

Abstract
Objective:
To compare the rate of change in cognition between glucocerebrosidase (GBA) mutation carriers and non-carriers with and without subthalamic nucleus deep brain stimulation (STN-DBS) in Parkinson’s disease (PD).
Methods:
Clinical and genetic data from 12 datasets were examined. Global cognition was assessed using the Mattis Dementia Rating Scale (MDRS). Subjects were examined for mutations in GBA and categorized as GBA carriers with or without DBS (GBA+DBS+, GBA+DBS−), and non-carriers with or without DBS (GBA−DBS+, GBA−DBS−). GBA mutation carriers were subcategorized according to mutation severity (risk variant, mild, severe). Linear mixed modeling was used to compare rate of change in MDRS scores over time among the groups according to GBA and DBS status and then according to GBA severity and DBS status.
Results:
Data were available for 366 subjects: 58 GBA+DBS+, 82 GBA+DBS−, 98 GBA−DBS+, and 128 GBA−DBS− subjects who were longitudinal followed (range 36 to 60 months after surgery). Using the MDRS, GBA+DBS+ subjects declined on average 2.02 points/year more than GBA−DBS− subjects (95% CI = −2.35, −1.69), 1.71 points/year more than GBA+DBS− subjects (95% CI = −2.14, −1.28), and 1.49 points/year more than GBA−DBS+ subjects (95% CI = −1.80, −1.18).
Interpretation:
Although non-randomized, this composite analysis suggests that the combined effects of GBA mutations and STN-DBS negatively impact cognition. We advise that DBS candidates be screened for GBA mutations as part of the pre-surgical decision-making process. We advise that GBA mutation carriers be counseled regarding potential risks associated with STN-DBS and alternative options may be considered.
Introduction:
Parkinson disease (PD) affects at least 1 million people in the U.S.1 with approximately 9,000 PD patients opting for deep brain stimulator (DBS) placement into the subthalamic nucleus (STN-DBS) annually.2 Studies have suggested that genetic subtyping of PD subjects may be useful in understanding cognitive and motor outcomes of DBS.3–5
Individuals with PD who carry mutations in the glucocerebrosidase (GBA) gene (PDGBA), are of particular interest, as they are at increased risk for cognitive impairment. PDGBA subjects have reduced activity of the glucocerebrosidase (GCase) enzyme resulting in disrupted sphingolipid metabolism6 with more rapid accumulation and spread of Lewy body pathology compared with non-mutation carriers.7 PDGBA is associated with earlier onset of disease and thus, these individuals may be more likely to pursue DBS.8 Up to 17% of PD subjects who received DBS carry GBA variants.3, 9 This is higher than the general PD population frequency since those who opt for DBS tend to be younger, have clear levodopa responsiveness, and troublesome clinical features that qualify the individual for DBS, such as dyskinesia and motor fluctuations.3, 9 Importantly, STN-DBS itself can impair cognition, with a negative impact on verbal fluency,10 executive control of action,11 and inhibitory control.12 Given the compact anatomy of the STN, there is potential for unintended current spread into adjacent associative and limbic subregions, adjacent fiber bundles13 or other nearby nuclei such as the substantia nigra.14 Imaging studies have also revealed concomitant activation of nonmotor frontal-striatal circuitry as a consequence of unintended current spread.15 Furthermore, a recent pilot study found differences in beta power comparing PDGBA vs. non-GBA subjects, suggesting that genotype may be associated with specific neurophysiologic changes.16 Whether these physiologic changes are responsible for heterogeneous outcomes of DBS remains to be explored.
In this study, we aimed to determine the combined effects of GBA and STN-DBS on global cognition. We examined longitudinal changes in cognition in four groups of patients: GBA carriers with or without DBS (GBA+DBS+, GBA+DBS−), and non-carriers with or without DBS (GBA−DBS+, GBA−DBS−). We hypothesized that GBA+DBS+ subjects would have the fastest rate of cognitive decline compared with the other groups.
Materials and Methods:
Datasets
Approval for the study was obtained from the Rush University Medical Center Institutional Review Board. Prospective and retrospective clinical and genetic data were pooled across 12 datasets from: Amsterdam University Medical Centers (Amsterdam, Netherlands), Columbia University (New York, NY, USA), Hôpital Pitié-Salpêtrière (Paris, France), National Institutes of Health (NIH, Bethesda, USA), Norwegian University of Science and Technology (Trondheim, Norway), Parkinson’s Progression Markers Initiative (PPMI, www.ppmi-info.org/data), Rush University (Chicago, IL, USA), Mount Sinai Beth Israel (New York, NY, USA), University College London (London, UK), and the Accelerating Medicines Partnership: Parkinson’s disease (AMP-PD, amp-pd.org). At all clinical sites (n = 8), DBS was performed awake, with microelectrode recording (MER) being used at all sites except one (University College London). Only 2 of 8 clinical sites did not make specific trajectory adjustments to avoid the caudate nucleus (Columbia University and Norwegian University of Science and Technology). All sites averaged between 1–3 tracks per side, with one site averaging 5 tracks per side (Norwegian University of Science and Technology). From the clinical sites, no cases of genetic testing were performed because of poor DBS outcome. Genetic testing was done as part of clinical and/or research efforts to offer genetic testing regardless of clinical status. Within AMP-PD, data were extracted from the LRRK2 Cohort Consortium, STEADY-PD, and the Parkinson’s Disease Biomarkers Program (PDBP), and only subjects were included where both GBA and DBS status were confirmed (present or absent).17 All DBS subjects had electrodes implanted bilaterally within the STN. Subjects were examined for mutations in GBA and then categorized as GBA carriers with or without DBS (GBA+DBS+, GBA+DBS−), and non-carriers with or without DBS (GBA−DBS+, GBA−DBS−). Four clinical sites (Columbia University, NIH, Norwegian University, Mount-Sinai Beth Israel) contributed only GBA+DBS+ subjects. Subjects from Rush University and PPMI contributed subjects to all four groups. Only GBA+DBS− subjects were drawn from AMP-PD given that details of DBS implantation were not collected in this dataset. The remaining three datasets (Hôpital Pitié-Salpêtrière, University College London, and University of Amsterdam), consisted of only DBS subjects. Data was checked for duplicate subjects based on demographics, mutations, and cognitive scores. Additional details for each dataset are described below and summarized in Supplementary Table 1.
University of Amsterdam (Amsterdam, Netherlands):
A total of 36 bilateral STN from the Netherlands SubThalamic and Pallidal Stimulation (NSTAPS) study18 had both clinical and genetic data available (2007–2011). Subjects were tested for the following GBA mutations: A456P, L444P, N370S, T369M, E326K, and D140H. Of the 36 STN subjects, 6 were GBA+DBS+ and 30 were GBA−DBS+. MDRS scores were available for 2 of 6 GBA+DBS+ subjects and 7 of 30 GBA−DBS+ subjects.
Columbia University (New York, NY, USA):
A total of 5 GBA+DBS+ subjects with clinical and genetic data were available in the Columbia University database (K02NS080915, 2006–2017). The GBA gene was fully sequenced as previously described.19 One subject was excluded who had a pre-DBS MDRS score < 130. Another subject carried the Q-8H mutation, a variant of unknown significance (VUS), was excluded.
Hôpital Pitié-Salpêtrière (Paris, France):
A total of 14 GBA+DBS+ subjects and 28 GBA−DBS+ subjects with clinical and genetic data were available for analysis (1998–2016). The GBA gene was fully sequenced as previously described.4
National Institutes of Health (NIH, Bethesda, USA):
A total of 5 GBA+DBS+ subjects with clinical and genetic data were available in the NIH Parkinson’s Clinic database (2012–2019). Genotyping was performed using a genotyping array (NeuroX or Neuro Consortium Array, Illumina, Inc., San Diego, CA) with custom content covering neurodegenerative disease-related variants. To identify SNPs from the genotyping array, Illumina GenomeStudio (v.2.0) was used cluster genotypes. After quality control, subjects with pathogenic GBA variants were included while VUS were excluded.
Norwegian University of Science and Technology (Trondheim, Norway):
A total of 3 GBA+DBS+ subjects with clinical and genetic data were available in the Norwegian University database (2002–2014). Subjects were tested for N370S and L444P mutations as previously described.20
Parkinson’s Progression Markers Initiative (PPMI):
The database was accessed February 1, 2021. First, all PD subjects with known DBS status (present/absent) were identified (n = 279). Then, genetic data were pulled for these PD subjects. Genotyping methods used in PPMI have been described extensively elsewhere.21 Subjects with VUS were excluded (2 patients without DBS, r44c, r39c). One subject carried a rare polymorphism, K(−27)R, and was excluded because this polymorphism has been observed to occur at a frequency of >5% in controls of African and Asian ancestry.22 One individual carried the I489L mutation, which has not been previously reported and, therefore, classified as a VUS and excluded. A total of 157 subjects remained: 2 GBA+DBS+, 40 GBA+DBS−, 6 GBA−DBS+, and 109 GBA−DBS− subjects.
Rush University (Chicago, IL, USA):
Retrospective data from consecutive PD subjects already implanted with bilateral STN-DBS were genotyped for GBA mutations (2003–2018). Additionally, consecutive PD subjects planning to undergo DBS and those without DBS were genotyped and followed prospectively (2017–2020). Subjects were fully sequenced for GBA as previously described.23 Data were available for a total of 102 subjects: 21 GBA+DBS+, 10 GBA+DBS−, 52 GBA−DBS+, 19 GBA−DBS− subjects.
Mount Sinai Beth Israel (New York, NY, USA):
A total of 4 GBA+DBS+ subjects with clinical and genetic data were available for review (2005–2019). Participants were genotyped for both LRRK2-G2019S and the 11 most common GBA mutations among Ashkenazim: N370S, 84GG, IVS2+1, V394L, D409G, L444P, A456P, RecNcil, R496H, E326K or T369M as previously described.24
University College London (London, UK):
Motor outcomes data from this cohort, which includes 32 subjects with bilateral STN-DBS, have been published previously.3 However, MDRS scores were available for only 10 subjects: 5 GBA+DBS+ subjects and 5 GBA−DBS+ subjects. Subjects were fully sequenced for GBA as previously described.3
AMP-PD:
This dataset includes the LRRK2 Cohort Consortium (LCC), STEADY-PD, and PDBP datasets. Whole genome sequencing methods for this cohort have been described elsewhere.17 There was one GBA+DBS+ subject in the PDBP dataset who was excluded since the specifics regarding unilateral vs. bilateral DBS and site of implantation were not collected. A total of 32 GBA+DBS− subjects were identified from AMP-PD: LCC (n = 2), STEADY-PD (n = 5), PDBP (n= 25).
GBA mutation carriers were further subcategorized according to mutation severity (risk variant, mild, severe) based on prior reports (Table 1).25 The specific mutations with their corresponding mutation severity categorization are shown in Table 2. Subjects who carried both GBA and LRRK2 mutations were excluded, since LRRK2 variants might be protective for cognitive decline in GBA mutation carriers.26, 27 Subjects with two different GBA mutations were categorized as compound heterozygotes.
Table 1.
GBA mutation carriers categorized according to mutation severity and DBS status25
| GBA mutation type | non-DBS (n=82) | DBS (n=58) | Total |
|---|---|---|---|
| Risk variant | 58 (71%) | 24 (41%) | 82 |
| Mild | 15 (18%) | 23 (40%) | 39 |
| Severe | 9 (11%) | 11 (19%) | 20 |
Table 2.
Specific GBA mutations according to severity
| Mutation Name | |
|---|---|
| Risk variant | E326K (53), E388K (1), T369M (28) |
| Mild | N370S (33), R120W (3), L279P (1), S364N (1) |
| Severe | L444P (5), RecNcil (3), IVS2+1 G>A (2), A456P (1), A456P, RecNcil (1), L444P/A456P (1), E326K/L444P (1), G115R/G193E (1), R463C/R463C (1), H255Q (1), L29AFs*18 (1), N370S/N370S (1), R131C (1) |
Demographic and clinical data
The following demographic and clinical data were collected: baseline age, age at disease onset, sex, date of DBS, family history of first-degree PD relative, Unified Parkinson’s Disease Rating Scale (UPDRS) or MDS revision of the UPDRS (MDS-UPDRS), Mini-Mental State Exam (MMSE), Montreal Cognitive Assessment (MoCA), and Mattis Dementia Rating Scale (MDRS). UPDRS scores were converted to MDS-UPDRS scores.28 The number of cognitive test points/subject and the interval between cognitive testing (months) were also collected. Cognitive assessments were performed in the subject’s native language where possible (Netherlands, France, Norway). Baseline age for DBS subjects was defined as the age of the subject pre-DBS. MMSE and MoCA scores were converted to MDRS scores.29 All subjects were required to have a baseline MDRS score of 130 or greater to be included in the analysis per typical pre-operative cognitive function recommendations for DBS implantation.30 GBA+DBS+ subjects were included if their baseline score was missing but their 1-year post-DBS MDRS score was 130 or greater (n = 4 of 366 subjects). No imputation was used to handle this missing data since it was relevant for only 4/366 subjects (1% of data).
Statistical analysis
Demographic characteristics were compared using one-way ANOVA or Kruskal-Wallis test as appropriate. Post-hoc Bonferroni correction was performed for p < 0.05. Post-DBS UPDRS-III scores (ON medication and ON stimulation vs. pre-DBS OFF medication) and levodopa equivalent daily dosing (LEDD)31 reduction (pre vs. post-DBS) were stratified by GBA status with available data within 2 years of DBS implantation. LEDD reduction was used as a surrogate marker for DBS efficacy since OFF medication/ON stimulation UPDRS scores were not available. Linear mixed modeling with random intercept was used to compare rate of change in MDRS scores over time among the groups according to GBA and DBS status and then according to GBA severity and DBS status. For DBS subjects, the time at which pre-DBS MDRS (baseline) assessments were conducted was defined as time zero. The model was adjusted for age, age at onset, sex, and study site (random factor). Given the non-random group assignment of our subjects, we also performed the mixed model analysis with propensity score weighting technique. Propensity score was estimated with the same fixed variables adjusted in the unweighted analysis including age, age at onset and sex. Finally, we also performed an analysis looking at a three-way interaction (DBS*GBA*time) while adjusting for sex, baseline age, age of onset, and study site.
Results:
Baseline characteristics
Data were available for 366 subjects: 58 GBA+DBS+, 82 GBA+DBS−, 98 GBA−DBS+, and 128 GBA−DBS− subjects across 12 datasets (Table 3). DBS subjects, regardless of GBA status, had significantly lower MDRS scores pre-DBS compared with GBA−DBS− subjects. GBA+DBS+ subjects had significantly worse cognition at baseline with GBA−DBS− subjects (p = 0.017). Based on the data available, the number of cognitive test points/subject, the interval between cognitive testing (months), and median follow-up time for cognitive assessments was significantly different between the four groups (Table 3). The number of subjects from each dataset are shown in Supplementary Table 1.
Table 3.
Demographic and baseline characteristics
| GBA+DBS+ (n=58) | GBA+DBS− (n=82) | GBA−DBS+ (n=98) | GBA−DBS− (n=128) | p-value | |
|---|---|---|---|---|---|
| Age at baseline, Mean (SD) | 57.19 (7.48) | 60.42 (9.55)2 | 58.28 (8.31) | 61.44 (9.50) | 0.01 |
| Age of onset, Mean (SD) | 46.76 (7.76) | 53.80 (10.36)9 | 46.71 (9.27) | 53.52 (10.95) | <0.0001 |
| Sex, % women | 20 (34.48%) | 37 (46.25%)2 | 32 (32.65%) | 48 (37.50%) | 0.28 |
| Family history, % with first degree relative | 20 (39.22%)7 | 15 (30.00%)32 | 18 (20.22%)9 | 31 (24.22%) | 0.08 |
| Baseline MDRS (Mattis), Mean (SD) | 139.15 (3.63)4 | 139.78 (3.53)2 | 139.18 (3.83) | 141.00 (2.72) | 0.0002 |
| Number of cognitive test points/subject (mean, IQR, range) | 2.5, 2, 2–5 | 4, 3, 2–6 | 2, 1, 2–6 | 6, 1, 2–6 | <0.0001 |
| Months between testing (mean, IQR, range) | 20, 40, 8.5–148 | 12, 0, 12–36 | 19.83, 30, 5.33–144 | 12, 0, 12–36 | <0.0001 |
| Follow-up time in months, Median (IQR) | 53.00 (91.00) | 36.00 (24.00) | 37.50 (82.00) | 60.00 (0.00) | 0.001 |
Superscript indicates number of subjects with missing values
DBS subjects
Of 156 subjects with DBS, 58 were GBA mutation carriers while 98 were non-carriers. There was no difference in the number of years from motor symptom onset to DBS implantation or number of years that subjects had DBS (p > 0.05, Table 4) with respect to GBA status. There was no difference in baseline MDRS scores in DBS subjects based on GBA status (p = 1.0). Pre-operative UPDRS-III OFF/ON scores, percent change in UPDRS-III OFF/ON medication, and pre-DBS LEDD were not significantly different based on GBA status. Similarly, post-operative UPDRS-III scores (ON medication/ON stimulation) and percent reduction in LEDD were not significantly different based on GBA status.
Table 4.
Characteristics of DBS subjects
| GBA with DBS (n=58) | Non-GBA with DBS (n=98) | p | |
|---|---|---|---|
| Years from motor onset to DBS | 10.54 (5.17) | 11.54 (5.79) | 0.28 |
| Years of DBS | 4.95 (3.96) | 4.39 (4.21) | 0.41 |
| Pre-DBS | |||
| MDRS | 139.184 (3.60) | 139.18 (3.83) | 1.0 |
| UPDRS-III (OFF medication) | 35.4815 (13.37) | 33.5912 (14.41) | 0.47 |
| UPDRS-III (ON medication) | 14.278 (8.51) | 12.8712 (9.66) | 0.39 |
| % change UPDRS-III OFF vs. ON medication | 58.2215 (30.20) | 63.5913 (22.26) | 0.25 |
| LEDD | 1079.9730 (461.88) | 1191.4639 (474.35) | 0.30 |
| Post-DBS motor function | |||
| UPDRS-III (ON medication/ON stimulation) | 12.8830 (10.78) | 11.7135 (9.71) | 0.61 |
| % change UPDRS-III (pre-DBS OFF medication vs. post-DBS ON medication/ON stimulation) | 57.9236 (41.49) | 61.2339 (32.78) | 0.71 |
| years post-DBS (UPDRS) | 1.6930 (1.54) | 1.5932 (1.23) | 0.74 |
| Post-DBS medication burden | |||
| LEDD reduction | 496.9233 (316.49) | 458.8746 (382.38) | 0.66 |
| % change LEDD pre vs. post-DBS | 49.4633 (40.79) | 60.9648 (27.24) | 0.15 |
| Years post-DBS | 1.5433 (1.24) | 1.4846 (1.13) | 0.84 |
Superscript indicates number of subjects with missing values
Effects of GBA and DBS on cognition
GBA mutation carriers with DBS (GBA+DBS+) had the fastest cognitive decline among the four groups based on change in MDRS scores per year (Figure 1). GBA+DBS+ had the worst cognition at baseline and declined on average 2.02 points/year (SE = 0.17) more than GBA−DBS− subjects, 1.71 points/year (SE = 0.22) more than GBA+DBS− subjects, and 1.49 points/year (SE = 0.16) more than GBA−DBS+ subjects (all p<0.0001, Table 5). Similar results were found with propensity score weighting technique (Table 5). The analysis testing for a three-way interaction including the term DBS*GBA*time was also statistically significant (p < 0.0001).
Figure 1.
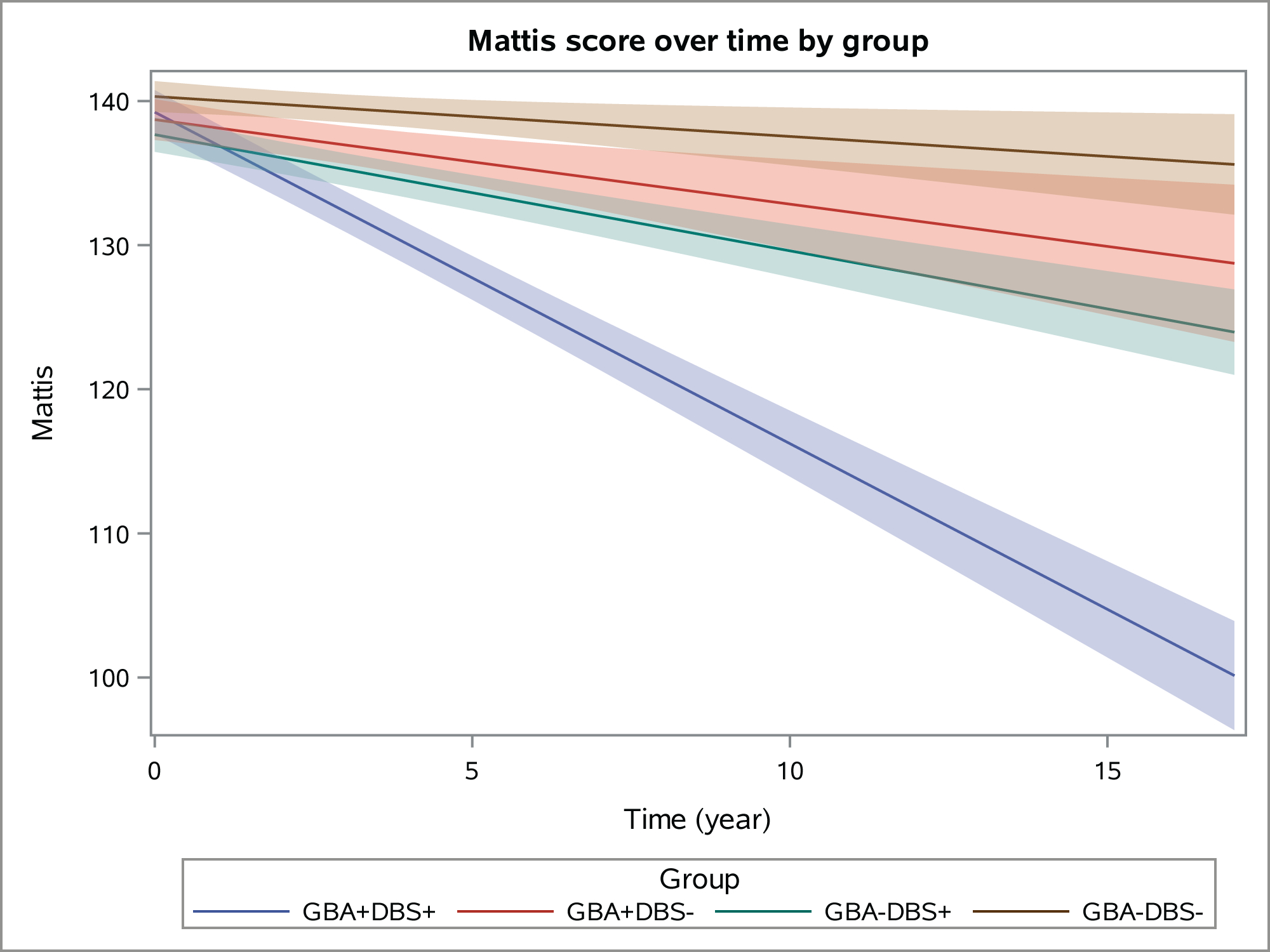
Linear fit (with 95% confidence interval bands) showing change in MDRS scores over time according to GBA and DBS status. For subjects with DBS, time zero equates to pre-DBS assessment (< 1 year prior to DBS). Median follow-up time ranges from 36.0–60.0 months.
Table 5.
Pairwise comparison of change in MDRS slope over time: estimate points/year (95% CI, p-value)
| ref | GBA+ DBS− | GBA−DBS+ | GBA+ DBS+ | |||
|---|---|---|---|---|---|---|
| unweighted | weighted | unweighted | weighted | unweighted | weighted | |
| GBA−DBS− | −0.31 (−0.72, 0.1), 0.15 |
−0.36 (−0.75, 0.03), 0.07 |
−0.53 (−0.82, −0.24), 0.0004 |
−0.46 (−0.75, −0.17), 0.002 |
−2.02 (−2.35, −1.69), <0.0001 |
−1.93 (−2.26, −1.60), <0.0001 |
| GBA+ DBS− | −0.22 (−0.61, 0.17), 0.28 |
−0.09 (−0.48, 0.30), 0.64 |
−1.71 (−2.14, −1.28), <0.0001 |
−1.56 (−1.99, −1.13), <0.0001 |
||
| GBA− DBS+ | −1.49 (−1.80, −1.18), <0.0001 |
−1.47 (−1.80, −1.14), <0.0001 |
||||
Effects of DBS on cognition in GBA subjects according to mutation severity
GBA+DBS+ subjects, subcategorized according to mutation severity, declined faster in the MDRS than their non-DBS counterparts with the same mutation severity (Figure 2, Table 6). Subjects with a GBA variant and DBS declined 1.15 points/year (SE = 0.29) faster than those with the equivalent GBA variant and no DBS (p < 0.0001). Subjects with mild GBA mutations and DBS declined 2.08 points/year (SE = 0.52) faster than their non-DBS counterparts (both p<0.0001). Subjects with severe GBA mutations and DBS declined 1.13 points/year (SE = 0.53) faster than their non-DBS counterparts (p = 0.03) with the mixed model analysis, but there was no difference between the groups using propensity score weighting technique (p = 0.11, Table 6).
Figure 2.

Linear fit (with 95% confidence interval bands) showing change in MDRS scores over time based on DBS status and according to GBA mutation severity. Panel A compares GBA variant mutation carriers with and without DBS. Panel B compares GBA mild mutation carriers with and without DBS. Panel C compares GBA severe mutation carriers with and without DBS. Panel D compares GBA subjects with neuronopathic (mild and severe) vs. non-neuronopathic mutations (variant), with and without DBS. Median follow-up time ranges from 36.0–60.0 months.
Table 6.
Pairwise comparison of changes in MDRS slope over time according to mutation severity: estimate points/year (95% CI), p-value
| ref | GBA variant, DBS+ | GBA mild, DBS+ | GBA severe, DBS+ | GBA−DBS+ | ||||
|---|---|---|---|---|---|---|---|---|
| unweighted | weighted | unweighted | weighted | unweighted | weighted | unweighted | weighted | |
| GBA variant, DBS− | −1.15 (−1.72, −0.58), <0.0001 |
−1.16 (−1.71, −0.61), <0.0001 |
−2.46 (−3.07, −1.85), <0.0001 |
−2.26 (−2.93, −1.59), <0.0001 |
−2.33 (−2.92, −1.74), <0.0001 |
−2.52 (−3.13, −1.91), <0.0001 |
−0.44 (−0.89, 0.01), 0.06 |
−0.36 (−0.79, 0.07), 0.11 |
| GBA mild, DBS− | −0.77 (−1.75, 0.21), 0.12 |
−0.86 (−1.76, 0.04), 0.06 |
−2.08 (−3.1, −1.06), <0.0001 |
−1.97 (−2.95, −0.99), <0.0001 |
−1.95 (−2.95, −0.95), 0.0001 |
−2.23 (−3.17, −1.29), <0.0001 |
−0.06 (−1.04, 0.92), 0.91 |
−0.07 (−0.91, 0.77), 0.88 |
| GBA severe, DBS− | 0.05 (−0.97, 1.07), 0.92 |
0.53 (−0.47, 1.53), 0.30 |
−1.26 (−2.32, −0.2), 0.02 |
−0.58 (−1.64, 0.48), 0.28 |
−1.13 (−2.17, −0.09),0.03 |
−0.84 (−1.88, 0.2), 0.11 |
0.77 (−0.19, 1.73), 0.12 |
1.32 (0.38, 2.26), 0.01 |
| GBA−DBS− | −1.25 (−1.7, −0.8), <0.0001 |
−1.25 (−1.68, −0.82), <0.0001 |
−2.55 (−3.06, −2.04), <0.0001 |
−2.36 (−2.93, −1.79), <0.0001 |
−2.43 (−2.9, −1.96), <0.0001 |
−2.62 (−3.13, −2.11), <0.0001 |
−0.53 (−0.82, −0.24), 0.0004 |
−0.46 (−0.75, −0.17), 0.002 |
Effects of mutation severity on cognition in DBS subjects
Amongst those with DBS, subjects with mild or severe GBA mutations (neuronopathic) declined 1.31 points/year (SE = 0.31, p<0.0001) and 1.18 points/year (SE = 0.29, p<0.0001) faster than subjects in the GBA variant group (non-neuronopathic) in their MDRS scores. Those with neuronopathic GBA mutations (mild or severe mutations combined) declined 1.26 points/year (SE = 0.25, p<0.0001) faster than GBA subjects with non-neuronopathic mutations. There was no difference in the rate of decline comparing subjects with mild GBA mutations vs. severe mutations though the sample size was limited.
Discussion:
This is the first study to demonstrate that the combined effects of GBA mutations and STN-DBS in PD negatively impact cognition. We also demonstrate that GBA mutation carriers, when stratified according to mutation severity, also show a graded cognitive outcome after STN-DBS compared with their non-DBS counterparts. Also, amongst subjects with DBS, those with neuronopathic mutations have faster cognitive decline than those with non-neuronopathic mutations. Other studies have shown that PDGBA subjects with STN-DBS have more severe decline in cognition, lower health related quality of life, and a greater burden of non-motor symptoms compared with non-mutation carriers with STN-DBS.4, 5, 32 However, because none of these studies included PDGBA subjects without DBS as a comparator group, it was unknown whether these suboptimal cognitive outcomes related to the natural disease trajectory of GBA carriers or related to the combination of both GBA and STN-DBS. We have filled this knowledge gap by our 2 × 2 design comparing four groups of subjects, PDGBA and non-GBA subjects, with and without bilateral STN-DBS. Based on our results, we would advise that DBS candidates for GBA mutations as part of the pre-surgical decision-making process. Further, we advise counseling of all patients, and particularly those with GBA mutations, regarding the potential cognitive risks of STN-DBS over time. There are slight differences in cognition even at baseline, which are statistically significant, but these differences become clearer over time as shown in Figures 1 and 2.
It is well established that cognitive impairment is more frequent and more severe in PDGBA patients compared to non-GBA patients.33 Clinically, PDGBA patients also develop dementia faster than non-GBA patients.7 This has been attributed to deficiency of the GCase enzyme which leads to more rapid accumulation and spread of α-synuclein, which in turn further lowers GCase, and this process continues in a bidirectional positive feedback loop.8, 34, 35 STN-DBS has been associated with a negative impact on timed tasks such as verbal fluency,10 executive control of action,11 and inhibitory control.12 Further studies are needed to determine whether these cognitive processes or other specific domains are affected in those with STN-DBS and GBA mutations. Additional studies are needed to determine the GBA related mechanisms that may interact with the potential deleterious effect of DBS on cognition over time.
Regarding benefits of DBS, in some cases, LEDD reduction in GBA+DBS+ has been shown to be less compared with other monogenic forms of PD.36 In our study, GBA+DBS+ subjects had a significant reduction in total LEDD compared with GBA−DBS+ subjects, suggesting a similar response to DBS in both groups. Indeed, the significant short-term37 and long-term38 motor benefits of STN-DBS on quality of life are well-established. However, the anticipated motor benefits of STN-DBS surgery need to be carefully weighed against the potential long term cognitive adverse effects of DBS specifically in PDGBA patients. We acknowledge that our findings can only be referenced to bilateral STN-DBS and not to other surgical procedures. DBS options, such as unilateral DBS,39 combined STN-DBS with contralateral GPi-DBS, or bilateral GPi-DBS, may provide a different risk/benefit profile than bilateral STN-DBS and deserve comprehensive evaluation.40 Additional device-aided therapies, such as apomorphine continuous subcutaneous infusion, levodopa duodenal gel or subcutaneous infusion, remain to be explored in this population as well.41
Although studies have shown that GPi-DBS may result in less cognitive decline compared with STN-DBS42 (without considering genotype), no data are available on cognitive outcomes of GPi-DBS in PDGBA patients. One of the key reasons for potential differences in outcomes based on target may simply be the size of the targets - GPi is nearly three times the size of STN (approximately 450 mm3 vs. 150 mm3 respectively).43–45 Given the compact anatomy of STN compared with GPi, the likelihood of unintended current spread is higher into adjacent STN subregions and nearby structures.13, 14 Imaging studies have revealed the likely region of accidental activation is that of nonmotor medial prefrontal-striatal circuitry in those with STN-DBS.15 Future studies can potentially examine the effects of GPi-DBS in this cognitively vulnerable population. Prior studies, such as the VA cooperative study by Weaver et al.46 which demonstrated faster cognitive decline with STN vs. GPi-DBS did not consider GBA status. Whether GBA status was driving the differences between targets remains unknown and since many of those subjects are now deceased (personal communication, Francis Weaver), this question is unlikely to be answered using retrospective data. In fact, we searched widely for a dataset with this focus (GBA+GPi) and did not locate sufficient cases with gene testing and the needed cognitive assessments or follow-up, so we are not optimistic that such data will exist without a specific prospective study. Interestingly, subjects with bilateral STN-DBS declined by approximately 2.0 points/year over 26 months in the Weaver et al. study46 and by 1.8 points at 6 months post-STN-DBS in another study,47 which are comparable to the degree of decline found in our study. To our knowledge, there is no minimal clinically important difference published for the MDRS, thus the clinical context of these annual changes remains unknown.
Of note, most of the GBA+DBS+ subjects in this study had risk variants or mild mutations, with the smallest group being those with severe mutations. Variants such as E326K are the most common abnormality in GBA, followed by mild (N370S) and severe (L444P) mutations,48 which is consistent with our results. It is possible that individuals with severe GBA mutations are “screened out” during neuropsychological testing as part of the DBS pre-operative evaluation, potentially due to higher risk of dementia earlier in the disease course, which may also contribute to the higher frequency of subjects with risk variants or mild mutations. Also, our results were largely consistent using the unweighted variable-adjusted mixed effects model and weighted analysis with propensity score weighting technique, except when examining those with severe GBA mutations with and without DBS (n = 11 and n = 9, respectively). This discrepancy could be secondary to the small sample size in these groups, a scenario where the utility of the propensity score weighting technique may be limited.49
Strengths of our study include a large sample size, international collaboration, long duration of follow-up, and use of mixed effects model analysis. Limitations include lack of randomization of DBS subjects, information regarding death and dropouts in each group, DBS lead location, details of actual surgery (awake vs. asleep, number of brain penetrations, etc.), programming parameters, lack of in-depth neuropsychological and motor assessments in the OFF medication/ON DBS state, heterogeneity of assessments, and variability of individual site contribution to GBA mutation carriers and non-carriers. Lastly, cognitive testing was performed without blinding for genotype or DBS status, and the analysis is categorically retrospective, and culled from multiple centers with no prescribed protocol at the time of data collection. PDGBA patients who are cognitively well enough to qualify for DBS may represent outliers in the PDGBA population given the higher risk of dementia in this group.7, 8, 20, 25, 33 As such, the decline in cognition seen after STN-DBS for these patients may represent a regression to the mean. However, the mild slope of decline in the MDRS scores per year argues against this (Figure 1, Table 5) and suggests rather that STN-DBS compounds the risk of cognitive decline associated with GBA mutations. Furthermore, we have on average greater than 2 time points per subject which reduces the likelihood of regression to the mean.50 Therefore, additional studies are needed to confirm our results and their impact on quality of life.
In terms of future directions, GBA mutations are a known risk factor for cognitive decline,8, 33 and STN-DBS appears to accelerate this decline. Our data permit us to advise that GBA status be part of pre-operative evaluations and that gene positive patients be counseled appropriately regarding the potential cognitive risks, benefits, and alternatives to STN-DBS prior to implantation. Determining the cost effectiveness of genetic testing as part of the pre-operative evaluation also should be considered through health-economic studies, though genetic testing is becoming increasingly accessible through free testing programs such as PD GENEration (NCT04057794) and currently no interventions exist to slow cognitive decline in PD. The influence of additional genes that are associated with cognitive decline, such as apolipoprotein E,51 also warrant examination in future studies. Integration of genetic data along with DBS status, target, DBS lead location, and details of surgery (awake vs. asleep, number of brain penetrations, etc.), should be considered for integration into large national and international datasets for further investigation. Ongoing efforts to genotype all PD patients through PD GENEration (NCT04057794) and 23andMe52 are critical to expediting studies that link genotype with outcomes of clinical interventions.
Supplementary Material
Summary for social media if published:
@GianPal4
Glucocerebrosidase (GBA) mutations and subthalamic nucleus deep brain stimulation (STN-DBS) may independently have a negative effect on cognition in Parkinson’s disease
The combined effects of GBA mutations and STN-DBS on cognition is unknown
The results of the present study suggest that the combined effects of GBA mutations and STN-DBS negatively impact cognition over time
It would be advised to screen deep brain stimulation candidates for GBA mutations. Furthermore, we advise counseling patients with GBA mutations regarding the potential risks and benefits of surgery and to consider alternative treatment options.
Acknowledgements:
We would like to thank our patients who inspire us daily and have contributed to this research. This research was supported by the Parkinson Disease Foundation and the National Institute of Neurological Disorders and Stroke K23-NS097625-05. Collection from Mount Sinai was supported by U01-NS094148-01 and U01-NS107016-01A1. The French collection was supported by the program “Investissements d’Avenir” ANR-10-IAIHU-06. Data from the NIH Parkinson’s Clinic was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke. Rob de Bie received research grants from Medtronic and Lysosomal Therapeutics, paid to the institution.
Data used in the preparation of this article were obtained from the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease (AMP PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. The AMP® PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson’s (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson’s Research; Pfizer Inc.; Sanofi US Services Inc.; and Verily Life Sciences.
ACCELERATING MEDICINES PARTNERSHIP and AMP are registered service marks of the U.S. Department of Health and Human Services.
Clinical data used in preparation of this article were obtained from the Michael J. Fox Foundation (MJFF) LRRK2 Cohort Consortium (LCC), the NINDS Parkinson’s disease Biomarkers Program (PDBP), and the NINDS Study of Isradipine as a Disease Modifying Agent in Subjects With Early Parkinson Disease, Phase 3 (STEADY-PD3).
The LRRK2 Cohort Consortium is coordinated and funded by The Michael J. Fox Foundation for Parkinson’s Research. Data used in preparation of this article were obtained from the MJFF-sponsored LRRK2 Cohort Consortium (LCC). The LCC Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit https://www.michaeljfox.org/biospecimens.
Parkinson’s Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript.
The Study of Isradipine as a Disease Modifying Agent in Subjects With Early Parkinson Disease, Phase 3 (STEADY-PD3) is funded by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health with support from the Michael J. Fox Foundation and the Parkinson Study Group. For additional study information, visit https://clinicaltrials.gov/ct2/show/study/NCT02168842. The STEADY-PD3 investigators have not participated in reviewing the data analysis or content of the manuscript.
Footnotes
Potential conflicts of interest: Nothing to report.
References:
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006. Jun;5(6):525–35. [DOI] [PubMed] [Google Scholar]
- 2.Ponce FA, Lozano AM. Deep brain stimulation state of the art and novel stimulation targets. Prog Brain Res. 2010;184:311–24. [DOI] [PubMed] [Google Scholar]
- 3.Angeli A, Mencacci NE, Duran R, et al. Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov Disord. 2013. Sep;28(10):1370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mangone G, Bekadar S, Cormier-Dequaire F, et al. Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat Disord. 2020. Jul;76:56–62. [DOI] [PubMed] [Google Scholar]
- 5.Artusi CA, Dwivedi AK, Romagnolo A, et al. Association of Subthalamic Deep Brain Stimulation With Motor, Functional, and Pharmacologic Outcomes in Patients With Monogenic Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Netw Open. 2019. Feb 1;2(2):e187800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015. Sep;138(Pt 9):2648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winder-Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain. 2013. Feb;136(Pt 2):392–9. [DOI] [PubMed] [Google Scholar]
- 8.Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology. 2012. May 1;78(18):1434–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pal GD, Hall D, Ouyang B, et al. Genetic and Clinical Predictors of Deep Brain Stimulation in Young-Onset Parkinson’s Disease. Mov Disord Clin Pract. 2016. Sep-Oct;3(5):465–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parsons TD, Rogers SA, Braaten AJ, Woods SP, Troster AI. Cognitive sequelae of subthalamic nucleus deep brain stimulation in Parkinson’s disease: a meta-analysis. Lancet Neurol. 2006. Jul;5(7):578–88. [DOI] [PubMed] [Google Scholar]
- 11.Jahanshahi M Effects of deep brain stimulation of the subthalamic nucleus on inhibitory and executive control over prepotent responses in Parkinson’s disease. Front Syst Neurosci. 2013. Dec 25;7:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jahanshahi M, Obeso I, Rothwell JC, Obeso JA. A fronto-striato-subthalamic-pallidal network for goal-directed and habitual inhibition. Nat Rev Neurosci. 2015. Dec;16(12):719–32. [DOI] [PubMed] [Google Scholar]
- 13.Coenen VA, Honey CR, Hurwitz T, et al. Medial forebrain bundle stimulation as a pathophysiological mechanism for hypomania in subthalamic nucleus deep brain stimulation for Parkinson’s disease. Neurosurgery. 2009. Jun;64(6):1106–14; discussion 14–5. [DOI] [PubMed] [Google Scholar]
- 14.Blomstedt P, Hariz MI, Lees A, et al. Acute severe depression induced by intraoperative stimulation of the substantia nigra: a case report. Parkinsonism Relat Disord. 2008;14(3):253–6. [DOI] [PubMed] [Google Scholar]
- 15.Stefurak T, Mikulis D, Mayberg H, et al. Deep brain stimulation for Parkinson’s disease dissociates mood and motor circuits: a functional MRI case study. Mov Disord. 2003. Dec;18(12):1508–16. [DOI] [PubMed] [Google Scholar]
- 16.David FJ, Munoz MJ, Shils JL, et al. Subthalamic Peak Beta Ratio Is Asymmetric in Glucocerebrosidase Mutation Carriers With Parkinson’s Disease: A Pilot Study. Frontiers in Neurology. 2021. 2021-September-30;12(1661). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwaki H, Leonard HL, Makarious MB, et al. Accelerating Medicines Partnership: Parkinson’s Disease. Genetic Resource. Mov Disord. 2021. May 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odekerken VJ, van Laar T, Staal MJ, et al. Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease (NSTAPS study): a randomised controlled trial. Lancet Neurol. 2013. Jan;12(1):37–44. [DOI] [PubMed] [Google Scholar]
- 19.Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007. Sep 18;69(12):1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009. Oct 22;361(17):1651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nalls MA, Keller MF, Hernandez DG, et al. Baseline genetic associations in the Parkinson’s Progression Markers Initiative (PPMI). Mov Disord. 2016. Jan;31(1):79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuang D, Leverenz JB, Lopez OL, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012. Nov 6;79(19):1944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols WC, Pankratz N, Marek DK, et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. 2009. Jan 27;72(4):310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moran EE, Bressman SB, Ortega RA, et al. Cognitive Functioning of Glucocerebrosidase (GBA) Non-manifesting Carriers. Front Neurol. 2021;12:635958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol. 2016. Nov;80(5):662–73. [DOI] [PubMed] [Google Scholar]
- 26.Yahalom G, Greenbaum L, Israeli-Korn S, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: Risk estimates and genotype-phenotype correlations. Parkinsonism Relat Disord. 2019. May;62:179–84. [DOI] [PubMed] [Google Scholar]
- 27.Ortega RA, Wang C, Raymond D, et al. Association of Dual LRRK2 G2019S and GBA Variations With Parkinson Disease Progression. JAMA Netw Open. 2021. Apr 1;4(4):e215845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hentz JG, Mehta SH, Shill HA, Driver-Dunckley E, Beach TG, Adler CH. Simplified conversion method for unified Parkinson’s disease rating scale motor examinations. Mov Disord. 2015. Dec;30(14):1967–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Steenoven I, Aarsland D, Hurtig H, et al. Conversion between mini-mental state examination, montreal cognitive assessment, and dementia rating scale-2 scores in Parkinson’s disease. Mov Disord. 2014. Dec;29(14):1809–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Defer GL, Widner H, Marie RM, Remy P, Levivier M. Core assessment program for surgical interventional therapies in Parkinson’s disease (CAPSIT-PD). Mov Disord. 1999. Jul;14(4):572–84. [DOI] [PubMed] [Google Scholar]
- 31.Smith C Levodopa dose equivalency: A systematic review [Powerpoint Slides] 2010. [cited 2021 04/11/2021]; Available from: https://silo.tips/download/levodopa-dose-equivalency-a-systematic-review.
- 32.Lythe V, Athauda D, Foley J, et al. GBA-Associated Parkinson’s Disease: Progression in a Deep Brain Stimulation Cohort. J Parkinsons Dis. 2017;7(4):635–44. [DOI] [PubMed] [Google Scholar]
- 33.Brockmann K, Srulijes K, Pflederer S, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. 2015. Mar;30(3):407–11. [DOI] [PubMed] [Google Scholar]
- 34.Manning-Bog AB, Schule B, Langston JW. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology. 2009. Nov;30(6):1127–32. [DOI] [PubMed] [Google Scholar]
- 35.Zunke F, Moise AC, Belur NR, et al. Reversible Conformational Conversion of alpha-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron. 2018. Jan 3;97(1):92–107 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Oliveira LM, Barbosa ER, Aquino CC, Munhoz RP, Fasano A, Cury RG. Deep Brain Stimulation in Patients With Mutations in Parkinson’s Disease-Related Genes: A Systematic Review. Mov Disord Clin Pract. 2019. Jun;6(5):359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deuschl G, Schade-Brittinger C, Krack P, et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2006. Aug 31;355(9):896–908. [DOI] [PubMed] [Google Scholar]
- 38.Bove F, Mulas D, Cavallieri F, et al. Long-term Outcomes (15 Years) After Subthalamic Nucleus Deep Brain Stimulation in Patients With Parkinson Disease. Neurology. 2021. Jun 2. [DOI] [PubMed] [Google Scholar]
- 39.Okun MS, Fernandez HH, Wu SS, et al. Cognition and mood in Parkinson’s disease in subthalamic nucleus versus globus pallidus interna deep brain stimulation: the COMPARE trial. Ann Neurol. 2009. May;65(5):586–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Wang L, Hu W, et al. Combined Unilateral Subthalamic Nucleus and Contralateral Globus Pallidus Interna Deep Brain Stimulation for Treatment of Parkinson Disease: A Pilot Study of Symptom-Tailored Stimulation. Neurosurgery. 2020. May 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salles PA, Mata IF, Fernandez HH. Should we start integrating genetic data in decision-making on device-aided therapies in Parkinson disease? A Point of View. Parkinsonism & Related Disorders. 2021. 2021/05/18/. [DOI] [PubMed] [Google Scholar]
- 42.Follett KA, Weaver FM, Stern M, et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2010. Jun 3;362(22):2077–91. [DOI] [PubMed] [Google Scholar]
- 43.Richter EO, Hoque T, Halliday W, Lozano AM, Saint-Cyr JA. Determining the position and size of the subthalamic nucleus based on magnetic resonance imaging results in patients with advanced Parkinson disease. J Neurosurg. 2004. Mar;100(3):541–6. [DOI] [PubMed] [Google Scholar]
- 44.Williams NR, Foote KD, Okun MS. STN vs. GPi Deep Brain Stimulation: Translating the Rematch into Clinical Practice. Mov Disord Clin Pract. 2014. Apr 1;1(1):24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirza S, Yazdani U, Dewey Iii R, et al. Comparison of Globus Pallidus Interna and Subthalamic Nucleus in Deep Brain Stimulation for Parkinson Disease: An Institutional Experience and Review. Parkinsons Dis. 2017;2017:3410820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weaver FM, Follett KA, Stern M, et al. Randomized trial of deep brain stimulation for Parkinson disease: thirty-six-month outcomes. Neurology. 2012. Jul 3;79(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Witt K, Daniels C, Reiff J, et al. Neuropsychological and psychiatric changes after deep brain stimulation for Parkinson’s disease: a randomised, multicentre study. Lancet Neurol. 2008. Jul;7(7):605–14. [DOI] [PubMed] [Google Scholar]
- 48.Malek N, Weil RS, Bresner C, et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J Neurol Neurosurg Psychiatry. 2018. Jul;89(7):702–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benedetto U, Head SJ, Angelini GD, Blackstone EH. Statistical primer: propensity score matching and its alternatives. Eur J Cardiothorac Surg. 2018. Jun 1;53(6):1112–7. [DOI] [PubMed] [Google Scholar]
- 50.Barnett AG, van der Pols JC, Dobson AJ. Regression to the mean: what it is and how to deal with it. Int J Epidemiol. 2005. Feb;34(1):215–20. [DOI] [PubMed] [Google Scholar]
- 51.Liu G, Locascio JJ, Corvol JC, et al. Prediction of cognition in Parkinson’s disease with a clinical-genetic score: a longitudinal analysis of nine cohorts. Lancet Neurol. 2017. Aug;16(8):620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019. Dec;18(12):1091–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.