the TBA (6) are displayed in Figure 4B, curve (g). The results demonstrate superior catalytic activities by the set of aptananozymes X–XIV as compared to the negligible catalytic activity revealed by the separated Cu2+ ion-functionalized C-dots and TBA constituents, curve (g). The catalytic activities of the aptananozymes relate to the structure of the TBA conjugated to the Cu2+ ion-functionalized C-dots. The catalytic features of the l-tyrosinamide oxygenation aptananozymes reveal some similarities to the catalytic functions of dopamine aptananozymes. That is, the linkage of the 5′-end of the TBA to the Cu2+ ion-functionalized C-dots, aptananozyme X, yields an aptananozyme of enhanced catalytic activity as compared to aptananozyme XI, where the 3′-end TBA is conjugated to the Cu2+ ion-functionalized C-dots. Similarly, the conjugation of the 5′-end TBA to the C-dots through spacers consisting of (TGTA) and (TGTA)2 bridges led to aptananozymes of enhanced catalytic performance XIII > XII, yet the longest spacer linking the 5′-end TBA to the Cu2+ ion-functionalized C-dots through the (TGTA)3 bridge, configuration XIV, yields the aptananozyme of the lowest catalytic activity. The order of catalytic activities of the l-tyrosinamide oxidizing aptananozymes is XIII > XII > X > XI > XIV. The entire set of l-tyrosinamide aptananozymes reveals Michaelis–Menten saturation curves that fit the experimental data with a Hill coefficient n ≈ 1. Thus, although four aptamer strands are associated with the catalytic core, no cooperative interactions between the aptamer binding domains exist. This result is attributed to the low coverage of the TBA units on the catalytic core. The Michaelis–Menten kinetic parameters derived for the different l-tyrosinamide aptananozymes are summarized in Table 2; the aptananozyme XIII reveals a 15-fold enhanced catalytic activity as compared to the separated Cu2+ ion-functionalized C-dots/TBA constituents. The oxidation of l-tyrosinamide to amidodopachrome in the presence of the H2O2/AA– mixture is selective to the Cu2+ ion-functionalized C-dots, and other metal ion-functionalized C-dots (Zn2+, Co2+, Mn2+, Cd2+, and Fe3+) did not show any catalytic activities, Figure S9B. (For further discussion on the stability of the aptananozymes, see Figure S8B and accompanying discussion.) Note that the functionalization of the C-dots with Cu2+ ions is essential to drive the catalytic oxidation of dopamine or tyrosinamide, and bare C-dots did not show any catalytic performance (For further discussion demonstrating the selective oxygenation and oxidation of tyrosinamide, see Figure S15 and the accompanying discussion).
Table 2. Kinetic Parameters Corresponding to the Aptananozymes X–XIV and Control Systemsa.
| aptananozyme | Vmax (μM min–1) | KM (μM) | kcat (10–4 s–1) | kcat/KM (s–1 M–1) | |
|---|---|---|---|---|---|
| XIII | 1.84 ± 0.08 | 375 ± 46 | 7.4 ± 0.2 | 1.97 | |
| XII | 1.51 ± 0.1 | 415 ± 71 | 6.1 ± 0.3 | 1.5 | |
| X | 1.32 ± 0.1 | 436 ± 62 | 5.3 ± 0.3 | 1.2 | |
| XI | 1.12 ± 0.06 | 460 ± 64 | 4.5 ± 0.2 | 0.98 | |
| XIV | 0.99 ± 0.05 | 531 ± 67 | 4.0 ± 0.1 | 0.75 | |
| scrambled | 0.6 ± 0.1 | 1202 ± 201 | 2.4 ± 0.3 | 0.2 | |
| separatedb | 0.12 | 0.48 |
All experiments were performed in a 50 mM phosphate buffer solution at pH 7.2, containing 100 mM NaCl and 5 mM MgCl2, in the presence of the respective aptananozymes (0.5 μg mL–1) or control catalyst and 5 mM H2O2, 5 mM AA–.
The separated Cu2+ ion-functionalized C-dots/TBA system shows pseudo-first-order kinetics: k = 0.48 × 10–4 s–1.
Table S6 summarizes the ITC-evaluated dissociation constants of the series of aptananozymes X-XIV. (For the experimental binding curves leading to the Kd values, derived from the binding curves and the respective control experiments, see Figure S16, panels i–vi, and the accompanying discussion.) The slightly lower catalytic activity of aptananozyme XI as compared to the catalytic activity of aptananozyme X can be attributed to the lower binding affinity of the l-tyrosinamide substrate to the aptananozyme XI (Kd (XI) = 1.24 ± 0.05 μM vs Kd (X) = 0.83 ± 0.01 μM). The Kd values of the aptananozymes X, XII, and XIII are very similar, indicating similar binding affinities toward l-tyrosinamide. Thus, the enhanced catalytic features of the aptananozymes XII and XIII are attributed, as before, to the enhanced flexibility of the (TGTA)- and (TGTA)2-spaced aptamers that lead to a spatial proximity between the l-tyrosinamide/TBA complexes and the catalyst Cu2+ ion-functionalized C-dots surface. As before, the long spaced (TGTA)3 aptananozyme, XIV, reveals a lower catalytic activity as compared to the reference aptananozyme X, despite the flexibility of the aptamer binding site. Also, in this case, we find that the dissociation constant of aptananozyme XIV is higher than the Kd value of the reference aptananozyme X (Kd (XIV) = 1.4 ± 0.1 μM vs Kd (X) = 0.83 ± 0.01 μM) (For further experiments addressing the enantioselective oxidation of L-/d-tyrosine by the aptananozyme X, see Figure S17 and the accompanying discussion, Supporting Information).
Finally, we addressed mechanistic aspects related to the reactive species generated by the Cu2+ ion-functionalized C-dots in the presence of H2O2 and, particularly, in the presence of the H2O2/AA– (ascorbate) mixture. In fact, Cu2+ enzymes or Fe3+ enzymes, such as catechol oxidase or cytochrome P-450, and methane monooxygenase act in nature as biocatalysts oxidizing catechol derivatives to quinones or biocatalysts mediating C–H bond oxygenation, and the native mechanisms associated with the biocatalysts attracted substantial research efforts.47 The aptamer-modified Cu2+-functionalized C-dots could, thus, be considered as nanozymes emulating these biological processes. Accordingly, we evaluated by electron-spin resonance (ESR) spectroscopy the formation of possible reactive oxygen species generated by the Cu2+-modified C-dots in the presence of H2O2 or H2O2/AA–, the reaction mixtures used in the catalyzed oxidation of dopamine to aminochrome or the C–H bond oxygenation of the l-tyrosinamide, respectively, Figure 5A. Subjecting the DBA-modified Cu2+ ion-functionalized C-dots to H2O2 yields the hydroxyl radical (·OH) as a reactive oxygen species, Figure S18. A treatment of the TBA-modified Cu2+ ion-functionalized C-dots with an aqueous H2O2 solution yields, similarly, to the formation of the hydroxyl radical (·OH), Figure 5A, Panel I. Subjecting the aptananozyme consisting of TBA-modified Cu2+ ion-functionalized C-dots to an AA– solution (in the absence of H2O2) yields to the formation of the ascorbate radical, AA·, Figure 5A, Panel II. Subjecting the TBA-modified Cu2+ ion-functionalized C-dots aptananozyme to the mixture of H2O2 and AA–, leads, however, to the formation of the mixture of ascorbate radical, AA·, and peroxy-radical ·OOH without the formation of the ·OH species, Figure 5A, Panel III. These results suggest that the reactive species in the oxidation of dopamine is the hydroxyl radical, ·OH, whereas the mixture of AA· and ·OOH species generated by the TBA-modified Cu2+ ion-functionalized C-dots in the AA–/H2O2 solution participate in the C–H bond oxygenation of l-tyrosinamide. These results imply that the formation of the ·OOH is intimately related to the formation of the AA· species. The addition of the l-tyrosinamide substrate to the mixture of H2O2/AA– leads, in the presence of the added TBA-modified Cu2+ ion-functionalized C-dots aptananozyme, to the formation of the two intermediates AA· and ·OOH species, Figure 5A, Panel IV, yet the content of the ·OOH is dampened while the content of the AA· in the reaction mixture is almost unaffected. These results indicate that the ·OOH is a reactive species in the C–H bond oxygenation. A possible mechanistic path for the ·OH-stimulated oxidation of the catechol dopamine residue to the quinone product is outlined in Figure S19. On the basis of previous reports suggesting the peroxy radical mediated the formation of bis-μ-oxo dicopper species in analogy to tyrosinase,48 we formulate the cycle presented in Figure 5B as a possible path for the oxygenation of the l-tyrosinamide aryl-H (Ar–H) bond into the catechol product. The set of reactions summarized in Figure S20, and the accompanying discussion, suggests a possible route for the formation of ·OOH by the Cu2+ ion-functionalized C-dots, in the presence of the H2O2/AA– mixture. In fact, XPS measurements indicated the presence of CuIII species generated upon treatment of the CuII-functionalized C-dots with the H2O2/AA– mixture, Figure S21, supporting the formation of the CuIII-μ-oxo-bridged intermediate. Also, no reactive oxygen species on the C-dots could be detected.
Figure 5.

(A) ESR spectra corresponding to Panel I, the ·OH generated by the aptananozyme X in the presence of H2O2. Panel II, the ·AA radical generated by the aptananozyme X in the presence of ascorbate. Panel III, the mixture of ·AA and peroxy radical ·OOH generated by the aptananozyme X in the presence of the mixture of AA–/H2O2. Panel IV, the mixture of ·AA and ·OOH generated by the aptananozyme X in the presence of the mixture of AA–/H2O2 with added l-tyrosinamide. (B) Schematic suggested mechanistic cycle for the oxygenation of the Ar–H bond of l-tyrosinamide to catechol product by the CuII-functionalized C-dots aptananozymes.
Conclusions
In conclusion, the present study has introduced the concept of aptananozymes, where sequence-specific aptamer strands are conjugated to nanosized heterogeneous catalysts as a means to enhance the catalytic activities of nanozymes. The binding of the reaction substrate at the catalyst core by means of aptamer-substrate complexes provides a means to concentrate the substrate in proximity to the catalytic interface, in analogy to the functions of native enzymes. This concept has been demonstrated by developing aptananozymes that catalyze the oxidation of dopamine to aminochrome and the oxygen insertion into aryl-H bonds of l-tyrosinamide to yield a catechol product that is further oxidized to amidodopachrome. By the structured engineering of the aptamer sequences, controlled catalytic functions of the aptananozymes were demonstrated. Note that, in contrast to previous reports demonstrating the similar oxidation process by homogeneous metal–organic complex-aptamer conjugates (nucleoapzymes),43 the present study extended the concept to heterogeneous catalysts. The present study introduces an important “missing” link into the area of hybrid catalytic nucleic acid nanostructures. The concept complements the “nucleoapzyme” and “photonucleoapzyme” paradigms where homogeneous catalysts or photosensitizers were conjugated to aptamer sequences to yield superior enzyme-like catalysts and photocatalysts. The approach introduced in this study paves a way for a versatile method to develop diverse aptananozymes by conjugating aptamers to other catalytic nanoparticles and, particularly, to conjugate aptamers to other metal-ion modified C-dots to drive other catalytic reactions. Furthermore, the study indicated the significance of bridging spacer units on the catalytic efficacy and substrate binding affinities of the aptananozymes. Further studies should be directed, however, to evaluate the effects of additional spacer lengths and base compositions on the performance of the aptananozymes. Particularly, the introduction of stimuli-responsive reconfigurable spacers, such as G-quadruplexes or light-responsive bridges, would be interesting to switch the catalytic activities of the aptananozymes.
Experimental Section
The C-dots were prepared according to the previously reported method.44
Sequences used in the study were commercially ordered (Integrated DNA Technologies, IDT):
(1) Amino-DBA for 5′-linked aptananozyme (I): 5′-NH2–CGACGCCAGTTTGAAGGTTCGTTCGCAGGTGTGGAGTGACGTCG-3′.
(1a) Amino-scrambled DBA for 5′-linked aptananozyme: 5′-NH2-GACTAGCGTGTGTGATGGGACCTTAGGCCGTCACGGGGCTTAGT-3′.
(2) Amino-DBA for 3′-linked aptananozyme (II): CGACGCCAGTTTGAAGGTTCGTTCGCAGGTGTGGAGTGACGTCG-NH2-3′.
(3) Amino-DBA with (TGTA) spacer for 5′-linked aptananozyme (III): 5′-NH2-TGTA-CGACGCCAGTTTGAAGGTTCGTTCGCAGGTGTGGAGTGACGTCG-3′.
(4) Amino-DBA with (TGTA)2 spacer for 5′-linked aptananozyme (IV): 5′-NH2-TGTATGTA-CGACGCCAGTTTGAAGGTTCGTTCGCAGGTGTGGAGTGACGTCG-3′.
(5) Amino-DBA with (TGTA)3 spacer for 5′-linked aptananozyme (V): 5′-NH2-TGTATGTATGTA-CGACGCCAGTTTGAAGGTTCGTTCGCAGGTGTGGAGTGACGTCG-3′.
(6) Amino-TBA for 5′-linked aptananozyme (X): 5′-NH2-TGTGGTGTGTGAGTGCGGTGCCC-3′.
(6a) Amino-scrambled TBA for 5′-linked aptananozyme: 5′-NH2-TGTGGTGTGTGAGTGCGGTGCCC-3′.
(7) Amino-TBA for 3′-linked aptananozyme (XI): TGTGGTGTGTGAGTGCGGTGCCC-NH2-3′.
(8) Amino-TBA with (TGTA) spacer for 5′-linked aptananozyme (XII): 5′-NH2-TGTA-TGTGGTGTGTGAGTGCGGTGCCC-3′.
(9) Amino-TBA with (TGTA)2 spacer for 5′-linked aptananozyme (XIII): 5′-NH2-TGTATGTA-TGTGGTGTGTGAGTGCGGTGCCC-3′.
(10) Amino-TBA with (TGTA)3 spacer for 5′-linked aptananozyme (XIV): 5′-NH2-TGTATGTATGTA-TGTGGTGTGTGAGTGCGGTGCCC-3′.
For the details of the conjugation of the aptamers to the C-dots, the characterization of the aptamer and C-dots conjugates, and the kinetic measurements see Supporting Information Pages S2–S5.
Acknowledgments
This research is supported by the Volkswagen Foundation, Germany, and by the Minerva Center for Biohybrid Complex Systems, The Hebrew University of Jerusalem.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03939.
Experimental procedures, quantification of aptamer and Cu2+ ion-functionalized C-dots, TEM image, XPS measurements, FTIR spectra, time-dependent absorbance changes upon the oxidation of different concentrations of dopamine or l-tyrosinamide, ITC measurements (PDF)
Author Contributions
§ Y.O. and Y.B. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Huang Y.; Ren J.; Qu X. Nanozymes: Classification, Catalytic Mechanisms, Activity Regulation, and Applications. Chem. Rev. 2019, 119, 4357–4412. 10.1021/acs.chemrev.8b00672. [DOI] [PubMed] [Google Scholar]; b Liang M.; Yan X. Nanozymes: from New Concepts, Mechanisms, and Standards to Applications. Acc. Chem. Res. 2019, 52, 2190–2200. 10.1021/acs.accounts.9b00140. [DOI] [PubMed] [Google Scholar]; c Ragg R.; Tahir M. N.; Tremel W. Solids Go Bio: Inorganic Nanoparticles as Enzyme mimics. Eur. J. Inorg. Chem. 2016, 2016, 1906–1915. 10.1002/ejic.201501237. [DOI] [Google Scholar]; d Wu J.; Wang X.; Wang Q.; Lou Z.; Li S.; Zhu Y.; Qin L.; Wei H. Nanomaterials with Enzyme-like Characteristics (Nanozymes): Next-Generation Artificial Enzymes (II). Chem. Soc. Rev. 2019, 48, 1004–1076. 10.1039/C8CS00457A. [DOI] [PubMed] [Google Scholar]; e Wang H.; Wan K.; Shi X. Recent Advances in Nanozyme Research. Adv. Mater. 2019, 31, 1805368. 10.1002/adma.201805368. [DOI] [PubMed] [Google Scholar]
- a Gao L.; Zhuang J.; Nie L.; Zhang B.; Zhang Y.; Gu N.; Wang H.; Feng J.; Yang L.; Perrett S.; Yan Y. Intrinsic Peroxidase-like Activity of Ferromagnetic Nanoparticles. Nat. Nanotechnol. 2007, 2, 577–583. 10.1038/nnano.2007.260. [DOI] [PubMed] [Google Scholar]; b Liang M. M.; Fan K. L.; Pan Y.; Jiang H.; Wang F.; Yang D.; Lu D.; Feng J.; Zhao J.; Yang L.; Yan X. Fe3O4 Magnetic Nanoparticle Peroxidase Mimetic-Based Colorimetric Assay for the Rapid Detection of Organophosphorus Pesticide and Nerve Agent. Anal. Chem. 2013, 85, 308–312. 10.1021/ac302781r. [DOI] [PubMed] [Google Scholar]; c Wang L.; Min Y.; Xu D.; Yu F.; Zhou W.; Cuschieri A. Membrane Lipid Peroxidation by the Peroxidase-Like Activity of Magnetite Nanoparticles. Chem. Commun. 2014, 50, 11147–11150. 10.1039/C4CC03082F. [DOI] [PubMed] [Google Scholar]; d He X.; Tan L.; Chen D.; Wu X.; Ren X.; Zhang Y.; Meng X.; Tang F. Fe3O4-Au@Mesoporous SiO2 Microspheres: An Ideal Artificial Enzymatic Cascade System. Chem. Commun. 2013, 49, 4643–4645. 10.1039/c3cc40622a. [DOI] [PubMed] [Google Scholar]
- Andre R.; Natalio F.; Humanes M.; Leppin J.; Heinze K.; Wever R.; Schroder H. C.; Muller W. E. G.; Tremel W. V2O5 Nanowires with An Intrinsic Peroxidase-Like Activity. Adv. Funct. Mater. 2011, 21, 501–509. 10.1002/adfm.201001302. [DOI] [Google Scholar]; b Natalio F.; Andre R.; Hartog A. F.; Stoll B.; Jochum K. P.; Wever R.; Tremel W. Vanadium Pentoxide Nanoparticles Mimic Vanadium Haloperoxidases and Thwart Biofilm Formation. Nat. Nanotechnol. 2012, 7, 530–535. 10.1038/nnano.2012.91. [DOI] [PubMed] [Google Scholar]
- Pirmohamed T.; Dowding J. M.; Singh S.; Wasserman B.; Heckert E.; Karakoti A. S.; King J. E. S.; Seal S.; Self W. T. Nanoceria exhibit redox state-dependent catalase mimetic activity. Chem. Commun. 2010, 46, 2736–2738. 10.1039/b922024k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragg R.; Natalio F.; Tahir M. N.; Janssen H.; Kashyap A.; Strand D.; Strand S.; Tremel W. Molybdenum Trioxide Nanoparticles with Intrinsic Sulfite Oxidase Activity. ACS Nano 2014, 8, 5182–5189. 10.1021/nn501235j. [DOI] [PubMed] [Google Scholar]
- a Wu Y. S.; Huang F.; Lin Y. W. Fluorescent Detection of Lead in Environmental Water and Urine Samples Using Enzyme Mimics of Catechin-Synthesized Au Nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 1503–1509. 10.1021/am3030454. [DOI] [PubMed] [Google Scholar]; b Liu Y.; Wang C.; Cai N.; Long S.; Yu F. Negatively Charged Gold Nanoparticles as An Intrinsic Peroxidase Mimic and Their Applications in the Oxidation of Dopamine. J. Mater. Sci. 2014, 49, 7143–7150. 10.1007/s10853-014-8422-x. [DOI] [Google Scholar]; c Zhu X.; Mao X.; Wang Z.; Feng C.; Chen G.; Li G. Fabrication of Nanozyme@DNA Hydrogel and Its Application in Biomedical Analysis. Nano Res. 2017, 10, 959–970. 10.1007/s12274-016-1354-9. [DOI] [Google Scholar]
- a Sun Z.; Zhang N.; Si Y.; Li S.; Wen J.; Zhu X.; Wang H. High-throughput Colorimetric Assays for Mercury (II) in Blood and Wastewater Based on the Mercury-Stimulated Catalytic Activity of Small Silver Nanoparticles in A Temperature-Switchable Gelatin Matrix. Chem. Commun. 2014, 50, 9196–9199. 10.1039/C4CC03851G. [DOI] [PubMed] [Google Scholar]; b Chen L.; Sha L.; Qiu Y. W.; Wang G. F.; Jiang H.; Zhang X. J. An Amplified Electrochemical Aptasensor Based on Hybridization Chain Reactions and Catalysis of Silver Nanoclusters. Nanoscale 2015, 7, 3300–3308. 10.1039/C4NR06664B. [DOI] [PubMed] [Google Scholar]
- a Jin L.; Meng Z.; Zhang Y.; Cai S.; Zhang Z.; Li C.; Shang L.; Shen Y. H. Ultrasmall Pt Nanoclusters as Robust Peroxidase Mimics for Colorimetric Detection of Glucose in Human Serum. ACS Appl. Mater. Interfaces 2017, 9, 10027–10033. 10.1021/acsami.7b01616. [DOI] [PubMed] [Google Scholar]; b Ye H.; Liu Y.; Chhabra A.; Lilla E.; Xia X. H. Polyvinylpyrrolidone (PVP)-Capped Pt Nanocubes with Superior Peroxidase-Like Activity. ChemNanoMat 2017, 3, 33–38. 10.1002/cnma.201600268. [DOI] [Google Scholar]
- Vázquez-González M.; Liao W. C.; Cazelles R.; Wang S.; Yu X.; Gutkin V.; Willner I. Mimicking Horseradish Peroxidase Functions Using Cu2+-Modified Carbon Nitride Nanoparticles or Cu2+-Modified Carbon Dots as Heterogeneous Catalysts. ACS Nano 2017, 11, 3247–3253. 10.1021/acsnano.7b00352. [DOI] [PubMed] [Google Scholar]
- Wang H.; Liu C. Q.; Liu Z.; Ren J. S.; Qu X. G. Specific Oxygenated Groups Enriched Graphene Quantum Dots as Highly Efficient Enzyme Mimics. Small 2018, 14, 1703710. 10.1002/smll.201703710. [DOI] [PubMed] [Google Scholar]
- a Pandey P. C.; Panday D. Tetrahydrofuran and Hydrogen Peroxide Mediated Conversion of Potassium Hexacyanoferrate into Prussian Blue Nanoparticles: Application to Hydrogen Peroxide Sensing. Electrochim. Acta 2016, 190, 758–765. 10.1016/j.electacta.2015.12.188. [DOI] [Google Scholar]; b Vazquez-Gonzalez M.; Torrente-Rodriguez R. M.; Kozell A.; Liao W.-C.; Cecconello A.; Campuzano S.; Pingarron J. M.; Willner I. Mimicking Peroxidase Activities with Prussian Blue Nanoparticles and Their Cyanometalate Structural Analogues. Nano Lett. 2017, 17, 4958–4963. 10.1021/acs.nanolett.7b02102. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ai K.; Ji X.; Askhatova D.; Du R.; Lu L.; Shi J. Comprehensive Insights into The Multi-Antioxidative Mechanisms of Melanin Nanoparticles and Their Application to Protect Brain from Injury in Ischemic Stroke. J. Am. Chem. Soc. 2017, 139, 856–862. 10.1021/jacs.6b11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mondloch J. E.; Katz M. J.; Isley W. C.; Ghosh P.; Liao P.; Bury W.; Farha O. K.; et al. Destruction of Chemical Warfare Agents Using Metal-Organic Frameworks. Nat. Mater. 2015, 14, 512–516. 10.1038/nmat4238. [DOI] [PubMed] [Google Scholar]; b Lopez-Maya E.; Montoro C.; Rodriguez-Albelo L. M.; Aznar Cervantes S. D.; Lozano-Perez A. A.; Cenis J. L.; Barea E.; Navarro J. A. R. Textile/Metal-Organic-Framework Composites as Self-Detoxifying Filters for Chemical-Warfare Agents. Angew. Chem., Int. Ed. 2015, 54, 6790–6794. 10.1002/anie.201502094. [DOI] [PubMed] [Google Scholar]; c Chen W. H.; Vázquez-González M.; Kozell A.; Cecconello A.; Willner I. Cu2+-Modified Metal-Organic Framework Nanoparticles: A Peroxidase-Mimicking Nanoenzyme. Small 2018, 14, 1703149. 10.1002/smll.201703149. [DOI] [PubMed] [Google Scholar]
- Li M.; Chen J.; Wu W.; Fang Y.; Dong S. Oxidase-like MOF-818 Nanozyme with High Specificity for Catalysis of Catechol Oxidation. J. Am. Chem. Soc. 2020, 142, 15569–15574. 10.1021/jacs.0c07273. [DOI] [PubMed] [Google Scholar]
- a Luo W.; Zhu C.; Su S.; Li D.; He Y.; Huang Q.; Fan C. Self-Catalyzed, Self-Limiting Growth of Glucose Oxidase-Mimicking Gold Nanoparticles. ACS Nano 2010, 4, 7451–7458. 10.1021/nn102592h. [DOI] [PubMed] [Google Scholar]; b Liu B.; Huang Z.; Liu J. Boosting the Oxidase Mimicking Activity of Nanoceria by Fluoride Capping: Rivaling Protein Enzymes and Ultrasensitive F– Detection. Nanoscale 2016, 8, 13562–13567. 10.1039/C6NR02730J. [DOI] [PubMed] [Google Scholar]; c Asati A.; Santra S.; Kaittanis C.; Nath S.; Perez J. M. Oxidase-Like Activity of Polymer-Coated Cerium Oxide Nanoparticles. Angew. Chem., Int. Ed. 2009, 48, 2308–2348. 10.1002/anie.200805279. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Comotti M.; Della Pina C.; Matarrese R.; Rossi M. The Catalytic Activity of “Naked” Gold Particles. Angew. Chem., Int. Ed. 2004, 43, 5812–5815. 10.1002/anie.200460446. [DOI] [PubMed] [Google Scholar]
- a Liu Y.; Purich D. L.; Wu C. C.; Wu Y.; Chen T.; Cui C.; Zhang L. Q.; Cansiz S.; Hou W. J.; Wang Y. Y.; Yang S. Y.; Tan W. H. Ionic Functionalization of Hydrophobic Colloidal Nanoparticles to Form Ionic Nanoparticles with Enzymelike Properties. J. Am. Chem. Soc. 2015, 137, 14952–14958. 10.1021/jacs.5b08533. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cai R.; Yang D.; Peng S. J.; Chen X. G.; Huang Y.; Liu Y.; Hou W. J.; Yang S. Y.; Liu Z. B.; Tan W. H. Single Nanoparticle to 3D Supercage: Framing for An Artificial Enzyme System. J. Am. Chem. Soc. 2015, 137, 13957–13963. 10.1021/jacs.5b09337. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang L.; Han L.; Hu P.; Wang L.; Dong S. TiO2 Nanotube Arrays: Intrinsic Peroxidase Mimetics. Chem. Commun. 2013, 49, 10480–10482. 10.1039/c3cc46163g. [DOI] [PubMed] [Google Scholar]
- Liang H.; Lin F.; Zhang Z.; Liu B.; Jiang S.; Yuan Q.; Liu J. Multicopper Laccase Mimicking Nanozymes with Nucleotides as Ligands. ACS Appl. Mater. Interfaces 2017, 9, 1352–1360. 10.1021/acsami.6b15124. [DOI] [PubMed] [Google Scholar]
- a Pirmohamed T.; Dowding J. M.; Singh S.; Wasserman B.; Heckert E.; Karakoti A. S.; King J. E. S.; Seal S.; Self W. T. Nanoceria Exhibit Redox State-Dependent Catalase Mimetic Activity. Chem. Commun. 2010, 46, 2736–2738. 10.1039/b922024k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Celardo I.; Pedersen J. Z.; Traversa E.; Ghibelli L. Pharmacological Potential of Cerium Oxide Nanoparticles. Nanoscale 2011, 3, 1411–1420. 10.1039/c0nr00875c. [DOI] [PubMed] [Google Scholar]
- Korsvik C.; Patil S.; Seal S.; Self W. T. Superoxide Dismutase Mimetic Properties Exhibited by Vacancy Engineered Ceria Nanoparticles. Chem. Commun. 2007, 10, 1056–1058. 10.1039/b615134e. [DOI] [PubMed] [Google Scholar]
- a Katz M. J.; Mondloch J. E.; Totten R. K.; Park J. K.; Nguyen S. T.; Farha O. K.; Hupp J. T. Simple and Compelling Biomimetic Metal-Organic Framework Catalyst for the Degradation of Nerve Agent Simulants. Angew. Chem. 2014, 126, 507–511. 10.1002/ange.201307520. [DOI] [PubMed] [Google Scholar]; b Plonka A. M.; Wang Q.; Gordon W. O.; Balboa A.; Troya D.; Guo W.; Sharp C. H.; Senanayake S. D.; Morris J. R.; Hill C. L.; Frenkel A. I. In Situ Probes of Capture and Decomposition of Chemical Warfare Agent Simulants by Zr-Based Metal-Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 599–602. 10.1021/jacs.6b11373. [DOI] [PubMed] [Google Scholar]
- a Wang T. Y.; Zhu H. C.; Zhuo J. Q.; Zhu Z. W.; Papakonstantinou P.; Lubarsky G.; Lin J.; Li M. X. Biosensor Based on Ultrasmall MoS2 Nanoparticles for Electrochemical Detection of H2O2 Released by Cells at The Nanomolar Level. Anal. Chem. 2013, 85, 10289–10295. 10.1021/ac402114c. [DOI] [PubMed] [Google Scholar]; b Tao Y.; Lin Y. H.; Ren J. S.; Qu X. G. A Dual Fluorometric and Colorimetric Sensor for Dopamine Based on BSA-Stabilized Au Nanoclusters. Biosens. Bioelectron. 2013, 42, 41–46. 10.1016/j.bios.2012.10.014. [DOI] [PubMed] [Google Scholar]; c Qin L.; Wang X. Y.; Liu Y. F.; Wei H. 2D-Metal-Organicframework-Nanozyme Sensor Arrays for Probing Phosphates and Their Enzymatic Hydrolysis. Anal. Chem. 2018, 90, 9983–9989. 10.1021/acs.analchem.8b02428. [DOI] [PubMed] [Google Scholar]; d Wang X. Y.; Qin L.; Zhou M.; Lou Z. P.; Wei H. Nanozyme Sensor Arrays for Detecting Versatile Analytes from Small Molecules to Proteins and Cells. Anal. Chem. 2018, 90, 11696–11702. 10.1021/acs.analchem.8b03374. [DOI] [PubMed] [Google Scholar]; e Tian Z. M.; Li J.; Zhang Z. Y.; Gao W.; Zhou X. M.; Qu Y. Q. Highly Sensitive and Robust Peroxidase-like Activity of Porous Nanorods of Ceria and Their Application for Breast Cancer Detection. Biomaterials 2015, 59, 116–124. 10.1016/j.biomaterials.2015.04.039. [DOI] [PubMed] [Google Scholar]
- a Fan K. L.; Cao C. Q.; Pan Y. X.; Lu D.; Yang D. L.; Feng J.; Song L. N.; Liang M. M.; Yan X. Y. Magnetoferritin Nanoparticles for Targeting and Visualizing Tumour Tissues. Nat. Nanotechnol. 2012, 7, 459–464. 10.1038/nnano.2012.90. [DOI] [PubMed] [Google Scholar]; b Ragg R.; Schilmann A. M.; Korschelt K.; Wieseotte C.; Kluenker M.; Viel M.; Völker L.; Preiß S.; Herzberger J.; Frey H.; et al. Intrinsic Superoxide Dismutase Activity of Mno Nanoparticles Enhances Magnetic Resonance Imaging Contrast. J. Mater. Chem. B 2016, 4, 7423–7428. 10.1039/C6TB02078J. [DOI] [PubMed] [Google Scholar]; c Zhang T. W.; Cao C. Q.; Tang X.; Cai Y.; Yang C. Y.; Pan Y. X. Enhanced Peroxidase Activity and Tumour Tissue Visualization by Cobalt-Doped Magnetoferritin Nanoparticles. Nanotechnology 2017, 28, 045704 10.1088/1361-6528/28/4/045704. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Ni D.; Rosenkrans Z. T.; Huang P.; Yan X.; Cai W. Nanozyme: New Horizons for Responsive Biomedical Applications. Chem. Soc. Rev. 2019, 48, 3683–3704. 10.1039/C8CS00718G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang Z. L.; Liu H. Y.; Yang S. H.; Wang T.; Liu C.; Cao Y. C. Nanoparticle-Based Artificial RNA Silencing Machinery for Antiviral Therapy. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12387–12392. 10.1073/pnas.1207766109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu C. P.; Wu T. H.; Liu C. Y.; Chen K. C.; Chen Y. X.; Chen G. S.; Lin S. Y. Self-Supplying O2 through the Catalase-Like Activity of Gold Nanoclusters for Photodynamic Therapy Against Hypoxic Cancer Cells. Small 2017, 13, 1700278. 10.1002/smll.201700278. [DOI] [PubMed] [Google Scholar]; c Huo M.; Wang L.; Chen Y.; Shi J. Tumor-Selective Catalytic Nanomedicine by Nanocatalyst Delivery. Nat. Commun. 2017, 8, 1–12. 10.1038/s41467-017-00424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang Y.; Wang Z. Y.; Li X. J.; Wang L.; Yin M.; Wang L. H.; Chen N.; Fan C. H.; Song H. Y. Dietary Iron Oxide Nanoparticles Delay Aging and Ameliorate Neurodegeneration in Drosophila. Adv. Mater. 2016, 28, 1387–1393. 10.1002/adma.201503893. [DOI] [PubMed] [Google Scholar]; b Guan Y. J.; Li M.; Dong K.; Gao N.; Ren J. S.; Zheng Y. C.; Qu X. G. Ceria/Poms Hybrid Nanoparticles as A Mimicking Metallopeptidase for Treatment of Neurotoxicity of Amyloid-β Peptide. Biomaterials 2016, 98, 92–102. 10.1016/j.biomaterials.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Hao C. L.; Qu A. H.; Xu L. G.; Sun M. Z.; Zhang H. Y.; Xu C. L.; Kuang H. Chiral Molecule-Mediated Porous CuxO Nanoparticle Clusters with Antioxidation Activity for Ameliorating Parkinson’s Disease. J. Am. Chem. Soc. 2019, 141, 1091–1099. 10.1021/jacs.8b11856. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Wang Z.; Ren J.; Qu X. Enzyme Mimicry for Combating Bacteria and Biofilms. Acc. Chem. Res. 2018, 51, 789–799. 10.1021/acs.accounts.8b00011. [DOI] [PubMed] [Google Scholar]
- Mu J. S.; Li J.; Zhao X.; Yang E. C.; Zhao X. J. Cobaltdoped Graphitic Carbon Nitride with Enhanced Peroxidase-like Activity for Wastewater Treatment. RSC Adv. 2016, 6, 35568–35576. 10.1039/C6RA02911F. [DOI] [Google Scholar]
- Zhang Z.; Zhang X.; Liu B.; Liu J. Molecular Imprinting on Inorganic Nanozymes for Hundred-Fold Enzyme Specificity. J. Am. Chem. Soc. 2017, 139, 5412–5419. 10.1021/jacs.7b00601. [DOI] [PubMed] [Google Scholar]
- a Zhong Y.; Zhao J.; Li J.; Liao X.; Chen F. Advances of Aptamers Screened by Cell-SELEX in Selection Procedure, Cancer Diagnostics and Therapeutics. Anal. Biochem. 2020, 598, 113620. 10.1016/j.ab.2020.113620. [DOI] [PubMed] [Google Scholar]; b Darmostuk M.; Rimpelova S.; Gbelcova H.; Ruml T. Current Approaches in SELEX: An Update to Aptamer Selection Technology. Biotechnol. Adv. 2015, 33, 1141–1161. 10.1016/j.biotechadv.2015.02.008. [DOI] [PubMed] [Google Scholar]; c Ellington A. D.; Szostak J. W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]; d Tuerk C.; Gold L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Acteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- a You K. M.; Lee S. H.; Im A.; Lee S. B. Aptamers as Functional Nucleic Acids: In Vitro Selection and Biotechnological Applications. Biotechnol. Bioprocess Eng. 2003, 8, 64–75. 10.1007/BF02940259. [DOI] [Google Scholar]; b Pendergrast P. S.; Marsh H. N.; Grate D.; Healy J. M.; Stanton M. Nucleic Acid Aptamers for Target Validation and Therapeutic Applications. J. Biomol. Technol. 2005, 16, 224–234. [PMC free article] [PubMed] [Google Scholar]
- a Zhou W.; Jimmy Huang P. J.; Ding J.; Liu J. Aptamer-Based Biosensors for Biomedical Diagnostics. Analyst 2014, 139, 2627–2640. 10.1039/c4an00132j. [DOI] [PubMed] [Google Scholar]; b Famulok M.; Mayer G. Aptamer Modules as Sensors and Detectors. Acc. Chem. Res. 2011, 44, 1349–1358. 10.1021/ar2000293. [DOI] [PubMed] [Google Scholar]; c Du Y.; Li B.; Wang E. Fitting” Makes “Sensing” Simple: Label-Free Detection Strategies Based on Nucleic Acid Aptamers. Acc. Chem. Res. 2013, 46, 203–213. 10.1021/ar300011g. [DOI] [PubMed] [Google Scholar]; d Willner I.; Zayats M. Electronic Aptamer-Based Sensors. Angew. Chem., Int. Ed. 2007, 46, 6408–6418. 10.1002/anie.200604524. [DOI] [PubMed] [Google Scholar]
- a Michaud M.; Jourdan E.; Villet A.; Ravel A.; Grosset C.; Peyrin E. A DNA Aptamer as A New Target-Specific Chiral Selector for HPLC.. J. Am. Chem. Soc. 2003, 125, 8672–8679. 10.1021/ja034483t. [DOI] [PubMed] [Google Scholar]; b Kotia R. B.; Li L.; Mcgown L. B. Separation of Nontarget Compounds by DNA Aptamers. Anal. Chem. 2000, 72, 827–831. 10.1021/ac991112f. [DOI] [PubMed] [Google Scholar]; c Deng Q.; German I.; Buchanan D.; Kennedy R. T. Retention and Separation of Adenosine and Analogues by Affinity Chromatography with An Aptamer Stationary Phase. Anal. Chem. 2001, 73, 5415–5421. 10.1021/ac0105437. [DOI] [PubMed] [Google Scholar]
- a Famulok M.; Hartig J. S.; Mayer G. Functional Aptamers and Aptazymes in Biotechnology, Diagnostics, and Therapy. Chem. Rev. 2007, 107, 3715–3743. 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]; b Lao Y. H.; Phua K. K. L.; Leong K. W. Aptamer Nanomedicine for Cancer Therapeutics: Barriers and Potential for Translation. ACS Nano 2015, 9, 2235–2254. 10.1021/nn507494p. [DOI] [PubMed] [Google Scholar]
- a Vazquez-Gonzalez M.; Willner I. Aptamer-Functionalized Micro- And Nanocarriers for Controlled Release. ACS Appl. Mater. Interfaces 2021, 13, 9520–9541. 10.1021/acsami.0c17121. [DOI] [PubMed] [Google Scholar]; b Chen W. H.; Yu X.; Liao W. C.; Sohn Y. S.; Cecconello A.; Kozell A.; Nechushtai R.; Willner I. ATP-Responsive Aptamer-Based Metal-Organic Framework Nanoparticles (NMOFs) for The Controlled Release of Loads and Drugs. Adv. Funct. Mater. 2017, 27, 1702102. 10.1002/adfm.201702102. [DOI] [Google Scholar]; c Chen W. H.; Yang Sung S.; Fadeev M.; Cecconello A.; Nechushtai R.; Willner I. Targeted VEGF-Triggered Release of An Anticancer Drug from Aptamer-Functionalized Metal-Organic Framework Nanoparticles. Nanoscale 2018, 10, 4650–4657. 10.1039/C8NR00193F. [DOI] [PubMed] [Google Scholar]
- Steen Burrell K. A.; Layzer J.; Sullenger B. A. A Kallikrein-Targeting RNA Aptamer Inhibits the Intrinsic Pathway of Coagulation and Reduces Bradykinin Release. J. Thromb. Haemostasis 2017, 15, 1807–1817. 10.1111/jth.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ray P.; White R. R. Aptamers for Targeted Drug Delivery. Pharmaceuticals 2010, 3, 1761–1778. 10.3390/ph3061761. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cerchia L.; De Franciscis V. Targeting Cancer Cells with Nucleic Acid Aptamers. Trends Biotechnol. 2010, 28, 517–525. 10.1016/j.tibtech.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Meng H. M.; Liu H.; Kuai H.; Peng R.; Mo L.; Zhang X. B. Aptamer-Integrated DNA Nanostructures for Biosensing, Bioimaging and Cancer Therapy. Chem. Soc. Rev. 2016, 45, 2583–2602. 10.1039/C5CS00645G. [DOI] [PubMed] [Google Scholar]
- a Wang J.; Yue L.; Li Z.; Zhang J.; Tian H.; Willner I. Active Generation of Nanoholes in DNA Origami Scaffolds for Programmed Catalysis in Nanocavities. Nat. Commun. 2019, 10, 4963. 10.1038/s41467-019-12933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knudsen J. B.; Liu L.; Bank Kodal A. L.; Madsen M.; Li Q.; Song J.; Woehrstein J. B.; Wickham S. F. J.; Strauss M. T.; Schueder F.; et al. Routing of Individual Polymers in Designed Patterns. Nat. Nanotechnol. 2015, 10, 892–898. 10.1038/nnano.2015.190. [DOI] [PubMed] [Google Scholar]
- Golub E.; Albada H. B.; Liao W. C.; Biniuri Y.; Willner I. Nucleoapzymes: Hemin/G-Quadruplex DNAzyme-Aptamer Binding Site Conjugates with Superior Enzyme-Like Catalytic Functions. J. Am. Chem. Soc. 2016, 138, 164–172. 10.1021/jacs.5b09457. [DOI] [PubMed] [Google Scholar]
- Biniuri Y.; Albada B.; Wolff M.; Golub E.; Gelman D.; Willner I. Cu2+ or Fe3+ Terpyridine/Aptamer Conjugates: Nucleoapzymes Catalyzing the Oxidation of Dopamine to Aminochrome. ACS Catal. 2018, 8, 1802–1809. 10.1021/acscatal.7b03454. [DOI] [Google Scholar]
- Biniuri Y.; Shpilt Z.; Albada B.; Vázquez-González M.; Wolff M.; Hazan C.; Golub E.; Gelman D.; Willner I. A Bis-Zn2+-Pyridyl-Salen-Type Complex Conjugated to the ATP Aptamer: An ATPase-Mimicking Nucleoapzyme. ChemBioChem 2020, 21, 53–58. 10.1002/cbic.201900182. [DOI] [PubMed] [Google Scholar]
- Luo G. F.; Biniuri Y.; Vázquez-González M.; Wulf V.; Fadeev M.; Lavi R.; Willner I. Metal Ion-Terpyridine-Functionalized L-Tyrosinamide Aptamers: Nucleoapzymes for Oxygen Insertion into C-H Bonds and the Transformation of L-Tyrosinamide into Amidodopachrome. Adv. Funct. Mater. 2019, 29, 1901484. 10.1002/adfm.201901484. [DOI] [Google Scholar]
- Qu S.; Wang X.; Lu Q.; Liu X.; Wang L. A Biocompatible Fluorescent Ink Based on Water-Soluble Luminescent Carbon Nanodots. Angew. Chem. 2012, 124, 12381–12384. 10.1002/ange.201206791. [DOI] [PubMed] [Google Scholar]
- Sun S.; Chen Q.; Tang Z.; Liu C.; Li Z.; Wu A.; Lin H. Tumor Microenvironment Stimuli-Responsive Fluorescence Imaging and Synergistic Cancer Therapy by Carbon-Dot-Cu2+ Nanoassemblies. Angew. Chem. 2020, 132, 21227–21234. 10.1002/ange.202007786. [DOI] [PubMed] [Google Scholar]
- Nakatsuka N.; Yang K. A.; Abendroth J. M.; Cheung K. M.; Xu X.; Yang H.; Andrews A. M.; et al. Aptamer-field-effect Transistors Overcome Debye Length Limitations for Small-molecule Sensing. Science 2018, 362, 319–324. 10.1126/science.aao6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bogdanova A. Y.; Nikinmaa M. Reactive Oxygen Species Regulate Oxygen-Sensitive Potassium Flux in Rainbow Trout Erythrocytes. J. Gen. Physiol. 2001, 117, 181–190. 10.1085/jgp.117.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pradhan K.; Das G.; Kar C.; Mukherjee N.; Khan J.; Mahata T.; Barman S.; Ghosh S. Rhodamine-Based Metal Chelator: A Potent Inhibitor of Metal-Catalyzed Amyloid Toxicity. ACS Omega 2020, 5, 18958–18967. 10.1021/acsomega.0c02235. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cuypers A.; Plusquin M.; Remans T.; Jozefczak M.; Keunen E.; Gielen H.; Smeets K.; et al. Cadmium Stress: An Oxidative Challenge. BioMetals 2010, 23, 927–940. 10.1007/s10534-010-9329-x. [DOI] [PubMed] [Google Scholar]; d Timoshnikov V. A.; Kobzeva T. V.; Polyakov N. E.; Kontoghiorghes G. J. Redox Interactions of Vitamin C and Iron: Inhibition of the Pro-Oxidant Activity by Deferiprone. Int. J. Mol. Sci. 2020, 21, 3967. 10.3390/ijms21113967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hatcher L. Q.; Karlin K. D. Oxidant Types in Copper–Dioxygen Chemistry: the Ligand Coordination Defines the Cun-O2 Structure and Subsequent Reactivity. JBIC, J. Biol. Inorg. Chem. 2004, 9, 669–683. 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]; b Holland P. L.; Rodgers K. R.; Tolman W. B. Is the Bis (μ-oxo) Dicopper Core Capable of Hydroxylating an Arene?. Angew. Chem., Int. Ed. 1999, 38, 1139–1142. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.