

Abstract
Neurotransmitter release is mediated by proteins that drive synaptic vesicle fusion with the presynaptic plasma membrane. While SNAREs form the core of the fusion apparatus, additional proteins play key roles in the fusion pathway. Here, we report that the C-terminal amphipathic helix of the mammalian accessory protein, complexin (Cpx), exerts profound effects on membranes, including the formation of pores and the efficient budding and fission of vesicles. Using nanodisc-black lipid membrane electrophysiology, we demonstrate that the membrane remodeling activity of Cpx modulates the structure and stability of recombinant exocytic fusion pores. Cpx had particularly strong effects on pores formed by small numbers of SNAREs. Under this condition, Cpx increased the current through individual pores 3.5-fold, and increased the open time fraction from ~0.1 to ~1.0. We propose that the membrane sculpting activity of Cpx contributes to the phospholipid rearrangements that underlie fusion by stabilizing highly curved membrane fusion intermediates.
INTRODUCTION
Complexins/synaphins (Cpx) comprise a family of four small cytosolic proteins (134–160 residues) in vertebrates. Cpx-I and Cpx-II are the main isoforms expressed in brain, whereas Cpx-III and Cpx-IV are expressed at lower levels1. These two major isoforms play a crucial role in synaptic vesicle (SV) exocytosis by interacting with both membranes and SNARE complexes, yet their precise mechanism of action remains the subject of debate2–4. In D. melanogaster and C. elegans, Cpx functions as a fusion clamp that inhibits spontaneous SV exocytosis prior to Ca2+ influx, but the preponderance of data indicate that it does not function as a fusion clamp in mammalian synapses5–7; this is attributed to evolutionary divergence8. Despite its complicated role in spontaneous release, there is a clear consensus that Cpx plays a positive role in evoked SV release, in both invertebrates and vertebrates6, 9–12. Herein, we primarily focus on the ability of Cpx to promote fusion. A number of potential fusion-promoting mechanisms have been envisioned, including: Cpx and the Ca2+ sensor synaptotagmin-1 cooperate to prime trans-SNARE complexes for exocytosis13, 14, Cpx promotes the docking of vesicles by binding SNAREs via its accessory and central helices while binding membranes via a C-terminal amphipathic helix15–18, or Cpx couples SVs to Ca2+ channels12, 19. Here, we report robust membrane sculpting properties of Cpx that contribute to its ability to stimulate membrane fusion. We demonstrate that the Cpx C-terminus promotes membrane fusion by deforming phospholipid bilayers and cooperating with trans-SNARE complexes to stabilize the open state of fusion pores.
RESULTS
The Cpx amphipathic C-terminus can form pores in membranes
Recently, we developed a nanodisc-black lipid membrane (ND-BLM) planar lipid bilayer electrophysiology approach to interrogate individual SNARE-mediated fusion pores20. This assay was later expanded to incorporate synaptotagmin 1 (syt1), and to address the action of NSF and α-SNAP21. Given the flexibility and high temporal resolution of the ND-BLM system, it is ideally suited to examine the impact of additional accessory proteins, such as Cpx, on trans-SNARE complexes. Surprisingly, while performing SNARE-free control experiments, we unexpectedly found that Cpx-II (hereafter referred to as Cpx) alone created pores in the BLM (Fig. 1a). A titration revealed that 50 nM Cpx created brief openings (flickers) in the BLM (Fig. 1a, left panel). At 500 nM Cpx, pores were initially unstable but often evolved into stable, reproducible pores over time, yielding an average unitary current of 8±1 pA (Fig. 1a, right panel), and average opening and closing rates of 0.04 and 2.26 (1/ms), respectively. From the current-voltage plot, the Cpx-alone pores had an estimated diameter of 1.3 ± 0.1 nm (Fig. 1b).
Fig 1. Cpx forms pores in bilayers via its C-terminal amphipathic helix.
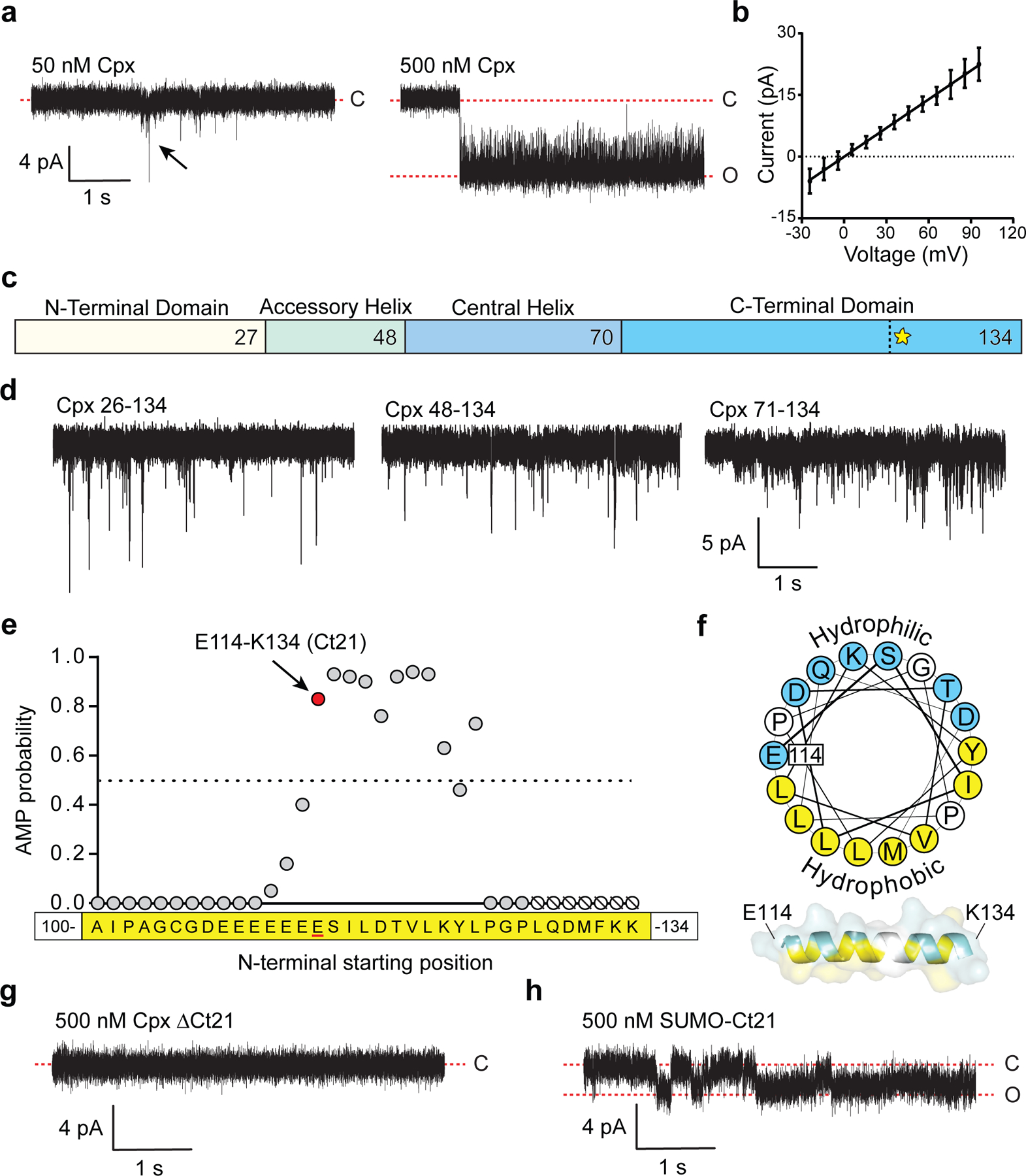
(a) Cpx-mediated pore formation in BLMs. Left panel: 50 nM Cpx forms transient pores. A transient pore is indicated by the black arrow. Right panel: 500 nM Cpx forms stable open pores that generate 8±1 pA current. (b) Current-Voltage (I/V) plot derived from three individual pores, formed by 500 nM Cpx. Error bars represent standard error of the mean. (c) Illustration depicting the four Cpx domains. The vertical dotted line and yellow star within the C-terminal domain denotes the beginning of the C-terminal amphipathic α-helix starting at residue E114. (d) Representative BLM recordings after treatment with N-terminally truncated (Δ1–25, Δ1–47 or Δ1–70) Cpx (500 nM). (e) Results of the antimicrobial peptide prediction analysis of the Cpx C-terminus, starting with peptide A100-K134. Each point represents a peptide that begins at the indicated residue, X-K134. The prediction algorithm requires at least eight residues, therefore residues 128–134 onward (shown as null symbols) could not be determined. See Table 1 for additional peptide comparisons. (f) Helical wheel projection of the Cpx amphipathic helix starting at E114. Hydrophobic residues are shown in yellow and hydrophilic residues are shown in blue. A model of Cpx residues E114-K134 as an amphipathic α-helix is shown below. (g) BLM recordings after addition of 500 nM Cpx ΔCt21. (h) Same as panel G, but using 500 nM recombinant SUMO-Ct21 fusion protein. The BLM recordings were repeated three to five times for each condition with consistent results. For all BLM experiments in this figure the lipid mixture was DOPC-DOPS (80:20).
Cpx comprises four distinct domains: N-terminal domain, accessory helix, central helix, and the C-terminal domain (Fig. 1c). By successively truncating each domain from the N-terminus, we found that the C-terminal domain alone (residues 71–134) was sufficient to create pores in the BLM (Fig. 1d). Interestingly, the Cpx C-terminus was previously reported to stimulate SNARE-mediated vesicle fusion in a reconstituted system22. To further narrow down the critical pore-forming residues, we used a machine-learning algorithm that predicts antimicrobial peptides (AMP)23. The algorithm assesses the ability of peptides to generate negative Gaussian curvature in lipid bilayers. By analyzing successively shorter Cpx C-terminal sequences, beginning with C-terminal peptide A100-K134, the last twenty-one residues of Cpx (E114 to K134) emerged as the putative pore forming motif (Fig. 1e). Indeed, the pore forming probability score for the Cpx C-terminus was comparable to other known amphipathic pore forming peptides, such as the bee venom peptide, melittin (Fig. 1e and Table 1). Scrambling these 21 residues, or removal of the two terminal lysines, reduces the pore forming probability from 0.83 to 0.07 and 0.43, respectively (Table 1). The propensity for Cpx to form pores is also supported by a helical wheel projection that illustrates how the C-terminus forms an amphipathic helix (Fig. 1f)24 upon insertion into membranes17, a characteristic property of a number of pore forming peptides25, 26. To validate the functional role of the last twenty-one residues from Cpx (Ct21), we purified a C-terminal truncation mutant (Cpx ΔCt21) and found that pore formation in the BLM was completely abolished (Fig. 1g). Importantly, we also found that a recombinant fusion protein, consisting of the Ct21 peptide fused to the C-terminus of a SUMO domain (SUMO-Ct21), was sufficient to form pores (Fig. 1h). These results were further corroborated by using both recombinant full length Cpx, as well as a synthetic Ct21 peptide, in an iGluSnFR-based optical assay that measures glutamate efflux from liposomes (Extended Data Fig. 1a), and an antimicrobial assay based on E. coli spheroplasts (Extended Data Fig. 1b).
Table 1. The Cpx Ct21 peptide has an AMP probability score similar to known pore forming peptides.
The listed peptides were queried for AMP probability using a machine-learning algorithm, described by Lee et al (2016).
| Peptide | Amino acid sequence | AMP probability |
|---|---|---|
| Cpx Ct21 (114–134) | ESILDTVLKYLPGPLQDMFKK | 0.83 |
| Ct21 scramble | YLQLKDLPKEVSGTPDLIKMF | 0.07 |
| Cpx Nt21 (1–21) | MDFVMKQALGGATKDMGKMLG | 0.11 |
| Cpx (114–132) | ESILDTVLKYLPGPLQDMF | 0.43 |
| Melittin | GIGAILKVLATGLPTLISWIKNKRKQ | 0.81 |
| Arenicin 3 | GFCWYVCYRNGVRVCYRRCN | 0.99 |
| α-synuclein (1–93) |
MDVFMKGLSKAKEGVVAAAEKTKQGVAEAAGK TKEGVLYVGSKTKEGVVHGVATVAEKTKEQVTN VGGAVVTGVTAVAQKTVEGAGSIAAATG |
0.99 |
| ARF1 (1–14) | MGNIFANLFKGLFG | 0.93 |
| Matrix protein 2 (48–63) | FKCIYRRFKYGLKRGP | 1.00 |
| Magainin 2 | GIGKFLHSAKKFGKAFVGEIMNS | 0.95 |
| δ-lysin | MAQDIISTIGDLVKWIIDTVNKFTKK | 0.99 |
| Buforin II | TRSSRAGLQFPVGRVHRLLRK | 0.89 |
| Perforin (22–42) | PCHTAARSECKRSHKFVPGAW | 0.91 |
MD simulations of pore formation by the Cpx Ct21 helix
To gain insights into the stoichiometry, structure, and dynamics of pores formed in bilayers by the Ct21 peptide, we conducted molecular dynamics (MD) simulations. We found that after initially positioning the Ct21 peptide adjacent to the membrane (Fig. 2a), it spontaneously inserted into the bilayer, beginning with L124 (Fig. 2b). Increasing the number of Ct21 peptides in the MD simulations resulted in a larger and more stable pore (Fig. 2c). As indicated by the movement of water molecules (shown in red) across the bilayer, a minimum number of nine peptide molecules were needed to occasionally form small, transient pores (Fig. 2d). A ten-peptide simulation showed the formation of a disordered toroidal pore, while a twelve-peptide simulation revealed the formation of a stable 2.6 ± 0.3 nm toroidal opening in the bilayer (Fig. 2c, 2e and 2f). Notably, the amphipathic Ct21 helix contains a proline-glycine kink (P125,G126); this AMP feature has been shown to favor toroidal pore formation27. After determining that twelve peptides are required to form a stable pore, we then assessed the ability of fewer than twelve Ct21 peptides to stabilize a pre-existing pore. This was done by initially forming a twelve-peptide pore, followed by sequentially removing peptides, one at a time, while monitoring the pore diameter (Fig. 2g). This analysis showed a sharp reduction in pore diameter by removal of a single peptide, resulting in an average pore diameter of 1.6 ± 0.3 nm when eleven peptides are present. Interestingly, as few as four peptides were sufficient to stabilize a pre-formed pore (Fig. 2g). Together, these results validate that the Cpx Ct21 helix can create pores at high copy number, while at low copy number, the Ct21 helix instead acts as a pore stabilizing molecule.
Fig 2. MD simulations of Cpx Ct21 peptides with lipid bilayers.
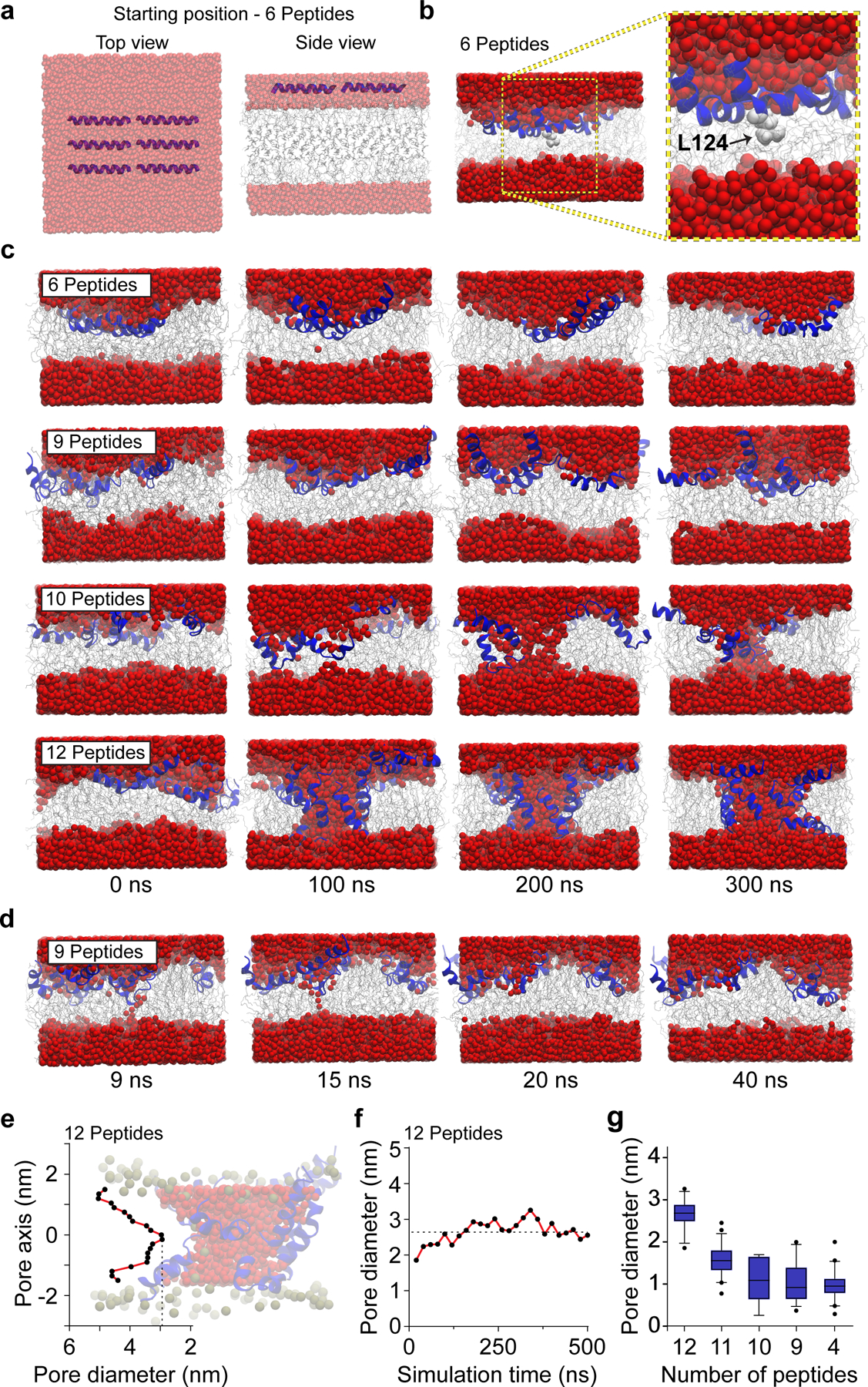
(a) HMMM model showing top and side views of six Ct21 peptides at the starting position of the MD simulation. Throughout the figure, water is shown as red spheres, lipids are grey, and Ct21 peptides are rendered in blue. Note: images in panel A are semi-transparent to aid in visualization of the Ct21 peptides within the layer of water molecules. (b) Six Ct21 peptide MD simulation snapshot and zoomed inset after 15 ns showing membrane insertion begins with L124 (light grey). (c) MD simulations snapshots between 0 and 300 ns with six, nine, ten or twelve Ct21 peptides. (d) MD simulation snapshots of nine Ct21 peptides in the bilayer, taken between 9 and 40 ns. Narrow transient pores, that water molecules occasionally traversed, were observed. (e) Quantification of the pore diameter, at a single point in the simulation, along the length of the twelve peptide Ct21 pore passing though the bilayer. Zero on the x-axis indicates the midpoint of the toroidal pore. A representative twelve peptide pore is also shown. (f) Quantification of the smallest diameter of the twelve peptide Ct21 pore throughout the 500 ns simulation. (g) Quantification of pore diameter from MD simulations with reducing Ct21 peptide number. A twelve-peptide pore was initially formed, followed by removal of single peptides, stepwise, to examine how reducing peptide number affects pore properties. The average pore diameter from 12, 11, 10, 9 and 4 peptides are 2.6 ± 0.3, 1.6 ± 0.3, 1.1 ± 0.5, 1.1 ± 0.4 and 1.0 ± 0.3 nm, respectively. The box and whisker plot was generated with 5th (min) and 95th (max) percentiles and an interquartile range representing the 25th and 75th percentiles. The center line represents the median.
Cpx dramatically remodels phospholipid bilayer structure
To further visualize the impact of Cpx on membranes, we developed a giant (~20 μm) unilamellar vesicle (GUV)-based assay (Fig. 3a). GUVs were loaded with recombinant HaloTag protein, and a fluorogenic, membrane impermeant HaloTag ligand (JF635i) was added to the media28 (Fig. 3a, Extended Data Fig. 1c and 1d). Initially, a fluorescent signal is absent since the JF635i ligand and HaloTag protein are separated by the intact GUV membrane (Fig. 3 and Extended Data Fig. 1e). Pore formation allows the JF635i dye to enter the vesicles and bind the HaloTag protein, causing an increase in luminal fluorescence, as exemplified using melittin (Fig. 3b). Consistent with the pore formation and leak assay data described above (Fig. 1 and Extended Data Fig. 1a), we found that Cpx created pores in the GUVs in a concentration dependent manner (Extended Data Fig. 1e). Surprisingly, this assay revealed a second, striking effect: Cpx and Ct21 dramatically remodel the bilayers as the concentration increased above 2 μM (Fig. 3c and Movie S1). Budding and fission of small vesicles from the surface of the GUVs was readily apparent. In contrast, Cpx ΔCt21 did not create pores or alter GUV morphology (Fig. 3c). We observed membrane remodeling activity when using mammalian Cpx-I, Cpx-II, C. elegans Cpx and D. melanogaster (DmCpx) Cpx-7B. Interestingly, DmCpx 7A, which lacks a prominent C-terminal amphipathic helix, did not form pores or cause bending and fission29 (Extended Data Fig. 1f). In agreement with these findings, the AMP score for DmCpx 7A and 7B were determined to be 0.43 and 0.99, respectively. The differential effects induced by DmCpx Cpx 7A and 7B in our GUV assay are also consistent with an in vivo report demonstrating that DmCpx 7B exhibited a two-fold increase in evoked neurotransmitter release, compared to DmCpx 7A43. It should be noted that DmCpx 7A is prenylated in vivo, and this post-translational modification will likely influence how this isoform interacts with membranes in vivo.
Fig 3. Cpx forms pores and remodels GUV membranes.
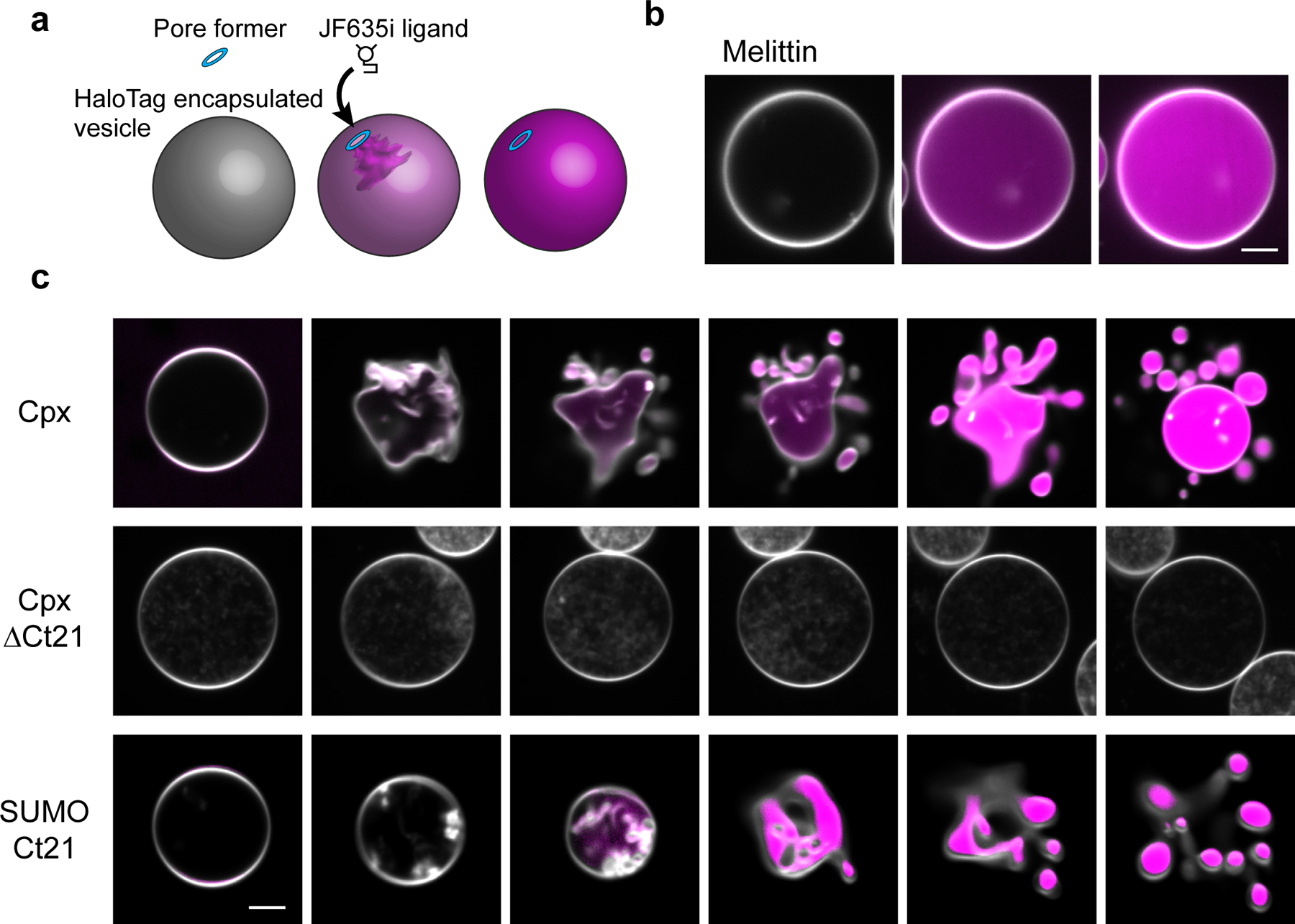
(a) Illustration of the GUV-pore formation assay. Recombinant HaloTag protein is encapsulated in the GUVs and the JF635i ligand is present in the media. Pore formation allows the ligand to enter the GUVs and bind the HaloTag protein, causing an increase in JF635i fluorescence (magenta). (b) Representative example of the GUV pore formation assay using melittin. GUV lipids are labeled with 0.1 % rhodamine-PE (white). Scale bar = 5 μm. (c) Progressive 5 second image series of GUVs after treatment of 5 μM Cpx, Cpx ΔCt21 or SUMO domain with the Cpx C-terminal helix, SUMO-Ct21. Pore formation and vesiculation was consistently observed in >10 repeated trials from >3 independent protein preparations. Scale bar = 5 μm.
Next, to further address the generality of the C-terminal amphipathic helix in regulating Cpx function, we appended the 26 residues that comprise melittin onto the end of Cpx ΔCt21 (Cpx ΔCt21-melittin). This construct restored pore forming (Extended Data Fig. 1f) and membrane remodeling activity (unpublished observations, Kevin C. Courtney, Edwin R. Chapman). We note that among the accessory factors the regulate fusion, this sculpting activity appears to be unique to Cpx, as myriad accessory proteins that are involved in SV exocytosis had no discernable effect on GUVs, including the cytoplasmic domain of syt1, which has been shown to bend membranes via electron microscopy (EM) (Extended Data Fig. 1g)30, 31.
To investigate the membrane sculpting activity of Cpx sub-optically, we performed EM on 100 nm large unilamellar vesicles (LUVs) with and without Cpx treatment (Fig. 4, Extended Data Fig. 2 and 3). Negative stain EM showed that a 10-minute incubation of both full-length Cpx and SUMO-Ct21 reduced the LUVs to ~30 nm nanoparticles (Fig. 4a); some LUVs were distorted into irregular shapes (Extended Data Fig. 2). Cpx ΔCt21, however, had no effect on the LUVs (Fig. 4a). Next, by rapid freezing (vitrification)32 directly after mixing, we captured intermediate stages of Cpx-mediated membrane remodeling via electron cryo-tomography (cryo-ET). This strategy allowed us to regularly observe small vesicles in the process of budding off the surface of LUVs (Fig. 4c and Extended Data Fig. 3b). It is noteworthy that these small vesicles produced by Cpx appear similar to the cryo-EM results reported by Malsam et al. (2012), albeit with an alternative interpretation33. In addition to simple vesiculation, we also found large networks of lipidic material emanating from the LUVs (Fig. 4d and Extended Data Fig. 3c). In another striking class of intermediates, Cpx apparently twisted the LUVs into highly curved structures (Fig. 4e and Extended Data Fig. 3d).
Fig 4. Cpx vesiculates, fragments and twists membranes.
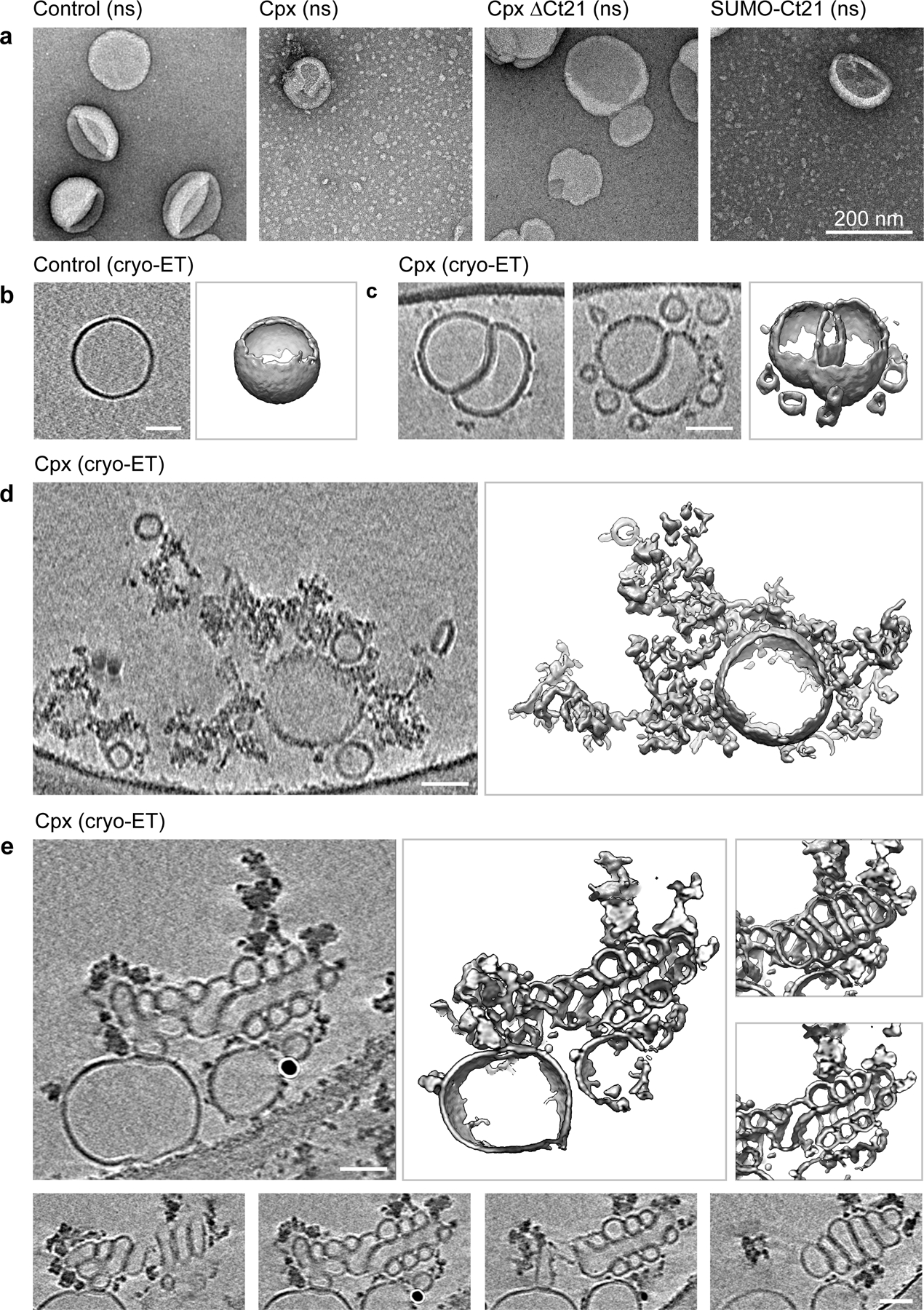
(a) Negative stain (ns) transmission electron microscopy of 100 nm LUVs following a ten-minute incubation with 10 μM Cpx, Cpx ΔCt21 or SUMO-Ct21; untreated LUVs served as a control. These experiments were repeated three times with consistent results. (b-e) Electron cryo-tomography (cryo-ET) of 100 nm LUVs with and without 10 μM Cpx. All panels show 2-nm thick virtual cross-sections of the reconstructed volumes on the right and surface representations of the segmented features on the left. All scale bars represent 50 nm. The experiment was repeated twice. (b) A typical LUV in the control samples (see Extended Data Fig. 3a for another example). (c-e) Typical features observed in LUVs after treatment with 10 μM Cpx. (c) Two slices 20 nm apart (left and center panels) and surface representation (right panel) of two interdigitated LUVs, budding off several smaller vesicular structures. Extended Data Fig. 3b shows more examples of this kind. (d) LUVs fracturing into membranous networks. Another example is shown in Extended Data Fig. 3c. (e) Formation of highly curved, twisted structures. The lower panels show successive slices through the relevant feature demonstrating the unusual, somewhat regular twist causing this membrane arrangement. Another example of this type of feature is shown in Extended Data Fig. 3d.
Cpx can form pores in the plasma membrane of mammalian cells
Next, we tested the ability of recombinant Cpx to form pores in the plasma membrane of mammalian cells. For this, we performed cell-attached patch-clamp electrophysiology on HEK-293T cells (Extended Data Fig. 4a). The patch pipette was first filled with a small volume of buffer, followed by backfilling the pipette with recombinant WT Cpx or Cpx ΔCt21 protein; this strategy provides time to establish a sealed patch on the membrane before Cpx diffuses to the cell surface34. In 30% of the trials, we found that WT Cpx caused positive current to flow through the plasma membrane, suggesting the formation of pores (Extended Data Fig. 4b). These large and reversible pores, preceded by brief openings (Extended data fig. 4c), could remain stably open for tens of seconds (Extended Data Fig. 4b). In line with the BLM data described above, we also found that exogenous Cpx ΔCt21 failed to form pores in the plasma membrane of HEK-293T cells (Extended Data Fig. 4b). The limited success rate for pore formation by WT Cpx in these experiments might be due to the presence of inhibitory factors or non-canonical interactions on the crowded surface of cells.
Regulation of Cpx membrane remodeling activity
Our data show that the membrane sculpting ability of Cpx is highly potent, so this activity is likely to be regulated in neurons. Possible regulatory mechanisms include phosphorylation and protein-protein interactions. It was previously shown that Cpx is phosphorylated within its C-terminal amphipathic helix at position S115 by casein kinase 2 in brain extracts4, 35. Cpx T119 is also strongly predicted to be a phosphorylation site for protein kinase C36, 37. In D. melanogaster, Cpx-7B was similarly shown to be phosphorylated in the C-terminal amphipathic helix at position S126 by protein kinase A29. Considering a charge substitution in the amphipathic helix (L117K) of Cpx was reported to inhibit membrane insertion24, and that an S115D substitution was found to block Cpx-mediated increases in SUV-SUV lipid-mixing22, C-terminal Cpx phosphorylation could be a dynamic mechanism to regulate SV release. Indeed, we found that a recombinant phosphomimetic Cpx mutant (S115, T119D) was much less effective at forming pores and deforming membranes (Extended Data Fig. 5a–5c). In addition to phosphorylation, we also found, as proof of principle, that a known amphipathic helix binding protein, calmodulin (CaM)38, binds the Cpx C-terminal helix in a Ca2+ promoted manner (Extended Data Fig. 5d and 5e). Accordingly, we found that CaM fails to protect against Cpx-mediated membrane leakage in EGTA; however, in the presence of Ca2+, CaM effectively blocked the pore forming properties of Cpx (Extended Data Fig. 5b and 5c). By regulating the activity of the C-terminal helix in vivo, phosphorylation and/or protein-protein interactions could enable Cpx to act specifically at the site of fusion, thus avoiding aberrant pore formation throughout the cell.
The Cpx stabilizes the open state of fusion pores
Finally, after characterizing how Cpx forms pores and deforms membranes, we revisited our initial objective to examine the effect of Cpx on the dynamics of nascent fusion pores using the ND-BLM system. We had initially concluded that Cpx was not compatible in this system because it created pores in the BLM by itself, thus confounding measurements of trans-SNARE fusion pores (Fig. 1a). Our MD simulations data suggested that we could overcome this issue by limiting the local number of Cpx molecules at the site of fusion (Fig. 2). To this end, we generated a recombinant transmembrane domain (TMD) anchored Cpx chimera (TMD-Cpx) that could be co-reconstituted with syb2 into NDs (Fig. 5a and 5b). The fusion protein is connected by a 30-residue linker that is expected to provide ample length and flexibility for Cpx to engage with trans-SNARE complexes (Fig. 5c). This strategy ensures that Cpx can only act upon the t-SNARE BLM at the site of a ND-BLM trans-SNARE complex, at a copy number that does not allow Cpx pore formation (Fig. 2). We note that the lack of pore formation by tethered Cpx was experimentally validated via BLM recordings (Extended Data Fig. 6b).
Fig 5. The Cpx C-terminal amphipathic helix stabilizes the open state of nascent fusion pores formed by trans-SNARE complexes.
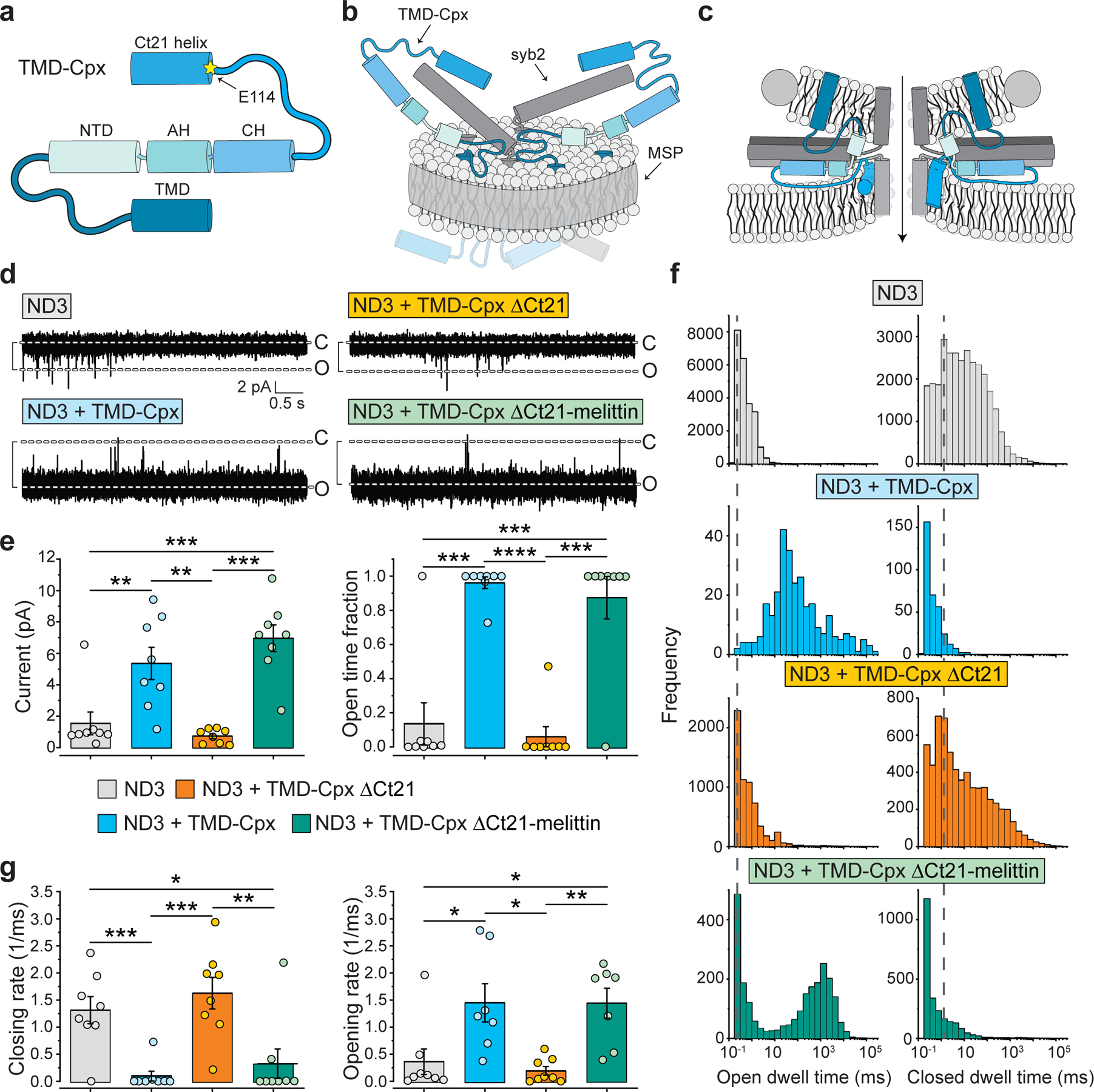
(a) Illustration of the TMD-Cpx fusion protein with each domain annotated. The protein comprises the TMD from CD4 and a 30-residue flexible linker, followed by: the N-terminal domain (NTD), the accessory helix (AH), the central helix (CH) and the C-terminal domain (CTD) of Cpx. The yellow asterisk indicates the location of the Ct21 truncation, beginning at residue E114. (b) Depiction of the 13 nm TMD-Cpx NDs used in this study. Cpx is shown in blue, syb2 shown in dark grey and the membrane scaffold protein (MSP) shown in light grey. Conditions include syb2 alone, syb2 + TMD-Cpx, syb2 + TMD-Cpx ΔCt21, and syb2 + TMD-Cpx ΔCt21-melittin; for all experiments in this figure, three copies of syb2 (ND3) and three copies of the indicated Cpx construct were co-reconstituted into the NDs. (c) Cross-section of the trans-SNARE complex formed between the TMD-Cpx ND and the t-SNARE BLM. Since the precise location of Cpx at the fusion pore site is unknown, a vertical (right) and horizontal (left) representation of the Cpx Ct21 is shown. A gradient of dark, medium, and light grey depicts SNAP-25B, syb2 and syntaxin1A, respectively. The MSP is shown as a grey circle. The black arrow indicates the ionic conduction path. (d) Representative open pore traces from ND-BLM recordings that were acquired with each type of ND. Seven to eight individual pores, each from independent experiments, were analyzed for each condition. (e) Left panel, quantification of the current (pA) through trans-SNARE ND-BLM fusion pores formed by ND3 alone (grey), ND3 + TMD-Cpx (blue), ND3 + TMD-Cpx ΔCt21 (orange) and ND3 + TMD-Cpx ΔCt21-melittin (green). Right panel, quantification of the fraction of time that the trans-SNARE ND-BLM fusion pores remained open for each condition. Color and label annotations also apply to panels f and g. (f) Open and closed dwell time distributions generated from trans-SNARE ND-BLM fusion pores for each condition. (g) Fusion pore opening and closing rates, respectively, derived from the closed and open dwell time analyses. **** denotes p-value < 0.0001; *** denotes p-value < 0.001; ** denotes p-value < 0.01; * denotes p-value < 0.05, determined from two-sided t-test analysis.
In line with our previous work20, 21, NDs with three copies of syb2 (ND3) formed small and transient pores with a current of approximately 1–2 pA, and an open time fraction of ~0.1 (Fig. 5d and 5e). The instability of ND3 pores make this condition especially sensitive to factors that affect their kinetic properties. Strikingly, incorporating three TMD-Cpx molecules into ND3 greatly increased the size of fusion pores, as evidenced by an increase in the average current to 6 pA (Fig. 5e, left panel). Under these conditions, TMD-Cpx also dramatically stabilized fusion pores in the open state, yielding an open time fraction of nearly 1.0 (Fig. 5e, right panel). Moreover, TMD-Cpx caused a significant shift in the open dwell time distribution, to >100-fold longer open times (Fig. 5f), and the closed dwell time distribution was shortened by 10-fold. Further kinetic analysis revealed that the opening and closing rates of pores with TMD-Cpx were significantly increased and decreased, respectively, as compared to the syb2 alone control (Fig. 5g). These findings indicate that TMD-Cpx has two effects on ND3 fusion pores, it lowers the energy barrier for opening, and increases the barrier for closing.
To determine whether Cpx-mediated stabilization of fusion pores involved Ct21-mediated membrane remodeling activity, we generated and reconstituted a TMD-anchored truncation mutant, lacking this motif (TMD-Cpx ΔCt21), into ND3. As described above, this truncation completely abolished the intrinsic pore forming properties of the soluble variant (Fig. 1, 3 and Extended Data Fig. 1). In the ND-BLM system, we found that the Ct21 truncation also eliminated the pore stabilizing activity of TMD-Cpx; all parameters that we analyzed were returned to the syb2-alone control levels (Fig. 5). Thus, Cpx does indeed stabilize nascent fusion pores through the Ct21 amphipathic helix (Fig. 5d–5g).
To further explore the importance of the amphipathic nature of the C-terminal tail of Cpx, we tested a TMD anchored version of the Cpx ΔCt21-melittin chimera (TMD-Cpx ΔCt21-melittin), described above, in the ND-BLM assay. Since melittin is a bona fide amphipathic pore-forming molecule on its own39, we first tested whether ND3 TMD-Cpx ΔCt21-melittin exhibited any SNARE-independent pore forming properties; pores were not observed in the absence of t-SNAREs in the BLM (Extended Data Fig. 6c). Hence, limiting the number of melittin molecules in the ND abrogated pore formation, as expected39. As a further control, t-SNAREs were subsequently incorporated into the BLM (in the presence of ND3 TMD-Cpx ΔCt21-melittin) and robust SNARE-mediated fusion pores were observed (Extended Data Fig. 6c). Quantitative analysis revealed that ND3, with three copies of TMD-Cpx ΔCt21-melittin, completely rescued the ΔCt21 defect (Fig. 5). TMD-Cpx ΔCt21-melittin significantly increased the fusion pore current and stabilized the pore in the open state (Fig. 5d–5g), similar to the levels using full-length TMD-Cpx. Together, these ND3 data demonstrate that WT Cpx stabilizes SNARE-mediated fusion pores via interactions between the Ct21 amphipathic helix and membranes.
Truncated Cpx destabilizes robust SNARE-mediated fusion pores
Although the ND3 studies are particularly useful for examining factors that stabilize nascent fusion pores, this condition is less well suited to study inhibitory factors. In the next series of experiments, we assessed the effect of Cpx on already-stable pores by increasing the copy number of syb2 to five (ND5). To maintain a 1:1 stoichiometry, TMD-Cpx was co-reconstituted at the same copy number. Under these conditions, ND5 alone yielded stable pores with large currents (Fig. 6), as previously reported20, 40. In contrast to ND3, incorporation of TMD-Cpx did not affect the current, open time fraction, or the closed dwell-time distribution (Fig. 6a–c) of ND5 pores, as compared to control. However, the peak of the open dwell-time distribution exhibited a large, 10-fold shift to the right (Fig. 6c). Further analysis revealed that the closing rate of ND5 TMD-Cpx pores was significantly slower (eight-fold) than the control (Fig. 6d). Hence, for both ND3 and ND5, TMD-Cpx stabilizes the open state of SNARE-mediated fusion pores. However, TMD-Cpx exerts a more dramatic effect when acting upon pores formed by a smaller number of SNAREs. In particular, TMD-Cpx affected both the opening and closing energy barriers of ND3 pores, while for ND5 pores, only the energy barrier for closing was affected. Interestingly, increasing either Syb2 or Cpx copy number (starting with the ND3 condition) had similar effects on these energy barriers, suggesting Cpx may compensate when the SNARE copy number is limited.
Fig. 6. Deletion of the C-terminal amphipathic helix converts Cpx to an inhibitor of stable fusion pores.
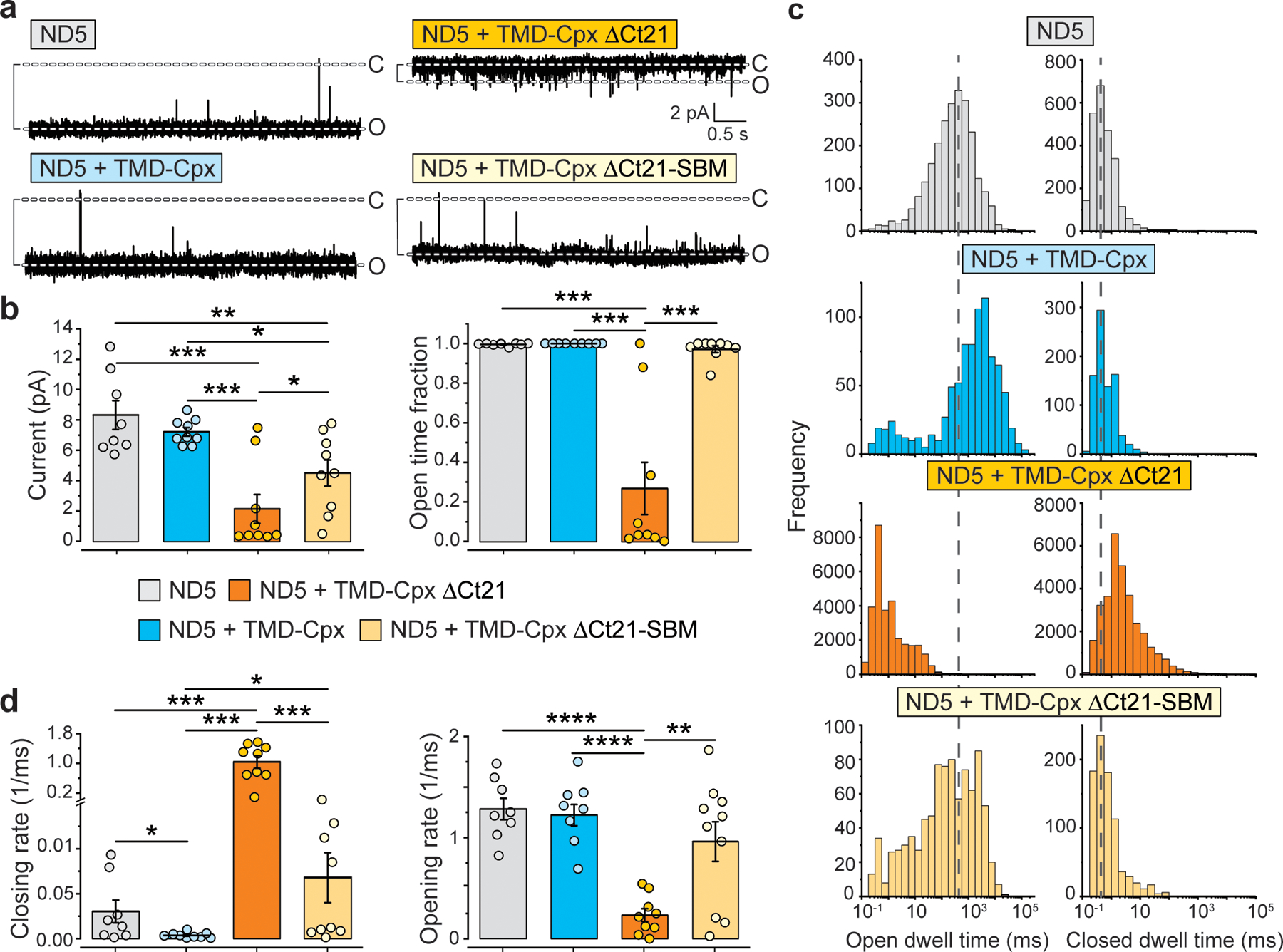
(a) Representative ND-BLM recording of fusion pores formed by the indicated NDs. The NDs were: syb2 alone, syb2 + TMD-Cpx, syb2 + TMD-Cpx ΔCt21, and TMD-Cpx ΔCt21-SBM (SNARE binding mutant, R48L,R59H). Importantly, in these experiments five copies of both syb2 (ND5) and the different Cpx constructs were co-reconstituted into NDs, yielding stable fusion pores, as compared to the ND3 condition in Fig. 5. Eight to ten individual pores, each from independent experiments, were analyzed under each condition. (b) Left panel, quantification of the current (pA) through trans-SNARE ND-BLM fusion pores formed by ND5 alone (grey), ND5 + TMD-Cpx (blue), ND5 + TMD-Cpx ΔCt21 (orange) and ND5 + TMD-Cpx ΔCt21-SBM (beige). The color and label annotations apply to all other panels in this figure. Right panel, quantification of the fraction of time that the trans-SNARE ND-BLM fusion pores remained open for each condition. (c) Open and closed dwell time distributions generated from trans-SNARE ND-BLM fusion pores for each condition. (d) Fusion pore opening and closing rates, respectively, derived from the closed and open dwell time analyses. **** denotes p-value < 0.0001; *** denotes p-value < 0.001; ** denotes p-value < 0.01; * denotes p-value < 0.05, determined from two-sided t-test analysis.
Another striking difference between the ND3 and ND5 conditions was observed when examining the Cpx C-terminal truncation mutant (TMD-Cpx ΔCt21). This construct dramatically reduced the size and stability of ND5 pores to values well below the syb2 alone control (Fig. 6a–d). So, in this ND5 context, TMD-Cpx ΔCt21 was strongly inhibitory. In contrast, when using ND3, deletion of the Ct21 helix served only to silence Cpx such that it had no apparent effect on pores. To ascertain if the reductions in pore size and stability in the ND5 TMD-Cpx ΔCt21 condition involved interactions with SNAREs, we made two additional substitutions (R48L,R59H) in the central helix of this Cpx variant (Fig. 5a). These residues align with aspartic acid residues on syb2 (Extended Data Fig. 6a); replacing them disrupts binding of Cpx to the SNARE complex41, 42. Importantly, these mutations completely abrogated the inhibitory activity of the truncated construct (Fig. 5b–5d), revealing that TMD-Cpx ΔCt21 inhibits fusion pores via direct interactions with SNARE proteins.
Our data suggest that when the full-length mammalian Cpx protein is intact, its fusion-promoting activity predominates. These conclusions were further validated by performing an in vitro liposome fusion assay (lipid mixing); when the above TMD-Cpx variants were reconstituted into syb2 containing vesicles, a virtually identical trend was observed. Namely, TMD-Cpx stimulated lipid mixing between v-SNARE and t-SNARE vesicles, TMD-Cpx ΔCt21 inhibited SNARE-catalyzed lipid mixing, and the TMD-Cpx ΔCt21-SBM construct was without effect (Extended Data Fig. 7b–7d). Together, these data indicate that Cpx promotes SV exocytosis by directly engaging the trans-SNARE complex and helping to stabilize nascent fusion pores through insertion of the C-terminal amphipathic helix into bilayers.
DISCUSSION
The literature describing the role of the Cpx C-terminal domain in regulating both spontaneous and evoked neurotransmitter release has been inconsistent43–48. Truncation of the Cpx C-terminus has been reported to either increase15, 43, 44, 48, 49, or have no effect45, 47, on spontaneous release. This might be expected, considering that the involvement of full-length Cpx in clamping spontaneous release is, itself, controversial5, 6, 8. Regarding the effect of C-terminal truncations on evoked release, there are conflicting reports that describe either defects in evoked release44, 46, 48 or no change15, 43, 45, 47. Interestingly, several of the studies that reported rescue of synchronous release in Cpx knockout or knockdown neurons, by expression of C-terminally truncated Cpx, share a common trend towards incomplete rescue15, 43, 45. These findings are also complicated by the potential for mislocalization of Cpx when the C-terminus has been removed44, 47. To date, a consensus regarding the role of the Cpx C-terminus in exocytosis has not been reached.
Here, we took a reductionist approach to examine how Cpx affects the membrane fusion reaction at the single fusion pore level. In doing so, we discovered that Cpx can form pores in bilayers and that this small soluble protein dramatically remodels membrane structure. This activity is highly specific for Cpx, occurs at low μM protein concentrations, and is robust across a variety of lipid compositions (Extended Data Fig. 1a). Although we do not believe Cpx alone forms pores in vivo, we propose that this membrane remodeling activity underlies at least part of the stimulatory effect of Cpx during evoked SV exocytosis, as membrane fusion requires profound changes in bilayer structure. We assign the membrane remodeling activity of Cpx to the last twenty-one residues that likely form an amphipathic helix upon insertion into membranes; this peptide length may be sufficient to completely span across a phospholipid bilayer50. We note that amphipathic helices are found in many other fusion machines, where they are thought to play key roles in the fusion reaction51, 52. We also note that membrane fusion and fission have been suggested to occur via similar intermediate structures53, 54. Thus, it is tempting to speculate that the budding and fission reactions reported here might correspond to intermediates that are formed during fusion.
When considering the dramatic membrane remodeling properties of Cpx, it is surprising that this protein can be overexpressed in neurons and can be abundantly purified from E. coli, without causing toxicity or generating pores. Interestingly, recombinant Cpx forms pores on mammalian cell membranes and lyses spheroplasts when applied exogenously (Extended Data Fig. 1b and 4). This suggests that the activity of the Cpx amphipathic helix must be regulated in neurons. Possible regulatory mechanisms include phosphorylation at S115 and T119D or interference by other intracellular proteins (Extended Data Fig. 5). Indeed, many pore-forming molecules are rendered innocuous by tight intracellular regulation55. Although the precise regulatory mechanism is currently unknown, we speculate that Cpx bound to trans-SNARE complexes is in an active form, while the unbound pool of Cpx is inactivated to prevent the unregulated formation of pores throughout the cell.
Biochemically, Cpx has the distinct property of preferentially binding to partially or fully assembled SNARE complexes as compared to isolated SNARE proteins2. Moreover, in cells it has been suggested that formation of trans-SNARE complexes serves to recruit Cpx, enabling it to facilitate fusion reactions56. Recently a cryo-ET study reported a symmetrical arrangement of six protein assemblies that connected docked SVs to the presynaptic plasma membrane57. If these protein assemblies are indeed trans-SNARE complexes, as proposed, this would serve to limit the number of Cpx molecules at the fusion site to six. Our MD simulations data suggest six Cpx molecules would be inefficient at creating pores on its own in vivo (Fig. 2c). Importantly, the MD simulations also showed that a low copy number of Ct21 peptides serves to stabilize a pre-formed pore (Fig. 2g). As such, rather than directly forming pores, the Cpx C-terminal helix likely promotes membrane fusion by contributing to the initiation and/or stabilization of highly curved intermediate structures (Fig. 5c). Since the Cpx C-terminus is clearly amphipathic (Fig. 1f), it is possible that Ct21 directly lines part of the pore, with one face towards the phospholipid acyl chains and the other face positioned towards the aqueous channel within the pore.
To restrict the pore forming activity of Cpx in our ND-BLM system, we tethered Cpx to syb2-containing NDs using the TMD of CD4. This strategy prevented the formation of spurious Cpx pores in the bilayer (Extended Data Fig. 6) and facilitated 1:1 stoichiometric control of Cpx with trans-SNARE complexes. When Cpx was specifically localized to the nascent fusion pore site, it significantly stabilized the open state in both ND3 and ND5 conditions (Fig. 5 and 6), with a greater stabilizing effect on ND3 pores. We propose that this stabilization effect is due to changes of the energy barrier for fusion pore formation via membrane insertion of the Cpx C-terminal amphipathic Ct21 helix, because: 1) the isolated Ct21 peptide has the intrinsic ability to form pores and remodel bilayers (Fig. 1 and 2), 2) this helix can be substituted for an alternative amphipathic helix, metlittin, with rescued function (Fig. 5), and 3) C-terminal truncation significantly destabilizes the fusion pore open state (Fig. 5 and 6). Indeed, truncation of the C-terminal amphipathic helix eliminated the fusion pore stabilizing activity of TMD-Cpx with ND3 (Fig. 5) and acted as a strong inhibitor with ND5 (Fig. 6).
By further mutating two charged residues (R48L, R59H) in the SNARE-binding interface of Cpx ΔCt21 in the ND5 condition, fusion pores were again large and stable. This result demonstrates that the observed inhibitory function of Cpx ΔCt21 requires SNARE binding activity. As outlined above, although Cpx is primarily considered to be a fusion-promoting factor in mammalian synapses, Cpx has been reported to clamp spontaneous SV fusion in invertebrates58 as well as in some in vitro studies33. However, we did not observe a clamping effect when using the full-length protein. Other studies have shown that the Ca2+ sensor, synaptotagmin 1 (syt1), is the dominant fusion clamp in mammals6, 7, 59, 60.
We also note that syt1 concurrently binds to SNARE complexes14 at a site adjacent to Cpx13. Upon binding Ca2+, four loops of syt1 penetrate the plasma membrane, and this step is essential for evoked release61–64. Although syt1 is widely considered a membrane bending molecule, our data demonstrate that Cpx is a comparatively more robust bilayer bender. In light of these observations, we propose that in response to Ca2+, syt1 undergoes structural changes65 that allow the Cpx amphipathic helix to encounter and perform work upon membranes, at the site of fusion, to help initiate and stabilize the pore. Alternatively, Cpx could also be poised to promote fusion by pre-insertion of the C-terminal helix into the bilayer adjacent to the TMDs of trans-SNARE complexes; this would act to lower the energy barrier for fusion, prior to the influx of Ca2+. Regardless, our data suggest that the C-terminal helix of Cpx engages directly with the nascent fusion pore to promote release. According to this view, Cpx and syt1 are both accessory proteins that, via their membrane insertion activity, enable SVs to rapidly and efficiently release neurotransmitters. Future studies will examine how Cpx and syt1 cooperate to regulate the dynamics of nascent fusion pores using the ND-BLM system.
METHODS
Reagents
1,2-diphytanoyl-sn-glycero-3-phosphocholine (DphPC), 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) were purchased from Avanti Polar Lipids (Alabaster, AL); the JF635i HaloTag ligand was synthesized as described 28. SM-2 absorbent Biobeads were purchases from Bio-Rad. All other reagents were purchased from Fisher Scientific.
Recombinant protein purification
All recombinant proteins were purified from BL21 (DE3) cells grown in LB media. E. coli were grown at 37 °C until an optical density of 0.6–1.2 was reached, depending on the protein being expressed, followed by induction with 250 μM IPTG for three to five hours. Bacteria were pelleted by centrifugation at 6500 RCF for fifteen minutes and resuspended in 50 mM Tris, 300 mM NaCl, 5% glycerol. The cells were lysed by sonication in the presence of a protease inhibitor cocktail tablet (Roche) plus 4 mM 2-mercaptoethanol in the buffer. Lysates were mixed with Triton X-100 (1% vol/vol final) for ~2 hours at 4 °C. Insoluble material was removed by centrifugation at 30,000 RCF for 45 minutes and the supernatant was collected. HaloTagged proteins were expressed as GST-fusion proteins and purified by affinity purification with glutathione Sepharose. GST-HaloTagged proteins were eluted with 20 mM reduced glutathione, followed by buffer exchange using a PD-10 desalting column (Cytiva). Each of the Cpx variants were expressed as SUMO fusion proteins and purified by binding to Ni-NTA or TALON metal affinity resin. The Cpx proteins were liberated from the resin by incubation with 0.5 μM of the SUMO protease, SENP2, at 4 °C overnight. The only exception was SUMO-Ct21, which was eluted by 300 mM imidazole to retain the SUMO domain. Each of the Cpx proteins were further purified by gel filtration through a Superdex 200 Increase 10/300 GL column. In the case of Cpx (L124C), the protein was labeled with a single NBD dye by thiol-reactive IANBD amide as previously described 66. The SNARE proteins synaptobrevin 2, syntaxin1A, SNAP-25B and accessory proteins synaptotagmin, Munc18, α-SNAP, NSF and α-synuclein were purified as previously described 21.
Liposome leakage assays
Giant unilamellar vesicles (GUVs) were prepared by electroformation from a phospholipid film on indium tin oxide (ITO) coated glass slides, as previously described 67. Stock solutions of lipids (1 mM) composed of DOPC/DOPS (80:20) plus 0.1% rhodamine-DOPE. Fifteen μl of the stock lipids were deposited dropwise onto three regions of two ITO slides under a stream of nitrogen gas. A greased PTFE o-ring was then placed around each of the 3 regions of deposited lipid and the o-rings were filled with 1 mM HEPES, 200 mM sucrose (pH 7.4) solution that also contained 10 μM recombinant HaloTag protein or Alexa 647 dye. The second ITO slide was placed on top to seal the o-rings between the two ITO slides. To initiate electroformation, the ITO slides were connected to a function generator by alligator clips and subjected to an alternating sine wave set to 10 Hz and 3 Vpp at 37 °C. After 2 hours, the frequency was changed to 0.5 Hz for 5 – 10 minutes to stimulate the release of vesicles from the glass surface. After electroformation, the ITO slides were separated and the GUV solution was recovered from within the o-rings. The collected GUVs were then washed with an iso-osmolar buffer composed of 20 mM HEPES, 100 mM KCl (pH 7.4) and gently filtered to purify GUVs greater than 3 μm in diameter. Finally, the GUVs were transferred to a Bioinert 35 mm μ-dish (Ibidi USA, Inc) for imaging.
Fluorescence microscopy of GUV fission in Fig. 3 was performed by dual camera imaging using two Hamamatsu Orca Flash 4 cameras on a Nikon Ti2 microscope with a Yokogawa CSU-W1 spinning disk module. The two-camera system facilitated simultaneous 300 msec acquisition of rhodamine-PE and JF635i. Images were acquired using Nikon Elements software. Supplemental GUV figures were acquired using a Zeiss 880 microscope and Zen software. For the encapsulated HaloTag experiments, 2 μM of JF635i HaloTag ligand was added to the media prior to imaging. For consistency, JF635i is displayed in magenta and Alexa 647 fluorescence is yellow throughout. Prior to examining the effect of Cpx, and various other protein controls, on GUV morphology, the proteins were passed through a PD-10 desalting column equilibrated with 20 mM HEPES, 100 mM KCl (pH 7.4).
For experiments with large unilamellar vesicles, the indicated lipids (10 mg) were dissolved in chloroform, dried under a stream of nitrogen and subjected to vacuum for 2 h. The dried lipid films were then hydrated in reconstitution buffer plus 50 mM glutamate at room temperature. Liposomes with encapsulated glutamate were then extruded through a 100 nm polycarbonate filter (Whatman) 50 times using the Mini-Extruder device (Avanti Polar Lipids) and purified through PD-10 desalting columns (GE Healthcare) equilibrated in reconstitution buffer. Purified liposomes were kept on ice and used immediately for leakage assays. The reactions were initiated by mixing with Cpx, Cpx ΔCt21 or synthetic Ct21 peptide, at the indicated concentrations, along with the glutamate sensor iGluSnFR (1 μM). Glutamate leakage from liposomes was determined by monitoring the fluorescence intensity of iGluSnFR using a Biotek Synergy plate reader for 30 mins. After reach run, 0.25% DDM was added to each reaction and data were collected for another 20 mins. The maximal fluorescence signal after the addition of detergent was used to normalize the percentage of glutamate leakage. Data were obtained from three independent trials. Synthetic Cpx peptide was custom synthesized by GenScript.
Spheroplast lysis
E. coli DH5α cells were grown until OD600 of ~0.5 and harvested at 1300 RCF for 10 min. Cells were resuspended in buffer A (20 mM Tris-HCl (pH 8), 100 mM NaCl, 18% sucrose, 2 mM EDTA) and converted into spheroplasts by addition of lysozyme (50 μg/ml) on ice for 1 h. The resulting spheroplasts were then sedimented by centrifugation at 400 RCF for 20 min at 4 °C and washed twice with buffer A. For the lysis assay, spheroplasts were diluted by 10-fold in buffer A and the absorbance at 500 nm was measured, in the presence and absence of the various Cpx treatments, after 30 min. Data were obtained from three independent trials.
Antimicrobial peptide prediction
The Cpx C-terminus was analyzed for antimicrobial activity using an antimicrobial peptide prediction machine-learning algorithm 23. Permission for using this tool was provided by A. Ferguson (University of Chicago). Progressively shorter C-terminal peptides were manually queried, stepwise, by removing one residue at time from the N-terminal side, starting with A100-K134, followed by I101-K134 etc. The probability of antimicrobial activity is displayed for each peptide; the score reflects the entire peptide that was queried. This iterative process was repeated until reaching P127-K134. The algorithm requires at least eight residues; therefore, the final seven data points, shown as null symbols, could not be determined.
Molecular dynamics simulations
A model C-terminal amphipathic helix (Cpx residues 114–134) was built using the UCSF Chimera software 68. Various (6, 9, 10 or 12) copies of the built peptide were placed on top of a lipid bilayer consisting of DOPC and DOPS lipids using CHARMM-GUI 69, 70, with a composition of DOPC:DOPS = 104:26. The lipid-peptide complex system was then solvated using a TIP3P 71 box of size ~9.0 9.0 11.5 nm3. Twenty-six Na+ ions were added to reach charge neutrality. Peptides, lipids, and ions were described using the CHARMM36 force field 72–74. The built structure was first energy minimized using the conjugate gradient method to remove any bad contacts between solvent and solute atoms; this was followed by a 30 ns long simulation in which the lipid bilayer was described using the HMMM model 75 to facilitate protein adsorption on the membrane surface and rapid equilibration of local lipid distribution. The equilibrated HMMM structures were then converted to full lipid models using CHARMM-GUI 76. These full lipid and peptide complex systems were simulated for 300 ns in restrain-free constant-pressure (1 bar) constant-temperature (303 K) simulations. The hydrogen atoms were constrained using the LINCS 77 algorithm to allow a 2 fs time step for integration. The temperature and pressure of the system in both the HMMM and full lipid models were controlled using the Nosè-Hoover thermostat 78, 79 and Parrinello-Rahman barostat 80, respectively. All simulations were performed using the GROMACS-2018.3 package 81.
First, we studied spontaneous pore formation by the peptides with various peptide to lipid ratios; for statistical significance, each peptide:lipid ratio was probed with multiple independent molecular dynamics simulations. A relatively high peptide to lipid ratio (10:130) (Fig. 2) was required to observe the spontaneous pore formation within the simulation time of several hundreds of nanoseconds. We then performed a few controlled simulations to investigate the minimum number of peptides required to stabilize a pore. For this, we gradually decreased the number of peptides from a preformed pore with 12 peptides and observed that a minimum of four peptides was required for the pore to remain stable for at least several hundreds of nanoseconds. Therefore, it is possible that a small number (<9) of peptides can also lead to the formation of membrane pore although the time scale is substantially longer than several hundreds of nanoseconds.
In cases where a membrane pore was observed, we estimated the diameter of the pore along the membrane normal direction, which was taken to be the z-axis. We first divided the water molecules enclosed in the membrane pore into bins of 1.5 Å thickness, and for each bin, we found the smallest circle that enclosed all water oxygens; the diameter of the circle was regarded as the pore diameter for that bin.
Negative stain electron microscopy
One-hundred nm large unilamellar vesicles composed of DOPC/DOPS (80:20) were generated, as described above. The liposomes, formed with 1 mM lipids, were ten-fold diluted and then incubated with or without 10 μM of the various Cpx proteins for one minute. The vesicles were imaged by negative stain electron microscopy as previously described 82.
Electron cryo-microscopy sample preparation and optimization
Various samples were screened to optimize sample concentration and distribution within the grid holes, vitrification quality, and fiducial size and distribution. Fresh samples were used within 1–2 days of preparation. In our hands, the best samples were generated when liposomes were mixed in solution with the complexin and vitrified within 15 minutes, generating four grids, with the 1st grid following a one minute incubation and the 4th one at 14 minutes. In general, we applied 4 μl of (100 μM) liposomes/(10 μM) complexin solution (25 mM Hepes pH 7.4, 100 mM KCl) and 1 μl 15-nm colloidal gold (Ted Pella) to plasma cleaned carbon grids with holes, incubated for 30 sec in a humidified chamber. Excess liquid was manually blotted and the samples were plunge-frozen in liquid nitrogen-cooled liquefied ethane using an in-house designed cryo-plunger. Plunge-frozen samples were stored in liquid nitrogen until they were used. Freezing quality, along with assessment of sample preservation and amenability for cryo-ET investigation, was conducted by an initial screening on a T12 Spirit equipped with a 4Kx4K Eagle camera (ThermoFisher Scientific), operated at a voltage of 120 kV. We screened samples vitrified on various Quantifoil, Protochips, and lacey grids. Through the use of lacey grids (EMS), we established a hole diameter range that best accommodates centering of the samples, leading us to focus on 200 mesh 1.2/1.3 Quantifoil grids for the incubated liposomes with complexin, and 200 mesh 2/2 Protochips for liposomes alone. These conditions allowed the features of interest to be centered within the holes for efficient tomography data collection. Whatman quantitative filter paper Grade 43 (SigmaAldrich), a medium to fast filter paper, with maximum 0.007% ash, provided high quality and homogenous ice. Micrographs were visually inspected for quality of samples, sample density, background, and the presence of fiducial gold.
Cryo-tomography data collection, reconstruction, and volume processing
The cryo-ET data was taken with a Titan Krios (ThermoFisher Scientific) equipped with a field emission gun (XFEG) and operated at 300kV for selected samples from the T12 screening sessions. Before imaging, the grids were incorporated into a AutoGrids grid sample carrier, which was then transferred via a Krios NanoCab transfer device (ThermoFisher Scientific) into the Krios Cryo AutoLoader, the automated sample loader/unloader system of the Titan Krios electron cryo-microscope. Within the Cryo AutoLoader, the AutoGrids were kept at liquid nitrogen temperature at all times. For the first steps in image acquisition after the AutoGrids were transferred into the AutoLoader, grids were screened for usability and quality. Although the sample preparation protocols were optimized, we had to screen for usable grids and grid squares manually due to damage introduced via clipping of the grids into the AutoGrids sample carrier. For the designated grids, tilt series were acquired on a Falcon 3EC direct detection imaging device (ThermoFisher Scientific) under minimal dose conditions using SerialEM automatic data collection software 83. The fidelity and quality of the data collection was monitored with real-time automatic reconstruction protocols implemented in the pyCoAn package (github.com/pyCoAn/distro), an extended python version of the CoAn package 84. Briefly, immediately after acquisition, tilt series were automatically aligned 85 and reconstructed using the Simultaneous Iterative Reconstruction Technique 86. Alignment and reconstruction statistics were used to determine quality scores that were provided in real time during data collection and later used to select the highest quality reconstructions for further analysis. Within each selected grid hole, one tilt series was acquired with 3° steps between −60° and +60°, with an average dose of 90–120 e−/Å2 and a defocus of 5–8 μm. The nominal magnification was set to 18,000 x, resulting in a calibrated pixel size of 0.48 nm in the reconstructions. A total of 242 tomograms from 7 grids from the different preparations were acquired. Segmentation was achieved using automated denoising with 20 rounds of iterative median filtering 87 on tomograms binned by two, followed by semi-automated watershed segmentation 88 within pyCoAn. Rendering was done using Chimera 89 and images of tomogram slices were generated with IMOD 90.
Cell culture and electrophysiology
HEK-293T cells (ATCC) were planted on 12 mm glass coverslips (Warner, Cat# 64–0732) that were coated with poly-D-lysine (Millipore Sigma, Cat# P0899–500MG). Cells were cultured in DMEM (4500 mg/L glucose, L-glutamine, sodium pyruvate and sodium bicarbonate) and 10% (v/v) fetal calf serum at 37 °C until to 80% confluency. Then cells were patched at room temperature using a Multiclamp 700B amplifier (Molecular Devices) as previously described 34. Recording pipettes were pulled from borosilicate glass (Sutter Instruments) and filled with ~1 μl the pipette internal solution composed of (in mM): 125 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 26 mM TEA-Cl, and 10 mM HEPES, (pH 7.3; 295 mOsm), and then back-filled with WT or ΔCt21 Cpx dissolved in the same buffer (the final concentration of aimed protein in pipette is ~10 μM), which gives a 3–5 ΜΩ resistance in a bath solution containing (in mM): 125 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES, pH 7.3 and 299 mOsm. On-cell patched cells were each held at 0 mV for 1–2 hours and data was collected by Digidata 1440A (Molecular Devices) and Clampex 10 software (Molecular Devices) at 5 kHz.
Nanodisc-Black lipid membrane electrophysiology
The Cpx alone BLM experiments were performed by painting 38 mM DOPC/DOPS (80:20) lipids, dissolved in n-decane, across a 150 μm diameter aperture; comparable results were observed with a DphPC/DOPE/DOPS (60:25:15) BLM (not shown). The cis chamber contained 25 mM HEPES pH 7.4, 100 mM KCl and the trans chamber contained 25 mM HEPES pH 7.4, 10 mM KCl. After establishing a capacitance of approximately 100 pS, the cis chamber received 50 nM, 500 nM or 5 μM WT Cpx-II, or 500 nM of either Cpx ΔCt21 or SUMO-Ct21 and the passage of current between cis and trans chambers was monitored over time. With the exception of generating the I/V plot, all experiments were conducted without externally applied voltage. The diameter of the Cpx-mediated pore was calculated as previously described 20, assuming a 5 nm path length.
The ND-BLM electrophysiology was performed and analyzed as previously described 91. Briefly, thirteen nm NDs were reconstituted with either five copies of syb2 or five copies of both syb2 and the various TMD-Cpx proteins. The BLM was formed by DphPC/DOPE/DOPS (60:25:15) lipids, dissolved in n-decane. The cis chamber contained 25 mM HEPES pH 7.4, 100 mM KCl and the trans chamber contained 25 mM HEPES pH 7.4, 10 mM KCl. After establishing a BLM bilayer with a capacitance greater than 100 pS, t-SNAREs were introduced into the BLM by incubation of fusogenic t-SNARE-containing vesicles composed of DOPE/POPG (75:25) in the cis chamber. The NDs were subsequently added into the cis chamber and the advent of current passing through the BLM was monitored over time.
Bulk SNARE-mediated lipid mixing assay
Lipid mixing between t-SNARE vesicles and v-SNARE vesicles, with or without each of the TMD-Cpx constructs, was monitored by the dequenching of Oregon green and Texas Red labeled phospholipids. The t-SNARE and v-SNARE vesicles were each reconstituted into SUVs composed of POPC/DOPE/DOPS (60:20:20) with an initial protein/lipid ratio of 1:400. The v-SNARE vesicles were also supplemented with 1% Oregon Green and Texas Red phosphatidylethanolamine lipids, while the t-SNARE proteoliposomes did not contain either fluorescent lipid. When lipids mix between the two vesicle populations, the fluorescent lipids in the v-SNARE vesicles are diluted in the unlabeled t-SNARE vesicles, thus dequenching the Oregon Green emission. The TMD-Cpx variants were co-reconstituted into the v-SNARE vesicles at the same concentration as syb2 (0.5 μM). The proteins and lipids were first mixed on ice in 25 mM HEPES pH 7.4, 100 mM KCl plus 0.9% CHAPS. The detergent was removed by two-fold diluting the samples in detergent-free buffer containing SM-2 Bio-Beads and incubated on a rotator at 4 ⁰C overnight. Proteoliposomes were then isolated by flotation of the vesicles using an Accudenz step gradient, as previously described 92. Subsequent lipid mixing experiments were performed by mixing t- and v-SNARE vesicles (10:1) in 25 mM HEPES pH 7.4, 100 mM KCl buffer and monitoring the Oregon Green emission over time using a Cytation 1 plate reader (Biotek).
Extended Data
Extended Data Fig. 1. The C-terminal alpha helix of Cpx creates pores in unilamellar vesicles and lyses spheroplasts.
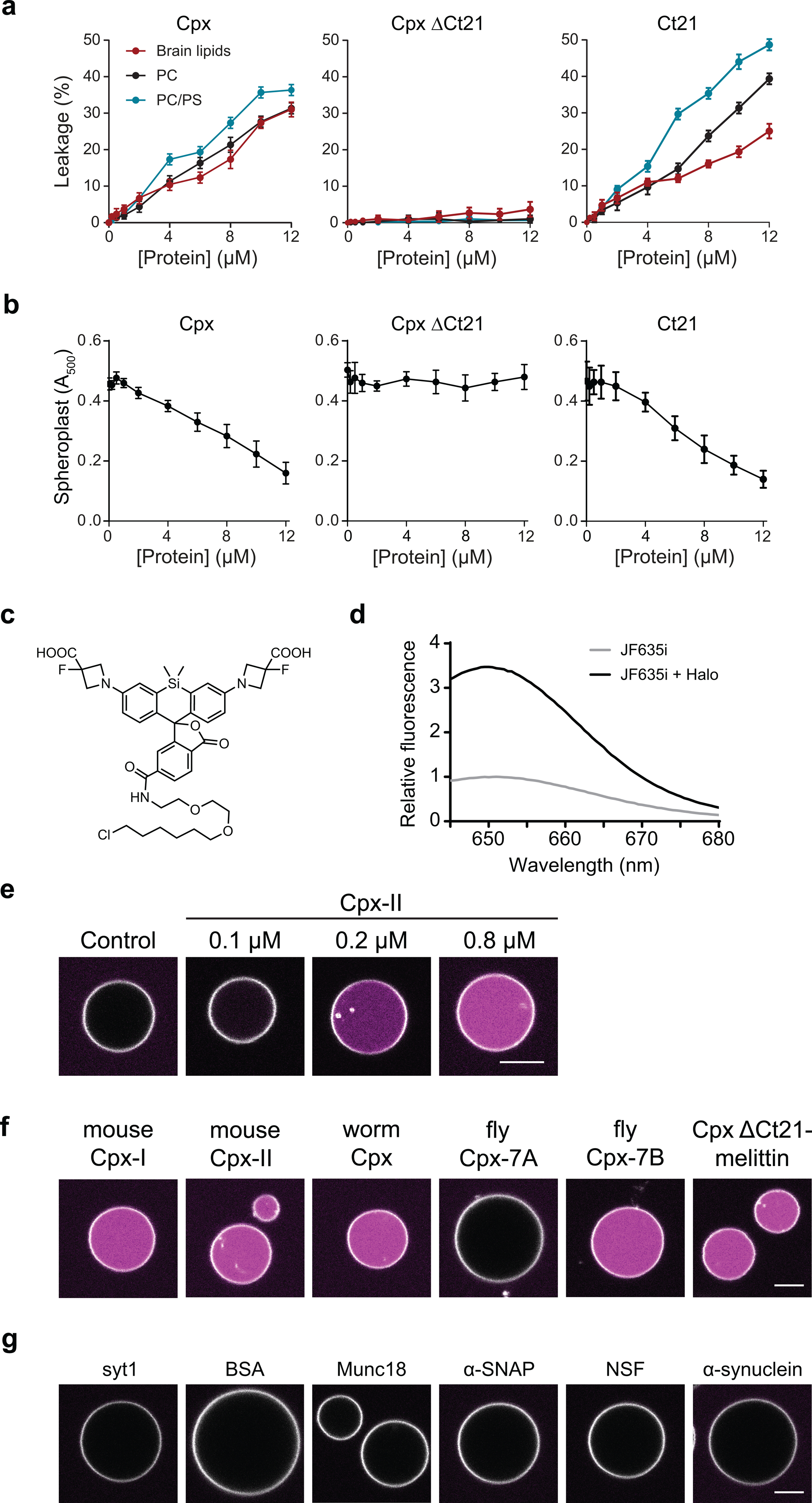
(a) Leakage of glutamate from LUVs composed of PC (black), PC/PS (blue) or total brain lipids (red) after treatment with increasing concentrations of Cpx, Cpx ΔCt21 and Ct21. Glutamate release was monitored via the glutamate sensor, iGluSnFR, in the media (1 μM). (b) Lysis of E. coli spheroplasts with increasing concentrations of Cpx, Cpx ΔCt21 and Ct21 peptide monitored via absorbance at 500 nm. Panels A and B represent the average of three independent experiments. Error bars represent standard error of the mean. (c) Image of the JF635i HaloTag ligand structure. (d) Representative fluorescence emission traces of 5 μM of the JF635i ligand, excited at 635 nm, with and without 5 μM of recombinant HaloTag protein. (e) GUVs labeled with rhodamine-PE (white), and with encapsulated HaloTag protein, before and after treatment with various concentrations of Cpx. JF635i (magenta) is present in the media; this dye strongly fluoresces upon entering GUVs and binding the HaloTag protein. (f) GUV leak assay, described in Figure 3, performed with 1 μM of multiple Cpx isoforms. (g) GUV leak assay performed with 2 μM of the cytoplasmic domain of synaptotagmin 1 (syt1), bovine serum albumin (BSA), Munc18, α-SNAP, NSF or α-synuclein. Each of the GUV conditions were repeated at least three times.
Extended Data Fig. 2. Negative-stain TEM images of miscellaneous structures of Cpx-treated LUVs.

(a) A zoom-out image with four distinct features highlighted by colored boxes that were consistently observed from three independent experiments. Each feature belongs to a category of relevant structures, and more representative cropped images are given in panels b-e: (b) mouth-like opening (pink); (c) ring-like attachment (green); (d) discoidal structures (blue); (e) potential vesicle budding intermediates (yellow). Scale bars: 50 nm.
Extended Data Fig. 3. Electron cryo-tomography of liposomes before and after Cpx treatment.
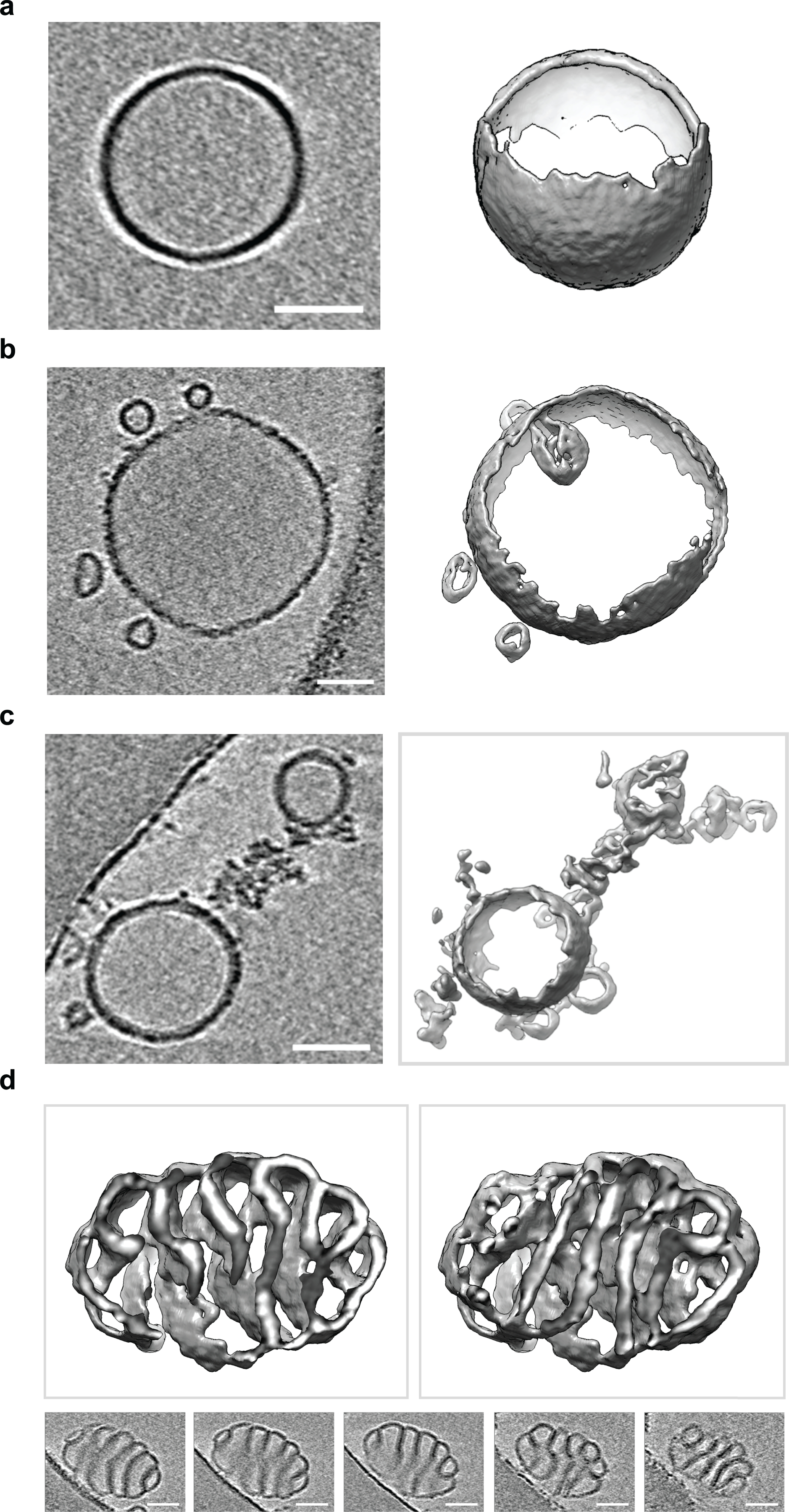
Electron cryo-tomography of 100 nm LUVs with or without treatment of 10 μM Cpx. Three-dimensional surface representations and the corresponding two-dimensional cross sections are shown. In all cases, the scale bar represents 50 nm. The experiment was repeated twice. (a) Representative electron cryo-tomography of untreated 100 nm LUVs. (b) Representative electron cryo-tomography of 100 nm LUVs showing Cpx treatment often induced vesiculation. (c) Representative electron cryo-tomography of 100 nm LUVs showing Cpx treatment commonly causes LUVs to fragment into membranous networks. (d) Representative electron cryo-tomography of 100 nm LUVs showing Cpx treatment can cause LUVs to twist into highly curved structures.
Extended Data Fig. 4. Recombinant Cpx forms pores in the plasma membrane of mammalian cells.
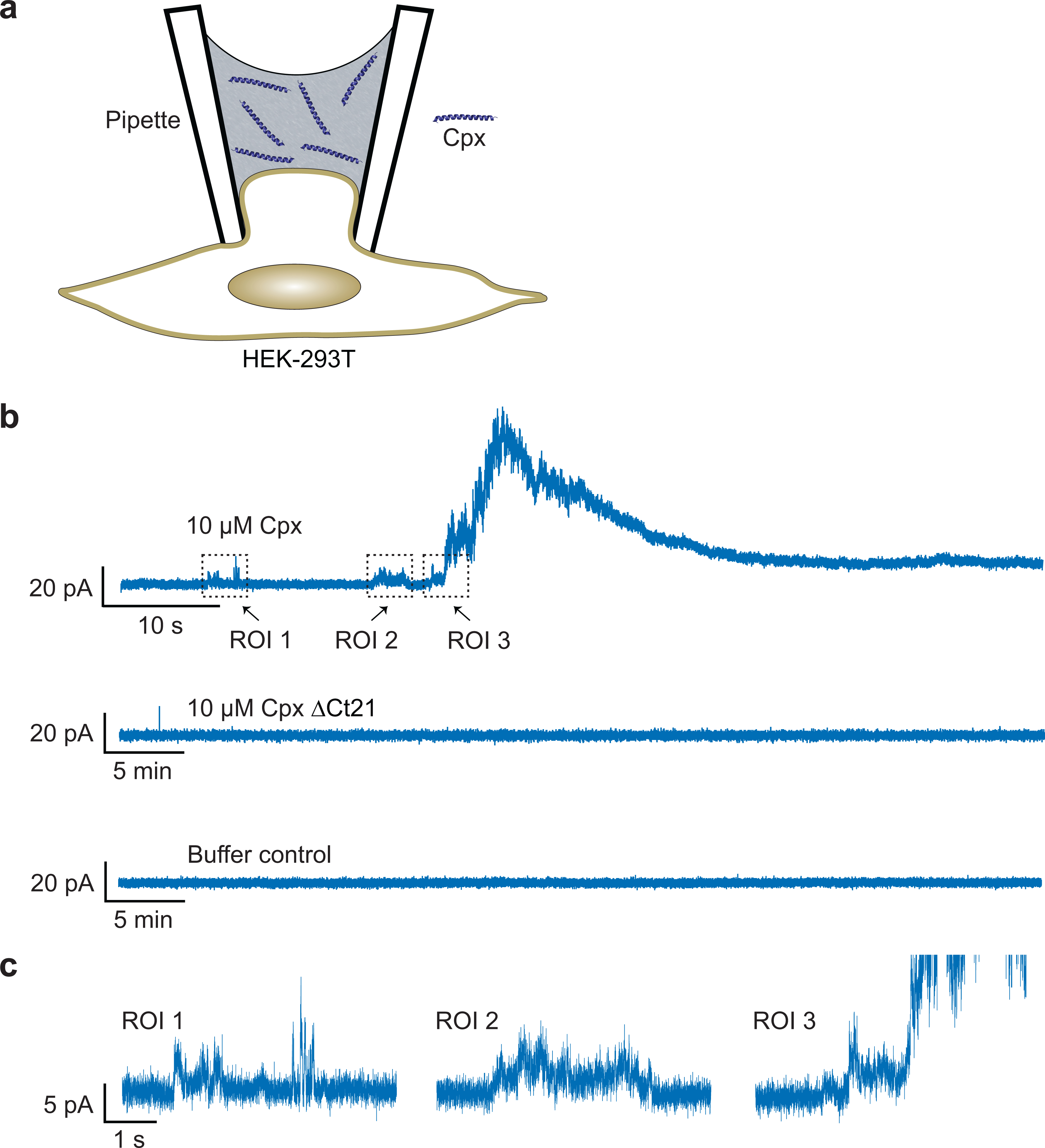
(a) Illustration of the HEK-293T cell patch-clamp setup. A typical cell-attached patch was formed on the surface of the cell, but using a pipette that contained WT full-length or ΔCt21 Cpx. (b) Representative electrophysiological recordings; in 7 out of 21 trials, WT Cpx generated positive currents. In the control or Cpx ΔCt21 conditions, no pores were observed in 5 and 8 trials, respectively. (c) Zoomed-in regions of interest (ROI), from the WT Cpx condition in panel b, showing transient positive currents that precede the large upswing in positive current.
Extended Data Fig. 5. Cpx activity can be regulated by phosphorylation and calmodulin.
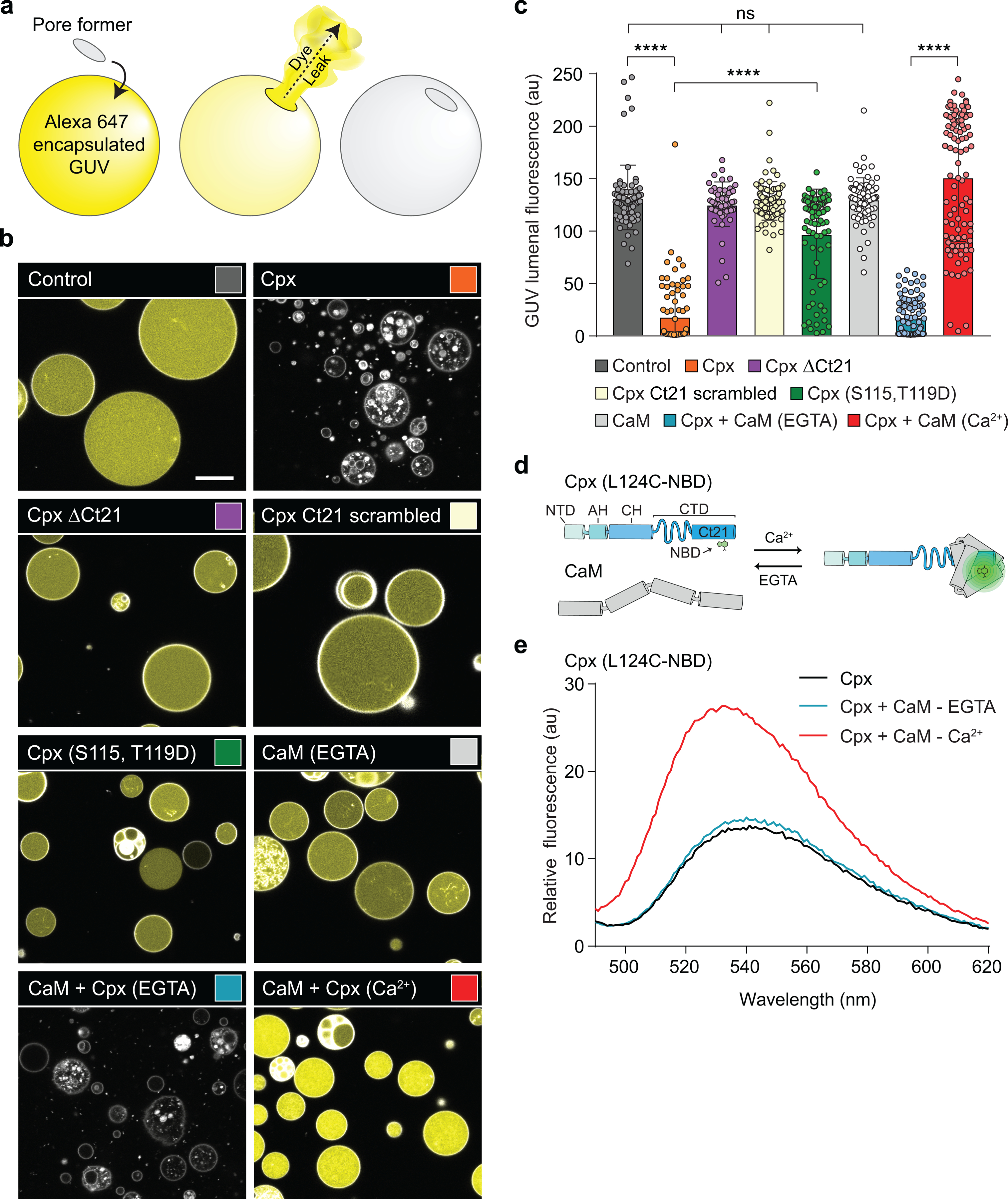
(a) Illustration of the GUV leakage assay using Alexa 647 dye. Rhodamine-PE labelled GUVs, pseudo-colored in white, were formed in the presence of 10 μM Alexa 647 dye, followed by iso-osmolar buffer exchange. Upon pore formation, the dye, pseudo-colored in yellow, escapes from the lumen of the GUVs. b) Representative images of Control GUVs and after treatment with 5 μM Cpx (orange), Cpx ΔCt21 (purple), Cpx Ct21 scrambled (beige), Cpx (S115, T119D) (green), 10 μM Calmodulin (CaM) (light grey) or CaM + Cpx in EGTA (blue) and Ca2+ (red). The indicated conditions were independently repeated >3 times with consistent results. The sequence for the Cpx Ct21 scramble is found in Table 1. The white scale bar represents 10 μm. (c) Quantification of GUV lumenal fluorescence after the treatments described in panel B. The data are pooled from three independent experiments and the errors bars represent standard deviation. **** denotes p < 0.0001 and ns denotes not significant, determined by ANOVA analysis and Tukey multiple comparisons test. (d) Illustration of the Cpx-CaM binding assay. A single cysteine variant (L124C) of Cpx was labeled with an environment-sensitive dye, NBD, and the fluorescence emission was monitored by fluorometer. In the presence of Ca2+, CaM binds the Cpx C-terminal helix, causing an increase in fluorescence (e) Representative fluorescence spectrum of Cpx (L124C-NBD). NBD fluorescence in 25 mM HEPES (pH 7.4), 100 mM KCl was monitored without CaM (black) and with CaM in EGTA (blue) or Ca2+ (red). The change in NBD fluorescence in the Ca2+ condition indicates CaM binds the Cpx amphipathic helix.
Extended Data Fig. 6. Five copies of TMD-Cpx and three copies of TMD-Cpx ΔCt21-melittin do not form SNARE-independent pores in the ND-BLM system.
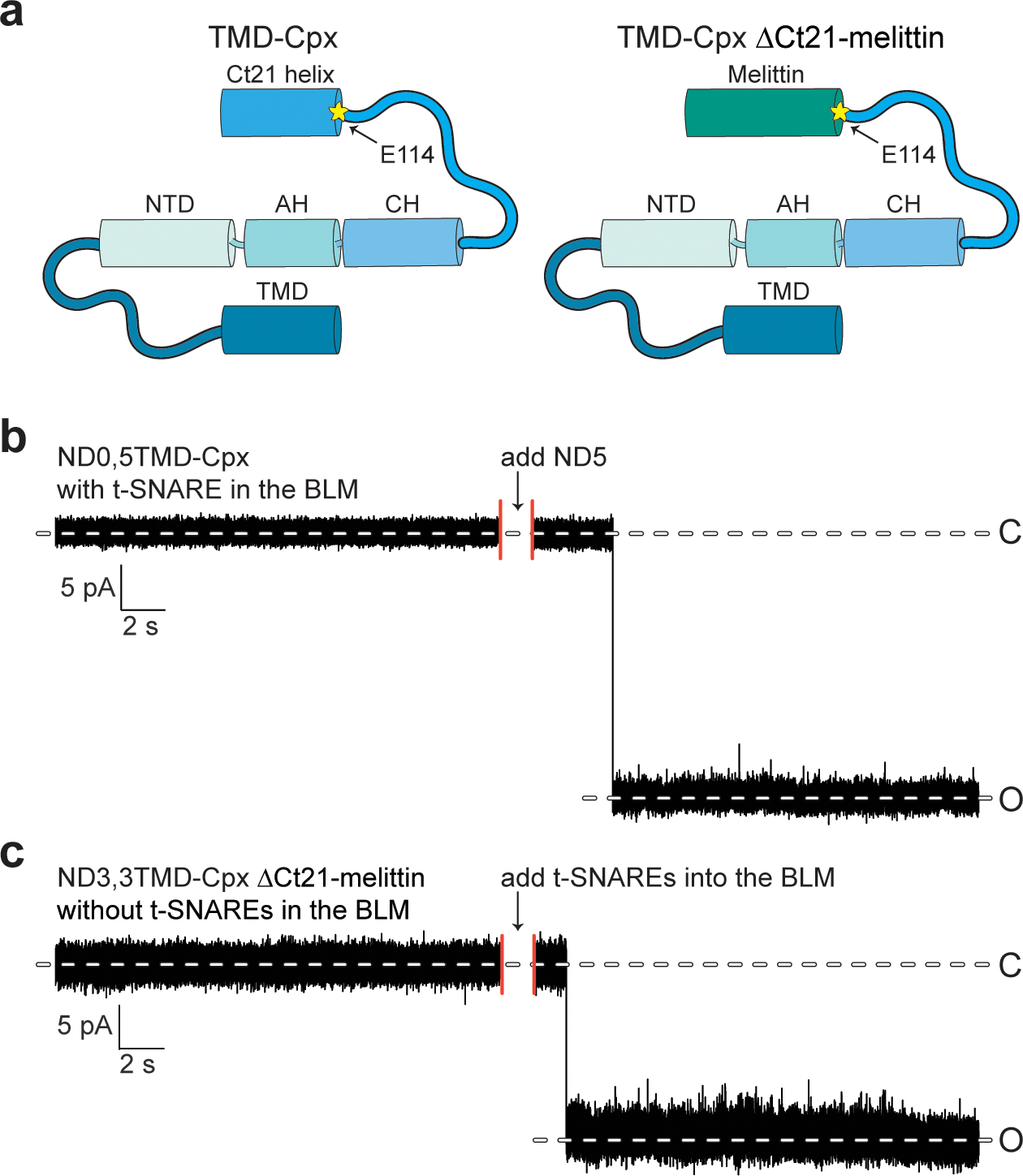
(a) Illustration for TMD-Cpx (left panel) and TMD- Cpx ΔCt21-melittin (right panel). The yellow asterisk indicates residue E114, the beginning of Ct21. (b) Sample trace from TMD-Cpx control experiments in the absence of syb2. Five copies of TMD-Cpx alone (no syb2) were reconstituted in the NDs. NDs with TMD-Cpx alone were added to the BLM system along with t-SNARE-liposomes, no pores formed, after 2 hrs. After NDs with syb2 were introduced, multiple pores opened. (c) Sample trace from TMD-Cpx ΔCt21-melittin experiments in the absence of t-SNAREs. Three copies of syb2 and three copies of TMD-Cpx ΔCt21-melittin were co-reconstituted in the NDs. NDs were introduced to the BLM system, no pore formed, after 2 hrs. After t-SNARE liposomes were added to the same system, multiple pores opened.
Extended Data Fig. 7. Truncation of the Cpx C-terminus causes fusion pore instability and reduces SUV lipid mixing.
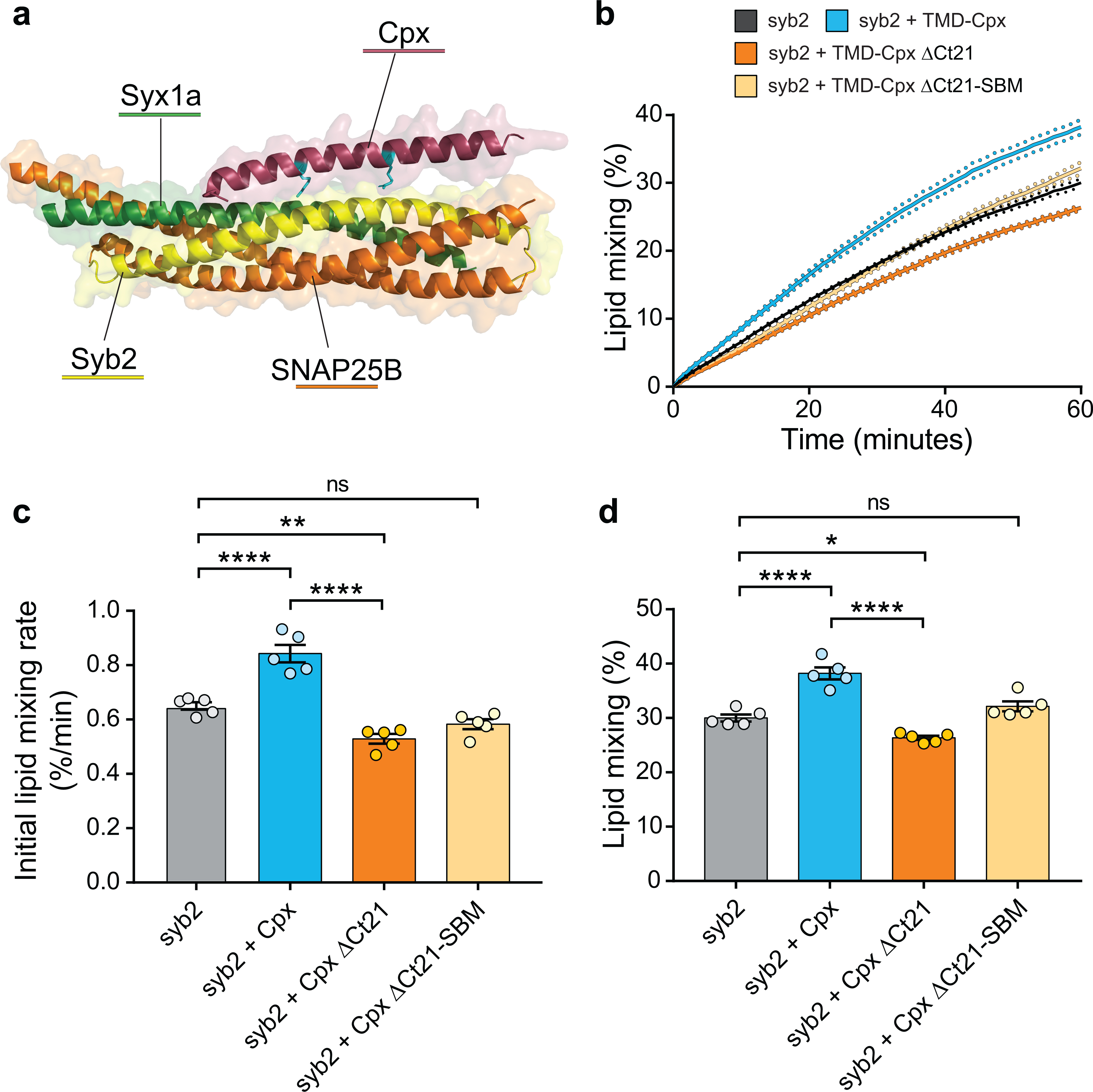
(a) Crystal structure from pdb 1KIL showing Cpx (shown in red) bound to the SNARE complex formed by syb2 (shown in yellow), SNAP25B (shown in orange) and syntaxin 1a (Syx1a) (shown in green). Cpx residues R48 and R59 are shown in cyan. (b) Lipid mixing assay performed with v-SNARE and t-SNARE containing vesicles over time. The t-SNARE SUVs remain constant, while the v-SNARE SUVs were reconstituted as syb2 alone (grey), syb2 + TMD-Cpx (blue), syb2 + TMD-Cpx ΔCt21 (orange) or syb2 + TMD-Cpx ΔCt21-SBM (beige). In addition to the syb2 alone condition, equimolar TMD-anchored Cpx variants were also separately co-reconstituted into the v-SNARE vesicles. The plot shows the average percent of lipid mixing over time from five individual experiments that each contained three technical replicates. The standard error of the mean from each condition is shown as dotted lines. (c) Initial lipid mixing rates for each of the listed conditions, derived from the plots in panel b. (d) Total lipid mixing for each condition after 60 minutes, derived from the plots in panel b. **** denotes p-value < 0.0001; *** denotes p-value < 0.001; ** denotes p-value < 0.01; * denotes p-value < 0.05 and error bars represent standard error of the mean.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the Chapman lab and M.B. Jackson for helpful discussions. We also thank A. Ferguson (University of Chicago) for providing access to the AMP prediction tool and S. Mishra for preliminary work performing HEK cell patch clamp recordings. This work was supported by Pew Charitable Trust Grant 864K625 (E.R.C., D.H.), National Institutes of Health grants MH061876 and NS097362 (E.R.C.), P01-GM121203 (N.V.) and DP2GM140920 (H.B.). Equipment for the cryo-CLEM and in-situ cellular tomography workflow employed in this work was funded by NIH Grants S10-OD012372 (D.H.), S10-OD026926 (D.H.), P01-GM121203 (N.V.), R01-AI132378 (N.V., D.H.) and Pew Charitable Trust grant 864K625. The computational component is supported by the grant NSF-DMS1661900 (Q.C.) Computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number OCI-1053575 (Q.C.), are greatly appreciated; computations are also supported in part by the Shared Computing Cluster, which is administered by Boston University’s Research Computing Services. E.R.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
CODE AVAILABILITY
No unique code was used in this study.
DATA AVAILABILITY
All graphed raw data are provided as a supplemental document. With the exception of RCSB PBD to access PDB: 1KIL, no specific databases or third party data were used in this study.
REFERENCES
- 1.Reim K et al. Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol 169, 669–680 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mcmahon HT, Missler M, Li C & Sudhof TC Complexins - Cytosolic Proteins That Regulate Snap Receptor Function. Cell 83, 111–119 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Reim K et al. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104, 71–81 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Ishizuka T, Saisu H, Odani S & Abe T Synaphin - A protein associated with the docking/fusion complex in presynaptic terminals. Biochemical and Biophysical Research Communications 213, 1107–1114 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Trimbuch T & Rosenmund C Should I stop or should I go? The role of complexin in neurotransmitter release. Nature reviews. Neuroscience 17, 118–125 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Murcia FJ, Reim K, Jahn O, Taschenberger H & Brose N Acute Complexin Knockout Abates Spontaneous and Evoked Transmitter Release. Cell Rep 26, 2521–2530 e2525 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Courtney NA, Bao H, Briguglio JS & Chapman ER Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin. Nature Communications 10, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wragg RT et al. Evolutionary Divergence of the C-terminal Domain of Complexin Accounts for Functional Disparities between Vertebrate and Invertebrate Complexins. Frontiers in Molecular Neuroscience 10, 24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai H et al. Complexin II plays a positive role in Ca2+-triggered exocytosis by facilitating vesicle priming. Proceedings of the National Academy of Sciences of the United States of America 105, 19538–19543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue M et al. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proceedings of the National Academy of Sciences of the United States of America 105, 7875–7880 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue M et al. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron 64, 367–380 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin MY et al. Complexin facilitates exocytosis and synchronizes vesicle release in two secretory model systems. J Physiol 591, 2463–2473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou QJ et al. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548, 420-+ (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chicka MC & Chapman ER Concurrent binding of complexin and synaptotagmin to liposome-embedded SNARE complexes. Biochemistry 48, 657–659 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin JA, Hu Z, Fenz KM, Fernandez J & Dittman JS Complexin has opposite effects on two modes of synaptic vesicle fusion. Current biology : CB 21, 97–105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diao J et al. Complexin-1 enhances the on-rate of vesicle docking via simultaneous SNARE and membrane interactions. Journal of the American Chemical Society 135, 15274–15277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snead D, Wragg RT, Dittman JS & Eliezer D Membrane curvature sensing by the C-terminal domain of complexin. Nature communications 5, 4955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snead D et al. Unique Structural Features of Membrane-Bound C-Terminal Domain Motifs Modulate Complexin Inhibitory Function. Frontiers in molecular neuroscience 10, 154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW & Littleton JT Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 18234–18245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao H et al. Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das D, Bao H, Courtney KC, Wu L & Chapman ER Resolving kinetic intermediates during the regulated assembly and disassembly of fusion pores. Nat Commun 11, 231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malsam J et al. The carboxy-terminal domain of complexin I stimulates liposome fusion. Proceedings of the National Academy of Sciences of the United States of America 106, 2001–2006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee EY, Fulan BM, Wong GCL & Ferguson AL Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proceedings of the National Academy of Sciences of the United States of America 113, 13588–13593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seiler F, Malsam J, Krause JM & Sollner TH A role of complexin-lipid interactions in membrane fusion. Febs Letters 583, 2343–2348 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel H & Jahnig F The structure of melittin in membranes. Biophysical Journal 50, 573–582 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams RW et al. Raman spectroscopy of synthetic antimicrobial frog peptides magainin 2a and PGLa. Biochemistry 29, 4490–4496 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Tuerkova A et al. Effect of helical kink in antimicrobial peptides on membrane pore formation. Elife 9, 38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jonker CTH et al. Accurate measurement of fast endocytic recycling kinetics in real time. J Cell Sci 133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho RW et al. Phosphorylation of Complexin by PKA Regulates Activity-Dependent Spontaneous Neurotransmitter Release and Structural Synaptic Plasticity. Neuron 88, 749–761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hui E, Johnson CP, Yao J, Dunning FM & Chapman ER Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell 138, 709–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martens S, Kozlov MM & McMahon HT How synaptotagmin promotes membrane fusion. Science 316, 1205–1208 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Dubochet J et al. Cryo-electron microscopy of vitrified specimens. Quarterly Reviews of Biophysics 21, 129–228 (1988). [DOI] [PubMed] [Google Scholar]
- 33.Malsam J et al. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J 31, 3270–3281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu ZY et al. Dilation of fusion pores by crowding of SNARE proteins. Elife 6, 26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shata A, Saisu H, Odani S & Abe T Phosphorylated synaphin/complexin found in the brain exhibits enhanced SNARE complex binding. Biochemical and Biophysical Research Communications 354, 808–813 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Wong YH et al. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res 35, W588–W594 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller ML & Blom N Kinase-specific prediction of protein phosphorylation sites. Methods in molecular biology 527, 299–310, x (2009). [DOI] [PubMed] [Google Scholar]
- 38.Chapman ER, Alexander K, Vorherr T, Carafoli E & Storm DR Fluorescence energy transfer analysis of calmodulin-peptide complexes. Biochemistry 31, 12819–12825 (1992). [DOI] [PubMed] [Google Scholar]
- 39.Lee MT, Sun TL, Hung WC & Huang HW Process of inducing pores in membranes by melittin. Proceedings of the National Academy of Sciences of the United States of America 110, 14243–14248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu L, Courtney KC & Chapman ER Cholesterol stabilizes recombinant exocytic fusion pores by altering membrane bending rigidity. Biophys J 120, 1367–1377 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue M et al. Distinct domains of complexin I differentially regulate neurotransmitter release. Nature Structural & Molecular Biology 14, 949–958 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen XC et al. Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397–409 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Kaeser-Woo YJ, Yang XF & Sudhof TC C-Terminal Complexin Sequence Is Selectively Required for Clamping and Priming But Not for Ca2+ Triggering of Synaptic Exocytosis. Journal of Neuroscience 32, 2877–2885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buhl LK et al. Differential regulation of evoked and spontaneous neurotransmitter release by C-terminal modifications of complexin. Molecular and Cellular Neuroscience 52, 161–172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang XF, Cao P & Sudhof TC Deconstructing complexin function in activating and clamping Ca2+-triggered exocytosis by comparing knockout and knockdown phenotypes. Proceedings of the National Academy of Sciences of the United States of America 110, 20777–20782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dhara M et al. Complexin synchronizes primed vesicle exocytosis and regulates fusion pore dynamics. Journal of Cell Biology 204, 1123–1140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong JH et al. C-terminal domain of mammalian complexin-1 localizes to highly curved membranes. Proceedings of the National Academy of Sciences of the United States of America 113, E7590–E7599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makke M et al. A mechanism for exocytotic arrest by the Complexin C-terminus. Elife 7, 25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wragg RT et al. Synaptic Vesicles Position Complexin to Block Spontaneous Fusion. Neuron 77, 323–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharpe HJ, Stevens TJ & Munro S A Comprehensive Comparison of Transmembrane Domains Reveals Organelle-Specific Properties. Cell 142, 158–169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu TY et al. Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proceedings of the National Academy of Sciences of the United States of America 109, E2146–2154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Podbilewicz B Virus and cell fusion mechanisms. Annual review of cell and developmental biology 30, 111–139 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Kozlov MM & Chernomordik LV The protein coat in membrane fusion: lessons from fission. Traffic 3, 256–267 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Daste F et al. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. Embo Reports 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krawczyk PA, Laub M & Kozik P To Kill But Not Be Killed: Controlling the Activity of Mammalian Pore-Forming Proteins. Front Immunol 11, 601405 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.An SJ, Grabner CP & Zenisek D Real-time visualization of complexin during single exocytic events. Nature Neuroscience 13, 577–U583 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radhakrishnan A et al. Symmetrical arrangement of proteins under release-ready vesicles in presynaptic terminals. Proc Natl Acad Sci U S A 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huntwork S & Littleton JT A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nature Neuroscience 10, 1235–1237 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Littleton JT, Stern M, Perin M & Bellen HJ Calcium dependence of neurotransmitter release and rate of spontaneous vesicle fusions are altered in Drosophila synaptotagmin mutants. Proc Natl Acad Sci U S A 91, 10888–10892 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vevea JD & Chapman ER Acute disruption of the synaptic vesicle membrane protein synaptotagmin 1 using knockoff in mouse hippocampal neurons. Elife 9, 24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chapman ER & Davis AF Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. Journal of Biological Chemistry 273, 13995–14001 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Wang P, Wang CT, Bai JH, Jackson MB & Chapman ER Mutations in the effector binding loops in the C2A and C2B domains of synaptotagmin I disrupt exocytosis in a nonadditive manner. Journal of Biological Chemistry 278, 47030–47037 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Bai J, Tucker WC & Chapman ER PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nature Structural & Molecular Biology 11, 36–44 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Liu HS et al. Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nature Neuroscience 17, 670-+ (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bai H et al. Different states of synaptotagmin regulate evoked versus spontaneous release. Nature Communications 7, 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bradberry MM et al. Molecular Basis for Synaptotagmin-1-Associated Neurodevelopmental Disorder. Neuron 107, 52–64 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu L et al. Beyond Amphiphilic Balance: Changing Subunit Stereochemistry Alters the Pore-Forming Activity of Nylon-3 Polymers. J Am Chem Soc (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Z et al. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 179, 269–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Jo S, Lim JB, Klauda JB & Im W CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophysical Journal 97, 50–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 72.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. The Journal of Physical Chemistry B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Best RB et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. Journal of Chemical Theory and Computation 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang J & MacKerell AD Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA & Tajkhorshid E Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J. 102, 2130–2139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qi Y et al. CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model. Biophys. J. 109, 2012–2022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hess B, Bekker H, Berendsen HJC & Fraaije JGEM LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- 78.Nos\’e S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984). [Google Scholar]
- 79.Hoover WG Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- 80.Parrinello M & Rahman A Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 81.Abraham MJ et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015). [Google Scholar]
- 82.Zhang Z & Chapman ER Programmable Nanodisc Patterning by DNA Origami. Nano Letters 20, 6032–6037 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Volkmann N & Hanein D Quantitative fitting of atomic models into observed densities derived by electron microscopy. Journal of Structural Biology 125, 176–184 (1999). [DOI] [PubMed] [Google Scholar]
- 85.Mastronarde DN & Held SR Automated tilt series alignment and tomographic reconstruction in IMOD. Journal of Structural Biology 197, 102–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agulleiro JI & Fernandez JJ Tomo3D 2.0-Exploitation of Advanced Vector eXtensions (AVX) for 3D reconstruction. Journal of Structural Biology 189, 147–152 (2015). [DOI] [PubMed] [Google Scholar]
- 87.van der Heide P, Xu XP, Marsh BJ, Hanein D & Volkmann N Efficient automatic noise reduction of electron tomographic reconstructions based on iterative median filtering. Journal of Structural Biology 158, 196–204 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Volkmann N A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. Journal of Structural Biology 138, 123–129 (2002). [DOI] [PubMed] [Google Scholar]
- 89.Goddard TD, Huang CC & Ferrin TE Software extensions to UCSF Chimera for interactive visualization of large molecular assemblies. Structure 13, 473–482 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Kremer JR, Mastronarde DN & McIntosh JR Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 91.Wu L, Courtney KC & Chapman ER Cholesterol stabilizes recombinant exocytic fusion pores by altering membrane bending rigidity. Biophys J (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tucker WC, Weber T & Chapman ER Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All graphed raw data are provided as a supplemental document. With the exception of RCSB PBD to access PDB: 1KIL, no specific databases or third party data were used in this study.