

Abstract
Heterozygous mutations in six tRNA synthetase genes cause Charcot-Marie-Tooth (CMT) peripheral neuropathy. CMT-mutant tRNA synthetases inhibit protein synthesis by an unknown mechanism. Here, we found that CMT-mutant glycyl-tRNA synthetases (GlyRS) bound tRNAGly, but failed to release it, resulting in tRNAGly sequestration. This sequestration potentially depleted the cellular tRNAGly pool, leading to insufficient glycyl-tRNAGly supply to the ribosome. Accordingly, we found ribosome stalling at glycine codons and activation of the integrated stress response (ISR) in affected motor neurons. Moreover, transgenic overexpression of tRNAGly rescued protein synthesis, peripheral neuropathy, and ISR activation in Drosophila and mouse CMT2D models. Conversely, inactivation of the ribosome rescue factor GTPBP2 exacerbated peripheral neuropathy. Our findings suggest a molecular mechanism for CMT2D, and elevating tRNAGly levels may thus have therapeutic potential.
One Sentence Summary:
tRNAGly sequestration by mutant glycyl-tRNA synthetase triggers Charcot-Marie-Tooth peripheral neuropathy.
Heterozygous mutations in six genes encoding cytoplasmic aminoacyl-tRNA-synthetases (aaRSs) cause axonal and intermediate forms of CMT (1–3). aaRSs are ubiquitously expressed enzymes which covalently attach amino acids to their cognate tRNAs (tRNA aminoacylation) (4, 5). Aminoacylated tRNAs are used by the ribosome for mRNA translation (6). Interestingly, some CMT-aaRS mutations do not affect aminoacylation activity (7–11), indicating that loss of aminoacylation activity is not a prerequisite for disease-causality. Rather, a gain-of-toxic-function mechanism may underlie CMT associated with GlyRS mutations (CMT2D) (9, 12). In vivo cell-type-specific visualization of newly synthesized proteins in Drosophila (13) by fluorescent non-canonical amino acid tagging (FUNCAT) (14) revealed that each of six GlyRS or tyrosyl-tRNA synthetase (TyrRS) mutants substantially inhibited global protein synthesis in motor or sensory neurons (9), implicating impaired mRNA translation in CMT-aaRS. Here, we investigated the molecular mechanism by which CMT-mutant GlyRS variants inhibit translation. Manipulation of upstream regulatory pathways or translation initiation did not rescue inhibition of translation (Fig. S1), suggesting that CMT-mutant GlyRS may interfere with translation elongation. We thus evaluated the effect of tRNAGly overexpression, by generating Drosophila carrying a BAC transgene containing five tRNAGly genes with GCC anticodon (tRNAGly-GCC) (Fig. 1A). Flies with 10 or 20 additional tRNAGly-GCC gene copies displayed ~13% and ~25% increased tRNAGly-GCC levels, respectively (Fig. S2A,B). The 10xtRNAGly-GCC transgene partially rescued the translation defect (Fig. 1B, Fig. S3A) and peripheral neuropathy-like phenotypes induced by three CMT-mutant GlyRS proteins (E71G, G240R, G526R) (9) (Table S1), including larval muscle denervation (Fig. 1C, Fig. S3F), developmental lethality (Fig. S3B–E), adult motor deficits (Fig. 1D), sensory neuron morphology defects (Fig. S3G,H), and reduced life span (Fig. S3I). In general, phenotypic rescue was more pronounced for G240R and G526R than for E71G. tRNAGly-GCC overexpression did not alter GlyRS protein levels (Fig. S4), and did not rescue peripheral neuropathy phenotypes induced by CMT-mutant TyrRS (Fig. S5), indicating that only the cognate tRNA can rescue. Transgenic lines containing 10 different tRNAGly-GCC genes (tRNAGly-GCC ‘scramble’, Fig. 1A) induced a dosage-dependent increase in tRNAGly-GCC level, more pronounced than the BAC transgene (~30% for 10xtRNAGly-GCC, Fig. S2C,D), and a more substantial rescue of muscle denervation and motor performance (Fig. 1E,F). Thus, the degree of rescue correlated with tRNAGly-GCC overexpression level.
Fig. 1. tRNAGly overexpression rescues inhibition of protein synthesis and peripheral neuropathy phenotypes in Drosophila CMT2D models.
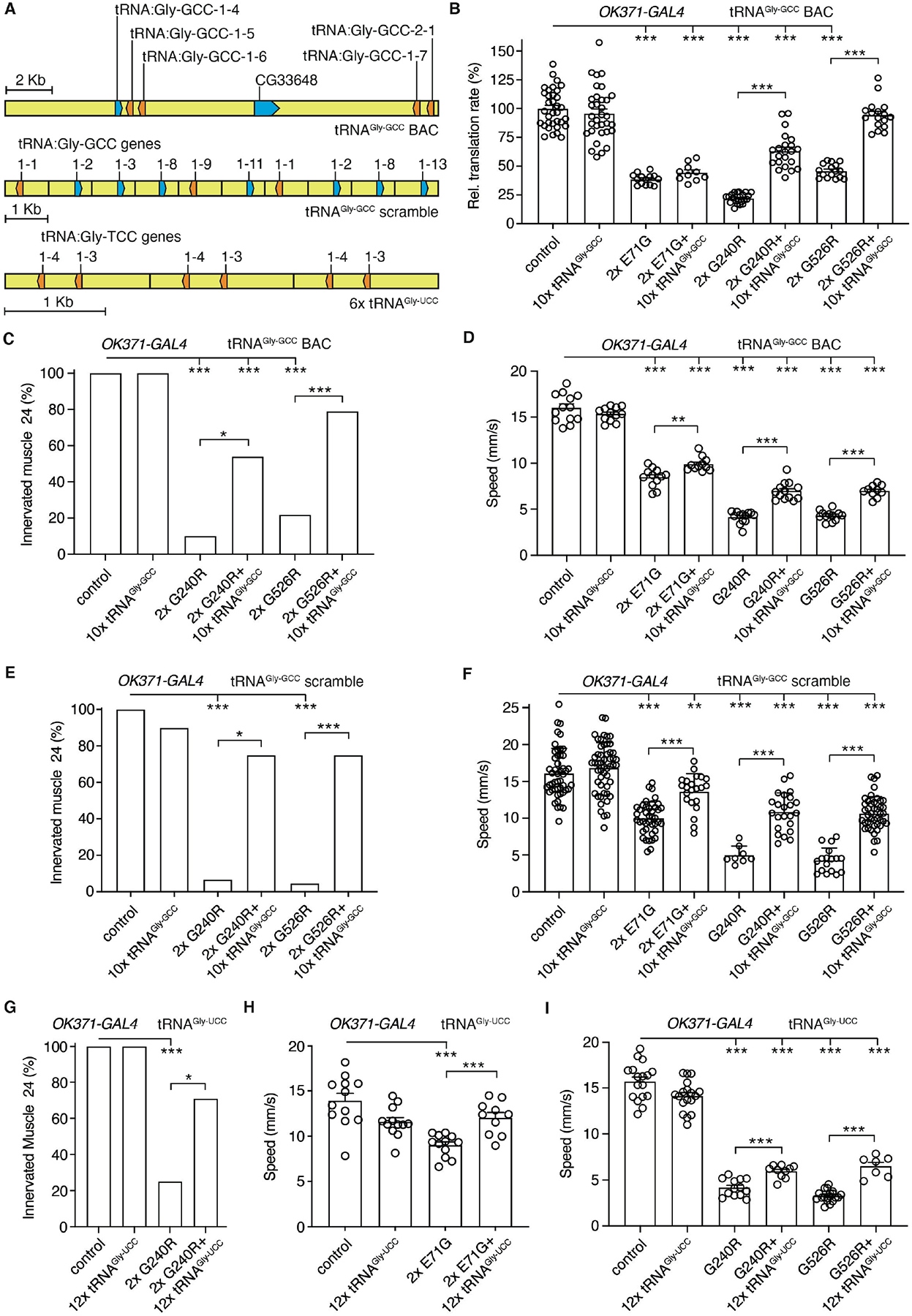
(A) Schematic of the transgenes used for tRNAGly-GCC or tRNAGly-UCC overexpression. (B) Relative translation rate as determined by FUNCAT in motor neurons (OK371-GAL4) of larvae expressing E71G, G240R, or G526R GlyRS (2x: two transgene copies), in the presence or absence of the tRNAGly-GCC BAC transgene (10xtRNAGly-GCC). n=10–34 animals per genotype; ***p<0.001 by Kruskal-Wallis test. (C,E,G) Percentage of larvae with innervated muscle 24. GlyRS transgenes were expressed in motor neurons (OK371-GAL4), in the presence or absence of 10xtRNAGly-GCC BAC (C), 10xtRNAGly-GCC scramble (E), or 12xtRNAGly-UCC (G). n=19–26 (C), 8–22 (E), 12–27 (G) animals per genotype; *p<0.05; ***p<0.005 by Fisher’s exact test with Bonferroni correction. (D,F,H,I) Negative geotaxis climbing speed of 7-day-old female flies expressing GlyRS transgenes in motor neurons (OK371-GAL4), in the presence or absence of 10xtRNAGly-GCC BAC (D), 10xtRNAGly-GCC scramble (F), or 12xtRNAGly-UCC (H,I). n=11–13 (D), 8–52 (F), 7–19 (H,I) groups of 10 flies per genotype; **p<0.01; ***p<0.005 by two-way ANOVA (D) or Brown-Forsythe and Welch ANOVA (F,H,I). Controls in (B-I) are driver-only. Graphs represent mean ± SEM.
We next generated transgenic lines overexpressing the other tRNAGly isoacceptor, tRNAGly-UCC (Fig. 1A). 12xtRNAGly-UCC flies displayed ~75% increased tRNAGly-UCC levels (Fig. S6A,B). For E71G and G240R, tRNAGly-UCC overexpression partially rescued developmental lethality (Fig. S6C–F), muscle denervation (Fig. 1G), motor deficits (Fig. 1H,I), and life span (Fig. S6I). For G526R, tRNAGly-UCC overexpression partially rescued motor performance (Fig. 1I), but aggravated sensory neuron morphology defects (Fig. S6G,H) and further reduced life span (Fig. S6I). Thus, for E71G and G240R, both tRNAGly-GCC and tRNAGly-UCC partially rescued peripheral neuropathy phenotypes, while for G526R, the rescue was isoacceptor-specific.
To strengthen the potential relevance for human CMT2D, we evaluated the effect of tRNAGly-GCC overexpression in CMT2D mouse models. We generated transgenic mice with ~27 (tRNAGly-high) or two (tRNAGly-low) copies of a genomic transgene containing two tRNAGly-GCC genes (Fig. 2A, Fig. S7A). In spinal cord (SC), tibialis anterior muscle and sciatic nerve of tRNAGly-high mice, tRNAGly-GCC levels were increased by ~90 to 150% (Fig. S7B–G). Targeted locus amplification (TLA) revealed integration of all transgene copies in Stk38 on Chr17, with a ~7kb deletion at the integration site, deleting exons 8–12 of Stk38 (Fig. S8). In both male and female GarsC201R/+ mice (15) of 3 to 6 weeks of age, tRNAGly-GCC overexpression fully rescued the reduced body weight (Fig. S9A,B) and motor deficits (Fig. S9C–F). Reduced nerve conduction velocity (NCV) and compound muscle action potential (CMAP) amplitude in GarsC201R/+ mice was also fully rescued (Fig. S9G–J). Thus, increasing tRNAGly-GCC levels completely prevented peripheral neuropathy in GarsC201R/+ mice, without affecting Gars mRNA and GlyRS protein levels (Fig. S10).
Fig. 2. tRNAGly-GCC overexpression rescues peripheral neuropathy in CMT2D mouse models.
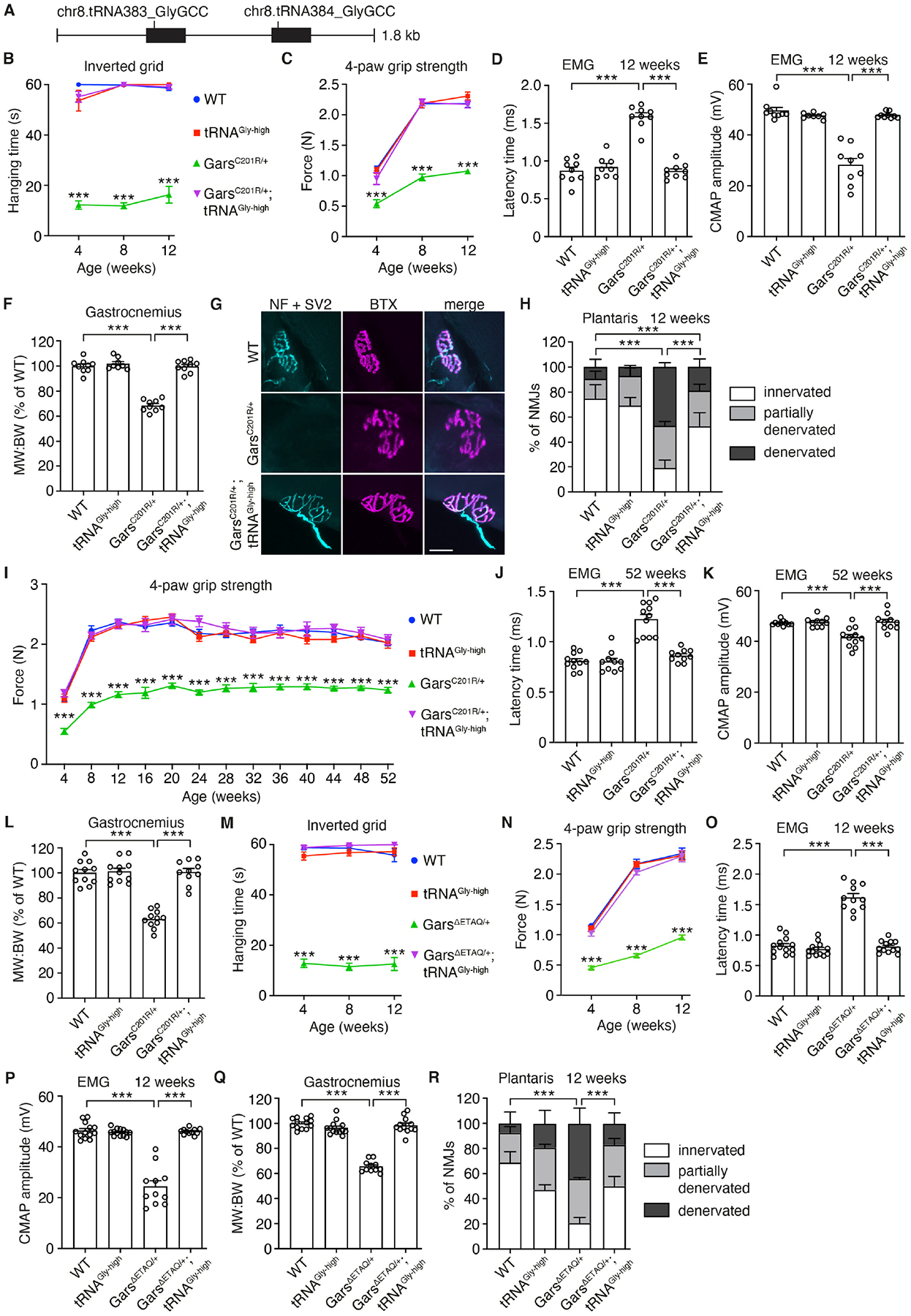
(A) Schematic of the genomic fragment used for generation of tRNAGly-GCC transgenic mice. (B,M) Hanging time in the inverted grid test of male GarsC201R/+ x tRNAGly-high (B) or GarsΔETAQ/+ x tRNAGly-high (M) mice. n=8–9 (B), 11–13 (M) mice per genotype; ***p<0.0001 by one-sample t-test and two-tailed unpaired t-test with Bonferroni correction per time point. (C,I,N) 4-paw grip strength as measured by dynamometer. n=8–9 (C), 10–11 (I), 11–13 (N) mice per genotype; ***p<0.001 by two-way ANOVA with Tukey’s multiple comparisons test per time point (C,I) or Brown-Forsythe and Welch ANOVA (N). (D,E,J,K,O,P) Electromyography (EMG) at 12 (D,E,O,P) or 52 (J,K) weeks of age. (D,J,O) Latency time between sciatic nerve stimulation at sciatic notch level and detection of a compound muscle action potential (CMAP) in the gastrocnemius muscle. n=8–9 (D), 10–11 (J), 11–13 (O) mice per genotype; ***p<0.0001 by two-way ANOVA with Tukey’s multiple comparisons test (D) or Brown-Forsythe and Welch ANOVA (J,O). (E,K,P) CMAP amplitude in the gastrocnemius muscle. n=8–9 (E), 10–11 (K), 11–13 (P) mice per genotype; ***p<0.0005 by Brown-Forsythe and Welch ANOVA (E,P) or two-way ANOVA with Tukey’s multiple comparisons test (K). (F,L,Q) Ratio of muscle weight to body weight (MW:BW, shown as % of WT) of the gastrocnemius at 12 (F,Q) or 52 (L) weeks of age. n=8–9 (F), 10–11 (L), 11–13 (Q) mice per genotype; ***p<0.0001 by two-way ANOVA with Tukey’s multiple comparisons test. (G,H,R) Representative images (G) and quantification (H,R) of NMJ innervation status in plantaris muscle. In (G), neurofilament (NF) and SV2 label presynaptic nerve endings, while TRITC-conjugated bungarotoxin (BTX) labels postsynaptic acetylcholine receptors. n=5 mice per genotype; ***p<0.005 by Fisher’s Exact test with Bonferroni correction. Scale bar: 25μm. Graphs represent mean ± SEM.
Follow-up of an independent cohort of GarsC201R/+ x tRNAGly-high mice from 4 to 12 weeks confirmed full rescue of motor performance (Fig. 2B,C) and neuromuscular transmission (Fig. 2D,E). At 12 weeks of age, tRNAGly-GCC overexpression fully rescued the reduced gastrocnemius muscle weight (Fig. 2F), and substantially mitigated muscle denervation (Fig. 2G,H). The rescuing effect persisted until 1 year of age in another cohort of GarsC201R/+ x tRNAGly-high mice. Body weight and motor performance were fully rescued from 4 to 52 weeks (Fig. 2I; Fig. S9K,L), as well as NCV, CMAP amplitude and gastrocnemius muscle weight (Fig. 2J–L). Thus, tRNAGly-GCC overexpression completely prevents peripheral neuropathy in GarsC201R/+ mice.
Finally, we crossed tRNAGly-high mice to another CMT2D mouse model carrying a patient mutation (245-248_delETAQ) in the mouse Gars gene (16). At 4, 8 and 12 weeks, tRNAGly-GCC overexpression fully rescued motor deficits, reduced NCV and CMAP amplitude, reduced gastrocnemius weight and muscle denervation (Fig. 2M–R). In tRNAGly-low mice, tRNAGly-GCC level was not altered (Fig. S11). GarsC201R/+; tRNAGly-low mice were indistinguishable from GarsC201R/+ mice for all parameters evaluated (Fig. S12), showing that tRNAGly-GCC overexpression and not the mere presence of the transgene is responsible for phenotypic rescue.
We next explored the molecular mechanism underlying the rescue of CMT2D phenotypes by tRNAGly overexpression. We hypothesized that CMT-mutant GlyRSs may exhibit altered kinetics of tRNAGly binding and release. Size-exclusion chromatography of various purified human GlyRS variants revealed that WT and E71G migrated predominantly as dimers, whereas L129P, C157R (≅mouse C201R), G240R, E279D and G526R partitioned between the monomer and dimer forms (Fig. 3A). All CMT-mutant GlyRS dimers bound tRNAGly-GCC (Kon) with a 2- to 10-fold lower affinity than WT dimers (Fig. 3B). L129P, C157R, G240R, E279D and G526R dimers displayed markedly slower tRNAGly-GCC release (Koff), with >80% of traces showing no tRNAGly-GCC release (Fig. 3B). In contrast, E71G dimers displayed tRNAGly-GCC release kinetics comparable to WT. L129P, C157R, G240R, E279D and G526R monomers bound tRNAGly-GCC with very low affinity, but once bound, the tRNAGly-GCC release was markedly inhibited (Fig. 3B). The tRNAGly-UCC isoacceptor displayed similar binding and release kinetics to GlyRS dimers and monomers (Table S2). The slow tRNAGly release by CMT-mutant GlyRS dimers and monomers suggests that mutant GlyRSs sequester a large fraction of cellular tRNAGly and thus deplete it for translation. To provide in vivo evidence for tRNAGly sequestration, we immunoprecipitated GlyRS from brains of GarsC201R/+ and WT littermate mice and quantified the amount of tRNAGly bound to GlyRS. The tRNAGly amount was ~65% higher in GarsC201R/+ versus WT (Fig. 3C, Fig. S13A), indicating stronger tRNAGly association with GlyRS-C201R. Because tRNAGly sequestration may lead to ribosome stalling at Gly codons, we performed ribosome profiling on SC extracts of GarsC201R/+ and WT littermate mice, revealing that Gly codons are more frequently found in the ribosomal A site in GarsC201R/+ SC relative to WT (cumulative increase of 79%, Fig. S13B).
Fig. 3. tRNAGly sequestration by CMT-mutant GlyRS induces ribosome stalling.
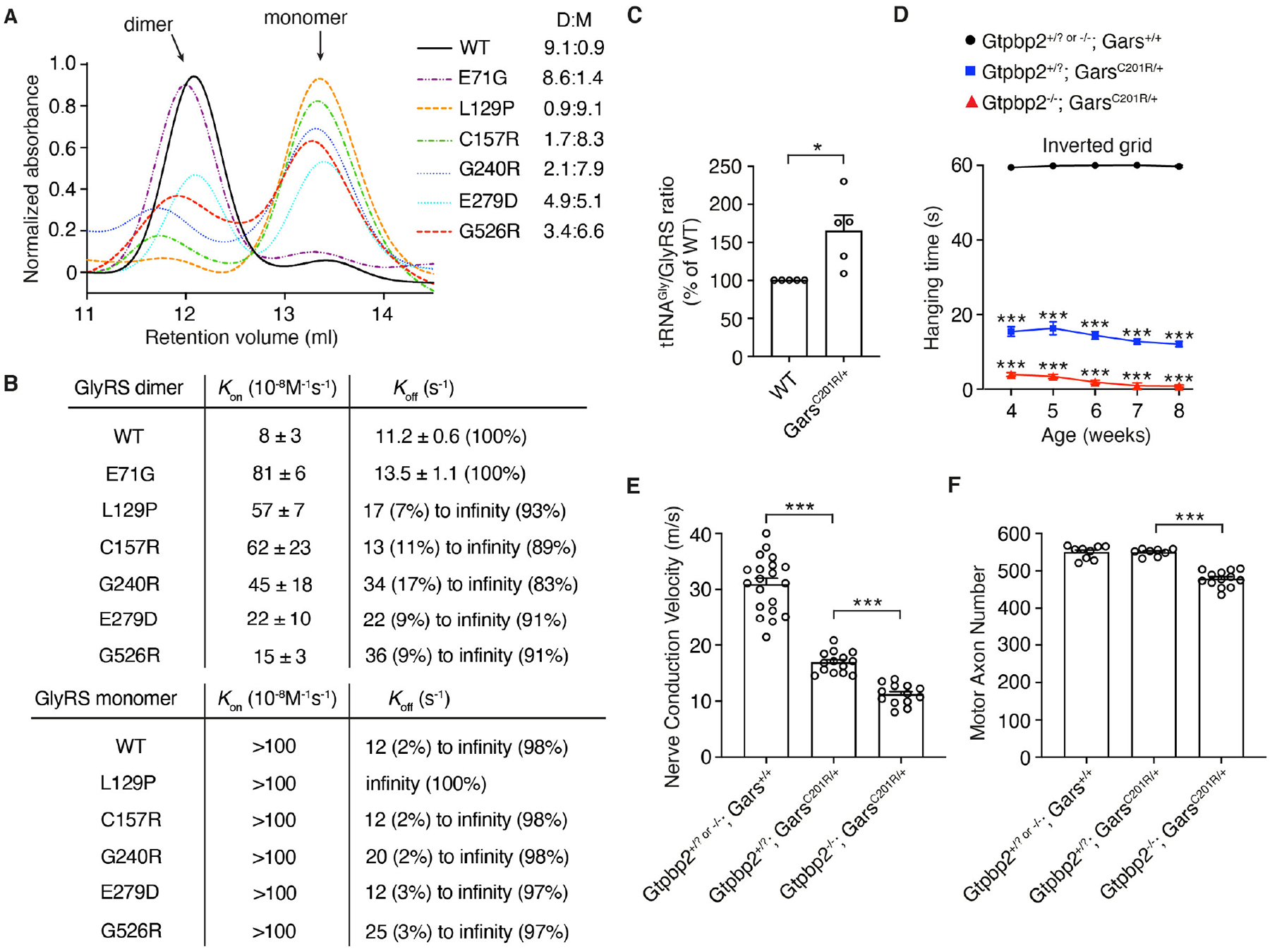
(A) Size-exclusion chromatography of purified recombinant human GlyRS proteins. D:M= dimer:monomer ratio. (B) Kon and Koff values of tRNAGly-GCC binding and release to dimer and monomer forms of the indicated GlyRS variants. The (percentage) denotes the frequency of a measured value. (C) Quantification of tRNAGly bound to GlyRS in tRNAGly:GlyRS complexes immunoprecipitated from whole brains of GarsC201R/+ and WT littermate control mice. tRNAGly/GlyRS ratio of WT is set as 100%; n=5 independent experiments; *p<0.05 by one-sample t-test. (D) Hanging time in the inverted grid test of male Gtpbp2+/? or −/−; Gars+/+ (control), Gtpbp2+/?; GarsC201R/+, and Gtpbp2−/−; GarsC201R/+ littermate mice at 4, 5, 6, 7 and 8 weeks of age. n=15–28 mice per genotype group; ***p<0.0005 by one-sample t-test and two-tailed unpaired t-test with Bonferroni correction per time point. (E) Nerve conduction velocity of the sciatic nerve at 8 weeks of age. n=13–20 mice per genotype group; ***p<0.0001 by Brown-Forsythe and Welch ANOVA. (F) Axon number in the motor branch of the femoral nerve at 8 weeks of age. n=8–13 per genotype group; ***p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. Graphs represent mean ± SEM.
Prolonged ribosome dwelling at codons is resolved by ‘ribosome rescue’ pathways (17–19), and because Gly codons are frequent, ribosome stalling in CMT2D may deplete ribosome rescue factors and inactivation of a rescue factor may aggravate the phenotype of CMT2D mice. Indeed, inactivation of Gtpbp2, encoding the ribosome rescue factor GTPBP2, does not induce peripheral neuropathy by itself (20), but substantially enhanced peripheral neuropathy in GarsC201R/+ mice (Fig. 3D–F, Fig. S14A,B). Thus, ribosome stalling causally contributes to CMT2D pathogenesis. Because stalled ribosomes may activate the integrated stress response (ISR) through GCN2 (21–23) and ISR activation was implicated in CMT2D (24), we evaluated ISR induction in CMT2D mice intercrossed with tRNAGly-high mice. tRNAGly-GCC overexpression fully rescued increased phosphorylated eIF2α immunostaining intensity (~75%) in spinal motor neurons of GarsΔETAQ/+ mice (Fig. 4A,B), as well as the strong induction of ATF4 target genes Gdf15, Adm2, B4galnt2 and Fgf21 in motor neurons of GarsC201R/+ mice (Fig. 4C–I). Thus, tRNAGly-GCC overexpression abrogates ISR activation in CMT2D mice, indicating that depletion of the cellular tRNAGly pool and consequent ribosome stalling is upstream of ISR activation. When Gtpbp2 is inactivated in GarsC201R/+ mice, the percentage of motor neurons showing ISR activation did not change, nor did additional cell types show ISR activation, despite widespread Gtpbp2 expression in SC (Fig. S15). This suggests that tRNAGly levels are only below a critical threshold in affected motor and sensory neurons, leading to ribosome stalling selectively in these cell types. This may explain the relatively modest increase in ribosome dwelling at Gly codons in GarsC201R/+ SC (Fig. S13B).
Fig. 4. tRNAGly-GCC overexpression prevents ISR activation in CMT2D mouse models.
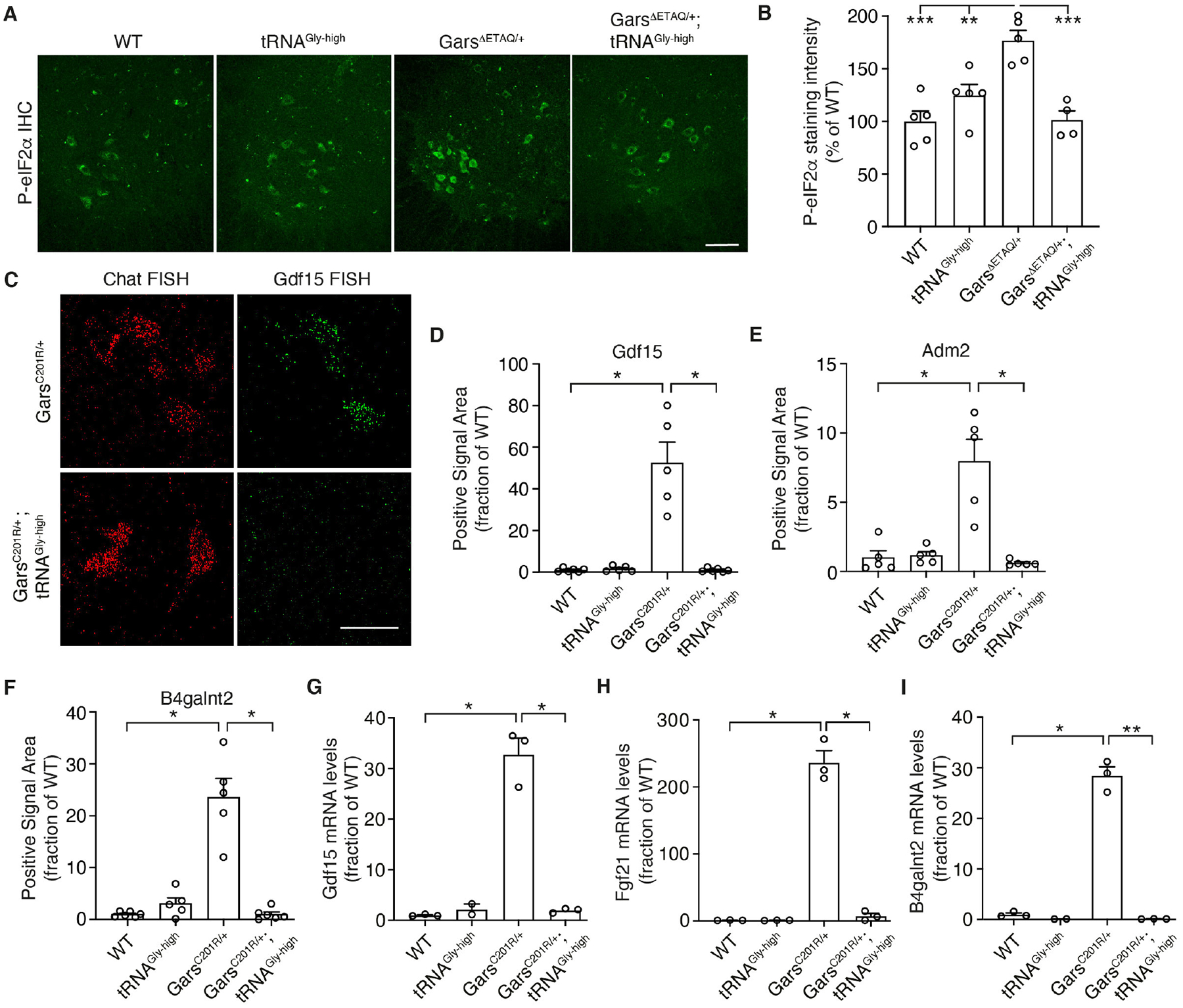
(A,B) Representative images (A) and quantification (B) of immunostaining intensity of phosphorylated eIF2α in motor neuron cell bodies in the spinal cord ventral horn of GarsΔETAQ/+ x tRNAGly-high mice. Scale bar: 100μm. n=4–5 mice per genotype; **p<0.01, ***p<0.0005 by two-way ANOVA with Tukey’s multiple comparisons test. (C-F) Representative images (C) and quantification of fluorescent in situ hybridization (FISH) for ATF4 target genes Gdf15 (D), Adm2 (E), and B4galnt2 (F). Scale bar: 50μm. n=5–6 mice per genotype; *p<0.05 by two-tailed Welch’s t-test with Bonferroni correction. (G-I) mRNA levels of ATF4 target genes Gdf15 (G), Fgf21 (H) and B4galnt2 (I) in spinal cord of GarsC201R/+ x tRNAGly-high mice. n=3 per genotype; *p<0.05, **p<0.01 by Brown-Forsythe and Welch ANOVA. Graphs represent mean ± SEM.
In all, our data propose a detailed molecular mechanism underlying CMT2D (Fig. S16). Beyond the seven CMT2D mutations studied here, this mechanism may apply to additional CMT-mutant GlyRS proteins, because 14 out of 25 reported CMT2D mutations result in net addition of positive charge (Table S3), which could alter binding and release kinetics of the negatively charged tRNAGly. Similarly, the majority of CMT-causing mutations in TyrRS and AlaRS also result in net addition of positive charge (Table S4). Finally, our data indicate that increasing tRNAGly level may constitute a therapeutic approach for CMT2D.
Supplementary Material
Acknowledgments:
We thank Xiang-Lei Yang for GlyRS expression plasmids and the General Instruments Facility (Faculty of Science, Radboud University) for advice on image acquisition and analysis.
Funding:
This work was supported by the Max Planck Society, the Donders Center for Neuroscience, the Muscular Dystrophy Association (MDA479773), the EU Joint Programme – Neurodegenerative Disease Research (JPND; ZonMW 733051075 (TransNeuro) and ZonMW 733051073 (LocalNMD)), the Radala Foundation, an ERC consolidator grant (ERC-2017-COG 770244), Deutsche Forschungsgemeinschaft (DFG, IG73/14-2) to Z.I., and NIH grants to R.W.B. (R01 NS054154, U54 OD020351).
Footnotes
Competing interests: Patent application 2024840 with E.S. as inventor was submitted to the Netherlands Patent Agency. E.L.S. and R.W.B. have a pending patent application ‘GCN2 inhibitors for treating peripheral neuropathy’. R.W.B. is a member of the Scientific Advisory Board of the Charcot-Marie-Tooth Association and the Hereditary Neuropathy Foundation.
Data and materials availability:
The ribosome profiling data are in GEO (accession number GSE160584). tRNAGly transgenic mice are available under a material transfer agreement.
References and Notes:
- 1.Kuo ME, Antonellis A, Ubiquitously Expressed Proteins and Restricted Phenotypes: Exploring Cell-Specific Sensitivities to Impaired tRNA Charging. Trends Genet, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei N, Zhang Q, Yang XL, Neurodegenerative Charcot-Marie-Tooth disease as a case study to decipher novel functions of aminoacyl-tRNA synthetases. The Journal of biological chemistry 294, 5321–5339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Storkebaum E, Peripheral neuropathy via mutant tRNA synthetases: Inhibition of protein translation provides a possible explanation. Bioessays 38, 818–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schimmel P, Aminoacyl tRNA synthetases: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annual review of biochemistry 56, 125–158 (1987). [DOI] [PubMed] [Google Scholar]
- 5.Ibba M, Soll D, Aminoacyl-tRNA synthesis. Annual review of biochemistry 69, 617–650 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Sonenberg N, Hinnebusch AG, Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonellis A et al. , Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J Neurosci 26, 10397–10406 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nangle LA, Zhang W, Xie W, Yang XL, Schimmel P, Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci U S A 104, 11239–11244 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niehues S et al. , Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nature communications 6, 7520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Storkebaum E et al. , Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc Natl Acad Sci U S A 106, 11782–11787 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Froelich CA, First EA, Dominant Intermediate Charcot-Marie-Tooth disorder is not due to a catalytic defect in tyrosyl-tRNA synthetase. Biochemistry 50, 7132–7145 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Motley WW et al. , Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet 7, e1002399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erdmann I et al. , Cell-selective labelling of proteomes in Drosophila melanogaster. Nature communications 6, 7521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dieterich DC et al. , In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci 13, 897–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achilli F et al. , An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech 2, 359–373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morelli KH et al. , Allele-specific RNA interference prevents neuropathy in Charcot-Marie-Tooth disease type 2D mouse models. J Clin Invest 129, 5568–5583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuller AP, Green R, Roadblocks and resolutions in eukaryotic translation. Nature reviews. Molecular cell biology 19, 526–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graille M, Seraphin B, Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nature reviews. Molecular cell biology 13, 727–735 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Joazeiro CAP, Ribosomal Stalling During Translation: Providing Substrates for Ribosome-Associated Protein Quality Control. Annu Rev Cell Dev Biol 33, 343–368 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Ishimura R et al. , RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inglis AJ et al. , Activation of GCN2 by the ribosomal P-stalk. Proc Natl Acad Sci U S A 116, 4946–4954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harding HP et al. , The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu CC, Peterson A, Zinshteyn B, Regot S, Green R, Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 182, 404–416 e414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spaulding EL et al. , The integrated stress response contributes to tRNA synthetase-associated peripheral neuropathy. Science, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han C, Jan LY, Jan YN, Enhancer-driven membrane markers for analysis of nonautonomous mechanisms reveal neuron-glia interactions in Drosophila. Proc Natl Acad Sci U S A 108, 9673–9678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venken KJ, He Y, Hoskins RA, Bellen HJ, P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314, 1747–1751 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Wang JW, Beck ES, McCabe BD, A modular toolset for recombination transgenesis and neurogenetic analysis of Drosophila. PLoS One 7, e42102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erdmann I et al. , Cell type-specific metabolic labeling of proteins with azidonorleucine in Drosophila. Bio-protocol 7, e2397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Vree PJ et al. , Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol 32, 1019–1025 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Scekic-Zahirovic J et al. , Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW, An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron 51, 715–726 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Scherer SS et al. , Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J Neurosci 25, 1550–1559 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corzo J, Time, the forgotten dimension of ligand binding teaching. Biochem Mol Biol Educ 34, 413–416 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Zhang G et al. , Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res 38, 4778–4787 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirchner S et al. , Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol 15, e2000779 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polte C et al. , Assessing cell-specific effects of genetic variations using tRNA microarrays. BMC Genomics 20, 549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tameire F et al. , ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nature cell biology 21, 889–899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amirbeigiarab S et al. , Invariable stoichiometry of ribosomal proteins in mouse brain tissues with aging. Proc Natl Acad Sci U S A 116, 22567–22572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo H, Ingolia NT, Weissman JS, Bartel DP, Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lareau LF, Hite DH, Hogan GJ, Brown PO, Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife 3, e01257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartholomaus A, Ignatova Z, Codon Resolution Analysis of Ribosome Profiling Data. Methods in molecular biology 2252, 251–268 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Magnuson B, Ekim B, Fingar DC, Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. The Biochemical journal 441, 1–21 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Zoncu R, Efeyan A, Sabatini DM, mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews. Molecular cell biology 12, 21–35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barcelo H, Stewart MJ, Altering Drosophila S6 kinase activity is consistent with a role for S6 kinase in growth. Genesis 34, 83–85 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Holcik M, Sonenberg N, Translational control in stress and apoptosis. Nature reviews. Molecular cell biology 6, 318–327 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Kong J, Lasko P, Translational control in cellular and developmental processes. Nat Rev Genet 13, 383–394 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Hinnebusch AG, The scanning mechanism of eukaryotic translation initiation. Annual review of biochemistry 83, 779–812 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Vichalkovski A et al. , NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr Biol 18, 1889–1895 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Stegert MR, Tamaskovic R, Bichsel SJ, Hergovich A, Hemmings BA, Regulation of NDR2 protein kinase by multi-site phosphorylation and the S100B calcium-binding protein. The Journal of biological chemistry 279, 23806–23812 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Cornils H et al. , Ablation of the kinase NDR1 predisposes mice to the development of T cell lymphoma. Sci Signal 3, ra47 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Elf J, Nilsson D, Tenson T, Ehrenberg M, Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300, 1718–1722 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Mohammad F, Green R, Buskirk AR, A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elliott DA, Brand AH, The GAL4 system : a versatile system for the expression of genes. Methods in molecular biology 420, 79–95 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Antonellis A et al. , Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72, 1293–1299 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dubourg O et al. , The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology 66, 1721–1726 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Xie W, Nangle LA, Zhang W, Schimmel P, Yang XL, Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc Natl Acad Sci U S A 104, 9976–9981 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rohkamm B et al. , Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome. J Neurol Sci 263, 100–106 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Forrester N et al. , Clinical and Genetic Features in a Series of Eight Unrelated Patients with Neuropathy Due to Glycyl-tRNA Synthetase (GARS) Variants. J Neuromuscul Dis 7, 137–143 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Yu X et al. , A Novel Mutation of GARS in a Chinese Family With Distal Hereditary Motor Neuropathy Type V. Front Neurol 9, 571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee HJ et al. , Two novel mutations of GARS in Korean families with distal hereditary motor neuropathy type V. J Peripher Nerv Syst 17, 418–421 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Liao YC et al. , Two Novel De Novo GARS Mutations Cause Early-Onset Axonal Charcot-Marie-Tooth Disease. PLoS One 10, e0133423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nan H et al. , Novel GARS mutation presenting as autosomal dominant intermediate Charcot-Marie-Tooth disease. J Peripher Nerv Syst 24, 156–160 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Argente-Escrig H et al. , Clinical, genetic and disability profile of pediatric distal hereditary motor neuropathy. Neurology, (2020). [DOI] [PubMed] [Google Scholar]
- 65.Yalcouye A et al. , A novel mutation in the GARS gene in a Malian family with Charcot-Marie-Tooth disease. Mol Genet Genomic Med 7, e00782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawakami N et al. , A novel mutation in glycyl-tRNA synthetase caused Charcot-Marie-Tooth disease type 2D with facial and respiratory muscle involvement. Rinsho shinkeigaku = Clinical neurology 54, 911–915 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Hamaguchi A, Ishida C, Iwasa K, Abe A, Yamada M, Charcot-Marie-Tooth disease type 2D with a novel glycyl-tRNA synthetase gene (GARS) mutation. J Neurol 257, 1202–1204 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Sun A et al. , A novel mutation of the glycyl-tRNA synthetase (GARS) gene associated with Charcot-Marie-Tooth type 2D in a Chinese family. Neurol Res 37, 782–787 (2015). [DOI] [PubMed] [Google Scholar]
- 69.James PA et al. , Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology 67, 1710–1712 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Sivakumar K et al. , Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain 128, 2304–2314 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Del Bo R et al. , Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology 66, 752–754 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Eskuri JM, Stanley CM, Moore SA, Mathews KD, Infantile onset CMT2D/dSMA V in monozygotic twins due to a mutation in the anticodon-binding domain of GARS. J Peripher Nerv Syst 17, 132–134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jordanova A et al. , Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet 38, 197–202 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Hyun YS et al. , Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clinical genetics 86, 592–594 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Gonzaga-Jauregui C et al. , Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep 12, 1169–1183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin KP et al. , The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 6, e29393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Motley WW et al. , A novel AARS mutation in a family with dominant myeloneuropathy. Neurology 84, 2040–2047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bacquet J et al. , Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation. BMJ Open 8, e021632 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weterman MAJ et al. , Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Hum Mol Genet 27, 4036–4050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Latour P et al. , A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 86, 77–82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McLaughlin HM et al. , A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat 33, 244–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bansagi B et al. , Genotype/phenotype correlations in AARS-related neuropathy in a cohort of patients from the United Kingdom and Ireland. J Neurol 262, 1899–1908 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao Z et al. , Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 78, 1644–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Safka Brozkova D et al. , Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain 138, 2161–2172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Royer-Bertrand B et al. , Peripheral neuropathy and cognitive impairment associated with a novel monoallelic HARS variant. Ann Clin Transl Neurol 6, 1072–1080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abbott JA et al. , Substrate interaction defects in histidyl-tRNA synthetase linked to dominant axonal peripheral neuropathy. Hum Mutat 39, 415–432 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li JQ, Dong HL, Chen CX, Wu ZY, A novel WARS mutation causes distal hereditary motor neuropathy in a Chinese family. Brain 142, e49 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Tsai PC et al. , A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain 140, 1252–1266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang B et al. , A novel WARS mutation (p.Asp314Gly) identified in a Chinese distal hereditary motor neuropathy family. Clinical genetics 96, 176–182 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Gonzalez M et al. , Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. Journal of neurology, neurosurgery, and psychiatry 84, 1247–1249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sagi-Dain L et al. , Whole-exome sequencing reveals a novel missense mutation in the MARS gene related to a rare Charcot-Marie-Tooth neuropathy type 2U. J Peripher Nerv Syst 23, 138–142 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Nam SH et al. , Identification of Genetic Causes of Inherited Peripheral Neuropathies by Targeted Gene Panel Sequencing. Mol Cells 39, 382–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ribosome profiling data are in GEO (accession number GSE160584). tRNAGly transgenic mice are available under a material transfer agreement.