

Abstract
Familial colorectal cancer type X (FCCTX) is a heterogeneous colorectal cancer predisposition syndrome that, although displays a cancer pattern similar to Lynch syndrome, is mismatch repair proficient and does not exhibit microsatellite instability. Besides, its genetic etiology remains to be elucidated. In this study we performed germline exome sequencing of 39 cancer-affected patients from 34 families at risk for FCCTX. Variant classification followed the American College of Medical Genetics and Genomics (ACMG) guidelines. Pathogenic/likely pathogenic variants were identified in 17.65% of the families. Rare and potentially pathogenic alterations were identified in known hereditary cancer genes (CHEK2), in putative FCCTX candidate genes (OGG1 and FAN1) and in other cancer-related genes such as ATR, ASXL1, PARK2, SLX4 and TREX1. This study provides novel important clues that can contribute to the understanding of FCCTX genetic basis.
Subject terms: Cancer, Genetics, Molecular biology, Molecular medicine, Oncology
Introduction
Hereditary cancer accounts for approximately 20–30% of colorectal cancer (CRC) cases1. Lynch syndrome (LS) is one of the main CRC predisposition syndromes. Familial colorectal cancer type X (FCCTX) can be considered a subgroup of LS. Both are quite similar in their clinical presentation and characterized by several early onset nonpolyposis hereditary colorectal cancer cases in patients and family members from different generations2. From a clinical perspective, both the FCCTX and Lynch families are characterized by the fulfilment of Amsterdam I clinical criteria3. However, FCCTX and LS differ at the molecular level, because LS is caused by mismatch repair genes (MMR) MLH1, MSH2, MSH6, and PMS2 malfunctioning, thus presenting microsatellite instability (MSI—high—high microsatellite instability). Conversely, FCCTX patients do not harbour any alteration in the MMR genes and, as a consequence, carry no alterations in microsatellites, being microsatellite stable (MSS)4. FCCTX tumours are also heterogeneous, occurring mainly as moderately differentiated adenocarcinomas in the rectum and sigmoid regions of the large intestine5. Cancer-related death is up to 10 years higher in FCCTX than in Lynch syndrome patients. Additionally, although the risk for developing a second CRC is higher for Lynch, the risk with FCCTX shows a higher proportional increase. Besides, patients with FCCTX have tumours with diagnosis in advanced ages than in Lynch and have fewer extracolonic tumours6.
The understanding of FCCTX molecular mechanisms is poorly explored. Some genes have already been reported to be potentially associated with FCCTX, namely BMPR1A7, RPS208, SEMA4A9, SETD610, BRCA211, OGG112 and FAN113. Besides, a review study suggested a possible association with CENPE, CHD18, GREM1, BCR, KIF24, GALNT12, ZNF367, HABP4, GABBR2, and BMP414. In addition, a review by Nejadtagui and collaborators15 pointed BRCA2, KRAS, APC, MGMT, BRAF, BMPR1A, RPS20, SEMA4A, and hypermethylation of at least one gene of the MMR system as potentially related to FCCTX. Despite these studies, no defined set of genes is conclusively associated with FCCTX. Therefore, we propose to perform a clinical and molecular characterization of a cohort of families fulfilling the clinical criteria for FCCTX syndrome.
Results
Clinical pathological information
The socio-demographic and histopathological data are summarized in Table 1. Twenty-eight patients were female (71.8%), with an average age of 49 years at diagnosis. Most of the primary tumours were colorectal (92.3%) and these ones were mostly located in the rectum (35.9%). Besides, three patients with colorectal cancer had other extra-colonic primary tumours (endometrium, prostate, and renal).
Table 1.
Socio-demographic and histopathological features.
| Amount (%) | |
|---|---|
| Sex | |
| Female | 28 (71.8) |
| Male | 11 (28.2) |
| Ethnicity (self-declared) | |
| White | 29 (74.3) |
| Brown | 5 (12.8) |
| Black | 3 (7.6) |
| Yellow | 1 (2.5) |
| Unavailable | 1 (2.5) |
| Diagnostic age (average; minimum and maximum) |
Years of age 49.4 (34–77; SD: 11.3) |
| Histology | |
| Adenocarcinoma (NOS*) | 37 (94.9) |
| Unavailable | 2 (5.1) |
| Primary tumour location** | |
| Colorectal | 36 (92.3) |
| Endometrium | 3 (7.7) |
| Location (colorectal) | |
| Rectum | 14 (38.8) |
| Sigmoid | 7 (19.4) |
| Right colon | 7 (19.4) |
| Left colon | 4 (11.1) |
| Transversal colon | 2 (5.5) |
| Unavailable | 2 (5.5) |
| CRC differentiation level | |
| Poorly differentiated | 9 (25) |
| Moderately differentiated | 24 (66.6) |
| Well differentiated | 0 |
| Unavailable | 3 (8.3) |
| CRC TNM stating | |
| I | 8 (22.2) |
| II (A, B, C) | 10 (27.7) |
| III (A, B, C) | 13 (36.1) |
| IV (A, B) | 2 (5.5) |
| Unavailable | 3 (8.3) |
| Status | |
| Follow up (following with no disease) | 28 (71.7) |
| Deceased | 7 (18) |
| Alive in treatment | 1 (2.5) |
| Loss of follow up | 3 (7.7) |
*Not otherwise specified.
**Three patients with colorectal cancer also had prostate, endometrium and renal tumors as second primary tumors.
Family history information
Details about the cancer history of the probands and their relatives are provided in Table 2. For all of the families analysed (n = 34), 22 fulfilled Amsterdam 1 criteria and 10 were quasi-Amsterdam, fulfilling three among the four required criteria (what we called for the purpose of the study as “Amsterdam-1” (minus one)), and two families fulfilled two out of the four criteria (here called “Amsterdam-2”). For three families, two relatives were analysed (ID 17, 23, 26), and for one family, three relatives were included (ID 21).
Table 2.
Family history.
| Family ID* | Criteria fulfilled | Index case cancer (sex, age at diagnosis) | Cancer family history Cancer type (sex, age at diagnosis) |
|---|---|---|---|
| 1 | Amsterdam | Colorectal M41 | Colorectal (M36, M34, F50, M?, M?), Hepatic (M49), Uterine (F50, F45), Urothelial (M?) |
| 2 | Amsterdam | Colorectal M38 | Colorectal (F40, F40) |
| 3 | Amsterdam-1 | Colorectal M35 | Colorectal (M68) |
| 4 | Amsterdam | Colorectal F41 | Colorectal (F45, M55, M40), Lung (M70, M?) |
| 5 | Amsterdam-1 | Colorectal F38 | Colorectal (M70), Breast (F50, F52) |
| 6 | Amsterdam-1 | Colorectal M54 | Colorectal (F42), Digestive (M50, M50, F42) |
| 7 | Amsterdam | Colorectal F46 | Colorectal (M68, M60) |
| 8 | Amsterdam | Colorectal F40 | Colorectal (M49, F?, M50, F69, M?), Gastric (F65), Uterine (F28) |
| 9 | Amsterdam | Colorectal M62 | Colorectal (M55, M67), Lung (M?), Sarcoma (M?), Bladder (M?), Central Nervous System (M?), Lymphoma (M?) |
| 10 | Amsterdam | Colorectal F34 | Colorectal (F28, M60), Polyps (F23), Ovary (F55), Bladder (M80) |
| 11 | Amsterdam | Colorectal F67 | Colorectal (M70, F62, F40), Oropharynx (M45) |
| 12 | Amsterdam | Endometrium F52 | Colorectal (M55, M30), “Cancer” (M?), Dysembryoplastic neuroepithelial tumour (M?), Langerhans cell histiocytosis (M?), Kidney (M50), Leukaemia (M68), Thyroid (F35, F35) |
| 13 | Amsterdam | Colorectal F37 | Colorectal (M54, F70), Central Nervous System (M45), Gastric (F60) |
| 14 | Amsterdam | Colorectal M54 | Colorectal (M43, F70), Skin (F55, M90) |
| 15 | Amsterdam-1 | Endometrium F57 | Colorectal (M54, F53, M74), Gastric (M?), Mouth (M?) |
| 16 | Amsterdam | Colorectal F74 | Colorectal (M70, M70, M81, F51, F?, F40), Renal (M30), Bladder (M81), Lung (M82), Skin (F74), Ovary (F?) |
| 18 | Amsterdam | Colorectal M56 | Colorectal (F30, F30), Kidney (M56), Breast (F63), “Cancer” (M16), Central Nervous System (M30, M42), maxillary sinus (F?) |
| 19 | Amsterdam | Colorectal F51 | Colorectal (M68, M32), Hodgkin Lymphoma (F55), “Cancer” (M58), Kidney (F66), Leukaemia (F7), Meningioma (F7) |
| 20 | Amsterdam-1 | Colorectal 68 | Colorectal (M90), Breast (F?), “Cancer” (F50), Thyroid (F25), Oesophageal (F60), |
| 22 | Amsterdam-1 | Colorectal F77 | Colorectal (F38), Lung (F47), Breast (F50) |
| 24 | Amsterdam | Colorectal M70 | Colorectal (M66, F45), Prostate (M70, M?) |
| 25 | Amsterdam-1 | Colorectal F55 | Colorectal (M50), Polyps (M?), Gastric (M52, F60), Epidermoid carcinoma (F55), Breast (F?, F38), Skin (F78, F?), “Cancer” (M60, M60), Prostate (M?), Uterine (F65) |
| 27 | Amsterdam | Colorectal F67 | Colorectal (F31, F78), Osteosarcoma (M?) |
| 28 | Amsterdam | Colorectal F46 | Colorectal (M24, F60) |
| 29 | Amsterdam-1 | Colorectal F41 | Liver (M51), Gastric (F?, F61, F?) |
| 30 | Amsterdam | Colorectal F40 | Colorectal (M58, M82) |
| 31 | Amsterdam | Colorectal F29 | Colorectal (M80, F42), Prostate (M80), Throat (M70, F?), Gastric (F?), Villous tubular adenoma (F?) |
| 32 | Amsterdam | Colorectal F37 | Colorectal (M50, F50), Polyps (F?), “Cancer” (M50) |
| 33 | Amsterdam | Colorectal M37 | Colorectal (F60, F?, M?), Basocellular (F60, F?), Breast (F50), Nasal (F?) |
| 34 | Amsterdam-2 | Colorectal M58 | Colorectal (M58, M60), Lymphoma (M80), Stomach (M80) |
| Families with multiple members analyzed | |||
| 17 | Amsterdam-1 | Colorectal M45 | Colorectal (M51 (17.1)), Bladder (M45), Polyps (F?, F?), “Cancer” (F?, F?, F?) |
| 21 | Amsterdam | Colorectal M49 | Colorectal (M50, M72 (21.1), F49 (21.2)), Cerebral (M65, M58), Breast (F?), Polyps (F?), Multiple Myeloma (F?), “Cancer” (F?, F?, F?) |
| 23 | Amsterdam-1 | Endometrium F58 | Colorectal (M33 (23.1), F?), Polyps (M?, M?, M?, F?), Myelodysplastic syndrome (M?), Breast (F70, F45, F50), Thyroid (F50), Melanoma (F50) |
| 26 | Amsterdam-2 | Colorectal F42 | Colorectal (F51 (26.1)), Endometrial (F47), Cervical (F42), “Cancer” (M?, M?) |
*Patient ID follows the family ID. Families with more than one individual analysed have a second number separated by a dot (.) (highlighted in bold); M = Male, F = Female; ? = Unknown age at diagnosis.
The families presented an average of six members with cancer. CRC was the most common type of cancer, with 76 cases in 33 families, median of 2.3 per family, diagnosed at an average age of 56.42 years old. CRC was followed by breast, gastric/digestive, and uterine tumours (uterus, endometrium, and cervix).
Molecular data
Germline variants
We identified 842 variants that passed the in silico filters (Fig. 2) and, following manual prioritization, 514 variants were eliminated for being considered benign or likely benign (ACMG class I or II), sequencing artefacts, or hypermutated gene regions. Of the 323 remaining variants, eight were classified as likely pathogenic or pathogenic. The remaining 315 variants were considered VUS (or class III) for lack of benign/pathogenic evidence. From these VUS, 70 were located in genes related to hereditary cancer, and 42 were in DNA repair genes (Supplementary Tables 2–6).
Figure 2.

Variant prioritization decision pipeline.
Likely pathogenic and pathogenic variants
Of the eight candidate variants (identified in six patients), three were classified as pathogenic (class V ACMG) and five as likely pathogenic (class IV ACMG). Three were missense, one frameshift insertion, three frameshift deletions, and one nonsense variant. The variant information is shown in Table 3, while the patient information is detailed in Table 4. The pedigrees are displayed in Supplementary Figs. 5 to 10.
Table 3.
Likely pathogenic and pathogenic variants information.
| Family ID | Gene | c | p | ClinVar* | Revel | M-CAP | ACMG | Function (hereditary syndrome)*** |
|---|---|---|---|---|---|---|---|---|
| 3 | ATR | c.3043C > T | p.Arg1015Ter | NF | NA | NA | V | FA (Familial cutaneous telangiectasia and cancer syndrome) |
| 5 | PARK2 | c.758G > A | p.Arg253His | NF | 0.752 | 0.203989 | IV | TSG |
| 5 | SLX4 | c.4259dupC | p.Pro1420fs | NF | NA | NA | IV | FA and HR |
| 6 | OGG1# | c.30dupC | p.Arg10fs | NF | NA | NA | IV | BER |
| 13 | TREX1 | c.506G > A | p.Arg169His | Pathogenic** | 0.828 | 0.376515 | V | MMR |
| 22 | ASXL1 | c.1927dupG | p.Gly642fs | Pathogenic | NA | NA | V | Chromatin regulation (leukaemia) |
| 33 | FAN1 | c.356_357del | p.Arg119fs | NF | NA | NA | IV | FA (Hereditary colorectal cancer) |
| 33 | CHEK2 | c.470T > C | p.Ile157Thr | Pathogenic** | 0.538 | NA | IV | Cell cycle regulation (Breast and colorectal cancer) |
NF = not found; NA = not available; *Consulted at 12/2019, 01/2020, 02/2020; **Although the official status is conflicting, we considered the pathogenic studies. ***Information from Das et al17. and GeneCard19. FA = Fanconi anaemia pathway, TSG = tumour suppressor gene, HR = homologous recombination, BER = base excision repair, MMR = mismatch repair; #alteration found in homozygosis.
Table 4.
Patient (harbouring class IV and V variants) information.
| Family | Criteria fulfilled | Proband tumour | Diagnostic age (in years) | Germline variant | Tumour location | Tumour differentiation |
|---|---|---|---|---|---|---|
| 3 | Amsterdam-2 | CRC | 34 | ATR:c.3043C > T | Rectum | Moderate |
| 5 | Amsterdam-2 | CRC | 38 | PARK2:c.758G > A | Left colon | Moderate |
| 5 | Amsterdam-2 | CRC | 38 | SLX4:c.4259dupC | Left colon | Moderate |
| 6 | Amsterdam-2 | CRC | 54 | OGG1:c.30dupC | Rectum | Poor |
| 13 | Amsterdam-1 | CRC | 37 | TREX1:c.506G > A | Rectum | Moderate |
| 22 | Amsterdam-2 | CRC | 77 | ASXL1:c.1927dupG | Rectum | Moderate |
| 33 | Amsterdam-1 | CRC | 37 | CHEK2:c.470T > C | Right colon | Moderate |
| 33 | Amsterdam-1 | CRC | 37 | FAN1:c.356_357del | Right colon | Moderate |
One of the potentially pathogenic variants found was the missense c.470T > C (p.Ile157Thr) on CHEK2 gene, a gene known for its association with CRC and breast cancer16. This patient (ID 33) also had a likely pathogenic frameshift variant in the FAN1 gene (c.356_357del; p.Arg119fs), a gene involved in the Fanconi anaemia pathway13,17 that interacts with MMR genes/proteins18. The patient had moderately different adenocarcinomas in the right colon. On the maternal side of the family, CRC and breast cancer cases have been reported (Supplementary Fig. 5).
A homozygous frameshift likely pathogenic alteration was found in the OGG1 gene (c.30dupC; p.Arg10fs) (ID6; Supplementary Fig. 6). OGG1 belongs to the base excision repair pathway17, and its protein is an enzyme (8-oxoguanine) that works repairing oxygen reactive DNA lesions (Source: MedlinePlus, National Library of Medicine). The frameshift insertion was located at the beginning of the gene upstream of the two protein domains, reinforcing its pathogenic effect. The patient (ID 6) was diagnosed at 54 years old with poorly differentiated adenocarcinoma located in the rectum and died at 60 years old. During the interview, the patient stated that a cousin and two uncles from the father’s side were diagnosed with gastric cancer, 14 deceased uncles also from the father’s side had “stomach problems”. Besides, his sister had a diagnosis of CRC at 42 years old.
Another frameshift variant identified is located in the ASLX1 gene (c.1927dupG; p.Gly642fs). The patient (ID 22) was diagnosed at 77 years old with moderately differentiated adenocarcinoma in the rectum. He had one daughter diagnosed with CRC at 38 years old (Supplementary Fig. 7). According to STRING18, ASXL1 works with several transcription factors and cell cycle regulators.
Two likely pathogenic variants were identified in patient ID 5: the missense variant c.758G > A (Arg253His) in the gene PARK2 (or PRKN2) and the frameshift insertion c.4259dupC (p.Pro1420fs) in the DNA repair gene SLX4. The patient was diagnosed at 38 years old with moderately differenced adenocarcinoma in the left colon (Supplementary Fig. 8). SLX4 works with several DNA repair pathways, nonhomologous end joining, homologous recombination, Fanconi anaemia, nucleotide excision repair, and some nonspecific pathways18.
A nonsense class V alteration in the ATR gene was identified in patient ID 3. The patient was diagnosed at 34 years old with moderately differenced adenocarcinoma in the rectum, and his father died with CRC (Supplementary Fig. 9). The ATR gene has an important role in several repair pathways and acts as a checkpoint activator during the cell cycle17,19,20. The c.3043C > T (p.Arg1015Ter) nonsense variant is located before all three protein domains21,22.
A pathogenic missense variant was identified in a patient with colorectal cancer at 37 years of age (ID 13). The c.506G > A (pArg169His) variant was identified in the TREX1 gene. The patient was diagnosed with moderately differenced adenocarcinoma in the rectum. Her family had two more CRC cases (Supplementary Fig. 10). The key function of TREX1 is digesting (degrade, metabolize) cytosolic ssDNA, which is stemmed from endogenous retroelements or abnormal replication intermediates, to suppress cell-intrinsic initiation of autoimmunity23. Besides, TREX1 encodes a repair exonuclease that acts on terminal mismatched regions17,19 .
Variants of unknown significance (VUS)
We identified three VUS in unrelated patients (ClinVar and by ACMG criteria) in the MMR genes (Table 5). All tumours had a normal IHC result and were MSS.
Table 5.
Variants in lynch syndrome genes.
| ID | Gene | c | p | Revel | M-CAP | ClinVar* | ACMG |
|---|---|---|---|---|---|---|---|
| 23 | MLH1 | c.2027T > C | p.Leu676Pro | 0.917 | 0.473622 | VUS | III |
| 30 | MSH6 | c.2177T > A | p.Phe726Tyr | 0.678 | 0.190663 | VUS | III |
| 34 | MSH2 | c.2785C > T | p.Arg929Ter | NA | NA | VUS | III |
*Consult in 03/2021.
NA not available.
In the four families with more than one person evaluated (families IDs 17, 21, 23, 26), we did not identify any likely pathogenic or pathogenic variant in these four families. Nonetheless, we identified 26 variants of unknown significance that segregated in every participating member: eight in family 17, seven in family 21, three in family 24 and eight in family 26. Beyond the likely pathogenic FAN1 variant identified in family ID33, we detected two more variants in the gene that segregated in family 24 and another in family 26. Despite the arising evidence showing a possible relationship between FAN1 and colorectal cancer or even with FCCTX, we had not enough evidence to categorize the variants in any pathogenic class. Nevertheless, a study shows that the variant FAN1:Met50Arg (which have a deleterious prediction by REVEL and was identified segregating in affected relatives from two of our families) impacts the repairing system causing genetic instability, possibly representing a cancer risk factor24. Although, according to the authors, it is not clear if the gene malfunctioning itself is enough to start the carcinogenesis. Another gene related to colorectal cancer segregating (in one family) was MSH5, but, despite having a deleterious score and segregating, the patients had no microsatellites instability and adding the lack of scientific information available for this variant, it remained with unknown significance. The pedigrees are displayed in Supplementary Figs. 1 to 4 and a detailed list of the variants in Table S6.
For most of the VUS identified, there is still insufficient evidence available to allow their classification. Sixty-five were located in genes related to hereditary syndromes, being FAN1, RASAL1, SDHA, ERCC2, and TRIM28 the genes with the higher number of VUS identified. Another 44 VUS were in repair pathway genes such as POLG, ATM, ERCC2, MSH6, PARP3, and POLL. The last group, the carcinogenesis related genes, contained 232 VUS, and the top mutated were TBP, POLG, BIRC6, EPHA8, MIB2 and WNK4.
Variants of unknown significance for several genes involved in DNA repair pathways were also identified in more than one family. Among them, we highlight POLG (with variants in eight unrelated families), POLE and POLH (with VUS in four unrelated families each), ATM (with variants in seven families). Moreover, FAN1, XPC and PARP3 genes harboured VUS in four unrelated families (for more details about the variants, see tables S2 to S4).
Variants of unknown significance identified can be found in Supplementary Tables 2 to 6.
Discussion
In this study, among the 34 families evaluated, we found that six of them (17.65%) presented a potentially pathogenic variant. Most of the genes were involved in DNA damage repair. One of them is the known CRC predisposing gene CHEK216,25,26. In addition, pathogenic/likely pathogenic variants were found in genes previously associated with FCCTX/hereditary CRC as OGG112,27 and FAN113. Furthermore, potentially pathogenic variants were identified in the ATR, TREX1, ASXL1, PARK2 and SLX4 genes. Although likely pathogenic and pathogenic variants were confirmed by Sanger sequencing, the association of the variants identified with the FCCTX family history requires further validation by segregation analysis, and in other familial cancer cohorts. Moreover, specific variants would also require in vitro analyses to investigate functional consequences for protein function.
Among the genes where a potentially pathogenic variant was identified, one of them was already associated with hereditary colorectal cancer, the CHEK2 gene16,26. The identified variant (c.470T > C) has already been associated with hereditary and sporadic CRC25. Suchy et al. identified this variant in Amsterdam I families negative for MMR alterations and concluded that it confers risk for hereditary CRC26. Liu et al. published a meta-analysis with 4029 cases and 13,844 controls and showed that the variant confers a moderate risk for hereditary CRC (odds ratio = 1.97, 95% CI 1.41–2.74, P < 0.001)25.
The CHEK2 mutated patient is also carrier of a frameshift deletion at FAN1, whose association with FCCTX has been described previously13. Segui and collaborators performed Exome sequencing of three family members (Amsterdam I and MSS) and 176 other families with a history of CRC and concluded that the malfunctioning of Fanconi anaemia pathway (due to FAN1 alteration) might predispose patients to CRC13. FAN1 works to support the MMR system18, which is already classically associated with hereditary CRC1. In addition, our group reported a missense FAN1 variant was identified in two families, one with a strong history of breast and CRC cases28. Considering the fact that both, CHEK2 and FAN1 can lead to a low to moderated increase in the CRC risk, we believe that a model with an additive effect of both variants can be feasible. This hypothesis should be further evaluated through segregation assays as well as with functional assays.
Other four families with loss of function pathogenic/likely pathogenic variants were identified. Among the frameshift alterations, c.30dupC in the OGG1 gene was identified in homozygosis, in a family where CRC cases were present in the proband and her sister. Besides, 19 other relatives with stomach problems/ “digestive” tumours were reported. Published studies have shown that missense alterations in OGG1 may confer a risk for early onset CRC12,29. Garre and collaborators evaluated 42 MSS-HNPCC families and identified a missense alteration in OGG1 that affects splicing. The alteration was detected in an Amsterdam I family and co-segregated with cancer27. A meta-analysis from Zhange & Mo with 5235 cases and 8438 controls also concluded that polymorphisms in OGG1 confer risk for CRC, especially in Caucasians30. Concerning the relation between OGG1 and gastric cancer, two case–control studies showed that polymorphisms in OGG1 confer risk for gastric cancer, as did a meta-analysis with 1180 cases, and 2444 controls31–33.
The other cancer susceptibility gene for which a potentially pathogenic frameshift variant was found is ASLX1, a classic leukaemia-associated gene related to Bohring-Optiz syndrome, which increases the risk for Wilms tumours34,35. To the best of our knowledge, there have been no previous associations with CRC. In the family carrying the ASLX1 pathogenic variant, no leukaemia or myelodysplasia cases have been reported, reinforcing the need for further studies evaluating the association of these genes with the phenotypes observed in the family.
One family carrying two likely pathogenic variants (at SLX4 and PARK2 genes) was identified in our cohort. Frameshift variants at SLX4 gene have already been detected in sporadic CRC36, and the gene is known for its association with hereditary breast and gastric cancer37. Regarding the PARK2 gene, it has no clear function to date. Ikeuchi et al. detected tumour suppressor activity by negatively regulating the cell cycle in colorectal cancer cell lines38. Pologiannis et al. detected PARK2 copy number alterations in CRC and found that APC-mutated tumours with malfunctioning PARK2 have a rapidly evolving carcinogenesis39.
The ATR gene, found mutated in a patient with colorectal cancer at very early ages, is considered as a hallmark of cancer40 and a component of the Fanconi Anaemia repair pathway17. Malfunctioning of the Fanconi anaemia pathway has been cited as a predisposing factor for CRC13. Interestingly, the ATM gene, which works together with ATR to maintain chromosome integrity and genome stability, has been considered as a moderately penetrant germline CRC predisposing gene41, suggesting that pathogenic alterations in ATR could also lead to a moderate CRC increased risk. Additionally, germline variants in ATR have already been associated with oropharyngeal cancer, as investigated by Tanaka et al. in a family with an ATR-related syndrome affecting 24 members from five generations42.
A missense alteration at TREX1 also called our attention. The missense alteration identified was classified as pathogenic according to the ACMG criteria. Few studies related TREX1 to cancer. Prati and collaborators detected an upregulation of TREX1 in HPV-transformed cell lines in precancerous lesions, carcinomas, and adenocarcinomas43. The authors also found that TREX1 silencing could affect tumour growth by upregulating p53, indicating a possible contribution to tumour development. Dong and collaborators detected TREX1 alterations in pancreatic adenocarcinomas and concluded that TREX1 might have a role in its carcinogenesis44,45. However, to the best of our knowledge, there is no studies associating TREX1 to hereditary cancer. Further studies are needed in order to prove or discard this association.
It is important to highlight that variants located at splicing consensus regions in ATM, GSDMA, PTPRE and RAD51B genes were identified and, although classified as VUS in this manuscript considering the current evidences available, should be closely monitored as more evidences in the literature can lead to an upgrade on the classification from VUS to likely pathogenic or pathogenic.
This study has some limitations. The initial investigation in our cohort was based on the protein function, prioritizing the analysis of a virtual panel of cancer-associated genes. Although this strategy may have restricted the results obtained it allowed the identification of potentially pathogenic variants in six unrelated families among the 34 evaluated. Besides, alterations in the number of copies were not evaluated by our analysis pipeline.
Additionally, polygenic risk factors were not evaluated in this study, which might justify the cases with a family history suggestive of a cancer predisposition syndrome that did not present pathogenic/likely pathogenic variants. Candidate genes identified in this work require further cohort validations. Moreover, LOH analysis in tumor tissue, affected families-segregation analysis and functional studies should be addressed in order to investigate its role in oncogenesis. In spite of that, this is the biggest Brazilian study evaluating patients at high-risk for hereditary colorectal cancer fulfilling criteria for the FCCTX and allowed the identification of pathogenic/likely pathogenic variants in about twenty percent of the patients evaluated.
Patients and methods
Institutional review board statement and informed consent statement
The study was reviewed and approved by Barretos Cancer Hospital’s Research Ethics Committee (approval numbers: 53417916.5.0000.5437 and 56164716.9.0000.5437). All research was performed in accordance with the Brazilian CEP/CONEP-system regulation. All participants were de-identified and provided their written informed consent to participate in this study.
Patient selection
Thirty-nine patients from 34 families identified at the Oncogenetics Department of Barretos Cancer Hospital46 were included in the study after signing the informed consent. All participants were negative for LS, which diagnostic is composed by immunohistochemistry (IHC) for MLH1, MSH2, MSH6, and PMS2 proteins; BRAF (V600E) sequencing and microsatellite instability (MSI) evaluation by fragment analysis47. Besides, gene panel sequencing by NGS is performed for all patients with High MSI or altered IHC. For the purpose of the present study, only patients with tumours presenting normal IHC results and microsatellite stability (MSS) were invited to participate.
From a clinical point of view, all families have at least two colorectal tumours in the family and 47% of them fulfilled the Amsterdam I criteria3. Clinical, sociodemographic, and histopathological information were extracted from the patients´ medical charts. Family history was obtained through the Oncogenetics chart, and all the pedigrees were drawn using Progeny software (https://pedigree.progenygenetics.com/).
Exome sequencing
Genomic DNA was extracted from peripheral blood using the QIAmp DNA Blood Mini Kit with the automated QIAcube platform (QIAGEN—Germany). All procedures were performed following the manufacturer’s instructions. DNA concentration was determined using Qubit dsDNAHS Assay Kit (Thermo Fisher Scientific—United States).
Exome sequencing (ES) was performed with the SOPHiA Whole Exome Solution version 1 kit (203,058 targeted regions, 40,907,213 base pairs, and 19,682 genes) (Sophia Genetics SA, Saint Sulpice, Switzerland) on a NovaSeq platform (Illumina San Diego, Canada) by SOPHiA Genetics (Sophia Genetics SA, Saint Sulpice, Switzerland).
Variant calling
Sequence reads were mapped to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA, version 0.7.17)48. Alignment files were pre-processed and single nucleotide and indel germline variants were called by the Genome Analysis Toolkit (GATK version 4.0.4.0)49. Variant files were filtered to exclude variants covered by < 10 reads or with variant allele fraction < 25%.
Genes analysed
A virtual panel consisting of 2389 genes involved directly or indirectly in carcinogenesis was selected for analysis. This panel was extracted from the Catalogue of Somatic Mutations in Cancer50 (Cosmic: using the keyword terms “cancer”, “tumour-suppressor gene”, “proto-oncogene”, and “oncogene”), Universal Protein Resource (UniProt)51, and DISEASE study52. Additionally, a second “subpanel” consisting of 228 genes involved in DNA repair was extracted from the study by Das et al17. Finally, a third “subpanel” consisting of 260 genes related to hereditary cancer syndromes extracted from commercial panels and revised in the literature (GeneCard19 and Genetics Home Reference—Source: MedlinePlus, National Library of Medicine) was employed. The interactions of the gene panels are shown in Fig. 1 and the gene list evaluated is available in Table S1.
Figure 1.
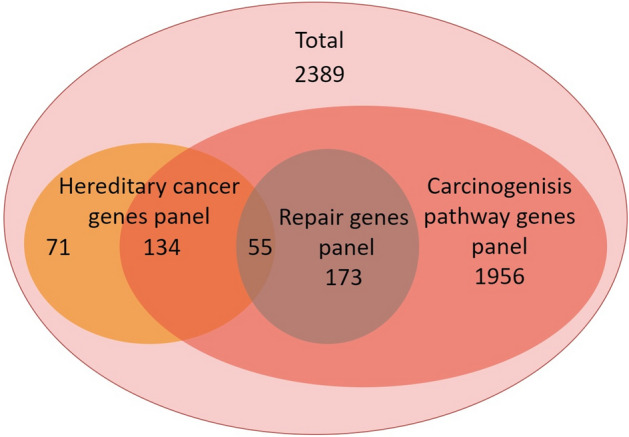
Venn diagram of the genes used in this study.
Variant annotation and classification
To variant classification we developed a pipeline (Fig. 2) of decisions based on criteria proposed by the American College of Medical Genetics and Genomics (ACMG)53. Functional and populational frequency annotation of variants was performed using ANNOVAR54.
All variants covered by ≥ 10 reads, variant allele fraction ≥ 25%, and present in any gene of the three panels were first filtered by their populational frequency using two databases, ABraOM55 and gnomAD56: only variants with a minor allele frequency (MAF) < 1% were considered, with the exception of families with consanguinity, where we analysed variants with MAF ≥ 1%, although no class III, IV, or V variants were detected. Variants considered “variants of unknown significance” (VUS) according to the ACMG criteria were then filtered using the in silico pathogenicity prediction tool Rare Exome Variant Ensemble Learner (REVEL—score: 0.7)57 or Mendelian Clinically Applicable Pathogenicity (M-CAP—score: 0.025)58 for missense variants and Human Splicing Finder (HSF)59 for splicing variants. Additionally, Eigen (score: 0.7)60 and GenoCanyon (0.7)61 were used as complementary scores when the above tools were not available or as tiebreaker criteria.
Variant manual review
Following the described automated prioritization steps, all variants classified as class III, IV, or V were manually curated in an independent way by two researchers (F.A.O.G and E.I.P). For this process, the ACMG criteria conferred by the Intervar62 and Varsome63, the GeneCard19 database, scientific literature (through PubMed—https://pubmed.ncbi.nlm.nih.gov/), and evidence deposited in the Clinvar64 database were reviewed. To avoid false positives, the variants were verified in the original BAM file using the Integrative Genome Viewer (IGV)65. For all analyses, we used the canonical transcript available at Locus Reference Genomic (LRG)66 or, if not available, the first one at Ensembl (http://www.ensembl.org/index.html).
Conventional (Sanger) sequencing
Variants classified as likely pathogenic (ACMG class IV) or pathogenic (ACMG class V) were confirmed by bidirectional Sanger sequencing. For this process, we amplified the patients’ genomic DNA by PCR and purified the reaction with ExoSAP-IT (USB) enzyme and later with the BigDye Terminator kit (Thermo Fisher Scientific—United States). The sequencing itself was performed bidirectionally using the X-terminator kit v3.1 (Thermo Fisher Scientific—United States) on an automated sequencer model 3500 (Applied Biosystem—Thermo Fisher Scientific—United States).
Conclusion
In conclusion, the present study represents the largest ES study involving Brazilian families at risk for FCCTX that allowed the identification of new candidate genes for FCCTX syndrome. In our cohort, 17.65% of our families were carriers of pathogenic/likely pathogenic variants that could explain cancer’s personal and family history. Our findings suggest that several cancer-associated genes may have a role in FCCTX, such as the known hereditary cancer gene CHEK2, the previously FCCTX candidate genes OGG1 and FAN1 and, other cancer-related genes such as ATR, ASXL1, PARK2, SLX4 and TREX1, bringing novel insights into the genetic risk factors for familial colorectal cancer type X. Nevertheless, more studies (in vitro and/or in vivo), such as functional assays, segregation, and loss of heterozygosity, are necessary to ascertain more conclusive hypotheses about their role in FCCTX predisposition.
Supplementary Information
Acknowledgements
We thank the medical doctors Luis Gustavo Romagnolo and Maximiliano Cadamuro (Oncogenetics—Barretos Cancer Hospital) for caring for our patients. We thank André Escremin de Paula, MSc, Gustavo Noriz Berardinelli, Ph.D., and Gabriela Carvalho Fernandes, MSc (Molecular Diagnostic Center—Barretos Cancer Hospital) for the routine molecular processing of our patient’s samples (microsatellite instability and genetic testing).
Author contributions
Conceptualization: F.A.O.G., E.I.P.; Methodology: F.A.O.G., E.S.A., A.F.E., I.V.V.S., A.E.P., E.I.P.; Software: F.A.O.G., E.S.A.; Validation: F.A.O.G., A.E.P., E.I.P.; Formal Analysis: F.A.O.G.; Investigation: F.A.O.G., H.C.R.G., C.S.S., A.E.P., N.C.; Resources: M.E.M., R.M.V.R., E.I.P.; Data Curation: F.A.O.G., E.S.A., A.F.E.; Writing—Original Draft Preparation: F.A.O.G.; Writing—Review and Editing: all authors; Visualization: F.A.O.G.; Supervision: E.I.P.; Project Administration: R.M.V.R., E.I.P.; Funding Acquisition: M.E.M., R.M.V.R., E.I.P.
Funding
This project was funded through grants from the National Oncology Care Support Program (PRONON, Grant number (25000.056766/2015-64)) from the Ministry of Health and from donations from the Cotemig group. EIP, MEM, and RMR are recipients of National Council for Scientific and Technological Development (CNPq) productivity fellowships. The study sponsors had no involvement in the study design, collection, analyses or interpretation of data.
Data availability
Class III, IV, and V variants identified in the 2389 evaluated genes shall be available in the ClinVar database; otherwise, the datasets presented in this article are not readily available due to privacy and ethical restrictions. Requests to access the datasets should be directed to the corresponding author.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-06782-8.
References
- 1.Lynch HT, Shaw TG. Practical genetics of colorectal cancer. Chin. Clin. Oncol. 2013;2:12. doi: 10.3978/j.issn.2304-3865.2013.03.04. [DOI] [PubMed] [Google Scholar]
- 2.Menahem B, Alves A, Regimbeau JM, Sabbagh C. Lynch syndrome: Current management In 2019. J. Visc. Surg. 2019;156:507–514. doi: 10.1016/j.jviscsurg.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Tome E, et al. Genome-wide linkage analysis and tumoral characterization reveal heterogeneity in familial colorectal cancer type X. J. Gastroenterol. 2015;50:657–666. doi: 10.1007/s00535-014-1009-0. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, et al. Comparison between familial colorectal cancer type X and Lynch syndrome: Molecular, clinical, and pathological characteristics and pedigrees. Front. Oncol. 2020;10:1603. doi: 10.3389/fonc.2020.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi YH, et al. Risks of colorectal cancer and cancer-related mortality in familial colorectal cancer type X and lynch syndrome families. J. Natl. Cancer Inst. 2019;111:675–683. doi: 10.1093/jnci/djy159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieminen TT, et al. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2011;141:e23–26. doi: 10.1053/j.gastro.2011.03.063. [DOI] [PubMed] [Google Scholar]
- 8.Nieminen, T. T. et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology147, 595–598 e595. 10.1053/j.gastro.2014.06.009 (2014). [DOI] [PMC free article] [PubMed]
- 9.Schulz E, et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat. Commun. 2014;5:5191. doi: 10.1038/ncomms6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin-Morales L, et al. SETD6 dominant negative mutation in familial colorectal cancer type X. Hum. Mol. Genet. 2017;26:4481–4493. doi: 10.1093/hmg/ddx336. [DOI] [PubMed] [Google Scholar]
- 11.Garre P, et al. BRCA2 gene: A candidate for clinical testing in familial colorectal cancer type X. Clin. Genet. 2015;87:582–587. doi: 10.1111/cge.12427. [DOI] [PubMed] [Google Scholar]
- 12.Kim IJ, et al. Mutational analysis of OGG1, MYH, MTH1 in FAP, HNPCC and sporadic colorectal cancer patients: R154H OGG1 polymorphism is associated with sporadic colorectal cancer patients. Hum. Genet. 2004;115:498–503. doi: 10.1007/s00439-004-1186-7. [DOI] [PubMed] [Google Scholar]
- 13.Segui N, et al. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology. 2015;149:563–566. doi: 10.1053/j.gastro.2015.05.056. [DOI] [PubMed] [Google Scholar]
- 14.Dominguez-Valentin M, Therkildsen C, Da Silva S, Nilbert M. Familial colorectal cancer type X: Genetic profiles and phenotypic features. Mod. Pathol. 2015;28:30–36. doi: 10.1038/modpathol.2014.49. [DOI] [PubMed] [Google Scholar]
- 15.Nejadtaghi M, Jafari H, Farrokhi E, Samani KG. Familial Colorectal Cancer Type X (FCCTX) and the correlation with various genes-A systematic review. Curr. Probl. Cancer. 2017;41:388–397. doi: 10.1016/j.currproblcancer.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Meijers-Heijboer H, et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am. J. Hum. Genet. 2003;72:1308–1314. doi: 10.1086/375121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das R, Ghosh SK. Genetic variants of the DNA repair genes from Exome Aggregation Consortium (EXAC) database: Significance in cancer. DNA Repair. (Amst.) 2017;52:92–102. doi: 10.1016/j.dnarep.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stelzer, G. et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform.54, 1 30 31–31 30 33. 10.1002/cpbi.5 (2016). [DOI] [PubMed]
- 20.Chakravarty, D. et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol.2017. 10.1200/PO.17.00011 (2017). [DOI] [PMC free article] [PubMed]
- 21.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao SS, et al. TREX1 as a potential therapeutic target for autoimmune and inflammatory diseases. Curr. Pharm. Des. 2019;25:3239–3247. doi: 10.2174/1381612825666190902113218. [DOI] [PubMed] [Google Scholar]
- 24.Lachaud C, et al. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science. 2016;351:846–849. doi: 10.1126/science.aad5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C, Wang QS, Wang YJ. The CHEK2 I157T variant and colorectal cancer susceptibility: A systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2012;13:2051–2055. doi: 10.7314/apjcp.2012.13.5.2051. [DOI] [PubMed] [Google Scholar]
- 26.Suchy J, et al. CHEK2 mutations and HNPCC-related colorectal cancer. Int. J. Cancer. 2010;126:3005–3009. doi: 10.1002/ijc.25003. [DOI] [PubMed] [Google Scholar]
- 27.Garre P, et al. Analysis of the oxidative damage repair genes NUDT1, OGG1, and MUTYH in patients from mismatch repair proficient HNPCC families (MSS-HNPCC) Clin. Cancer Res. 2011;17:1701–1712. doi: 10.1158/1078-0432.CCR-10-2491. [DOI] [PubMed] [Google Scholar]
- 28.Felicio PS, et al. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 2021;42:290–299. doi: 10.1002/humu.24158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai CY, et al. Association between polymorphisms of APE1 and OGG1 and risk of colorectal cancer in Taiwan. World J. Gastroenterol. 2016;22:3372–3380. doi: 10.3748/wjg.v22.i12.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang M, Mo R. Association of hOGG1 Ser326Cys polymorphism with colorectal cancer risk: An updated meta-analysis including 5235 cases and 8438 controls. Tumour Biol. 2014;35:12627–12633. doi: 10.1007/s13277-014-2586-5. [DOI] [PubMed] [Google Scholar]
- 31.Canbay E, et al. Possible associations of APE1 polymorphism with susceptibility and HOGG1 polymorphism with prognosis in gastric cancer. Anticancer Res. 2010;30:1359–1364. [PubMed] [Google Scholar]
- 32.Lu J, et al. The association analysis of hOGG1 genetic variants and gastric cancer risk in a Chinese population. Oncotarget. 2016;7:66061–66068. doi: 10.18632/oncotarget.11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni M, Qiu J, He W, Wang X. The functional Ser326Cys polymorphism in hOGG1 is associated with gastric cancer risk: evidence from 1180 cases and 2444 controls. Eur. J. Gastroenterol. Hepatol. 2012;24:683–687. doi: 10.1097/MEG.0b013e328351d1c0. [DOI] [PubMed] [Google Scholar]
- 34.Gelsi-Boyer V, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009;145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 35.Russell B, et al. Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am. J. Med. Genet. A. 2015;167A:2122–2131. doi: 10.1002/ajmg.a.37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JH, An CH, Kim MS, Yoo NJ, Lee SH. Rare frameshift mutations of putative tumor suppressor genes CSMD1 and SLX4 in colorectal cancers. Pathol. Res. Pract. 2018;214:325–326. doi: 10.1016/j.prp.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Shah S, et al. Assessment of SLX4 Mutations in Hereditary Breast Cancers. PLoS ONE. 2013;8:e66961. doi: 10.1371/journal.pone.0066961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikeuchi K, et al. Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced Parkin in human colorectal cancers. Int. J. Cancer. 2009;125:2029–2035. doi: 10.1002/ijc.24565. [DOI] [PubMed] [Google Scholar]
- 39.Poulogiannis G, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang D, et al. CHG: A systematically integrated database of cancer hallmark genes. Front. Genet. 2020;11:29. doi: 10.3389/fgene.2020.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.AlDubayan SH, et al. Inherited DNA-repair defects in colorectal cancer. Am. J. Hum. Genet. 2018;102:401–414. doi: 10.1016/j.ajhg.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka A, et al. Germline mutation in ATR in autosomal-dominant oropharyngeal cancer syndrome. Am. J. Hum. Genet. 2012;90:511–517. doi: 10.1016/j.ajhg.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prati B, et al. Three Prime Repair Exonuclease 1 (TREX1) expression correlates with cervical cancer cells growth in vitro and disease progression in vivo. Sci. Rep. 2019;9:351. doi: 10.1038/s41598-018-37064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong X, et al. Significant associations of mismatch repair gene polymorphisms with clinical outcome of pancreatic cancer. J. Clin. Oncol. 2009;27:1592–1599. doi: 10.1200/JCO.2008.20.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong X, Li Y, Hess KR, Abbruzzese JL, Li D. DNA mismatch repair gene polymorphisms affect survival in pancreatic cancer. Oncologist. 2011;16:61–70. doi: 10.1634/theoncologist.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmero EI, et al. Oncogenetics service and the Brazilian public health system: The experience of a reference Cancer Hospital. Genet. Mol. Biol. 2016;39:168–177. doi: 10.1590/1678-4685-GMB-2014-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Paula AE, et al. Clinicopathological and molecular characterization of Brazilian families at risk for Lynch syndrome. Cancer Genet. 2021;254–255:82–91. doi: 10.1016/j.cancergen.2021.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tate JG, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.UniProt C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pletscher-Frankild S, Palleja A, Tsafou K, Binder JX, Jensen LJ. DISEASES: Text mining and data integration of disease-gene associations. Methods. 2015;74:83–89. doi: 10.1016/j.ymeth.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 53.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naslavsky MS, et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017;38:751–763. doi: 10.1002/humu.23220. [DOI] [PubMed] [Google Scholar]
- 56.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ioannidis NM, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jagadeesh KA, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016;48:1581–1586. doi: 10.1038/ng.3703. [DOI] [PubMed] [Google Scholar]
- 59.Desmet FO, et al. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016;48:214–220. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu Q, et al. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 2015;5:10576. doi: 10.1038/srep10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 2017;100:267–280. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kopanos C, et al. VarSome: The human genomic variant search engine. Bioinformatics. 2019;35:1978–1980. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Landrum MJ, et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the integrative genomics viewer. Cancer Res. 2017;77:e31–e34. doi: 10.1158/0008-5472.CAN-17-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacArthur JA, et al. Locus Reference Genomic: reference sequences for the reporting of clinically relevant sequence variants. Nucleic Acids Res. 2014;42:D873–878. doi: 10.1093/nar/gkt1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Class III, IV, and V variants identified in the 2389 evaluated genes shall be available in the ClinVar database; otherwise, the datasets presented in this article are not readily available due to privacy and ethical restrictions. Requests to access the datasets should be directed to the corresponding author.