

SUMMARY
Tau (MAPT) drives neuronal dysfunction in Alzheimer disease (AD) and other tauopathies. To dissect the underlying mechanisms, we combined an engineered ascorbic acid peroxidase (APEX) approach with quantitative affinity purification mass spectrometry (AP-MS) followed by proximity ligation assay (PLA) to characterize Tau interactomes modified by neuronal activity and mutations that cause frontotemporal dementia (FTD) in human induced pluripotent stem cell (iPSC)-derived neurons. We established interactions of Tau with presynaptic vesicle proteins during activity-dependent Tau secretion and mapped the Tau-binding sites to the cytosolic domains of integral synaptic vesicle proteins. We showed that FTD mutations impair bioenergetics and markedly diminished Tau’s interaction with mitochondria proteins, which were downregulated in AD brains of multiple cohorts and correlated with disease severity. These multimodal and dynamic Tau interactomes with exquisite spatial resolution shed light on Tau’s role in neuronal function and disease and highlight potential therapeutic targets to block Tau-mediated pathogenesis.
Graphical Abstract
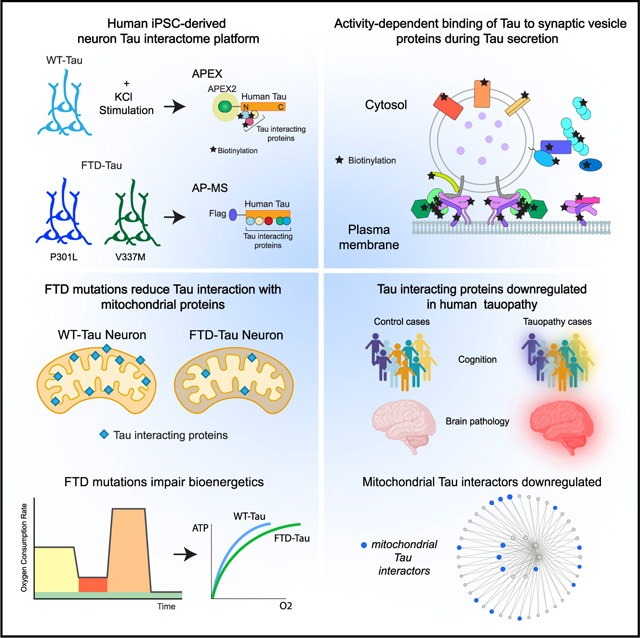
In brief
By combining APEX and AP-MS proteomic approaches, Tau interactome mapping reveals that Tau interactors are modified by neuronal activity and FTD mutations in human iPSC-derived neurons.
INTRODUCTION
Mutations in the microtubule-associated protein Tau (MAPT) gene are sufficient to cause familial frontotemporal dementia (FTD), and Tau is the major factor that drives neurodegeneration in other tauopathies including Alzheimer disease (AD), corticobasal syndrome, and progressive supranuclear palsy (PSP) syndrome. In healthy neurons, Tau interacts with and stabilizes microtubules (Santarella et al., 2004). More recent studies revealed that Tau participates in diverse processes important for neuronal pathophysiology including cytoskeletal dynamics (Bardai et al., 2018; Chang et al., 2021; DuBoff et al., 2012), axonal transport (Ishihara et al., 1999; Ittner et al., 2008), synaptic transmission (Hoover et al., 2010; Ittner et al., 2010; McInnes et al., 2018; Tracy et al., 2016; Yoshiyama et al., 2007), proteostasis (Myeku et al., 2016), nuclear transport (Eftekharzadeh et al., 2018), and axon initial segment function (Li et al., 2011; Sohn et al., 2016, 2019).
Direct interactions of Tau with specific proteins or protein complexes that can alter these cellular processes have been identified (Eftekharzadeh et al., 2018; Ittner et al., 2009, 2010; McInnes et al., 2018; Sohn et al., 2019; Vanderweyde et al., 2016), indicating that Tau impacts neuron function through microtubule-independent protein-protein interactions (Morris et al., 2011). Affinity purification mass spectrometry (AP-MS)-based characterizations of the Tau interactome have focused on mouse neurons in wild-type mice (Liu et al., 2016; Wang et al., 2017b) or tauopathy mice that express high levels of mutant Tau (Choi et al., 2020; Maziuk et al., 2018). Other studies examined Tau interactomes in SH-SY5Y neuroblastoma cells (Gunawardana et al., 2015) or a mixed population of neuroprogenitor-derived human ReN cells (Wang et al., 2019). However, no studies have directly compared the interactomes of wild-type Tau with those of FTD mutant Tau or captured how the Tau interactome is affected by neuronal activity in human neurons.
Neuronal activity triggers Tau secretion (Pooler et al., 2013; Wu et al., 2016; Yamada et al., 2014) and the translocation of Tau into synapses (Frandemiche et al., 2014). Secreted Tau can spread trans-neuronally among synaptically connected neurons (Wang et al., 2017c; Wu et al., 2016), and Tau spreads across functionally connected regions of the brain (Vogel et al., 2020). Depolarization of human AD brain synaptosomes triggers Tau release from presynaptic terminals (Sokolow et al., 2015), suggesting that Tau secretion is regulated by the presynaptic function and that yet-unknown Tau protein-protein interactions may be involved.
To characterize the Tau interactome in human neurons, we used both engineered ascorbic acid peroxidase (APEX) proximity-dependent mapping with mass spectrometry and AP-MS to reveal the dynamic and multifaceted interactome of Tau in human induced pluripotent stem cell (iPSC)-derived glutamatergic neurons. The APEX technique generates precise spatial and temporal maps of subcellular compartment proteomes (Loh et al., 2016; Rhee et al., 2013) and protein localization during intracellular trafficking (Lobingier et al., 2017). Here, APEX mapping enabled us to elucidate the spatial regulation of the Tau interactome during enhanced neuronal activity. AP-MS allowed us to quantitatively compare protein-protein interactions of wild-type Tau and FTD mutant Tau in human neurons. Together, these analyses of the Tau interactome provide a comprehensive mapping of protein-protein interactions, which could be targeted therapeutically to slow Tau-mediated disease progression.
RESULTS
Labeling of the Tau interactome in human iPSC-derived neurons
We developed a platform called i3Neurons via genetic editing of a human iPSC line (WTC11) to carry inducible expression of NGN2, allowing a simplified, scalable, and homogeneous differentiation into glutamatergic neurons (Wang et al., 2017a; Zhang et al., 2013). The i3Neurons exhibit dendrites and axons with functional axonal initial segments and synapses (Sohn et al., 2019; Wang et al., 2017a). To investigate Tau interactions in living neurons, we modified i3Neurons by integrating a transgene for doxycycline-inducible expression of human Tau tagged with a flag epitope and APEX2, a highly active version of the engineered ascorbate peroxidase (Lam et al., 2015) (Figure 1A). Proteolytic cleavage of Tau has been reported in cultured human neuron models, and the accumulation of Tau fragments is associated with neurodegenerative phenotypes (Ehrlich et al., 2015; Fong et al., 2013). To capture proteins interacting with N or C terminus truncated Tau, we generated two independent isogenic clonal iPSC lines expressing wild-type 2N4R Tau (Tau) tagged on either the N terminus or the C terminus with APEX2 and flag (Figure 1B). As a control for Tau-specific interactions, we made an isogenic iPSC clone expressing APEX2-tagged Tubulin α 1B (APEX-α Tubulin). All iPSC clones exhibit normal karyotypes (Figures S1A–S1C).
Figure 1. APEX-Tau-mediated proximity-dependent biotinylation in human iPSC-derived neurons.

(A) Transcription Activator-Like Effector Nucleases (TALEN)-mediated integration of NGN2 into one allele of the AAVS1 locus in human iPSCs, followed by integration of APEX-Tau into the second AAVS1 allele. Arrowheads represent transcription from the transgene.
(B) APEX-flag-tagged human Tau (2N4R) and Tubulin α 1B constructs. See also Figure S1.
(C) Workflow of i3Neuron differentiation and experiments.
(D) Western blot analysis of N- and C-terminal APEX-tagged Tau and APEX-α Tubulin expressed in i3Neurons.
(E) Images of APEX-Tau expression (green) and biotinylated proteins (red) in i3Neurons. Scale bars, 100 μm.
After neuronal differentiation and maturation, neurons were treated with doxycycline to activate the expression of APEX2-tagged proteins for 24 h (Figure 1C). Immunoblotting revealed comparable levels of full-length N-APEX Tau or C-APEX Tau, with some distinct N and C terminus fragments labeled by the flag epitope (Figure 1D). APEX-tagged Tau expression was lower than the expression of endogenous Tau in human neurons (Figure S1D). Biotin-phenol (BP) treatment and brief H2O2 exposure induced biotinylation of proteins associated with APEX-α Tubulin, N-APEX Tau, and C-APEX Tau at similar levels (Figure 1D). Biotinylated proteins were strongly colocalized with APEX2-tagged Tau in the soma and in neuronal processes (Figure 1E).
Spatially defined proteomic mapping of the Tau interactome
Neurons expressing APEX-Tau were treated with BP, followed by 1 min of H2O2 to catalyze production of biotin-phenoxyl radicals, which react with tyrosine residues in close proximity because tyrosine is an electron-rich amino acid. An antibiotin antibody was then used to enrich for biotin-modified peptides (Udeshi et al., 2017), which were detected by mass spectrometry enabling direct detection of biotinylated tyrosines in close proximity to APEX-Tau (Figure 2A). Proteins biotinylated by N-APEX Tau, C-APEX Tau, or APEX-α Tubulin were defined as those enriched in the majority of neuronal cultures expressing those constructs (Table S1).
Figure 2. Subcellular and subprotein domain Tau interactome in living human neurons identified by APEX-mediated biotinylation.

(A) Workflow for proximity-dependent identification of Tau-associated proteins.
(B) Venn diagram of biotinylated proteins detected in human neurons with N-APEX Tau (n = 7 cultures), C-APEX Tau (n = 9 cultures), or APEX-α Tubulin (n = 12 cultures). See also Table S1.
(C) N-APEX Tau, C-APEX Tau, and APEX-α Tubulin biotinylated proteins include components of the microtubule cytoskeleton. Only interconnected nodes based on the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database are shown. Function pathway analyses categories are grouped by color.
(D) ClueGO molecular function pathway enrichment of proteins biotinylated by both N-APEX Tau and C-APEX Tau but not APEX-α Tubulin. Node colors denote functionally grouped networks (kappa connectivity score ≥ 30%).
(E) Gene Set Enrichment Analysis (GSEA) on proteins biotinylated by both N-APEX and C-APEX Tau showing major cellular component categories.
(F and G) ClueGO biological processes pathway enrichment of proteins biotinylated only by N-APEX Tau (F) and only by C-APEX Tau (G).
(H and I) Subprotein-level APEX Tau associations identified that regulate autophagy (H) and proteasome degradation (I).
The majority of proteins enriched for association with α Tubulin were also detected in the Tau interactome, consistent with the close interaction of Tau and Tubulin on microtubules (Figure 2B). Indeed, pathway analyses revealed that most of the proteins associated with both α Tubulin and Tau were components of microtubules and cytoskeletal-binding proteins (Figure 2C), demonstrating the specific enrichment for proteins in close proximity using this method. To focus on Tau-specific interactions, we removed the 19 proteins coassociated with both Tau and α Tubulin from further analyses. Of the 246 putative Tau-interacting proteins, 136 were enriched in both N-APEX and C-APEX Tau neurons (Figure 2B). Gene Ontology (GO) enrichment analyses revealed that the molecular functions of these overlapping proteins largely comprised microtubule and actin cytoskeletal networks (Figure 2D), as well as established Tau-binding proteins including α-synuclein (SNCA) (Benussi et al., 2005), MAP1A (Alonso et al., 1997), HSP70 (Taylor et al., 2018), HSP90 (Weickert et al., 2020), and protein phosphatase 5 (PPC5C), which forms a complex with HSP90 (Silverstein et al., 1997) and Tau (Liu et al., 2005) (Table S1). The interactome snapshot also revealed an enrichment for components of SNARE complexes including SNAP29, Syntaxin 7 (STX7), SNAP25, ɣ-SNAP, and Munc18 (STXBP1), evidence of Tau’s localization at sites of membrane and vesicle fusion. Classifications of biotinylated proteins by cellular components included synapses, microtubule and actin cytoskeletons, secretory vesicles, and ribonucleoprotein granules (Figure 2E).
Strikingly, 45% of all interactions were uniquely detected in either N-APEX or C-APEX Tau neurons (Figure 2B). Enrichment analyses of the distinct N- and C-terminal Tau-associated proteins revealed networks involved in synaptic vesicle regulation (Figures 2F and 2G). While N-APEX Tau biotinylated proteins are mostly involved in active zone docking—including Dynamin 1 (DNM1), α-/β-SNAP (NAPA/B), RAB3GAP1, and Liprin α3 (PPFIA3)—the C-APEX Tau interacting proteins, such as Syntaxin 1A/1B (STX1A/1B), RAB3A, RIMS1, and Mint1 (APBA1), are important for the fusion of vesicles. This indicates that Tau is localized at sites of presynaptic vesicle fusion and could have domain-specific interactions with synaptic vesicle-associated proteins. Interestingly, the N-APEX Tau interactome also included proteins that regulate dendritic development and postsynaptic organization, including ABI2, cadherin-2 (CDH2), nectin (PVRL1), PAK3, 14-3-3ζ (YWHAZ), and GRIP1, indicating that the N terminus of Tau could impact both presynaptic and postsynaptic structures through multiple pathways (Figure 2F).
Tau interactomes also included proteins that modulate protein degradation. The N-APEX Tau interactome included YKT6, a SNARE protein on autophagosomes critical for lysosomal fusion (Matsui et al., 2018), that forms a complex with SNAP29 and lysosomal syntaxin 7, which were biotinylated by both N- and C-APEX Tau (Figure 2H), suggesting that Tau is localized to sites of autophagosome-lysosome fusion. In support of Tau degradation by the 26S proteasome (David et al., 2002), the regulatory component of the 19S cap complex of the proteosome, ADRM1, and three 19S subunits of the 26S proteasome—PSMC2, PSMC5, and PSMD10—were only enriched in the N-terminal Tau interactome (Figure 2I). This suggests that the N terminus is key for the interaction of Tau with the 26S proteasome in human neurons. USP14 and RAD23, involved in the ubiquitin-dependent proteolysis of Tau by proteasome, were identified in both N- and C-APEX Tau interactomes, suggesting that the interaction of Tau with ancillary factors involved in proteasome function is not domain-specific.
Biotinylation sites reveal Tau-interacting proteins at subcellular and amino acid levels
The antibiotin purification strategy enabled direct detection of >600 distinct biotinylated tyrosine residues on Tau- and tubulin-associated proteins (Figures 3A and 3B; Table S2). The fraction of common biotinylation sites between N-APEX and C-APEX Tau was 45% (280 out of 627 total sites, Figure 3B), and the fraction of common interacting proteins was 55% (136 out of 246 total proteins, Figure 2B). APEX-α Tubulin, N-APEX Tau, and C-APEX Tau biotinylated largely the same sites on α Tubulin, β Tubulin, MAP1B, and MAP2, supporting the close structural proximity of these microtubule cytoskeletal proteins (Figure 3C). Four tyrosine residues in Tau (2N4R) were biotinylated by both N- and C-APEX Tau; one in the N terminus was only biotinylated by N-APEX Tau (Figure 3C).
Figure 3. Mapping of biotinylation sites on Tau-associated proteins.

(A) Workflow used to enrich and identify biotinylated peptides following antibiotin pulldown of proteins biotinylated by APEX Tau.
(B) Venn analysis of the biotinylated tyrosines on peptides detected in human neurons expressing N-APEX Tau (n = 7 cultures), C-APEX Tau (n = 9 cultures), or APEX-α tubulin (n = 12 cultures). See also Table S2.
(C) Biotinylated and unlabeled tyrosines (Y) on proteins biotinylated by APEX-α Tubulin, N-, and C-APEX Tau. See also Figure S2.
(D) Biotinylation sites (stars) on SNARE complex and synaptic vesicle-associated proteins labeled by APEX Tau. Biotinylation was detected on the t-SNAREs, syntaxin, and SNAP25; however, the v-SNARE, synaptobrevin, was not detected.
(E) Tyrosine residues biotinylated by N- and C-APEX Tau mapped onto SNARE complex proteins adjacent to structural and functional domains that mediate vesicle fusion.
(F) Images of the PLA reaction (red) in human neurons with the Tau5 antibody alone (left) and with both the Tau5 and Munc18 antibodies (right). Scale bars, 20 μm.
(G) Quantification of PLA puncta fluorescence intensity in human neurons with and without the Munc18 antibody (n = 6 images/group, ***p < 0.001, unpaired Student’s t test). Values are given as means ± SEM. See also Figure S3.
(H) Biotinylated residues detected in relation to the topology of integral synaptic vesicle membrane proteins. Biotinylation sites were detected on the cytosolic, not the lumenal domains. Asterisks denote biotinylated proteins that were detected in less than half of the samples (VGLUT2, n = 3, and synaptogyrin-1, n = 2 out of 7 samples).
(I) Human neurons labeled with fixable FM 1–43 dye (green) and synapsin (red) following treatment with 50-mM KCl to enhance neuronal activity. FM 1–43 dye-labeled sites of vesicle fusion at presynaptic terminals (arrows). Scale bars, 5 μm.
(J) HT7 immunolabeling of Tau (red) colocalized with FM 1–43 uptake at sites of vesicle fusion in human neurons. Scale bars, 5 μm.
Nuclear localization of Tau was supported by the detection of four biotinylated residues on emerin (EMD) (Figure S2A), an integral component of the inner nuclear membrane (Berk et al., 2013), as well as RUVBL1 and RUVBL2 (Table S2), nuclear proteins critical for chromatin remodeling and transcription (Jha et al., 2008; Jónsson et al., 2004). Biotinylation sites were also detected on RNA- and DNA-binding proteins (Figure S2B). Consistent with perinuclear cytosolic association of Tau and Importin-β (KPNB1) (Lee et al., 2005; Zachariae and Grubmuller, 2008), both N- and C-APEX Tau biotinylated importin-β within the RanGTP binding site (Figure S2A), and N-APEX Tau biotinylated RANGAP1, which promotes the release of RanGTP from importin-β when it returns to the cytosol from the nucleus.
We detected domain-specific biotinylation sites on SNARE complex and synaptic vesicle proteins (Figure 3D; Table S2) and the known interaction between Tau and α-synuclein (Giasson et al., 2003). Biotinylation sites were detected on two of the three components in the assembled SNARE complex required for synaptic vesicle fusion, syntaxin-1A/1B and SNAP25 (Figure 3E). Six biotinylation sites were detected on Munc18 (Figure 3E), a protein essential for vesicle fusion that interacts directly with the SNARE complex via syntaxin-1 (Dulubova et al., 2007). The recruitment of Munc18 to sites of vesicle release is facilitated by its interaction with Mint1 (Biederer and Sudhof, 2000), which was biotinylated by C-APEX Tau. Munc18 colocalized with synapsin, a presynaptic terminal marker that overlaps with postsynaptic PSD-95 at synapses in human neurons (Figures S3A and S3B). A proximity ligation assay (PLA) revealed a stronger PLA signal in neurons stained with Tau and Munc18 antibodies than in a Tau antibody-alone control (Figures 3F and 3G), supporting the close proximity (<40 nm) of endogenous Tau and Munc18. The only biotinylation site on SV2A, a synaptic vesicle integral membrane protein, was on a cytosolic domain of the protein (Figure 3H). Two additional vesicular proteins, synaptogyrin-1 (SYNGR1) and VGLUT2 (SLC17A6), were biotinylated on their cytosolic domains by N-APEX Tau (Figure 3H). These results suggest that Tau is associated with the cytosolic surface of synaptic vesicles, as opposed to the vesicle lumen, and in proximity to assembled SNARE complexes at vesicle fusion sites in human neurons. Tau colocalized with the activity-driven uptake of a fixable FM 1–43 dye at the presynaptic terminals of human neurons, indicating that Tau is localized at sites of presynaptic vesicle fusion (Figures 3I and 3J).
Mapping the activity-dependent change in the Tau interactome
We next used APEX-Tau to obtain a snapshot of the dynamic changes in the interactome that occur when Tau is released from neurons during enhanced neuronal activity. Consistent with previous reports in mammalian neurons (Pooler et al., 2013; Schoch et al., 2016; Yamada et al., 2014), increasing the excitability of human neurons with a 30-min treatment of 50-mM KCl caused a significant rise in extracellular Tau levels, which was dependent on intracellular calcium (Figure 4A), but did not promote cell death (Figure 4B). Biotinylated proteins enriched in neurons treated with KCl (Table S3) were compared with biotinylated proteins enriched in unstimulated neurons (Figure 2B; Table S1). Most of the biotinylated proteins identified, 76% in N-APEX Tau neurons and 68% in C-APEX Tau neurons, were detected in both unstimulated and KCl-treated conditions (Figure 4C; Table S3), suggesting that the majority of the Tau interactome, including many cytoskeletal-associated proteins, did not change in response to activity. Of the biotinylated proteins affected by activity (Figure 4C; Table S3), most were detected only in neurons treated with KCl (17% in N-APEX and 28% in C-APEX Tau neurons) with fewer biotinylated proteins. Only 6% in N-APEX and 4% in C-APEX Tau neurons were detected exclusively in unstimulated neurons (Figures S4A and S4B; Table S3).
Figure 4. Activity-induced changes in the Tau interactome during activity-dependent Tau secretion from human neurons.

(A) Quantification of Tau secreted from human neurons treated with high KCl to enhance neuronal activity and BAPTA-AM, a membrane-permeable calcium chelator. Secreted Tau measured by ELISA was normalized to total Tau in the neuron culture (n = 8–9 cultures/group, *p < 0.05, one-way ANOVA, Bonferroni post hoc analyses). Values are given as means ± SEM.
(B) Lactate dehydrogenase (LDH) levels in the media collected from human neurons after 30 min with or without high KCl (n = 8–9 cultures/group). Values are given as means ± SEM.
(C) Venn analyses of the biotinylated proteins and the individual biotinylation sites labeled by N-APEX Tau and C-APEX Tau without stimulation (blue, data from Figure 2B, n = 7–9 cultures/group) and with enhanced neuronal activity (red, n = 7–9 cultures/group). See also Table S3.
(D and E) ClueGO biological processes pathway enrichment of proteins biotinylated by (D) N-APEX Tau or by (E) C-APEX Tau in neurons with KCl-induced activity. Analyses were performed on the 44 (D) and 70 (E) biotinylated proteins labeled in (C). See also Figure S4.
(F) Activity-induced biotinylated residues detected on synaptic vesicle-associated proteins.
(G) Images of PLA fluorescence with Tau5 and SYT1 antibodies in unstimulated and high-KCl-stimulated human neurons. Scale bars, 5 μm. See also Figure S3.
(H) Tau5 and SYT1 PLA puncta fluorescence intensity in human neurons with and without KCL-enhanced activity (n = 6–12 images/group, ***p < 0.001, one-way ANOVA, Bonferroni post hoc analyses). Values are given as means ± SEM.
Network analyses on the biotinylated proteins that were enriched only in the neurons treated with KCl revealed activity-dependent networks that regulate synaptic vesicle exocytosis, which were common to both N-APEX and C-APEX Tau-expressing neurons (Figures 4D and 4E). Activity-dependent biotinylated residues were detected on SNARE complex-associated proteins, synaptotagmin-1 (SYT1) and Mint1, and synaptic vesicle proteins SV2C and synapsin 1 (SYN1) (Figure 4F; Table S3). Further supporting the interaction of Tau with the cytosolic side of synaptic vesicle membranes, the biotinylated residues in SYT1 and SV2C were in the cytosolic, rather than lumenal, domains.
Two activity-dependent N-APEX Tau biotinylation sites were detected within the calcium-binding domains of SYT1, which colocalized with synapsin at presynaptic terminals of human neurons (Figure S3C). The intensity of the PLA signal with Tau and SYT1 antibodies significantly increased in neurons following treatment with KCl (Figures 4G and 4H). These data support a model in which Tau associates with proteins in SNARE complexes and synaptic vesicles at the presynaptic active zone and also show that activity-dependent Tau secretion from neurons could involve enhanced interaction with SYT1 during the fusion of synaptic vesicles at the presynaptic membrane.
Familial FTD mutations modify the Tau interactome
To identify Tau protein interactions altered by FTD-causing Tau mutations, we generated i3Neurons with inducible expression of either TauP301L or TauV337M with an N-terminal flag tag (Figure S5A) and normal karyotypes (Figures S5B and S5C). While the APEX approach was highly specific for the detection of Tau-interacting proteins, it can have limited sensitivity due to the short duration (1 min) of biotinylation. Instead, affinity purification mass spectrometry (AP-MS) was performed to detect steady-state Tau interactions that differ between wild-type Tau (TauWT) and Tau with familial FTD mutations (Figure 5A). Analyses of the TauWT interacting proteins identified by both APEX and AP-MS approaches revealed shared major classifications in cellular components including synapses, ribonucleotide complexes, secretory vesicles, mitochondria, and cytoskeleton (Figures S5D and S5E; Table S4). Importantly, the same levels of flag-tagged TauWT, TauP301L, and TauV337M proteins were immunoprecipitated and detected (Figure 5B).
Figure 5. Comparison of interactomes of wild-type Tau and FTD Tau mutants by AP-MS with PLA validation.

(A) Workflow for the affinity purification identification of Tau-associated proteins by mass spectrometry.
(B) Expression levels of flag-tagged TauWT, TauP301L, and TauV337M detected by mass spectrometry in the AP-MS experiments. See also Figure S5.
(C) Venn diagram indicating the number of proteins identified as differential interactors between TauWT (n = 7 cultures) and mutant Tau (TauV337M and TauP301L, n = 8 cultures/group). Proteins that preferentially interact with TauWT or FTD Tau are labeled in red and green, respectively. See also Tables S4 and S5.
(D and E) ClueGO cellular compartment pathway enrichment of (D) the 184 proteins that preferentially interact with TauWT with respect to TauV337M and of (E) the 108 proteins that preferentially interact with TauWT with respect to TauP301L and are labeled in (C). See also Figure S6.
(F) Illustration of TauWT-preferential interactors localized to the mitochondrial membrane. The TauWT-preferential interactors compared with TauV337M (green), TauP301L (purple), and both FTD Tau mutants (blue) are labeled. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane.
(G) Images of the PLA reactions between ATP5IF1, TOMM40, or TIMM13 and TauWT, TauV337M-Het, or TauV337M-Homo (Dako or HT7 antibodies) in 5-week-old human neurons. PLA reaction controls only contain either the antimitochondria or anti-Tau antibodies. PLA signal, red; DAPI, blue. Scale bars, 5 μm.
(H–J) Quantification of the area of the PLA reaction per neuron between (H) ATP5IF1 and Tau, (I) TOMM40 and Tau, and (J) TIMM13 and Tau (n > 150 neurons/group/experiment from 3 independent experiments with 2 technical replicates, ***p < 0.001, linear mixed-effects model with Tukey’s post hoc analyses). The center lines in boxplots (H–J) represent the median with the 25th and 75th percentiles marked by the box limits. The bars extend to the farthest data points and the dots outside of the bars are outliers.
We focused on interactions that were lost or gained with the TauV337M or TauP301L FTD mutations (Figures 5C and S6; Table S5). Compared with TauWT, TauV337M had increased interaction with 69 proteins (Figure S6A), enriched in spliceosomal complex subunits and synaptic vesicle membrane proteins (Figure S6C). Only 14 proteins showed enhanced interaction with TauP301L compared with TauWT (Figure S6B), including proteins involved in nucleoside metabolic processes (Figure S6D). The V337M and P301L mutations led to loss of interaction with 184 and 108 proteins, respectively, compared with TauWT (Figures S6A and S6B). Lost interacting proteins included ribosomal and mitochondrial proteins (Figures 5D and 5E).
The mitochondrial proteins that preferentially interact with TauWT are mostly located within the inner mitochondrial membrane (IMM), such as subunits of complex I, III, IV, and ATPase of the electron transport chain (ETC) (Figure 5F). Both TauV337M and TauP301L reduced the interaction with cytochrome C (CYCS). TauP301L exhibited weaker interactions with SLC25A4, SLC25A5, and SLC25A6, distinct adenine nucleotide translocators (ANT) that transport the ATP generated within the mitochondria to the cytoplasm (Figure 5F). Compared with TauV337M, TauWT interacts more strongly with HADHA and HADHB, two components of the mitochondrial trifunctional protein that catalyzes the beta oxidation of fatty acids into acetyl-CoA (Figure 5F). TauWT also showed increased interaction with several amino acid transporters compared with TauV337M, including SLC25A13 and MPC2, which control the mitochondrial levels of glutamate and pyruvate, respectively (Figure 5F), further supporting a role of Tau in mitochondrial metabolism.
We next validated the interaction of TauWT with mitochondria proteins and the effects of the V337M mutation using isogenic human iPSC-derived neurons carrying WT, V337M-Heterozygous (V337M-Het), or V337M-Homozygous (V337M-Homo) mutations at the endogenous MAPT locus (Sohn et al., 2019; Wang et al., 2017a). Three mitochondrial proteins were selected for validation: TOMM40, located at the outer mitochondrial membrane (OMM) where it regulates the import of proteins into mitochondria, TIMM13, located in the intermembrane space (IMS) where it serves as a chaperone for imported proteins, and ATP5IF1, which localizes to the IMM and constitutes an inhibitor of the ETC ATPase (Figure 5F). The interaction between TauWT and mitochondrial proteins was confirmed by PLA (Figures 5G–5J). Negative controls including only one antibody confirmed the specificity of the PLA signal (Figures 5G–5J). Quantification of the PLA signal showed that TauWT exhibited stronger interactions with TOMM40 (Figure 5H), TIMM13 (Figure 5I), and ATP5IF1 (Figure 5J) than TauV337M. Together, our results provide direct evidence that TauWT’s interaction with mitochondrial proteins is weakened by FTD mutations.
TauV337M neurons have altered mitochondrial bioenergetics
We next evaluated mitochondrial bioenergetics in isogenic human iPSC-derived neurons carrying WT, V337M-Het, or V337M-Homo Tau. Because the V337M mutation weakened interactions with mitochondria proteins in the ETC, we measured mitochondrial membrane potential (ΔΨm)-dependent tetramethylrhodamine methyl ester (TMRM) accumulation in the mitochondria of 4-week-old neurons (Figures 6A and 6B). V337M-Het and V337M-Homo neurons had reduced ΔΨm compared with that of WT neurons (Figure 6B), suggesting either a decrease of the activity of ETC complexes I–IVor an increased proton flux across the IMM of mutant Tau neuronal mitochondria. No differences in total mitochondrial content were observed among genotypes (Figure 6C).
Figure 6. TauV337M neurons display mitochondrial bioenergetic alterations.

(A) Images of 4-week-old TauWT, TauV337M-Het (VM-Het), and TauV337M-Homo (VM-Homo) neurons incubated with TMRM (red) and hoechst 33342 (blue). Scale bars, 5 μm.
(B) TMRM intensity analysis of TauWT, TauV337M-Het, and TauV337M-Homo neurons at baseline or treated with oligomycin (n > 30,000 mitochondria/replicate from two independent experiments and >8 internal replicates/genotype and treatment, ***p < 0.001, linear mixed-effects model with Tukey’s post hoc analyses).
(C) Quantification of mitochondria number per neuron from experiments depicted in (A).
(D) Representative OCR measurements obtained from Seahorse assays with TauWT, TauV337M-Het, and TauV337M-Homo neurons normalized to calcein intensity. Arrows indicate addition of oligomycin, FCCP, and rotenone+antimycin A (Rot/AA).
(E–H) Quantification of basal respiration (E), proton leak (F), ATP-linked respiration (G), and maximal respiration (H) from Seahorse experiments depicted in (D) with 2- and 4-week-old neurons (from 3 experiments and 8 technical replicates/condition, ***p < 0.001, **p < 0.01, *p < 0.05, linear mixed-effects model with Tukey’s post hoc analyses).
(I–K) Coupling efficiency (I), respiratory control ratio (J), and spare respiratory capacity (K) were calculated using the indicated parameters from the Seahorse experiments depicted in (D–F) (***p < 0.001, **p < 0.01, *p < 0.05, linear mixed-effects model with Tukey’s post hoc analyses). The center lines in boxplots (B–K) represent the median with the 25th and 75th percentiles marked by the box limits. The bars extend to the farthest data points and the dots outside of the bars are outliers.
To further dissect the effects of V337M mutation on ETC function, we measured the oxygen consumption rate (OCR) in 2- and 4-week-old neurons upon exposure to different stressors (Figure 6D) (Brand and Nicholls, 2011). A dramatic increase in basal (Figure 6E) and maximal respiration (Figure 6H) was observed when comparing 4- and 2-week-old neurons, consistent with a metabolic shift from aerobic glycolysis to oxidative phosphorylation and/or an increase in mitochondria number associated with neuronal differentiation and maturation (Zheng et al., 2016). V337M-Homo neurons showed higher levels of basal respiration than WT neurons at both ages (Figure 6E). While ATP-linked respiration was not significantly different among genotypes in 4-week-old neurons (Figure 6G), V337M-Homo neurons showed significantly elevated proton leak not associated with energy generation, supporting elevated uncoupling of V337M-Homo mitochondria (Figure 6F). Upon depolarization of the IMM with FCCP, V337M-Homo neurons had a small but significant increase in maximal respiration compared with WT neurons (Figure 6H), suggesting that the ETC from V337M-Homo mitochondria can work as efficiently as that from WT mitochondria in the setting of acute energy demand.
V337M-Homo mitochondria exhibited diminished coupling efficiency (Figure 6I) and a significant reduction in the respiratory control ratio of V337M-Homo mitochondria (Figure 6J), indicating that V337M-Homo mitochondria used a higher fraction of the basal respiration to oxidize substrates not destined to drive ATP synthesis. Moreover, the spare respiratory capacity, which indicates how close a cell is to its bioenergetic limit, was significantly reduced in V337M-Homo mitochondria compared with WT mitochondria (Figure 6K). Altogether, these functional analyses showed that the reduced mitochondrial interactions caused by the V337M mutation could compromise a neuron’s ability to sustain prolonged energy demand.
Levels of Tau interactors modified by FTD mutation correlate with disease severity in human AD, FTD, and tauopathy brains
A multiomics approach on multiple cohorts from the Accelerating Medicines Partnership—Alzheimer’s Disease (AMP-AD) consortium identified six co-expressed protein modules that significantly associate with AD (Swarup et al., 2020). These protein co-expression modules represent robust biological processes that are differentially regulated in dementia involving Tau, including FTD and PSP. We leveraged this dataset to probe the pathophysiological relevance of the proteins that show reduced interaction with FTD mutant Tau in human brain. Interestingly, 29 out of the 108 proteins and 48 out of the 184 proteins that preferentially interact with TauWT compared with TauP301L and TauV337M, respectively, were represented in the modules associated with AD.
Analyses of the proteomic data from the Banner cohort revealed a significant reduction in the levels of TauWT-preferential interactors in AD patients compared with those in control, asymptomatic, and mild-cognitive impairment (MCI) patients (Figure 7A). The levels of these Tau-interacting proteins were negatively correlated with two distinct neuropathological scores: the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) (Figure 7B), and the Braak scores (Figure 7C). Similar correlations were observed in the Baltimore longitudinal study of aging (BLSA) cohort (Figures S7A–S7C). Altogether, these data suggest that proteins that normally interact with TauWT whose interaction is reduced by FTD Tau may play an important role in AD pathogenesis, where reduced levels correlate with disease severity.
Figure 7. Decreased expression of TauWT-preferential interactors correlates with human AD progression.

(A–C) Plots showing TauWT-preferential interactor eigenprotein trajectory with AD diagnosis (A), CERAD (B), and BRAAK (C) scores in the Banner cohort. TauWT-preferential interactors as compared with TauV337M and P301L are colored as green and pink, respectively (***p < 0.005). See also Figure S7.
(D) Co-expression graph of the first 50 hub proteins of the C2-mitochondrial module showing TauWT-preferential interactors compared with TauV337M (green), P301L (pink), or both mutants (blue).
(E) Plots showing C2-mitochondrial module TauWT-preferential interactor eigenprotein trajectory with AD, FTD, and PDP-CBS diagnosis in the UPenn cohort. TauWT-preferential interactors as compared with TauV337M and P301L are colored as green and pink, respectively (***p < 0.005).
(F–H) Plots showing C2-mitochondrial module TauWT-preferential interactor eigenprotein trajectory with AD diagnosis (F), CERAD (G), and BRAAK (H) scores in Banner cohort. TauWT-preferential interactors as compared with TauV337M and P301L are colored as green and pink, respectively. ***p < 0.005. See also Figure S7. The center lines in boxplots (A), (F), and (E) represent the median with the 25th and 75th percentiles marked by the box limits. The bars extend to the farthest data points and the dots outside of the bars are outliers.
Given the abundance of mitochondrial proteins that were TauWT-preferential interactors, we further explored the dementia-associated AMP-AD neuronal-specific C2 module, which is enriched for mitochondrial ETC subunits. We found that 25 and 13 TauWT-preferential interactors, compared with TauV337M and TauP301L, respectively, are components of the C2 module (Figure 7D). The total protein levels of these C2-TauWT-mitochondrial interactors were reduced in frontal cortical samples from patients with clinically diagnosed AD, FTD, and PSP with corticobasal degeneration (PSP-CBD) in a quantitative proteomic dataset from the University of Pennsylvania (UPenn) Brain Bank (Figure 7E). The protein levels of these C2-TauWT-mitochondrial interactors were also reduced in AD patients compared with those in control, asymptomatic, and MCI patients in the Banner (Figure 7F) and BLSA cohort (Figure S7D), where they negatively correlated with CERAD and Braak scores (Figures 7G, 7H, and S7D–S7F). Using western blot, we showed that levels of CYCS, a key mitochondrial protein within the C2 module that exhibits reduced interaction with mutant Tau, were reduced in AD brains compared with those in control brains (Table S6; Figures S7G and S7H). These data suggest that the C2-TauWT-mitochondrial interactors identified in vitro using human neurons are downregulated in both primary and secondary tauopathies and are associated with both cognitive decline and the accumulation of pathological Tau in the brain.
DISCUSSION
Our proteomic analyses reveal the multimodal and dynamic Tau interactome with unprecedented spatial resolution in human neurons. Our APEX approach utilized living neurons with intact cellular compartments, maintaining endogenous subcellular distribution of the Tau interactome. The detection of numerous presynaptic vesicle-associated proteins at the active zone suggests that Tau is closely associated with docked vesicles at presynaptic terminals. Consistent with studies showing that Tau can bind directly to synaptic vesicles (McInnes et al., 2018; Zhou et al., 2017), we identified biotinylation on 5 integral synaptic vesicle proteins that span the vesicle membrane. The tyrosines biotinylated by APEX-Tau were all located on the cytosolic domain of the vesicle proteins rather than the lumenal domains, supporting that the Tau protein-protein interaction occurred on the cytosolic side of the vesicle. The interaction of the N-terminal of Tau with synaptogyrin-3 inhibits presynaptic vesicle release (McInnes et al., 2018). We did not detect synaptogyrin-3 in our Tau interactome; however, synaptogyrin-1, which is found on the same vesicles as synaptogyrin-3 (Belizaire et al., 2004), was biotinylated by N-APEX Tau.
Our APEX mapping revealed distinct interactions with the N- versus C-terminal domains of Tau, which may be mediated in part by Tau fragments or by domain-specific interactions. It is estimated that APEX2 biotinylates proteins up to 10–20 nm away (Hung et al., 2014; Rhee et al., 2013), whereas the length of the Tau protein is approximately 63–70 nm (Hagestedt et al., 1989; Ruben et al., 1991), thus allowing for the spatial resolution of interactions in proximity to the N or C terminus. It is also possible that the presence of the APEX tag on the N or C terminus of Tau blocks a protein from binding to Tau, resulting in an apparent domain-specific interaction. The interactions detected here are different from those reported in a study on fragments of the N-terminal and C-terminal domains of Tau in SH-SY5Y cells (Gunawardana et al., 2015), possibly because these cells do not have the physiological and morphological features of human neurons. Proteins can be quantified in subcellular compartments of neurons using mass spectrometry together with super-resolution microscopy (Helm et al., 2021), and this approach may be useful to investigate the subcellular localization of tau-interacting proteins.
Our finding that neuronal activity enhanced the interaction of Tau with SNARE and synaptic vesicle proteins suggests that presynaptic vesicle fusion machinery could regulate activity-dependent Tau release through a direct protein-protein interaction. Depolarization of presynaptic terminals causes a local influx of calcium that binds to SYT1, which interacts with syntaxin1/SNAP25 in the SNARE complex to force the plasma membrane to break for vesicle fusion. Activity-induced Tau release from neurons is inhibited by tetanus toxin that disrupts SNARE complex function (Pooler et al., 2013) and supports a key role of the SNARE complex in Tau release. Another proposed mechanism for Tau release involves exosome-mediated release of Tau and uptake across synaptic connections (Wang et al., 2017c). In human neuron cultures, the majority of secreted Tau is free-floating, with a smaller amount detected in extracellular vesicles (Guix et al., 2018), indicating that there could be multiple mechanisms contributing to Tau release in cell culture. Whether this holds true for Tau secretion in the brain and to what extent Tau release at presynaptic terminals involves vesicular or vesicle-free routes remain to be determined.
Our AP-MS results identified a broader set of protein-protein interaction networks for Tau in human neurons. AP-MS in Tau transgenic mice and Tau-overexpressing cell lines identified RNA-binding proteins as Tau interactors (Maziuk et al., 2018) and revealed regulators of ubiquitination and protein degradation that bind to Tau (Choi et al., 2020; Gunawardana et al., 2015; Thompson et al., 2012; Wang et al., 2017b). We also found that Tau in human neurons interacts with RNA-binding proteins, although TauV337M and TauP301L weakened interactions with some ribosomal proteins. Indeed, others have shown that mutant Tau has reduced interaction with ribosomes (Maziuk et al., 2018), resulting in decreased protein synthesis, including ribosomal subunits, in different FTD mouse models (Evans et al., 2019). However, Tau showed increased affinity to ribosomes in microsomes from AD patients compared with control patients (Meier et al., 2016). While these discrepancies might be explained by the differing models and techniques used to characterize the interactions, cumulatively the data point toward an important role of Tau in ribonucleoprotein organization and ribosomal functions that might contribute to pathogenesis.
We established that tau interacts extensively with proteins involved in mitochondrial bioenergetics, including many subunits of the ETC and the ATP/ADP transporter SLC25A4, which interacts with Tau in synaptic mitochondria of AD patients (Amadoro et al., 2012). Although the interaction between Tau and mitochondrial proteins has been reported in mouse models (Liu et al., 2016; Wang et al., 2017b), our combined AP-MS study and PLA assays demonstrate that FTD-linked mutations diminished these interactions in human neurons. The diminished interactions in V337M neurons were associated with alterations in mitochondria bioenergetics with decreased coupling efficiency, which could impact their ability to maintain ATP levels under prolonged energetic stress. Indeed, tauopathy mice expressing P301L mutant Tau exhibit reduced respiratory control ratio and impaired ATP synthesis with aging (David et al., 2005), and P301L-overexpressing SH-SY5Y cells exhibit decreased mitochondrial ATP levels and increased susceptibility to oxidative stress (Schulz et al., 2012). Importantly, we showed that levels of TauWT-preferential interactors negatively correlated with clinical and pathological disease progression in two cohorts representing control, asymptomatic AD, MCI, and AD patients. The reduced levels of mitochondrial TauWT-preferential interactors in patients with AD, FTD, and PSP-CBS from the UPenn cohort suggest that these interactions represent a converging mechanism in tauopathy. TauWT-preferential interactors might play an important role in disease development, and it is possible that either a reduction of the levels of these proteins or their decreased association with mutant Tau adversely impact cellular bioenergetics.
Despite the broad physiological consequences of pathogenic Tau, development of Tau-targeted therapies has been limited by lack of understanding of how Tau directly mediates these processes. Our work provides an extensive resource for possible mechanisms by which Tau directly influences cellular functions through protein-protein interactions in human neurons, which paves the way for effective tau-targeted strategies to counteract neurodegenerative processes.
Limitations of the study
The APEX approach allows identification of proteins in proximity to Tau with very high confidence through direct detection of biotinylated peptides; however, the enrichment of such peptides is not efficient (<5% specificity). This limits sensitivity; thus, there are likely many additional interacting proteins that we did not detect. It is also important to note that APEX allows for detection of stable and transient interactions, as well as proteins simply in close proximity that may not physically interact with Tau. Our AP-MS results showed that changes in the Tau interactome are associated with mitochondrial dysfunction, but given the complexity of mitochondrial bioenergetics, it could be difficult to identify which interactions may be responsible for the oxidative phosphorylation deficits observed in FTD neurons. Lastly, our study provides an atlas of the Tau interactome in human neurons in cultured conditions without other brain cell types. There could be additional changes in the Tau interactome in human brain that contribute to disease pathogenesis that we did not identify in this study.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Li Gan (lig2033@med.cornell.edu)
Materials availability
The plasmids and cell lines generated in this study will be made available on request upon the completion of a Material Transfer Agreement (MTA).
Data and code availability
Mass spectrometry data files (raw and search results) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with dataset identifier: PXD026306. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication (Mendeley Data: https://doi.org/10.17632/hcsn98z4y3.1). Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell cultures
We used human iPSCs described in a previous study (Wang et al., 2017a), that were engineered for inducible expression of Ngn2 from a transgene integrated in the AAVS1 locus of WTC11 cells from a male with a wild-type genetic background (Miyaoka et al., 2014). The iPSC clone used contained one copy of the Ngn2 cassette integrated in a single allele of the AAVS1 locus. Cre recombinase treatment was used to excise the puromycin resistance gene from the iPSC clone carrying the Ngn2 transgene. This permitted a second puromycin selection to isolate iPSC clones after the APEX-tagged human Tau transgene was incorporated into the second AAVS1 allele using Transcription Activator-Like Effector Nucleases (TALENs). The sequence for APEX2 with a flag tag was cloned into either the N- or C-terminus of wild-type human Tau (2N4R) as well as the N-terminus of Tubulin α 1b, and the N-termini of TauP301L and TauV337M (2N4R). The N-APEX Tau, C-APEX Tau, N-APEX TauP301L and N-APEX TauV337M sequences were each subcloned into a pUCM vector with AAVS1 homology arms and the Tet-On 3G tetracycline-inducible expression system as described (Wang et al., 2017a). The pUCM donor vector together with TALEN pairs (kind gifts from Dr. Bruce Conklin, Gladstone Institutes) were used for targeted integration of each cassette into the second allele of the AAVS1 locus. The Ngn2 integrated iPSCs were first grown in E8 media, dissociated with Accutase and then transfected with one of the Tau or Tubulin pUCM vectors along with AAVS1 TALEN pairs using a Human Stem Cell Nucleofector Kit 1 (Lonza) with the Nucleofector 2b Device (Lonza). Puromycin (0.1–0.3 ug/mL) was added 24–48 hours later to select for cells with genetic integration. E8 media with puromycin was regularly replenished until isolated and established colonies were formed. An EVOS FL microscope (Invitrogen) was used to pick colonies for individual clones. Clones were then tested for APEX-tagged Tau or Tubulin expression by immunoblotting and immunocytochemistry. All iPSC cultures were maintained in E8 media in a cell culture incubator with 5% CO2 at 37°. The use of iPSCs in this study was approved by the Committee on Human Research at the University of California, San Francisco (15–15798).
Pre-differentiation of iPSCs into neurons was initiated by plating 2 ×106 iPSCs in each well of matrigel-coated 6-well plate with Knockout DMEM/F-12 media containing doxycycline (2 ug/mL), N2 supplement, non-essential amino acids, brain-derived neurotrophic factor (10 ng/mL, Peprotech), neurotrophin-3 (10 ng/mL, Peprotech) and ROCK inhibitor (Y-27632, Cayman chemicals). The media was replaced the next day without ROCK inhibitor, and pre-differentiation was maintained for a total of three days in a cell culture incubator with 5% CO2 at 37°. On day 0 the pre-differentiated precursor cells were dissociated with Accutase and plated onto matrigel coated coverslips or tissue culture plates for the growth of neuron cultures in Neurobasal-A media containing B27 supplement, Glutamax, BDNF (10 ng/mL) and NT3 (10 ng/mL) with doxycycline (2ug/mL). For proteomics experiments, 8 × 106 precursor cells were plated in a 10 cm tissue culture plate for each replicate and cultures were maintained in a cell culture incubator with 5% CO2 at 37°. Rat astrocytes were added to the neurons on day 1, and half of the media was replaced on day 5 with supplemented Neurobasal-A media containing cytosine β-D-arabinofuranoside (Ara-C) but lacking doxycycline. On day 10, half of the media was removed and three times the remaining volume in the culture was replenished with MEM media containing glucose (27.7mM), NaHCO3 (2.4mM), B-27 supplement, L-glutamine, Ara-C) and fetal bovine serum (5%). One third of the supplemented MEM media was replaced every week thereafter. At 5–7 weeks of age, doxycycline (2 ug/mL) was added to the neurons 24 hours before experiments were performed to express the APEX2-flag tagged Tau or Tubulin proteins. For experiments performed to assess mitochondrial function, human iPSC-derived neurons were differentiated from wild-type (WTC11) iPSCs carrying the NGN2 transgene and isogenic iPSCs that were edited by CRISPR/Cas9 to generate heterozygous or homozygous V337M mutations as previously described (Sohn et al., 2019).
Human subjects
The tissues used for this study were the mid-frontal cortices from brains of age-matched patients with AD (n=16, 6 females and 10 males) and controls (n=8, 3 females and 5 males). Samples were obtained from the University of Pennsylvania brain bank. All brains were donated after consent from the next-of-kin or an individual with legal authority to grant such permission. Brain tissues of University of Pennsylvania brain bank used in this study are not considered identified “human subjects” and not subjected to IRB oversight. The institutional review board has determined that clinicopathologic studies on de-identified postmortem tissue samples are exempt from human subject research according to Exemption 45 CFR 46.104(d)(2). Additional information about the donors can be found in Table S6.
Label-free quantitative proteomic data from postmortem human brain tissues were downloaded for the Baltimore Longitudinal Study of Aging (BLSA) at Johns Hopkins University and Banner Sun Health Research Institute (Banner), and University of Pennsylvania School of Medicine Brain Bank (UPenn) from the Accelerating Medicines Partnership – Alzheimer’s Disease (AMP-AD) consortium (https://adknowledgeportal.synapse.org/) (Hodes and Buckholtz, 2016; Logsdon et al., 2019). The BLSA samples consisted of 97 samples from the dorsolateral prefrontal cortex (BA9 area) representing 15 controls, 15 AsymAD and 20 AD cases (Seyfried et al., 2017). The Banner samples were from prefrontal cortex of 30 controls, 28 mild cognitive impairment, 33 AsymAD and 98 confirmed AD cases (Swarup et al., 2020). The UPenn samples were from prefrontal cortex consisting of 43 controls, 48 PSP-CBD, and 29 FTD. The PSP-CBD included 25 PSP, 15 CBD, 2 Pick’s disease, 2 FTD with parkinsonism linked to chromosome 17, and 4 tangle predominant senile dementia. The FTD included 27 frontotemporal lobar degeneration with transactive response DNA binding protein-43 inclusions (FTD-TDP43), 1 frontotemporal lobar degeneration with FUS inclusions (FTD-FUS), and 1 frontotemporal lobar degeneration with ubiquitin inclusions (Swarup et al., 2020).
METHOD DETAILS
Western blot
Following APEX stimulation and quenching, human iPSC-derived neuron cultures were washed with cold PBS, then homogenized in cold RIPA buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Nonidet P-40, 1mM EDTA, 1 mM phenylmethyl sulfonyl fluoride, protease inhibitor cocktail, phosphatase inhibitor cocktail. Lysates were sonicated, centrifuged at 18,000 g at 4°C for 15 min, and the supernatant was collected. Protein concentration was determined by Bradford assay (Bio-Rad). Equal amounts of protein for each sample were loaded and run on a 4–12% SDS-PAGE gel (Invitrogen). A nitrocellulose membrane (GE Healthcare) was used for transfer of proteins, and blots were blocked with 5% milk, and incubated with primary antibodies for mouse anti-flag (Sigma), HT7 (Thermo Fisher), GAPDH (Millipore), rabbit anti-Tau (Agilent) and streptavidin horseradish peroxidase conjugated (HRP, Thermo Fisher). Secondary HRP antibodies (Millipore) and chemiluminescence (Pierce) were used for detection of immunoblotting.
Human brains were homogenized in RIPA buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA (ThermoFisher Scientific), cOmplete protease inhibitor cocktail (Roche), and Halt’s phosphatase inhibitor cocktail (ThermoFisher Scientific). After sonication, brain lysates were centrifuged at 20,000 g at 4°C for 15 min. Supernatants were collected and protein concentrations were measured with the Pierce BCA Protein Assay Kit (ThermoFisher Scientific). The same amount of protein was loaded onto a 4–12% SDS-PAGE gel (Invitrogen), transferred to PVDF membranes (BioRad), blocked with 5% milk, and immunoblotted in 1% milk. Bands in immunoblots were visualized by enhanced chemiluminescence (BioRad) and quantified by intensity with ImageLab (BioRad). The antibodies used for western blot were anti-cytochrome c (1:1000, ProteinTech, #10993-1-AP) and anti-GAPDH (1:10000, GeneTex, GTX627408). Immunoreactivity was detected with goat anti-rabbit HRP (1:5000, ThermoFisher Scientific).
APEX proximity-dependent labeling and enrichment of biotinylated proteins
To start proximity-dependent labeling by APEX, the neuronal media was removed and cells were washed with extracellular solution warmed to 37°C containing (in mM): 140 NaCl, 5 KCl, 2.5 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose at pH 7.4. Then neurons were incubated in extracellular solution with biotin-phenol (500 μM, Adipogen) in a 37°C cell culture incubator for 30 minutes. The neurons were next treated with 1 mM hydrogen peroxide (H2O2) for 1 minute and quickly washed once with Dulbecco’s phosphate buffered saline (DPBS) followed by two washes with quenching solution consisting of 1 × PBS with 10 mM sodium azide, 10 mM sodium ascorbate, and 5 mM Trolox. The quenching solution was removed, and the neurons were either fixed for immunocytochemistry or lysed to extract proteins. The neurons were lysed at 4°C in buffer containing 50mM Tris-HCl pH 7.4, 500 mM NaCl, 0.2% SDS, 2% Triton, 1 mM DTT, a protease inhibitor tablet (Pierce), phosphatase inhibitor cocktails (Sigma), 10 mM sodium azide, 10 mM sodium ascorbate, and 5 mM Trolox. The samples were sonicated and then centrifuged at 16,500 g for 10 minutes at 4°C. The collected supernatant was then subjected to dialysis to remove excess free biotin using a Slide-A-Lyzer MINI dialysis device (Thermo Scientific). Proteins were precipitated by adding 9 parts methanol to 1 part lysate. The supernatant was discarded, and the precipitated protein pellet was washed once with methanol and then dried by vacuum centrifugation. Protein was resuspended in 8M urea, 100 mM ammonium bicarbonate, then sonicated with a probe sonicator for 1 minute to re-solubilize the proteins. A Bradford Assay was performed to determine protein concentration and 1 mg of protein was taken out to be processed further. TCEP was added to 4 mM and incubated for half an hour at room temperature. Each sample was treated with 10 mM iodoacetamide, then excess iodoacetamide was quenched with 10 mM DTT. The samples were diluted 4-fold with 100 mM ammonium bicarbonate. Trypsin (Promega) was added in a 1:100 enzyme:substrate ratio and samples were incubated overnight at 37°C with agitation. The digested samples were desalted on a 50 mg SepPak column and eluted peptides were dried thoroughly via vacuum centrifugation. Peptides were dissolved in IAP buffer containing 50 mM MOPS, 10 mM HNa2PO4, 50 mM NaCl at pH 7.5. Anti-biotin beads (Immune Chem Pharmaceuticals) were washed twice in IAP buffer before the samples were added to the beads for incubation on a rotator for two hours at 4°C. The beads were washed twice with IAP buffer and twice with H2O (HPLC-grade). The biotinylated peptides were eluted from the beads by 0.15% TFA with vortexing followed by a 10 minute incubation. The elution was repeated twice and the supernatants were collected for desalting on NEST C18 tips then drying by vacuum centrifugation.
Flag pulldown of Tau
Neuron cultures were washed three times with DPBS before cold lysis buffer was added containing 50mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP40, a protease inhibitor tablet (Pierce), and phosphatase inhibitor cocktails (Sigma) at pH 7.4. The samples were lysed by 2 freeze/thaw cycles and then centrifuged at 16,500 g for 15 minutes at 4°C. Magnetic anti-flag beads (Sigma) were washed twice with IP buffer containing 50mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA (pH 7.4). Lysates were added to the beads and incubated with rotation at 4°C for 2 hours. The beads were then washed twice times with 50mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.05% NP40 (pH 7.4) and then with two additional washes using IP buffer. To elute proteins from the beads a 3xFLAG peptide (100 ug/mL, Sigma) with 0.05% RapiGest in IP buffer was added to the beads for 15 minutes at room temperature with agitation. The elution was repeated a second time and the supernatants were combined with the addition of 1.7M urea, 50 mM Tris, and 1 mM DTT. The samples were incubated at 60°C for 15 minutes, then 3mM iodoacetamide was added for 45 minutes at room temperature. After adding 3mM DTT the samples were digested with trypsin (Promega) overnight at 37°C. Trypsinized samples were acidified with 0.5% TFA then desalted on a C18 stage tip and dried by vacuum centrifugation.
Mass spectrometry data acquisition
Samples were resuspended in 4% formic acid, 4% acetonitrile solution, separated by a reversed-phase gradient over a nanoflow column (360 μm O.D. × 75 μm I.D.) packed with 25 cm of 1.9 μm Reprosil C18 particles with (Dr. Maisch). Mobile phase A consisted of 0.1% FA and mobile phase B consisted of 80% ACN/0.1% FA. Biotinylated peptides were separated by an organic gradient from 12% to 22% mobile phase B over 35 minutes followed by an increase to 40% B over 40 minutes, then held at 100% B for 6 minutes at a flow rate of 300 nL/minute. Eluting peptides were directly injected into an Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo). Data was collected in positive ion mode with MS1 detection in profile mode in the orbitrap using 120,000 resolution, 350–1400 m/z scan range, 100 ms maximum injection, and an AGC target of 1e6. MS2 fragmentation was performed on charge states from 2–6 with a 30s dynamic exclusion, and all MS2 data was collected in centroid mode at a rapid scan rate in the ion trap with HCD (32% normalized collision energy), 15ms maximum injection time, 2e4 AGC.
Peptides resulting from FLAG-purified samples were separated by an organic gradient from 7% to 36% mobile phase B over 53 minutes followed by an increase to 95% B over 7 minutes, then held at 90% B for 6 minutes at a flow rate of 300 nL/minute. Eluting peptides were directly injected into a Q-Exactive Plus mass spectrometer (Thermo). Data was collected in positive ion mode with MS1 detection in profile mode in the orbitrap using 70,000 resolution, 350–1500 m/z scan range, 250 ms maximum injection, and an AGC target of 1e6. MS2 fragmentation was performed on charge states from 2–6 with automatic dynamic exclusion, and all MS2 data was collected in centroid mode in the orbitrap at 17,500 resolution with HCD (26% normalized collision energy), 60ms maximum injection time, 5e4 AGC. Mass spectrometry data files (raw and search results) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with dataset identifier PXD026306 (Vizcaíno et al., 2016).
Enrichment and network analyses
Gene Ontology (GO) term enrichment analyses were performed using either Gene Set Enrichment Analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005) or ClueGO version 2.5.4 in Cytoscape version 3.7.1 (Bindea et al., 2009; Shannon et al., 2003). Enrichment and network analyses were applied from three ontologies including GO Molecular Function, GO Biological Process, and GO Cellular Compartments.
Proximity ligation assay
Human iPSC-derived neurons were grown on glass coverslips and fixed at 5–7 weeks of age in 4% paraformaldehyde in PBS for 15 min then washed three times for 5 min with PBS. The DuoLink In Situ Fluorescence (Sigma) protocol was used to perform the proximity ligation assay (PLA) on the neurons. The cultures were incubated in a humidified chamber with Duolink Blocking Solution for 1 hour at 37°C, then the blocking solution was removed and replace with the primary antibodies diluted in Duolink Antibody Diluent for 1 hour at room temperature. The primary antibodies used for PLA included Dako-Tau (Agilent), HT7 (Thermo Fisher), Munc18 (BD Biosciences), Synaptotagmin-1 (Synaptic Systems), ATP5IF1 (ThermoFisher, Cat# A-21355), TOMM40 (ProteinTech, Cat# 18409–1-AP), TIMM13 (ThermoFisher, Cat# PA5-61856). The coverslips were washed twice for 5 min with Wash Buffer A (Sigma) then the Duolink In Situ PLA Probe Anti-Rabbit PLUS and Duolink In Situ PLA Probe Anti-Mouse MINUS PLA probes (Sigma) diluted in Duolink Antibody Diluent were applied. The coverslips were incubated in a humidified chamber with PLA probe solution for 1 hour at 37°C. Coverslips were washed twice with Wash Buffer A followed by the addition of the ligation solution for 30 min at 37°C, then another two washing steps, and incubation in amplification solution for 100 min at 37°C. The coverslips were washed twice for 10 minutes in Wash Buffer B (Sigma) and mounted onto slides with ProLong Gold Mountant (Thermo Fisher).
Human Tau ELISA assay
Human iPSC-derived neurons were differentiated from wild-type (WTC11) NGN2 iPSCs and matured for six weeks in culture. The cell culture media was removed and replaced with extracellular solution containing (in mM): 115 NaCl, 3.5 KCl, 10 HEPES, 1 MgCl2, 2.5 CaCl2 at pH 7.4 or with high KCl extracellular solution containing (in mM): 68.5 NaCl, 50 KCl, 10 HEPES, 20 Glucose, 1 MgCl2, 2.5 CaCl2 at pH 7.4. The neurons were incubated with extracellular solution with or without high KCl for 30 min at 37°C. Some neurons were treated with 100 μM BAPTA-AM (Sigma) for 30 min before and during stimulation with high KCl extracellular solution. The extracellular solution was collected for ELISA analysis, centrifuged at 24,000 × g for 10 min and the supernatant was transferred to a new, low-protein-binding tube and stored on ice. The neurons were lysed with cold RIPA buffer described above. Plates with 96-wells (Corning 3925 “high-binding”) were prepared for ELISAs by coating each well with 50ul of 1.5ug/ml HT7 monoclonal antibody (Thermo Fisher) the night before use. Plates were sealed and incubated at 4°C, rocking, for ~12 hr. The HT7 antibody was removed, wells were washed four times briefly (no incubation) with 300ul of DPBS, and then blocked in 300ul of 3% BSA/DPBS per well and incubated at room temperature, rocking, while samples were prepared as described above. A concentration series of seven Tau-441 protein standards were prepared for each plate. Each sample or standard (50uL) was added to individual blocked wells and incubated for 1 hr at room temperature with slow shaking. All samples and standards were run in triplicate. Then 50ul of Tau5-AP monoclonal antibody (1:500) in 0.3% BSA, 0.1% Tween-20, DPBS was added to each well and incubated overnight (~12 hr) at 4°C with slow shaking. The next day, the plates were brought to room temperature (~1hr). Wells were washed four times for 10 min in 300ul DPBS + 0.05% Tween-20 and tapped dry. Substrate CDP star was added (50ul per well) and the plates were incubated 30 min in the dark, followed by scanning on the plate reader. The CytoTox-ONE Membrane Integrity Assay (Promega) was performed according to the manufacturer’s instructions. The amount of secreted Tau was quantified from the percent of extracellular Tau detected out of the total amount of intracellular and extracellular Tau for each well of culture.
Immunocytochemistry and imaging
Human iPSC-derived neurons on coverslips were fixed in 4% paraformaldehyde in PBS for 15 min then washed three times for 5 min with PBS. The coverslips were then placed in blocking solution containing PBS, 0.1% Triton X-100, and 2% normal goat serum (NGS) for 1 hour at room temperature. Primary antibodies diluted in blocking solution were added to the coverslips for 1 hour at room temperature then washed three times for 5 min with PBS. Primary antibodies used included mouse anti-flag (Sigma), anti-Munc18 (BD Biosciences), anti-Synaptotagmin 1 (Synaptic Systems), anti-PSD95 (Thermo Fisher), anti-tau (HT7, Thermo Fisher) and rabbit anti-Synapsin (Cell Signaling Technology). The secondary antibodies diluted in blocking solution, including Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 546 anti-Rabbit (Life Technologies), were added to the coverslips for 1 hour at room temperature. Cy3-streptavidin (Jackson ImmunoResearch) was used for fluorescent labeling of biotinylated proteins. The coverslips were washed three more times for 5 min with PBS before mounting onto a slide with ProLong Gold Mountant (Thermo Fisher). Human neurons treated with FM1–43FX (5 μg/mL, Thermo Fisher) in extracellular solution for 5 min at room temperature were then washed once with extracellular solution lacking calcium and fixed with 4% paraformaldehyde for 15 min at room temperature. After fixation, cells were washed three times for 5 min with PBS. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 3 minutes then washed two times with PBS for 10 min. Cells were blocked with 2% NGS in PBS for one hour and immuolabeling was performed in 2% NGS in PBS. Images were acquired with a laser scanning confocal microscope (LSM 700, Zeiss) using a 63x oil objective. The settings used for image acquisition were selected to keep the majority of brightest pixel intensities from reaching saturation. Maximum intensity projections were generated from confocal z-sections. Fluorescence intensity measurements were quantified using ImageJ software.
Mitochondrial respiration
Oxygen Consumption Rate (OCR) from live human iPSC-derived neurons was measured using a Seahorse XFe-96 Analyzer (Agilent). 30,000 human pre-differentiated neurons were seeded per well in Poly-D-Lysine + Laminin-coated 96-well Seahorse plate and differentiated for the indicated time (2 or 4 weeks) before performing the OCR measurements. 1 hour before the Seahorse experiment, neurons were washed and incubated with the assay media (Agilent Seahorse XF Base Medium (Agilent, Cat# 103335-100) supplemented with 20mM Glucose, 1.2mM Glutamine, 1mM Sodium Pyruvate, pH=7.4) in a non-CO2 incubator. Oligomycin (final concentration=1uM, Fisher Scientific, Cat# 49-545-510MG), FCCP (final concentration=1uM, Fisher Scientific, Cat# 04-531-0) and a mixture of Rotenone (final concentration=0.5uM, Fisher Scientific, Cat# 36-165-0) and Antimycin A (final concentration=0.5uM, MilliporeSigma, Cat# A8674) were sequentially injected into the media to modulate distinct components of the ETC and reveal key parameters of mitochondrial bioenergetics. After the OCR measurements were obtained, neurons were incubated with Calcein-AM (Invitrogen, Cat# C3099) for 30 min, and imaged using ImageXpress® Pico Automated Cell Imaging System (Molecular Devices). OCR was normalized to live neurons using Calcein-AM staining intensity.
Mitochondrial membrane potential and mitochondria number measurements using TMRM
iPSC-derived neurons were cultured in PDL-coated 96-well plate (Cat#354640, Corning) for 4 weeks. The day of the experiment, neurons were washed twice in assay medium containing 50% phenol-free Neurobasal-A media plus 50% phenol-free DMEM/F12 supplemented with N2 supplement (0.5X), B27 supplement (0.5X), Glutamax (0.5X), Non-Essential Aminoacids (1X), BDNF (10 ng/mL) and NT3 (10 ng/mL). Neurons were incubated with assay media containing 10nM Tetramethylrhodamine, methyl ester (TMRM) (Cat# I34361, Invitrogen) and 1ug/ml Hoechst 33342 (Cat# H3570, Invitrogen) at 37°C for 30 minutes. Neurons were washed three times with assay media before being imaged at 37°C and 5% CO2 using the KEYENCE BZ-X700 microscope. Images were segmented using ImageJ to identify mitochondria number per neuron and TMRM intensity per mitochondria.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
The results from imaging, ELISA, and western blot experiments are reported as mean ± SEM. Quantifications in Figures 5, 6, 7, and S7 are represented as boxplots, where the center line represents the median with the 25th and 75th percentiles marked by the box limits. The bars extend to 1.5 times the interquartile range from the median, and dots represent outliers. The type of statistical analyses performed and the value of n for each experiment was described in the corresponding figure legends. Statistical analyses were performed using SigmaPlot (Systat Software) and RStudio, and included student’s t-test, one-way ANOVA with Bonferroni post-hoc analyses, linear mixed-effects model with Tukey’s post-hoc analyses, and Mann Whitney test. A statistically significant outlier (p < 0.05) was identified in Figure S7H using the Grubb’s test in GraphPad, and the outlier was excluded from further analyses. The statistics used in ClueGO analyses revealed enriched networks with p-values < 0.05 from right-sided hypergeometric testing with Bonferroni correction.
Analyses of TauWT-preferential interactors in postmortem human tauopathy and neurodegenerative disease brain
Recent work identified six co-expressed protein modules (C1, C2, C3, C4, C8, and C10) that were significantly associated with Alzheimer disease progression using a multi-omics approach on multiple cohorts from the Accelerating Medicines Partnership – Alzheimer’s Disease (AMP-AD) consortium (Swarup et al., 2020). 29 out of the 108 proteins and 48 out of the 184 proteins, which preferentially interact with TauWT compared to TauP301L and TauV337M, respectively, were also in the six modules associated with AD. We determined if the number of intersecting proteins was significantly higher than expected using a permutation test (Efron and Gong, 1983). We considered all unique proteins identified by AP-MS from TauWT, TauP301L and TauV337M as the background set of proteins (N=1740). We randomly sampled 108 and 184 proteins, for TauP301L and TauV337M, respectively, from the background set of proteins and determined how many proteins overlapped with the six AD disease associated modules to create the null distribution. The p-value was calculated from the percentile of the null distribution that was ≥ 29 and ≥ 48 for TauP301L and TauV337M, respectively.
The label free quantitation intensities were log2 transformed and assessed for effects from biological covariates (diagnosis, age, gender) and technical variables (batch, brain bank). We used a linear regression model accounting for biological and technical covariates. The final model used was implemented in R version 3.6.1 (R Core Team, 2019) as follows:
We evaluated the proteins that were TauWT preferential interactors compared to TauP301L (TauWT>TauP301L) and TauV337M (TauWT>TauV337M). We also evaluated the TauWT>TauP301L proteins and TauWT>TauV337M proteins that intersected with the mitochondrial C2 proteomic module (TauWT>TauP301L:C2 and TauWT>TauV337M:C2) from previous work (Swarup et al., 2020). In this work, the C2 proteomic module was significantly downregulated in Alzheimer disease compared to controls. For these protein sets, we considered their eigenprotein as the first principal component of their protein expression (Langfelder and Horvath, 2007; Zhang and Horvath, 2005).
We determined if the eigenprotein for TauWT>TauP301L, TauWT>TauV337M, TauWT>TauP301L:C2 and TauWT>TauV337M:C2 was significantly different from disease diagnosis (AD, mild cognitive impairment, AsymAD, PSP-CBD, FTD) versus control using the Wilcoxon rank sum test (Wilcoxon, 1945). Eigenproteins were correlated with neuropathological scores (CERAD, Braak) using Pearson correlation (Pearson, 1931).
Mass spectrometry analyses
All raw MS data were searched with MaxQuant against the human proteome (Uniprot canonical protein sequences downloaded March 21, 2018 for anti-biotin samples, or July 21, 2017 for FLAG-purified samples). Peptides, proteins, and PTMs were filtered to 1% false discovery rate in MaxQuant (Cox and Mann, 2008). MaxQuant default parameters were used with the exception that label-free quantification was turned on with match between runs set to 1 min, and for anti-biotin purified samples a variable mass addition of biotin-phenol (361.14601 Da) on Y residues was considered. Biotinylated proteins identified in APEX experiments that were detected in fewer than half of the neuronal culture replicates (<4/7 replicates for N-APEX Tau unstimulated or 50mM KCl groups, <5/9 replicates for C-APEX Tau unstimulated or 50mM KCl groups, or <6/12 replicates for APEX-α tubulin) were removed from analyses (Tables S1 and S3). To classify Tau interactors identified by AP-MS (Table S4), proteins detected in fewer than half (<4/7 for TauWT and <4/8 for TauV337M and TauP301L) of the neuronal culture replicates for each group were removed to reduce non-specific binders. To classify proteins as TauWT, TauV337M or TauP301L preferential interactors (Table S5), proteins needed to meet one of the two following criteria: 1) To be identified in more than 50% of the replicates in both groups of the comparison and to be significantly different (adjusted P value <0.05) with a detection fold change bigger than 1.5 between genotypes, or 2) to be consistently detected in one group (more than 50% of replicates) and in 3 times more replicates than in the other genotype in the comparison (i.e. 75% of replicates in genotype A and 25% replicates of genotype B).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| Mouse anti-Tau (HT7) | Thermo Fisher | Cat#MN1000; RRID:AB_2314654 |
| Mouse anti-GAPDH | Millipore | Cat#MAB 374; RRID:AB_2107445 |
| Rabbit anti-Tau (Dako) | Agilent | Cat#A0024; RRID:AB_10013724 |
| Mouse anti-FLAG | Sigma-Aldrich | Cat#F3165; RRID:AB_259529 |
| Goat Anti-Mouse IgG, H & L Chain Antibody, Peroxidase Conjugated | Millipore | Cat#401253; RRID:AB_437779 |
| Goat Anti-Rabbit IgG, H & L Chain Specific Peroxidase Conjugate antibody | Millipore | Cat#401393; RRID:AB_437797 |
| Anti-FLAG M2 Magnetic Beads antibody | Sigma-Aldrich | Cat#M8823; RRID:AB_2637089 |
| Mouse anti-Munc18 | BD Biosciences | Cat#610336; RRID:AB_397726 |
| Mouse anti-Synaptotagmin-1 | Synaptic Systems | Cat#105 011C2; RRID:AB_2619760 |
| Mouse anti-PSD95 | Thermo Fisher | Cat#MA1-046; RRID:AB_2092361 |
| Rabbit anti-Synapsin | Cell Signaling Technology | Cat#5297; RRID:AB_2616578 |
| Rabbit anti-Cytochrome c | ProteinTech | Cat#10993-1-AP; RRID:AB_2090467 |
| Mouse anti-GAPDH | GeneTex | Cat#GTX627408; RRID:AB_11174761 |
| Biotin Antibody Agarose | Immune Chem Pharmaceuticals | Cat#ICP0615; RRID:AB_2893170 |
| Rabbit anti-TOMM40 | Proteintech | Cat#18409-1-AP; RRID:AB_2303725 |
| Mouse anti-ATPIF1 | Thermo Fisher Scientific | Cat#A-21355; RRID:AB_2535841 |
| Rabbit anti-TIMM13 | Thermo Fisher Scientific | Cat#PA5-61856; RRID:AB_2648474 |
|
| ||
| Biological samples | ||
|
| ||
| Human brain tissue | University of Pennsylvania brain bank | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 3xFLAG peptide | Sigma-Aldrich | Cat#F4799 |
| Cy3-Streptavidin | Jackson Immuno Research Labs | Cat#016-160-084; RRID:AB_2337244 |
| Streptavidin, horseradish peroxidase conjugate | Thermo Fisher | Cat#S911 |
| Brain-derived neurotrophic factor (BDNF) | Peprotech | Cat#450-02 |
| Neurotrophin-3 (NT3) | Peprotech | Cat#450-03 |
| Y-27632 | Cayman chemicals | Cat#10005583 |
| Biotin-phenol (Biotinyl tyramide) | Adipogen | Cat#CDX-B0270 |
| FM1-43FX | Thermo Fisher | Cat#F35355 |
| Oligomycin | Millipore Sigma | Cat#495455 |
| FCCP | Fisher Scientific | Cat#04-531-0 |
| Rotenone | Fisher Scientific | Cat#36-165-0 |
| Antimycin A | MilliporeSigma | Cat#A8674 |
| ReprosilPur 1.9 mm particles | ESI SOURCE SOLUTIONS | Cat#R119 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Duolink In Situ Detection Reagents Red | Sigma-Aldrich | Cat#DUO92008 |
| Duolink In Situ PLA Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat#DUO92002 |
| Duolink In Situ PLA Probe Anti-Mouse MINUS | Sigma-Aldrich | Cat#DUO92004 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat#PI-23225 |
| Bio-Rad Protein Assay Dye Reagent Concentrate | Bio-Rad | Cat#500-0006 |
| Human Stem Cell Nucleofector Kit 1 | Lonza | Cat#VPH-5012 |
| CytoTox-ONE Homogeneous Membrane Integrity Assay | Promega | Cat#G7890 |
| Image-iT TMRM Reagent | Invitrogen | Cat#I34361 |
| Hoechst 33342 | Invitrogen | Cat#H3570 |
| Calcein-AM | Invitrogen | Cat#C3099 |
|
| ||
| Deposited data | ||
|
| ||
| Proteomics Data | This paper | PRIDE ProteomeXchange Consortium: PXD026306 |
| Immunoblot Data | This paper | https://doi.org/10.17632/hcsn98z4y3.1 |
| Banner Sun Health Research Institute (Banner) proteomic data | Swarup et al., 2020 | https://doi.org/10.7303/syn7170616 |
| University of Pennsylvania (UPenn) proteomic data | Swarup et al., 2020 | https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn21411742 |
| Baltimore Longitudinal Study of Aging (BLSA) proteomic data | Seyfried et al., 2017 | https://doi.org/10.7303/syn3606086 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| human WTC11 iPSC with Ngn2 transgene integration | Wang et al., 2017a | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pUCM-N-APEX-flag TauWT | This paper | N/A |
| pUCM-C-APEX-flag TauWT | This paper | N/A |
| pUCM-N-APEX-flag TauV337M | This paper | N/A |
| pUCM-N-APEX-flag TauP301L | This paper | N/A |
| AAVS1 A TALEN | Bruce Conklin | N/A |
| AAVS1 B TALEN | Bruce Conklin | N/A |
| pUCM-APEX-flag Tubulin α 1b | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GSEA | Mootha et al., 2003 | https://www.gsea-msigdb.org/gsea/msigdb/annotate.jsp |
| ClueGO | Bindea et al., 2009; Shannon et al., 2003 | https://apps.cytoscape.org/apps/cluego |
| Rstudio | RStudio: Integrated Development for R. RStudio | http://www.rstudio.com/ |
| MaxQuant, 1.6.1.0 | PMID: 19029910 | https://www.maxquant.org |
| Cytoscape, version 3.7.1 | PMID: 14597658 | https://cytoscape.org |
| WGCNA R package | Langfelder and Horvath, 2007 | https://cran.r-project.org/web/packages/WGCNA/index.html |
Highlights.
APEX-mapped Tau interactome at subcellular and amino acid levels in human neurons
Activity-dependent binding of Tau to synaptic vesicle proteins during Tau secretion
FTD mutations reduce Tau binding to mitochondria proteins and impair bioenergetics
Tau interactors modified by FTD mutation are downregulated in human tauopathy
ACKNOWLEDGMENTS
We thank Dr. Bruce Conklin (Gladstone Institutes) for the WTC11 iPSCs, pUCM vector, and TALENS; Connor Ludwig for technical support; Kathryn Claiborn for editorial support; and Nathan Basisty for analysis advice. This work was supported by the NIH (U54NS100717 to L.G. and N.J.K., R01AG054214 to L.G., K01 AG057862 to T.E.T., R25NS065723), Tau Consortium and JPB Foundation (to L.G.), and Fundación Ramón Areces (to J.M.-P.). The results published here are based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). Dr. Levey (Emory University) provided BLSA data from brain tissue collected through the NIA’s BLSA, UPenn data from brain tissue collected through the University of Pennsylvania, Emory data from brain tissue collected through Emory Alzheimer’s Disease Research Center Brain Bank, and Banner data generated in part from samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona, supported by the NINDS (U24 NS072026), the NIA (P30 AG19610), the Arizona Department of Health Services (contract 211002), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05–901, and 1001 to the Arizona Parkinson’s Disease Consortium), and the Michael J. Fox Foundation for Parkinson’s Research.
Footnotes
DECLARATION OF INTERESTS
L.G. is a founder of Aeton Therapeutics. N.J.K. received research support from Vir Biotechnology and F. Hoffmann-La Roche; has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics, and Interline Therapeutics; is a shareholder in Tenaya Therapeutics, Maze Therapeutics, and Interline Therapeutics; and is a financially compensated Scientific Advisory Board Member for GEn1E Life sciences.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.12.041.
REFERENCES
- Alonso AD, Grundke-Iqbal I, Barra HS, and Iqbal K (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 94, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Atlante A, Florenzano F, Capsoni S, Bussani R, Mercanti D, and Calissano P (2012). Interaction between NH(2)-tau fragment and Abeta in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 33, 833, e1–833.25. [DOI] [PubMed] [Google Scholar]
- Bardai FH, Wang L, Mutreja Y, Yenjerla M, Gamblin TC, and Feany MB (2018). A conserved cytoskeletal signaling cascade mediates neurotoxicity of FTDP-17 tau mutations in vivo. J. Neurosci. 38, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belizaire R, Komanduri C, Wooten K, Chen M, Thaller C, and Janz R (2004). Characterization of synaptogyrin 3 as a new synaptic vesicle protein. J. Comp. Neurol. 470, 266–281. [DOI] [PubMed] [Google Scholar]
- Benussi L, Ghidoni R, Paterlini A, Nicosia F, Alberici AC, Signorini S, Barbiero L, and Binetti G (2005). Interaction between tau and alpha-synuclein proteins is impaired in the presence of P301L tau mutation. Exp. Cell Res. 308, 78–84. [DOI] [PubMed] [Google Scholar]
- Berk JM, Tifft KE, and Wilson KL (2013). The nuclear envelope LEM-domain protein emerin. Nucleus 4, 298–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, and Südhof TC (2000). Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J. Biol. Chem. 275, 39803–39806. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagè s F, Trajanoski Z, and Galon J (2009). ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics Oxf. Engl. 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, and Nicholls DG (2011). Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Shao E, and Mucke L (2021). Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371, eabb8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S, Kim J, Choi H, Song H, Lee CK, et al. (2020). Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 19, e13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Müller WE, et al. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. [DOI] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, and Spillantini MG (2002). Proteasomal degradation of Tau protein. J. Neurochem. 83, 176–185. [DOI] [PubMed] [Google Scholar]
- DuBoff B, Götz J, and Feany MB (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Südhof TC, and Rizo J (2007). Munc18–1 binds directly to the neuronal SNARE complex. Proc. Natl. Acad. Sci. USA 104, 2697–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, and Gong G (1983). A leisurely look at the bootstrap, the jackknife, and cross-validation. Am. Stat. 37, 36–48. [Google Scholar]
- Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, et al. (2018). Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99, 925–940.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Hallmann AL, Reinhardt P, Araúzo-Bravo MJ, Korr S, Röpke A, Psathaki OE, Ehling P, Meuth SG, Oblak AL, et al. (2015). Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant Tau protein. Stem Cell Rep 5, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HT, Benetatos J, van Roijen M, Bodea LG, and Götz J (2019). Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J 38, e101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, Leung L, Ring KL, Seeley WW, Karydas A, et al. (2013). Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep 1, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lanté F, and Buisson A (2014). Activity-dependent Tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 34, 6084–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, and Lee VM (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640. [DOI] [PubMed] [Google Scholar]
- Guix FX, Corbett GT, Cha DJ, Mustapic M, Liu W, Mengel D, Chen Z, Aikawa E, Young-Pearse T, Kapogiannis D, et al. (2018). Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int. J. Mol. Sci. 19, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardana CG, Mehrabian M, Wang X, Mueller I, Lubambo IB, Jonkman JE, Wang H, and Schmitt-Ulms G (2015). The human tau interactome: binding to the ribonucleoproteome, and impaired binding of the proline-to-leucine mutant at Position 301 (P301L) to chaperones and the proteasome. Mol. Cell. Proteomics 14, 3000–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagestedt T, Lichtenberg B, Wille H, Mandelkow EM, and Mandelkow E (1989). Tau protein becomes long and stiff upon phosphorylation: correlation between paracrystalline structure and degree of phosphorylation. J. Cell Biol. 109, 1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm MS, Dankovich TM, Mandad S, Rammner B, Jähne S, Salimi V, Koerbs C, Leibrandt R, Urlaub H, Schikorski T, and Rizzoli SO (2021). A large-scale nanoscopy and biochemistry analysis of postsynaptic dendritic spines. Nat. Neurosci. 24, 1151–1162. [DOI] [PubMed] [Google Scholar]
- Hodes RJ, and Buckholtz N (2016). Accelerating medicines partnership: Alzheimer’s disease (AMP-AD) knowledge portal Aids Alzheimer’s drug discovery through open data sharing. Expert Opin. Ther. Targets 20, 389–391. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, and Ting AY (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, and Lee VM (1999). Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24, 751–762. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Fath T, Ke YD, Bi M, van Eersel J, Li KM, Gunning P, and Götz J (2008). Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. USA 105, 15997–16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, and Götz J (2009). Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 284, 20909–20916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Shibata E, and Dutta A (2008). Human Rvb1/Tip49 is required for the histone acetyltransferase activity of Tip60/NuA4 and for the downregulation of phosphorylation on H2AX after DNA damage. Mol. Cell. Biol. 28, 2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson ZO, Jha S, Wohlschlegel JA, and Dutta A (2004). Rvb1p/Rvb2p recruit Arp5p and assemble a functional Ino80 chromatin remodeling complex. Mol. Cell 16, 465–477. [DOI] [PubMed] [Google Scholar]
- Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2007). Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Matsuura Y, Liu SM, and Stewart M (2005). Structural basis for nuclear import complex dissociation by RanGTP. Nature 435, 693–696. [DOI] [PubMed] [Google Scholar]
- Li X, Kumar Y, Zempel H, Mandelkow EM, Biernat J, and Mandelkow E (2011). Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J 30, 4825–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Song X, Nisbet R, and Götz J (2016). Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J. Biol. Chem. 291, 8173–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, and Gong CX (2005). Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer’s disease. J. Biol. Chem. 280, 1790–1796. [DOI] [PubMed] [Google Scholar]
- Lobingier BT, Hüttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, and Krogan NJ (2017). An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 169, 350–360.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon BA, Perumal TM, Swarup V, Wang M, Funk C, Gaiteri C, Allen M, Wang X, Dammer E, Srivastava G, et al. (2019). Meta-analysis of the human brain transcriptome identifies heterogeneity across human AD coexpression modules robust to sample collection and methodological approach. bioRxiv. https://www.biorxiv.org/content/10.1101/510420v1.abstract. [Google Scholar]
- Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, et al. (2016). Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166, 1295–1307.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, and Mizushima N (2018). Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 217, 2633–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maziuk BF, Apicco DJ, Cruz AL, Jiang L, Ash PEA, da Rocha EL, Zhang C, Yu WH, Leszyk J, Abisambra JF, et al. (2018). RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes J, Wierda K, Snellinx A, Bounti L, Wang YC, Stancu IC, Apóstolo N, Gevaert K, Dewachter I, Spires-Jones TL, et al. (2018). Synaptogyrin-3 mediates presynaptic dysfunction induced by tau. Neuron 97, 823–835.e8. [DOI] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H 3rd., et al. (2016). Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J. Neurosci. 36, 1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So PL, and Conklin BR (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 11, 291–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, and Mucke L (2011). The many faces of tau. Neuron 70, 410–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, and Duff KE (2016). Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson ES (1931). The test of significance for the correlation coefficient. J. Am. Stat. Assoc. 26, 128–134. [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, and Hanger DP (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2019). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, and Ting AY (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben GC, Iqbal K, Grundke-Iqbal I, Wisniewski HM, Ciardelli TL, and Johnson JE Jr. (1991). The microtubule-associated protein tau forms a triple-stranded left-hand helical polymer. J. Biol. Chem. 266, 22019–22027. [PubMed] [Google Scholar]
- Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow EM, Mandelkow E, and Hoenger A (2004). Surface-decoration of microtubules by human tau. J. Mol. Biol. 339, 539–553. [DOI] [PubMed] [Google Scholar]
- Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, Dawson HN, Bennett CF, Rigo F, and Miller TM (2016). Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron 90, 941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Müller WE, and Leuner K (2012). A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 46, 205–216. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. (2017). A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst 4, 60–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, and Pratt WB (1997). Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J. Biol. Chem. 272, 16224–16230. [DOI] [PubMed] [Google Scholar]
- Sohn PD, Huang CT, Yan R, Fan L, Tracy TE, Camargo CM, Montgomery KM, Arhar T, Mok SA, Freilich R, et al. (2019). Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104, 458–470.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, and Gan L (2016). Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol. Neurodegener. 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolow S, Henkins KM, Bilousova T, Gonzalez B, Vinters HV, Miller CA, Cornwell L, Poon WW, and Gylys KH (2015). Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 133, 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup V, Chang TS, Duong DM, Dammer EB, Dai J, Lah JJ, Johnson ECB, Seyfried NT, Levey AI, and Geschwind DH (2020). Identification of conserved proteomic networks in neurodegenerative dementia. Cell Rep 31, 107807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IR, Ahmad A, Wu T, Nordhues BA, Bhullar A, Gestwicki JE, and Zuiderweg ERP (2018). The disorderly conduct of Hsc70 and its interaction with the Alzheimer’s-related Tau protein. J. Biol. Chem. 293, 10796–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AD, Scaglione KM, Prensner J, Gillies AT, Chinnaiyan A, Paulson HL, Jinwal UK, Dickey CA, and Gestwicki JE (2012). Analysis of the tau-associated proteome reveals that exchange of Hsp70 for Hsp90 is involved in tau degradation. ACS Chem. Biol. 7, 1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, et al. (2016). Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes Tauopathy-related memory loss. Neuron 90, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Pedram K, Svinkina T, Fereshetian S, Myers SA, Aygun O, Krug K, Clauser K, Ryan D, Ast T, et al. (2017). Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat. Methods 14, 1167–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, et al. (2016). Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, Iturria-Medina Y, Strandberg OT, Smith R, Levitis E, Evans AC, and Hansson O; Alzheimer’s Disease Neuroimaging Initiative, and Swedish BioFinder Study (2020). Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 11, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, et al. (2017a). Scalable production of iPSC-derived human neurons to identify tau-lowering compounds by high-content screening. Stem Cell Rep 9, 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Joberty G, Buist A, Vanoosthuyse A, Stancu IC, Vasconcelos B, Pierrot N, Faelth-Savitski M, Kienlen-Campard P, Octave JN, et al. (2017b). Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta neuropathol 133, 731–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Williams D, Müller I, Lemieux M, Dukart R, Maia IBL, Wang H, Woerman AL, and Schmitt-Ulms G (2019). Tau interactome analyses in CRISPR-Cas9 engineered neuronal cells reveal ATPase-dependent binding of wild-type but not P301L Tau to non-muscle myosins. Sci. Rep. 9, 16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, et al. (2017c). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert S, Wawrzyniuk M, John LH, Rüdiger SGD, and Drescher M (2020). The mechanism of Hsp90-induced oligomerizaton of Tau. Sci. Adv. 6, eaax6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxon F (1945). Individual comparisons by ranking methods. Biom. Bull. 1, 80–83. [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgräfe K, Mandelkow EM, et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, and Lee VM (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- Zachariae U, and Grubmüller H (2008). Importin-beta: structural and dynamic determinants of a molecular spring. Structure 16, 906–915. [DOI] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article 17. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, and Hunter T (2016). Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 5, e13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, Wang YC, Swerts J, Beyens J, Miskiewicz K, et al. (2017). Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 8, 15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data files (raw and search results) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with dataset identifier: PXD026306. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication (Mendeley Data: https://doi.org/10.17632/hcsn98z4y3.1). Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.