

Summary
Stresses such as heat shock trigger formation of protein aggregates and induction of a disaggregation system composed of molecular chaperones. Recent work reveals that several cases of apparent heat-induced aggregation, long thought to be the result of toxic misfolding, instead reflect evolved, adaptive biomolecular condensation, with chaperone activity contributing to condensate regulation. Here, we show that the yeast disaggregation system directly disperses heat-induced biomolecular condensates of endogenous poly(A)-binding protein (Pab1) orders of magnitude more rapidly than aggregates of the most commonly used misfolded model substrate, firefly luciferase. Beyond its efficiency, heat-induced condensate dispersal differs from heat-induced aggregate dispersal in its molecular requirements and mechanistic behavior. Our work establishes a bona fide endogenous heat-induced substrate for long-studied heat shock proteins, isolates a specific example of chaperone regulation of condensates, and underscores needed expansion of the proteotoxic interpretation of the heat shock response to encompass adaptive, chaperone-mediated regulation.
Introduction
In all cellular life, a sudden increase in temperature—heat shock—causes formation of intracellular aggregates and production of heat shock proteins, many of which act as molecular chaperones (Parsell and Lindquist, 1993). An early and long-standing interpretation of these observations, which follow a wide range of so-called “proteotoxic stresses,” is that molecular chaperones are produced to protect cells from the toxic effects of stress-induced misfolded proteins and their damaged aggregates (Lindquist, 1986; Vogel et al., 1995; Morimoto, 2008; Vabulas et al., 2010).
Supporting this view, the molecular disaggregation system, which includes molecular chaperones Hsp100, Hsp70, and Hsp40, disperses aggregates of model substrates such as heat-misfolded firefly luciferase, restoring their function in vitro (Glover and Lindquist, 1998; Goloubinoff et al., 1999). Decades of biochemical studies on these model substrates have uncovered the general mechanism of disaggregation as follows (reviewed in detail by Mogk et al. (2018)): J-domain proteins such as Hsp40 target and promote binding of Hsp70 to specific substrates while simultaneously stimulating Hsp70’s ATPase activity (Laufen et al., 1999; Lu and Cyr, 1998; Jiang et al., 2019; Faust et al., 2020). Substrate-bound Hsp70 then recruits and activates the AAA+ disaggregase Hsp100 (Rosenzweig et al., 2013; Carroni et al., 2014; Seyffer et al., 2012; Haslberger et al., 2007), which threads the substrate delivered by Hsp70 through its central channel to extract the substrate from aggregates (Haslberger et al., 2008; Gates et al., 2017; Avellaneda et al., 2020).
Alongside nascent polypeptides (Baler et al., 1992; Masser et al., 2019), heat-induced aggregates of misfolded mature proteins are considered major substrates of the disaggregation system. However, no eukaryotic endogenous mature protein has yet been identified to misfold in response to physiological heat shock—i.e., nonlethal elevated temperature fluctuations encountered in nature to which cells have adapted. Yeast prion fibers are an endogenous substrate of the disaggregation system (Shorter and Lindquist, 2004; Inoue et al., 2004), but they do not form in response to acute heat shock in wild-type cells (Franzmann et al., 2018; Tyedmers et al., 2008). As a consequence, a central element in our understanding of the heat shock response—that molecular chaperones directly engage and disperse endogenous aggregates induced by heat shock—has remained untested.
Recent work suggests that the proteotoxicity model, and the view that physiological heat shock induces widespread protein misfolding, must be expanded. A proteome-wide study in budding yeast showed that a specific set of mature proteins form fully reversible aggregates in response to heat shock (Wallace et al., 2015). Closer inspection revealed that several cases of this apparent aggregation reflect evolved, adaptive biomolecular condensation (Riback, Katanski et al., 2017; Iserman et al., 2020). For example, physiological heat shock temperature and pH changes cause poly(A)-binding protein (Pab1), an abundant and broadly conserved eukaryotic RNA-binding protein, to phase-separate and form gel-like condensates in vitro. Suppressing Pab1 condensation reduces cell fitness during prolonged heat stress, indicating that condensation is adaptive (Riback, Katanski et al., 2017).
Here we will use the term biomolecular condensates to refer to endogenous putatively adaptive membraneless structures of concentrated biomolecules (Banani et al., 2017) regardless of the condensation mechanism, and the term aggregates to refer to amorphous clumps of misfolded proteins, commonly deleterious to cells (Geiler-Samerotte et al., 2011).
Substantial in vivo evidence indicates that endogenous heat-induced condensates interact with the disaggregation system. All members of the yeast disaggregation system (Hsp104/Hsp70/Hsp40) co-localize with stress granules, which contain Pab1 (Cherkasov et al., 2013; Walters et al., 2015; Kroschwald et al., 2015, 2018). Deletion or inhibition of any member of the system, or the Hsp70 nucleotide exchange factor Hsp110 (Sse1/2), delays dissolution of stress granules during recovery (Cherkasov et al., 2013; Walters et al., 2015; Kroschwald et al., 2015, 2018). Interestingly, dispersal of endogenous stress granules precedes that of exogenously expressed misfolded protein aggregates (Figure S1A–C) (Cherkasov et al., 2013; Kroschwald et al., 2015) and only the former correlates with the resumption of translation activity and the cell cycle (Cherkasov et al., 2013; Kroschwald et al., 2018).
We and others have hypothesized that heat-induced biomolecular condensates are major endogenous substrates of molecular chaperones (Wallace et al., 2015; Riback, Katanski et al., 2017; Kroschwald et al., 2018; Yoo et al., 2019; Triandafillou et al., 2020; Begovich et al., 2020; Snead et al., 2019). However, the questions of whether heat shock proteins directly engage heat-induced condensates, and whether and how functional engagement differs between adaptive condensates and aggregates of model misfolded substrates, have remained unanswered.
Here, we address these major open questions by reconstituting in vitro the dispersal of heat-induced Pab1 condensates by their cognate disaggregation system. Comparative studies of Pab1 condensates and aggregates of misfolded luciferase reveal four key differences. First, and most strikingly, chaperones which show slow and incomplete dispersal of luciferase aggregates disperse Pab1 condensates rapidly and completely. Second, unlike luciferase (Cashikar et al., 2005), Pab1 does not require co-condensation with small heat shock protein Hsp26 for efficient dispersal. Third, unlike luciferase for which type I (Ydj1) and type II (Sis1) Hsp40 show synergistic activity (Nillegoda et al., 2015, 2017), Pab1 condensate dispersal depends only on Sis1 and is antagonized by Ydj1. Fourth, we show that unlike luciferase, Pab1 is only partially threaded by Hsp104 and readily regains its function upon dispersal.
Finally, we investigate the dispersal system’s puzzling dependence on excess Hsp70 for optimal activity, which we find also applies to Pab1 condensate dispersal. Using a kinetic model, we show that the requirement of multiple accessible Hsp70s for Hsp104 recruitment and activation suffices to render the disaggregation system sensitive to the relative Hsp70 level.
Our results establish heat-induced biomolecular condensates of Pab1 as direct endogenous substrates of the disaggregation system, and reveal that conclusions drawn from studying heat-induced aggregates of “model” misfolded proteins do not generalize to these endogenous condensates. Further study of how chaperones engage with these and other adaptive, endogenous substrates, and how this engagement differs from foreign or proteotoxic substrates, appears likely to yield substantial insights into the mechanistic features and biological roles of this ancient molecular system.
Results
Heat shock causes Pab1 condensation, which is not spontaneously reversible
In budding yeast, Pab1 forms RNase-resistant, sedimentable condensates after heat shock (Wallace et al., 2015; Riback, Katanski et al., 2017). Condensates of mature, endogenous proteins disperse to their pre-stress soluble form within an hour, without degradation, as cells recover from stress (Wallace et al., 2015; Cherkasov et al., 2013).
Consistent with previous results, 20 minutes of heat shock at 42°C caused a roughly six-fold increase in the proportion of RNase resistant, large sedimentable Pab1 (P8) compared to the pre-shock level (Figure 1A–C). This fraction decreased as the cells recovered at 30°C and reached the pre-shock level by 60 minutes. Pab1 is not significantly induced during heat shock, and cells recovered in the presence of cycloheximide (CHX) to block new protein synthesis still solubilized most of Pab1 condensates by 60 minutes, with some reduction likely due to suppression of chaperone synthesis by CHX (Figure S1D–E). These results confirm that under these conditions Pab1 rapidly forms stable RNase-resistant condensates in vivo.
Figure 1. Heat shock causes Pab1 condensation, which is not spontaneously reversible.
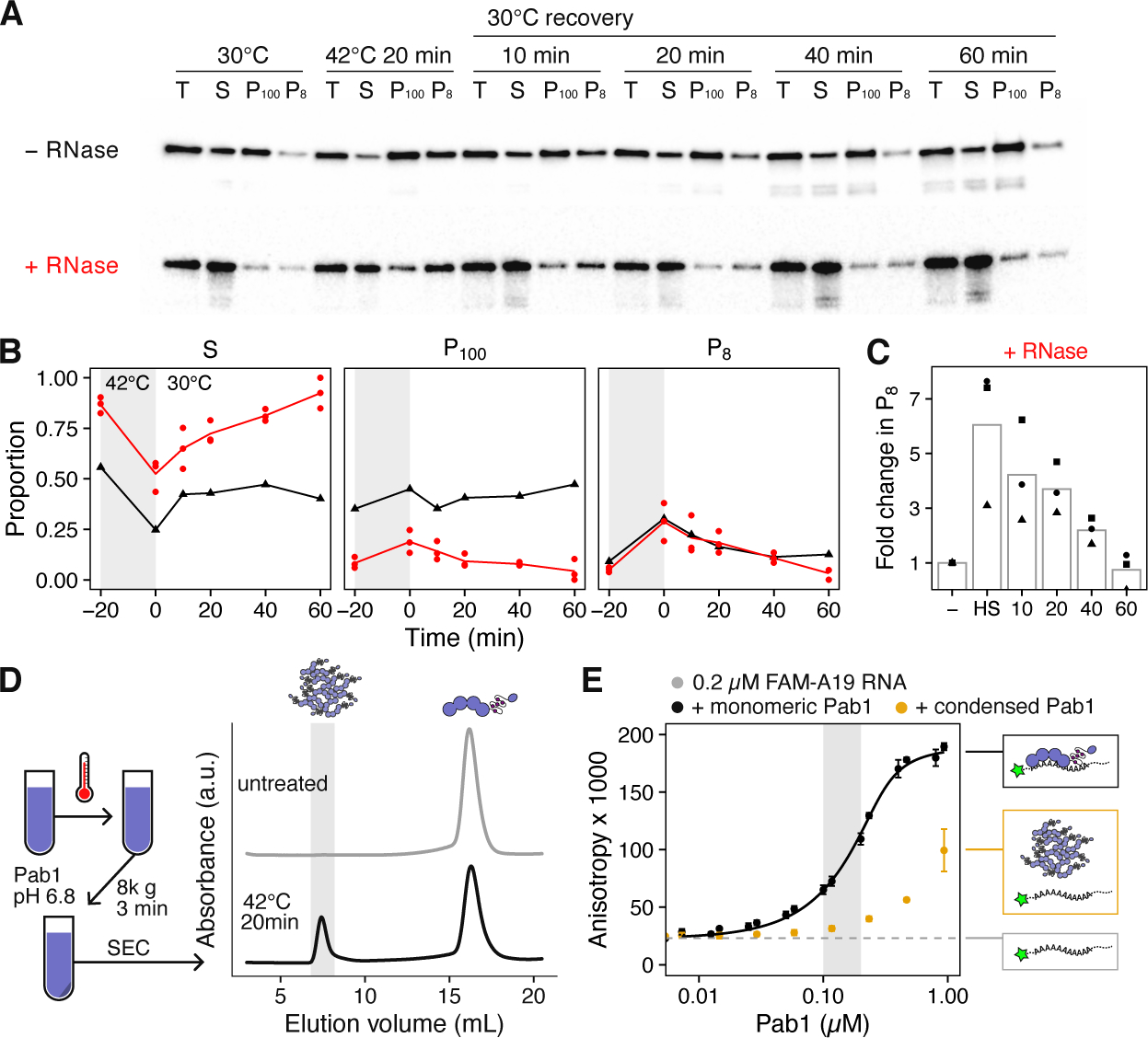
(A) Western blot against Pab1 before and after heat shock, and during recovery. Cell lysates were incubated with or without RNase and centrifuged to separate the supernatant (S) from the pellet (P8 and P100). (B) Mean fold change in P8, normalized to pre-stress level. Red and black colors correspond to the RNase- or mock-treated sample, respectively. (C) Relative change in the fraction of RNase resistant, large sedimentable Pab1 compared to pre-shock level. Shapes represent different biological replicates. (D) Schematic description of in vitro Pab1 condensate preparation and the representative SEC traces for untreated and heat shocked Pab1. Pab1 condensates elute in the void volume shaded in gray. (E) Fluorescence anisotropy of labeled A19 RNA in the presence of increasing amount of either monomeric or condensed Pab1. The range of condensate concentrations used in in vitro dispersal assays are shaded in gray. Data from at least two independent experiments were used to compute mean and SD.
To reconstitute Pab1 condensates in vitro, we treated purified Pab1 at 42°C for 20 minutes at pH 6.8, which is approximately the pH of the yeast cytoplasm during the same heat shock (Triandafillou et al., 2020) (Figure 1D). Pab1 condensates formed in vitro by heat shock or pH drop remained stable and did not dissolve after dilution in a high-salt buffer (Figure S1F). When analyzed by size-exclusion chromatography (SEC), about 25% of heat shocked recombinant Pab1 eluted in the void volume, corresponding to Pab1 condensates larger than 5,000 kDa (Figure 1D). Because a previous study indicated that misfolded proteins can nucleate stress granule formation in vivo (Kroschwald et al., 2015), we tested whether heat-shocking Pab1 in the presence of thermolabile firefly luciferase can further promote condensation. Indeed, we found that the presence of 100-fold lower amount of luciferase, but not thermostable BSA, allows Pab1 condensation at a lower temperature and also increases the condensation yield by about two-fold (Figure S2A–C).
Condensation requires Pab1’s folded RNA recognition motifs (RRMs) and excess RNA inhibits Pab1 condensation in vitro (Riback, Katanski et al., 2017), suggesting a competition between Pab1’s condensation and RNA-binding activity. Consistent with this, heat shock reduces the association of Pab1 with RNA in vivo (Bresson et al., 2020). We measured the RNA-binding capacity of Pab1 condensates isolated from SEC using fluorescence anisotropy and electrophoretic mobility shift assay (EMSA). For 1:1 binding of Pab1 to RNA, we used fluorescently labeled 19-mer poly(A) RNA (A19). Pab1 condensates showed specific but significantly reduced binding to A19 compared to monomers (Figure 1E and Figures S2D–E). The condensates remained stable at neutral pH and room temperature, consistent with a requirement for cellular disaggregation machinery as repeatedly indirectly demonstrated.
Hsp104, Hsp70, and type II Hsp40 are necessary and sufficient for rapid, complete dispersal of Pab1 condensates in vitro
To monitor the dispersal Pab1 condensates into functional monomers, we developed a fluorescence anisotropy-based assay in which the increase in fluorescence anisotropy of A19 indicates RNA binding by Pab1 monomers (Figure 2A). We mixed Pab1 condensates and A19 with molecular chaperones Hsp104, Ssa2 (Hsp70), Ydj1 and Sis1 (type I and II Hsp40, respectively), and Sse1 (Hsp110). The concentrations of Pab1 condensates and A19 were chosen to maximize the assay’s sensitivity to Pab1 monomers without saturating the signal (Figure 1E). In the absence of ATP, no change in fluorescence anisotropy was observed. In contrast, in the presence of ATP, fluorescence anisotropy quickly increased and reached a plateau after about 5 minutes, marking the completion of Pab1 dispersal (Figure 2A).
Figure 2. Hsp104, Hsp70, and Sis1 are necessary and sufficient for complete dispersal of Pab1 condensates in vitro.

(A) Schematic representation of Pab1 condensate, A19 RNA, and chaperones used in the fluorescence anisotropy assay. A representative data fitted to the logistic equation is shown on the right. (B) Fluorescence anisotropy of A19 in the presence of Pab1 condensates and chaperones. Experiments shown in the top row included all chaperones shown in (A) except the component specified at the top of each column. The same experiment repeated in the absence of Sse1 is shown in the bottom row. Fitted data from two independent experiments are shown. Concentrations of chaperones are indicated on the right. Hsp40 and Hsp104 concentrations are for dimers and hexamers, respectively. (C) Western blot of Pab1 after sedimentation. Hsp104 is present at a low level and does not show up in the total protein gel shown at the bottom. CK stands for creatine kinase. Asterisk indicates unknown contaminant. (D) Quantification of Pab1 sedimentation results. Mean and SD were computed from at least three experiments. (E) FSEC profiles of Pab1-Clover. The dashed lines, from left to right, mark the peaks corresponding to condensed, RNA-bound monomeric, and free monomeric Pab1-Clover.
We next tested which set of molecular chaperones are necessary and sufficient for complete Pab1 dispersal in vitro (Figure 2B–C). Condensate dispersal in the absence of Sse1 absolutely required ATP, Ssa2, Sis1, and Hsp104 with both substrate-binding pore-loop tyrosine residues (Figure 2B and Figure S3A). Condensates formed in the presence of luciferase were dispersed by chaperones as well as homogeneous condensates (Figure S2F). Interestingly, in the former case, chaperones also refolded luciferase but luciferase refolding continued after the completion of Pab1 condensate dispersal (Figure S2F–G). When Sse1 was added, the rate of Pab1 condensate dispersal increased by 2.5-fold (Figure S3B) and removal of Hsp104 led to only partial dispersal of condensates (Figure 2B). These results are consistent with the weak disaggregation activity of Hsp110/70/40 observed against amorphous aggregates and amyloid fibrils (Shorter, 2011), and with continued albeit strongly delayed dispersal of Pab1 condensates in vivo in cells lacking Hsp104 (Cherkasov et al., 2013).
The same chaperone requirement pattern was observed when we repeated the assay with Ssa1 and Ssa4, which are respectively the constitutively expressed and heat-inducible paralogs of Ssa2 (Figure S3C–G). The overall dispersal rate was slower with Ssa4 than with Ssa2 (Figure S3C), which is consistent with the weaker activity of stress-inducible human Hsp70 observed against amyloid fibrils (Gao et al., 2015; Scior et al., 2018).
We verified our fluorescence anisotropy results using two independent methods. First, we examined solubilization of Pab1 using sedimentation. About half of Pab1 condensates isolated from SEC were too small to pellet, but incubating Pab1 condensates with the minimal disaggregation system (Hsp104, Ssa2, Sis1) completely solubilized Pab1 (Figure 2C–D). In contrast, Pab1 solubility remained unchanged from background levels when the condensates were incubated with an incomplete disaggregation system (Figure 2C–D).
Next, we prepared Pab1-Clover condensates and examined their size distribution by fluorescence-detection SEC (FSEC). Pab1-Clover condensates remained stable when incubated for an hour at 30°C in the absence of a complete disaggregation system (Figure 2E). After incubation with the complete disaggregation system, however, the condensate peak disappeared and a new peak corresponding to RNA-bound Pab1-Clover appeared. A similar experiment performed with unlabeled Pab1 using SEC and western blot confirmed these results (Figure S3E–G).
In summary, the results from three independent methods consistently indicate that Hsp104, Hsp70, and type II Hsp40 Sis1 are necessary and sufficient for rapid, complete dispersal of Pab1 condensates in vitro. These results are consistent with the in vivo observations that deletion or inhibition of Hsp104, Hsp70, or Hsp40 delays dispersal of stress granules marked by Pab1 (Cherkasov et al., 2013; Walters et al., 2015; Kroschwald et al., 2015, 2018).
Pab1 condensates and misfolded protein aggregates exhibit different chaperone dependence for dispersal
Substrate dispersal by the disaggregation system has been extensively studied using non-native model substrates such as firefly luciferase, which readily misfolds in vitro at elevated temperatures and whose light-producing enzymatic activity offers a precise measure of the extent of protein refolding. To determine whether and how Pab1 condensate dispersal differs from luciferase aggregate disaggregation, we first focused on two chaperone-related features of luciferase disaggregation which we could recapitulate: the dependence of dispersal on small heat shock protein Hsp26, and on type I and type II Hsp40s.
As reported previously, co-aggregation with excess Hsp26 promotes luciferase disaggregation and reactivation by several-fold (Cashikar et al., 2005) (Figure 3A). To investigate the effect of Hsp26 on Pab1, we subjected Pab1 to a more severe heat shock (46°C for 20 minutes at pH 6.4) in the absence or presence of increasing concentrations of Hsp26. Hsp26 suppressed Pab1 condensation and sedimentation in a concentration-dependent manner (Figure S4A–B). Dynamic light scattering (DLS) also revealed that Hsp26 suppresses nucleation of Pab1 (Figure S4C–D). When added after Pab1 condensation, free Hsp26 neither affected Pab1’s RNA binding activity nor further accelerated the dispersal rate (Figure S4E–G). Thus, unlike luciferase which readily co-aggregates with Hsp26 upon heat shock, heat-induced Pab1 condensation is suppressed by Hsp26. Most importantly, unlike luciferase aggregates, Pab1 condensates formed in the absence of Hsp26 are rapidly and completely dispersed by the disaggregation system (Figure 2).
Figure 3. Pab1 condensates and misfolded protein aggregates exhibit different chaperone dependence for dispersal.

(A) Fraction of reactivated luciferase after a two-hour incubation of luciferase aggregates, formed in the absence or presence of Hsp26, with the disaggregation system. (B) Reactivation kinetics of aggregated luciferase. Mean and SD were calculated from two independent experiments with duplicates in each experiment. (C) Pab1 dispersal using either Sis1 (black) or a combination of Sis1 and Ydj1 (blue) as co-chaperones. (D) Titration of either Sis1 (left) or Ydj1 (right) to reactions containing 0.2 μM Pab1 condensates, 0.05 μM Hsp104, 0.5 μM Ssa2, and 0.25 μM Sis1. The amount of additional Hsp40 added was 0.25, 0.5, 1, and 2 μM. (E) The average rate of dispersal and SD quantified from three independent titration experiments, one of which is shown in (D). (F) Reactivation kinetics of Pab1 condensates or luciferase aggregates. The inset shows zoomed-in refolding kinetics of luciferase over three hours.
We confirmed that type I and II Hsp40s, Ydj1 and Sis1, act synergistically in luciferase aggregate dispersal (Nillegoda et al., 2015, 2017) (Figure 3B). We next investigated whether Pab1 dispersal is accelerated in the presence of both Ydj1 and Sis1. Ydj1 is a type I Hsp40 which has a highly conserved N-terminal J domain followed by a G/F-rich region, a zinc-finger domain, C-terminal domains, and a dimerization domain (Kampinga and Craig, 2010). Type II Sis1 largely resembles the architecture of Ydj1 but lacks the zinc-finger domain. The rate of dispersal did not improve when both Sis1 and Ydj1 were added to Pab1 condensates compared to when only Sis1 was added (Figure 3C). Instead, Ydj1 slightly inhibited Pab1 dispersal in a concentration-dependent manner (Figure 3D–E). These results indicate that, unlike luciferase aggregates for which Sis1 and Ydj1 show synergistic activity, Sis1 and Yd1j show antagonistic activity for Pab1 condensates.
The disaggregation system restores Pab1 condensates far more efficiently than misfolded protein aggregates
The poor activity of the disaggregation system against aggregates of model substrates has been observed since the first biochemical reconstitution of the system (Glover and Lindquist, 1998; Goloubinoff et al., 1999). Even with co-aggregation with five-fold excess Hsp26, less than half of luciferase activity is regained after a two-hour incubation with 37.5-fold excess Hsp104 and Ssa2 (Figure 3B). Indeed, the standard in vitro disaggregation protocol requires the use of 10- to 100-fold excess molecular chaperones over substrates to obtain moderate to good yield (Figure 6C and Table S1). We found that when sub-stoichiometric concentration of Hsp104 and closer to stoichiometric concentrations of Ssa2 and Sis1 are used, Pab1 dispersal still completes within 20 minutes while less than 1% of luciferase is reactivated after an hour (Figure 3F).
Figure 6. Cooperative binding of Hsp70 labels condensates for disaggregation.

(A) Rate of Pab1 dispersal in the presence (black) or absence (orange) of Sse1. Solid lines in Hsp104, Sis1, and Ssa2 panels represent logistic fit to the data. Solid line in Sse1 panel is the smoothing line. The baseline concentrations of the chaperones are indicated on the right. (B) Relative chaperone concentration at half-maximal dispersal rate. Standard errors around the estimated parameter are shown. (C) Summary of disaggregation data in the literature. Maximal yield of disaggregation experiments and the relative amount of Hsp104 and Hsp70 used are shown. Color and size of each data point correspond to the yield. Circles represent studies with wild-type Hsp104. The one square data point in the sub-stoichiometric Hsp70 area is from a study using a hyperactive variant of Hsp104. (D) Schematic comparison of the cooperative and non-cooperative models. (E) Pab1 dispersal monitored by fluorescence anisotropy (left) and the simulated Pab1 dispersal results from the cooperative model (right). (F) Quantitative comparison of Pab1 dispersal data (black) to the cooperative (blue) and non-cooperative (orange) model simulation results. Fraction restored at the end of the experiment (left) and the rate of dispersal (right) were used for comparison. (G) Representative Sse1 titration Pab1 dispersal data. (H) Simulated Pab1 condensate dispersal with different ADP nucleotide exchange rates.
What causes this large difference in restoration efficiency between Pab1 and luciferase? To gain insight into the potential sources of this discrepancy, we turned to computational kinetic modeling.
Higher disaggregation rate and partition coefficient lead to more efficient substrate restoration in silico
We synthesized existing simulation studies (Powers et al., 2012; De Los Rios and Barducci, 2014; Nguyen et al., 2017; Xu, 2018; Goloubinoff et al., 2018; Assenza et al., 2019; Wentink et al., 2020) to build what we call a cooperative model of the disaggregation system (Figure 4A and Figure S5A). The cooperative model captures the current model of Hsp104 regulation by Hsp70, in which binding of more than one Hsp70 is required to activate Hsp104 (Seyffer et al., 2012; Carroni et al., 2014). Many of the rate parameters involved have been measured using bacterial chaperones and model substrates or peptides (Table S2). We assumed that these parameters are generally consistent in the eukaryotic system, and that the same model architecture can be used for both luciferase and Pab1. For details of this ordinary differential equation model, see Methods.
Figure 4. Higher disaggregation rate and folding partition coefficient lead to more efficient substrate restoration in silico.

(A) Cooperative model of the disaggregation system. For more details, see Methods and Figure S5A. (B) Summary of model output in terms of fraction substrate restored at two hours as a function of the partition coefficient. Dashed lines indicate the partition coefficient used to simulate Pab1 (black) and luciferase (light blue) results in (C). (C) Simulated substrate dispersal kinetics with either high (black) or low (light blue) partition coefficients. Simulation results (solid line) are overlaid on top of Pab1 and luciferase experimental data from Figure 3F. (D) Simulated fraction substrate restored as a function of disaggregation rate. (E) Simulated fraction substrate restored as a function of Hsp70(ATP) substrate affinity. Dashed lines in (D) and (E) indicate the default value used in the simulation experiments.
We examined how varying each of the following parameters affected the substrate restoration yield: 1) rate of disaggregation by Hsp104, 2) efficiency of the released substrate from regaining its native structure, which we define as the partition coefficient, and 3) substrate affinity for Hsp70. Modulation of each parameter over 1–2 orders of magnitude substantially affected the restoration yield, measured from 0 to 1 (Figure 4B, D, E). A large difference in partition coefficient alone reproduced the Pab1 and luciferase dispersal data (Figure 4C). The simulation also revealed Hsp70 affinity as a potential factor which can contribute to the observed difference in dispersal efficiency.
Because Hsp104 is associated with both the disaggregation rate and the partition coefficient of a substrate, e.g., through complete threading vs. partial threading of a substrate, we decided to experimentally investigate and compare how Hsp104 engages with Pab1 condensates and luciferase aggregates.
Pab1 is partially threaded by Hsp104
Substrate threading through the central channel of Hsp104 is a common mechanism for protein unfolding and disaggregation (Tessarz et al., 2008). To probe the folding state of Pab1 and luciferase during their release from Hsp104, we first attempted to use a mutated version of a bacterial chaperonin, GroEL-D87K, which traps unfolded protein (Weber-Ban et al., 1999; Siegers et al., 1999; Mogk et al., 2003; Motojima et al., 2012) (Figure 5A).
Figure 5. Pab1 is partially threaded by Hsp104.
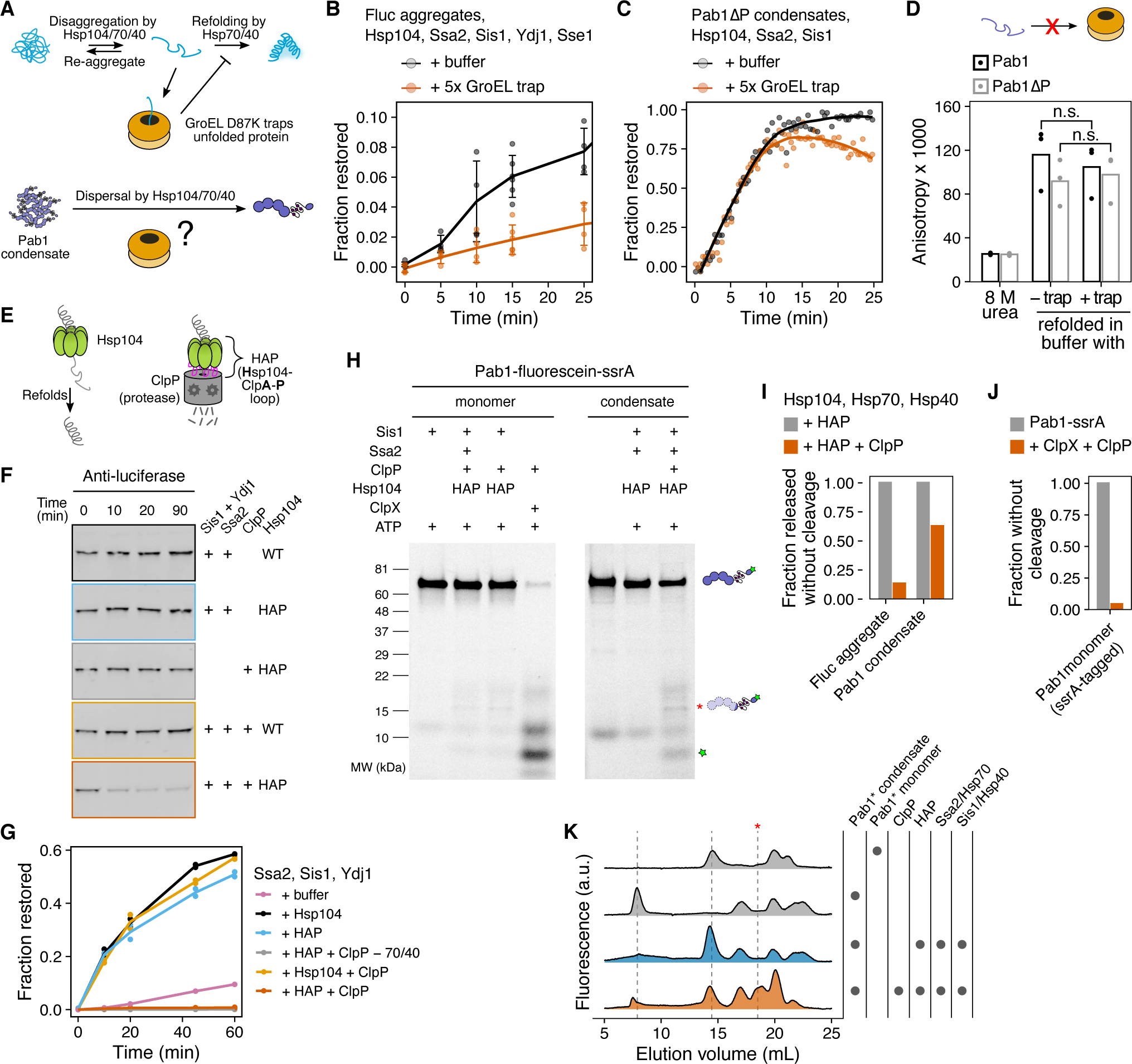
(A) Schematic description of GroEL trap system. (B) Luciferase disaggregation in the absence or presence of excess GroEL trap. Mean and SD were calculated from three independent experiments. (C) Pab1ΔP dispersal in the absence or presence of excess GroEL trap. Solid lines represent smoothed data. The decrease in the signal in the presence of GroEL trap after 10 min is due to RNA degradation. (D) Refolding of urea-denatured Pab1 (black) and Pab1ΔP (gray) in buffer containing no or 10-fold excess GroEL trap. (E) Schematic description of HAP/ClpP system. (F) Chemically aggregated luciferase was incubated with the indicated components and the extent of luciferase degradation was visualized by western blot. (G) Refolding kinetics of chemically aggregated luciferase. (H) SDS-PAGE gels of Pab1-fluorescein-ssrA monomers or condensates after an hour incubation with the indicated components. Asterisk indicates HAP/ClpP-specific band. (I) Fraction of full-length protein released from luciferase aggregates or Pab1 condensates, normalized to HAP control. The restoration yield of 60% was used for both luciferase and labeled Pab1 based on the disaggregation and FSEC results, respectively. (J) Fraction of full-length Pab1-ssrA monomers after incubation with ClpX and ClpP. (K) FSEC traces of Pab1-fluorescein-ssrA condensates after an hour incubation with the indicated components. Dashed lines indicate peaks corresponding to Pab1 condensates (8 mL), full-length monomers (14.5 mL), and HAP/ClpP-specific C-terminal fragment (18.5 mL). Unmarked peaks in the control reactions are unknown contaminants.
Although GroEL trap prevented folding of luciferase after disaggregation (Figure 5B), the trap did not engage with full-length Pab1 (64 kDa) nor Pab1ΔP (55 kDa) even after full denaturation by urea (Figure 5C–D), indicating that this well-used system is not suitable for investigating Pab1. Pab1ΔP lacks the disordered P domain but retains the ability to condense (Riback, Katanski et al., 2017). Regardless of GroEL trap presence, Pab1ΔP condensates were still readily dispersed by the disaggregation system, although the rate of dispersal was slightly lower compared to full-length Pab1 condensates (Figures S6J–K).
To circumvent the limitation of the GroEL trap, we turned to the HAP/ClpP system (Tessarz et al., 2008). HAP (Hsp104-ClpA-P loop) is an engineered Hsp104 that interacts with the bacterial peptidase ClpP to form a proteolytic system (Figure 5E). HAP behaved like wild-type Hsp104 and quickly degraded luciferase in the presence of ClpP (Figures 5F–G), consistent with a previous report that luciferase is fully threaded and degraded by HAP/ClpP (Haslberger et al., 2008). In the absence of ClpP, HAP also dispersed Pab1 condensates without degradation like wild-type Hsp104 (Figures S6A–B).
If complete threading of Pab1 were required for condensate dispersal, we would expect to see complete degradation of Pab1 by HAP/ClpP. A control experiment using Pab1-fluorescein tagged with the ssrA degradation tag and ClpX, which recognizes the ssrA tag and unfolds the substrate for ClpP, confirmed that ClpP can degrade Pab1 (Figure 5H and Figure 5J). We made condensates using this Pab1 construct and examined the degradation pattern after dispersal with HAP/ClpP using SDS-PAGE (Figure 5H and Figures S6C–D) and FSEC (Figure 5K). A mixed group of full-length and degraded Pab1 populations were observed after dispersal. The appearance of full-length Pab1 monomers suggested partial translocation of Pab1 by HAP and release before Pab1 enters the proteolytic chamber of ClpP (Figure 5I and Figure 5K). Specific degradation fragments appeared upon incubation of HAP/ClpP and chaperones with Pab1 condensates in an ATP-dependent manner, but also to a lesser extent with Pab1 monomers, suggesting a basal level of interaction between Pab1 and HAP/ClpP (Figure 5H and Figure S6I). Similar C-terminal fragments containing a part of the P domain and the C-terminal domain of Pab1 appeared for Pab1-Clover and Pab1-fluorescein without the ssrA tag (Figure S6E and Figure S6H). However, much less full-length monomer appeared for Pab1-Clover than Pab1-fluorescein-ssrA (compare Figure 5K with Figure S6F), suggesting that a fluorescent label can affect the processing by HAP/ClpP. We also examined C-terminal fragments of Pab1ΔP-fluorescein and N-terminal fragments of fluorescein-Pab1 and fluorescein-Pab1ΔP (Figures S6G and S6L). HAP/ClpP-specific fragments were observed for fluorescein-Pab1 and Pab1ΔP-fluorescein while fluorescein-Pab1ΔP showed a smear indicative of continued translocation and degradation.
Together, these results show that, unlike luciferase which requires complete threading and unfolding by HAP for disaggregation, partial threading of Pab1 still leads to condensate dispersal. This is consistent with the partial threading mechanism proposed for proteins with a mixture of misfolded and folded domains (Haslberger et al., 2008; Sweeny et al., 2015) and the lack of major secondary structure changes in Pab1 at condensation temperature (Riback, Katanski et al., 2017).
Cooperative binding of Hsp70 targets condensates for dispersal
How does the disaggregation system recognize its substrates? To address this question, we first performed a series of fluorescence anisotropy Pab1 dispersal assays with varying chaperone concentrations and quantified the rate of dispersal (Figures 6A–B). Pab1 condensate dispersal was most robust to the Hsp104 concentration, showing half-maximal dispersal rate at 1:10 Hsp104:Pab1 ratio. Excess Sse1 was inhibitory and Sse1 worked most optimally at sub-stoichiometric level, consistent with previous observations (Kaimal et al., 2017; Wentink et al., 2020). Pab1 condensate dispersal was most sensitive to the concentrations of Sis1 and Ssa2. In particular, the rate of dispersal plummeted as the Ssa2 concentration approached the stoichiometric level (Figure 6A).
Indeed, a survey of in vitro disaggregation studies showed a consistent requirement for super-stoichiometric Hsp70 (Figure 6C), but the reason for this dependence has been a long-standing puzzle (Goloubinoff et al., 1999; Ben-Zvi et al., 2004).
We decided to investigate why excess Hsp70 over substrate is needed for what is still a catalytic series of reactions. We titrated Ssa2 over a narrow window around the stoichiometric Hsp70:Pab1 ratio and monitored Pab1 dispersal (Figure 6E). We also simulated the cooperative model (Figure 4A) using the same chaperone concentrations used in the in vitro experiment (Figure 6E). The cooperative model recapitulated the disaggregation system’s Hsp70-sensitive Pab1 dispersal activity (Figure 6F). This model reflects the results from recent studies which indicate that interaction with more than one Hsp70 is required for activation of Hsp104 (Carroni et al., 2014; Seyffer et al., 2012). Indeed, simulation of the non-cooperative model, in which a single Hsp70 is sufficient to recruit and activate Hsp104, resulted in high Pab1 dispersal activity even with sub-stoichiometric Hsp70 (Figure 6D and 6F).
The cooperative model was also able to recapitulate the general trend seen in the Hsp104 and Ssa2 titration experiments (Figure 6A and Figure S5B). Although Sis1 and Sse1 were not explicitly included in the model, modulating the ATP hydrolysis rate and ADP exchange rate allowed us to mimic the effect of titrating Sis1 and Sse1, respectively (Figure S5B). Interestingly, although we were able to recapitulate the inhibitory effect of Sse1 with high ADP exchange rate, modulating ADP exchange rate was not enough to recapitulate the facilitative effect of sub-stoichiometric Sse1 (Figures 6G–H).
These results converge on a picture in which the presence of multiple Hsp70 molecules accessible to a single Hsp104 molecule on the surface of condensates provide a molecular marker labeling condensates for Hsp104-dependent dispersal, as proposed in the bacterial disaggregation system by Seyffer et al. (2012). Our simulation results indicate that a cooperative Hsp70 effect on Hsp104 binding and activation suffices to explain the disaggregation system’s intrinsic sensitivity to the level of Hsp70 (Figure 7).
Figure 7. Model of condensate dispersal by the Hsp104/Hsp70/Hsp40 disaggregation system.

Super-stoichiometric levels of Hsp70, which should produce clusters of Hsp70 on condensate and aggregate surfaces, are required for rapid dispersal of both structures. Consistent with a previous hypothesis (Seyffer et al., 2012), we propose that Hsp70 clusters act as endogenous condensate markers and serve to activate the Hsp104 disaggregase machinery. Luciferase is fully threaded by Hsp104 and released as unfolded protein (Haslberger et al., 2008), resulting in slow and inefficient refolding because unfolded protein is more prone to misfolding and reaggregation. In contrast, Pab1 is released after partial threading by Hsp104 and readily refolds into native structure, resulting in faster and more efficient folding compared to luciferase. The schematic of Hsp104 with repressed (red) and activated (green) protomers is adapted from Carroni et al. (2014).
Discussion
Heat-induced biomolecular condensates are endogenous substrates of the molecular disaggregation system
In yeast, heat-induced biomolecular condensates including stress granules adopt a solid-like characteristic (Kroschwald et al., 2015; Riback, Katanski et al., 2017). Timely dispersal of endogenous condensates depends on molecular chaperones (Cherkasov et al., 2013; Walters et al., 2015; Kroschwald et al., 2015, 2018), and the timing of dispersal correlates with resumption of active cellular translation and growth (Cherkasov et al., 2013; Kroschwald et al., 2015, 2018). These in vivo observations strongly suggest heat-induced biomolecular condensates are the endogenous substrates of the molecular disaggregation system. However, direct biochemical evidence for chaperone-mediated condensate dispersal has been missing.
In this study, we show using in vitro reconstitution that the Hsp104/70/40 disaggregation system directly engages and disperses heat-induced condensates of Pab1. The contrast between Pab1 condensates and luciferase aggregates on multiple dimensions demonstrates that luciferase, and by extension other similarly behaving “model” misfolded proteins, have limitations as models of endogenous heat-induced chaperone substrates. Whether the authentic substrate Pab1 is itself a suitable model for other endogenous substrates remains an important open question.
We also show that Hsp110, Hsp70, and Hsp40 can disperse Pab1 condensates. This weaker alternative disaggregation system is conserved in animals (Shorter, 2011; Wentink et al., 2020), which lack cytosolic Hsp104 (Erives and Fassler, 2015), suggesting a potential evolutionary and biochemical bridge to chaperone-mediated condensate dispersal in animals.
Co-aggregation of luciferase with Hsp26 facilitates aggregate dispersal and refolding (Cashikar et al., 2005; Ungelenk et al., 2016; Żwirowski et al., 2017). However, Hsp26 is almost undetectable in cells pre-stress (Cashikar et al., 2005) and therefore unlikely to be involved in condensate regulation during the initial exposure to stress. We showed that Hsp26 prevents condensation of Pab1 in vitro, similar to suppression of FUS phase separation by human small heat shock protein Hsp27 (Liu et al., 2020). Post-stress accumulation of Hsp26 in cells may be involved in desensitization of the cells to sustained or repeated stress by modulating the phase boundary of endogenous mature proteins.
The canonical type I (Ydj1; DNAJA2 in humans) and type II Hsp40 (Sis1; DNAJB1) chaperones show synergistic activity toward luciferase aggregates (Nillegoda et al., 2015, 2017). This synergistic activity stems from the preference of type I and II Hsp40 chaperones for small (200–700 kDa) and large (>5,000 kDa) aggregates, respectively (Nillegoda et al., 2017), although how aggregates of different sizes can be distinguished at the molecular level remains unclear. Sis1 and Ydj1 also exhibit different amino acid sequence preference (Jiang et al., 2019) and different mode of binding Hsp70 (Faust et al., 2020). The inability of Ydj1 to support Pab1 dispersal could be due to many factors including the condensate size, the lack of Ydj1 binding sites among the exposed region of Pab1 in condensates, dependence on the Hsp70 binding mode unique to type II Hsp40, or any combination of these.
The antagonistic effect of Ydj1 on Sis1 suggests a competition between the two co-chaperones for Hsp70. In cells, stress-induced phosphorylation of Hsp70 can reprogram Hsp70’s Hsp40-binding specificity, for example, by preventing Hsp70 from interacting with Ydj1 (Truman et al., 2012). Because Ydj1 localizes to stress granules (Walters et al., 2015), Ydj1 may be involved in dispersal of other stress-granule constituents by engaging with a different pool of Hsp70 from Sis1. We anticipate that the in vitro system introduced here will substantially enable future work on how the disaggregation system’s substrate specificity is regulated.
Hsp104 functions by threading substrate through its central channel (Gates et al., 2017). However, our results indicate that Pab1 is partially threaded by Hsp104. Partial threading activity of Hsp104 and its bacterial homolog ClpB has been reported previously, where both proteins selectively thread misfolded moiety of the substrate while leaving the natively folded domains intact (Haslberger et al., 2008; Sweeny et al., 2015), and we now report this for an endogenous heat-induced eukaryotic substrate. The disordered P domain of Pab1 is dispensable for both condensation and dispersal. Thus, we hypothesize that partial threading of the locally unfolded region of Pab1, possibly the same or near the region mediating Pab1 condensation interaction, allows for condensate dispersal without substantial protein unfolding.
Hsp70 clusters are a potential condensate-specific marker for Hsp104
Efficient dispersal of Pab1 condensates depends on the presence of excess Hsp70. Nearby Hsp70s, which would be rare on monomers but common on condensates, both increase Hsp104 binding and stimulate additional Hsp104 activity. In this sense, consistent with an insightful proposal from Seyffer et al. (2012) working in the homologous bacterial system, Hsp70 clusters provide an active label for engagement and activation of dispersal machinery only in spatial proximity to condensed substrates.
Cooperative action of Hsp70 in substrate unfolding has been proposed to explain the requirement of excess Hsp70 during glucose-6-phosphate dehydrogenase (G6PDH) disaggregation (Ben-Zvi et al., 2004). We find by simulation that cooperative action of Hsp70 in the recruitment and activation of Hsp104 is sufficient to reproduce the in vitro Hsp70 titration data.
We also found that, in the cooperative model, modulating the ADP exchange rate alone was not enough to reproduce the facilitative effect of Sse1 (Figure 6H). A recent study by Wentink et al. (2020) uncovered an additional function of human Hsp110 in promoting local clustering of Hsp70 on the substrate surface. A similar function in the yeast Hsp110 may explain the discrepancy between our model and the data.
Biomolecular condensates in the cellular heat shock response
Engagement of Hsp70 with heat-induced biomolecular condensates provides a tangible means to explain how yeast cells sense temperature. The yeast transcriptional heat shock response is triggered when Hsp70 is titrated away from the transcription factor Hsf1 by heat-induced substrates (Zheng et al., 2016; Krakowiak et al., 2018; Peffer et al., 2019; Masser et al., 2019; Feder et al., 2021). Suppression of protein synthesis is not sufficient to suppress the transcriptional heat shock response, implying the existence of mature substrates which also depend on stress-associated intracellular acidification for formation (Triandafillou et al., 2020). Condensation of Pab1 and other heat-sensitive proteins is strongly pH-sensitive (Riback, Katanski et al., 2017; Iserman et al., 2020; Kroschwald et al., 2018). And here we show that heat-induced condensates of Pab1 are authentic chaperone substrates which depend on Hsp70 for dispersal. In short, Pab1—and by extension presumably others of the dozens of previously identified heat-condensing proteins (Wallace et al., 2015; Cherkasov et al., 2015), including more than a dozen which condense in response to a 37°C heat shock—now appears to have all the characteristics needed to act as an inducer of the transcriptional heat shock response.
Molecular chaperones as biomolecular condensate remodelers
In this work, we show that molecular chaperones can regulate biomolecular condensates by acting as dispersal factors. This expands the list of known condensate dispersal factors, which currently includes the dual-specificity kinase DYRK3 (Wippich et al., 2013) and nuclear-import receptor karyopherin-β2 (Guo et al., 2018). The functional repertoire of molecular chaperones in biomolecular condensate regulation is likely to be much broader than just dispersal. For example, Hsp104, Hsp70, and Hsp40 in yeast are required for condensate formation of SNF1 kinase activator Std1 during fermentation (Simpson-Lavy et al., 2017). Illumination of the roles of molecular chaperones as facilitators, remodelers, and dispersers of biomolecular condensates—and the mechanisms and biological consequences of this regulation—presents an enormous opportunity for expanding our understanding of these ancient molecules.
Limitations of the study
It remains to be determined to what extent Pab1 condensates we use in in vitro experiments fully recapitulate submicroscopic condensates that form in vivo. We use the recovery of Pab1’s RNA-binding activity as a readout for the release of functional Pab1 from condensates, and have not determined to what extent other known functions of Pab1 are restored after condensate dispersal. In addition, as with many studies of stress-induced condensation, relevant phenotypes are an important unrealized goal.
In the molecular engagement of Pab1 with the disaggregation machinery, the lack of a consistent fragmentation pattern from different labeled Pab1 condensates after dispersal by HAP/ClpP limits our ability to identify the precise stopping point for Pab1 threading. We also cannot rule out the possibility that wild-type Hsp104 processes unlabeled Pab1 differently from what we observe with HAP and labeled Pab1 constructs.
Our kinetic model relies on two key assumptions: 1) the parameters measured using bacterial chaperones and model misfolded proteins and peptides are generally consistent with the yeast system, and 2) the same model architecture can be applied for both luciferase aggregates and Pab1 condensates. Although Hsp70 from bacteria and yeast exhibit similar affinity towards model peptides (Xu et al., 2012), more studies will be necessary to carefully examine the validity of these assumptions. The model was designed to be minimal. Some of the missing interactions, such as the potential role of yeast Hsp110 in promoting Hsp70 clustering (Wentink et al., 2020) and the role of Hsp70 in triggering substrate release from Hsp104 (Durie et al., 2018), may become important in certain conditions and the model architecture may need to be modified in those cases. An extension of this model allowing spatial segregation of aggregated substrates would allow investigating the effect of aggregate shape or size on the dispersal efficiency. Lastly, instead of relying on parameters derived from external studies, the model can be fitted directly to experimental data to estimate parameters (Hong et al., 2020).
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, D. Allan Drummond (dadrummond@uchicago.edu).
Materials Availability
All unique and stable reagents generated in this study are available upon request.
Data and Code Availability
All raw and processed data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOI is listed in the key resources table.
All custom scripts for data processing, analysis, and figure generation have been deposited at Mendeley Data and are publicly available as of the date of publication. The original python code for simulation is available on GitHub. The git address and DOI are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Pab1p | EnCor Biotechnology | Cat#MCA-1G1; RRID: AB_2572370; Lot#020407 |
| Mouse monoclonal anti-Pgk1 | Thermo Fisher | Cat#459250 |
| Mouse monoclonal anti-GFP | Proteintech | Cat#66002-1-Ig |
| Rabbit polyclonal anti-GFP | Thermo Fisher | Cat#A-11122 |
| Rabbit polyclonal anti-luciferase | MilliporeSigma | Cat#L0159 |
| IRDye 800CW Donkey anti-rabbit IgG | LI-COR | Cat#92532213 |
| IRDye 800CW Donkey anti-mouse IgG | LI-COR | Cat#92532212 |
| Bacterial and virus strains | ||
| Subcloning Efficiency™ DH5α Competent Cells | Thermo Fisher | Cat#18265017 |
| BL21(DE3) Competent Cells | MilliporeSigma | Cat#69450 |
| Chemicals, peptides, and recombinant proteins | ||
| SUPERase·In™ RNase Inhibitor | Thermo Fisher | Cat#AM2694 |
| Creatine phosphokinase | MilliporeSigma | Cat#C3755 |
| Creatine phosphate | GoldBio | Cat#C-323 |
| Protease Inhibitor Cocktail Set IV | MilliporeSigma | Cat#539136 |
| Pierce™ Protease Inhibitor Tablets, EDTA-free | Thermo Fisher | Cat#A32965 |
| Polyadenylic acid (Poly(A)) | MilliporeSigma | Cat#3 |
| RNase If | NEB | Cat#M0243S |
| 5-FAM-HHHHHHLPETGG peptide | Biomatik | custom |
| GGGK(FAM) peptide | Biomatik | custom |
| GGGK(FAM)AANDENYALAA peptide | Biomatik | custom |
| BSA | MilliporeSigma | Cat#12659 |
| Critical commercial assays | ||
| ONE-HOUR Western™ Basic Kit (Mouse) | GenScript | Cat#L00205 |
| Clarity™ Western ECL Substrate | Bio-Rad | Cat#1705060 |
| Luciferase assay system | Promega | Cat#E1500 |
| Deposited data | ||
| Genome of S288C (SGD: R64-2-1) | Saccharomyces Genome Database | https://www.yeastgenome.org/ |
| Raw and processed experimental data | This paper | doi:10.17632/wvztspxbbd.1 |
| Simulation data | This paper | doi:10.17632/wvztspxbbd.1 |
| Experimental models: Organisms/strains | ||
| Saccharomyces cerevisiae strain BY4741: background strain S288C; genotype: MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | ATCC; Brachmann et al., 1998 | ATCC 201388 |
| BY4741 yGFP-luciferase:URA | Cherkasov et al., 2013 | VCY245 |
| BY4741 Δhsp104 yGFP-luciferase:URA | Cherkasov et al., 2013 | VCY325 |
| Oligonucleotides | ||
| Fluorescein-A19 RNA: 5’ 6-FAM-AAAAAAAAAAAAAAAAAAA | IDT | N/A |
| Atto550-A19 RNA: 5’ ATTO550-AAAAAAAAAAAAAAAAAAA | IDT | N/A |
| Kozak RNA: 5’ 6-FAM-ACCUCUGCCGCCGCCAUGG | IDT | N/A |
| Recombinant DNA | ||
| Plasmid: 6xHis-(TevC)-Pab1-clover in pET28a backbone | Riback and Katanski et al., 2017 | pESN07 |
| Plasmid: 8xHis-(TevC)-Pab1 in pET28a backbone | Riback and Katanski et al., 2017 | pJAR006 |
| Plasmid: 8xHis-(TevC)-Pab1ΔP in pET28a backbone | Riback and Katanski et al., 2017 | pJAR015 |
| Plasmid: 6xHis-(SUMO)-Ssa2 in pET28a backbone | This manuscript | pHY024 |
| Plasmid: 6xHis-(SUMO)-Ssa4 in pET28a backbone | This manuscript | pHY025 |
| Plasmid: GroEL D87K in pDMI.1 backbone | Axel Mogk; Weibezahn et al., 2003 | N/A |
| Plasmid: 6xHis-(SUMO)-GroEL D87K in pET28a backbone | This manuscript | pHY044 |
| Plasmid: 6xHis-(SUMO)-Photinus pyralis Luciferase in pET28a backbone | This manuscript | pHY045 |
| Plasmid: 6xHis-(SUMO)-Ssa1 in pET28a backbone | This manuscript | pEP051 |
| Plasmid: 6xHis-(SUMO)-Sse1 in pET28a backbone | This manuscript | pEP053 |
| Plasmid: 6xHis-(SUMO)-Hsp104 in pET28a backbone | This manuscript | pEP055 |
| Plasmid: 6xHis-(SUMO)-Ydj1 in pET28a backbone | This manuscript | pEP056 |
| Plasmid: 6xHis-(SUMO)-Sis1 in pET28a backbone | This manuscript | pEP057 |
| Plasmid: 6xHis-(SUMO)-Hsp26 in pET28a backbone | This manuscript | pEP065 |
| Plasmid: ClpP-6xHIS in pQE70 backbone | Martin et al., 2005 | N/A |
| Plasmid: 6xHIS-ClpXΔN- ClpXΔN- ClpXΔN | Martin et al., 2005 | N/A |
| Plasmid: 6xHis-(SUMO)-Hsp104(G739I S740G K741F T746A) in pET28a backbone | This manuscript | pJB548 |
| Plasmid: 6xHis-(SUMO)-Hsp104 Y257A in pET28a backbone | This manuscript | pJB551 |
| Plasmid: 6xHis-(SUMO)-Hsp104 Y662A in pET28a backbone | This manuscript | pJB552 |
| Plasmid: srtA-6xHIS in pET28a backbone | Guimaraes et al. 2013 | N/A |
| Plasmid: srtAheptamutant-6xHIS in pET30b backbone | Hirakawa et al., 2015 | N/A |
| Plasmid: 8xHis-(TevC)-GG-Pab1 in pET28a backbone | This manuscript | pJB545 |
| Plasmid: 8xHis-(TevC)-Pab1-srtA-strep in pET28a backbone | This manuscript | pJB547 |
| Plasmid: 8xHis-(TevC)-GG-Pab1ΔP in pET28a backbone | This manuscript | pJB774 |
| Plasmid: 8xHis-(TevC)-Pab1ΔP-srtA-strep in pET28a backbone | This manuscript | pJB775 |
| Software and algorithms | ||
| Image Lab ver. 6.1.0 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Image Studio Lite ver. 5.2.5 | LI-COR | https://www.licor.com/bio/image-studio-lite/ |
| RStudio ver. 1.2.1335 | RStudio Team, 2018 | http://www.rstudio.com/. |
| Jupyter Notebook ver. 6.0.3 | Kluyver et al., 2016 | https://jupyter.org/ |
| DYNAMICS ver. 7.1 | Wyatt | N/A |
| SparkControl™ ver. | TECAN | N/A |
| UNICORN ver. 5.31 | GE Healthcare | N/A |
| Custom R (version 3.5.2) scripts for data processing, analysis, and figure generation | This paper | doi:10.17632/wvztspxbbd.1 |
| Custom Python (version 3.7.7) code for simulation | This paper | https://github.com/haneulyoo/sim_disagg_2021 |
| Other | ||
| NBS™ 384 well microplates (white; flat bottom) for luciferase assays | Corning | Cat#CLS3574 |
| NBS™ 384 well microplates (black; flat bottom) for anisotropy assays | Corning | Cat#CLS3575 |
| Nunc™ Sealing Tapes for microplates | Thermo Fisher | Cat# 235307 |
| Whatman® Anotop® 10 syringe filter for DLS experiments | MilliporeSigma | Cat# WHA68091002 |
Experimental Model and Subject Details
Yeast strain and growth conditions
All yeast strains listed in the Key Resources Table were cultured in yeast extract peptone dextrose (YPD) media in shaking baffled flasks at 30°C.
Bacteria strain and growth conditions
All recombinant proteins used in this work are expressed in and purified from E. coli BL21(DE3) using the plasmids listed in the Key Resources Table. Cells were first grown overnight in Luria broth (LB) at 37°C and then inoculated to 1–2 L Terrific Broth (TB) media. Specific growth condition used for each recombinant protein is described in Method Details.
Method Details
Physiological heat shock
By ‘physiological heat shock’, we mean nonlethal elevated temperature fluctuations encountered in nature to which cells have adapted during their evolutionary history. We have in previous published work discussed how physiological heat shock serves as an important environmental signal, as opposed to damaging stress, for immune cells and both pathogenic and non-pathogenic fungi (Triandafillou et al., 2020). Briefly, for budding yeast, ingestion and dispersal by passerine birds, whose body temperature averages around 42°C is an ecologically established and physiologically relevant heat shock condition for budding yeast (Triandafillou et al., 2020).
Purification of Pab1 and Pab1 variants
Protein expression and purification protocols were adapted with modification from (Riback, Katanski et al., 2017). Cells transformed with an expression plasmid for N-terminally 8xHis-tagged Pab1 were grown in TB until the optical density at 600 nm (OD600) reached between 0.4 and 0.6 and the flask was moved into a 30°C incubator. After 30 minutes, IPTG was added to the final concentration of 0.2 mM to induce protein expression. Cells were harvested after four hours and lysed by sonication in His binding buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, 20 mM imidazole, 10 % glycerol, and 1 mM BME) supplemented with a Pierce protease inhibitor tablet listed in the Key Resources Table. Lysate was cleared at 20,000 g for 15 minutes. Cleared lysate was loaded onto a HisTrap FF column (Thermo Fisher 17525501) equilibrated with His binding buffer on an AKTA FPLC system. Protein was eluted with a 20 mL gradient from 0 to 100 % His elution buffer (His binding buffer with 400 mM imidazole). Fractions containing Pab1 were buffer exchanged into a Q binding buffer (20 mM HEPES pH 7.3, 50 mM KCl, 2.5 mM MgCl2, 10 % glycerol, and 1 mM DTT) and loaded onto a HiTrap Heparin HP column (GE Healthcare 17040701) to remove nucleic acids. Nucleic acid-free protein was eluted over a 20 mL gradient from 0 to 100 % Q elution buffer (Q binding buffer with 1 M KCl). Fractions of interest were combined with an aliquot of a homemade tobacco etch virus (TEV) protease and dialyzed against a liter of His binding buffer overnight. On the next day, the dialyzed solution was loaded again onto a HisTrap FF column and the flow-through which contains the cleaved protein was collected. The protein was concentrated and loaded onto a Superose 6 10/300 GL column (GE Healthcare) equilibrated with SEC/Storage buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT). Monomeric proteins were pooled together, further concentrated if necessary, and stored at −80°C. Protein concentration was measured using Bradford assay.
Purification of Hsp70 chaperones
We adapted with minor modifications the protocol provided by Zachary March in James Shorter’s group. Hsp70 proteins tagged with an N-terminal 6xHis-SUMO were expressed at 18°C overnight. Cells were harvested after 14 – 18 hours of induction and lysed by sonication in Hsp70 His binding buffer (50 mM HEPES pH 7.3, 750 mM KCl, 5 mM MgCl2, 20 mM imidazole, 10 % glycerol, 1 mM BME, and 1 mM ATP) supplemented with Pierce protease inhibitor tablet listed in the Key Resources Table. Cleared lysate was loaded onto a HisTrap FF column equilibrated with Hsp70 His binding buffer on an AKTA FPLC system. After loading, the column was washed with more Hsp70 His binding buffer until the UV reading returned to the baseline level. The column was further washed with about 20 mL of high ATP buffer (Hsp70 His binding buffer with 20 mM ATP) and incubated in this buffer for 30 minutes. The high ATP buffer was washed out with Hsp70 His binding buffer and the protein was eluted with a 20 mL gradient from 0 to 100 % Hsp70 His elution buffer (Hsp70 His binding buffer with 400 mM imidazole). Fractions containing Hsp70 were combined and dialyzed against a liter of Hsp70 His binding buffer for at least 2 hours to remove excess imidazole. An aliquot of homemade SUMO protease Ulp1 was added to the dialysis bag. Dialysis was continued overnight at 4°C. Next day, the cleaved protein was recovered by running the dialyzed solution through His column and collecting flow-through. Flow-through fractions containing tag-free Hsp70 proteins were combined, diluted in Hsp70 Q binding buffer (20 mM HEPES pH 7.3, 50 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, and 1 mM ATP), and loaded onto an equilibrated HiTrap Q HP anion exchange column (GE Healthcare 17115401). Hsp70 was eluted over a 50 mL gradient from 0 to 100 % Hsp70 Q elution buffer (Hsp70 Q binding buffer with 1 M KCl). Fractions containing Hsp70 were determined by SDS-PAGE. We observed a peak with a left shoulder or two closely overlapping peaks around 25 mS/cm. Both peaks contained Hsp70, but only the later peak fractions exhibited activity in luciferase and Pab1 disaggregation assays. We combined the fractions corresponding to the second peak, concentrated, and buffer exchanged the protein into Hsp70 storage buffer (50 mM HEPES pH 7.3, 150 mM KCl, 5 mM MgCl2, 10 % glycerol, 2 mM DTT, and 1 mM ATP). Protein concentration was measured using Bradford assay. Protein aliquots were snap-frozen in liquid nitrogen and stored at −80°C.
Purification of sortase A enzymes
Wild-type (used for N-terminal labeling) and heptamutant sortase A (used for C-terminal labeling) were purified using the same protocol. Descriptions of these constructs and the sources are given in the Key Resources Table. Constructs were expressed with 0.5 mM IPTG overnight at 18°C. The cells were harvested in His binding buffer supplemented with homemade protease inhibitors and Pierce Universal Nuclease (Thermo Scientific PI88702). Cells were lysed by sonication, clarified by centrifugation at 20,000 g for 30 minutes, then bound to 5 mL of Ni-NTA resin (Thermo Scientific 88222) for one hour at 4°C. The resin was washed with 100 mL of His binding buffer, then the protein was eluted in 20 mL of His elution buffer (His binding buffer with 250 mM imidazole). The protein was concentrated in a spin concentrator, then loaded onto a Superdex 200 16/60 column (GE Healthcare) equilibrated in buffer (20 mM HEPES pH 7.5, 150 mM KCl, 2.5 mM MgCl2, 10 % glycerol, and 0.5 mM TCEP). Fractions corresponding to the monomeric protein were pooled together, concentrated, and aliquoted for storage at −80 °C.
Purification of ClpX and ClpP
The purification of ClpXΔN and ClpP were done as previously described (Martin et al., 2005). Specific information about the plasmid is listed in the Key Resources Table. A plasmid encoding a linked trimer of ClpXΔN with an N-terminal 6xHis affinity tag was transformed and protein production was induced with 0.5 mM IPTG. Cells were harvested after 4 hours at 37°C and resuspended in His binding buffer (20 mM HEPES pH 7.5, 100 mM KCl, 400 mM NaCl, 20 mM imidazole, 10% glycerol, and 1 mM BME), supplemented with protease inhibitors and Pierce Universal Nuclease (Thermo Scientific PI88702). Cells were lysed by sonication, clarified by centrifugation at 20,000 g for 30 minutes, then bound to 5 mL of Ni-NTA resin (Thermo Scientific 88222) for an hour at 4°C. The resin was washed with 100 mL of His binding buffer, then the protein was eluted in 20 mL of His elution buffer (His binding buffer with 250 mM imidazole). The protein was concentrated in a spin concentrator, then loaded onto a Superdex 200 16/60 column (GE Healthcare) equilibrated in buffer (20 mM HEPES pH 7.5, 300 mM KCl, 0.1 mM EDTA, 10 % glycerol and 1 mM DTT). Fractions corresponding to the monomeric protein were pooled together, concentrated and aliquoted for storage at −80°C. Protein concentration was determined by measuring A280.
A plasmid encoding ClpP with a C-terminal 6xHis affinity tag was transformed and protein production was induced with 0.5 mM IPTG. Cells were harvested after 4 hours at 37°C and resuspended in His binding buffer supplemented with Pierce Universal Nuclease. Protease inhibitors were omitted. Cells were lysed by sonication, clarified by centrifugation at 20,000 g for 30 minutes, then bound to 5 mL of Ni-NTA resin for an hour at 4°C. The resin was washed with 100 mL of His binding buffer, then the protein was eluted in 20 mL of His elution buffer. The protein was then bound to a HiTrap MonoQ column equilibrated in low salt buffer (50 mM Tris-HCL pH 8.0, 50 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 10% glycerol, and 1 mM BME), washed with 20 mL of low salt buffer and then eluted using a 100 mL gradient between low salt buffer and high salt buffer (low salt buffer with 200 mM KCl). The protein was concentrated in a spin concentrator, then loaded onto a Superdex 200 16/60 column (GE Healthcare) equilibrated in buffer (20 mM HEPES pH 7.5, 200 mM KCl, 0.1 mM EDTA, 10 mM MgCl2, 10 % glycerol and 1 mM DTT). Fractions corresponding to the monomeric protein were pooled together, concentrated and aliquoted for storage at −80°C. Protein concentration was determined by measuring A280.
Purification of remaining recombinant proteins
The rest of the recombinant proteins used in this paper were expressed with an N-terminal 6xHis-SUMO tag and purified as described for Pab1, but using a HiTrap MonoQ anion exchange column instead of a heparin column.
Fluorescein labeling of Pab1
Fluorescein labeling of Pab1 termini was done using sortase catalyzed ligation of labeled peptides. For N-terminal labeling, 6xHis-TEV-GGG-Pab1 was purified using the same protocol described for wild-type Pab1 above. The labeling was done in SEC buffer (20 mM HEPES 7.3, 150 mM KCl, and 2.5 mM MgCl2) as a 500 μL reaction with 100 μM Pab1, 20 μM wild-type sortase A, 0.5 mM TCEP, 10 mM CaCl2 and 0.5 mM 5-FAM-HHHHHHLPETGG peptide. After an hour incubation at room temperature, labeled protein was separated from free peptide on a HiTrap Desalting column equilibrated in aggregation buffer (20 mM HEPES pH 6.8, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT). The C-terminally labeled Pab1 was prepared similarly, but the labeling reaction was done with the following condition: 100 μM Pab1, 20μM heptamutant sortase A, 0.5 mM TCEP, and 0.5 mM GGGK(FAM)AANDENYALAA or GGGK(FAM) peptide. More information on the labeled peptides can be found in the Key Resources Table.
In vivo Total/Soluble/Pellet (TSP) assay
Yeast cells were grown at 30°C in 200 mL YPD until OD600 reached between 0.2 and 0.3. A pre-shock sample was collected by spinning 30 mL of the cell culture in a 50 mL conical tube at 3,000 g for 5 minutes at room temperature (RT). The cell pellet was resuspended in 1 mL of cold soluble protein buffer (SPB; 20 mM HEPES pH7.3, 120 mM KCl, 2 mM EDTA, 0.2 mM DTT, and 1:500 protease inhibitors cocktail IV (MilliporeSigma 539136; listed in the Key Resources Table), transferred to a pre-chilled 1.5 mL microcentrifuge tube, and centrifuged again at 5,000 g for 30 seconds at RT. Supernatant was removed and the pellet was resuspended in 170 μL SPB. Two 100 μL aliquots from the resuspended sample were snap-frozen in a safe-lock tube in liquid nitrogen. The remaining cell culture was vacumm filtered and the cells stuck on the filter membrane was resuspended in 30 mL YPD in a 50 mL conical tube. The membrane was removed and the resuspended cells were pelleted again by spinning the tube at 3,000 g for 30 seconds. After decanting the supernatant, the 50 mL conical tube containing cell pellet was placed in a heated water bath. After heat shock, cells were resuspended in 170 mL of pre-warmed 30°C YPD media. Heat shock sample was collected by taking 30 mL of this culture. The remaining cells were recovered at 30°C in a shaking 1 L baffled flask. Recovery samples were collected at different time points and processed in the same way as the pre-shock sample. Cells were lysed by cryomilling and fractionated as described in Wallace et al. (2015), with minor modifications on spin conditions. Briefly, cell lysates were cleared at 3,000 g for 30 seconds. Unlysed cells and membrane-associated proteins were removed during this step. 150 μL of the cleared lysate was transferred to a new 1.5 mL tube. RNase If (NEB M0243S; see Key Resources Table) was added to the final concentration of 0.3 units/μL and the sample was incubated at RT for 30 minutes. 50 μL of the sample was transferred to a new tube, mixed with Total protein buffer (TPB; 20 mM HEPES pH 7.3, 150 mM NaCl, 5 mM EDTA, 3% SDS, 1:100 PMSF, 2 mM DTT, and 1:500 protease inhibitors IV (MilliporeSigma 539136; see Key Resources Table), and spun at 6,000 g for one minute to collect Total protein sample. Pellet samples were collected by spinning the remaining 100 μL sample at 8,000 g for 5 minutes (P8) and at 100,000 g for 20 minutes (P100) at 4°C, and resuspending the pellet in Insoluble protein buffer (IPB; TPB with 8 M urea). Supernatant after the 100,000 g spin was collected as the Soluble protein sample. Western blots were performed as described in (Wallace et al., 2015) using a 1:10,000 dilutions of mouse monoclonal antibodies for Pab1, Pgk1, and GFP, and a 1:10,000 dilution of secondary antibody. The sources of the antibodies are listed in the Key Resources Table. The gels and blots were visualized using either ChemiDoc (Bio-rad) or Odyssey CLx (LI-COR) using their associated software listed in the Key Resources Table. For cycloheximide treatment, post-shock pellet was resuspended in 300 mL YPD and, after collecting 30 mL heat shock sample, the culture was split in half. Cycloheximide in DMSO was added to one culture at the final concentration of 100 μg/mL and DMSO was added to another culture as control.
In vitro Total/Soluble/Pellet (TSP) assay
Reaction mixture containing Pab1 condensates and molecular chaperones were prepared and incubated at 30°C for an hour either in the absence or presence of 5 mM ATP. The reaction mixture (Table S3) were centrifuged at 100,000 g for 20 minutes at 4°C. Supernatant was collected as the soluble fraction sample. Buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, 0.01 % Triton X-100, 0.5 mg/mL BSA, and 1 mM DTT) was added to the pellet and the sample was centrifuged again under the same condition. After removing the supernatant, the pellet was directly resuspended in 1x Laemmli buffer. Pab1 in each sample was visualized by SDS-PAGE and western blot using ONE-HOUR Western Basic Kit and Clarity Western Enhanced Chemiluminescence (ECL) Substrate (see Key Resources Table for catalog numbers).
Preparation of Pab1 condensates
Pab1 monomers were buffer exchanged into aggregation buffer (20 mM HEPES pH 6.8, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) and diluted to make a 500 μL sample of 25 μM Pab1. The sample was sometimes supplemented with 0.25 μM firefly luciferase to increase yield. The sample was incubated in a 42°C water bath for 20 minutes at pH 6.8. Under this heat shock condition, the solution remained clear and minimal protein pelleting was observed after a 3 minute centrifugation at 8,000 g. N-terminally fluorescein labeled Pab1 aggregates were prepared at 39°C. The supernatant was loaded onto a Superose 6 10/300 GL column (GE Healthcare) equilibrated with storage buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT). Void fractions containing small Pab1 assemblies (>5,000 kDa) were collected. Concentration of Pab1 condensates in each void fraction was measured using Bradford assay and/or SDS-PAGE with Pab1 standards of known concentrations. Protein aliquots were snap-frozen in liquid nitrogen and stored at −80°C.
Condensate reversibility assay
The protocol was adapted from Kroschwald et al. (2018) and modified for Pab1. Pab1 condensates were formed at 30 μM by either pH drop (pH 5 at 30 °C) or heat shock (pH 6.4 at 46 °C for 10 minutes) in buffered solution containing 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT. The formed condensates were diluted in control buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, 1 mM DTT) or in buffer with high salt (20 mM HEPES pH 7.3, 1 M KCl, 2.5 mM MgCl2, 1 mM DTT). After 30 minute incubation at room temperature, absorbance at 340 nm (turbidity) was measured in Spark microplate reader using its software (see Key Resources Table).
Preparation of A90
To prepare A90, 7.5 mg of polyadenylic acid (MilliporeSigma 10108626001; see Key Resources Table) was digested with 70 units of RNase I (Thermo Fisher AM2294) for 25 minutes at 37°C in 500 μL reaction containing 10 mM HEPES pH 7.5 and 20 mM KCl. Digested RNA was then purified by two rounds of phenol extraction in the presence of 1% SDS and ethanol precipitated. RNA was dissolved in 300 μL water and applied to Superose 6 10/300 GL column in buffer (20 mM HEPES pH 7.5, 400 mM KCl). RNA species eluted in 14.6–15.4 mL volume were collected, precipitated with ethanol, dissolved in water and further re-applied to Superose 6 column. The same elution fractions containing polyA RNA were collected, ethanol precipitated, dissolved in 100 μL water, and stored at −80°C. RNA concentration was determined using NanoDrop and the average RNA size was evaluated by running the RNA on a 15% TBE-Urea Criterion Precast Gel (Bio-Rad) with an RNA ladder.
Monitoring Pab1 dispersal using fluorescence anisotropy
Because Pab1 binds 12-mer poly(A) with full affinity and has a binding footprint of roughly 25 nucleotides (Sachs et al., 1987), we used 19-mer poly(A) RNA to get 1:1 binding of Pab1 to RNA. Pab1 condensates, molecular chaperones, and 5’ labeled A19 RNA (FAM-A19 or Atto550-A19) were diluted to desired concentrations in disaggregation buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, 0.5 mg/mL BSA, 0.01 % Triton X-100, and 1 mM DTT). The reaction mixture (Table S3) was supplemented with 5 mM ATP, and 8 mM creatine phosphate (CP) and 1 μM creatine kinase (CK) for ATP regeneration. The reaction mixture was also supplemented with 2 % SUPERase RNase Inhibitor (Thermo Fisher AM2694). The final reaction volume was 15 μL. Calibration samples were prepared by adding known concentrations of monomeric Pab1 to the same concentration of FAM-A19 used in the reaction samples in disaggregation buffer supplemented with 5 mM ATP. A new calibration curve was made each time an experiment was performed. Concentrations of Pab1 condensates and RNA were chosen so that a plateau indicates the completion of condensate dispersal, not signal saturation (Figure 1E).
The reaction mixtures were added to a black, non-binding surface 384-well plate (Corning CLS3575). The plate wells were sealed with a plate sealer (Thermo Fisher 235307) to prevent liquid evaporation. Fluorescence anisotropy was measured every 20 second in Spark microplate reader (TECAN) using excitation/emission wavelengths of 485 nm/535 nm, each with a bandwidth of 20 nm, at 30°C. Disaggregation buffer was used as blank. G factor was calibrated with a solution of free 6-iodoacetamidofluorescein to produce a fluorescence polarization reading of 20 mP. Descriptions and catalog information of all key reagents are listed in the Key Resources Table.
Fluorescence-detection size-exclusion chromatography (FSEC)
Fluorescent Pab1 condensates were prepared and mixed with chaperones, ATP, ATP regeneration system, and SUPERase RNase Inhibitor as described above for wild-type Pab1. The reaction samples (120 μL per run) were incubated at 30°C for an hour. 120 μL of the sample was loaded to a Superdex 200 10/300 GL column (GE Healthcare) equilibrated with filtered running buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) using Akta system. Fluorescence was measured by a fluorescence detector (JASCO FP-2020 Plus) connected to the Akta system.
Luciferase reactivation assay
Recombinant firefly luciferase was aggregated by incubating 2 μM of luciferase with 10 μM Hsp26 in aggregation buffer (20 mM HEPES pH 6.8, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) at 42°C for 20 minutes. After cooling on ice for 2 minutes, the aggregates were diluted to 0.2 μM in the disaggregation buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, 0.5 mg/mL BSA, 0.01 % Triton X-100, and 1 mM DTT) supplemented with 5 mM ATP, 8 mM CP, 1 μM CK, and specified concentrations of chaperones (Table S3). The mixed sample was incubated at 30°C. At each time point, 1.5 μL of the reaction sample was added to 13.5 μL of luciferin mix (Promega E1500; see Key Resources Table) in a white 384-well plate (Corning CLS3574; see Key Resources Table), and luminescence was measured using Spark microplate reader (TECAN) with integration time of 1 second.
To make luciferase aggregates that are more amenable to disaggregation (Figure 3B), 33.8 nM luciferase was heat shocked in the presence of 169 nM Hsp26 at 42°C for 20 minutes in low-salt aggregation buffer (25 mM HEPES pH 7.3, 50 mM KCl, 0.1 mM EDTA, and 1 mM DTT). Luciferase aggregates were diluted to the final concentration of 20 nM in the disaggregation buffer containing chaperones. Luminescence was measured as described above. For disaggregation of chemically aggregated luciferase using HAP/ClpP system (Figure 5H and 5I), luciferase was diluted to 5 μM in low-salt urea buffer (25 mM HEPES pH 7.3, 50 mM KCl, 0.1 mM EDTA, 1 mM DTT, and 8 M urea) and incubated at 30°C for 30 minutes. Aggregation was induced by diluting luciferase 100-fold into the disaggregation buffer containing chaperones and HAP/ClpP (Table S3). Western blot samples were collected at the specified time points and stained using luciferase antibody (MilliporeSigma L0159; see Key Resources Table). Blots were visualized using Odyssey CLx (LI-COR). Luminescence was measured as described above.
Dynamic light scattering (DLS)
DLS measurements were performed using DynaPro NanoStar (Wyatt) with its software (see Key Resources Table). Sample acquisition was done as described in (Riback, Katanski et al., 2017). All experiments, unless noted otherwise, were performed with 10 μM Pab1 in buffer (20 mM HEPES pH 6.8, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) filtered with a 0.02 μm Anotop syringe filter (listed in the Key Resources Table). All protein samples used for DLS experiments were dialyzed against buffer overnight at 4°C and cleared of aggregates by spinning at 20,000 g for 20 minutes.
Electrophoretic mobility shift assay (EMSA)
The assay was performed using agarose gels as described in a previous study (Ream et al., 2016) with little modifications. Briefly, 10 nM of fluorescein-labeled A19 RNA was incubated with increasing concentrations of either monomeric or condensed Pab1 for at least 10 minutes at room temperature. The same concentration of kozak sequence RNA (5’ACCUCUGCCGCCGCCAUGG; see Key Resources Table) was used as a negative control. The samples were loaded to a 2.5% agarose gel and the gel was ran for 45 minutes at 30 V/cm in 0.5x Tris/Boric Acid/EDTA (TBE) buffer (Bio-rad 1610733). Fluorescence from the labeled RNA was visualized using a ChemiDoc (Bio-rad).
GroEL trap assay
Pab1 dispersal and luciferase reactivation assays were performed as described above, but in the presence of five-fold excess GroEL trap. For refolding experiment, 5 μM Pab1 was denatured in 8 M urea buffer (20 mM HEPES pH 6.8, 150 mM KCl, 2.5 mM MgCl2, 1 mM DTT, and 8 M urea) and incubated at 30°C for 30 minutes. Pab1 was first diluted to 0.5 μM in refolding buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) containing no or 10-fold excess GroEL trap, and then to 0.1 μM in the same respective refolding buffer with 0.1 μM FAM-A19. Pab1’s RNA-binding activity was measured by fluorescence anisotropy.
Gel analysis of HAP/ClpP degradation
Digestion by the HAP/ClpP system was done in disaggregation buffer (20 mM HEPES pH 7.3, 150 mM KCl, 2.5 mM MgCl2, 0.5 mg/mL BSA, 0.01 % Triton X-100, and 1 mM DTT) with 0.2 μM Pab1 condensate or monomer, 1.5 μM ClpP, 1 μM Hsp104 (WT or HAP), 0.5 μM Sis1, 1–2 μM Ssa2, 5 mM ATP, and 8 mM CP and 1 μM CK for ATP regeneration. Reactions were run for an hour at 30°C, then quenched with Laemmli buffer and run on a Bio-rad TGX 4–20% SDS-PAGE gel. Fluorescent gels were imaged using a ChemiDoc (Bio-rad) and western blots were performed using a 1:5000 dilution of Rabbit anti-GFP antibody and a 1:20,000 dilution of Donkey anti-rabbit secondary antibody (see Key Resources Table for more descriptions and catalog numbers). Blots were visualized using Odyssey CLx (LI-COR).
The ClpXP digestion reaction was done for 30 minutes at 30°C with 0.2 μM Pab1-FAM monomer, 0.1 μM ClpX and 1 μM ClpP in disaggregation buffer.
Broad Range Blue Prestained Protein Standard (NEB P7718L) was ran alongside the protein samples in a 4–20% Criterion TGX Stain-Free Protein Gel. Prestained standards run differently from their true molecular weight due to the dyes. To more accurately determine the true molecular weight of the degraded product, we adjusted the apparent molecular weight of the prestained standards based on the calibration reported in the NEB website and our own calibration comparing prestained and unstained ladders. The molecular weights shown next to the gels in Figures 5H and S6 reflect the adjusted molecular weight values.
Disaggregation data from the literature
The following data from 18 different studies (Nillegoda et al., 2017; Doyle et al., 2015; Rampelt et al., 2012; Yu et al., 2015; Żwirowski et al., 2017; Martín et al., 2014; Kłosowska et al., 2016; Reidy et al., 2014; Rosenzweig et al., 2013; DeSantis et al., 2012; Shorter, 2011; Haslberger et al., 2008; Glover and Lindquist, 1998; Ratajczak et al., 2009; Cashikar et al., 2005; Goloubinoff et al., 1999; Sielaff and Tsai, 2010; Duennwald et al., 2012) were compiled for comparison (Table S1): 1) substrate identity; 2) concentrations of substrate and molecular chaperones used; 3) maximum yield observed within the experimental time; 4) the names of molecular chaperones; and 5) reference to the source paper with DOI. Only the results from in vitro experiments were recorded. Studies reporting fold-change relative to negative control were omitted because yield cannot be determined from the given information for comparison. For a study which reports multiple disaggregation results with the same substrate, the concentrations of substrate and chaperones which give the highest maximal yield were recorded.
Modeling and simulation
In our model, free substrates exist in one of the following states: folded (Sf), unfolded (Su), misfolded (Sm), and aggregated (Sa). Free Hsp70 exists either in an ATP-bound state (Hsp70ATP) or an ADP-bound state (Hsp70ADP), and each state can bind certain free substrates to form a complex (e.g., Hsp70ATP:Sa). ATP hydolysis of Hsp70ATP in Hsp70ATP:Sm complex results in substrate unfolding, a step we call “priming”. For an aggregated substate, the same sequence of events do not result in any state transition but instead primes the complex (Hsp70ATP:Sap) for interaction with Hsp104. In the cooperative model, a second Hsp70ATP can bind Hsp70ADP:Sap complex to form a ternary complex (Hsp70ATP,ADP:Sap) and only the ternary complex with both Hsp70s in the ADP-bound state (Hsp70ADP,ADP:Sap) can engage with Hsp104 for disaggregation. For simplicity, we did not allow unfolding of a natively folded substrate. The parameter values used for the simulation are listed in Table S2.
The time evolution of the concentrations of all distinct species in the cooperative model was described using the following ODEs:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
Quantification and Statistical Analysis
Quantification of sedimentation data
The western blot band intensities were quantified using either Image Lab (Figures 1A, 2C, and S7) or Image Studio (Figures S1) software. The band intensity from soluble (S), P8, or P100 fraction was divided by the sum of those values to compute proportion soluble, P8, and P100, respectively, in R. The statistical details of data shown in Figures 1C and 2D can be found in the respective figure legends. For Figure 1C, RNase-treated samples from three independent biological replicates were used. The western blots and total protein gels for the biological replicates are shown in Figures 1A, S1E, and S7A–C.
Fluorescence anisotropy data fitting and analysis
The kinetic data were fitted with the following logistic equation:
| (20) |
where d, m, a, b, and c are fitting parameters. The negative linear term accounts for the chaperone concentration-dependent signal decay, which comes from RNA-degrading contaminants co-purified with chaperones.
To extract maximal rate of Pab1 dispersal, the fluorescence anisotropy values were first converted to the concentration of RNA-binding competent monomeric Pab1 using a calibration curve (Figure 1E) fitted with the following equation:
| (21) |
Min and max refer to the fluorescence anisotropy values of the calibration samples with no or saturating amount of monomeric Pab1, respectively. The values of d and n extracted from the calibration fit were used to convert fluorescence anisotropy values in the reaction samples to concentrations of Pab1 with this rearranged equation (21):
| (22) |
The converted data were fitted again with equation (20). Maximal rate of dispersal was calculated by computing the extracted fit parameters to the derivative of equation (20) when x = b:
| (23) |
| (24) |
To convert the y-axis from Pab1 concentration to fraction restored Pab1, we first subtracted background signal using the negative control data (no ATP). Background-subtracted data were divided by the total concentration of Pab1, which was approximated by taking the mean of highest data points, i.e., data points in the plateau region of the positive control. Total Pab1 concentration in the reaction had to be approximated this way for more accurate quantification because we noticed that Pab1 condensates adhere to plastic, causing loss of about 30–50% substrate during transfer. To compensate for this, 1.5 to 2-fold excess Pab1 was added to aim for, e.g., the final concentration of 0.2 μM Pab1 in the reaction. The same total Pab1 concentration was used to normalize reactions prepared from the same master mix. The presence of ATP slightly lowered the fluorescence anisotropy baseline compared to the no ATP control, and this led to negative starting values for all ATP-containing reactions after background subtraction. All traces were shifted upward by the same amount to make the positive control reactions to start around the value of zero.
The rate data in Figure 6A were fitted with a logistic equation:
| (25) |
with a, b, and c as fitting parameters. We used the total Pab1 concentration to calculate the ratio of chaperone to substrate.
All of the statistical details of data shown in Figures 1E, 3E, and 6A–B can be found in the respective figure legends.
Luciferase disaggregation
Reactivation yield was computed in R by normalizing luminescence of aggregated luciferase to that of mock-treated luciferase at each time point. The statistical details of data shown in Figures 3A–B can be found in the respective figure legends.
Dynamic light scattering (DLS)
Temperature at which Pab1’s hydrodynamic radius doubles (Tdouble in Figure S2A–B) or crosses 20 nm (T20 in Figure S4C–D) was calculated using custom scripts written in R. The script for computing Tdouble was originally written in Mathematica and published in Riback, Katanski et al., 2017. Quantification was done for the representative data shown in Figures S2A and S4C.
Hsp26 titration experiment
Protein bands in the Coomassie-stained gel (Figure S4A) was quantified using Image Lab. Proportion soluble Pab1 shown in Figure S4B was computed in R by dividing the soluble fraction band intensity by the sum of both soluble and pelleted fraction band intensities. Quantification was done for the representative data shown in Figures S4A.
Electrophoretic mobility shift assay (EMSA)
Fluorescence of free RNA (Figure S2D–E) was quantified using Image Studio. Fraction bound was calculated in R by normalizing to the RNA-alone control. A19 and Pab1 monomer titration data were fitted with a quadratic binding equation. The statistical details can be found in the figure legends.
Analysis of Pab1 refolding
RNA-binding activity of refolded Pab1 quantified by fluorescence anisotropy was normalized to that of urea-denatured Pab1 (Figure 5D). Statistical significance was determined with Welch t-test using R’s built-in t-test function, using a p-value of 0.05 as the cutoff for significance.
Turbidity analysis
Turbidity values of Pab1 condensates in Figure S1F were normalized by dividing by the turbidity of buffer. Statistical significance was determined with Welch t-test using R’s built-in t-test function, using a p-value of 0.05 as the cutoff for significance.
Quantification of protein degradation by HAP/ClpP
Full-length protein band intensities were quantified from luciferase and Pab1 western blots (Figures 5F and H) using Image Studio. Because chemically-denatured luciferase showed about 60% restoration after an hour (Figure 5G) and the FSEC traces of Pab1-fluorescein-ssrA also showed around 60% dispersal under the same condition (Figure 5K), we assumed 60% of the full-length band corresponds to the protein released from aggregates without degradation by HAP/ClpP. The quantified value was normalized to either HAP alone control (for HAP/ClpP experiments in Figure 5I) or Sis1 alone control (for ClpX/ClpP experiment in Figure 5J). Quantification was done for the representative data shown in Figure 5.
Supplementary Material
Literature data (Excel file), Related to Figure 6
Acknowledgements
We thank the members of the Drummond lab, Tobin Sosnick, and Ruofan Chen for helpful discussions and comments on the manuscript. We also thank Zachary March and James Shorter for the original protocol for Hsp70 purification, Axel Mogk and Bernd Bukau for GroEL trap materials, Andreas Martin for ClpX/P materials, David Pincus and Michael Rust for feedback on modeling, Eduardo Perozo’s group for FSEC instrument use, and Elena Solomaha at the Biophysics Core for assistance with DLS experiments. Research reported in this publication was supported by the National Institute of General Medical Sciences and the National Institute of Environmental Health Sciences of the National Institutes of Health (NIH) through awards to HY (award numbers T32GM007183 and F31ES030697). JAMB acknowledges fellowship support from the Helen Hay Whitney Foundation. DAD acknowledges support from the NIH (award numbers R01GM126547 and R01GM127406) and the US Army Research Office (W911NF-14-1-0411). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Assenza S, Sassi AS, Kellner R, Schuler B, De Los Rios P and Barducci A (2019), ‘Efficient conversion of chemical energy into mechanical work by Hsp70 chaperones’, Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avellaneda MJ, Franke KB, Sunderlikova V, Bukau B, Mogk A and Tans SJ (2020), ‘Processive extrusion of polypeptide loops by a hsp100 disaggregase’, Nature 578(7794), 317–320. [DOI] [PubMed] [Google Scholar]
- Baler R, Welch WJ and Voellmy R (1992), ‘Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor’, J. Cell Biol. 117(6), 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA and Rosen MK (2017), ‘Biomolecular condensates: organizers of cellular biochemistry’, Nat. Rev. Mol. Cell Biol. 18(5), 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begovich K and Wilhelm JE (2020), ‘An in vitro assembly system identifies roles for rna nucleation and atp in yeast stress granule formation’, Molecular Cell. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi A, De Los Rios P, Dietler G and Goloubinoff P (2004), ‘Active solubilization and refolding of stable protein aggregates by cooperative unfolding action of individual hsp70 chaperones’, J. Biol. Chem. 279(36), 37298–37303. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P and Boeke JD (1998), ‘Designer deletion strains derived from saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications’, Yeast 14(2), 115–132. [DOI] [PubMed] [Google Scholar]
- Bresson S, Shchepachev V, Spanos C, Turowski T, Rappsilber J and Tollervey D (2020), Stress-induced translation inhibition through rapid displacement of scanning initiation factors. [DOI] [PMC free article] [PubMed]
- Carroni M, Kummer E, Oguchi Y, Wendler P, Clare DK, Sinning I, Kopp J, Mogk A, Bukau B and Saibil HR (2014), ‘Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with hsp70 in protein disaggregation’, Elife 3, e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashikar AG, Duennwald M and Lindquist SL (2005), ‘A chaperone pathway in protein disaggregation. hsp26 alters the nature of protein aggregates to facilitate reactivation by hsp104’, J. Biol. Chem. 280(25), 23869–23875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasov V, Grousl T, Theer P, Vainshtein Y, Glässer C, Mongis C, Kramer G, Stoecklin G, Knop M, Mogk A and Bukau B (2015), ‘Systemic control of protein synthesis through sequestration of translation and ribosome biogenesis factors during severe heat stress’, FEBS Lett. 589(23), 3654–3664. [DOI] [PubMed] [Google Scholar]
- Cherkasov V, Hofmann S, Druffel-Augustin S, Mogk A, Tyedmers J, Stoecklin G and Bukau B (2013), ‘Coordination of translational control and protein homeostasis during severe heat stress’, Curr. Biol. 23(24), 2452–2462. [DOI] [PubMed] [Google Scholar]
- De Los Rios P and Barducci A (2014), ‘Hsp70 chaperones are non-equilibrium machines that achieve ultra-affinity by energy consumption’, Elife 3, e02218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN and Shorter J (2012), ‘Operational plasticity enables Hsp104 to disaggregate diverse amyloid and nonamyloid clients’, Cell 151(4), 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SM, Shastry S, Kravats AN, Shih Y-H, Miot M, Hoskins JR, Stan G and Wickner S (2015), ‘Interplay between e. coli DnaK, ClpB and GrpE during protein disaggregation’, J. Mol. Biol. 427(2), 312–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duennwald ML, Echeverria A and Shorter J (2012), ‘Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans’, PLoS Biol. 10(6), e1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durie CL, Duran EC and Lucius AL (2018), ‘Escherichia coli DnaK allosterically modulates ClpB between high- and Low-Peptide affinity states’, Biochemistry 57(26), 3665–3675. [DOI] [PubMed] [Google Scholar]
- Erives AJ and Fassler JS (2015), ‘Metabolic and chaperone gene loss marks the origin of animals: Evidence for hsp104 and hsp78 chaperones sharing mitochondrial enzymes as clients’, PLoS One 10(2), e0117192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust O, Abayev-Avraham M, Wentink AS, Maurer M, Nillegoda NB, London N, Bukau B and Rosenzweig R (2020), ‘HSP40 proteins use class-specific regulation to drive HSP70 functional diversity’, Nature 587(7834), 489–494. [DOI] [PubMed] [Google Scholar]
- Feder ZA, Ali A, Singh A, Krakowiak J, Zheng X, Bindokas VP, Wolfgeher D, Kron SJ and Pincus D (2021), ‘Subcellular localization of the j-protein sis1 regulates the heat shock response’, J. Cell Biol. 220(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann TM, Jahnel M, Pozniakovsky A, Mahamid J, Holehouse AS, Nüske E, Richter D, Baumeister W, Grill SW, Pappu RV, Hyman AA and Alberti S (2018), ‘Phase separation of a yeast prion protein promotes cellular fitness’, Science 359(6371). [DOI] [PubMed] [Google Scholar]
- Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda NB, Szlachcic A, Guilbride DL, Saibil HR, Mayer MP and Bukau B (2015), ‘Human hsp70 disaggregase reverses Parkinson’s-Linked α-Synuclein amyloid fibrils’, Mol. Cell 59(5), 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, Buell CE, Sweeny EA, Mack KL, Chuang E, Torrente MP, Su M, Shorter J and Southworth DR (2017), ‘Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase hsp104’, Science 357(6348), 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL and Drummond DA (2011), ‘Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast’, Proc. Natl. Acad. Sci. U. S. A. 108(2), 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisler SM, Pierpaoli EV and Christen P (1998), ‘Catapult mechanism renders the chaperone action of hsp70 unidirectional’, J. Mol. Biol. 279(4), 833–840. [DOI] [PubMed] [Google Scholar]
- Glover JR and Lindquist S (1998), ‘Hsp104, hsp70, and hsp40: a novel chaperone system that rescues previously aggregated proteins’, Cell 94(1), 73–82. [DOI] [PubMed] [Google Scholar]
- Goloubinoff P, Mogk A, Zvi AP, Tomoyasu T and Bukau B (1999), ‘Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network’, Proc. Natl. Acad. Sci. U. S. A. 96(24), 13732–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P, Sassi AS, Fauvet B, Barducci A and De Los Rios P (2018), ‘Chaperones convert the energy from ATP into the nonequilibrium stabilization of native proteins’, Nat. Chem. Biol. 14(4), 388–395. [DOI] [PubMed] [Google Scholar]
- Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AEM and Ploegh HL (2013), ‘Site-specific c-terminal and internal loop labeling of proteins using sortase-mediated reactions’, Nat. Protoc. 8(9), 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, Zhang ZC, Coughlin M, Sweeny EA, DeSantis ME, Jackrel ME, Rodell CB, Burdick JA, King OD, Gitler AD, Lagier-Tourenne C, Pandey UB, Chook YM, Taylor JP and Shorter J (2018), ‘Nuclear-Import receptors reverse aberrant phase transitions of RNA-Binding proteins with prion-like domains’, Cell 173(3), 677–692.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslberger T, Weibezahn J, Zahn R, Lee S, Tsai FTF, Bukau B and Mogk A (2007), ‘M domains couple the ClpB threading motor with the DnaK chaperone activity’, Mol. Cell 25(2), 247–260. [DOI] [PubMed] [Google Scholar]
- Haslberger T, Zdanowicz A, Brand I, Kirstein J, Turgay K, Mogk A and Bukau B (2008), ‘Protein disaggregation by the AAA+ chaperone ClpB involves partial threading of looped polypeptide segments’, Nat. Struct. Mol. Biol. 15(6), 641–650. [DOI] [PubMed] [Google Scholar]
- Hirakawa H, Ishikawa S and Nagamune T (2015), ‘Ca2+ -independent sortase-a exhibits high selective protein ligation activity in the cytoplasm of escherichia coli’, Biotechnol. J. 10(9), 1487–1492. [DOI] [PubMed] [Google Scholar]
- Hong L, Lavrentovich DO, Chavan A, Leypunskiy E, Li E, Matthews C, LiWang A, Rust MJ and Dinner AR (2020), ‘Bayesian modeling reveals metabolite-dependent ultrasensitivity in the cyanobacterial circadian clock’, Mol. Syst. Biol. 16(6), e9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoglu R, Balchin D, Hayer-Hartl M and Hartl FU (2020), ‘Bacterial hsp70 resolves misfolded states and accelerates productive folding of a multi-domain protein’, Nat. Commun. 11(1), 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Taguchi H, Kishimoto A and Yoshida M (2004), ‘Hsp104 binds to yeast sup35 prion fiber but needs other factor(s) to sever it’, J. Biol. Chem. 279(50), 52319–52323. [DOI] [PubMed] [Google Scholar]
- Iserman C, Desroches Altamirano C, Jegers C, Friedrich U, Zarin T, Fritsch AW, Mittasch M, Domingues A, Hersemann L, Jahnel M, Richter D, Guenther U-P, Hentze MW, Moses AM, Hyman AA, Kramer G, Kreysing M, Franzmann TM and Alberti S (2020), ‘Condensation of ded1p promotes a translational switch from housekeeping to stress protein production’, Cell 181(4), 818–831.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Rossi P and Kalodimos CG (2019), ‘Structural basis for client recognition and activity of hsp40 chaperones’, Science 365(6459), 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimal JM, Kandasamy G, Gasser F and Andréasson C (2017), ‘Coordinated hsp110 and hsp104 activities power protein disaggregation in saccharomyces cerevisiae’, Mol. Cell. Biol. 37(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH and Craig EA (2010), ‘The HSP70 chaperone machinery: J proteins as drivers of functional specificity’, Nat. Rev. Mol. Cell Biol. 11(8), 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kłosowska A, Chamera T and Liberek K (2016), ‘Adenosine diphosphate restricts the protein remodeling activity of the hsp104 chaperone to hsp70 assisted disaggregation’, Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowiak J, Zheng X, Patel N, Feder ZA, Anandhakumar J, Valerius K, Gross DS, Khalil AS and Pincus D (2018), ‘Hsf1 and hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response’, Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, Mateju D, Malinovska L, Nüske E, Poser I, Richter D and Alberti S (2015), ‘Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules’, Elife 4, e06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Munder MC, Maharana S, Franzmann TM, Richter D, Ruer M, Hyman AA and Alberti S (2018), ‘Different material states of pub1 condensates define distinct modes of stress adaptation and recovery’, Cell Rep. 23(11), 3327–3339. [DOI] [PubMed] [Google Scholar]
- Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J and Bukau B (1999), ‘Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones’, Proc. Natl. Acad. Sci. U. S. A. 96(10), 5452–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist S (1986), ‘The heat-shock response’, Annu. Rev. Biochem. 55, 1151–1191. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhang S, Gu J, Tong Y, Li Y, Gui X, Long H, Wang C, Zhao C, Lu J, He L, Li Y, Liu Z, Li D and Liu C (2020), ‘Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation’, Nat. Struct. Mol. Biol. 27(4), 363–372. [DOI] [PubMed] [Google Scholar]
- Lu Z and Cyr DM (1998), ‘Protein folding activity of hsp70 is modified differentially by the hsp40 co-chaperones sis1 and ydj1’, J. Biol. Chem. 273(43), 27824–27830. [DOI] [PubMed] [Google Scholar]
- Martin A, Baker TA and Sauer RT (2005), ‘Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines’, Nature 437(7062), 1115–1120. [DOI] [PubMed] [Google Scholar]
- Martín I, Celaya G, Alfonso C, Moro F, Rivas G and Muga A (2014), ‘Crowding activates ClpB and enhances its association with DnaK for efficient protein aggregate reactivation’, Biophys. J. 106(9), 2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser AE, Kang W, Roy J, Mohanakrishnan Kaimal J, Quintana-Cordero J, Friedländer MR and Andréasson C (2019), ‘Cytoplasmic protein misfolding titrates hsp70 to activate nuclear hsf1’, Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Schröder H, Rüdiger S, Paal K, Laufen T and Bukau B (2000), ‘Multistep mechanism of substrate binding determines chaperone activity of hsp70’, Nat. Struct. Biol. 7(7), 586–593. [DOI] [PubMed] [Google Scholar]
- McCarty JS, Buchberger A, Reinstein J and Bukau B (1995), ‘The role of ATP in the functional cycle of the DnaK chaperone system’, J. Mol. Biol. 249(1), 126–137. [DOI] [PubMed] [Google Scholar]
- Mogk A, Bukau B and Kampinga HH (2018), ‘Cellular handling of protein aggregates by disaggregation machines’, Mol. Cell 69(2), 214–226. [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Friedrich KL, Schönfeld H-J, Vierling E and Bukau B (2003), ‘Refolding of substrates bound to small hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK’, J. Biol. Chem. 278(33), 31033–31042. [DOI] [PubMed] [Google Scholar]
- Morimoto RI (2008), ‘Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging’, Genes Dev. 22(11), 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motojima F, Motojima-Miyazaki Y and Yoshida M (2012), ‘Revisiting the contribution of negative charges on the chaperonin cage wall to the acceleration of protein folding’, Proc. Natl. Acad. Sci. U. S. A. 109(39), 15740–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B, Hartich D, Seifert U and Rios PDL (2017), ‘Thermodynamic bounds on the ultra- and infra-affinity of hsp70 for its substrates’, Biophys. J. 113(2), 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, Guilbride DL, Wade RC, Morimoto RI, Mayer MP and Bukau B (2015), ‘Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation’, Nature 524(7564), 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Stank A, Malinverni D, Alberts N, Szlachcic A, Barducci A, De Los Rios P, Wade RC and Bukau B (2017), ‘Evolution of an intricate j-protein network driving protein disaggregation in eukaryotes’, Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA and Lindquist S (1993), ‘The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins’, Annu. Rev. Genet. 27, 437–496. [DOI] [PubMed] [Google Scholar]
- Peffer S, Gonçalves D and Morano KA (2019), ‘Regulation of the hsf1-dependent transcriptome via conserved bipartite contacts with hsp70 promotes survival in yeast’, J. Biol. Chem. 294(32), 12191–12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ET, Powers DL and Gierasch LM (2012), ‘FoldEco: a model for proteostasis in e. coli’, Cell Rep. 1(3), 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI and Bukau B (2012), ‘Metazoan hsp70 machines use hsp110 to power protein disaggregation’, EMBO J. 31(21), 4221–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak E, Zietkiewicz S and Liberek K (2009), ‘Distinct activities of escherichia coli small heat shock proteins IbpA and IbpB promote efficient protein disaggregation’, J. Mol. Biol. 386(1), 178–189. [DOI] [PubMed] [Google Scholar]
- Ream JA, Lewis LK and Lewis KA (2016), ‘Rapid agarose gel electrophoretic mobility shift assay for quantitating protein: RNA interactions’, Anal. Biochem. 511, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy M, Sharma R, Shastry S, Roberts B-L, Albino-Flores I, Wickner S and Masison DC (2014), ‘Hsp40s specify functions of hsp104 and hsp90 protein chaperone machines’, PLoS Genet. 10(10), e1004720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback JA, Katanski CD, Kear-Scott JL, Pilipenko EV, Rojek AE, Sosnick TR and Drummond DA (2017), ‘Stress-triggered phase separation is an adaptive, evolutionarily tuned response’, Cell 168(6), 1028–1040.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR and Kay LE (2013), ‘Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction’, Science 339(6123), 1080–1083. [DOI] [PubMed] [Google Scholar]
- Russell R, Jordan R and McMacken R (1998), ‘Kinetic characterization of the ATPase cycle of the DnaK molecular chaperone’, Biochemistry 37(2), 596–607. [DOI] [PubMed] [Google Scholar]
- Sachs AB, Davis RW and Kornberg RD (1987), ‘A single domain of yeast poly (a)-binding protein is necessary and sufficient for RNA binding and cell viability’, Mol. Cell. Biol. 7(9), 3268–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Baici A, Gehring H and Christen P (1994), ‘Kinetics of molecular chaperone action’, Science 263(5149), 971–973. [DOI] [PubMed] [Google Scholar]
- Scior A, Buntru A, Arnsburg K, Ast A, Iburg M, Juenemann K, Pigazzini ML, Mlody B, Puchkov D, Priller J, Wanker EE, Prigione A and Kirstein J (2018), ‘Complete suppression of htt fibrilization and disaggregation of htt fibrils by a trimeric chaperone complex’, EMBO J. 37(2), 282–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, Sourjik V, Bukau B and Mogk A (2012), ‘Hsp70 proteins bind hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces’, Nat. Struct. Mol. Biol. 19(12), 1347–1355. [DOI] [PubMed] [Google Scholar]
- Sharma SK, De los Rios P, Christen P, Lustig A and Goloubinoff P (2010), ‘The kinetic parameters and energy cost of the hsp70 chaperone as a polypeptide unfoldase’, Nat. Chem. Biol. 6(12), 914–920. [DOI] [PubMed] [Google Scholar]
- Shorter J (2011), ‘The mammalian disaggregase machinery: Hsp110 synergizes with hsp70 and hsp40 to catalyze protein disaggregation and reactivation in a cell-free system’, PLoS One 6(10), e26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J and Lindquist S (2004), ‘Hsp104 catalyzes formation and elimination of self-replicating sup35 prion conformers’, Science 304(5678), 1793–1797. [DOI] [PubMed] [Google Scholar]
- Siegers K, Waldmann T, Leroux MR, Grein K, Shevchenko A, Schiebel E and Hartl FU (1999), ‘Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin–GimC system’, EMBO J. 18(1), 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sielaff B and Tsai FTF (2010), ‘The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner’, J. Mol. Biol. 402(1), 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson-Lavy K, Xu T, Johnston M and Kupiec M (2017), ‘The Std1 activator of the Snf1/AMPK kinase controls glucose response in yeast by a regulated protein aggregation’, Mol. Cell 68(6), 1120–1133.e3. [DOI] [PubMed] [Google Scholar]
- Snead WT and Gladfelter AS (2019), ‘The control centers of biomolecular phase separation: How membrane surfaces, PTMs, and active processes regulate condensation’, Mol. Cell 76(2), 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, Gupta K and Shorter J (2015), ‘The hsp104 n-terminal domain enables disaggregase plasticity and potentiation’, Mol. Cell 57(5), 836–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, Mogk A and Bukau B (2008), ‘Substrate threading through the central pore of the hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation’, Mol. Microbiol. 68(1), 87–97. [DOI] [PubMed] [Google Scholar]
- Theyssen H, Schuster HP, Packschies L, Bukau B and Reinstein J (1996), ‘The second step of ATP binding to DnaK induces peptide release’, J. Mol. Biol. 263(5), 657–670. [DOI] [PubMed] [Google Scholar]
- Triandafillou CG, Katanski CD, Dinner AR and Drummond DA (2020), ‘Transient intracellular acidification regulates the core transcriptional heat shock response’, eLife 9, e54880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, Perrett S, Prodromou C, Jones GW and Kron SJ (2012), ‘CDK-dependent hsp70 phosphorylation controls G1 cyclin abundance and cell-cycle progression’, Cell 151(6), 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Madariaga ML and Lindquist S (2008), ‘Prion switching in response to environmental stress’, PLoS Biol. 6(11), e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungelenk S, Moayed F, Ho C-T, Grousl T, Scharf A, Mashaghi A, Tans S, Mayer MP, Mogk A and Bukau B (2016), ‘Small heat shock proteins sequester misfolding proteins in near-native conformation for cellular protection and efficient refolding’, Nat. Commun. 7(1), 13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M and Hartl FU (2010), ‘Protein folding in the cytoplasm and the heat shock response’, Cold Spring Harb. Perspect. Biol. 2(12), a004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JL, Parsell DA and Lindquist S (1995), ‘Heat-shock proteins hsp104 and hsp70 reactivate mRNA splicing after heat inactivation’, Curr. Biol. 5(3), 306–317. [DOI] [PubMed] [Google Scholar]
- Wallace EWJ, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, Airoldi EM, Pan T, Budnik BA and Drummond DA (2015), ‘Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress’, Cell 162(6), 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RW, Muhlrad D, Garcia J and Parker R (2015), ‘Differential effects of ydj1 and sis1 on hsp70-mediated clearance of stress granules in saccharomyces cerevisiae’, RNA 21(9), 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Ban EU, Reid BG, Miranker AD and Horwich AL (1999), ‘Global unfolding of a substrate protein by the hsp100 chaperone ClpA’, Nature 401(6748), 90–93. [DOI] [PubMed] [Google Scholar]
- Weibezahn J, Schlieker C, Bukau B and Mogk A (2003), ‘Characterization of a trap mutant of the AAA+ chaperone ClpB’, J. Biol. Chem. 278(35), 32608–32617. [DOI] [PubMed] [Google Scholar]
- Wentink AS, Nillegoda NB, Feufel J, Ubartaitė G, Schneider CP, De Los Rios P, Hennig J, Barducci A and Bukau B (2020), ‘Molecular dissection of amyloid disaggregation by human HSP70’, Nature 587(7834), 483–488. [DOI] [PubMed] [Google Scholar]
- Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R and Pelkmans L (2013), ‘Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling’, Cell 152(4), 791–805. [DOI] [PubMed] [Google Scholar]
- Xu H (2018), ‘Cochaperones enable hsp70 to use ATP energy to stabilize native proteins out of the folding equilibrium’, Sci. Rep. 8(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sarbeng EB, Vorvis C, Kumar DP, Zhou L and Liu Q (2012), ‘Unique peptide substrate binding properties of 110-kda heat-shock protein (hsp110) determine its distinct chaperone activity’, J. Biol. Chem. 287(8), 5661–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H, Triandafillou C and Drummond DA (2019), ‘Cellular sensing by phase separation: Using the process, not just the products’, J. Biol. Chem. 294(18), 7151–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HY, Ziegelhoffer T, Osipiuk J, Ciesielski SJ, Baranowski M, Zhou M, Joachimiak A and Craig EA (2015), ‘Roles of intramolecular and intermolecular interactions in functional regulation of the hsp70 j-protein co-chaperone sis1’, J. Mol. Biol. 427(7), 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Krakowiak J, Patel N, Beyzavi A, Ezike J, Khalil AS and Pincus D (2016), ‘Dynamic control of hsf1 during heat shock by a chaperone switch and phosphorylation’, Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żwirowski S, Kłosowska A, Obuchowski I, Nillegoda NB, Piróg A, Ziętkiewicz S, Bukau B, Mogk A and Liberek K (2017), ‘Hsp70 displaces small heat shock proteins from aggregates to initiate protein refolding’, EMBO J. 36(6), 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Literature data (Excel file), Related to Figure 6
Data Availability Statement
All raw and processed data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOI is listed in the key resources table.
All custom scripts for data processing, analysis, and figure generation have been deposited at Mendeley Data and are publicly available as of the date of publication. The original python code for simulation is available on GitHub. The git address and DOI are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Pab1p | EnCor Biotechnology | Cat#MCA-1G1; RRID: AB_2572370; Lot#020407 |
| Mouse monoclonal anti-Pgk1 | Thermo Fisher | Cat#459250 |
| Mouse monoclonal anti-GFP | Proteintech | Cat#66002-1-Ig |
| Rabbit polyclonal anti-GFP | Thermo Fisher | Cat#A-11122 |
| Rabbit polyclonal anti-luciferase | MilliporeSigma | Cat#L0159 |
| IRDye 800CW Donkey anti-rabbit IgG | LI-COR | Cat#92532213 |
| IRDye 800CW Donkey anti-mouse IgG | LI-COR | Cat#92532212 |
| Bacterial and virus strains | ||
| Subcloning Efficiency™ DH5α Competent Cells | Thermo Fisher | Cat#18265017 |
| BL21(DE3) Competent Cells | MilliporeSigma | Cat#69450 |
| Chemicals, peptides, and recombinant proteins | ||
| SUPERase·In™ RNase Inhibitor | Thermo Fisher | Cat#AM2694 |
| Creatine phosphokinase | MilliporeSigma | Cat#C3755 |
| Creatine phosphate | GoldBio | Cat#C-323 |
| Protease Inhibitor Cocktail Set IV | MilliporeSigma | Cat#539136 |
| Pierce™ Protease Inhibitor Tablets, EDTA-free | Thermo Fisher | Cat#A32965 |
| Polyadenylic acid (Poly(A)) | MilliporeSigma | Cat#3 |
| RNase If | NEB | Cat#M0243S |
| 5-FAM-HHHHHHLPETGG peptide | Biomatik | custom |
| GGGK(FAM) peptide | Biomatik | custom |
| GGGK(FAM)AANDENYALAA peptide | Biomatik | custom |
| BSA | MilliporeSigma | Cat#12659 |
| Critical commercial assays | ||
| ONE-HOUR Western™ Basic Kit (Mouse) | GenScript | Cat#L00205 |
| Clarity™ Western ECL Substrate | Bio-Rad | Cat#1705060 |
| Luciferase assay system | Promega | Cat#E1500 |
| Deposited data | ||
| Genome of S288C (SGD: R64-2-1) | Saccharomyces Genome Database | https://www.yeastgenome.org/ |
| Raw and processed experimental data | This paper | doi:10.17632/wvztspxbbd.1 |
| Simulation data | This paper | doi:10.17632/wvztspxbbd.1 |
| Experimental models: Organisms/strains | ||
| Saccharomyces cerevisiae strain BY4741: background strain S288C; genotype: MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | ATCC; Brachmann et al., 1998 | ATCC 201388 |
| BY4741 yGFP-luciferase:URA | Cherkasov et al., 2013 | VCY245 |
| BY4741 Δhsp104 yGFP-luciferase:URA | Cherkasov et al., 2013 | VCY325 |
| Oligonucleotides | ||
| Fluorescein-A19 RNA: 5’ 6-FAM-AAAAAAAAAAAAAAAAAAA | IDT | N/A |
| Atto550-A19 RNA: 5’ ATTO550-AAAAAAAAAAAAAAAAAAA | IDT | N/A |
| Kozak RNA: 5’ 6-FAM-ACCUCUGCCGCCGCCAUGG | IDT | N/A |
| Recombinant DNA | ||
| Plasmid: 6xHis-(TevC)-Pab1-clover in pET28a backbone | Riback and Katanski et al., 2017 | pESN07 |
| Plasmid: 8xHis-(TevC)-Pab1 in pET28a backbone | Riback and Katanski et al., 2017 | pJAR006 |
| Plasmid: 8xHis-(TevC)-Pab1ΔP in pET28a backbone | Riback and Katanski et al., 2017 | pJAR015 |
| Plasmid: 6xHis-(SUMO)-Ssa2 in pET28a backbone | This manuscript | pHY024 |
| Plasmid: 6xHis-(SUMO)-Ssa4 in pET28a backbone | This manuscript | pHY025 |
| Plasmid: GroEL D87K in pDMI.1 backbone | Axel Mogk; Weibezahn et al., 2003 | N/A |
| Plasmid: 6xHis-(SUMO)-GroEL D87K in pET28a backbone | This manuscript | pHY044 |
| Plasmid: 6xHis-(SUMO)-Photinus pyralis Luciferase in pET28a backbone | This manuscript | pHY045 |
| Plasmid: 6xHis-(SUMO)-Ssa1 in pET28a backbone | This manuscript | pEP051 |
| Plasmid: 6xHis-(SUMO)-Sse1 in pET28a backbone | This manuscript | pEP053 |
| Plasmid: 6xHis-(SUMO)-Hsp104 in pET28a backbone | This manuscript | pEP055 |
| Plasmid: 6xHis-(SUMO)-Ydj1 in pET28a backbone | This manuscript | pEP056 |
| Plasmid: 6xHis-(SUMO)-Sis1 in pET28a backbone | This manuscript | pEP057 |
| Plasmid: 6xHis-(SUMO)-Hsp26 in pET28a backbone | This manuscript | pEP065 |
| Plasmid: ClpP-6xHIS in pQE70 backbone | Martin et al., 2005 | N/A |
| Plasmid: 6xHIS-ClpXΔN- ClpXΔN- ClpXΔN | Martin et al., 2005 | N/A |
| Plasmid: 6xHis-(SUMO)-Hsp104(G739I S740G K741F T746A) in pET28a backbone | This manuscript | pJB548 |
| Plasmid: 6xHis-(SUMO)-Hsp104 Y257A in pET28a backbone | This manuscript | pJB551 |
| Plasmid: 6xHis-(SUMO)-Hsp104 Y662A in pET28a backbone | This manuscript | pJB552 |
| Plasmid: srtA-6xHIS in pET28a backbone | Guimaraes et al. 2013 | N/A |
| Plasmid: srtAheptamutant-6xHIS in pET30b backbone | Hirakawa et al., 2015 | N/A |
| Plasmid: 8xHis-(TevC)-GG-Pab1 in pET28a backbone | This manuscript | pJB545 |
| Plasmid: 8xHis-(TevC)-Pab1-srtA-strep in pET28a backbone | This manuscript | pJB547 |
| Plasmid: 8xHis-(TevC)-GG-Pab1ΔP in pET28a backbone | This manuscript | pJB774 |
| Plasmid: 8xHis-(TevC)-Pab1ΔP-srtA-strep in pET28a backbone | This manuscript | pJB775 |
| Software and algorithms | ||
| Image Lab ver. 6.1.0 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Image Studio Lite ver. 5.2.5 | LI-COR | https://www.licor.com/bio/image-studio-lite/ |
| RStudio ver. 1.2.1335 | RStudio Team, 2018 | http://www.rstudio.com/. |
| Jupyter Notebook ver. 6.0.3 | Kluyver et al., 2016 | https://jupyter.org/ |
| DYNAMICS ver. 7.1 | Wyatt | N/A |
| SparkControl™ ver. | TECAN | N/A |
| UNICORN ver. 5.31 | GE Healthcare | N/A |
| Custom R (version 3.5.2) scripts for data processing, analysis, and figure generation | This paper | doi:10.17632/wvztspxbbd.1 |
| Custom Python (version 3.7.7) code for simulation | This paper | https://github.com/haneulyoo/sim_disagg_2021 |
| Other | ||
| NBS™ 384 well microplates (white; flat bottom) for luciferase assays | Corning | Cat#CLS3574 |
| NBS™ 384 well microplates (black; flat bottom) for anisotropy assays | Corning | Cat#CLS3575 |
| Nunc™ Sealing Tapes for microplates | Thermo Fisher | Cat# 235307 |
| Whatman® Anotop® 10 syringe filter for DLS experiments | MilliporeSigma | Cat# WHA68091002 |