

Abstract
Cells employ intracellular signaling pathways to sense and respond to changes in their external environment. In recent years, live-cell biosensors have revealed complex pulsatile dynamics in many pathways, but studies of these signaling dynamics are limited by the necessity of live-cell imaging at high spatiotemporal resolution. Here, we describe an approach to infer pulsatile signaling dynamics from a single measurement in fixed cells using a pulse-detecting gene circuit. We computationally screened for circuits with the capability to selectively detect signaling pulses, revealing an incoherent feedforward topology that robustly performs this computation. We implemented the motif experimentally for the Erk signaling pathway using a single engineered transcription factor and fluorescent protein reporter. Our ‘recorder of Erk activity dynamics’ (READer) responds sensitively to spontaneous and stimulus-driven Erk pulses. READer circuits open the door to permanently labeling transient, dynamic cell populations to elucidate the mechanistic underpinnings and biological consequences of signaling dynamics.
Introduction
The textbook view of many cell signaling pathways is that of an amplifier or an off-to-on switch. A stimulus (e.g., an extracellular ligand, small-molecule nutrient, or toxic stress) is received by the cell and stimulates activation of a signaling pathway, which amplifies the input to trigger a potent gene expression response. However, the recent development of live cell signaling biosensors has revealed that many signaling pathways implement more complex functions, including pulses, oscillations or even traveling waves of pathway activity. Examples include the signaling pulses observed from the tumor suppressor p53, the mitogen associated protein kinase (MAPK) Erk, and the immune signaling transcription factor NF-κB (Hoffmann et al., 2002; Nelson et al., 2004; Purvis et al., 2012; Purvis and Lahav, 2013; Shankaran et al., 2009). Pulses of Erk activity have been observed in vivo in the early mouse embryo (Pokrass et al., 2020; Simon et al., 2020) and in tumors (Gerosa et al., 2020), and self-organize into propagating waves from sites of epithelial injury in both mouse (Hiratsuka et al., 2015) and zebrafish (De Simone et al., 2021; Mayr et al., 2018). The breadth of biological systems exhibiting signaling dynamics suggests that they may play important functional roles, but the molecular underpinnings and downstream cellular functions of signaling pulses remain poorly understood.
In nearly every context, dynamics are studied exclusively using time-lapse microscopy in single living cells. This granularity of measurement is crucial for accurately inferring dynamics. For example, to determine whether a cell has pulsed, one must perform at least three measurements to observe a succession of low, high, and low signaling states. However, the requirement for live-cell microscopy also puts severe constraints on the type and scale of experimental study that can be performed. Live cell biosensors must often be imaged at high spatial and temporal resolution, limiting experimental throughput and presenting an obstacle to large-scale chemical or genetic screens. Microscopy also restricts study to tissues that are compatible with single-cell imaging, limiting our ability to determine the functional role of dynamics in vivo. The ability to measure signaling dynamics without time-lapse imaging could be transformational for studying both the molecular mechanisms and biological consequences of dynamic signaling (Figure 1A). This capability would enable workflows whereby dynamic cells could be isolated and subjected to high-throughput analyses (e.g., RNA sequencing or proteomic studies), or entire tissues could be fixed and examined for the spatial distribution of dynamically active cells.
Figure 1. A computational screen for pulse-detecting gene circuits.
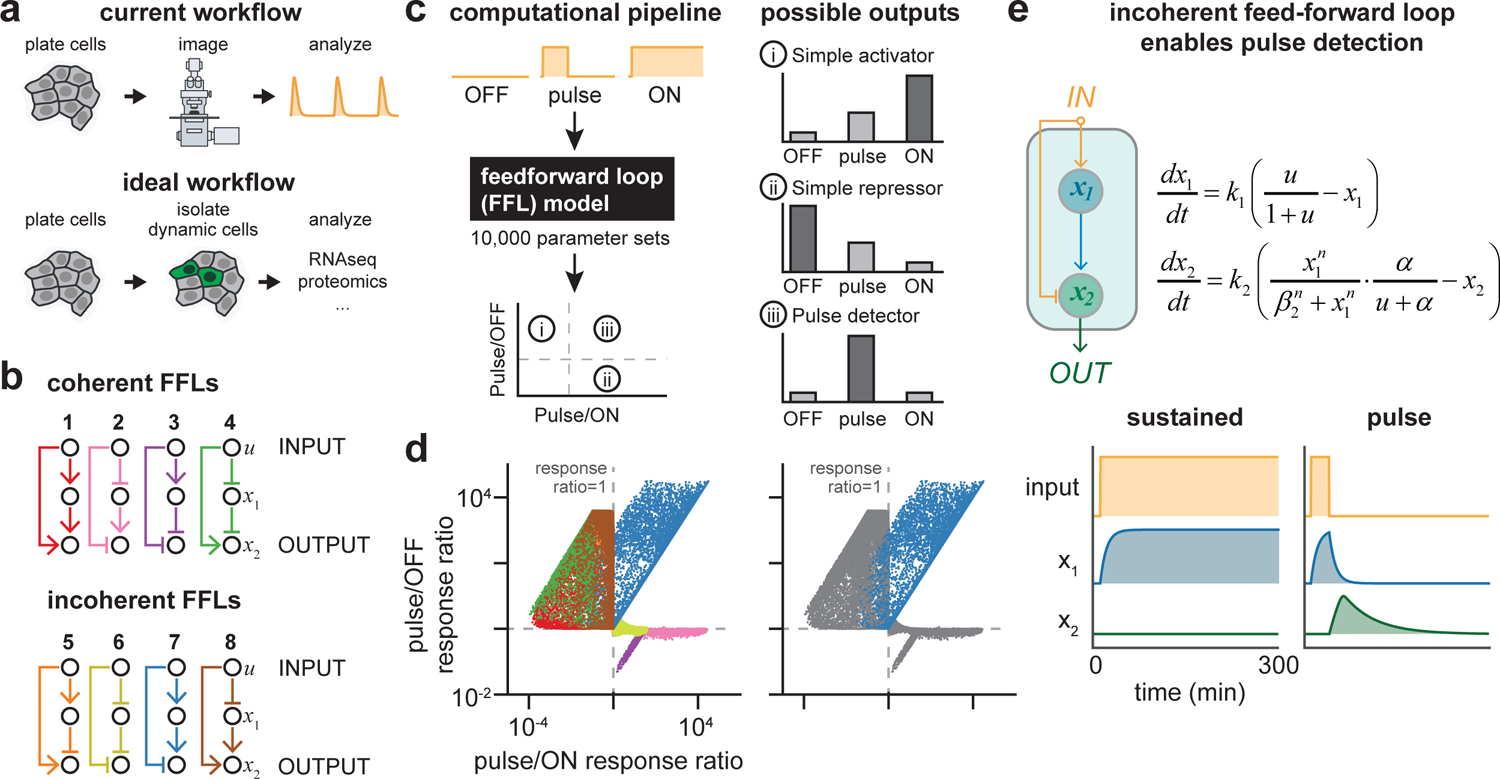
(a) Workflows for studying signaling dynamics. Currently, signaling dynamics are typically obtained from live-imaging of individual cells, limiting throughput. In an alternative workflow, dynamics could be inferred from a single fluorescence measurement, enabling rapid and scalable isolation of dynamic cells for downstream analyses. (b) Coherent and incoherent feed-forward loop (FFL) topologies screened for pulse detection. (c) Computational screen workflow: constant-off, constant-on and pulsed inputs were applied to 10,000 random parameterizations of each network shown in b. Circuits exhibiting pulse detection would lie in quadrant 3, where responses to pulsed inputs exceed those of either ON or OFF stimuli. (d) Responses of all circuits from the computational screen. Each point represents a single parameterization of an FFL circuit colored as in b (left), or with only Circuit 7 colored (right). (e) Representative time course of Circuit 7 simulated with either a constant-on (left) or pulsed input (right), along with the model equations representing Circuit 7.
Motivated by these challenges, we set out to develop methods to infer a cell’s dynamic history from a single fixed measurement. Our strategy centers on the development of synthetic gene circuits that selectively respond to signaling pulses, filtering out constant-high and constant-low states. We first performed a computational screen for pulse-detecting gene circuits, revealing an incoherent feedforward loop that robustly performs this computation. We then implemented this motif experimentally for the Erk signaling pathway using just two components: an engineered transcription factor and a fluorescent protein reporter. Our ‘recorder of Erk activity dynamics’ (READer) circuit responds sensitively to both stimulus-driven and spontaneous Erk pulses. READer circuits thus open the door to permanently labeling transient, dynamic cell populations to elucidate the mechanistic underpinnings and biological consequences of signaling dynamics.
Results
A computational screen for biochemical circuits that perform pulse detection
Our first goal was to identify circuit topologies that might serve as pulse detectors, selectively responding to dynamics while filtering out and ignoring constant high or low signaling states. We focused our attention on feedforward loops (FFLs), a class of network topologies that repeatedly arise in the search for motifs that can generate and process dynamic information (Alon, 2007; Gerardin et al., 2019; Mangan and Alon, 2003). The defining feature of FFLs is that two paths link input to output, with the difference in timescale between these two paths typically acting as the source of dynamic filtering. Classic studies on FFL networks have defined variants that can generate pulses, produce a temporally-ordered sequence of outputs, or detect fold-changes in input signals (Basu et al., 2004; Csikasz-Nagy et al., 2009; Goentoro et al., 2009); whether such networks might also selectively detect an input pulse remains unknown. FFLs are classified as either coherent or incoherent based on whether the two paths connecting input and output have the same or different signs (Figure 1B). We devised a simple, modular 2-equation model to represent all 8 FFLs with AND logic at the output node (Mangan and Alon, 2003) (Figure S1A, Supplementary Information), and in each case simulated 10,000 random parameter sets with 3 input conditions: sustained on, sustained off and a pulse of activation.
We assessed the performance of each circuit by calculating integrated output over time in response to all three inputs. We then plotted the ratio of the pulse-induced response to both the constant-on and constant-off cases (Figure 1C). By definition, a pulse detector circuit should show stronger induction in response to a pulse than either constant stimulus, leading to high values of both ratios and enrichment in the upper-right quadrant of such a plot, whereas simple activators (circuits that induce gene expression in proportion to the quantity of input signal) would appear in the lower-right quadrant and simple repressors would appear in the upper-left quadrant (Figure 1C). Analysis of all 8 FFL topologies revealed that only a single topology, Circuit 7, could perform pulse detection (Figure 1D, left). Pulse detection also appeared to be a robust feature of the Circuit 7 FFL, with 96% of simulations showing a stronger response to pulsed stimuli than either high or low constant inputs (Figure 1D, right). We also tested all 8 FFL topologies with OR logic at the output node (Figure S1B). While none exhibited pulse-specific activation, one OR-FFL circuit did exhibit pulse-specific repression and can be understood as the logical inverse of our pulse-detecting Circuit 7 FFL (Figure S1C, Supplementary Information). These results suggest that pulse detection is yet another function enabled by FFLs.
Examining simulation trajectories provided further insight into the operation of the Circuit 7 FFL (Figure 1E, Figure S2). Application of a stimulus (“input”) rapidly results in production of an intermediate node (x1), but also immediately blocks the ability for x1 to activate an output node (x2). Only upon removal of the stimulus is repression quickly relieved, enabling x1 to trigger output. Constant-on inputs are unable to trigger a response because input permanently blocks output, whereas constant-off inputs fail because the essential activator x1 is not produced. It is important to note that this FFL design also places constraints on the timescales of the two paths between input and output, in line with prior studies of FFL dynamics (Alon, 2007; Mangan and Alon, 2003). Repression must be achieved faster than activation, so that output remains off in response to a constant-on stimulus. Conversely, repression must be relieved more rapidly than x1 is degraded so that output production can be triggered as the stimulus is switched off. Overall, our simulations reveal an intuitive and logical relationship between the Circuit 7 FFL topology and pulse detection, demonstrating that pulse detection can arise quite generally out of this FFL architecture.
Experimental implementation of pulse detection for the mammalian Ras/Erk pathway
We next set out to implement our pulse detector circuit in the context of a dynamic signaling pathway in mammalian cells: the Erk mitogen-activated protein kinase (MAPK) pathway. Erk is a canonical example of a dynamically regulated signaling protein, exhibiting complex pulses and traveling waves in many cellular contexts (Albeck et al., 2013; Aoki et al., 2013; De Simone et al., 2021; Hiratsuka et al., 2015). Our implementation centered around a single synthetic transcription factor that is regulated by Erk in two opposing ways (Figure 2A). For the forward activation path (e.g., Erk input activating x1 which then activates x2) we envisioned a two-step transcriptional cascade: an Erk-responsive promoter to drive expression of a synthetic Gal4-VP64 transcription factor, which then induces GFP expression from a Gal4-responsive UAS promoter. To match the Circuit 7 FFL topology, our synthetic transcription factor must also be rapidly and reversibly inhibited by Erk (so that the Erk input also directly inhibits x2 production). We conjectured that fusion with an Erk “kinase translocation reporter” (ErkKTR) would be ideal for implementing this stimulus-dependent inhibition of the engineered transcription factor (Regot et al., 2014). Because the ErkKTR is exported from the nucleus in response to Erk activity, an ErkKTR-transcription factor fusion protein would be precluded from encountering DNA and expressing a target gene if the pathway remained active.
Figure 2. Experimental implementation of pulse detection for the Erk signaling pathway.
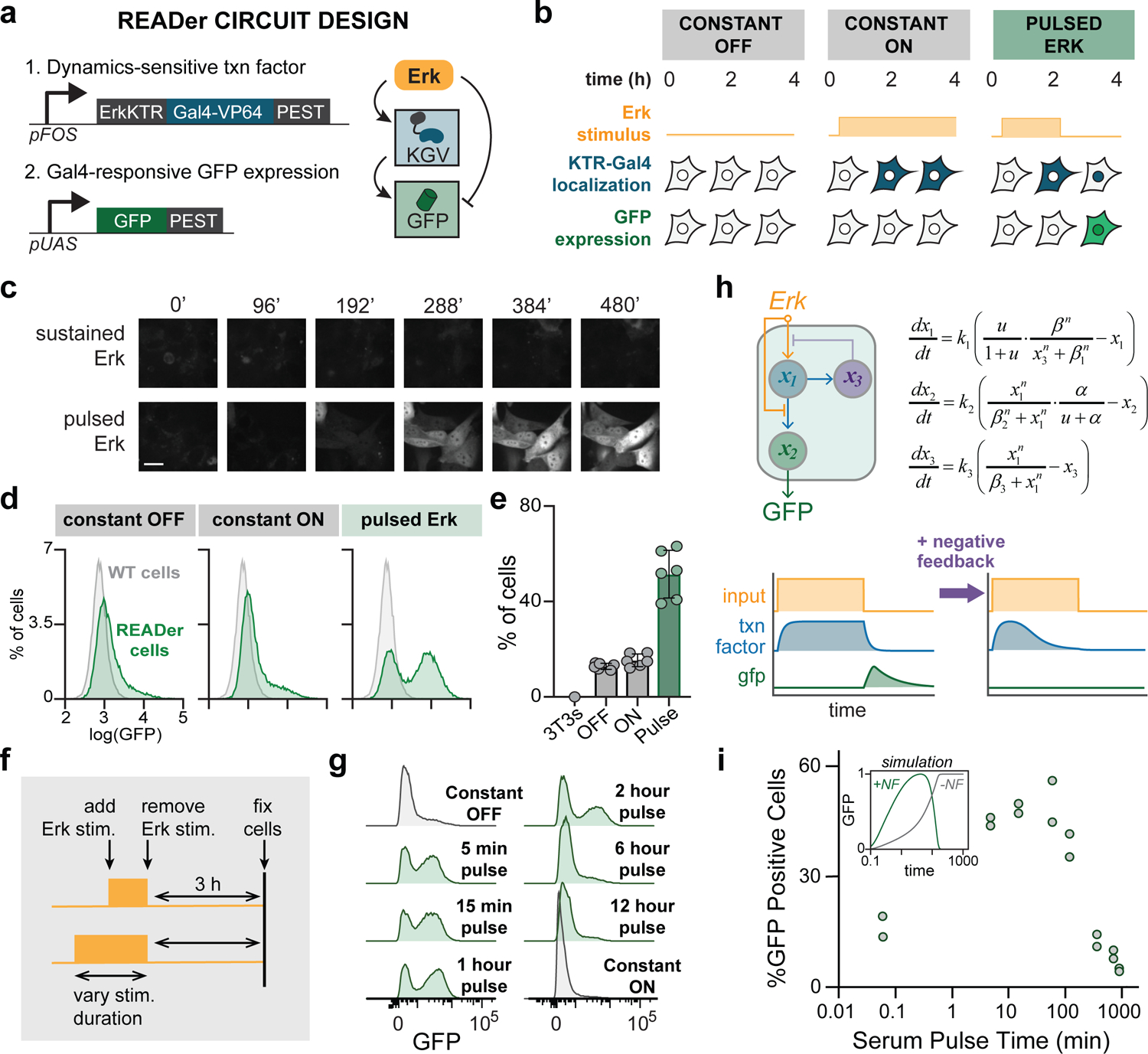
(a) Schematic of the Recorder of Erk Activity Dynamics (READer) circuit. An Erk-responsive promoter drives expression of a KTR-Gal4-VP64 (KGV) fusion protein, which in turn triggers expression of a GFP reporter. Destabilization (PEST) sequences are added to both components to ensure rapid degradation and ‘resetting’ in the absence of stimulus. (b) Illustration of READer circuit logic. Under constant-off stimuli, KGV and GFP levels remain low. A constant-on stimulus drives KGV expression and nuclear export, preventing GFP production. In response to an input pulse, KGV is first expressed and then imported into the nucleus, leading to high GFP expression. (c) Images of NIH3T3 READer cells exposed to constant serum or a 1 h serum pulse. Scale bar: 20 μm. (d) Flow cytometry histograms of GFP from parental NIH3T3 (gray) or READer cells (green) incubated in growth factor free media (constant OFF), 10% serum (constant ON) or a one-hour pulse of 10% serum. (e) Quantification of data from d in all three conditions. Data is quantified from 2 wells each for 3 independent replicates. Error bars represent standard deviation. (f) Mapping how pulse duration affects READer circuit output. Serum inputs of varying duration were applied to cells, which were fixed 3 h after the end of the pulse. (g) GFP histograms obtained by flow cytometry for the experimental workflow in f. (h) An extended mathematical model of the READer circuit incorporating negative feedback on Erk target gene induction. An input u (gold) stimulates intermediate node x1 (blue), which produces a negative regulator x3 (purple) that inhibits the production of x1. (i) Quantification of flow cytometry data in g, showing data from 2 experimental replicates. Inset shows simulated results from the model from h, with (green) or without (gray) negative feedback.
To realize this design experimentally we expressed a KTR-Gal4-VP64 synthetic transcription factor (abbreviated throughout as KGV) downstream of the Erk-responsive FOS promoter (PFOS). We then used a standard reporter construct, the Gal4-responsive UAS promoter driving destabilized GFP, to record the circuit’s output. Only in response to a pulse of Erk would KGV be first expressed and then shuttled into the nucleus, resulting in GFP production (Figure 2B). We termed our circuit – comprising a dynamics-sensitive transcription factor and reporter gene – a Recorder of Erk Activity Dynamics, or READer. We transduced NIH3T3 cells with a lentiviral PUAS-dGFP reporter and transfected them with a PiggyBAC transposase-integrable PFOS-KGV Erk-responsive transcription factor, based on our prior data showing that the PiggyBAC system can be used to integrate immediate-early gene promoters that potently induce gene expression upon Erk activation (Ravindran et al., 2020). We reasoned that the use of a destabilized GFP variant could be useful to “reset” cells to a low GFP state prior to the start of an experiment, eliminating background signal from detection of any Erk dynamics in the hours or days prior to study. We then sorted clonal cell lines harboring stable integration of both the KGV and dGFP components (Figure S3A; see STAR Methods).
We next tested whether cells expressing the READer circuit were able to discriminate between pulsed and constant signaling inputs as had been observed in our simulations. We switched between growth factor-free (GF-free) and serum-containing media to deliver dynamic Erk stimuli to cells after confirming that both serum addition and withdrawal led to rapid changes in ErkKTR biosensor localization on a similar ~10 min timescale (Figure S3B–C). We cultured READer cells overnight in media lacking growth factors (GF-free media) and monitored GFP induction by time-lapse microscopy in continued GF-free media (‘constant off’), addition of 10% serum (‘constant on’) or a 1 h pulse of serum followed by a return to GF-free media (‘serum pulse’). Performing confocal imaging for GFP induction in each case revealed that the serum pulse led to strong GFP induction within the 6 h imaging period, whereas constant-on and constant-off stimuli each led to minimal GFP accumulation (Figure 2C; Movie S1). Varying the serum pulse width revealed similar GFP induction for pulses of 30 min, 1 h, and 2 h duration, with a time shift in GFP induction corresponding to the different pulse durations (Figure S3D). We also noted that small subpopulation of cells in the constant-off and constant-on conditions displayed strong GFP induction, potentially suggesting underlying pulsatile Erk dynamics in a subpopulation of NIH3T3 cells, a prediction that we tested further below.
A key advantage of a pulse detection circuit is that it can be used to infer prior signaling dynamics from a single measurement in fixed cells, eliminating the requirement for live-cell imaging. To demonstrate this capability, we again exposed NIH3T3 READer cells to constant-off, constant-on and pulsed serum inputs, fixed cells 4 h after the start of stimulation and performed flow cytometry to measure GFP accumulation at a single, final timepoint (Figure 2D). We found that GFP remained low in most cells exposed to constant-on and constant-off conditions, again noting a small ‘tail’ of GFP-high cells that will be discussed in detail below. In contrast, a pulse of serum induced strong GFP induction over the following 3–12 hours in approximately 50% of cells (Figure 2E; Figure S4A–C). To further ensure that READer specifically reported on Erk activity, rather than other consequences of serum stimulation, we confirmed that GFP induction was blocked by 2 h pretreatment with 1 μM cobimetinib (MEKi), a small-molecule inhibitor of the Erk-activating kinase MEK (Figure S4D–E). Together, these data indicate that the READer pulse-detecting circuit can indeed be used to infer the Erk dynamics triggered by externally supplied mitogenic stimuli.
We were intrigued by the fractional response of the READer circuit, where only a subset of cells expresses GFP in response to the same pulsatile stimulus. We and others have previously found that serum and optogenetic stimulation can result in uniform Erk activation across a cell population (Mackeigan et al., 2005; Toettcher et al., 2013), yet trigger endogenous immediate-early gene (IEG) expression in only a subset of these cells (Jena et al., 2021; Mackeigan et al., 2005). We reasoned that the READer system’s fractional response may also reflect a non-genetic cell state, possibly tied to its incorporation of an Erk-responsive IEG promoter. We confirmed that the fractional response was non-genetic in origin by sorting cells from the GFP-high or GFP-low population and repeating the pulsed stimulus experiment. Indeed, cells sorted from only the GFP-high or GFP-low populations generated the same bimodal response upon a second stimulus challenge (Figure S5A–B). We next examined DNA content from the GFP-high and GFP-low subpopulations to test whether Erk-triggered gene expression might depend on cell cycle phase. We found similar DNA content distributions regardless of GFP status, arguing against cell cycle control over READer induction (Figure S5C–D). These data confirm that even isogenic cells exhibit fractional READer responses, reminiscent of the fractional response observed for endogenous IEGs and suggesting that READer reports on Erk dynamics in the subset of cells that are permissive to Erk-dependent gene expression.
The scalability of flow cytometry enabled us to rapidly scan additional stimulus conditions to characterize the READer circuit’s response in more detail. We first tested how selective the circuit was to changes in the pulse duration. Endogenous Erk pulses are typically observed to be less than 1 h in length, with sustained responses lasting for multiple hours (Albeck et al., 2013; Aoki et al., 2013; Bugaj et al., 2018; Gerosa et al., 2020; Goglia et al., 2020; Santos et al., 2007). We applied pulses of different durations ranging from 5 min to 12 h, then incubated cells for an additional 3 h to allow GFP to accumulate prior to fixation (Figure 2F). Pulses from 5 min to 2 h resulted in similar profiles of GFP expression, but longer pulses were filtered and ignored by the circuit (Figure 2G). While this long pulse rejection was not a prediction from our original “Circuit 7” model, it can be readily understood based on prior reports that even a sustained Erk stimulus only drives a transient, 30 min pulse of gene expression from the PFOS promoter due to multiple sources of negative regulation (e.g. Erk-induced expression of DUSP phosphatases; auto-repression by Fos protein), after which subsequent expression is suppressed (Kholodenko et al., 1999; Nakakuki et al., 2010; Wilson et al., 2017). Even a long input pulse would thus be expected to only transiently induce KGV synthesis. This transient pulse of KGV synthesis would be followed by a period in which Erk remains active but KGV RNA/protein levels drop, leaving little KGV protein to return to the nucleus to drive GFP expression when the Erk pulse eventually terminates (Figure 2H). We verified that our transposase-integrated PFOS promoter indeed produced a transient pulse of expression, in agreement with this conceptual model and our prior data on endogenous fos expression (Figure S6) (Wilson et al., 2017). Moreover, implementing transient PFOS-driven expression in our computational model was sufficient to match the duration-based filtering that we observed experimentally (Figure 2I, inset).
Optogenetic stimuli reveal that READer responds to a wide range of pulsatile Erk signals
To test for READer system function across a broader range of dynamic Erk inputs, we turned to our OptoSOS optogenetic system (Johnson et al., 2017; Toettcher et al., 2013). Light inputs can be delivered in complex sequences that are cumbersome to achieve by repeated changes in soluble growth factor concentration. We first transduced READer cells with a single-vector, blue-light-sensitive OptoSOS system that we previously showed can potently activate Ras/Erk signaling (Figure 3A) (Goglia et al., 2020). Next, we validated that our OptoSOS READer cell line was able to trigger rapid, potent Erk activity changes in response to light stimuli (Figure 3B). We then confirmed whether optogenetic stimulation recapitulated our previous single-pulse detection results. We quantified the fraction of GFP-positive cells in response to various light pulse durations by fitting a mixture of Gaussians to the GFP distribution in each case (Figure 3C,D), revealing that a 10–60 min pulse of blue light triggered maximal GFP expression (Figure 3E,F), just as in the case of serum (Figure 2I). We did notice lower overall GFP expression level when light was used as the stimulus, suggesting that maximal immediate-early gene expression may depend on both Erk and other parallel pathways downstream of growth factor receptors.
Figure 3. OptoSOS enables characterization of READer dynamic filtering.
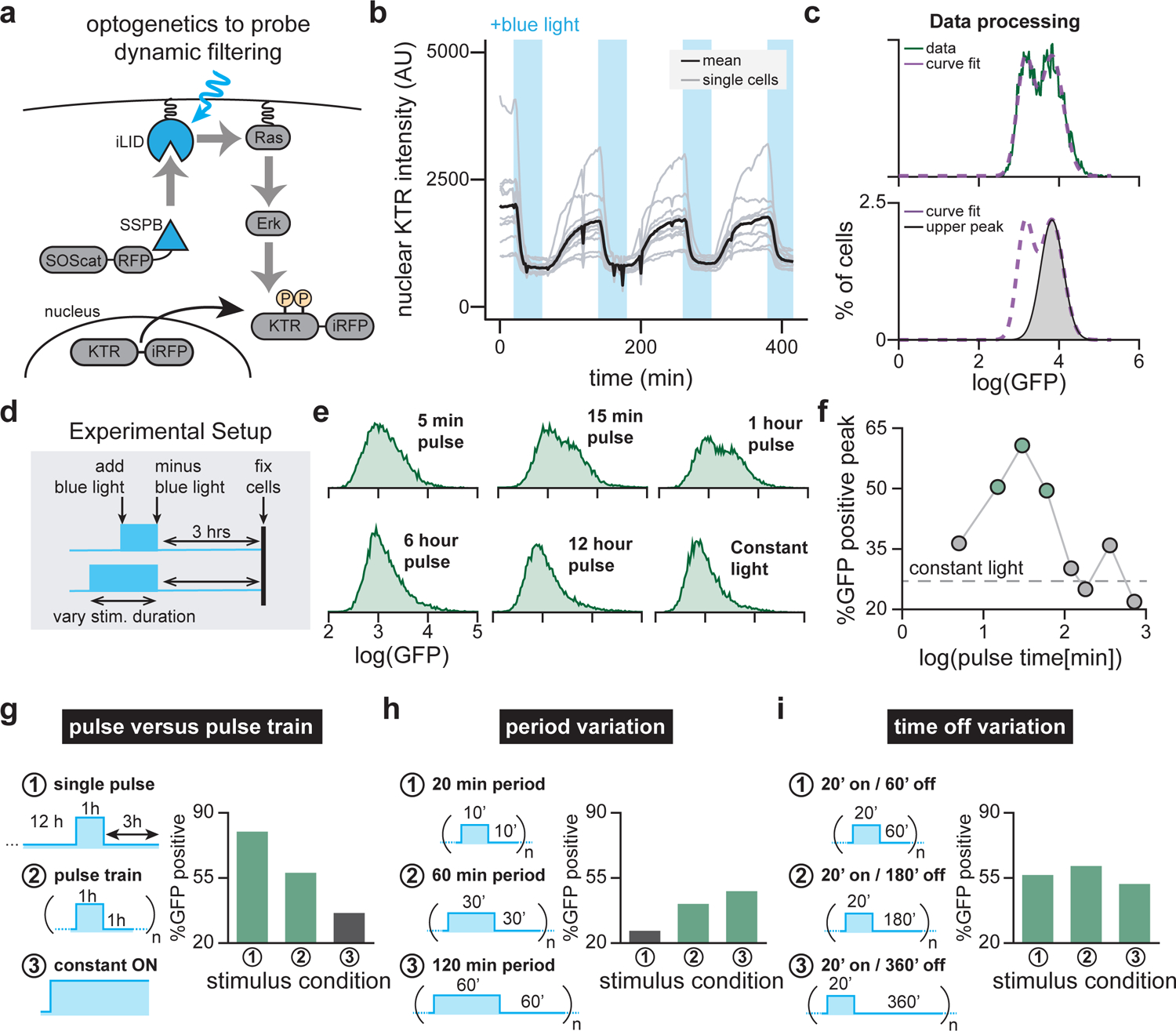
(a) Diagram of the OptoSOS system and ErkKTR biosensor. Blue light drives heterodimerization between tagRFP-SSPB-SOScat and membrane-localized iLID-CAAX. Upon recruitment to the membrane, SOScat activates Ras/Erk signaling. Phosphorylation of the ErkKTR-irFP biosensor by Erk leads to its export from the nucleus. (b) Single-cell traces showing nuclear ErkKTR-iRFP during cycles of optogenetic stimulation. Blue bars indicate periods of optogenetic illumination. Curves represent single cells (gray lines, n = 11 cells) and the mean of all cells (black line) from a single experiment. (c) Data processing pipeline for all READer optogenetic experiments. The GFP histogram is fit to a sum of two Gaussians (top) and then the area under the curve of each Gaussian is used to estimate the fraction of GFP-high cells (bottom). (d) Mapping how optogenetic pulse stimulation affects READer circuit output. Light inputs of varying duration were applied to cells, which were fixed 3 h after the end of the pulse. (e) GFP histograms for OptoSOS READer cells subjected to the experimental workflow shown in d. (f) Quantification of flow cytometry data in e (points) as well as constant-on control (gray dotted line). See Figure 4H for results from an independently derived clonal cell line. (g) READer responses to a single pulse versus oscillatory stimulus. Cells were exposed to (1) a single 1 h light pulse followed by 3 h in darkness before fixation, (2) a pulse train of alternating 1 h on/1 h off periods for 16 h or (3) constant light for 16 h. (h) READer responses at various oscillation periods. Cells were given pulse trains at various periods T, each at 50% duty cycle, for 12 h: (1) T = 20 min, (2) T = 60 min or (3) T = 120 min. (i) READer responses to various times between pulses. Repeated 20 min pulses were delivered every 60 min (case 1), 180 min (case 2) or 360 min (case 3) for a total of 12 h, with each leading to comparable GFP responses. For g-i, data is shown from one of two independent replicates.
We next set out to characterize READer responses to oscillating Erk inputs, dynamics that have been reported for the Erk pathway (Albeck et al., 2013; Aoki et al., 2013; Goglia et al., 2020; Shankaran et al., 2009) but that are difficult to implement by growth factor washes. We stimulated OptoSOS READer cells with a blue LED panel in a tissue-culture incubator, using an Arduino-microcontroller to drive programmable time-varying inputs. We first compared a train of eight 1 h-on, 1 h-off pulses to either 16 h constant illumination or a single light pulse delivered after 12 h in GF-free media (Figure 3G, left). Similar GFP induction was observed in cells stimulated with the pulse train and the single pulse, indicating that the READer system is broadly responsive to both forms of dynamic Erk activity (Figure 3G, right). We next tested duty-cycle matched inputs of varying periods from 20–120 min, each applied for the same 16 h total duration (Figure 3H, left). We found that the READer did not detect pulses in the 20 min period condition, likely because the fast switching timescale led the input was perceived as a constant-on stimulus, but both slower-varying stimuli led to a GFP response (Figure 3H, right). Finally, we tested how the time between pulses affected READer system activation for a fixed 20 min pulse duration and 16 h total stimulus time (Figure 3I, left). We found potent GFP induction as the time between pulses was varied from 1–6 h (Figure 3I). Together, these data indicate that READer is permissive to a wide range of pulsatile inputs: both pulse trains and single pulses elicit a GFP response if cycles are not so rapid that they are mis-perceived as a constant stimulus. This broad permissiveness is ideal for studies aiming to cast a wide net by capturing all cells undergoing a dynamic Erk response. Nevertheless, future variants of the READer system should be designed for more selective dynamic filtering, such as the ability to discriminate single pulses from pulse trains or to detect specific windows of on-time, off-time, or duty cycle.
Defining READer circuits with altered signal processing capabilities
What parameters define the signaling dynamics that are transmitted to GFP accumulation in the READer circuit? Answering this question would enable the design of alternative READer circuits with shifted band-pass capabilities, enabling researchers to selectively detect pulse durations of interest. To address this question, we queried our mathematical model for parameter sets with different band-pass filtering capabilities (Supplementary Text). We first defined three features to characterize the band-pass filter (Figure 4A): the maximum amplitude of GFP accumulation (‘height’), the pulse length at maximum GFP accumulation (“position”), and the difference between maximum and minimum pulse lengths leading to half-maximal GFP accumulation (“width”) and measured each feature as a single parameter was varied (Figure 4B). We observed that four parameters (k1, β1, β2, and k3) were capable of strongly shifting the position of the band-pass filter (Figure 4B–C), and tuning three of these parameters (k1, β2, and k3) in conjunction could shift the model’s band-pass peak over a broad range (Figure 4D–E; see Supplementary Text). These computational results indicate that the READer circuit, and “Circuit 7” with negative feedback more broadly, might indeed be capable of tunable dynamic filtering.
Figure 4. Altering band-pass filtering by tuning READer circuit parameters.
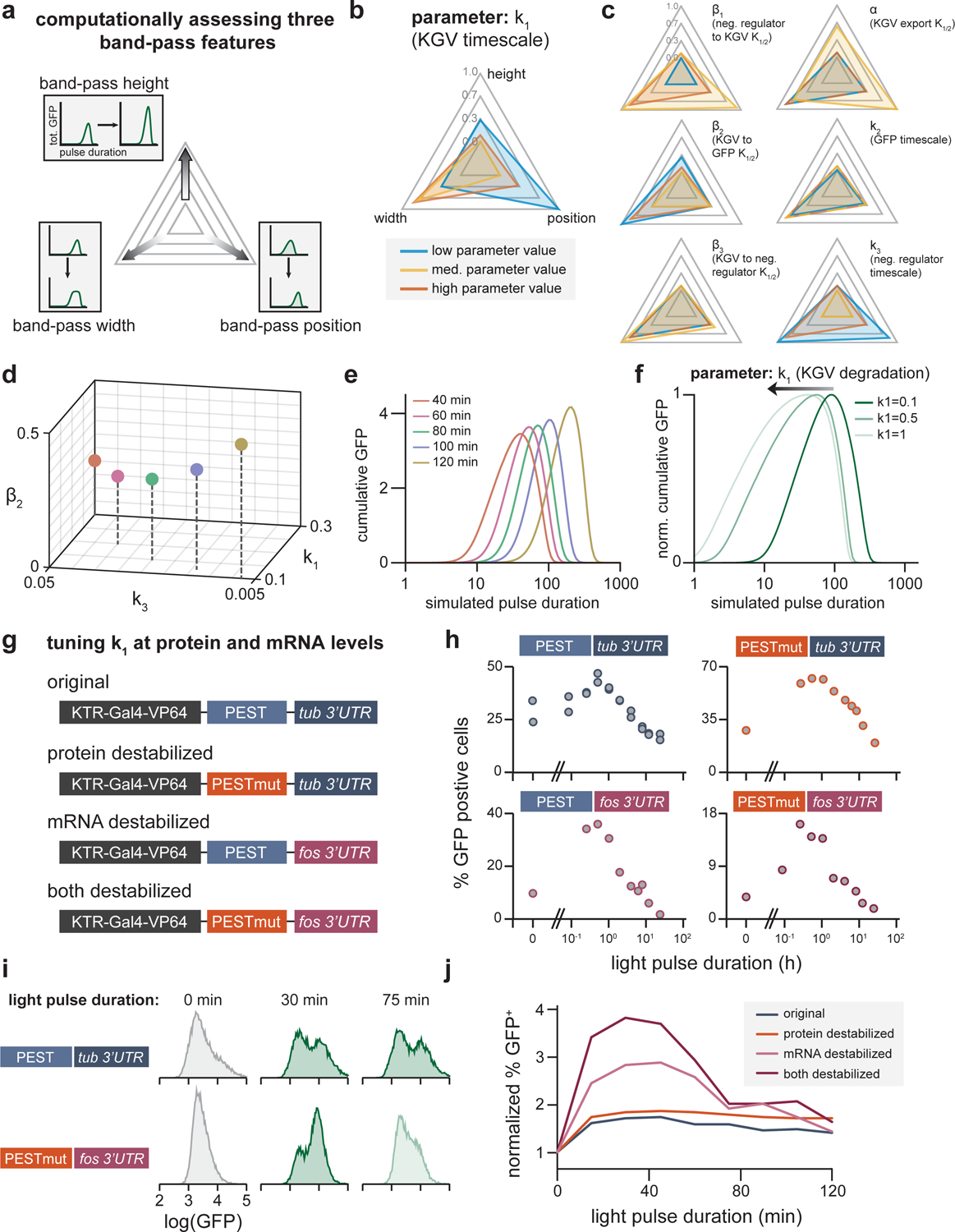
(a) Schematic of plots showing band-pass features as parameters are varied. For the mathematical model described in Figure 2H, each parameter was varied from 100-fold up and down the baseline value. The resulting band-pass curves were then analyzed for three features: band-pass height (maximum GFP response across all pulse durations), band-pass width (the span of durations achieving half-maximal GFP response) and band-pass position (the pulse duration that results in maximal GFP). (b) Example radar chart showing the results of the parameter scan for the relative changes in all three band-pass features. The lowest, baseline and maximum values of the parameter are shown in blue, yellow, and red, respectively. The further away from the center, the higher the value a particular simulation has for that feature. (c) Radar charts for remaining parameters as displayed in b. (d-e) Simultaneously tuning k1, k3, and β2 enables more complete control over band-pass filtering. For the altered parameter values shown (in d), band-pass responses exhibited shifted positions but similar widths and amplitudes (in e). (f) Simulated band pass curves (GFP output for pulses of different duration) produced for different values of the x1 node’s stability (parameter k1). (g) Schematic of altered READer circuits implementing enhanced mRNA or protein degradation. (h) Experimentally measured band-pass responses for all four READer circuit variants after stimulation with light pulses as in Figure 3. (i) Representative GFP histograms for the “original” and “both destabilized” READer variants incubated in constant dark conditions, 30 minutes of blue light or 75 minutes of blue light, indicating that the destabilized circuit rejects the 75 min pulse but responds to the 30 min pulse. (j) Quantification of fold-change in the GFP+ fraction between unstimulated and pulse-stimulated conditions for all four READer circuit variants in response to optogenetic pulses of the indicated lengths. Combined mRNA/protein destabilization results in narrow band-pass filtering and a high fold-change between unstimulated and pulsed conditions.
We next sought to confirm these computational results experimentally but recognized that simultaneously varying multiple model parameters in an experimental system can be extremely challenging. Fortunately, we found that even varying just a single experimentally addressable parameter (the KGV transcription factor stability, k1) resulted in some control over band-pass filtering (Figure 4F). We thus sought to define alternative READer circuits in which the KGV transcription factor was destabilized at either the mRNA or protein level. We replaced the tubulin 3’UTR with the less-stable FOS 3’UTR to tune mRNA stability and identified a more potent PEST sequence to decrease protein stability (Li et al., 1998; Shyu et al., 1989) and constructed new optogenetic READer cell lines incorporating either change or both changes together (Figure 4G). Indeed, cells harboring destabilized KGV variants exhibited a narrower band-pass response (Figure 4H–J). We found that incorporation of both modifications led to GFP induction only in response to pulses of 1 h or less (Figure 4I) as well as the highest fold change in the fraction of GFP+ cells between constant-off and pulsed conditions (Figure 4J). Taken together, our simulations and experiments converge on an intuitive result: the READer circuit acts as a band-pass filter whose pulse detection characteristics can be further tuned by modulating the mRNA/protein stability of the engineered transcription factor.
The READer circuit detects endogenous Erk signaling pulses
We have seen that the READer circuit responds selectively to externally supplied stimulus pulses; can it also detect spontaneous, naturally occurring Erk pulses? We previously had noticed that a small subpopulation of READer-expressing fibroblasts expressed high levels of GFP even when cultured in constant GF-free or growth media (Figure 2D; Movie S1), raising the possibility that this sub-population may undergo spontaneous Erk pulses that are then detected by the READer circuit. To directly confirm whether such a pulsatile sub-population exists, we transduced NIH3T3 fibroblasts with a fluorescent ErkKTR-irFP biosensor and imaged them for 48 hours in continuous growth media or immediately after the switch to GF-free media. Indeed, we observed that a subpopulation of cells underwent Erk pulses in growth media and began to pulse spontaneously after approximately 12 h of culture in GF-free conditions (see example trajectories in Figure S7A; Movie S2). These data would be consistent with the appearance of a GFP-positive population being driven by spontaneous pulses.
To directly compare endogenous Erk pulses to READer-based GFP accumulation, we next transduced our READer clonal cell line with the ErkKTR-mScarlet fluorescent biosensor to monitor both Erk and READer dynamics in the same live cells (Figure 5A). We also tested a third variation on long-term culture conditions for observing spontaneous Erk pulses: overnight incubation in GF-free media followed by a shift to long-term culture in growth media, to avoid the cytotoxicity associated with long-term imaging in GF-free conditions. We hypothesized that this protocol would also elicit spontaneous Erk pulses on a similar timescale as the switch to GF-free conditions, a phenomenon we had previously observed in spontaneously pulsing keratinocyte cells (Goglia et al., 2020). Model simulations confirmed that an acute switch from GF-free to growth media would indeed prevent READer system activation during a phase of high, constant Erk activity and elicit a sharp rise in GFP induction upon a shift to stochastic Erk pulses (Figure 5B). Subsequent experiments confirmed this picture: serum stimulation first drove a constant-on Erk state and ErkKTR nuclear export, with cells spontaneously initiating Erk activity pulses ~15 hours after serum stimulation (Figure 5C; Movie S3). These cells also exhibited a rapid rise in GFP levels that appeared to be correlated with the shift to a pulsatile state.
Figure 5. READer detects endogenous Erk pulses.
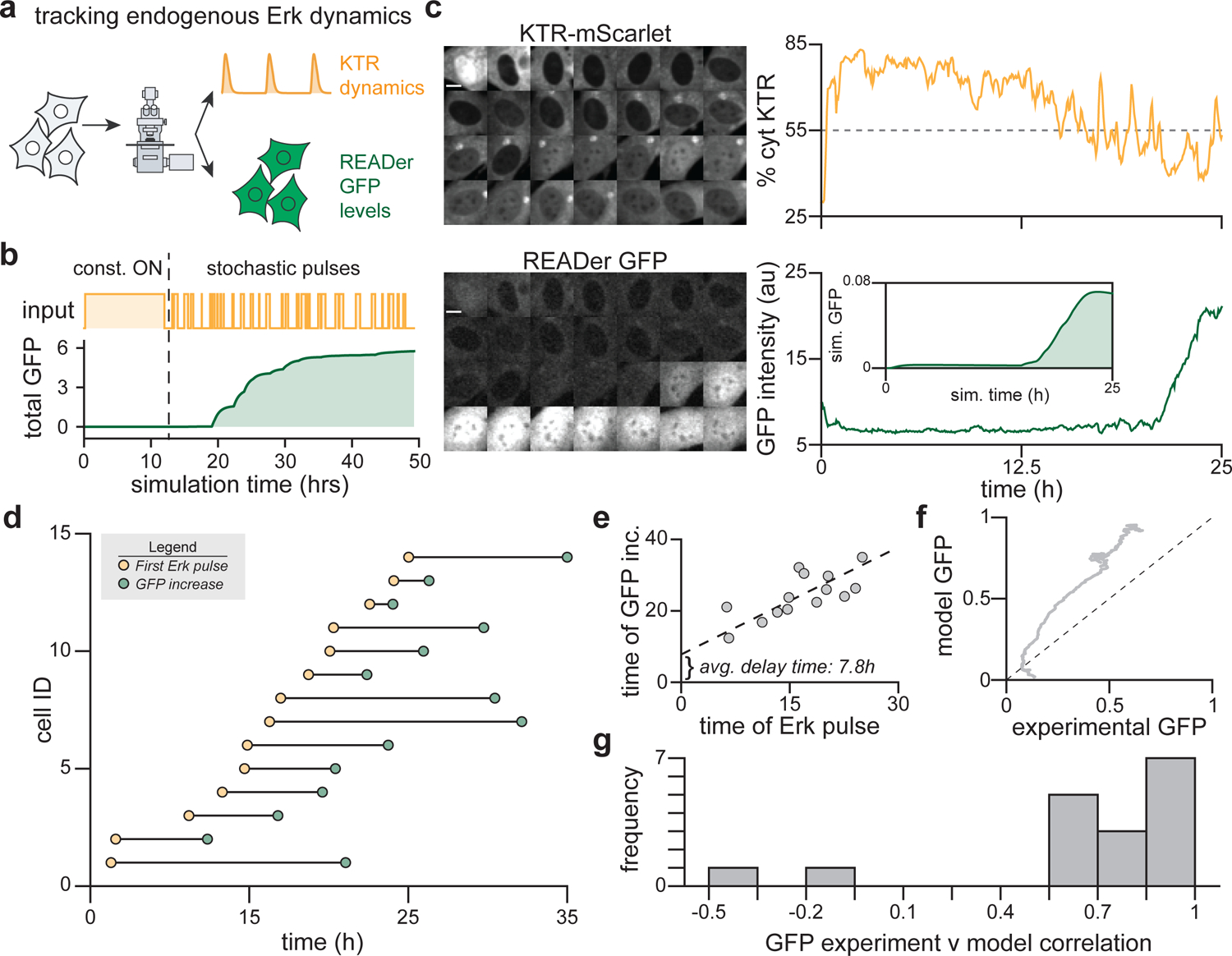
(a) NIH3T3 READer cells transduced with ErkKTR-mScarlet can be used to simultaneously visualize Erk activity dynamics and READer GFP output in single cells. (b) Simulated READer response during the switch from constant-on to stochastic pulses of Erk activity using the model from Figure 2H. (c) Images and quantification from a representative READer cell expressing ErkKTR-mScarlet stimulated with serum at time 0 and imaged for 2 days; see Figure S7 for full trajectories of all cells. Confocal images and quantification of the ErkKTR-mScarlet cytosolic fraction and GFP intensity are shown. Inset shows the simulated GFP response when the same pulsatile ErkKTR-mScarlet trace is used as a model input (see Supplementary Information). Scale bars: 10 μm. (d) Quantification of all cell trajectories (n = 17 cells from a representative experiment) for the time at which the cytoplasmic ErkKTR fraction first falls below 55% (yellow points) and when GFP intensity rises 25% above its initial level (green points). Note that 3 cells fail to cross at least one of these thresholds during the entire time course and are thus excluded from this plot. (e) Plot of data from d with best-line fit, revealing a delay time of ~7.8 h between initiation of Erk pulses and the GFP increase. (f) Comparison of overall experimental GFP induction to modeled GFP output for each cell; see Figure S7 for all individual experimental and simulated trajectories. The mean GFP induction is shown at each timepoint for all simulated and experimental data; dotted line shows identical values. (g) Histogram of correlation coefficients between modeled and experimental GFP induction for each cell as in f.
We next set out to quantitatively analyze the relationship between Erk dynamics and GFP responses across cells. We found that the time of the switch to pulsatile Erk (which we estimated across all cells as the first time point where the ErkKTR C/N ratio dropped below 55%) correlated well with the time of GFP induction (estimated as the first time point where GFP rose 25% above its initial baseline) (Figure 5D). From these data we calculated the time delay between the shift to Erk pulsing and the GFP increase, revealing a ~7.8 h delay (Figure 5E) that was within the 3–12-hour period of GFP accumulation previously observed for serum pulses (Figure S4A–C). Finally, we binarized each cell’s Erk activity trace (using the 55% C/N ratio as the cutoff between Erk-OFF and Erk-ON states) and used it as the input for our mathematical model to predict GFP induction. We found that modeled and experimental GFP correlated strongly across all cells (Figure 5F–G; see Figure S7B for additional cells and simulations), indicating that our feedforward loop can explain GFP responses to experimentally observed Erk dynamics. Taken together, these data demonstrate that the READer circuit can indeed sense spontaneous, naturally occurring Erk pulses.
READer provides distinct dynamic information from the Erk target gene c-Fos
As a final test of the READer biosensor, we set out to compare its response to staining for classic Erk target genes. To our knowledge, no endogenous Erk target genes have been identified that specifically sense pulsatile stimuli, but the Fos immediate-early gene product has long been used as a marker to identify cells exhibiting sustained levels of Erk pathway activity (Murphy et al., 2004). Fos staining has been particularly useful in neuroscience, as it labels cells that have recently experienced high levels of neuronal activity (Hunt et al., 1987; Kim et al., 2015). We thus wondered whether READer and Fos might provide overlapping or distinct information about the shared Erk pathway by responding to transient and sustained stimuli, respectively (Figure 6A).
Figure 6. READer provides distinct information from the canonical Erk target gene Fos.

(a) Although both READer and the canonical Erk target gene Fos report on the shared Ras/Erk pathway, they may be predicted to respond to distinct (transient vs sustained) dynamics. (b) Representative images of NIH3T3 READer cells that were fixed after overnight incubation in growth factor free media (off), after a 20 min pulse of serum followed by 180 min of GF-free media (transient), or after 180 min after stimulation with constant 10% serum (sustained). Cells were stained for Fos protein (red) and imaged for GFP (green). Scale bar: 20 μm. (c) Quantification of Fos immunofluorescence (left) and GFP fluorescence (right) from cells stimulated as in b and incubated for the indicated times in continuous growth media (sustained) or after a 20 min pulse of 10% serum (transient). Points represent the mean normalized intensity across all cells for sustained (square points) and transient (circles) stimuli; lines represent overall means across two biological replicates. See Figure S8 for full READer/Fos joint distributions.
To directly compare Fos and READer activation in response to different dynamic stimuli, we incubated cells overnight in GF-free media, and switched them to either sustained growth media or a 20 min pulse of growth media. We then fixed cells at various time points post-stimulus and monitored Fos protein by immunofluorescence and READer-induced GFP fluorescence in the same cells. We observed rapid, strong induction of Fos at early time points regardless of stimulus duration, demonstrating that simply measuring Fos cannot be used to discriminate pulsatile from sustained signaling. In contrast, the READer circuit only triggered GFP expression in response to a pulse but not constant stimulation (Figure 6B–C; see Figure S8 for full joint READer/Fos distributions). These results confirm that the dynamic information using READer cannot be obtained by staining for classic Erk target genes like Fos.
Discussion
Here we report the discovery and characterization of a simple gene network that can selectively and robustly differentiate between pulsatile and constant signaling states. Our network is based on an incoherent feedforward loop with slow activation and fast repression (Figure 1). Incoherent feedforward loops have been studied extensively for their dynamic filtering capabilities, including pulse generation and temporal ordering (Alon, 2007; Csikasz-Nagy et al., 2009; Mangan and Alon, 2003); our work adds highly selective pulse detection to this list of capabilities. We also report a simple, flexible implementation of this network architecture for mammalian signaling, centered on the pathway-regulated expression of a transcription factor that is fused to a kinase translocation reporter (Figure 2). Although we have focused on Erk signaling in this work, we believe this report provides the roadmap to the development of a suite of new reporters that capture the previous dynamic history for many dynamic signaling pathways (i.e. p53, Wnt, NFkB, etc.) (Hoffmann et al., 2002; Nelson et al., 2004; Purvis et al., 2012; Sonnen et al., 2018; Tsiairis and Aulehla, 2016). Kinase translocation reporters are available for a growing number of proteins and pathways (Miura et al., 2018; Sampattavanich et al., 2018) and other forms of fast negative regulation (e.g., signal-induced protein degradation) would be expected to work with similar efficacy.
While we have developed a single-measurement reporter of signaling dynamics, there are some important limitations to the system that should be addressed in future studies. Most importantly, we built the READer circuit using a combination of synthetic components (e.g., KTR; Gal4) and natural ones (e.g., the cell’s transcriptional machinery for transducing Erk activity to FOS promoter activation). Such a ‘hybrid’ system (Toettcher et al., 2010) greatly simplified our design, as the FOS promoter already implements potent and rapid Erk-responsive transcription from a low initial state. However, the inclusion of natural components also raises the possibility of regulation from additional biological sources. For example, we have seen that the current READer system only triggers GFP expression in a subset of cells that receive an Erk pulse (Figure 2, Figure S5) and responds more strongly to serum than optogenetic Ras stimulation. These results match analogous observations from the endogenous FOS gene (Jena et al., 2021) and are likely related to the underlying biology of cellular control over immediate-early gene expression. In future studies it would thus be highly desirable to engineer a fully synthetic READer circuit, bypassing the use of endogenous circuit elements to avoid potential crosstalk and trigger GFP induction in all Erk-pulsing cells.
A second challenge for future synthetic dynamics-detection circuits is that of recording timescale. The current READer system reports only on cells’ recent history, detecting pulses that occurred during the 3–12 hours prior to measurement (Figure S4). While this is suitable for some scenarios (e.g., performing high-throughput genetic screens), a longer memory would be useful for some applications, such as relating signaling dynamics to cell fate choices that may occur over days. Such a long-lived READer system could be achieved by replacing the GFP reporter with a genome modification step (e.g., CRISPR editing or a recombinase-based reporter).
Finally, while the READer circuit is broadly sensitive to pulsatile signaling activity (Figure 3), it is ill-suited for distinguishing between closely related dynamics (e.g., short versus long pulses; single pulses versus pulse trains). Initial studies suggest that narrow, tunable dynamic detection can be achieved by altering specific circuit parameters (Figure 4), and we look forward to future variants of the READer design that can perform even more complex filtering.
Overall, biosensors like the READer circuit could be transformative for mapping signaling dynamics in vivo, for large-scale genetic screens to identify the biochemical networks that generate pulses, and for tracing the lineages and eventual fates of pulsing cells. Such circuits may shed new light on the roles played by signaling dynamics in diverse contexts from disease to development.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jared Toettcher (toettcher@princeton.edu).
Materials availability
There are no restrictions on material availability. Plasmids are available from Addgene (#176530 and #178531; see www.addgene.org/Jared_Toettcher), and all cell lines produced in this study will be made available upon request.
Data and code availability
There are no restrictions on data availability. All data generated or analyzed during this study are included in this published article and its supplementary files or will be shared by the lead contact upon request.
MATLAB code for simulating the computational model is available on the laboratory GitHub page (doi:10.5281/zenodo.5482931). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line maintenance
All cells (NIH 3T3s and HEK 293Ts) were grown in DMEM plus 10% FBS in Thermo Fischer Nunc Cell Culture Tissue Flasks with filter caps at 37C and 5% CO2.
METHOD DETAILS
Plasmid Construction
We cloned all of our constructs/synthetic gene circuits into the pHR lentiviral expression plasmid (Naldini et al., 1996) or into Piggybac synthetic immediate early gene (synIEG) plasmid (Ravindran et al., 2020; Yusa et al., 2011). All linear DNA fragments were prepared by PCR, using GXL polymerase for fragments > 7500 bp and HiFi polymerase (Takara Bio) for shorter fragments. After PCR, Dpn1 digestion was used to remove template DNA. PCR products were cut from agarose gels and purified using the Nucleospin gel purification kit (Takara Bio). Final plasmids were constructed using Infusion assembly and amplified in Stellar chemically competent E. coli (Takara Bio). DNA was extracted by miniprep (Qiagen). All plasmid verification was done by restriction enzyme digestion and Sanger sequencing (Genewiz).
To create the KTR-Gal4-VP64 synthetic transcription factor plasmid, we took the original synIEG plasmid (pFos – destabilized GFP – Tubulin 3’UTR – pCMV – BFP) and replaced the GFP with KTR-iRFP (Addgene #111510) (Dine et al., 2018), keeping the PEST destabilizing element at the end of the coding region. In a second round of cloning, the iRFP was replaced with a Gal4-VP64 fusion protein amplified from pHR_pGK_LaG17_synNotch_Gal4VP64 (Addgene number: 79127, a gift from the Lim lab) (Morsut et al., 2016). For the reporter gene, we started pHR_Gal4UAS_IRES_mC_pGK_tBFP (Addgene #79123) (Morsut et al., 2016). First, the tBFP was replaced with iRFP (Addgene# 111510) (Dine et al., 2018). In a second round of cloning, we amplified destabilized GFP from the original synIEG plasmid and cloned it in place of the IRES-mCherry element.
READer cell line generation
Lentivirus production
Lenti-X 293T cells (Clontech) were plated in a 6 well plate at ~40% confluency at least 12 hours before transfection. The cells were then co-transfected with 1.5 ug of the pHR reporter vector along with 1.33 ug and 0.17 ug of CMV and pMD packaging plasmids, respectively, using Fugene HD (Promega). Virus was collected after 48 hours post-transfection and filtered through a 0.45 μm filter. To the ~2 mL of viral media, 2 μL of polybrene and 40 μL of 1 M HEPES were added. Cells to be infected were plated at 40% confluency in a 6 well plate at least 12 hours before infection and then 200–500 μL of viral media was added the cells. 24 hours post transduction virus containing media was replaced with fresh media.
PiggyBac Integration
NIH 3T3s that were infected with the reporter construct and were to be integrated with KGV TF PiggyBAC plasmid were plated 24 hours prior to transfection. 2.08ug of the plasmid to be integrated along with 0.41 μg of the PiggyBAC helper plasmid were co-transfected into the cells using Lipofectamine LTX with Plus reagent. Cells were then selected using FACS for dual BFP/iRFP expression after 3 days post-transfection. Cells underwent single cell sorting to isolate clonal cell lines.
Variants on cell line
To perform optogenetic experiments, pHR OptoSOS-tgRFP was introduced into clonal cell line generated above. To do so, lentivirus was generated from pHR SFFVp tgRFP-SSPB-SOScat-P2A-ILID-CAAX as described above. READer clonal cells were plated at ~40% confluency in 6-well plates and infected with virus. To perform simultaneous visualization of Erk pulsatile activity and READer output, we lentivirally transduced the original READer clonal cell line with pHR SFFVp KTR-mScarlet.
To develop the versions on the READer circuit with different degradation properties, we first generated a clonal cell line containing the OptoSOS construct as well as the reporter plasmid (both introduced by lentiviral transduction). This line was then validated to have photoswitchable Erk activation by introducing KTR-iRFP using lentivirus (Goglia et al., 2020) and applying cycles of light. After this, 4 different variants of the READer circuit (combinations of PEST/PESTmut and tubulin/fos 3’UTR) were introduced using PiggyBAC integration method described above, and cells were subjected to another round of clonal cell line generation.
Microscopy
Preparation
Cells to be imaged were plated into InVitro Scientific’s 96 well, black-walled, 0.17mm high performance glass bottom plates. 10 μg / mL of fibronectin diluted in PBS was placed on the wells, washed off and then cells were plated in DMEM with 10% FBS for at least 12 h. 12 h prior to imaging, cells were placed in growth factor free media (DMEM with 0.00476 mg / mL HEPES). 50 μL of mineral oil was pipetted onto the wells right before placing onto the scope to prevent media from evaporating.
Imaging
Cells were maintained at 37C with 5% CO2 for the duration of all imaging experiments. Confocal microscopy was performed on a Nikon Eclipse Ti microscope with a Prior linear motorized stage, a Yokogawa CSU-X1 spinning disk, an Agilent laser line module containing 405, 488, 561 and 650 nm lasers, a 60X oil or 20X air objective and an iXon DU897 EMCCD camera.
Preparation of fixed cell samples
Cells to be analyzed via flow cytometry were plated at ~60% confluency in 6-well plates. 36 hours post-plating and 12 hours before the experiment began, cells were switched into growth-factor free media (DMEM with 0.00476mg/mL HEPES). Serum additions (up to 10%) were then given and removed depending on the stimulus. After stimulation protocol was applied, media on cells was aspirated and cells were washed with PBS. After trypsinizing cells, neutralizing the trypsin with media and spinning cells down, the supernatant was removed, and the cell pellet was resuspended in a 2% PFA solution (50:50 mixture of BD CytoFix solution and PBS). After leaving the samples at 4 °C for 20 minutes, cells were spun down and then resuspended in cold PBS. Samples were run on LSRII Flow cytometer between 24 and 48 hours after fixation. Gating strategy is described in the supplemental text.
Pulse experiments
Simple, single pulse experiment
READer clonal cells were plated into 6 well plates 24 hours before the start of stimulation and placed into growth-factor free media 12 hours prior to stimulation. For the constant off condition, cells were given 200 μL of growth-factor free media and both the constant on and pulsed conditions were given 200 μL of FBS (to a final concentration of 10% v/v). After 1 hour, the media on the cells of the pulsed and constant off condition was removed, and fresh growth-factor free media was added while the media for the cells in the constant on condition was switched for regular growth media (containing growth factors). After 3 hours, the cells underwent the fixation protocol described above.
Bandpass pulse experiment – serum
READer clonal cells were plated into 6 well plates 24 hours before the start of stimulation and placed into growth-factor free media 12 hours prior to stimulation. After this 12-hour starvation period, the constant on condition was given serum to a final concentration of 10% v/v, and the longest duration of pulse (typically 12 hours) was given serum to the same final concentration. Then, working backwards from longest pulse duration to shortest, serum was added to the appropriate well. After the shortest pulse duration, all wells that were receiving pulsed inputs, as well as the constant off condition, were placed in growth-factor free media while the constant ON condition was placed in full growth media for 3 hours. After this wait time, cells underwent the fixation protocol described above.
For the optogenetic bandpass experiments described in Figure 3, READer clonal cells transduced with the OptoSOS system were plated in individual 35mm dishes 24 hours before the start of the experiments and, upon being placed into growth factor free media 12 hours prior to stimulation, were placed in a box covered in foil to protect the cells from the light. As described above, in order from longest pulse duration to shortest, the individual dishes of cells were placed in a light box stimulated with blue light. After all of the cells were placed in the box for the appropriate amount of time, the light plate was turned off for 3 hours. After this wait time, cells underwent the fixation protocol described above.
Off-time analysis
READer clonal cells were plated into 6-well plates 24 hours prior to the start of stimulation and placed into growth-factor free media 12 hours prior to stimulation. Cells were stimulated with a 15-minute pulse of serum and switched to growth-factor free media at staggered time points, so that all conditions were harvested together but experienced different intervals between the serum pulse and harvesting. From there, the cells underwent the fixation protocol described above.
Pulse train experiment
For pulse train experiments involving optogenetic stimuli, READer cells expressing the OptoSOS system were plated into 6 well plates 36 hours prior to experimentation. 12 hours prior to the beginning of the experiment, cells were placed in growth-factor free media and wrapped in foil to prevent light exposure. Light was delivered using custom-printed circuit boards of blue 450 nm light-emitting diodes (LEDs). During light stimulation, cells were maintained in an incubator at 37 °C in separate foil-wrapped boxes covered with separate blue LED boards delivering different patterns of light inputs. Each LED board was connected to a separate constant-current LED driver, all of which were controlled using an Arduino MEGA 2560 microcontroller board. The Arduino was programmed with open-source IDE software to deliver different dynamic light input regimes to each circuit board. To minimize phototoxicity, light inputs were delivered in cycles of 20 sec ON and 10 sec OFF which allowed us to minimize light exposure while still delivering a constant stimulus to cells by taking advantage of the slow (~0.5–1 min) dark decay rate of iLID activation (Guntas et al., 2015). After 16 hours in the light box, the cells underwent the fixation protocol described above.
Fos immunofluorescence staining
READer clonal cells were plated in wells of a 96-well plate. 24 hours post plating, and 12 hours prior to stimulation began, cells were placed into growth-factor free media. 20 minutes prior to each timepoint, the pulse condition was given a 20-minute pulse of serum by adding 10ul (10% by volume final concentration) of FBS and then placed back into growth-factor free media. At the endpoint of the 20-minute pulse, the corresponding ‘sustained-input’ well for that timepoint was given 10 μL of FBS (10% by volume final concentration). After all of the timepoints were completed, all cells were fixed by removing the media, adding 75 μL of CytoFix solution and incubating at room temperature for 10 minutes. CytoFix was then removed and an additional 100 μL of PBS was added and then dumped out to further wash out remaining CytoFix. Cells were then permeabilized by adding 100 μL of ice-cold 90% methanol to each well and incubating at −20C for 10 minutes. Methanol was then removed, and the cells were washed with 100 μL of PBS. Cells were then incubated in IF buffer (PBS + 10% FBS + 2mM EDTA) at room temperature for 1 hour. After blocking, the cells were then placed in 75 μL of primary antibody (Fos antibody Rabbit mAb – 9F6) at a 1:3000 dilution in IF-T buffer (50 mL of IF buffer + 150 μL Trixon X-100) and incubated overnight at 4 C. After primary incubation, cells were washed with IF-T buffer three times with 5-minute incubations between washes. Cells were then placed in 70 μL of secondary antibody that was diluted 1:500 in IF-T buffer and were incubated at room temperature for 1 hour. After 3 more washes using IF-T buffer with 5-minute incubations, DAPI was added to stain nuclei, and PBS was added for long term storage at 4 °C. Images were acquired with 24 hours of staining.
Cell cycle analysis
READer clonal cells were plated into T75 flask at 40% confluency. After 36 hours, the media was switched out for growth-factor free media for 12 hours. Serum was added for a final concentration of 10% by volume for 1 hour and then switched back to growth-factor free media. Cells were then fixed using the protocol delineated above. After 24 hours, the cells were sorted into two tubes, one for GFP-positive and the other for GFP-negative cells using the FACS Aria. The two populations were then spun down and re-suspended in FACS staining buffer (0.1% Triton X-100 in PBS). After a mild vortex to get single-cell suspension, PI was added to a final concentration of 20 μg/mL and RNase A was added to a final concentration of 200 μg/mL. After a mild vortex, the cells were incubated at room temperature for 30 minutes and then run on LSRII flow cytometer. FSC Express 7 was then used to perform cell-cycle analysis.
Mathematical Modeling
0. Basic simulation details
Computational screen
u(t) - pathway input (Erk activity) took on values of either 0 or 10. Inputs were either 0 simulation minutes (.0001 minutes for computational ease), 40 minutes or 400 minutes for a total simulation time of 500 minutes. Inputs represent constant OFF, pulsed and constant ON respectively.
x1(t) - intermediate node/TF and x2(t) – output node/GFP. Values were determined using ode15s and equations delineated in Figure S1.
For each simulation, random parameter values were drawn from a logarithmic scale and plugged into the respective model. For each input condition, after ode15s was run, GFP (x2) traces were cut off at the 400 minute mark for all inputs such that the 400 minute input simulation could represent constant ON. The MATLAB trapz() function was then used to find the area under the curve (AUC) of the resulting GFP traces under each input condition. Response ratio was calculated by dividing the AUC values of the pulse condition by either constant ON (Pulse versus ON) or constant OFF (Pulse versus OFF).
Pulse-scan experiment
u(t) - pathway input (Erk activity) took on values of either 0 or 10. 100 different inputs were used ranging from 1 simulation minute to 1400 simulation minutes, increasing in a log scale.
x1(t) - intermediate node/TF, x2(t) – output node/GFP, and x3(t) – negative feedback. Values were determined using ode15s and equations delineated in equations 28–30 of supplementary information.
For each input time, model was simulated in 3 parts: (1) from time 0 to 10, input was off (value of 0), (2) from time 10 to 10+pulse duration, input was on (value of 10), (3) from the end of (2) until that plus 1000 minutes, input was off. The final part was done to ensure that any GFP that could be made would be allowed to do so. The MATLAB trapz() function was used to calculate the area under the curve of the GFP trace and saved in a vector the same length as the space of pulse durations.
1. Computational screen for pulse detecting feed forward loops
1.1. Goal: identification of synthetic gene circuit to respond selectively to pulsatile signaling
Our objective in the report was to find a network architecture that could implement pulse detection in the hopes that we could use this architecture to make a synthetic gene to respond to pulsatile signaling events. With the advent of live-cell biosensors, it has become clear that many signaling pathways do not follow the textbook dogma of activating to a high constant manner. Instead, signaling pathways in mammalian cells display ornate time-varying patterns of activation. More recent advances in optogenetics and microfluidics have shown that different cellular fates rely upon these time-varying inputs to drive specific cell fates. Although connections between dynamic signaling pathways and their consequences are beginning to be revealed, there are still major unanswered questions about where such dynamics may exist in vivo and what genetic networks enable such pulsatile activity. As one needs high resolution, long-term imaging to capture dynamic signaling activity, live-cell biosensors are still limited in addressing such questions as they are relatively low-throughput and many endogenous contexts are inaccessible to such imaging. If one had a gene that selectively responds to such pulses, one could stain for such a gene to perform higher-throughput and in vivo fixed assays. Because no genes with selective pulse detection have been identified/characterized, we decided to engineer such a gene.
To build such a synthetic gene, we first had to identify networks that could enable robust pulse detection. We decided to focus on the feed-forward loop (FFL) network architecture class. FFLs are a pattern of genes in which an initial input acts an intermediate node and both then go on to act upon the final output node. If both the direct connection and the indirect connection from the stimulus to the output node have the same sign (both activating or both inactivating) the motif is deemed coherent; in the opposite scenario it is considered incoherent8,9. In the past, detailed characterization of these network motifs has revealed their wide range of filtering capabilities. For instance, the coherent FFL type 1 has been shown to enable persistence detection – sustained but not pulsatile inputs lead to expression of the final output10. Incoherent FFLs have been implicated in enabling fold-change detection, specifically in the case of WNT signaling11. Finally, it has been shown that many of the incoherent FFLs can respond to pulses, however their selectivity over sustained on or off signals has yet to be determined12.
1.2. Computational screen using AND logic
To determine which FFL architecture could implement pulse detection, we decided to first assay all eight of the possible combinations for a FFL with three connections using both AND and OR logic for the final output node. To build these models we started from a simple set of two equations in which the intermediate node (x1) is made in proportion to the amount of input and the output node (x2) is made by integrating the amounts of x1 and input. As an example, below is the derivation for Circuit 2 (Figure 1B):
| (1) |
| (2) |
In Eq. 1, u represents the signaling input and has values of either 0 uM when the input is off, and a value of 10 uM when the input is on. Species x1 is produced in a manner proportional to the parameter V1, and in proportion to one Hill function. In this Hill function, u acts as an inhibitor and C1 is the activation coefficient. As u surpasses C1 the fraction approaches 0 making the production term go to 0. On the other hand, when u is less than C1, the Hill function is close to 1, making the production term a positive number. The Hill coefficient n1 govern the steepness of this response. Species x1 degrades at a rate proportional to itself and parameter k1. The structure of Eq. 2 resembles the structure of Eq. 1, except there are now two hill functions integrated in AND logic. The activator in the first Hill function is x1 and the repressor in the second Hill function is u.
Next, we nondimensionalize Eq. 1–2 using the identities in Eq. 3–5 to focus in on the characteristic dynamics of the READer system.
| (3) |
| (4) |
| (5) |
We also define the following nondimensional parameters:
| (6) |
| (7) |
Inserting these nondimensional identities and parameters into Eq. 1–2 produces the equations used for Circuit 2 in the computational screen (Figure 1D, Figure S1–2):
| (8) |
| (9) |
From this derivation one can appreciate that a general form for the FFL loops is as follows:
| (10) |
| (11) |
In these equations, the positive (activating) form for the hill functions f, g, or h uses the form:
| (12) |
And the negative (repressive) form of the hill function uses the form:
| (13) |
For equations 12–13, in the case of functions f and h in equations 10–11, u would take the place of x and would take the place of .
In the following table, we delineate all of the equations used in the computational screen for each incoherent feedforward circuit with AND logic:
| Circuit 1 |
|
| Circuit 2 |
|
| Circuit 3 |
|
| Circuit 4 |
|
| Circuit 5 |
|
| Circuit 6 |
|
| Circuit 7 |
|
| Circuit 8 |
|
To perform the computational screen, we applied three separate inputs: 0.00001 (constant off), 4 (pulsed), and 40 simulation time units (constant on) to a randomly chosen set of parameters for each circuit for a total simulation time of 40 time unites. The area under the curve (AUC) of the x2 curve for each simulation was used to calculate the output under each input condition. By computing the ratio of the AUC of pulsed to those of the other constant inputs, we could identify simulations that enabled selective pulse detection. This was done 10,000 times for each circuit (Figure 1C–D). From this analysis, only Circuit 7 showed any robust level of pulse detection, with >90% of simulations resulting in greater output in the pulsed condition compared to the constant on and off inputs.
To ensure that our screen recapitulated known features of FFL, we looked at each individual circuit under the same parameter values. From this analysis, it was clear that Circuit 7 did provide pulse detection while the other circuits did not (Figure 1E, Figure S2). We also saw that Circuit 1, also known as Coherent FFL type 1, responded most in the sustained constant on input case (Figure S2A). This is in line with previous literature that suggests that this network motif enables persistence detection10.
1.3. Computational screen using OR logic
In the previous computational screen, we computed the final output node (x2) using AND logic. By this we mean that both input and x1 need to be positively acting on x2 at the same time to produce x2; if either input or x1 is a repressor, then their absence is require for the production of x2. We next wanted to perform the same screen except using OR logic for x2 when it integrates the levels of both x1 and input. To do this, we changed the form of equation 11 to the following:
| (14) |
In this setup, let us say that both g and h are activating functions (eq. 12). In the case when either x1 or u is greater than their respective threshold value, then, the inner (1- ) or ) go to 0 making the production term 1. Only in the case that both g(x1) and h(u) are approaching 0 does the production term go to 0. Using this logic, we arrive at the following 8 sets of equations for incoherent feedforward networks using OR logic:
| Circuit 1 |
|
| Circuit 2 |
|
| Circuit 3 |
|
| Circuit 4 |
|
| Circuit 5 |
|
| Circuit 6 |
|
| Circuit 7 |
|
| Circuit 8 |
|
We then perform the same computational screen that we performed for the AND gate logic. From this analysis, it was clear that none of the OR gate logics performed pulse detection as few simulations ever landed in the upper right hand quadrant (Figure S1B). However, one circuit seemed to display pulse specific repression – Circuit 5 (Figure S1C). Interestingly, this circuit is the logical inverse of Circuit 7 with AND logic. Circuit 7 with AND logic can be represented as:
| (15) |
If x2 is pulse detection and we are looking for pulse specific repression, we are essentially searching for (NOT x2). When we perform this NOT operation over eq. 15, we get:
| (16) |
The logical operation represented by eq. 16 is the one performed in Circuit 5 with OR logic.
1.4. Conclusion
Overall, we have successfully performed a computational screen over all FFL networks using both AND and OR logic for the final output node to search for pulse detecting circuits. Validating our approach, Circuit 1 (which has previously been identified as a persistence detection circuit) robustly responded to sustained signals more strongly than pulsed and off inputs. Using this screen, we identified one circuit topology – Circuit 7 – which provides pulse detection. Interestingly, our OR gate screen revealed that the logical inverse of Circuit 7 with AND gate logic, Circuit 5, allowed for pulse-specific repression, further validating the approach. This work, in conjunction with studies10,13–15, demonstrates the utility of computational screens for identifying biological networks that enable specific filtering capabilities.
2. Characterization of 3-node network that enables band-pass filtering
2.1. Goal: Understand and experimentally tune parameters controlling band-pass filtering
To our knowledge, this is the first network motif that has been described to possess such selective dynamic band-pass filtering capabilities at the protein level, providing the first insight into how cells may interpret different frequencies. However, how can different genes respond to different frequencies? What parameters do cells alter to change the selectivity and placement of the optimal response pulse? To address these questions, we used both mathematical modeling and experiments to understand what affects properties of the band-pass such as peak position, peak height and band-pass width (selectivity). To computationally assay the system, we took our model (Figure 2H) that has three equations for the three species (transcription of KTR-Gal4, GFP and negative regulator) with its 11 parameters and varied each parameter independently 100-fold down and up from the values previous used that seemed to recapitulate experimental data (Figure 2I). For each parameter set, we simulated a band-pass experiment in which we initiate pulses of different lengths and then wait a fixed amount of simulated time. Each resulting band-pass was then analyzed for the peak position, peak height, and width (selectivity). From this analysis, it was clear that there were 3 key parameters that affected these features: the timescale on which the KTR-Gal4 is made, the timescale that the negative regulator is made, and the affinity that Gal4 protein has for the GFP promoter (Figure 4A–F). By simply altering these three parameters, we could define band-pass curves of similar selectivity and peak height, for different peak positions (Figure 4G–J).
2.2: Mathematical model for parameter scan
Equations 17–19 describe the dynamics of the READer system. For simplicity, we do not represent every component of the READer system with an equation. Instead, we choose the minimal number of equations necessary to reproduce the most salient features of the READer system dynamics: pulse detection and band-pass filtering. Species x1 can be thought of as the transcription of KTR-Gal4, both in the active and inactive form. Species x2 can be thought of as the amount of nuclear KTR-Gal4 protein. Species x3 can be thought of as the amount of ERK negative regulator in cells. At t = 0 min, x1 = x2 = x3 = 0 uM.
| (17) |
| (18) |
| (19) |
In Eq. 17, u represents the ERK signaling input, which is on in the presence of growth factors or optogenetic stimulation and off when growth factors and optogenetic stimulation are absent. When off, u has a value of 0 μM, and when on u has a value of 10 μM. Species x1 is produced in a manner proportional to the parameter V1, and in proportion to two Hill functions. In the first Hill function, u acts as an activator and C1 is the activation coefficient. In the second Hill function, x3 acts as a repressor and C2 is the repression coefficient. The Hill coefficients n1 and n2 govern the steepness of the first and second Hill functions, respectively. Species x1 degrades at a rate proportional to itself and parameter k1. The structure of Eq. 18 resembles the structure of Eq. 17, except the activator in the first Hill function is x1 and the repressor in the second Hill function is u. Eq. 19 only contains a single Hill function with activator x1. We nondimensionalize Eq. 17–19 using the identities in Eq. 20–23 to focus in on the characteristic dynamics of the READer system.
| (20) |
| (21) |
| (22) |
| (23) |
We also define the following nondimensional parameters:
| (24) |
| (25) |
| (26) |
| (27) |
Inserting Eq. 20–27 nondimensional identities and parameters into Eq. 17–19 produces the nondimensionalized equations used in the main text (Figure 2H):
| (28) |
| (29) |
| (30) |
As discussed in the main text, the READer system serves as a bandpass filter that discriminates among different durations of ERK signaling. Using the set of equations from 28–30, we can reproduce all of the important dynamic filtering capabilities: (1) detection of a single short pulse of input (Figure 2D–G), (2) ignoring of long pulses (Figure 2G), and (3) band-pass filtering depending on Eq. 30 (negative feedback), meaning that both very short pulses and very long pulses do not result in maximum output and that an intermediate pulse results in the optimal output of the system (Figure 2I).
| Parameter | Value [units] |
|---|---|
| n1 | 1 |
| n2 | 5 |
| n3 | 5 |
| n4 | 5 |
| n5 | 5 |
| k1 | 0.1 [1/min] |
| k2 | 0.02 [1/min] |
| k3 | 0.025 [1/min] |
| β1 | 0.2 |
| β2 | 0.1 |
| β3 | 0.01 |
| α | 0.5 |
For the model, the values above were picked by hand to reflect the time scales of KTR and GFP production and degradation observed in the time course data (Figure S3). The variable corresponds to the amount GFP transcription induced by nuclear KTR-Gal4. We can generate a computational bandpass curve from Equations 28–30 by plotting the area under the curve of the trace for different values of pulse length where:
| (31) |
The total simulation time for each τ is τ +100 time units to allow for GFP to be made. The bandpass curve for the parameter values listed in Table 1 is centered around (i.e. has the highest value for) an ERK signaling duration of t = 41.8 min.
2.3: Parameter scan for variables that control bandpass filtering
To begin to understand what controls bandpass filtering we decide to perform a parameter scan for each parameter in eq. 28–30. We sought to better under how changing parameter values changes the bandpass curve’s peak location, peak amplitude, and selectivity. A higher width corresponds to a lower selectivity. Peak location is the pulse length τ that allows for the highest GFP and peak amplitude is the resulting GFP from this optimal pulse length. We define the selectivity of the bandpass as the width of the curve at half the maximum amplitude for each bandpass curve. The results of this analysis would shed light on how the physical properties of the molecules that compose READer and the native ERK-dependent processes it interacts with relate to the dynamic filtering features of READer.
The parameters in Table 1 relate to physical properties of the molecules that READer interacts with and is composed of. Parameters k1, k2, and k3 dictate the time scale of KTR-Gal4, GFP, and ERK negative regulator production, respectively, with low k values resulting in slow production and high k values resulting in fast production. In Equations 17–19, k1, k2, and k3 appear as the parameters governing the degradation of , and , respectively. The value of k1 can be altered through manipulation of the KTR-Gal4 degradation tag, and the value of k2 can be altered through manipulation of the GFP degradation tag. The value of k3 would be difficult to alter, as it corresponds to the degradation rate of the unknown ERK negative regulator native to the cells into which READer has been introduced. In all cases, a high k value would correspond to a protein with fast degradation.
β1, β2, and β3 are the threshold values that help determine whether , and are produced, respectively. Large amounts of KTR-Gal4 can only be produced when the amount of ERK negative regulator is less than β1, GFP can only be produced in large amounts when the amount of KTR-Gal4 exceeds β2, and negative regulator can only be produced in large amounts when the amount of active ERK exceeds β3. While β1 and β3 are properties native to the cells into which READer has been introduced, β2 can be altered by strengthening or weakening the UAS sequence that governs GFP production. A stronger UAS would lower the value of β2 while a weaker UAS would raise the β2 value.
Finally, α is the ratio of the u threshold C4 to the u threshold C1. When u > C1, large amounts of KTR-Gal4 can be produced. When u < C4, large amounts of GFP can be produced. Because u can only take on two values, 0 for uoff or 10 mM for uon, the ratio α = C4/C1 dictates whether or not a high level of GFP production is possible. If uon > C1 and C1 > C4, i.e. a < 1, KTR-Gal4 can be produced, but it is also excluded from the nucleus and thus cannot induce GFP production while ERK signaling is still present. If uon > C1 and C1 < C4, i.e. α > 1, then GFP can be produced while the ERK signal remains on. For READer to function well as a pulse detector, α must be less than 10. The value of α can be altered by changing the nuclear export sequence (NES) on the KTR-Gal4. A stronger NES would lower the value of α while a weaker NES would raise it.
To better understand how to achieve high-selectivity, high-amplitude bandpass filtering at various ERK signaling durations, we varied each parameter’s value from one hundredth its Table 1 value to one hundred times the Table 1 value. The radar plots show how peak position, peak selectivity, and peak amplitude vary in response to variations in k1, k2, k3, β1, β2, β3, and α (Figure 4B–C). The parameters that show the greatest variation in one or more of the bandpass filtering metrics, as shown by the greatest difference along one of the spider plot axes between the yellow (low parameter value) and blue (high parameter value) marker, are k1, k3, β1 and β2. After additional analysis we do find that β1 seems to break bandpass filtering at various parameters. This is intuitive as β1 represents the threshold where negative feedback acts on x1 so with high values of this parameter we break the negative feedback and bandpass filtering. Thus we focus further analysis on k1, k3, and β2. Although varying each parameter changes the value of multiple metrics, each parameter controls each metric to a different extent. k3 primarily changes the peak position: increasing k3 moves peak position to a higher ERK signaling duration. Moving the peak position to a higher ERK signaling duration by varying k3 also mildly increases the amplitude and selectivity. To center the bandpass peak around a shorter ERK signaling duration, k1 and β2 must be varied to maintain high amplitude and selectivity. Increasing β2 does nothing to the peak position, but it increases amplitude and decreases selectivity. Increasing k1 changes all three metrics: the peak position moves to a higher signaling duration, selectivity decreases, and amplitude increases. Notably, only two of the important parameters we identified have values that can be engineered in one direction or another. We predict k1 can be altered by changing the degron tag attached to the KTR-Gal4, while we predict β2 can be altered by changing the strength of the UAS sequence to which the KTR-Gal4 binds to produce GFP. k3, however, the parameter that most directly corresponds to the peak position, is dictated by the degradation rate of the unknown ERK negative regulator native to the system.
Because each of these three parameters control the three bandpass metrics to different extents, the values of the parameters can be adjusted in concert to achieve high-selectivity, high-amplitude bandpass filters for any ERK signaling duration. Plotting the parameters values that allowed us to achieve high-selectivity, high-amplitude bandpass filtering for ERK signaling durations of 40, 60, 80, 100, and 120 min, it becomes apparent that a curve can be drawn through 3D parameter space to select for bandpass filters of any duration (Figure 4D–E).
2.4: Experimental tuning of band-pass
To experimentally tune the READer system, we decided to try and change the degradation of the KTR-Gal4 protein and thus the timescale of the KTR-Gal4 (k1 in our model). Intuitively, if the KTR-Gal4 is degraded faster then fewer of the longer pulse durations will make it to GFP output. To test this, we needed a way to quickly turn on and off signaling on demand in clones that express the reporter GFP gene at equivalent levels. For the first, we used an optogenetic Ras/Erk activator previously created in our lab in which the catalytic domain of SOS is recruited to the membrane via the blue-light dimerization system, iLID and SSPB. We took cells expressing this system and infected them with the reporter GFP construct and sorted clones. To ensure that a clone had functional optogenetic Erk activation, we infected clones with ErkKTR-iRFP, placed them on the scope and subjected the cells to cycles of blue light and dark (Figure 3). From this analysis we chose one clone that had a functional optogenetic actuator and the reporter.
With this clone in hand we could now test variants of the READer circuit. To increase the degradation of the circuit at the protein level, we either added a modified PEST sequence that decreases the half-life even further than the original PEST sequence. At the transcriptional level we used either the original TUBULIN 3’ UTR or the one from FOS, which has been shown to have a short-lived mRNA. All combinations of these PEST sequences and 3’ UTRs were cloned into the READer PiggyBAC construct (Figure 4G). These variants were transfected into the test-bed clone and single-cell clones were then derived. When we did a full band-pass experiment with pulse durations ranging from 5 minutes to 12 hours, we found that all 4 READer circuits do implement band-pass filtering (Figure 4H). However, when we did a more fine-tuned experiment where we did every pulse duration in increments of 15 minutes from no stimulation to 2 hour pulses, we find that READer constructs harboring the FOS 3’ UTR have markedly increased selectivity (smaller band-widths) when compared to those that have the longer-lived TUBULIN 3’ UTR (Figure 4I–J).
2.6: Conclusion
Overall, we have successfully developed a minimal mathematical system of equations that recapitulates the bandpass filtering experimentally observed in the READer system. By performing a parameter scan for all of the major components of the system, we find that there are three parameters (k1, k3 and β2) that control the selectively, peak position and peak amplitude of the bandpass. Because each of these parameters control more than one of these features, we tune all three simultaneously to derive bandpass curves of specific peak position, height and selectivity. Experimentally we show that by tuning the k1 parameter (degradation of the KTR-Gal4) we are able to change the filtering capabilities of the READer system. This analysis points to the key knobs that nature may alter to specify which pulses may be read out by downstream targets of pulsatile signaling activity. This also provides us a framework with which to develop more synthetic systems to create more tunable and orthogonal dynamic channels.
3. Extending the model to our experimental implementation of the READer circuit
While our initial modeling was intended as a general, flexible implementation of different feedforward architectures, we also seek to model the operation of a particular experimental implementation of the READer circuit (e.g. in comparing modeled and experimental single-cell GFP responses to various ErkKTR-mScarlet dynamics; Figure 5). We thus sought to derive a model that retained the simplicity of our initial system while also appropriately representing Erk-triggered nuclear export of the KTR-Gal4-VP64 (KGV) transcription factor and its resulting effect on GFP production.
We first modeled nuclear KGV as partitioning between nucleus and cytosol, under the simplifying assumption that the two compartments are of equal volume. We can write:
| (32) |
where kcn and knc are the rate of movement into the nucleus and out of the nucleus, respectively – rates that could in principle be influenced by Erk (u). We denote the amounts of KGV in the nucleus and cytosol as xn and xc, respectively, and the total KGV protein as x1, the intermediate node produced by Erk-triggered gene expression in our generic feedforward model.
| (33) |
By assuming KGV nuclear/cytosolic transport equilibrates rapidly relative to changes in protein concentration and Erk activity, we manipulate Equations 32 and 33 to obtain:
| (34) |
We make two simplifying assumptions on the Erk-driven rates of nuclear import and export. First, we assume that Erk exerts its effect primarily through increasing KGV nuclear export. Second, we assume that KGV is primarily nuclear in the absence of Erk activity, an assumption that reasonably matches prior observations of strong nuclear ErkKTR localization in Erk-OFF conditions. Together, these allow us to write:
| (35) |
Plugging 35 into 34 and re-arranging gets us:
| (36) |
For an appropriate constant K, we thus obtain
| (37) |
A simple model of gene expression would hold that GFP production depends only on nuclear KGV concentration by a standard Hill production term, allowing us to write:
| (38) |
| (39) |
To compare our model to experimental data, we also included an additional integrator variable (x4) to represent total GFP protein produced from mRNA (x2). This leads to a full, 4-equation model:
| (40) |
| (41) |
| (42) |
| (43) |
| (44) |
QUANTIFICATION AND STATISTICAL ANALYSIS
Microscopy Data Analysis
Initial READer cell experiment analysis
Raw images were collected as ND2 files (Nikon Elements) and were subsequently imported into ImageJ. The ImageJ “Measure” tool was used to manually quantify mean intensity of the nuclei of cells of interest. These files were saved and then imported into R for subsequent statistical analysis and plotting. The code for this analysis can be found in Supplementary Code from (Ravindran et al., 2020).
Simultaneous KTR and READer analysis
ND2 files from Nikon Elements software were imported into ImageJ. The measure tool was used to quantify mean intensity of the nuclei and cytoplasmic intensity for cell of interest in the KTR channel (mScarlet). These text files were then saved and imported into MATLAB to capture nuclear intensity for READer channel (GFP) and to produce figures. Specifically, this code first reads in the two text files that provide the nuclear and cytoplasmic intensities of the KTR channel as well as provides the details for the position of the nucleus. From here, the percent nuclear KTR is calculated by dividing the nuclear intensity by the sum of the nuclear and cytoplasmic intensity and multiplying by 100. The details of the ellipse over time used to quantify the nuclear intensity is then read out (specifically the x and y coordinates, the width and the height of the ellipse). These are then used to create a mask that is then applied on a tif file version of the GFP images such that all intensity values are NaN except for those within the mask. The average intensity is then used to calculate the average GFP intensity in the nucleus over time. Return to a pulsatile state was defined as a drop in cytoplasmic intensity to 55% and an increase in GFP was defined as a 25% increase in GFP intensity over initial fluorescence intensity.
To feed the KTR intensities into our mathematical model, we first binarized the traces by taking any value above 45% nuclear KTR (55% cytoplasmic) to mean an Erk off state (input = 0) and any value above below 45% nuclear KTR (55% cytoplasmic) to mean an Erk on state (input =10). These traces were then fed into the model and the resulting GFP curve (variable x2) was plotted. Both the resulting model GFP traces and experimental GFP traces were min-max normalized and then interpolation was used on experimental GFP such that both traces had an equivalent number of points. Code is available upon reasonable request.
Fos analysis
ND2 files from Nikon were imported into ImageJ and saved as ‘.tif’ files. These were then imported into MATLAB and custom code was written to analyze the cells. Essentially, masks were created using DAPI image for each condition and then this mask was applied to the Fos and READer images. The mean nuclear intensity from each cell was then used for downstream analysis. Code is available upon reasonable request.
Transcriptional Burst Analysis
Protocol was adapted from (Wilson et al., 2017). Briefly, 7 z-stack slices spanning 4.5 um (0.8um between z-slices) which was centered on the middle of the nucleus. This z-stack was max projected to allow all the bursts to be visualized on a single plane. Positional information was tracked using the measure tool in Fiji. MATLAB code was used to take in the positional information, fit a 2-dimensional Gaussian to the identified region and finally calculated the integrated area under the fitted Gaussian as the burst intensity. The code for this analysis can be found in Supplementary Code from (Ravindran et al., 2020).
Statistical Test
For supplementary figure 3, statistical testing was done using a two-sided student’s t-test where p-values < 0.05 were considered significant.
Supplementary Material
Movie S1. Time-lapse imaging of NIH3T3 READer cells in response to different dynamic stimuli. TagRFP (white) is uniformly expressed in READer cells and can be used to identify cells. GFP (green) reports on READer system activation. Cells were imaged with a 60X oil objective every 6 min for 5 h. Panels show three stimuli: constant GF-free media (constant OFF; left), constant 10% serum (constant ON; middle), and a 10% serum pulse for 1 hour followed by GF-free media (pulse; right). Related to Figure 2.
Movie S2: Long-term imaging of NIH3T3 cells expressing ErkKTR-iRFP reveals spontaneous pulses. Cells were imaged using a 20X air objective every 3 min for 23 h in either growth media containing 10% serum (left) or were switched to GF-free media immediately prior to imaging (right). Related to Figure 5.
Movie S3: Time-lapse imaging of representative NIH3T3 READer cell expressing ErkKTR-mScarlet. Cells were imaged using a 20X air objective every 3.5 min for 48 h. After having been placed in GF-free media overnight, cells were stimulated with constant 10% serum at the beginning of imaging. Images show ErkKTR-mScarlet (left) and GFP (right), which report on Erk dynamics and READer system activation, respectively. Quantification of the images are shown below each, with the fraction of nuclear KTR shown on the left, and the GFP fluorescence intensity shown on the right. Related to Figure 5.
Acknowledgements
We thank all members of the Toettcher lab for helpful comments. We also thank Katherine Rittenbach and Dr. Christina DeCoste of the Princeton Molecular Biology Flow Cytometry Resource Center for cell sorting. This work was supported by NIH grant DP2EB024247 (to J.E.T.) and T32GM007388 (to R.H.T.), the Hertz Foundation Fellowship and NSF GRFP Fellowship (to S.M.) and the Lidow Independent Work 2019 Research Grant (to P.T.R.).
Footnotes
Declaration of Interests
J.E.T. is a member of the advisory board of Cell Systems. The authors have no other competing interests to declare.
References
- Adler M, Szekely P, Mayo A & Alon U Optimal Regulatory Circuit Topologies for Fold-Change Detection. Cell Syst. 4, 171–181.e8 (2017). [DOI] [PubMed] [Google Scholar]
- Albeck JG, Mills GB, and Brugge JS (2013). Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 49, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U (2007). Network motifs: theory and experimental approaches. Nat Rev Genet 8, 450–461. [DOI] [PubMed] [Google Scholar]
- Aoki K, Kumagai Y, Sakurai A, Komatsu N, Fujita Y, Shionyu C, and Matsuda M (2013). Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol Cell 52, 529–540. [DOI] [PubMed] [Google Scholar]
- Basu S, Mehreja R, Thiberge S, Chen MT, and Weiss R (2004). Spatiotemporal control of gene expression with pulse-generating networks. Proc Natl Acad Sci U S A 101, 6355–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaj LJ, Sabnis AJ, Mitchell A, Garbarino JE, Toettcher JE, Bivona TG, and Lim WA (2018). Cancer mutations and targeted drugs can disrupt dynamic signal encoding by the Ras-Erk pathway. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau AH, Walter JM, Gerardin J, Tang C & Lim WA Designing synthetic regulatory networks capable of self-organizing cell polarization. Cell 151, 320–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csikasz-Nagy A, Kapuy O, Toth A, Pal C, Jensen LJ, Uhlmann F, Tyson JJ, and Novak B (2009). Cell cycle regulation by feed-forward loops coupling transcription and phosphorylation. Mol Syst Biol 5, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone A, Evanitsky MN, Hayden L, Cox BD, Wang J, Tornini VA, Ou J, Chao A, Poss KD, and Di Talia S (2021). Control of osteoblast regeneration by a train of Erk activity waves. Nature 590, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dine E, Gil AA, Uribe G, Brangwynne CP, and Toettcher JE (2018). Protein Phase Separation Provides Long-Term Memory of Transient Spatial Stimuli. Cell Syst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerardin J, Reddy NR, and Lim WA (2019). The Design Principles of Biochemical Timers: Circuits that Discriminate between Transient and Sustained Stimulation. Cell Syst 9, 297–308 e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerosa L, Chidley C, Frohlich F, Sanchez G, Lim SK, Muhlich J, Chen JY, Vallabhaneni S, Baker GJ, Schapiro D, et al. (2020). Receptor-Driven ERK Pulses Reconfigure MAPK Signaling and Enable Persistence of Drug-Adapted BRAF-Mutant Melanoma Cells. Cell Syst 11, 478–494 e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goentoro L, Shoval O, Kirschner MW, and Alon U (2009). The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol Cell 36, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goglia AG, Wilson MZ, Jena SG, Silbert J, Basta LP, Devenport D, and Toettcher JE (2020). A Live-Cell Screen for Altered Erk Dynamics Reveals Principles of Proliferative Control. Cell Syst 10, 240–253 e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G, Hallett RA, Zimmerman SP, Williams T, Yumerefendi H, Bear JE, and Kuhlman B (2015). Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci U S A 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka T, Fujita Y, Naoki H, Aoki K, Kamioka Y, and Matsuda M (2015). Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. Elife 4, e05178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, and Baltimore D (2002). The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 298, 1241–1245. [DOI] [PubMed] [Google Scholar]
- Hunt SP, Pini A, and Evan G (1987). Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature 328, 632–634. [DOI] [PubMed] [Google Scholar]
- Jena SG, Yu C, and Toettcher JE (2021). Dynamics and heterogeneity of Erk-induced immediate-early gene expression. bioRxiv, 2021.2004.2030.442166. [Google Scholar]
- Johnson HE, Goyal Y, Pannucci NL, Schupbach T, Shvartsman SY, and Toettcher JE (2017). The Spatiotemporal Limits of Developmental Erk Signaling. Dev Cell 40, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko BN, Demin OV, Moehren G, and Hoek JB (1999). Quantification of short term signaling by the epidermal growth factor receptor. J Biol Chem 274, 30169–30181. [DOI] [PubMed] [Google Scholar]
- Kim Y, Venkataraju KU, Pradhan K, Mende C, Taranda J, Turaga SC, Arganda-Carreras I, Ng L, Hawrylycz MJ, Rockland KS, et al. (2015). Mapping social behavior-induced brain activation at cellular resolution in the mouse. Cell Rep 10, 292–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, and Kain SR (1998). Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273, 34970–34975. [DOI] [PubMed] [Google Scholar]
- Ma W, Trusina A, El-Samad H, Lim WA & Tang C Defining Network Topologies that Can Achieve Biochemical Adaptation. Cell 138, 760–773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackeigan JP, Murphy LO, Dimitri CA, and Blenis J (2005). Graded mitogen-activated protein kinase activity precedes switch-like c-Fos induction in mammalian cells. Mol Cell Biol 25, 4676–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, and Alon U (2003). Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 100, 11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr V, Sturtzel C, Stadler M, Grissenberger S, and Distel M (2018). Fast Dynamic in vivo Monitoring of Erk Activity at Single Cell Resolution in DREKA Zebrafish. Front Cell Dev Biol 6, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Kondo Y, Matsuda M, and Aoki K (2018). Cell-to-Cell Heterogeneity in p38-Mediated Cross-Inhibition of JNK Causes Stochastic Cell Death. Cell Rep 24, 2658–2668. [DOI] [PubMed] [Google Scholar]
- Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, and Lim WA (2016). Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LO, MacKeigan JP, and Blenis J (2004). A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol 24, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakakuki T, Birtwistle MR, Saeki Y, Yumoto N, Ide K, Nagashima T, Brusch L, Ogunnaike BA, Okada-Hatakeyama M, and Kholodenko BN (2010). Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 141, 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, and Trono D (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267. [DOI] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, et al. (2004). Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306, 704–708. [DOI] [PubMed] [Google Scholar]
- Pokrass MJ, Ryan KA, Xin T, Pielstick B, Timp W, Greco V, and Regot S (2020). Cell-Cycle-Dependent ERK Signaling Dynamics Direct Fate Specification in the Mammalian Preimplantation Embryo. Dev Cell 55, 328–340 e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, and Lahav G (2012). p53 dynamics control cell fate. Science 336, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, and Lahav G (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran PT, Wilson MZ, Jena SG, and Toettcher JE (2020). Engineering combinatorial and dynamic decoders using synthetic immediate-early genes. Commun Biol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampattavanich S, Steiert B, Kramer BA, Gyori BM, Albeck JG, and Sorger PK (2018). Encoding Growth Factor Identity in the Temporal Dynamics of FOXO3 under the Combinatorial Control of ERK and AKT Kinases. Cell Syst 6, 664–678 e669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Verveer PJ, and Bastiaens PI (2007). Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol 9, 324–330. [DOI] [PubMed] [Google Scholar]
- Shankaran H, Ippolito DL, Chrisler WB, Resat H, Bollinger N, Opresko LK, and Wiley HS (2009). Rapid and sustained nuclear-cytoplasmic ERK oscillations induced by epidermal growth factor. Mol Syst Biol 5, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu AB, Greenberg ME, and Belasco JG (1989). The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev 3, 60–72. [DOI] [PubMed] [Google Scholar]
- Simon CS, Rahman S, Raina D, Schroter C, and Hadjantonakis AK (2020). Live Visualization of ERK Activity in the Mouse Blastocyst Reveals Lineage-Specific Signaling Dynamics. Dev Cell 55, 341–353 e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen KF, Lauschke VM, Uraji J, Falk HJ, Petersen Y, Funk MC, Beaupeux M, Francois P, Merten CA, and Aulehla A (2018). Modulation of Phase Shift between Wnt and Notch Signaling Oscillations Controls Mesoderm Segmentation. Cell 172, 1079–1090 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Mock C, Batchelor E, Loewer A, and Lahav G (2010). A synthetic-natural hybrid oscillator in human cells. Proc Natl Acad Sci U S A 107, 17047–17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Weiner OD, and Lim WA (2013). Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiairis CD, and Aulehla A (2016). Self-Organization of Embryonic Genetic Oscillators into Spatiotemporal Wave Patterns. Cell 164, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MZ, Ravindran PT, Lim WA, and Toettcher JE (2017). Tracing Information Flow from Erk to Target Gene Induction Reveals Mechanisms of Dynamic and Combinatorial Control. Mol Cell 67, 757–769 e755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, and Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 108, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Time-lapse imaging of NIH3T3 READer cells in response to different dynamic stimuli. TagRFP (white) is uniformly expressed in READer cells and can be used to identify cells. GFP (green) reports on READer system activation. Cells were imaged with a 60X oil objective every 6 min for 5 h. Panels show three stimuli: constant GF-free media (constant OFF; left), constant 10% serum (constant ON; middle), and a 10% serum pulse for 1 hour followed by GF-free media (pulse; right). Related to Figure 2.
Movie S2: Long-term imaging of NIH3T3 cells expressing ErkKTR-iRFP reveals spontaneous pulses. Cells were imaged using a 20X air objective every 3 min for 23 h in either growth media containing 10% serum (left) or were switched to GF-free media immediately prior to imaging (right). Related to Figure 5.
Movie S3: Time-lapse imaging of representative NIH3T3 READer cell expressing ErkKTR-mScarlet. Cells were imaged using a 20X air objective every 3.5 min for 48 h. After having been placed in GF-free media overnight, cells were stimulated with constant 10% serum at the beginning of imaging. Images show ErkKTR-mScarlet (left) and GFP (right), which report on Erk dynamics and READer system activation, respectively. Quantification of the images are shown below each, with the fraction of nuclear KTR shown on the left, and the GFP fluorescence intensity shown on the right. Related to Figure 5.
Data Availability Statement
There are no restrictions on data availability. All data generated or analyzed during this study are included in this published article and its supplementary files or will be shared by the lead contact upon request.
MATLAB code for simulating the computational model is available on the laboratory GitHub page (doi:10.5281/zenodo.5482931). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.