

Abstract
Uterine serous carcinoma (USC) is a highly aggressive endometrial cancer subtype with limited therapeutic options and a lack of targeted therapies. While mutations to PPP2R1A, which encodes the predominant protein phosphatase 2A (PP2A) scaffolding protein Aα, occur in 30–40% of USC cases, the clinical actionability of these mutations has not been studied. Using a high-throughput screening approach, we showed that mutations in Aα results in synthetic lethality following treatment with inhibitors of ribonucleotide reductase (RNR). In vivo, multiple models of Aα mutant uterine serous tumors were sensitive to Clofarabine, an RNR inhibitor. Aα mutant cells displayed impaired checkpoint signaling upon RNRi treatment and subsequently accumulated more DNA damage than wild type cells. Consistently, inhibition of PP2A activity using LB-100, a catalytic inhibitor, sensitized wild type USC cells to RNRi. Analysis of TCGA data indicated that inactivation of PP2A, through loss of PP2A subunit expression, was prevalent in USC, with 88% of USC patients harboring loss of at least one PP2A gene. In contrast, loss of PP2A subunit expression was rare in uterine endometrioid carcinomas. While RNRi are not routinely used for uterine cancers, a retrospective analysis of patients treated with gemcitabine as a second or later line therapy revealed a trend for improved outcomes in USC patients treated with RNRi gemcitabine compared to patients with endometrioid histology. Overall, our data provide experimental evidence to support the use of ribonucleotide reductase inhibitors for the treatment of USC.
Introduction
Uterine cancer is the most common gynecologic malignancy and fourth most common cause of new cancer diagnoses in women, with over 65,000 new cases diagnosed in the United States in 2020 (1). The majority of uterine cancers are endometrial carcinomas (EMCA), malignancies of the endometrial epithelium lining. There are distinct subtypes of EMCA with markedly different prognoses: endometrioid-type carcinomas are most common (~80% of EMCA cases), frequently involve deregulated hormone signaling, and exhibit favorable outcomes; serous (USC), clear cell, and undifferentiated carcinomas do not share this etiology, rarely respond to hormone therapy, and are typically high-grade and invasive at time of diagnosis, resulting high rate of recurrence and a relatively poor prognosis (2). For recurrent cancers, after progression on upfront platinum-based chemotherapy and immunotherapy with lenvatinib and pembrolizumab viable therapeutic options remain limited. While other cancers have benefited from the development of targeted therapeutic strategies, the lack of well- characterized targetable disease drivers for USC and other high-grade EMCA tumors has generated very limited opportunities for targeted therapy to date. Progress in elucidating disease driver mechanisms will therefore be imperative to advancing new treatment options and patient outcomes.
Previous work by our group and others have identified a heterozygous mutational hotspot within PPP2R1A which includes two recurrent mutations, P179R and S256F, which almost exclusively exist within the high-grade subtypes (3–9). Additional analysis of matched primary and metastatic tumors revealed that these mutations were trunk-biased suggesting that they constitute early events in the development of endometrial carcinoma (10). PPP2R1A encodes the gene for the Aα scaffolding subunit of the protein phosphatase 2A (PP2A), a heterotrimeric serine/threonine phosphatase and tumor suppressor (11–18). The active PP2A holoenzyme is composed of a scaffolding “A” subunit, catalytic “C” subunit, and one substrate determining “B” subunit (19,20). We have recently shown that the P179R or S256F Aα mutations result in altered assembly of the PP2A holoenzyme, specifically by disrupting the ability of PP2A B subunits and/or the catalytic C subunit to bind and contribute to uterine tumorigenesis through the inactivation of PP2A’s tumor suppressive activities (3,21,22).
Here, we investigated whether the two hotspot mutations, P179R or S256F, in PPP2R1A would result in the identification of a druggable target. We screened 3,200 bioactive compounds and measured the cell viability of mutant and wild type patient-derived isogenic serous endometrial cancer cells to determine synthetic lethal targets in Aα mutant cells. From this screen, we identified that cells with either recurrent mutation displayed synthetic lethality to ribonucleotide reductase (RNR) inhibitors. Furthermore, the synthetic lethality was specific to RNR inhibition, where other inducers of DNA damage, showed no differences in drug sensitivity between wild type and mutant Aα cells. Using xenograft studies in vivo, we demonstrated that Aα mutant tumors were also sensitive to Clofarabine given orally. Analysis of mutant and wild type treated USC cells showed that Aα mutant cells displayed impaired checkpoint signaling in response to Clofarabine treatment, and subsequently accumulated more DNA damage. Analysis of the TCGA revealed that loss or altered PP2A expression was common among all USC, and inhibitors of PP2A’s catalytic activity, LB-100, sensitized PP2A wild type cells to RNR inhibition, indicating the identified synthetic lethality was PP2A dependent. Finally, retrospective analysis of a cohort of endometrial cancer patients given gemcitabine revealed that despite the expected poor outcomes, patients with USC had a trend for longer time to next treatment and overall survival when given gemcitabine when compared to those with endometrioid histology. This was in contrast to analysis of the TCGA data, where patients with recurrent uterine serous carcinomas had a worse overall survival compared to those with recurrent endometrioid carcinomas. Overall, our findings provide rationale for the use of the FDA approved class of RNR inhibitors for USC treatment, allowing for the near-term clinical translation of these findings to patients suffering from this particularly lethal subtype of endometrial cancer.
Materials and Methods
Cell Lines and Culture:
UT42 and UT89 were generated from primary recurrent uterine serous tumors in the laboratory of Dr. Analisa DiFeo and described previously (3,23). UT89 Aα knockout cells were generated and described previously (23). OV17R was purchased from Sigma Aldrich through the European Collection of Authenticated Cell Cultures (ECACC 9602076, RRID# CVCL_2672). AN3CA and KLE were purchased from ATCC (HTB-111 (CVCL_0028) and CRL-1622 (CVCL_1329), respectively). HEC50B were purchased from the JCRB Cell Bank (CVCL_2929). Mutational status of PPP2R1A in all cells were determined by Sanger sequencing. UT42, UT89, and UT185 were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. OV17 was cultured in DMEM F12 media supplemented with 5% FBS, 0.4 μg/mL hydrocortisone, 10 μg/mL insulin, and 1% penicillin/streptomycin. KLE cells were cultured in DMEM F12 supplemented with 10% FBS and 1% penicillin/streptomycin. AN3CA cells were cultured in Eagle’s Minimum Essential Medium supplemented with 10% FBS and 1% penicillin/streptomycin. HEC50B cells were cultured in Eagle’s Minimum Essential Medium supplemented with 15% FBS and 1% penicillin/streptomycin. All cells were grown in a humidified atmosphere containing 5% CO2 at 37°C. All cell lines underwent monthly testing for mycoplasma contamination and identity was confirmed by STR profiling.
Constructs and Lentivirus Production:
pLX304: PPP2R1A plasmids (HsCD00444402) were purchased from DNASU and were part of the ORFome collaboration and described previously (24). Lentivirus was generated in collaboration with the Vector Core at the University of Michigan. Following lentiviral production, virus was incubated on cells in penicillin/streptomycin free media for 24 hours, when media was replaced with normal media. After 72 hours, cells were selected in 16 μg/mL Blasticidin (Invivogen) to generate stable cell lines.
High-Throughput Compound Screening:
Compound screening was completed by the Small Molecule Drug Development (SMDD) Core at Case Western Reserve University. UT42 cells, expressing EGFP or wild type Aα protein, were seeded into 384 well plates and incubated for 24 hours. After incubation, cells were treated with the Bioactives Compound Library (combination of the Selleck Chemical Library and the Sigma LOPAC Library), consisting of 3,200 compounds, at 10 μM for 72 hrs. After incubation, cell viability was determined using CellTiter-Glo, where luminescence signal is proportional to the amount of ATP present. The luminescence signal was normalized to control wells for each cell line. The normalized signal for each cell lines was graphed along the x-axis (EGFP) and y-axis (WT) to determine viability differences between the two groups. One biological replicate of the compound screening was performed, and potential hits were validated using subsequent assays and cell lines.
Compounds and Reagents:
Clofarabine, Cladribine, Gemcitabine HCl, Triapine, Nelarabine, Cisplatin, and LB-100 were purchased from Selleck Chemical. BS-181 and PHA-767491 were purchased from MedChem Express. In vitro use: All compounds except for LB-100 and Cisplatin were reconstituted in DMSO, aliquoted, and stored at −80°C until use. LB-100 was reconstituted to 10 mM in sterile water, aliquoted, and stored at −20°C until use. Cisplatin was reconstituted to 3 mM in sterile saline and stored at 4°C until use. MTT was purchased from Research Products International and reconstituted to 5 mg/mL in sterile PBS, aliquoted, and stored at −20°C until use. In vivo formulation: Clofarabine was prepared for in vivo xenografts in 25% polyethylene glycol 400 (PEG400) (Sigma Aldrich 06855) in 0.9% sterile saline (USP Sterile Grade, Fisher Scientific Z1376).
Cell Viability Assays:
MTT: 2,000 cells per 96 well were plated in 100 μL of media and allowed to adhere for 24 hours at 37°C, following incubation cells were treated with increasing 2X concentrations of the appropriate compound in 100 μL to give a final concentration of 1X. After the specified incubation time at 37°C, 20 μL of 5 mg/mL MTT was added to each well and incubated at 37°C for an additional 2 hours. Following MTT incubation, media was aspirated, and cells were dissolved in 100 μL of N-propanol. Plates were analyzed on a spectrophotometer at 570 nM and 650 nM and cell viability was calculated and EC50 values were graphed and analyzed using Prism (SCR_005375). Annexin/PI: Staining for flow cytometry was completed using the APC Annexin V Apoptosis Detection Kit with PI per protocol (BioLegend, 640932), and analyzed by flow cytometry at the University of Michigan Flow Cytometry Core and FlowJo software (SCR_008520). Synergy calculations: For calculations of synergy, Compusyn Software was used (25).
Knockdown experiments:
esiRNAs for RRM1, RRM2, or RLUC (control) were purchased from Sigma Aldrich and transfected at a concentration of 3000 ng esiRNA per 10 cm plate using Oligofectamine 2000 (ThermoFisher Scientific). After transfection, cells were incubated for 72 hrs. at 37 °C and subsequently harvested for protein and cell viability and knockdown was analyzed by immunoblot.
Antibodies and Immunoblotting:
All antibodies used in the described studies can be found in Supplementary Table 3. Proteins from whole cells were lysed in RIPA buffer (ThermoFisher Scientific) supplemented with protease and phosphatase inhibitors (Roche). Protein concentrations of cell extracts were determined using the Pierce BCA Protein Assay kit (ThermoFisher) and equal quantities of protein were separated by SDS/PAGE 12% polyacrylamide gels (Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad). Primary antibodies were detected with goat anti-mouse (Abcam) or donkey anti-rabbit (GE Healthcare) conjugated to horseradish peroxidase using the Bio-Rad ChemiDoc XRS+ or the Bio-Rad ChemiDoc MP using chemiluminescence. Densitometry quantification was performed within the Bio-Rad Image Lab software (SCR_014210).
In vivo xenografts:
UT42 cells: 10–20 million UT42 cells, expressing EGFP or wild type Aα protein, were injected subcutaneously in 5 mg/mL Matrigel (Corning 354234) into the flank of severely immunocompromised SRG rats (Sprague Dawley Rag2−/− Il2rg −/− rats from Hera BioLabs, Lexington, KY). After tumor growth to >5,000 mm3, tumors were aseptically harvested, sectioned into 2 × 2 × 2 mm fragments and implanted subcutaneously into the flank of NSG (The Jackson Laboratory; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, (IMSR_JAX:00557)) or NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac; Taconic) mice using a trocar. Upon tumor growth to 150–250 mm3, mice were treated with either Vehicle Control or Clofarabine at 30 mg/kg once daily (QD) by oral gavage. In a subset of animals, tumors were allowed to grow to 500 mm3, then dosed for 3 days once daily with Vehicle Control or Clofarabine at 30 mg/kg by oral gavage before harvesting tissue for molecular analysis. Body weight was recorded three times weekly. Tumor volume was calculated as (LxW2/2), where length and width were measured with digital calipers three times weekly. After euthanasia, the tumor was collected, and half was fixed in neutral buffered formalin and half was flash frozen in liquid nitrogen. UT89 cells: Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Michigan. Animal use and care was in strict compliance with institutional guidelines and all experiments conformed to the relevant regulatory standards by The University of Michigan. 1 million UT89 cells were injected subcutaneously into the flanks of 6–8 week old female NCI nude mice (IMSR_JAX: 002019) in 50% Matrigel (Corning 354234). Tumor volumes were assessed by caliper measurement (LxW2/2). Upon tumor growth to 150–250 mm3, mice were randomized and given Vehicle Control or Clofarabine at 30 mg/kg once daily (QD) by oral gavage. Tumor tissue was both formalin-fixed and snap frozen in liquid nitrogen for analysis. OV17R cells: 5 million and 10 million cells were injected subcutaneously into the flanks of 6–8 week old female Nod Scid Gamma (ISMR_JAX: 001303) mice in 50% Matrigel (Corning 354234). No tumors formed, so this cell line was not used for in vivo studies.
Immunocytochemistry:
Cells were plated on 4 chamber cell culture slides (CellTreat 229164) and treated with control or compound containing media for specified times. Prepared slides were imaged at the University of Michigan Microscopy Core, using the Zeiss Apotome. Quantification of the images was performed using Image J, scale bar on images represents 50 μm.
TCGA Data Analysis:
TCGA PanCancer Atlas data was accessed through cBioPortal. Loss of PP2A subunit expression or mutation of PP2A subunits were calculated from TCGA data where copy number and mutation data were available (n=109, Uterine Serous Carcinomas; n=399 Endometrioid Uterine Carcinomas, analyzed on cBioPortal). Kaplan-Meier analysis of overall surviaval in Serous or Endometrioid uterine carcinoma patients with recurrent disease were downloaded and analyzed via Prism (SCR_005375), Log-rank (Mantel-Cox) test was used to calculate p-value.
MSKCC Cohort Analysis:
Patient selection: Patients with recurrent endometrial cancer who received gemcitabine from December 2010 to December 2019 at Memorial Sloan Kettering Cancer Center (MSKCC) were retrospectively analyzed and followed until April 20, 2020. Patient clinical characteristics including histology, tumor grade, stage at diagnosis, treatment history including prior chemotherapy, tumor genomic profiling results, and outcomes were abstracted from the medical record. The study was approved by the Institutional Review Board at MSKCC. Statistical analysis: Baseline clinical and disease characteristics were summarized as medians and ranges for continuous variables and as numbers and percentages for categorical variables. Fisher’s exact test or Mann-Whitney U test was used for analysis as appropriate. A two-tailed p-value of less than 0.05 was considered statistically significant. Kaplan-Meier survival analysis was used to determine time to next treatment (TNT) and gemcitabine-specific survival. Time was calculated from initiation of gemcitabine to start of next therapy or hospice for TNT and from initiation of gemcitabine to death from any cause for gemcitabine-specific survival. For patients that received gemcitabine more than once, their first course was used for analysis. All statistical analyses were performed using SPSS (version 14.0; SPSS, Inc, Chicago, Ill, USA) (SCR_002865).
Results
PP2A Aα-P179R mutation sensitizes serous endometrial cancer cells to RNR inhibitors
To evaluate the relevance of PP2A Aα P179R mutations, the most prevalent Aα mutation in USC, to drug response, we used high-throughput screening (HTS) approach to test the drug sensitives and resistance of 3,200 bioactive compounds from the Sigma LOPAC and Selleck Chemical libraries. We used a patient derived cellular model, UT42, which harbors a heterozygous Aα-P179R mutation (UT42Aα-P179R) and compared viability changes of UT42Aα-P179R cells expressing EGFP (control) or wild type (WT) Aα (mutational correction), described previously (Figure 1A and B) (3). To compare viability changes between EGFP and Aα-WT expressing UT42Aα-P179R cells, the viability of +EGFP cells (mutant) were plotted on the x-axis and the viability of +Aα-WT cells were plotted on the y-axis, where each dot represents a different compound (Figure 1C, Supplemental Figure 1A and B, Supplemental Table 1 and 2). Of particular interest were compounds falling above the line, indicating increased drug sensitivity in the Aα mutant expressing cells compared to the WT cells, which included the ribonucleotide reductase (RNR) inhibitor class of compounds, highlighted in red (Figure 1C, Supplemental Table 1 and 2). We performed dose response curves of multiple RNR inhibitors identified in the screen, including Cladribine, Clofarabine, Gemcitabine and Hydroxyurea, and independently confirmed the original screening results (Figure 1D–F, Supplemental Figure 1C–E) (26,27). Due to the extensive difference in EC50 with Clofarabine, and the ability to use an oral preparation for this compound in in vivo studies, we focused our future experiments on this RNR inhibitor (28). Apoptosis was measured by Annexin V/PI staining and similarly showed a higher percentage of apoptotic cells in the mutant cells compared to the Aα-WT expressing cells when treated with Clofarabine (Supplemental Figure 2A&B). While the class of RNR inhibitors validated as a positive hit in the screen, other compounds did not, including BS-181, a CDK7 inhibitor, which showed a dramatic difference between mutant and WT viability (60% and 125% respectively) and PHA-767491, a CDK9 inhibitor. When dose response curves were performed for these compounds, there were no significant differences in viability between the mutant and WT cells, highlighting RNR inhibitors as a positive hit (Supplemental Figure 3A–C).
Figure 1. High throughput screening identifies PP2A A⍺ mutations sensitize cancer cells to RNR inhibitors.
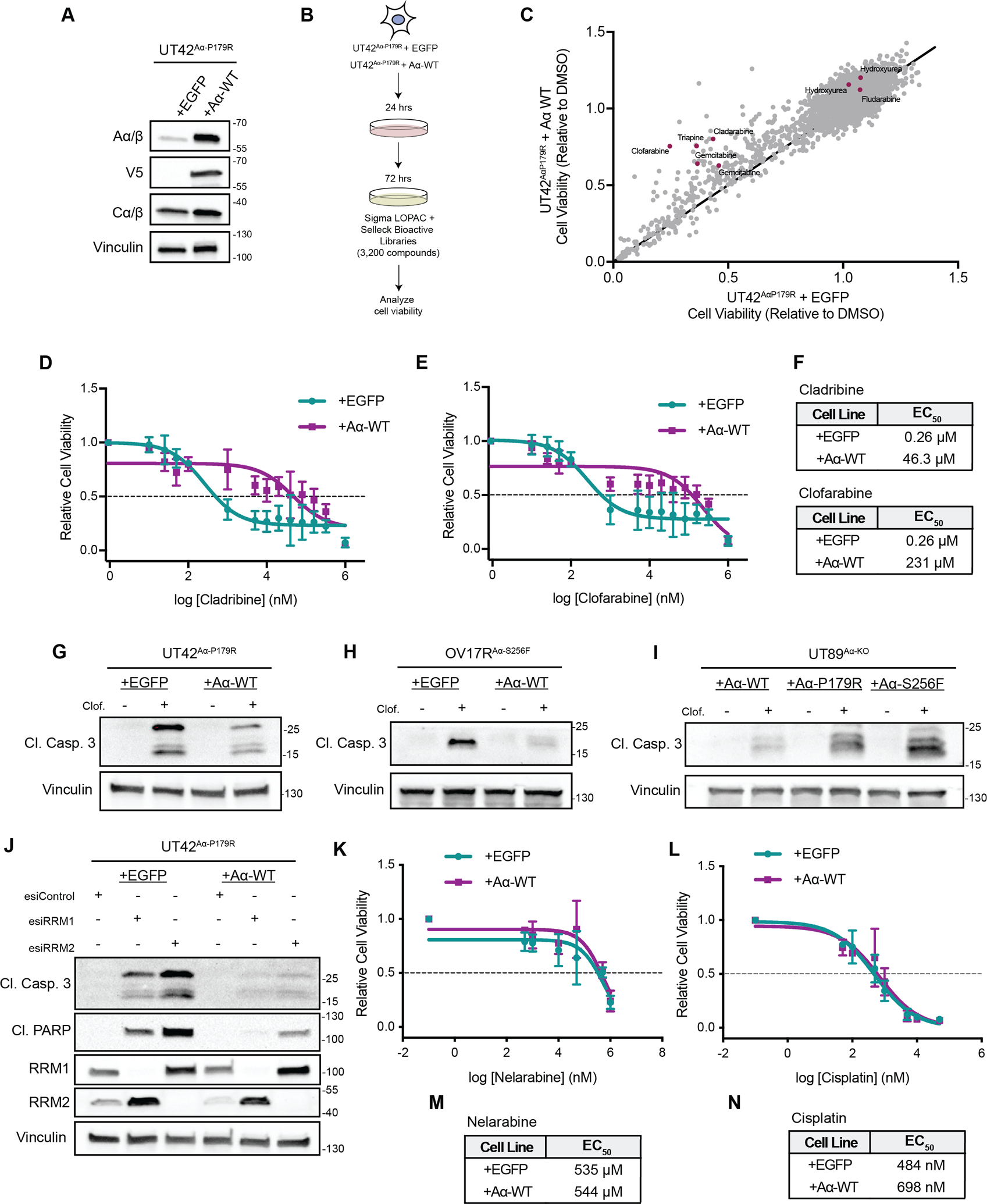
A, Representative western blot of UT42Aα-P179R isogenic cells expressing EGFP or WT Aα protein demonstrating overexpression. B, Schematic of the high-throughput screening workflow. C, Overview of the viability results from all 3,200 compounds, with the viability of the EGFP expressing cells on the x-axis and the viability of the WT Aα protein expressing cells on the y-axis. Compounds included in the screen which are classified to harbor activity for ribonucleotide reductase from the high throughput screen were highlighted in red. D&E, Dose response curves of UT42 isogenic cells treated with Cladribine (D) or Clofarabine (E) as measured by MTT, n=3 biological replicates, error bars ± SD. F, Calculated EC50 values from the MTT assays in D and E (calculated from the average of all biological replicates). G-I, Isogenic UT42Aα-P179R (G), OV17RAα-S256F (H) or UT89 CRISPR Aα-KO cells (I) were treated with Clofarabine and representative immunoblots of the apoptotic marker Cleaved Caspase 3 is shown, n=3 biological replicates. Quantification from the immunoblots shown is represented in Supplemental Figure 3. J, Representative immunoblot of apoptotic markers Cleaved Caspase 3, Cleaved PARP, or RRM1 and RRM2 in isogenic UT42Aα-P179R cells with knockdown of RRM1, RRM2, or RLUC (Control) at 72 hrs., n=3 biological replicates. K&L, Dose response curves of UT42 isogenic cells treated with Nelarabine (K) or Cisplatin (L) as measured by MTT, n=3 biological replicates, error bars ± SD. M&N, Calculated EC50 values from the MTT assays in K and L (calculated from the average of all biological replicates).
Clofarabine treatment results in increased apoptosis of P179R and S256F mutant cells
To validate whether these recurrent scaffold mutations were driving the differential response to RNR inhibitors, we sought to correct these mutations using CRISPR/Cas9, but were unable to do so, likely because of low efficiency of homologous recombination and the dependency of this cell line on this mutation. Therefore, to expand our model systems and validate our results, we used two additional cell models (Supplemental Figure 4A–C). The first was OV17R cells, which harbor a heterozygous Aα-S256F mutation (OV17RAα-S256F). OV17RAα-S256F cells were stably transduced to express EGFP (control) or wild type Aα (mutational correction) (Supplemental Figure 4A). Additionally, we used UT89 cells, a patient derived serous endometrial cancer cell line which is wild type for PP2A Aα, and knocked out the Aα subunit, which we have described previously (UT89AαKO) (3,23). UT89AαKO cells were transduced to stably express V5 tagged Aα-WT, Aα-P179R or Aα-S256F and the levels of the A subunit, C subunit and V5 was determined by western blot (Supplemental Figure 4B and C).
All three cell models were treated with Clofarabine for 72 hours, and consistent with the cell viability assays, the mutant cells showed significantly more cleaved caspase 3, indicating higher amounts of apoptosis in the mutant cells upon Clofarabine treatment compared to the Aα-WT expressing cells (Figure 1G–I and Supplemental Figure 4D–F).
The synthetic lethality between RNRi and PP2A is dependent on ribonucleotide reductase
We investigated whether the synthetic lethality was specific to RNR inhibition or also true for other nucleoside analogues or DNA damaging agents. First, we used pooled siRNAs to knock down the two main subunits of RNR, RRM1 and RRM2 in the isogenic UT42Aα-P179R cells and measured markers of apoptosis including cleaved PARP and cleaved caspase 3 following knockdown (Figure 1J and Supplemental Figure 5A and B). Consistent with the inhibitor data, siRNAs targeting RRM1 or RRM2 resulted in more apoptosis in the mutant Aα cells as compared to the wild type cells. Additionally, we treated the isogenic UT42 and OV17R cells with Nelarabine, a nucleoside analogue sharing high chemical similarity to Clofarabine or Cladribine but with minimal to no RNR inhibitory properties (Figure 1K and M, Supplemental Figure 6A) (29). These experiments showed no differences in viability between the mutant and wild type Aα cells, further indicating that the synthetic lethality is specific to RNR inhibition. Finally, treatment of WT and mutant cells with Cisplatin, a compound showing equal response in the HTS, showed equal response in a dose response curve, further supporting the screening results and specificity to RNR (Figure 1L and N, Supplemental Figure 6B). Finally, because response to RNR inhibitors could be impacted by cellular proliferation rates, we measured the proliferation of the isogenic UT42 and OV17R cells and found no differences in the proliferation rates between the mutant and WT cells, indicating that the increased sensitivity in the mutant cells was not due to increased proliferation (Supplemental Figure 7A and B).
Taken together, these data identified a potential synthetic lethal interaction between inhibition of ribonucleotide reductase in Aα mutant cells. Further analysis of additional cell models confirmed that the Aα-P179R cells are more sensitive to RNR inhibitors, in particular Clofarabine, and also show that this sensitivity profile is consistent in cells expressing other recurrent gynecological specific mutations, including Aα-S256F.
PP2A Aα mutated tumors are sensitive to Clofarabine treatment in vivo
To determine if PP2A Aα mutant serous endometrial tumors were sensitive to Clofarabine treatment in vivo, we performed multiple independent xenograft studies. We have previously published a long latency period for UT42Aα-P179R tumors (3). To limit the latency, we grew the cells subcutaneously in immunocompromised SRG rats, and subsequently sectioned and implanted tumor fragments into immunocompromised mice for treatment (30,31). These UT42Aα-P179R tumors were randomized and treated with 30 mg/kg Clofarabine or vehicle control once per day by oral gavage, as this dose and formulation was recently reported to have anti-tumor effects in a Ewing Sarcoma xenograft model (28). Consistent with cell-based data, treatment of Clofarabine resulted in a significant tumor growth inhibition in this model (Figure 2A and B). Further, we injected the UT89Aα-KO cells expressing mutant Aα-P179R or S256F subcutaneously into immunocompromised mice and tumors were subsequently randomized and treaded with vehicle control or 30 mg/kg Clofarabine once per day by oral gavage. In these xenograft studies, the mutant tumors also responded to Clofarabine treatment (Figure 2C and D, Figure 2E and F).
Figure 2. PP2A A⍺ mutant tumors are sensitive to Clofarabine in vivo.
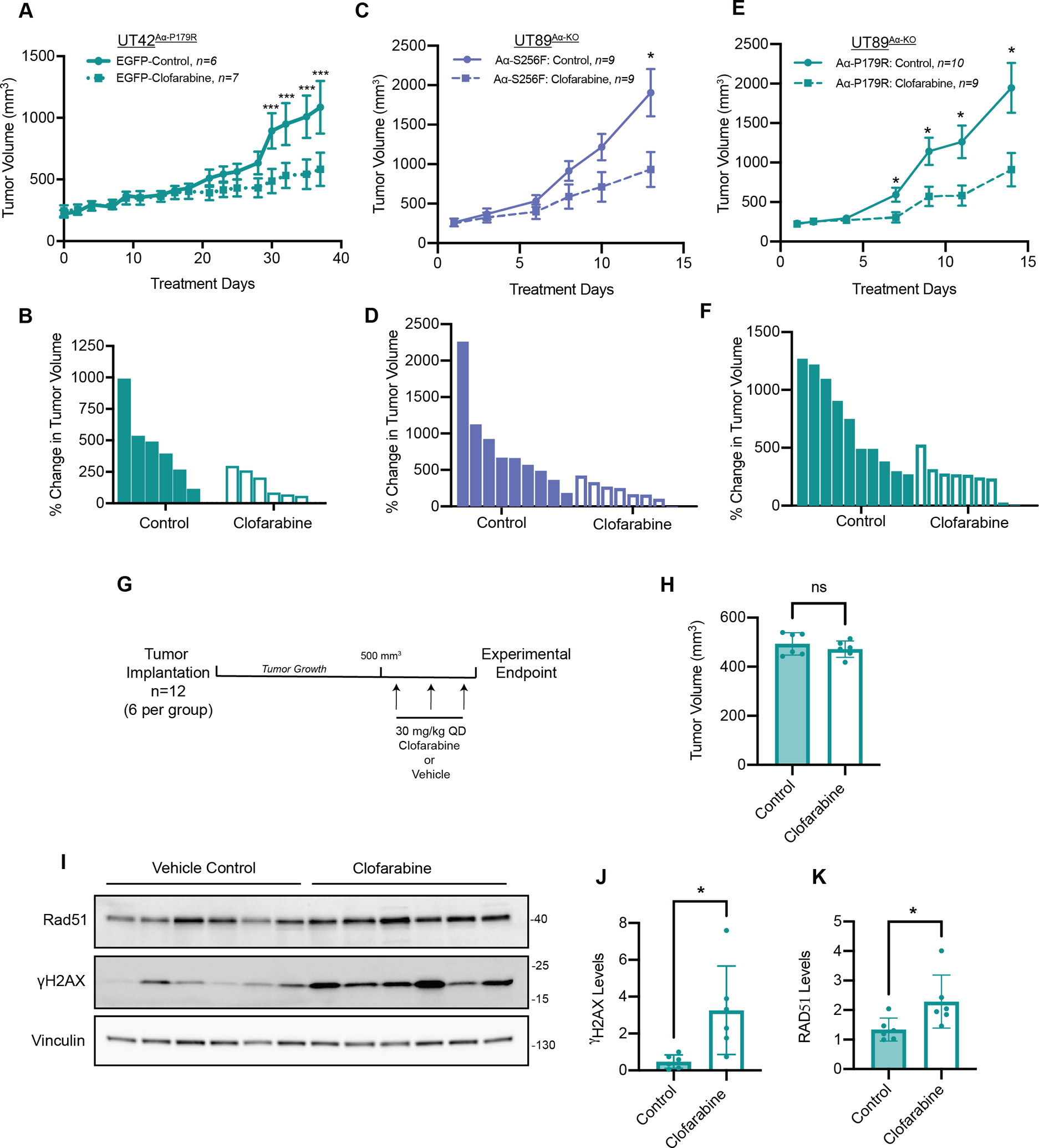
A, Subcutaneous xenograft growth of control UT42Aα-P179R tumors treated with vehicle (solid line) or 30 mpk Clofarabine (dashed line), error bars ± SEM, (multiple T-tests, p-values: *** < 0.001). B, Waterfall plot of UT42Aα-P179R tumors treated with Control or Clofarabine showing the percent change in tumor volume, corresponding to (A). C, Subcutaneous xenograft growth of control UT89Aα-KO tumors expressing Aα-S256F treated with vehicle (solid line) or 30 mpk Clofarabine (dashed line), error bars ± SEM, (multiple T-tests, p-values: * < 0.05). D, Waterfall plot of UT89Aα-KO tumors expressing Aα-S256F tumors treated with Control or Clofarabine showing the percent change in tumor volume, corresponding to (B). E, Subcutaneous xenograft growth of control UT89Aα-KO tumors expressing Aα-P179R treated with vehicle (solid line) or 30 mpk Clofarabine (dashed line), error bars ± SEM, (multiple T-tests, p-values: * < 0.05). F, Waterfall plot of UT89Aα-KO tumors expressing Aα-P179R tumors treated with Control or Clofarabine showing the percent change in tumor volume, corresponding to (C). G, Schematic of the in vivo pharmacodynamic (PD) study. H, Quantification of the tumor volumes in the Control (n=6) and Clofarabine (n=6) treatment groups, indicating no difference in tumor volume at the imitation of the PD study. I, Lysates from Control and Clofarabine treated UT42Aα-P179R PD xenograft tumors were analyzed by western blot for γH2AX and Rad51, with Vinculin as a housekeeping protein. J and K, Quantification of γH2AX levels (J) and Rad51 (K) all tumors normalized relative to the average of the two control tumors. Error bars ± SD, (Students T-test, p-values * < 0.05).
Ribonucleotide reductase inhibitors, including Clofarabine, result in a depletion of the dNTP pools necessary for DNA replication, leading to DNA damage (32). To evaluate whether treatment of Clofarabine was leading to an accumulation of DNA damage in vivo, we lysed tumor samples and analyzed for γH2AX, a marker of dsDNA breaks, by immunoblot. Paradoxically, analysis of the tumors for all three studies showed either a significant reduction or no change in γH2AX levels in all three in vivo studies (Supplemental Figure 8A–F), leading us to hypothesize that by the terminal endpoint the treated cells remaining in the tumor had become resistant to Clofarabine. To test this, we performed a pharmacodynamic (PD) xenograft study, where UT42Aα-P179R tumor fragments were implanted into immunocompromised mice, randomized when tumors reached 500 mm3, and treated with three doses of Clofarabine or vehicle control by oral gavage over three days (Figure 2G and H). Analysis of this PD study showed a significant increase in multiple DNA damage markers including γH2AX and Rad51, a marker of ssDNA breaks (Figure 2I–K), further supporting the resistance acquired upon completion of the terminal efficacy studies as well as confirming Clofarabine activity at this dose in vivo.
Combined, these studies show that PP2A Aα mutant cells are sensitive to Clofarabine in vivo and tumors treated with Clofarabine result in an accumulation of DNA damage, highlighting the potential therapeutic benefit of using these compounds for the treatment of USC, a subtype of cancer with limited therapeutic options.
PP2A Aα mutations impair checkpoint signaling and increase DNA damage upon replication stress induced by Clofarabine treatment
Ribonucleotide reductase inhibitors impede the progression of replication forks and activates replication checkpoint kinases (32). To understand why PP2A Aα mutant cells were preferentially sensitive to Clofarabine treatment, we analyzed the phosphorylation and activation of checkpoint kinases in wild type PP2A Aα and mutant cells by western blot (Supplemental Figure 9A–L). Analysis of phosphorylated/total ratios of the checkpoint kinases ATR, CHK1, ATM, and CHK2 showed lower levels of activation in the mutant cells compared to the wild type cells in both the UT42Aα-P179R and OV17Aα-S256F cell models following 3 and 6 hours of Clofarabine treatment (Supplemental Figure 9A–L).
To determine if the impaired checkpoint signaling and control in the PP2A Aα mutant cells was in turn resulting in DNA damage, we analyzed γH2AX foci by immunofluorescent microscopy in both the UT42Aα-P179R and OV17Aα-S256F cell models following Clofarabine treatment (Figure 3A–D). This revealed an increase in the amount of accumulated dsDNA damage in the PP2A Aα mutant cells, consistent with a lack of checkpoint control. These findings were further confirmed by measuring γH2AX levels by western blot (Figure 3E–H). Interestingly, these analyses also revealed increased γH2AX in both mutant cells at baseline, indicating a higher level of DNA damage in these cells in the absence of RNR inhibition. To ensure that expression levels of the A subunit was not contributing to the effects seen, γH2AX levels were also measured in the UT89 isogenic model following Clofarabine treatment (Figure 3I). Supportive of the other findings, in this model of matched A subunit expression, the mutant cells expressed more γH2AX in response to Clofarabine treatment than the WT cells (Figure 3I).
Figure 3. Aα mutations impair checkpoint signaling and checkpoint control leading to increased accumulation of DNA damage following Clofarabine treatment.
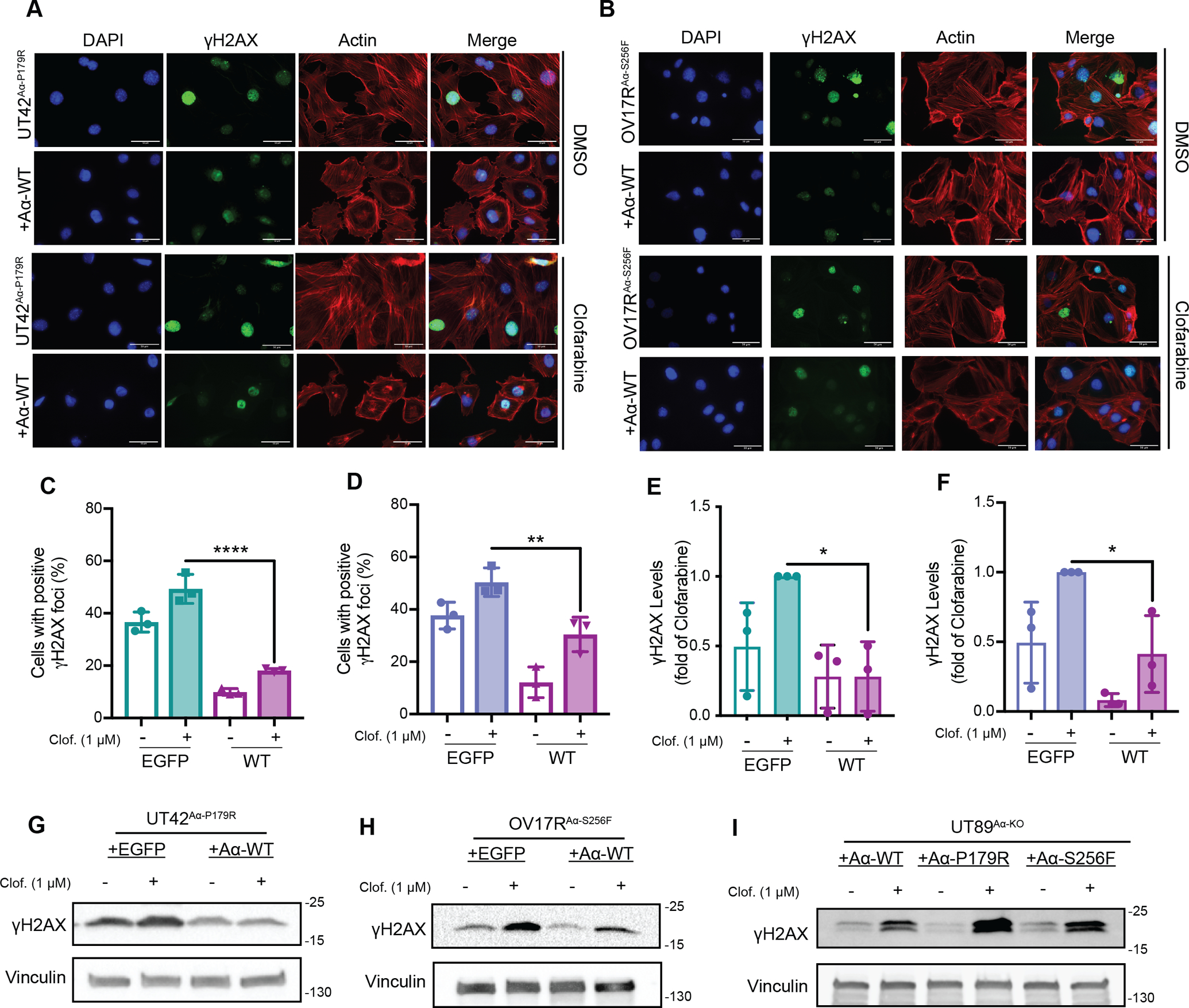
A Representative immunofluorescence images of γH2AX, DAPI, and Actin in isogenic UT42Aα-P179R cells treated with DMSO control (top) or 1 μM Clofarabine (bottom) for 3 hrs., n=3 biological replicates. Scale bars = 50 microns. B, Representative immunofluorescence images of γH2AX, DAPI, and Actin in isogenic OV17RAα-S256F cells treated with DMSO control (top) or 1 μM Clofarabine (bottom) for 3 hrs., n=3 biological replicates. Scale bars = 50 microns. C and D, Quantification of immunofluorescence images of UT42Aα-P179R isogenic cells (A) and OV17RAα-S256F isogenic cells (B), n=3 biological replicates, error bars ± SD, (One-way ANOVA relative to EGFP Clofarabine with Dunnett’s correction for multiple comparisons, p-values **, <0.01, **** < 0.0001). E and F, Quantification of γH2AX levels by immunoblot of UT42Aα-P179R isogenic cells (e) and OV17RAαS256F isogenic cells (f), n=3 biological replicates, error bars ± SD, (One-way ANOVA relative to EGFP Clofarabine with Dunnett’s correction for multiple comparisons, p-values * < 0.05). G and H, Representative immunoblot of γH2AX in isogenic UT42Aα-P179R (G) and OV17RAα-S256F (H), treated with 1 μM Clofarabine for 3 hrs., n=3 biological replicates, quantification in (E and F). I, Representative immunoblot of γH2AX in isogenic UT89Aα-KO cells treated with 1 μM Clofarabine for 1 hr., n=3 biological replicates.
Taken together, these data indicate that the inability of PP2A mutant cells to initiate the replication checkpoint results in an accumulation of dsDNA damage in response to the replicative stress induced by Clofarabine treatment both in vitro and in vivo.
Inhibition of PP2A is common in USC and mediates the synthetic lethality with Clofarabine
Given the high prevalence and tumorigenic nature of Aα mutations in USC (2,3,5,10,33,34), we postulated that dysregulation or inactivation of PP2A might be a more widespread phenomenon in this highly aggressive uterine cancer subtype. To explore this, we analyzed the prevalence of expression loss of PP2A family genes, specifically within high-grade serous uterine cancer samples in The Cancer Genome Atlas (TCGA) and found that heterozygous loss of either catalytic subunit Cα/β (PPP2CA/B) or B55α (PPP2R2A) to be the most commonly lost PP2A subunit genes, at a rate ranging from 50–75%, (n=109) (Figure 4A). Interestingly, analysis of endometrioid endometrial carcinomas did not share the same result, with very low frequencies of PP2A subunit loss or mutation (Supplemental Figure 10). This is consistent with the idea that PP2A inhibition through Aα mutation seem to be an early driver event for USC specifically (9,10). Importantly, further analysis of this data showed a correlation between mRNA and copy number, indicating that the heterozygous loss called by the TCGA did in fact correlate to decreased mRNA expression (Supplemental Figure 11). Testing for mutual exclusivity revealed that 88% of USC patients harbored at least one alteration in these genes (96/109), with a significant co-occurrence between loss of PPP2CB and PPP2R2A (Supplemental Figure 12). Our group and others have previously shown that mutations to the Aα subunit of PP2A cause structural defects resulting in the inability to form active PP2A heterotrimers, including loss of binding of the C subunit, resulting in proteosome mediated degradation and decreased C subunit expression (3–5,21,23).
Figure 4: Loss of PP2A subunit expression is common in USC and inactivation of PP2A sensitizes to Clofarabine.
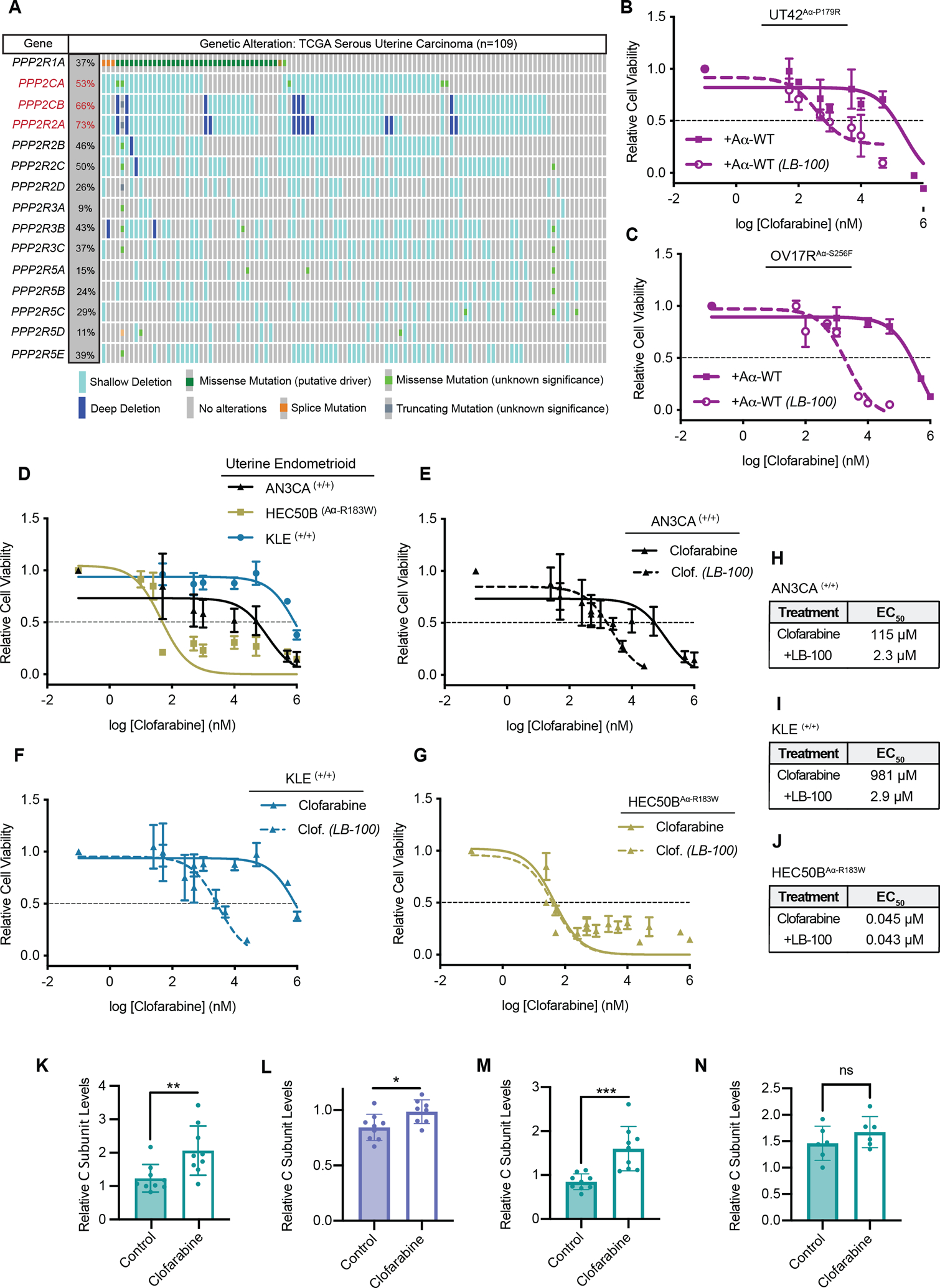
A, Analysis of heterozygous and homozygous loss of canonical PP2A subunits in uterine serous carcinoma samples from the TCGA. Subunits with loss at greater than 50% are highlighted in red. In aggregate, 101 of 109 USC patients harbor some alteration to PP2A. B and C, Dose response curve for UT42Aα-P179R cells expressing WT Aα (B) or OV17RAα-S256F cells expressing WT Aα (C) were treated with Clofarabine with or without LB-100 in increasing doses held at a constant ratio, n=3 biological replicates, error bars ± SD. D, Dose response curves of AN3CA, HEC50B, and KLE cells treated with Clofarabine as measured by MTT at 72 hrs, n=3 biological replicates, error bars ± SD. Clofarabine alone data from figure D included in panels E-G. E-G, Dose response curve for AN3CA (E), KLE (F) or HEC50B (G) cells treated with Clofarabine with or without LB-100 in increasing doses held at a constant ratio, 72hrs, n=3 biological replicates, error bars ± SD. H-J, Calculated EC50 values from the MTT assays in E-G (calculated from the average of all biological replicates). K, Quantification of total C Subunit levels in UT42Aα-P179R terminal efficacy xenograft tumors as measured by western blot. All tumors normalized to the average of two control tumors. Error bars ± SD, (Students T-test, p-values *** < 0.001). L, Quantification of total C Subunit levels in UT89Aα-KO + S256F terminal efficacy xenograft tumors as measured by western blot. All tumors normalized to the average of two control tumors. Error bars ± SD, (Students T-test, p-values * < 0.05). M, Quantification of total C Subunit levels in UT89Aα-KO + P179R terminal efficacy xenograft tumors as measured by western blot. All tumors normalized to the average of two control tumors. Error bars ± SD, (Students T-test, p-values *** < 0.001). N, Quantification of total C Subunit levels in UT42Aα-P179R pharmacodynamic xenograft tumors from Figure 2G. All tumors normalized to the average of two control tumors. Error bars ± SD, (Students T-test, p-values *** < 0.001). Westerns for (K-N) can be found in Supplemental Figure 8.
This data, combined with the highly prevalent loss of the PP2A C subunit isoforms, led us to hypothesize that loss of catalytic subunit expression may also be predictive marker of ribonucleotide reductase sensitivity, and the synthetic lethality identified here could occur in the majority of USC patients. To test this, we used LB-100, a catalytic site inhibitor of PP2A currently being used in clinical trials. Treatment with LB-100 in combination with Clofarabine showed synergy in both UT42Aα-P179R and OV17Aα-S256F cells expressing WT Aα, showing that the inhibition of PP2A’s catalytic activity could phenocopy the effects of a mutant Aα, sensitizing cells to Clofarabine treatment (Figure 4B and C, Supplemental Figure 13A and B). Further, we also tested the effects of LB-100 in combination with Clofarabine in UT42Aα-P179R and OV17Aα-S256F cells expressing EGFP. Interestingly, inhibition of PP2A catalytic activity showed synergy at some doses, consistent with the heterozygous nature of these mutations (Supplemental Figure 13C–F). However, unlike the WT cell lines, only the mutant lines displayed Clofarabine/LB-100 ratios that were antagonistic (Supplemental Figure 13C–F).
To expand on these findings, we wanted to extend these investigations into cell lines of endometrioid lineage. Mutations to Aα are less frequent in uterine endometrioid carcinomas, occurring in 11% of patients compared to 30–40% of USC. Additionally, many of these mutations are not recurrent hotspot mutations. We tested AN3CA and KLE, both of which are WT for PPP2R1A, and HEC50B, which harbors a heterozygous A R183W mutation. Aα-R183 is a hotspot mutation and the most commonly mutated residue of PPP2R1A across cancer, although less prevalent than P179R and S256F in uterine cancer, and we and others have previously shown that this mutation also inhibits normal PP2A function (5,21,35). We hypothesized that the mutant endometrioid line, HEC50BAα-R183W would be more sensitive to Clofarabine than the WT, AN3CA(+/+) and KLE(+/+) cells. To test this, we treated these cells with increasing doses of Clofarabine and performed an MTT cell viability assay and found that the mutant HEC50B cells were the most sensitive to Clofarabine (Figure 4D). Additionally, when all three cell lines were tested with the combination of Clofarabine with LB-100, only the WT cell lines, AN3CA and KLE, showed synergy (Figure 4E–J). γH2AX levels were also measured by western blot in these lines following Clofarabine treatment, and consistent with the USC data, the HEC50BAα-R183W accumulated more DNA damage than either WT cell line (Supplemental Figure 14A and B). Finally, because proliferation rates can also influence response to RNRi, proliferation was measured in the HEC50B, AN3CA, and KLE cell lines (Supplemental Figure 14C). Similar to our isogenic systems, we found no correlation to proliferation rate and RNRi response.
Finally, we were interested in exploring the expression levels of PP2A subunits in the control tumor samples and those treated with Clofarabine (Figure 2). When PP2A C subunit levels were analyzed in these studies by western blot, we found that the Clofarabine treated samples in the terminal efficacy studies had significantly more PP2A C subunit expression than the vehicle control treated tumors, again suggestive of acquired resistance in these models (Figure 4K–M, Supplemental Figure 5A–F). Additionally, this data supports the notion that PP2A activity, through increased C subunit expression, may be a resistance mechanism to RNRi, supportive of PP2A mediating synthetic lethality we identified with RNR inhibitors. Finally, in contrast to the increase in PP2A C subunit levels in the terminal efficacy studies, analysis of the pharmacodynamic UT42Aα-P179R samples showed no changes in PP2A C subunit expression (Figure 4N).
Together, this data suggests that PP2A is essential for a cells ability to respond to replicative stress. In the event of a PP2A scaffolding mutation, a common event in USC, the C subunit is unable to bind and is degraded (3,23). When these cells are then treated with Clofarabine or other ribonucleotide reductase inhibitors, they are unable to effectively activate the replicative checkpoint signals, accumulate DNA damage, and undergo cell death as a result. Further, impairment of PP2A’s catalytic activity is common across the majority of patients with USC, and renders these cells sensitive to Clofarabine treatment. Additionally, the use of LB-100 as a sensitizer to RNR inhibition could broaden the potential clinical and translational impact of these findings.
Uterine serous carcinomas are more responsive to gemcitabine than uterine endometrioid carcinomas
Given the prevalence of PP2A subunit loss in USC, we were interested in exploring if RNR inhibition could be a potential therapeutic strategy for this subtype of uterine cancer. The serous subtype of uterine carcinoma is highly aggressive, and the overall prognosis is typically worse in this subtype due to high risk of recurrence. To determine whether USCs also exhibit inferior prognosis in the setting of recurrent disease, patients with recurrent disease were selected from The Cancer Genome Atlas (TCGA) (36,37), and, when stratified by histology, 55 patients had endometrioid carcinoma and 31 patients had serous carcinoma. Overall survival (from time of diagnosis) of this study showed that overall survival of the endometrioid cohort was 38.1 months vs 31.4 months for the serous cohort, although this did not meet statistical significance likely due to small sample size (p=0.18) (Figure 5A).
Figure 5: PP2A inactivation predicts sensitivity and response to Gemcitabine treatment in a cohort of patients.
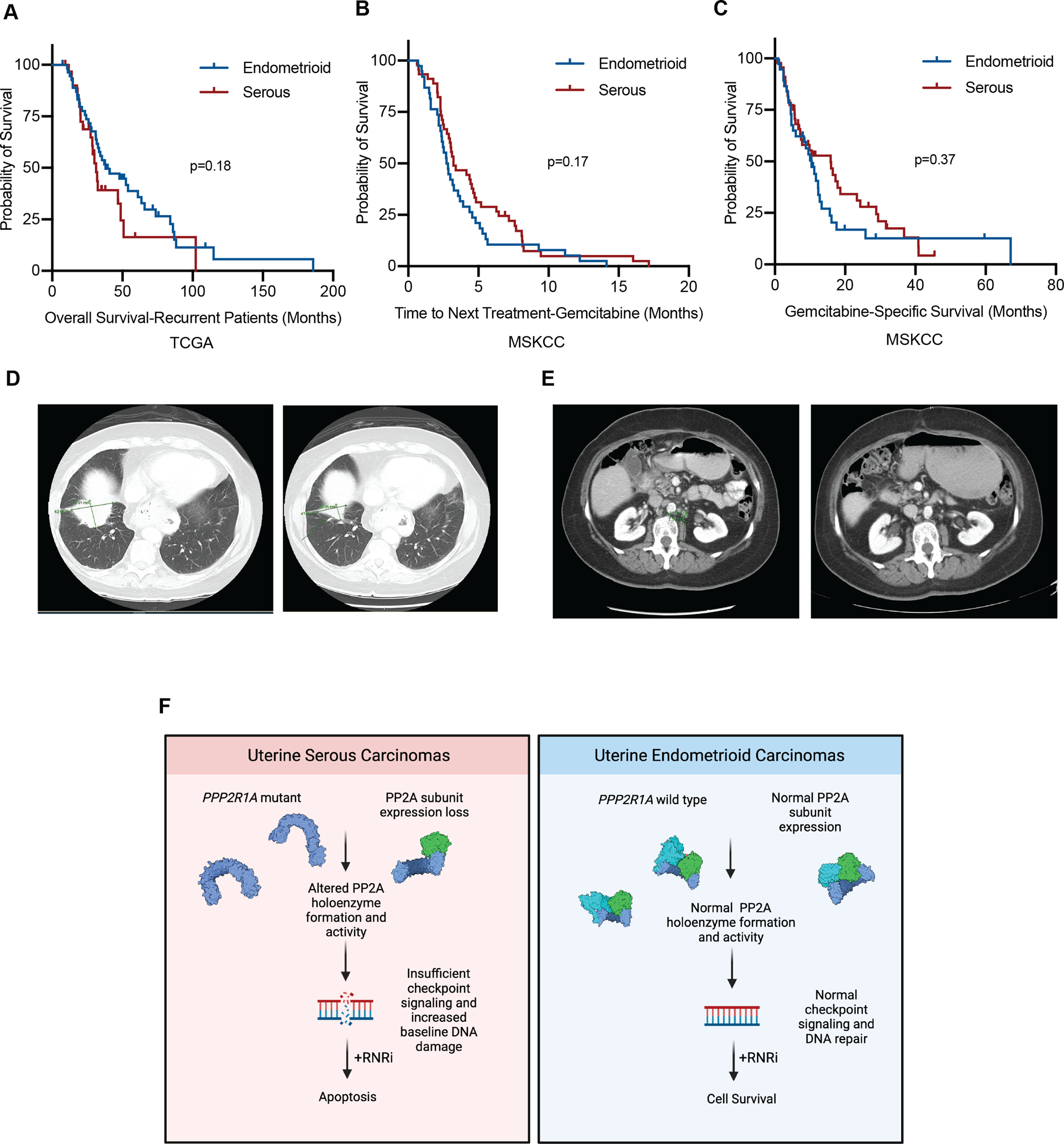
A-C, Kaplan-Meier estimates of survival as stratified by histology. Blue line represents endometrioid or mixed endometrioid (not serous) histology, red line represents serous or mixed serous histology. (A) Overall survival of patients with recurrent disease from the TCGA (B) Time to next treatment following initiation of gemcitabine. (C) Initiation of gemcitabine to date of death or last follow-up. D, CT images of the first patient before initiation of gemcitabine (left) and after (right). E, CT images of the second patient before the initiation of gemcitabine (left) and after (right). F, Schematic of the working model of the proposed studies. In USC (left) PP2A subunit expression is decreased, or PP2A Aα mutations are present, leading to a reduction in PP2A subunit expression. This in turn leads to altered PP2A activity, causing inefficient checkpoint signaling and increased DNA damage, leading to a synthetic lethal interaction with RNR inhibition. Conversely, in UEC PP2A Aα mutations and subunit expression loss are infrequent, allowing for normal checkpoint signaling and DNA damage repair. Under challenge with RNR inhibition, these cells are more capable of dealing with the DNA damage caused by these agents, and cells are more likely to survive. Created with BioRender.com.
While RNR inhibitors are not routinely used for uterine cancers, we have identified a cohort of patients with recurrent disease treated with gemcitabine at MSKCC as a second or later line therapy. A total of 83 patients were identified from this unique patient cohort (Supplemental Figure 15). Included patients were those with serous, endometrioid, or mixed endometrial adenocarcinoma histology that received gemcitabine as monotherapy or in combination with a platinum-based agent during the study period. Patients were excluded if they received less than two doses of gemcitabine, received gemcitabine for an unrelated malignancy, or had lack of adequate follow-up information. Prior to initiation of gemcitabine, the median number of prior lines of therapy was 3 (range 0–11) and 98% (n=81) received a prior platinum agent. When stratified by histology, 45 patients had serous or mixed adenocarcinoma with serous features and 38 patients had endometrioid or mixed adenocarcinoma without serous features. In the serous cohort, 38% (n=17) patients received combination gemcitabine + carboplatin while 26% (n=10) of the endometrioid cohort received combination gemcitabine + carboplatin. There was no difference between receipt of combination vs. single-agent gemcitabine in either cohort (p=0.35) and no difference in number of prior lines of therapy (p=0.6). The median time to next treatment (TNT) for the serous cohort was 3.2 months (95% CI 1.8–4.6) vs. 2.7 months (95% CI 2.2–3.2) in the endometrioid cohort (p=0.17) (Figure 5B). There was a trend for increased median gemcitabine-specific survival in the serous cohort (15.9 months (95% CI 7.2–24.5) vs. 10.4 (95% CI 7.3–13.5) in the endometrioid cohort), but it did not reach statistical significance (p=0.37) (Figure 5C). While these results did not reach statistical significance (likely in part due to small sample size), given the expected inferior overall survival of the USC subtype compared to the endometrioid subtype in the TCGA (Figure 4H), these data nevertheless suggest that gemcitabine may improve outcomes for the patients with serous as opposed to endometrioid histology.
Of all patients analyzed in this cohort, the two patients with the longest TNT were of serous histology and derived significant clinical benefit from gemcitabine with TNT > 16 months. The first patient was diagnosed with stage IV serous endometrial cancer in November 2016. Prior to treatment initiation, PET imaging revealed pulmonary metastasis measuring 5.2 × 3.2cm with significant thoracic and abdominopelvic adenopathy. She was treated with six cycles of carboplatin + paclitaxel with excellent response and near-resolution of her pulmonary disease. She had a relatively short disease-free interval and developed disease recurrence eight months later with multiple pleural metastases, the largest of which measured 6.2 × 4.3cm (Figure 5D left). Gemcitabine + carboplatin was initiated with decreasing size of pleural lesion to 4.8 × 2.6 on post-cycle four imaging and continued improvement to 2.4 × 2.3cm after cycle eight (Figure 5D right). Treatment was discontinued at that time with ongoing observed response on subsequent surveillance imaging. Recurrent peritoneal disease was diagnosed a total of eleven months after discontinuation of gemcitabine + carboplatin, at which point she was re-challenged with gemcitabine + carboplatin with observed response. She is currently alive with disease on her fifth line of therapy.
A second patient was diagnosed with stage IV serous endometrial cancer in December 2013. At time of diagnosis, imaging demonstrated pelvic ascites, extensive peritoneal carcinomatosis with omental involvement, adnexal metastases, and subhepatic implant. She was treated with three cycles of neoadjuvant carboplatin + paclitaxel and underwent resection of residual disease followed by three additional cycles of carboplatin + docetaxel. Imaging at completion of therapy demonstrated persistent low-volume peritoneal carcinomatosis. First recurrence was diagnosed eight months later in the form of abdominopelvic adenopathy and multiple peritoneal implants, the largest of which was 1.6 × 1.4cm in the left anterior mid-abdomen (Figure 5E left). Gemcitabine + carboplatin was initiated at that time with resolution of adenopathy and decreasing size of peritoneal implants with resolution of prior left anterior mid-abdominal lesion on post-cycle four imaging (Figure 5E right). She received a total of eight cycles with resolution of all implants on imaging. Recurrence was observed in the form of hepatic metastases, abdominopelvic adenopathy, and carcinomatosis nine months later, at which time gemcitabine + carboplatin was repeated with observed response. She ultimately received five total lines of therapy with progression of disease and death 4.5 years after diagnosis. These cases highlight examples where therapy with gemcitabine achieved higher therapeutic efficacy than upfront standard platinum/taxane combination.
In summary, we propose the following model where in USC, PP2A subunit expression is decreased, or PP2A Aα mutations are present, leading to a reduction in PP2A subunit expression. This in turn leads to altered PP2A activity, causing inefficient checkpoint signaling and increased DNA damage, resulting in a synthetic lethal interaction with RNR inhibition. Conversely, in UEC, PP2A Aα mutations and subunit expression loss are infrequent, allowing for normal checkpoint signaling and DNA damage repair. Under challenge with RNR inhibition, these cells are more capable of dealing with the DNA damage caused by these agents, and cells are more likely to survive (Figure 5F). Collectively, these data highlight the critical role of PP2A signaling in pathogenesis of uterine serous carcinoma and support repurposing as well as development of new RNR inhibitors for the treatment of this aggressive histological subtype of endometrial cancer.
Discussion
The intention of this work was to elucidate whether highly recurrent mutations to PPP2R1A, the scaffolding subunit of PP2A, present in 40% of USC could be targeted using approved drugs. Here, we demonstrated that, indeed, mutations to the Aα subunit were predictive of sensitivity to ribonucleotide reductase (RNR) inhibitors. We further showed that patient-derived mutant cell models of USC were sensitive to RNR inhibition in vivo and the synthetic lethality could be phenocopied by inhibiting PP2A catalytic activity, and that dysregulation of PP2A was common in serous uterine tumors compared to those with endometrioid histology. Additionally, upregulation of PP2A catalytic subunit was detected in terminal xenograft tumors samples, potentially indicating that PP2A could be a regulator of RNRi therapy resistance. Finally, we presented that in a small cohort of uterine carcinoma patients treated with gemcitabine, serous patients tended to do better overall compared to endometrioid patients, in marked contrast to what is typically seen in this disease.
Our analysis of the TCGA indicated that PP2A subunit expression loss was common in USC. Interestingly, B55α (PPP2R2A) expression loss was frequent in USC, occurring in almost 73% of samples. It has not previously been explored as to whether loss of B55α is a driver event for USC tumorigenesis, but given the high frequency of genetic alteration to this subunit, further studies are warranted. Additionally, in other cancer types, B55α expression loss has been recently been implicated to render cells and tumors more sensitive to both PARP inhibitors and ATR inhibitors (38,39), and we have recently shown that the half-life and stability of B55α is tightly regulated by its binding to the Aα subunit scaffold (3,23). Specific to this study, we have shown that the Aα−P179R mutation disrupts binding of B55α, ultimately resulting in decreased expression of the regulatory subunit (3). While not tested here, it would be important to understand if there is a particular PP2A regulatory subunit responsible for RNR sensitivity, and if mutations to Aα would also render USC cells more sensitive to PARP and ATR inhibitors.
The study of the effects of PP2A Aα subunit mutations on signaling in response to Clofarabine treatment was limited to the checkpoint pathways involved in the regulation of replicative stress. While identification of a single PP2A substrate responsible for the observed changes in sensitivity to RNR inhibition is a compelling area of research inquiry, we have previously published on our findings of global phosphorylation changes upon expression of a single scaffolding subunit mutation using phosphoproteomics and identified hundreds of altered phospho-peptides (21). Given the broad biological role of PP2A holoenzymes the regulation of cellular signaling cascades, combined with the extensive disruption of regulatory subunits each scaffolding mutation causes, identification of one substrate responsible for the drug response may not be possible. Likely, there is an aggregate effect of signaling changes which ultimately render the PP2A mutant, or catalytically inactivated, cells sensitive to RNR inhibition.
Perhaps most importantly, our work provides clinical evidence that RNR inhibition, through the use of gemcitabine, could be beneficial to USC patients. Of all patients analyzed in our cohort, the two patients with the most durable gemcitabine response had tumors of serous origin. While advances have been made for the treatment of uterine carcinomas, new treatment options, including immunotherapy are not effective strategies for tumors of the serous subtype, and this remains a highly aggressive and devastating disease (2). Beyond USC, PPP2R1A mutations have been identified in undifferentiated endometrial carcinomas and PP2A dysregulation is a common event in human cancer, opening the possibility of using modulators of replicative stress in a precision medicine approach for a broad range of tumor types. Collectively, the findings presented here could have immediate translational impact and alter the treatment trajectory for cancer patients.
Supplementary Material
Statement of Significance:
A drug repurposing screen identifies synthetic lethal interactions in PP2A-deficient uterine serous carcinoma, providing potential therapeutic avenues for treating this deadly endometrial cancer.
Acknowledgements:
We would like to thank Dr. Shirish Shenolikar for his constructive feedback. Funding for this work was provided by NIH/NCI grants to G. Narla (R01 CA-181654 and R01 CA-240993) and CM O’Connor (T32 CA-009676). Funding for this work was also provided by the Rogel Cancer Center at the University of Michigan.
Disclosure of Conflict of Interest:
C.M. O’Connor and G. Narla are named inventors on a US provisional patent application concerning compositions and methods for treating high grade subtypes of uterine cancer. C.M. O’Connor, T.K. Suhan, K.P. Zawacki, and J. Sangodkar are consultants for RAPPTA Therapeutics. G. Narla is chief scientific officer at RAPPTA Therapeutics, is an SAB member at Hera BioLabs, reports receiving commercial research support from RAPPTA Therapeutics, and has ownership interest (including patents) in RAPPTA Therapeutics. D. Zamarin reports research support to his institution from Astra Zeneca, Plexxikon, and Genentech; personal/consultancy fees from Synlogic Therapeutics, GSK, Genentech, Xencor, Memgen, Immunos, Celldex, Calidi, and Agenus. D. Zamarin is an inventor on a patent related to use of oncolytic Newcastle Disease Virus for cancer therapy.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30 doi 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Urick ME, Bell DW. Clinical actionability of molecular targets in endometrial cancer. Nat Rev Cancer 2019;19(9):510–21 doi 10.1038/s41568-019-0177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor SE, O’Connor CM, Wang Z, Shen G, Song H, Leonard D, et al. The highly recurrent PP2A Aalpha-subunit mutation P179R alters protein structure and impairs PP2A enzyme function to promote endometrial tumorigenesis. Cancer research 2019. doi 10.1158/0008-5472.can-19-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeong AL, Han S, Lee S, Su Park J, Lu Y, Yu S, et al. Patient derived mutation W257G of PPP2R1A enhances cancer cell migration through SRC-JNK-c-Jun pathway. Scientific reports 2016;6:27391 doi 10.1038/srep27391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haesen D, Abbasi Asbagh L, Derua R, Hubert A, Schrauwen S, Hoorne Y, et al. Recurrent PPP2R1A Mutations in Uterine Cancer Act through a Dominant-Negative Mechanism to Promote Malignant Cell Growth. Cancer research 2016;76(19):5719–31 doi 10.1158/0008-5472.can-15-3342. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn E, Wu RC, Guan B, Wu G, Zhang J, Wang Y, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst 2012;104(19):1503–13 doi 10.1093/jnci/djs345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shih Ie M, Panuganti PK, Kuo KT, Mao TL, Kuhn E, Jones S, et al. Somatic mutations of PPP2R1A in ovarian and uterine carcinomas. The American journal of pathology 2011;178(4):1442–7 doi 10.1016/j.ajpath.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McConechy MK, Anglesio MS, Kalloger SE, Yang W, Senz J, Chow C, et al. Subtype-specific mutation of PPP2R1A in endometrial and ovarian carcinomas. J Pathol 2011;223(5):567–73 doi 10.1002/path.2848. [DOI] [PubMed] [Google Scholar]
- 9.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497(7447):67–73 doi 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson WJ, Hoivik EA, Halle MK, Taylor-Weiner A, Cherniack AD, Berg A, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet 2016;48(8):848–55 doi 10.1038/ng.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. The FEBS journal 2016;283(6):1004–24 doi 10.1111/febs.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Connor CM, Perl A, Leonard D, Sangodkar J, Narla G. Therapeutic Targeting of PP2A. The international journal of biochemistry & cell biology 2018;96:182–93 doi 10.1016/j.biocel.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer research 2005;65(18):8183–92 doi 10.1158/0008-5472.can-05-1103. [DOI] [PubMed] [Google Scholar]
- 14.Sablina AA, Hector M, Colpaert N, Hahn WC. Identification of PP2A complexes and pathways involved in cell transformation. Cancer research 2010;70(24):10474–84 doi 10.1158/0008-5472.can-10-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 2004;5(2):127–36. [DOI] [PubMed] [Google Scholar]
- 16.Sablina AA, Hahn WC. The Role of PP2A A Subunits in Tumor Suppression. Cell Adh Migr. Volume 12007. p 140–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson JB, Pallas DC. Circumventing Cellular Control of PP2A by Methylation Promotes Transformation in an Akt-Dependent Manner1. Neoplasia (New York, NY). Volume 142012. p 585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, et al. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 1990;60(1):167–76. [DOI] [PubMed] [Google Scholar]
- 19.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007;445(7123):53–7 doi 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y Serine/threonine phosphatases: mechanism through structure. Cell 2009;139(3):468–84 doi 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 21.O’Connor CM, Leonard D, Wiredja D, Avelar RA, Wang Z, Schlatzer D, et al. Inactivation of PP2A by a recurrent mutation drives resistance to MEK inhibitors. Oncogene 2020;39(3):703–17 doi 10.1038/s41388-019-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruediger R, Zhou J, Walter G. Mutagenesis and expression of the scaffolding Aalpha and Abeta subunits of PP2A: assays for measuring defects in binding of cancer-related Aalpha and Abeta mutants to the regulatory B and catalytic C subunits. Methods in molecular biology (Clifton, NJ) 2007;365:85–99 doi 10.1385/1-59745-267-x:85. [DOI] [PubMed] [Google Scholar]
- 23.O’Connor CM, Hoffa MT, Taylor SE, Avelar RA, Narla G. Protein phosphatase 2A Aalpha regulates Abeta protein expression and stability. The Journal of biological chemistry 2019. doi 10.1074/jbc.RA119.007593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Boehm JS, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 2011;8(8):659–61 doi 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006;58(3):621–81 doi 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 26.Wisitpitthaya S, Zhao Y, Long MJ, Li M, Fletcher EA, Blessing WA, et al. Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition. ACS Chem Biol 2016;11(7):2021–32 doi 10.1021/acschembio.6b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aye Y, Stubbe J. Clofarabine 5’-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc Natl Acad Sci U S A 2011;108(24):9815–20 doi 10.1073/pnas.1013274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Celik H, Sciandra M, Flashner B, Gelmez E, Kayraklioglu N, Allegakoen DV, et al. Clofarabine inhibits Ewing sarcoma growth through a novel molecular mechanism involving direct binding to CD99. Oncogene 2018;37(16):2181–96 doi 10.1038/s41388-017-0080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker WB. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chemical Reviews 2009;7(109):2880–93 doi 10.1021/cr900028p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noto FK, Adjan-Steffey V, Tong M, Ravichandran K, Zhang W, Arey A, et al. Sprague Dawley Rag2 null rats created from engineered spermatogonial stem cells are immunodeficient and permissive to human xenografts. Mol Cancer Ther 2018;17(11):2481–9 doi 10.1158/1535-7163.mct-18-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noto FK, Sangodkar J, Adedeji BT, Moody S, McClain CB, Tong M, et al. The SRG rat, a Sprague-Dawley Rag2/Il2rg double-knockout validated for human tumor oncology studies. PloS one 2020;15(10):e0240169 doi 10.1371/journal.pone.0240169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 2015;34(16):2011–21 doi 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- 33.Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A 2013;110(8):2916–21 doi 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leskela S, Perez-Mies B, Rosa-Rosa JM, Cristobal E, Biscuola M, Palacios-Berraquero ML, et al. Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma. Cancers (Basel) 2019;11(7) doi 10.3390/cancers11070964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perl AL, O’Connor CM, Fa P, Mayca Pozo F, Zhang J, Zhang Y, et al. Protein phosphatase 2A controls ongoing DNA replication by binding to and regulating cell division cycle 45 (CDC45). The Journal of biological chemistry 2019;294(45):17043–59 doi 10.1074/jbc.RA119.010432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173(2):400–16 e11 doi 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018;33(4):690–705 e9 doi 10.1016/j.ccell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu Z, Fa P, Liu T, Prasad CB, Ma S, Hong Z, et al. A Genome-Wide Pooled shRNA Screen Identifies PPP2R2A as a Predictive Biomarker for the Response to ATR and CHK1 Inhibitors. Cancer research 2020;80(16):3305–18 doi 10.1158/0008-5472.CAN-20-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalev P, Simicek M, Vazquez I, Munck S, Chen L, Soin T, et al. Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer research 2012;72(24):6414–24 doi 10.1158/0008-5472.CAN-12-1667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.