

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a solid malignant tumor with an extremely poor prognosis. Gemcitabine (GEM)-based chemotherapy remains one of the most important treatment choices for PDAC. However, either as monotherapy or as a part of the combination chemotherapy, GEM achieved only limited success in improving the survival of patients with advanced PDAC, primarily due to GEM resistance. PDAC is characterized by an extensive desmoplasia in the tumor microenvironment (TME). Increasing evidence indicates that this fibrotic TME not only actively participates in the tumor growth and spread of PDAC but also contributes to the induction of GEM resistance. Here we review the current advances of how TME components are involved in the induction of GEM resistance.
Keywords: pancreatic ductal adenocarcinoma, tumor microenvironment, gemcitabine, chemoresistance
Introduction
Pancreatic cancer is the solid malignant tumor with the worst prognosis in humans, and histologically, approximately 90% of cases are pancreatic ductal adenocarcinoma (PDAC). Despite continued advances in PDAC treatment, the survival rate has barely improved in the past 40 years, and the 5-year survival rate is still below 10% (1). At present, surgical resection remains the only possible cure for PDAC. However, due to a lack of clear clinical symptoms or signs in the early stage, the diagnosis of PDAC is extremely difficult, and only 15–20% of patients are eligible for surgical resection(2). Therefore, for borderline-resectable, advanced or metastatic PDAC, systemic chemotherapy (including neoadjuvant therapy) is the most important or the only treatment option. Although the modified FOLFIRINOX regimen (oxaliplatin, leucovorin, irinotecan, and 5-fluorouracil) has been shown to be superior to gemcitabine (GEM) for the treatment of PDAC, higher toxicity limits its application(3). Therefore, monotherapy or combined therapy with GEM is still the mainstay of current PDAC chemotherapy. As a nucleoside analog of deoxycytidine, GEM enters PDAC cells and undergoes a series of precise phosphorylation processes. Its derivatives are incorporated into DNA strands and interferes with DNA replication, ultimately leading to apoptosis (4) (Figure 1). However, the overall response rate of pancreatic cancer to GEM treatment is less than 20%(5). The median progression-free survival for GEM monotherapy for advanced PDAC is only 3.7 months(6). GEM resistance is one of the most critical determinants of chemotherapy efficacy and is also the basis of poor prognosis. Therefore, overcoming GEM resistance is an urgent objective for PDAC treatment(7). Compared with other malignant tumors, an extensive and dense fibrous matrix is a defining characteristic of PDAC. Hyperplastic connective tissue surrounding PDAC cells accounts for approximately 90% of the total tumor volume and has a significant impact on the pancreatic tissue structure(8). The hyperplastic connective tissue is mostly composed of stromal cells and extracellular matrix (ECM), which together with PDAC cells form the tumor microenvironment (TME). The interaction between PDAC cells and the TME is the primary driving force behind desmoplasia(9). Mounting evidence shows that the TME not only actively participates in the proliferation and metastasis of PDAC but also potentially contributes to inducing GEM resistance(10). Existing literature has extensively reviewed how the intracellular metabolism and molecular regulation are altered in PDAC cells and confer GEM resistance. In contrast, this review focuses on the major cellular and molecular mechanisms of GEM resistance through various components of the TME. Identification of potential therapeutic targets for GEM resistance facilitates the development of new strategies for chemotherapy sensitization and improves the overall prognosis of patients with PDAC.
Figure 1. The mechanism of action of GEM in PDAC treatment.
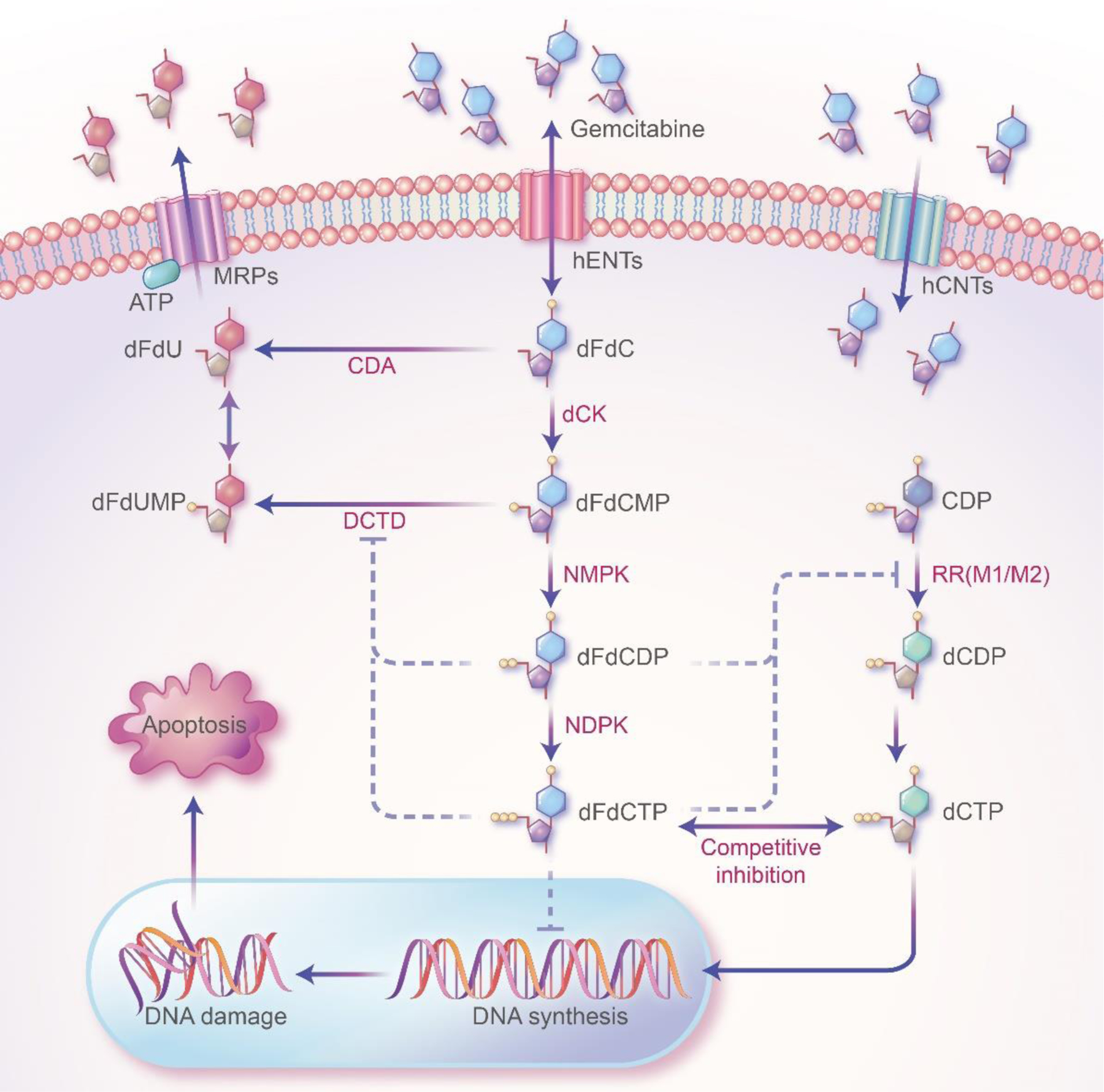
GEM (2’,2’-difluorodeoxycytidine, dFdC) crosses the cell membrane and is continuously phosphorylated by the phosphorylation rate-limiting enzyme deoxycytidine kinase (dCK) to form GEM monophosphate (dFdCMP), GEM diphosphate (dFdCDP), and GEM triphosphate (dFdCTP). As a substrate for DNA polymerase, dFdCTP competes with deoxycytidine triphosphate (dCTP), a raw material for DNA synthesis. DNA polymerase stochastically incorporates dFdCTP into DNA during replication, which terminates DNA strand elongation and ultimately leads to apoptosis. Two active metabolites, dFdCDP and dFdCTP, can inhibit ribonucleotide reductase (RR). RR is the rate-limiting enzyme (RRM1 and RRM2) in the DNA synthesis pathway and is mainly responsible for converting ribonucleotides into deoxyribonucleoside triphosphates (dNTPs), which are essential for DNA assembly and repair. In addition, dFdCDP and dFdCTP can inhibit cytidine deaminase (CDA) or deoxycytidylate deaminase (DCTD), which can inactivate dFdCMP.
Review of the current literature
A comprehensive literature search was conducted in the PubMed, Embase, and Web of Science databases. Only articles in English from 2000 to 2021 were included. Search terms included pancreatic cancer; pancreatic ductal adenocarcinoma; tumor stroma; tumor microenvironment; gemcitabine; and chemoresistance. We present the following article in accordance with the Narrative Review reporting checklist.
GEM resistance and hypoxia
Desmoplasia and a paucity of blood vessels reduces tissue perfusion of PDAC, resulting in a hypoxic TME(11). Adaptive mechanisms to the hypoxic TME confer GEM resistance to PDAC cells (Figure 2). Hypoxia promotes epidermal-mesenchymal transition (EMT) and activates glycolytic pathway through hypoxia inducible factor-1α (HIF-1α) in PDAC cells, resulting in GEM resistance(12, 13). In vitro experiments have also confirmed that the downregulation of HIF-1α increases GEM sensitivity(12, 14). In addition, GEM has been shown to induce stemness in PDAC cells, which can be further enhanced by hypoxia and promote chemoresistance through the protein kinase B/Notch homolog 1 (Akt/Notch1) signaling pathway in vitro (15). Therefore, hypoxia in the TME confers resistance to GEM. Unfortunately, the results of a phase III clinical trial (NCT01746979) with GEM combined with a cytotoxic drug (TH-302) that targets the hypoxic TME for the treatment of advanced PDAC did not show a significant difference in overall survival (OS)(16). The heterogeneity of PDAC itself and its associated TME can complicate the mechanism of hypoxia-induced GEM resistance, which may be one of the reasons why clinical trials failed. Hypoxia-induced acidosis is another important feature of the TME. HIF-1α-mediated glycolytic lactate production and carbonic anhydrase (CA)-mediated carbonic acid formation are the main sources of extracellular H+ in PDAC(17). McDonald et al. showed that in PDAC, hypoxia-induced HIF-1α promoted glycolysis and changes in intra- and extracellular pH and enhanced GEM resistance by upregulating carbonic anhydrase 9 (CA9) expression in vitro. In contrast, silencing or inhibiting CA9 reversed this process(18). In addition, an acidic TME may mediate GEM resistance by inducing EMT(19). Therefore, targeted correction of extracellular pH may become a new strategy to restore GEM sensitivity (20). In addition, TME stress can reshape cell metabolism, but the direct relationship between its metabolic effects and GEM resistance remains to be further elucidated (7).
Figure 2. Crosstalks between PDAC cells and TME and their contribution to GEM resistance.
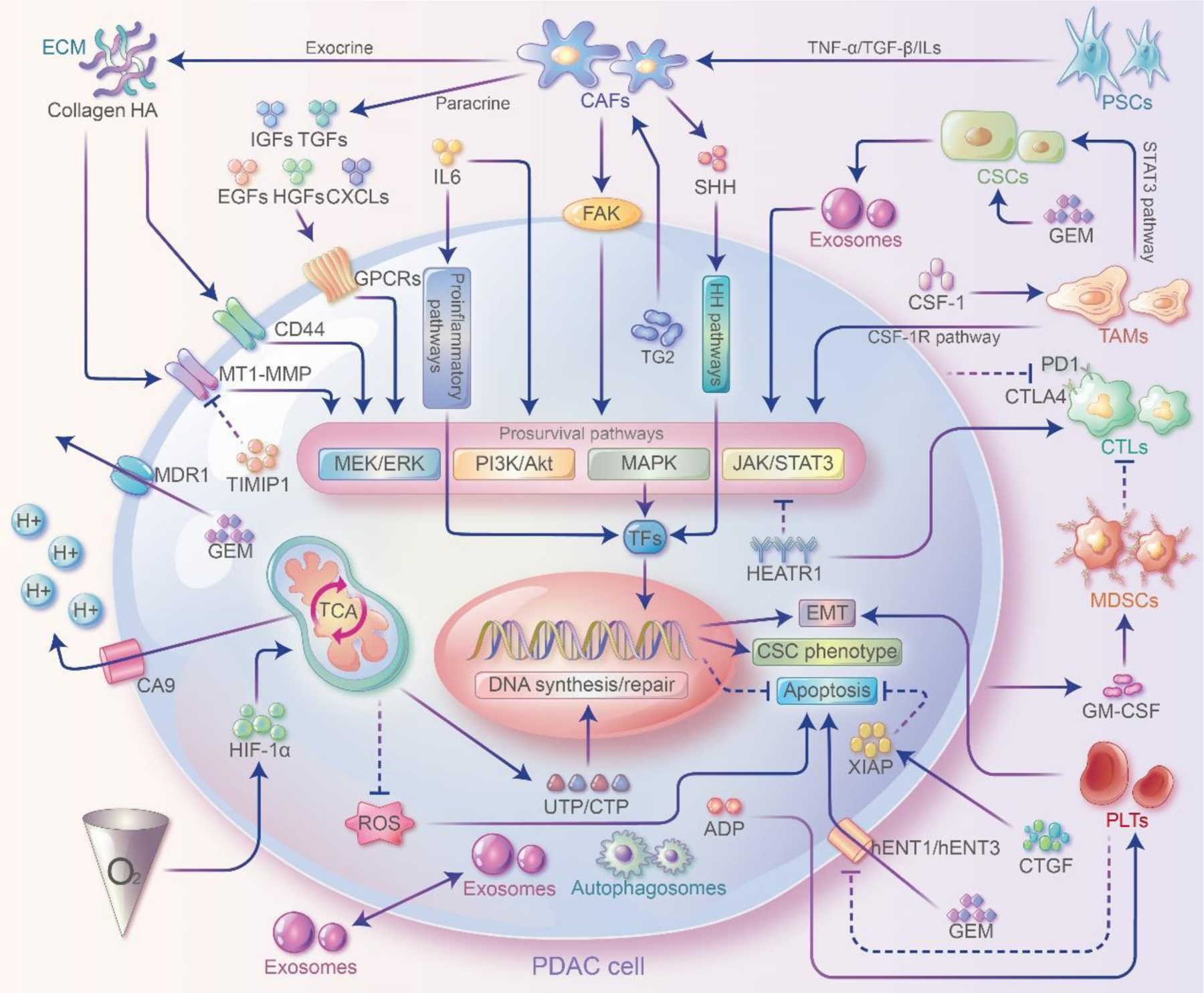
Stromal cells interact with PDAC cells through direct cell-cell contact and paracellular pathways to modulate GEM resistance. Stromal cells can secrete ECM proteins, cytokines or chemokines and interact with PDAC cells by activating prosurvival pathways to promote EMT and cell stemness or inhibit apoptosis and ultimately induce GEM resistance. In addition to secreting cytokines such as ILs that bind directly to receptors on PDAC cell surface, immune cells can also exert immunosuppression through proinflammatory pathways and induce GEM resistance. In addition, exosomes, autophagosomes play essential roles in signaling and substance exchange. Abbreviations: PDAC, pancreatic ductal adenocarcinoma; GEM, gemcitabine; ECM, extracellular matrix; TME, tumor microenvironment; CSCs, cancer stem cells; PSCs, pancreatic stellate cells; CAFs, cancer-associated fibroblasts; MDSCs, Myeloid-derived suppressor cells; TAMs, tumor-associated macrophages; CTLs, TFs, transcription factors; cytotoxic T lymphocytes; PLTs, Platelets; MSCs mesenchymal stem cells; EMT, epithelial-mesenchymal transition; HA, hyaluronic acid; HEATR1, HEAT repeat-containing protein 1; ADP, adenosine diphosphate; GPCRs, G protein-coupled receptors; HOTAIR, HOX transcript antisense RNA; CTLA-4, cytotoxic T lymphocyte associated antigen-4; PD-1, programmed death-1; XIAP, X-linked inhibitor of apoptosis protein X; FAK, focal adhesion kinase; TG2, transglutaminase 2; TIMP1, tissue inhibitor of metalloproteinases 1; MT1-MMP, membrane type 1-matrix metalloproteinase; CA9, carbonic anhydrase 9; MDR1, multidrug resistance protein 1.
GEM resistance and ECM
Numerous dense matrix components are present in the TME of PDAC. Dense fibrous tissue blocks blood vessels in tumor tissue and prevents GEM from entering into the tissue; furthermore, the interaction between matrix components and PDAC cells promotes GEM resistance (Figure 2).
Collagen
Type I collagen is the most abundant type of collagen in the ECM. The normal subtype of type I collagen is a heterotrimer that can be degraded by collagenase. However, PDAC cells can secrete unique homotrimers, which are resistant to all collagenolytic matrix metalloproteinases (MMPs)(21). Preclinical studies confirmed that matrix collagen forms a physical barrier that impedes GEM infiltration and the therapeutic response(10). In addition, collagen upregulates the expression of membrane type 1-matrix metalloproteinase (MT1-MMP) in PDAC, increasing the phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 and further upregulating high mobility group A2 (HMGA2) expression (22). HMGA2 plays a role in DNA end-joining repair; it has purine/pyrimidine cleavage enzyme activity and removes small, damaged bases from DNA, which can impair the efficacy of GEM in a collagen-rich TME (23). In addition, HMGA2 overexpression promotes the upregulation of histone acetyltransferases (HATs) in PDAC cells; increases the acetylation of histones, including histone H3 lysine 9 (H3K9) and histone H3 lysine 27 (H3K27); promotes chromatin relaxation and final DNA repair; and mediates GEM resistance (24). Thus, targeting the MT1-MMP/HMGA2/HAT signaling pathway may be a new GEM sensitization strategy. However, no clinical trials with broad-spectrum MMP inhibitors have shown a prognostic advantage over GEM monotherapy so far. The lack of selective MMP inhibitors might be one of the reasons for trial failure (25). Studies by D’Costa et al. show that tissue inhibitor of metalloproteinase 1 (TIMP1) promotes GEM resistance by activating the PI3K/Akt signaling pathway and that this process can be reversed by downregulating TIMP1 expression (26). Therefore, PDAC matrix collagen may play a bidirectional role in the mechanism of GEM resistance, suggesting that TIMP1 may be a potential target of GEM sensitization.
Hyaluronic acid (HA)
HA is a nonsulfated glycosaminoglycan and is regulated by a dynamic balance between synthesis and degradation under physiological conditions(27). HA is a predominant component in the ECM and plays a key role in GEM resistance, and high HA expression is an independent prognostic marker for PDAC patients (28). Because HA is highly water absorptive, its accumulation significantly increases interstitial fluid pressure (IFP) and impairs convection in tumors. Consequently, the reduced solute flux in perfusing vessels lead to tumor hypoperfusion, which is a hydrodynamic mechanism of GEM resistance (29). Notably, HA binds to cell surface receptors such as cluster of differentiation 44 (CD44) and mediates chemoresistance through a tyrosine kinase receptor-mediated signaling pathway, and CD44 downregulation can reverse GEM resistance (30). In addition, the HA-CD44 axis can promote upregulation of multidrug resistance protein 1 (MDR1); increase GEM efflux by enhancing the phosphorylation of Nanog, a biomarker for cancer stem cells (CSCs); or induce EMT to mediate GEM resistance via the Wnt signaling pathway through β-catenin(31, 32). Therefore, targeting the HA-CD44 signaling axis may be a promising strategy for GEM sensitization. In this regard, Serri et al. designed a novel liposome nanoparticle loaded with GEM that targeted CD44 and confirmed its efficacy in in vitro experiments(33). Pegvorhyaluronidase alfa (PEGPH20) is a novel PEGylated recombinant human hyaluronidase that can effectively scavenge matrix HA and achieve GEM sensitization in a PDAC animal model(34). Recently, two clinical trials for the treatment of advanced PDAC (NCT01839487 and NCT02715804) involving PEGPH20 combined with GEM have reported positive results(35, 36). Two recent studies have shown that dynamic contrast-enhanced (DCE)-MRI can identify perfusion changes in tumor microvasculature (37) and that SPECT/CT can identify novel fluorescent molecular labeled HA-CD44 tumor cells (38). These technologies will facilitate direct evaluation of responses to HA-targeted PDAC therapeutics in the future. Nevertheless, a phase III clinical trial of metastatic PDAC (HALO 109–301) recently suggested that PEGPH20 in combination with GEM did not have any additional advantages but had significantly increased toxic side effects(39). This finding further raises the question about the effectiveness of antistromal treatment.
Laminin (LN)
LN, another key component of the ECM, binds to the integrin family of cell adhesion molecules expressed on the PDAC cell surface, and its expression is significantly correlated with poor patient prognosis(40). The adhesion of PDAC cells to LNs and subsequent activation of signaling pathways form the basis of GEM resistance. Focal adhesion kinase (FAK) is a key intracellular molecule that transmits ECM signals to cells. By inducing FAK/Akt phosphorylation, LN promotes survivin expression and resistance to GEM-induced cytotoxicity and apoptosis(41). In addition, a study in a xenotransplantation model showed that transglutaminase 2 (TG2) secreted by PDAC cells stimulates LN-1 secretion by cancer-associated fibroblasts (CAFs), which in turn increases GEM resistance and that the downregulation of TG2 reverses this trend(42). Therefore, TG2/LN/FAK may be a potential target for GEM sensitization. However, due to the lack of specific inhibitors, only a limited number of relevant studies have been reported.
Fibronectin (FN)
FN, another main component of the ECM, can interact with integrins to transmit signals inside and outside the cell and play an important role in GEM resistance (43). In addition to forming stromal barriers like other ECM proteins, FN also mediates GEM resistance by inducing ERK1/2 phosphorylation to confer resistance to GEM-induced apoptosis. In an in vitro study, ERK inhibition restored response to GEM(44). Therefore, targeting FN/ERK may be a potential strategy to overcome GEM resistance in PDAC, but relevant studies are very limited so far (44, 45).
Cytokines and chemokines
Cytokines and chemokines are regulatory mediators between PDAC cells and the ECM and play a key role in GEM resistance. Among them, connective tissue growth factor (CTGF) promotes proliferation of stromal fibroblasts and directly inhibits apoptotic vesicle formation initiated by caspase-3/7/9 by inducing the phosphorylation of X-linked inhibitor of apoptosis protein (XIAP), thereby conferring GEM resistance(46). In vitro and xenogeneic model studies have shown that the targeted inhibition of CTGF (FG-3019) or XIAP (AZD5582) reverses GEM resistance by inducing PDAC cell apoptosis(46, 47). Therefore, the CTGF/XIAP axis is a potential target to overcome GEM resistance. Currently, an ongoing clinical trial (NCT02210559) is evaluating the efficacy of a monoclonal antibody (pamrevlumab) against CTGF combined with GEM for the treatment of locally advanced PDAC. Preliminary results show that the combination regimen can increase the R0 resection rate and is well tolerated(48). Transforming growth factor-β (TGF-β) is another cytokine critically involved in PDAC. Its overexpression in PDAC induces the expression of cysteine-rich angiogenic inducer 61 (CYR61) through the classical TGF-β/ALK5/Smad2/3 signaling pathway. CYR61 negatively regulates the nucleoside transporters human equilibrative nucleoside transporters 1 (hENT1) and human concentrative nucleoside transporter 3 (hCNT3) (Figure 1) and increases the cellular uptake of GEM, and the targeted inhibition of TGF-β/CYR61 can result in GEM sensitization(49–51). In addition to regulating genes in the TGF-β/Smad pathway, TGF-β also promotes EMT and mediates GEM resistance through epigenetic changes, such as methylation of the Vav guanine nucleotide exchange factor 1 (VAV1) gene, and targeted inhibition of the TGF-β/VAV1 axis can significantly enhance the efficacy of GEM (52). Therefore, targeting TGF-β/CYR61/VAV1 is also a potential strategy for GEM sensitization.
The epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) pathways play key roles in cancer initiation and progression (53). Among them, EGFR is a member of the epidermal growth factor family of receptor tyrosine kinases (ErbBs) and expressed in 40–60% of PDAC cases. Preclinical studies have shown that GEM itself can induce adaptive resistance to GEM in PDAC by activating the HAb18G/CD147-EGFR-signal transducer and activator of transcription 3 (STAT3) prosurvival signaling pathway(54, 55). Targeted inhibition of HAb18G (CD147) or EGFR/STAT3 signaling reverses GEM resistance(55, 56). Therefore, the HAb18G (CD147)/EGFR/STAT3 axis is a potential target for overcoming GEM resistance. Nevertheless, compared with GEM monotherapy, combination therapies of EGFR tyrosine kinase inhibitors (EGFR-TKIs) and GEM did not improve disease-free survival (DFS) or OS in resectable or advanced PDAC clinical trials (57–59). In addition, phase III clinical trials (NCT00471146) of VEGFR-TKIs combined with GEM failed(60). TKI-induced fast compensation mechanisms in cells may be one of the reasons for clinical trial failures(61).
Binding of insulin-like growth factor-1 (IGF-1) to its receptor IGF-1R can activate the PI3K/Akt/mechanistic target of rapamycin (mTOR) and MEK/ERK pathways, promote PDAC proliferation and induce GEM resistance(62). The targeted inhibition of IGF-1R can enhance the antitumor effect of GEM in PDAC xenografts(63). Monoclonal antibodies against human IGF-1R have been used to treat several cancers, but a phase III clinical trial (NCT01231347) of anti-IGF-1R monoclonal antibodies combined with GEM for the treatment of metastatic PDAC was not successful(64). A recent phase I/II clinical trial (NCT00769483) conducted by Abdel-Wahab et al. in advanced PDAC showed that compared with GEM + EGFR-TKIs, IGF-1R inhibitors combined with GEM + EGFR-TKIs significantly improved OS(65). Platelet-derived growth factor-D (PDGF-D) is critically involved in inducing interstitial fibrosis and increasing IFP in PDAC and promotes GEM resistance, presumably by triggering EMT via the PI3K/Akt and mTOR, nuclear factor kappa B (NF-κB), ERK, mitogen-activated protein kinase (MAPK), and Notch pathways(66). Although PDGF receptor (PDGFR) inhibitors combined with GEM showed efficacy in preclinical studies (67), the most recent clinical trials failed(68–70).
In addition to cytokines, the binding of CXC ligand (CXCL) to its receptor (CXC chemokine receptor, CXCR) can induce GEM resistance through the paracrine action of PDAC stromal cells and autocrine signal transduction by cancer cells. CXCL12/CXCR4 is the crucial nexus in this pathway (71). CXCR4 is a G-protein-coupled receptor localized on the cell surface. It is highly expressed in PDAC cells and is closely linked to poor patient prognosis(72). The CXCL12/CXCR4 axis can activate the FAK, ERK, and Akt prosurvival pathways, enhance the transcriptional activity of β-catenin and NF-κB, promote survivin expression, and induce GEM resistance(73). Preclinical studies have shown that CXCR4 antagonists can significantly enhance the efficacy of GEM(73–75). In recent years, novel CXCR pathways have been revealed, and the inhibition of related pathways in vitro has shown similar effects(76, 77). Therefore, CXCR appears to be an attractive target of intervention in future studies on GEM sensitization.
GEM resistance and stromal cells
Stromal cells are an important component of the TME. They interact with PDAC cells via direct intercellular contact and through paracrine (ECM protein) pathways to confer GEM resistance (Figure 2).
Pancreatic stellate cells (PSCs) and CAFs
Quiescent PSCs characterized by stellate morphology and enriched in vitamin A are the main effector cells in PDAC desmoplasia. However, the origin of PSCs is still being debated, and mesenchymal, endodermal, and neuroectodermal origins are suggested (78). Quiescent PSCs can be activated from a static phenotype to CAFs by tumor necrosis factor α (TNF-α), TGF-β, interleukin 1 (IL-1), IL-2, IL-10, and PDGF (79). CAFs secrete ECM proteins that form fibrous matrix and hypoxic TME, promoting GEM resistance(79, 80). PSCs can also induce GEM resistance through direct contact with PDAC cells and paracrine cytokine signaling. In vitro coculture studies confirmed that PSCs activate the Notch signaling pathway by increasing Hes1 expression in PDAC cells. The Notch signaling pathway induces GEM resistance by promoting EMT and the CSC phenotype. PSC-induced chemoresistance can be effectively reversed when Hes1 gene expression or the Notch pathway is inhibited(81). Therefore, targeting the Hes/Notch signaling pathway may be an effective strategy to reverse GEM resistance. Most current in vitro studies use two-dimensional (2D) monolayer culture models, which cannot simulate the TME in vivo, such as cell-cell and cell-matrix interactions. The latest three-dimensional (3D) culture models can better simulate TME conditions in vivo; therefore, 3D models have become important tools for cancer research. An in vitro 3D coculture study by Firuzi et al. showed that PSCs can bind to the receptor tyrosine kinase c-Met in PDAC cells and induce c-Met phosphorylation by secreting hepatocyte growth factor (HGF). c-Met induces GEM resistance by activating the PI3K/Akt/mTOR prosurvival pathway (82). Preclinical studies of a c-Met-targeting inhibitor combined with GEM reported significantly improved efficacy(82, 83), and a phase I clinical trial (NCT00874042) also confirmed that the combination of these two had good safety and tolerability(84). Therefore, targeted inhibition of the HGF/c-Met pathway may be a promising strategy for GEM sensitization. Recently, a study by Dalin et al. showed that PSCs protect PDAC from GEM toxicity by secreting deoxycytidine (dC), but the specific mechanism remains unclear (85). This process might be related to the competition with deoxycytidine kinase (dCK) in GEM-treated cells (Figure 1). Therefore, stromal dC may also be a potential target for GEM sensitization.
In PDAC, most CAFs are derived from the activation of PSCs; a small portion are derived from resting fibroblasts, mesenchymal stem cells, or the EMT of tumor cells (86). Activated CAFs are characterized by the expression of α-smooth muscle actin (α-SMA) and fibroblast activation protein (FAP). They are naturally resistant to GEM and may acquire drug resistance through various mechanisms (87). However, studies in a transgenic mouse model showed that targeted depletion of α-SMA in CAFs did not enhance the efficacy of GEM in PDAC but upregulated the expression of cytotoxic T lymphocyte associated antigen-4 (CTLA-4); notably, antibody therapy targeting CTLA-4 can improve OS(88). A recent study by Principe et al. showed that pretreatment with GEM enhanced the expression of programmed death-1 (PD-1) and subsequently elevated sensitivity to combination PD-1 immunotherapy(89). Therefore, GEM might increase the expression of immune targets by remodeling of the TME in PDAC. Combination immunotherapy may be a potential second-line treatment of GEM-resistant PDAC. Although the targeted inhibition of FAP improved the efficacy of chemotherapy in a preclinical model (90), a phase II clinical trial by Nugent et al. showed that the efficacy of combination therapy with a FAP inhibitor and GEM for the treatment of metastatic PDAC was very limited(91).
In addition, the Hedgehog (HH) signaling pathway and one of its ligands, sonic hedgehog (SHH), are potent regulators of CAF activation and promote proliferation and repair of PDAC cells; however, their role in the induction of GEM resistance remains controversial (25). Although preclinical studies demonstrated that small-molecule inhibitors of the SHH pathway sensitized tumors to GEM (92, 93), subsequent phase II clinical trials of GEM combination therapy for the treatment of advanced PDAC failed(5, 94). In addition, genomic analyses indicated that inflammatory mediators such as IL-6 were overexpressed in GEM-resistant CAFs and that drug resistance was positively correlated with IL-6 levels(95), suggesting that CAF-induced GEM resistance might be linked to proinflammatory pathways(96). In addition, IL-6 can also induce GEM resistance through the classical IL-6/JAK/STAT3 pathway, and targeted inhibition of the JAK/STAT3 axis improves drug delivery and upregulates CDA via stromal remodeling, ultimately sensitizes tumors to GEM in both in vitro and ex vivo models (97, 98). Therefore, targeting the IL-6/JAK/STAT3 axis may be an effective strategy to reverse GEM resistance. Interestingly, Ohlund et al. used a 3D coculture platform to characterize CAFs and revealed that two subtypes of CAFs with different levels of α-SMA and IL-6 expression have opposite effects in PDAC. This study emphasized the heterogeneity of stromal CAFs(99), which may account for the failure of the above clinical trial(87). Recently, studies by Richards et al. have shown that GEM-resistant CAFs release significantly more extracellular vesicles known as exosomes. These exosomes may promote EMT and ultimately induce GEM resistance by activating the mesenchymal transcription factor Snail(100). miRNA-106b in exosomes may play an important role in this process(101). The treatment of GEM-resistant CAFs with exosomal release inhibitors or miRNA-106b inhibitors significantly reduced the survival rate of cocultured PDAC cells(100, 101), indicating that exosomes might be potential targets for overcoming GEM resistance.
CSCs
CSCs are a special group of cells that have characteristics of PDAC stem cells, which maintain tumor formation and growth. The role of CSCs in GEM resistance has gradually attracted attention, but the exact cellular and molecular mechanisms have not been fully elucidated(102). In vitro studies have shown that CSCs may escape the cytotoxic effect of GEM by EMT (103). HOX transcript antisense RNA (HOTAIR) is a class of long noncoding RNAs (lncRNAs) produced by GEM-induced CSCs; HOTAIR can induce GEM resistance by promoting proliferation and inhibiting apoptosis(104). Recently, Yang et al. showed that CSCs can regulate miR-210 expression in noncoding microRNA (miRNA) and increase the release of exosomal miR-210, which further promotes GEM resistance in PDAC cells by activating the prosurvival PI3K/Akt/mTOR pathway(105). Therefore, targeting CSCs and related noncoding RNAs (ncRNAs) is expected to become a new GEM sensitization strategy.
GEM resistance and immune cells
Inflammatory cell infiltration and immunosuppression are characteristic of PDAC(106). Immune cells in PDAC are not innocent bystanders of GEM resistance but are important participants in this process (Figure 2).
Myeloid-derived suppressor cells (MDSCs)
MDSCs are heterogeneous immune cell populations derived from myeloid progenitor cells, and the granulocytic myeloid-derived suppressor cell (G-MDSC) is the most widely distributed subtype in tumors(107). MDSCs can accelerate tumor progression by inhibiting T cell immunity and promoting angiogenesis(108). Therefore, MDSCs are also known as T cell-inhibiting neutrophils. An in vitro study by Takeuchi et al. showed that through activation of the MAPK and NF-κB pathways, GEM stimulates the release of granulocyte-macrophage colony-stimulating factor (GM-CSF), a major cytokine associated with KRAS mutations, which can further enhance MDSC differentiation in PDAC and promote drug resistance. Targeted neutralization of GM-CSF using antibodies can effectively reduce the proportion of MDSCs, help restore T cell function, and reverse GEM resistance(109). Notably, GM-CSF can also induce or activate antitumor T cell immunity by stimulating dendritic cells; a GM-CSF transduced pancreatic cancer vaccine (GVAX) was designed based on this principle(110). Regardless, GM-CSF may function both positively and negatively in the TME of PDAC, which may be one of the reasons for the failure of a phase III clinical trial (ISRCTN4382138) testing the treatment of advanced PDAC with GM-CSF + telomerase vaccine (GV1001) with or without GEM(111). It should be noted that due to the lack of specific markers, MDSCs cannot be phenotypically identified. Therefore, the comparability and reliability of relevant studies need to be further confirmed(107, 112).
Tumor-associated macrophages (TAMs)
The phagocytosis of apoptotic cells is the main function of TAMs. In addition, M2-polarized TAMs can directly enhance the tumor initiation ability of CSCs by activating the transcription factor STAT3(113) and inhibit GEM-induced apoptosis by downregulating the caspase-3 pathway(114) to promote GEM resistance. In vitro studies have shown that the targeted inhibition of colony-stimulating factor-1 receptor (CSF-1R) or C-C motif chemokine receptor 2 (CCR2) can selectively deplete TAMs and restore the efficacy of GEM(113, 114). Moreover, activated STAT3 can also promote GEM resistance by upregulating the expression of cytidine deaminase (CDA; Figure 1) and promoting matrix remodeling(97, 114). Studies in animal models have shown that when combined with specific STAT3 inhibitors, the efficacy of GEM significantly improves(97). Therefore, targeting TAMs/STAT3 may be a potential GEM sensitization strategy(115, 116). Notably, TAMs have high plasticity and can be polarized into not only M2 macrophages, which promote tumorigenesis and confer chemotherapy resistance, but also M1 macrophages, which are antitumorigenic and confer chemotherapy sensitivity (117). Therefore, directed polarization of TAMs is another important pathway of GEM sensitization in addition to the depletion of TAMs(118, 119).
Tumor-associated neutrophils (TANs)
TANs are key regulators of inflammation and immune status in PDAC(120). Similar to TAMs, TANs can also be polarized into antitumor (N1) or tumorigenic (N2) phenotypes by different chemokines in the TME(121). Although the number and proportion of TANs are recognized as prognostic biomarkers for solid tumors including PDAC(122), the relationship between TANs and GEM resistance remains unknown. Two recent studies have indicated that the binding of CXCL2/5 to its receptor CXCR2 is a key to promote N2 differentiation of TANs in in vitro and in vivo models of PDAC. Targeted inhibition of the CXCL2/CXCR2 axis can activate NF-κB prosurvival pathways, promote functional T cell infiltration, and enhance antitumor immunity and chemotherapy (FOLFIRINOX) sensitivity(123, 124). Nevertheless, CXCR2 is not a TAN-specific marker, which may be an enormous obstacle to current clinical trials. Therefore, it is necessary to identify a definitive TAN-specific phenotype(125). Nonetheless, studies on the plasticity of TANs have gradually become a hot area of investigation(112, 125), and TANs may become a potential target for GEM sensitization.
Tumor infiltrating lymphocytes (TILs)
TILs include cytotoxic T lymphocytes (CTLs), helper T cells (Th), regulatory T cells (Tregs), and many other subtypes, which are key mediators in both antitumor and regulatory immunity. Among them, PD-1 and CTLA-4 are suppressive immune checkpoint receptors expressed on TILs and can negatively regulate their antitumor functions(126). The high level of CTL infiltration is positively correlated with the favorable prognosis in patients with PDAC(127), providing the theoretical basis of targeted blockade of immune checkpoints. However, single-inhibitor therapy targeting the PD-1/L1 and CTLA-4 immune checkpoints has not been shown to be effective for PDAC(128). A heterologous mouse pancreatic tumor model study by Winograd et al. showed that GEM increased the sensitivity to PD-1 and CTLA-4 inhibitors by inducing T cell immunity and that GEM combined with immune checkpoint inhibitors improved the survival rate of patients with PDAC(129). However, the actual efficacy in patients still needs validation in clinical trials (130). Another potential target is HEAT repeat-containing protein 1 (HEATR1). HEATR1 suppresses the Akt signaling pathway, which exerts antitumor immune effects by inducing CTLs. However, HEATR1 is generally downregulated in PDAC and closely associated with GEM resistance(131). Zhou et al. showed that downregulation of HEATR1 activated nuclear factor erythroid-2-related factor 2 (Nrf2) signal transduction, which promotes the transcription of downstream antioxidative and cytoprotective genes, ultimately leading to GEM resistance (132). Therefore, HEATR1/Nrf2 may be a potential target for GEM chemotherapy sensitization.
Platelets (PLTs)
The risk of blood hypercoagulation and venous thromboembolism (VTE) is one of the characteristics of PDAC(133). PDAC cells can induce PLT activation and aggregation, while PDGF secreted by PLTs contributes to GEM resistance(134–137). Adenosine diphosphate (ADP) is an important PLT agonist in PDAC and has hypoxic characteristics. Recently, ADP-P2Y12 receptors on PLTs have attracted increasing attention (138). P2Y12 is a P2 purinergic receptor, a class of G-protein-coupled receptors (GPCRs). In PLTs, binding of ADP to the P2Y12 receptor can upregulate the expression of EMT-related transcription factors such as Slug and zinc finger E-box-binding homeobox 1 (ZEB1) and downregulate the expression of the GEM transporter hENT1 and the GEM-metabolizing enzyme CDA, resulting in GEM resistance(136, 137). Preclinical studies have confirmed that ADP-P2Y12 axis inhibitors (ticagrelor) significantly synergize with GEM in the treatment of PDAC but not as monotherapy (137). A clinical trial (NCT02404363) of the treatment of PDAC with clopidogrel (P2Y12 inhibitor) combined with GEM is ongoing, and the results are promising. In addition, the inhibition of P2Y12 signaling significantly enhances the efficacy of anti-EGFR therapy(137). This process may also be related to matrix remodeling, suggesting that P2Y12 inhibitors + GEM combined with EGFR-TKIs might be a potential strategy for the treatment of PDAC.
Conclusions and perspectives
The TME, which is characterized by extensive connective tissue proliferation in PDAC, not only promotes tumor proliferation and metastasis but also plays a key role in the induction of GEM resistance (Figure 3). Unfortunately, most therapies targeting TME-related molecular pathways have not achieved satisfactory results (139)(Table 1), and even the physical barrier function of the stroma has been questioned(140). The key reasons for this disappointing outcome may include the heterogeneity of PDAC cells and their TME, possible bidirectional effects of the TME, a lack of effective selective inhibitors, and the rapid upregulation of alternative compensatory pathways. Recent technological advances in single-cell analysis have enabled analysis of cell heterogeneity in tumor samples. Single-cell analysis can reveal not only the behavior of individual cells but also interactions between cells in the TME, and this approach has been employed to investigate intratumoral heterogeneity, to identify cell subsets of PDAC, and to probe mechanisms of chemotherapy resistance (141). It should also be pointed out that most of the currently available evidence is from in vitro experimental models. Although there have been advances in in vitro modeling, the latest 3D or organoid culture models still fall short of fully reproducing the complex biological interactions between PDAC cells and the TME in preclinical studies (142). Similarly, inherent differences between species and discordances between murine xenotransplant models should also be noted (143, 144). In short, the results of preclinical studies in in vitro culture or in vivo animal models should be interpreted with great caution. Therefore, the issues listed above should be the key to enhance the efficacy of GEM chemotherapy and improve the overall prognosis of PDAC in the future. In-depth studies of the effect of GEM on TME remodeling are expected to guide multitarget combination therapy, which may eventually improve the future treatment model for patients with PDAC.
Figure 3. Schematic diagram of the mechanism of TME-induced resistance to GEM in PDAC cells.
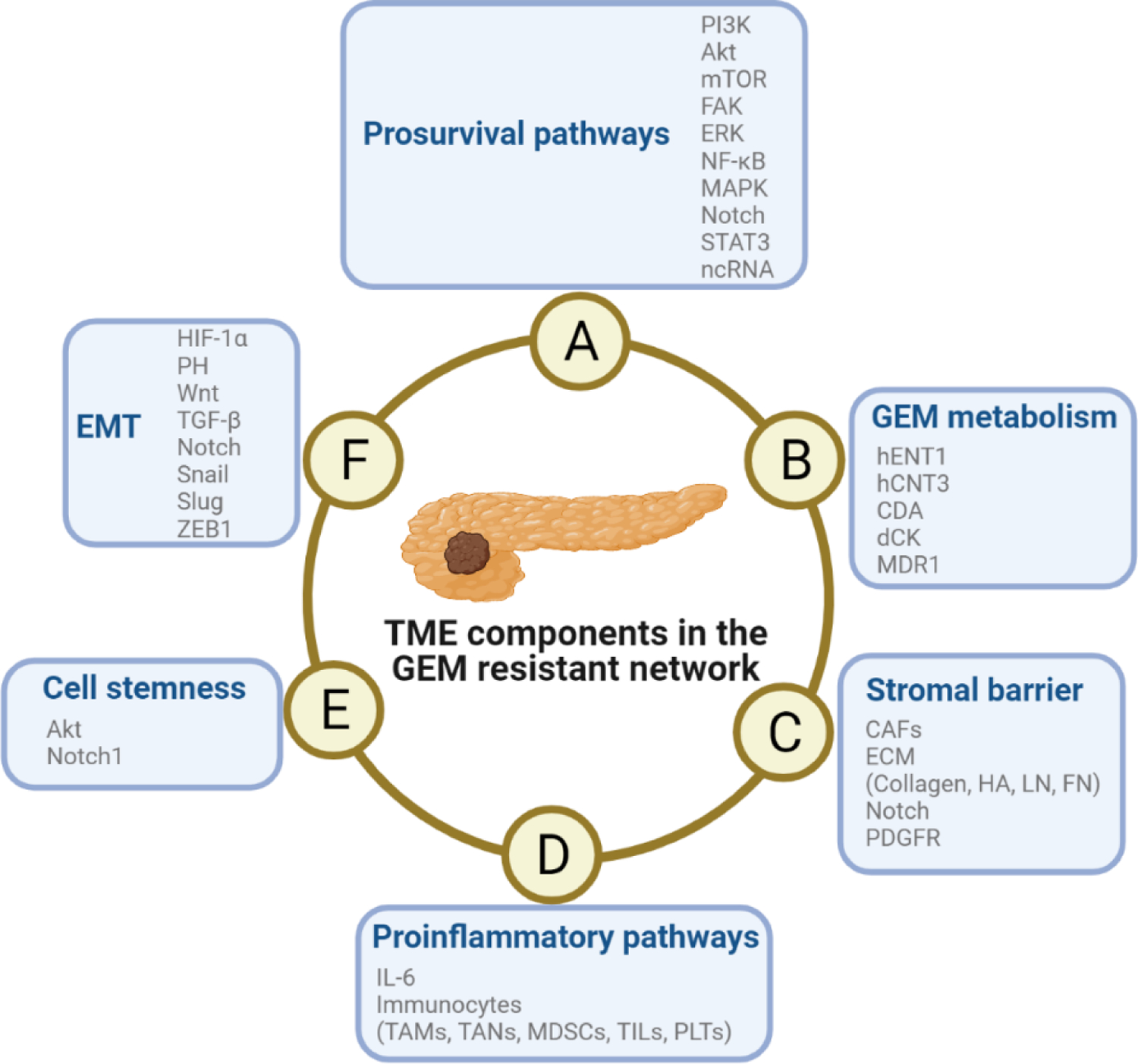
The pathways are not independent from each other, but they engage in crosstalk and promote the development of GEM resistance. Abbreviations: EMT, epidermal-mesenchymal transition; GEM, gemcitabine; TME, tumor microenvironment.
Table 1.
Potential targets and mechanisms of TME-induced resistance to GEM in PDAC cells.
| Potential target | Drug resistance mechanism | Preclinical models | Clinical trials | Literature |
|---|---|---|---|---|
| Hypoxia/HIF-1α | Induce the EMT; increase glycolysis and dCTP levels; activate the Akt/Notch1 pathway; induce the CSC phenotype | Cell lines/mouse models | NCT01746979 | [12-16] |
| CA9/Acidosis | Increase glycolysis; change intracellular and extracellular pH; induce the EMT | Cell lines/mouse models | NA | [18-20] |
| MT1-MMP/HMGA2/HATs | Stromal barrier; increase H3K9 and H3K27 acetylation; repair damaged DNA | Cell lines/mouse models | NA | [10,22-25] |
| TIMP1 | Activate the PI3K/Akt l pathway | Cell lines/mouse models | NA | [26] |
| HA/CD44 | Increase IFP; upregulate MDR1 expression; induce the EMT | Cell lines/mouse models |
NCT01839487 NCT02715804 HALO109–301 |
[29-36,39] |
| TG2/LN/FAK | Stromal barrier; activate the PI3K/Akt and NF-κB pathways | Cell lines/mouse models | NA | [41,42] |
| FN/ERK | Stromal barrier; activate the ERK pathway; inhibit apoptosis | Cell lines/mouse models | NA | [44,45] |
| CTGF/XIAP | Inhibit the formation of caspase-3/7/9-mediated apoptotic vesicles | Cell lines/mouse models | NCT02210559 | [46-48] |
| TGF-β/CYR61/VAV1 | Activate the TGF-β/ALK5/Smad2/3 pathway; downregulate hENT1 and hCNT3 expression; induce the EMT | Cell lines/mouse models | NA | [49-52] |
| HAb18G/EGFR/STAT3 | Activate the EGFR/STAT3 pathway | Cell lines/mouse models | CONKO005 ISRCTN96397434 NCT00471146 |
[54-60] |
| IGF-1R | Activate the PI3K/Akt/mTOR and MEK/ERK pathways | Cell lines/mouse models |
NCT00769483
NCT01231347 |
[62-65] |
| PDGFR | Increase IFP; activate the PI3K/Akt, mTOR, NF-κB, ERK, MAPK, and Notch pathways; induce the EMT | Cell lines/mouse models | BAYPAN | [66-70] |
| CXCL12/CXCR4 | Activate the FAK, ERK, and Akt pathways; upregulate survivin expression | Cell lines | NA | [73-75] |
| Hes1/Notch | Stromal barrier; activate the Notch pathway; induce the EMT and CSC phenotype | Cell lines/mouse models | NA | [79-81] |
| HGF/c-Met | Activate the PI3K/Akt/mTOR pathway | Cell lines/mouse models | NCT00874042 | [82-84] |
| dC | Intracellular dCK competition | Organoid cell culture | NA | [85] |
| α-SMA-CAFs | Increase hypoxia; induce the EMT and CSC phenotype; stromal barrier | Mouse models | NA | [87,88] |
| FAP-CAFs | Remodel the ECM; stromal barrier | Cell lines/mouse models | *Ref. [91] | [90,91] |
| SHH | Activate the HH pathway | Cell lines/mouse models |
NCT01064622
NCT01088815 |
[5,92-94] |
| IL-6 | Activate the IL-6/JAK/STAT3 pathway; promote proinflammatory pathways | Cell lines/mouse models | NA | [95-98] |
| Exosome | Activate Snail; induce the EMT | Cell lines | NA | [100,101] |
| CSCs/ncRNA | Induce the EMT; inhibit apoptosis; activate the PI3K/Akt/mTOR pathway | Cell lines | NA | [103-105] |
| GM-CSF | Activate the MAPK and NF-κB pathways; promote MDSCs differentiation | Cell lines/mouse models | ISRCTN4382138 | [109-111] |
| TAMs/STAT3 | Activate STAT3; induce the CSC phenotype; inhibit apoptosis; upregulate CDA expression; M2 polarization | Cell lines/mouse models | NA | [113-119] |
| CXCL2,5/CXCR2 | Activate NF-κB pathway; promote functional T cell infiltration | Cell lines/mouse models | NA | [123,124] |
| PD-1/L1, CTLA-4 | Inhibit the cytotoxicity of CTLs | Cell lines/mouse models | *Ref. [130] | [129,130] |
| HEATR1/Nrf2 | Negatively regulate the Akt pathway; activate effector CTLs; negatively regulate the transcription of downstream antioxidative and cytoprotective genes | Cell lines/mouse models | NA | [131,132] |
| ADP-P2Y12 | Upregulate Slug and ZEB1 expression; induce the EMT; downregulate hENT1 and CDA expression; activate the EGFR pathway | Cell lines/mouse models | NCT02404363 | [136,137] |
Modified from our previous work (139) with permission. *Registration number cannot be retrieved from the website “clinicaltrials.gov“. NA, Not Applicable. Abbreviations: HIF-1α, hypoxia-inducible factor-1α; CA9, carbonic anhydrase 9; MT1-MMP, membrane type 1-matrix metalloproteinase; HMGA2, high mobility group A2; HATs, histone acetyltransferases; TIMP1, tissue inhibitor of metalloproteinases 1; HA, hyaluronic acid; TG2, transglutaminase 2; LN, laminin; FAK, focal adhesion kinase; FN, fibronectin; ERK, extracellular regulated protein kinases; CTGF, connective tissue growth factor; XIAP, X-linked inhibitor of apoptosis protein X; TGF-β, transforming growth factor β; CYR61, cysteine-rich angiogenic inducer 61; VAV1, Vav guanine nucleotide exchange factor 1; EGFR, epidermal growth factor receptor; STAT3, signal transducer and activator of transcription 3; IGF-1R, insulin-like growth factor 1 receptor; PDGFR, platelet-derived growth factor receptor; CXCL, CXC ligand; CXCR, CXC chemokine receptor; HGF, hepatocyte growth factor; dC, deoxycytidine; dCK, deoxycytidine kinase; α-SMA, α-smooth muscle actin; FAP, fibroblast activation protein; SHH, sonic hedgehog pathway; ncRNA, non-coding RNA; GM-CSF, granulocyte-macrophage colony-stimulating factor; PD-1, programmed death-1; CTLA-4, cytotoxic T lymphocyte associated antigen-4; HEATR1, HEAT repeat-containing protein 1; Nrf2, nuclear factor erythroid-2-related factor 2; ADP, adenosine diphosphate.
Acknowledgments
We thank American Journal Experts (AJE) for assisting in the preparation of this manuscript.
Funding
This work was supported by NSFC 81972314 and SC1 GM135050–06.
Footnotes
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest statement
All authors have completed the ICMJE uniform disclosure form. The authors have declared no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: a cancer journal for clinicians 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Mizrahi JD, Surana R, Valle JW, et al. Pancreatic cancer. Lancet 2020;395(10242):2008–20. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. New England Journal of Medicine 2018;379(25):2395–406. [DOI] [PubMed] [Google Scholar]
- 4.Saif MW, Lee Y, Kim R. Harnessing gemcitabine metabolism: a step towards personalized medicine for pancreatic cancer. Ther Adv Med Oncol 2012;4(6):341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catenacci DV, Junttila MR, Karrison T, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol 2015;33(36):4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369(18):1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin C, Yang G, Yang J, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer 2020;19(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apte MV, Park S, Phillips PA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 2004;29(3):179–87. [DOI] [PubMed] [Google Scholar]
- 9.Neesse A, Algul H, Tuveson DA, et al. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 2015;64(9):1476–84. [DOI] [PubMed] [Google Scholar]
- 10.Neesse A, Bauer CA, Ohlund D, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut 2019;68(1):159–71. [DOI] [PubMed] [Google Scholar]
- 11.Lohse I, Lourenco C, Ibrahimov E, et al. Assessment of hypoxia in the stroma of patient-derived pancreatic tumor xenografts. Cancers (Basel) 2014;6(1):459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang R, Cheng L, Xia J, et al. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1α in pancreatic cancer cells. Current Cancer Drug Targets 2014;14(4):407–17. [DOI] [PubMed] [Google Scholar]
- 13.Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017;32(1):71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou XF, Li S, Wu C, et al. Effects of obatoclax combined with gemcitabine on the biological activity of pancreatic cancer cells under hypoxic conditions. Mol Med Rep 2018;18(1):495–501. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Han H, Rong Y, et al. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J Exp Clin Cancer Res 2018;37(1):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cutsem EV, Lenz H-J, Furuse J, et al. MAESTRO: A randomized, double-blind phase III study of evofosfamide (Evo) in combination with gemcitabine (Gem) in previously untreated patients (pts) with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma (PDAC). Journal of Clinical Oncology 2016;34(15_suppl):4007-. [Google Scholar]
- 17.Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer 2017;17(10):577–93. [DOI] [PubMed] [Google Scholar]
- 18.McDonald PC, Chafe SC, Brown WS, et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia. Gastroenterology 2019;157(3):823–37. [DOI] [PubMed] [Google Scholar]
- 19.Riemann A, Rauschner M, Giesselmann M, et al. Extracellular Acidosis Modulates the Expression of Epithelial-Mesenchymal Transition (EMT) Markers and Adhesion of Epithelial and Tumor Cells. Neoplasia 2019;21(5):450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ling Q, Kalthoff H. Transportome Malfunctions and the Hallmarks of Pancreatic Cancer. Rev Physiol Biochem Pharmacol 2020. [DOI] [PubMed] [Google Scholar]
- 21.Makareeva E, Han S, Vera JC, et al. Carcinomas contain a matrix metalloproteinase-resistant isoform of type I collagen exerting selective support to invasion. Cancer Res 2010;70(11):4366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dangi-Garimella S, Krantz SB, Barron MR, et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res 2011;71(3):1019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Summer H, Li O, Bao Q, et al. HMGA2 exhibits dRP/AP site cleavage activity and protects cancer cells from DNA-damage-induced cytotoxicity during chemotherapy. Nucleic Acids Res 2009;37(13):4371–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dangi-Garimella S, Sahai V, Ebine K, et al. Three-dimensional collagen I promotes gemcitabine resistance in vitro in pancreatic cancer cells through HMGA2-dependent histone acetyltransferase expression. PLoS One 2013;8(5):e64566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol 2020;17(9):527–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Costa Z, Jones K, Azad A, et al. Gemcitabine-Induced TIMP1 Attenuates Therapy Response and Promotes Tumor Growth and Liver Metastasis in Pancreatic Cancer. Cancer Res 2017;77(21):5952–62. [DOI] [PubMed] [Google Scholar]
- 27.Liang C, Shi S, Meng Q, et al. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: where we are and where we are going. Exp Mol Med 2017;49(12):e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakazawa H, Yoshihara S, Kudo D, et al. 4-methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemother Pharmacol 2006;57(2):165–70. [DOI] [PubMed] [Google Scholar]
- 29.Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer 2013;108(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao S, Chen C, Chang K, et al. CD44 Expression Level and Isoform Contributes to Pancreatic Cancer Cell Plasticity, Invasiveness, and Response to Therapy. Clin Cancer Res 2016;22(22):5592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong SP, Wen J, Bang S, et al. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer 2009;125(10):2323–31. [DOI] [PubMed] [Google Scholar]
- 32.Skandalis SS, Karalis TT, Chatzopoulos A, et al. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell Signal 2019;63:109377. [DOI] [PubMed] [Google Scholar]
- 33.Serri C, Quagliariello V, Iaffaioli RV, et al. Combination therapy for the treatment of pancreatic cancer through hyaluronic acid-decorated nanoparticles loaded with quercetin and gemcitabine: A preliminary in vitro study. J Cell Physiol 2019;234(4):4959–69. [DOI] [PubMed] [Google Scholar]
- 34.Wong KM, Horton KJ, Coveler AL, et al. Targeting the Tumor Stroma: the Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr Oncol Rep 2017;19(7):47. [DOI] [PubMed] [Google Scholar]
- 35.Thompson B, Lee J, Clift R, et al. PO-262 Remodelling of the tumour microenvironment by pegvorhyalurondiase alfa (PEGPH20): a novel, first-in-class biologic that enzymatically degrades tumour hyaluronan (HA) to improve anti-tumour efficacy. ESMO Open 2018;3(Suppl 2):A329–A30. [Google Scholar]
- 36.Hingorani SR, Zheng L, Bullock AJ, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol 2018;36(4):359–66. [DOI] [PubMed] [Google Scholar]
- 37.Cao J, Pickup S, Clendenin C, et al. Dynamic Contrast-enhanced MRI Detects Responses to Stroma-directed Therapy in Mouse Models of Pancreatic Ductal Adenocarcinoma. Clin Cancer Res 2019;25(7):2314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubey RD, Klippstein R, Wang JT, et al. Novel Hyaluronic Acid Conjugates for Dual Nuclear Imaging and Therapy in CD44-Expressing Tumors in Mice In Vivo. Nanotheranostics 2017;1(1):59–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Cutsem E, Tempero MA, Sigal D, et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J Clin Oncol 2020;38(27):3185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi S, Hasebe T, Oda T, et al. Cytoplasmic expression of laminin gamma2 chain correlates with postoperative hepatic metastasis and poor prognosis in patients with pancreatic ductal adenocarcinoma. Cancer 2002;94(6):1894–901. [DOI] [PubMed] [Google Scholar]
- 41.Huanwen W, Zhiyong L Fau - Xiaohua S, Xiaohua S Fau - Xinyu R, et al. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol Cancer 2009(8):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J, Yakubov B, Ivan C, et al. Tissue Transglutaminase Activates Cancer-Associated Fibroblasts and Contributes to Gemcitabine Resistance in Pancreatic Cancer. Neoplasia 2016;18(11):689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Topalovski M, Brekken RA. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett 2016;381(1)(1872–7980 (Electronic)):252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amrutkar M, Aasrum M, Verbeke CS, et al. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019;19(1):596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xavier CPR, Castro I, Caires HR, et al. Chitinase 3-like-1 and fibronectin in the cargo of extracellular vesicles shed by human macrophages influence pancreatic cancer cellular response to gemcitabine. Cancer Lett 2021;501:210–23. [DOI] [PubMed] [Google Scholar]
- 46.Neesse A, Frese KK, Bapiro TE, et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci U S A 2013;110(30):12325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moon JH, Shin JS, Hong SW, et al. A novel small-molecule IAP antagonist, AZD5582, draws Mcl-1 down-regulation for induction of apoptosis through targeting of cIAP1 and XIAP in human pancreatic cancer. Oncotarget 2015;6(29):26895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picozzi VJ, Rocha FG, Helton S, et al. Randomized, open-label trial of gemcitabine/nab-paclitaxel (G/NP) ± pamrevlumab (P) as neoadjuvant chemotherapy in locally advanced, unresectable pancreatic cancer (LAPC). Journal of Clinical Oncology 2017;35(4_suppl):365-. [Google Scholar]
- 49.Hesler RA, Huang JJ, Starr MD, et al. TGF-beta-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016;37(11):1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xian G, Zhao J, Qin C, et al. Simvastatin attenuates macrophage-mediated gemcitabine resistance of pancreatic ductal adenocarcinoma by regulating the TGF-beta1/Gfi-1 axis. Cancer Lett 2017;385:65–74. [DOI] [PubMed] [Google Scholar]
- 51.Pei Y, Chen L, Huang Y, et al. Sequential Targeting TGF-beta Signaling and KRAS Mutation Increases Therapeutic Efficacy in Pancreatic Cancer. Small 2019;15(24):e1900631. [DOI] [PubMed] [Google Scholar]
- 52.Huang PH, Lu PJ, Ding LY, et al. TGFβ promotes mesenchymal phenotype of pancreatic cancer cells, in part, through epigenetic activation of VAV1. Oncogene 2017;36(16):2202–14. [DOI] [PubMed] [Google Scholar]
- 53.Tortora G, Ciardiello F, Gasparini G. Combined targeting of EGFR-dependent and VEGF-dependent pathways: rationale, preclinical studies and clinical applications. Nat Clin Pract Oncol 2008;5(9):521–30. [DOI] [PubMed] [Google Scholar]
- 54.Dosch AR, Dai X, Reyzer ML, et al. Combined Src/EGFR Inhibition Targets STAT3 Signaling and Induces Stromal Remodeling to Improve Survival in Pancreatic Cancer. Mol Cancer Res 2020;18(4):623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu BQ, Fu ZG, Meng Y, et al. Gemcitabine enhances cell invasion via activating HAb18G/CD147-EGFR-pSTAT3 signaling. Oncotarget 2016;7(38):62177–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkatasubbarao K, Peterson L, Zhao S, et al. Inhibiting signal transducer and activator of transcription-3 increases response to gemcitabine and delays progression of pancreatic cancer. Mol Cancer 2013;12(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinn M, Bahra M, Liersch T, et al. CONKO-005: Adjuvant Chemotherapy With Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients After R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J Clin Oncol 2017;35(29):3330–7. [DOI] [PubMed] [Google Scholar]
- 58.Evans TRJ, Van Cutsem E, Moore MJ, et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann Oncol 2017;28(2):354–61. [DOI] [PubMed] [Google Scholar]
- 59.Middleton G, Palmer DH, Greenhalf W, et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): a prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol 2017;18(4):486–99. [DOI] [PubMed] [Google Scholar]
- 60.Kindler HL, Ioka T, Richel DJ, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol 2011;12(3):256–62. [DOI] [PubMed] [Google Scholar]
- 61.Lakkakula B, Farran B, Lakkakula S, et al. Small molecule tyrosine kinase inhibitors and pancreatic cancer-Trials and troubles. Semin Cancer Biol 2019;56:149–67. [DOI] [PubMed] [Google Scholar]
- 62.Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011;3(3):192–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Camblin AJ, Pace EA, Adams S, et al. Dual Inhibition of IGF-1R and ErbB3 Enhances the Activity of Gemcitabine and Nab-Paclitaxel in Preclinical Models of Pancreatic Cancer. Clin Cancer Res 2018;24(12):2873–85. [DOI] [PubMed] [Google Scholar]
- 64.Fuchs CS, Azevedo S, Okusaka T, et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trial. Ann Oncol 2015;26(5):921–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abdel-Wahab R, Varadhachary GR, Bhosale PR, et al. Randomized, phase I/II study of gemcitabine plus IGF-1R antagonist (MK-0646) versus gemcitabine plus erlotinib with and without MK-0646 for advanced pancreatic adenocarcinoma. J Hematol Oncol 2018;11(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Q, Hou X, Xia J, et al. Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat Rev 2013;39(6):640–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kozono S, Ohuchida K, Eguchi D, et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res 2013;73(7):2345–56. [DOI] [PubMed] [Google Scholar]
- 68.Goncalves A, Gilabert M, Francois E, et al. BAYPAN study: a double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann Oncol 2012;23(11):2799–805. [DOI] [PubMed] [Google Scholar]
- 69.Moss RA, Moore D, Mulcahy MF, et al. A Multi-institutional Phase 2 Study of Imatinib Mesylate and Gemcitabine for First-Line Treatment of Advanced Pancreatic Cancer. Gastrointest Cancer Res 2012;5(3):77–83. [PMC free article] [PubMed] [Google Scholar]
- 70.Kindler HL, Wroblewski K, Wallace JA, et al. Gemcitabine plus sorafenib in patients with advanced pancreatic cancer: a phase II trial of the University of Chicago Phase II Consortium. Invest New Drugs 2012;30(1):382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sleightholm RL, Neilsen BK, Li J, et al. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol Ther 2017;179:158–70. [DOI] [PubMed] [Google Scholar]
- 72.Marechal R, Demetter P, Nagy N, et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer 2009;100(9):1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh S, Srivastava SK, Bhardwaj A, et al. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer 2010;103(11):1671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morimoto M, Matsuo Y, Koide S, et al. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: effect of CXCR4 antagonists. BMC Cancer 2016;16:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Costa MJ, Kudaravalli J, Ma JT, et al. Optimal design, anti-tumour efficacy and tolerability of anti-CXCR4 antibody drug conjugates. Sci Rep 2019;9(1):2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee S, Heinrich EL, Li L, et al. CCR9-mediated signaling through beta-catenin and identification of a novel CCR9 antagonist. Mol Oncol 2015;9(8):1599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Timaner M, Letko-Khait N, Kotsofruk R, et al. Therapy-Educated Mesenchymal Stem Cells Enrich for Tumor-Initiating Cells. Cancer Res 2018;78(5):1253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Erkan M, Adler G, Apte MV, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 2012;61(2):172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol 2016;22(9):2678–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu SL, Cao SG, Li Y, et al. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol Lett 2019;17(2):2057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cao F, Li J, Sun H, et al. HES 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncology Reports 2015;33(4):1883–9. [DOI] [PubMed] [Google Scholar]
- 82.Firuzi O, Che PP, El Hassouni B, et al. Role of c-MET Inhibitors in Overcoming Drug Resistance in Spheroid Models of Primary Human Pancreatic Cancer and Stellate Cells. Cancers (Basel) 2019;11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu Z, Pang TCY, Liu AC, et al. Targeting the HGF/c-MET pathway in advanced pancreatic cancer: a key element of treatment that limits primary tumour growth and eliminates metastasis. Br J Cancer 2020;122(10):1486–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pant S, Saleh M, Bendell J, et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann Oncol 2014;25(7):1416–21. [DOI] [PubMed] [Google Scholar]
- 85.Dalin S, Sullivan MR, Lau AN, et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res 2019;79(22):5723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nair N, Calle AS, Zahra MH, et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Scientific reports 2017;7(1):6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang H, Brekken RA. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am J Physiol Cell Physiol 2020;319(2):C233–C43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2015;28(6):831–3. [DOI] [PubMed] [Google Scholar]
- 89.Principe DR, Narbutis M, Kumar S, et al. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. Cancer research 2020;80(15):3101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li M, Li M, Yin T, et al. Targeting of cancerassociated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol Med Rep 2016;13(3):2476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nugent FW, Cunningham C, Barve MA, et al. Phase 2 study of talabostat/gemcitabine in Stage IV pancreatic cancer. Journal of Clinical Oncology 2007(25):4616.17925557 [Google Scholar]
- 92.Chun SG, Zhou W, Yee NS. Combined targeting of histone deacetylases and hedgehog signaling enhances cytoxicity in pancreatic cancer. Cancer Biol Ther 2009;8(14):1328–39. [DOI] [PubMed] [Google Scholar]
- 93.Khan MA, Srivastava SK, Zubair H, et al. Co-targeting of CXCR4 and hedgehog pathways disrupts tumor-stromal crosstalk and improves chemotherapeutic efficacy in pancreatic cancer. J Biol Chem 2020;295(25):8413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Jesus-Acosta A, Sugar EA, O’Dwyer PJ, et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer 2020;122(4):498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neumann CCM, von Horschelmann E, Reutzel-Selke A, et al. Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobiliary Pancreat Dis Int 2018;17(5):461–72. [DOI] [PubMed] [Google Scholar]
- 96.Toste PA, Nguyen AH, Kadera BE, et al. Chemotherapy-Induced Inflammatory Gene Signature and Protumorigenic Phenotype in Pancreatic CAFs via Stress-Associated MAPK. Mol Cancer Res 2016;14(5):437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagathihalli NS, Castellanos JA, Shi C, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015;149(7):1932–43 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018;15(4):234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214(3):579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Richards KE, Zeleniak AE, Fishel ML, et al. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017;36(13):1770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fang Y, Zhou W, Rong Y, et al. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp Cell Res 2019;383(1):111543. [DOI] [PubMed] [Google Scholar]
- 102.Gzil A, Zarębska I, Bursiewicz W, et al. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Molecular Biology Reports 2019;46(6):6629–45. [DOI] [PubMed] [Google Scholar]
- 103.Yin T, Wei H, Gou S, et al. Cancer stem-like cells enriched in Panc-1 spheres possess increased migration ability and resistance to gemcitabine. Int J Mol Sci 2011;12(3):1595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang L, Dong P, Wang W, et al. Gemcitabine treatment causes resistance and malignancy of pancreatic cancer stem-like cells via induction of lncRNA HOTAIR. Exp Ther Med 2017;14(5):4773–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang Z, Zhao N, Cui J, et al. Exosomes derived from cancer stem cells of gemcitabine-resistant pancreatic cancer cells enhance drug resistance by delivering miR-210. Cell Oncol (Dordr) 2020;43(1):123–36. [DOI] [PubMed] [Google Scholar]
- 106.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140(6):883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bergenfelz C, Leandersson K. The Generation and Identity of Human Myeloid-Derived Suppressor Cells. Front Oncol 2020;10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology 2009;9(3):162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takeuchi S, Baghdadi M, Tsuchikawa T, et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res 2015;75(13):2629–40. [DOI] [PubMed] [Google Scholar]
- 110.Nemunaitis J. Vaccines in cancer: GVAX, a GM-CSF gene vaccine. Expert Rev Vaccines 2005;4(3):259–74. [DOI] [PubMed] [Google Scholar]
- 111.Middleton G, Silcocks P, Cox T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. The Lancet Oncology 2014;15(8):829–40. [DOI] [PubMed] [Google Scholar]
- 112.Moses K, Brandau S. Human neutrophils: Their role in cancer and relation to myeloid-derived suppressor cells. Seminars in Immunology 2016;28(2):187–96. [DOI] [PubMed] [Google Scholar]
- 113.Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013;73(3):1128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weizman N, Krelin Y, Shabtay-Orbach A, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014;33(29):3812–9. [DOI] [PubMed] [Google Scholar]
- 115.Beltraminelli T, De Palma M. Biology and therapeutic targeting of tumour-associated macrophages. J Pathol 2020;250(5):573–92. [DOI] [PubMed] [Google Scholar]
- 116.Zou S, Tong Q, Liu B, et al. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer 2020;19(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ireland L, Santos A, Ahmed MS, et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Research 2016;76(23):6851–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bulle A, Dekervel J, Deschuttere L, et al. Gemcitabine Recruits M2-Type Tumor-Associated Macrophages into the Stroma of Pancreatic Cancer. Transl Oncol 2020;13(3):100743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yao L, Wang M, Niu Z, et al. Interleukin-27 inhibits malignant behaviors of pancreatic cancer cells by targeting M2 polarized tumor associated macrophages. Cytokine 2017;89:194–200. [DOI] [PubMed] [Google Scholar]
- 120.Lianyuan T, Gang L, Ming T, et al. Tumor associated neutrophils promote the metastasis of pancreatic ductal adenocarcinoma. Cancer Biol Ther 2020;21(10):937–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009;16(3):183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shen M, Hu P, Donskov F, et al. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS ONE 2014;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res 2016;4(11):968–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018;67(6):1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jaillon S, Ponzetta A, Di Mitri D, et al. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 2020;20(9):485–503. [DOI] [PubMed] [Google Scholar]
- 126.Zhou Q, Tao X, Xia S, et al. T Lymphocytes: A Promising Immunotherapeutic Target for Pancreatitis and Pancreatic Cancer? Front Oncol 2020;10:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Cancer Research 2013;73(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gong J, Hendifar A, Tuli R, et al. Combination systemic therapies with immune checkpoint inhibitors in pancreatic cancer: overcoming resistance to single-agent checkpoint blockade. Clin Transl Med 2018;7(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 2015;3(4):399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kamath SD, Kalyan A, Kircher S, et al. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020;25(5):e808–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu T, Fang Y, Zhang H, et al. HEATR1 Negatively Regulates Akt to Help Sensitize Pancreatic Cancer Cells to Chemotherapy. Cancer Res 2016;76(3):572–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou Y, Wang K, Zhou Y, et al. HEATR1 deficiency promotes pancreatic cancer proliferation and gemcitabine resistance by up-regulating Nrf2 signaling. Redox Biol 2020;29:101390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Delluc A, Rousseau A, Delluc C, et al. Venous thromboembolism in patients with pancreatic cancer: implications of circulating tissue factor. Blood Coagul Fibrinolysis 2011;22(4):295–300. [DOI] [PubMed] [Google Scholar]
- 134.Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res 2009;69(19):7775–83. [DOI] [PubMed] [Google Scholar]
- 135.Battinelli EM, Markens BA, Italiano JE Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood 2011;118(5):1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Elaskalani O, Falasca M, Moran N, et al. The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance. Cancers (Basel) 2017;9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Elaskalani O, Domenichini A, Abdol Razak NB, et al. Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells. Cancers (Basel) 2020;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ballerini P, Dovizio M, Bruno A, et al. P2Y12 Receptors in Tumorigenesis and Metastasis. Front Pharmacol 2018;9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.ZT G, ZZ L, CF W. Advances in research of extracellular mechanisms underlying gemcitabine resistance in pancreatic cancer. Shijie Huaren Xiaohua Zazhi 2021;29(8):421–34 (in Chinese). [Google Scholar]
- 140.Ramu I, Buchholz SM, Patzak MS, et al. SPARC dependent collagen deposition and gemcitabine delivery in a genetically engineered mouse model of pancreas cancer. EBioMedicine 2019;48:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ligorio M, Sil S, Malagon-Lopez J, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019;178(1):160–75 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Frappart PO, Hofmann TG. Pancreatic ductal adenocarcinoma (Pdac) organoids: The shining light at the end of the tunnel for drug response prediction and personalized medicine. Cancers 2020;12(10):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mazumdar C, Driggers EM, Turka LA. The Untapped Opportunity and Challenge of Immunometabolism: A New Paradigm for Drug Discovery. Cell Metab 2020;31(1):26–34. [DOI] [PubMed] [Google Scholar]
- 144.Kettler B, Trauzold A, Roder C, et al. Topology impacts TRAIL therapy: Differences in primary cancer growth and liver metastasis between orthotopic and subcutaneous xenotransplants of pancreatic ductal adenocarcinoma cells. Hepatobiliary Pancreat Dis Int 2021. [DOI] [PubMed] [Google Scholar]