

Abstract
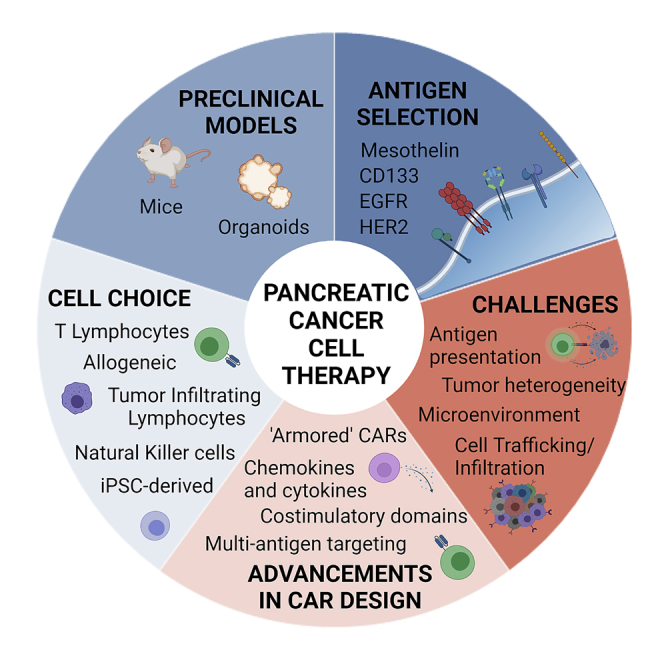
Pancreatic cancer is an aggressive disease that is predicted to become the second leading cause of cancer-related death worldwide by 2030. The overall 5-year survival rate is around 10%. Pancreatic cancer typically presents late with locally advanced or metastatic disease, and there are limited effective treatments available. Cellular immunotherapy, such as chimeric antigen receptor (CAR) T cell therapy, has had significant success in treating hematological malignancies. However, CAR T cell therapy efficacy in pancreatic cancer has been limited. This review provides an overview of current and ongoing CAR T cell clinical studies of pancreatic cancer and the major challenges and strategies to improve CAR T cell efficacy. These strategies include arming CAR T cells; developing off-the-shelf allogeneic CAR T cells; using other immune CAR cells, like natural killer cells and tumor-infiltrating lymphocytes; and combination therapy. Careful incorporation of preclinical models will enhance management of affected individuals, assisting incorporation of cellular immunotherapies. A multifaceted, personalized approach involving cellular immunotherapy treatment is required to improve pancreatic cancer outcomes.
Keywords: pancreatic cancer, cellular immunotherapy, adoptive T cell therapy, CAR T cell therapy, tumor microenvironment, checkpoint blockade, combination therapy, preclinical models, organoids
Graphical Abstract
Pancreatic cancer remains intractable to modern therapies, with a 5-year survival rate of around 10%. Here we summarize current advances toward cellular immunotherapies using chimeric antigen receptor (CAR) T cells in pancreatic cancer. Challenges are outlined along with potential solutions designed to improve outcomes.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease accounting for over 90% of pancreatic malignancies. The overall 5-year survival rate is around 10%. In 2021, it represented the fourth most common cause of cancer-related deaths in the United States, with 60,430 predicted new cases diagnosed and 48,220 deaths.1 Based on current trends, with no significant improvements in survival, it is expected to become the second leading cause of cancer-related death by 2030.2
The majority of individuals with PDAC present late at diagnosis with locally advanced or metastatic disease. There is a lack of early specific symptoms and, outside of research protocols, no early detection test exists. Diagnosis is mainly based on imaging, which is complicated by the deep anatomical location of the pancreas. Surgical resection remains the only potentially curative treatment option for individuals with pancreatic cancer, but less than 20% are suitable at diagnosis.3 Systemic chemotherapy (FOLFIRINOX, a combination of folinic acid, fluorouracil (5-FU), irinotecan, and oxaliplatin, or a combination of gemcitabine and nab-paclitaxel) is commonly used as first-line treatment for advanced individuals.4, 5, 6 However, response rates are less than 32%, and chemotherapy-related toxicities may reduce their wider utility.7
Advancements in next-generation and single-cell sequencing have greatly improved our understanding of the underlying genetic and biological mechanisms of PDAC.8 Although this has not translated to significant overall clinical improvements, it has facilitated greater precision in providing a personalized medicine approach where a molecular signature or biomarker is used to match individuals to a targeted therapy. For example, individuals with BRCA-mutated PDAC (up to 25% of PDAC) have been found to be responsive to PARP inhibitors followed by platinum-based chemotherapy,9 and immune checkpoint inhibitors are approved for use in individuals with PDAC with high microsatellite instability (1%–2% of PDAC).10 The Know Your Tumor program found that individuals with PDAC who were given a tailored therapy had longer median survival than those who were not, in retrospective analyses.11 However, it was found that only 26% had actionable mutations in this program, a limited subset of individuals, highlighting the need for further therapeutic options to improve the treatment and outcomes in individuals with PDAC.
Cellular immunotherapy
Cellular immunotherapy has transformed the landscape of therapeutic oncology. One approach that has garnered significant attention is chimeric antigen receptor (CAR) T cell therapy, a technology where an individual’s T cells are collected and genetically engineered to express a CAR that recognizes and attacks cancer cells.12,13 CAR T cells are typically infused systemically to target tumor cells and exert antitumor activity.13,14
The CAR consists of an extracellular antigen-binding domain connected to endodomain(s), responsible for downstream signaling.15,16 To date, five generations of CARs have been developed (Figure 1). The first-generation CAR consists of the antigen-binding domain derived from the single-chain variable fragment (scFv) of an antibody. This is connected to a CD3ζ chain, acting as the transmembrane signaling domain to mediate antigen-dependent activation.17,18 The second-generation CAR adds a co-stimulatory molecule (CD28, 4-1BB, OX40, CD27, or ICOS) to enhance T cell response.19 CD28 and 4-1BB are the most commonly employed co-stimulatory domains.19,20 To improve antitumor efficacy, the third generation includes two co-stimulatory domains to the CD3ζ chain.19,20 Further research to enhance CAR efficacy resulted in fourth-generation CARs or TRUCKs (T cells redirected for antigen-unrestricted cytokine-initiated killing), which can encode secretion of cytokines.21, 22, 23 This improves CAR T cell function and regulation of the innate immune cell response. The term “armored CAR” has been used to describe this strategy of genetic engineering to encode for secretion of cytokines, modulation of cytokine function, or secretion of antibody-like proteins to enhance CAR efficacy.24,25 Another armored CAR and potential concept for a fifth-generation CAR has been developed recently to enhance the proliferation and antitumor activity of CAR T cells by insertion of interleukin (IL)-2Rβ, inducing antigen-dependent activation of the JAK-STAT pathway.26
Figure 1.

Schematic of the different CAR generations
Chimeric antigen receptors (CARs) are receptors composed of an extracellular single-chain variable fragment (scFv) comprising a variable light (VL) and variable heavy (VH) chain fused to a transmembrane domain. The intracellular signaling domain varies across generations for T cell activation. First-generation CARs contain a CD3ζ chain. Second- and third-generation CARs contain one or two co-stimulatory domains, respectively. Fourth-generation CARs, also known as TRUCKs, have an interleukin (IL) inducer, which leads to release of cytokines to improve CAR T cell function. Fifth-generation CARs are based on the second-generation CAR with an additional IL-2Rβ domain to induce JAK/STAT antigen-dependent signaling pathways for enhanced proliferation and antitumor activity.
As of July 2021, the US Food and Drug Administration (FDA) has approved five CAR T cell therapies. Yescarta® (axicabtagene ciloleucel)27 and Tecartus™ (brexucabtagene autoleucel),28 which utilize second-generation CD19-directed CARs with a CD28 co-stimulatory domain, were approved in 2017 and 2020, respectively. Kymriah® (tisagenlecleucel),27 approved in 2017, and Breyanzi® (lisocabtagene maraleucel),29 approved in 2021, utilize a second-generation CD19-directed CAR with a 4-1BB co-stimulatory domain. Recently, the FDA approved an anti-B cell maturation antigen (BCMA)-directed CAR, Abecma® (idecabtagene vicleucel).30 These therapies have been very successful in treatment of hematological malignancies, with one study reporting up to 90% of individuals achieving remission.31,32 So far, no CAR T cell therapy is approved for solid tumors. Early indications in solid tumor clinical trials have failed to replicate the same success observed in hematological malignancies. Completed PDAC CAR T cell clinical trials to date have demonstrated limited efficacy (Table 1). Rates of stable disease were achieved in a limited subset of individuals where 44% achieved stable disease. However, the majority only exhibited short-term responses or disease progression.
Table 1.
Completed and published CAR T cell therapy clinical trials in PDAC
| Trial number | Phase | Target | Stimulatory domain | Number of individuals with PDAC (total) | Preconditioning | Cell infusion number; treatment frequency | Efficacy in PDAC (total) | Reference |
|---|---|---|---|---|---|---|---|---|
| NCT02541370 | I | CD133 | anti-CD133 scFv-CD137-CD3z | 7 (23) | nab-paclitaxel (150 mg/m2) cyclophosphamide (30 mg/kg) |
0.5–2 × 10⁶/kg cells; 2–4 cycles | PR 2 (3) SD 3 (14) PD 2 (6) |
Wang et al.44 |
| NCT01869166 | I | EGFR | anti-EGFR scFv-CD8a-CD137-CD3z | 16 (16) | nab-paclitaxel (100–200 mg/m2) cyclophosphamide (15–35 mg/kg) |
3.48 × 10⁶/kg; 25 cycles/6 months | PR 4 (4) SD 8 (8) PD 2 (2) |
Liu et al.45 |
| NCT01935843 | I | HER2 | anti-HER2 scFv-CD8a-CD137-CD3z | 2 (11) | nab-paclitaxel (100–200 mg/m2) cyclophosphamide (15–35 mg/kg) |
2.1 × 10⁶/kg, 1–2 cycles | PR 0 (1) SD 0 (5) PD 2 (5) |
Feng et al.49 |
| NCT01897415 | I | MSLN | anti-MSLN scFv-4-1BB-CD3z | 6 (6) | N/A | NA; 3 cycles 3 times/week for 3 weeks | SD 2 (2) PD 1 (1) Unknown 3 (3) |
Beatty et al.39 |
| NCT02159716 | I | MSLN | anti-MSLN scFv-4-1BB-CD3z | 5 (15) | with or without cyclophosphamide (1.5 g/m2) | 1–3 × 10⁷ or 1–3 × 10⁸ cells; 1 cycle | SD not specified (11) | Haas et al.40 |
PR, partial response; SD, stable disease; PD, progressive disease; MSLN, mesothelin; EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; PDAC, pancreatic ductal adenocarcinoma, N/A, not available.
Current PDAC CAR T cell clinical trials
Mesothelin
Mesothelin (MSLN) is one of the most examined target antigens for immunotherapies in PDAC. MSLN is normally expressed at limited levels on the surface of cells by the mesothelial tissues of the body (pleura, peritoneum, and pericardium).33,34 However, the protein is overexpressed in many solid tumors, including PDAC, ovarian adenocarcinoma, mesothelioma, and lung adenocarcinoma.35,36
Based on promising preclinical mouse studies, where administered MSLN-targeted CAR T cells reduced tumor burden and prolonged survival,37,38 a phase 1 clinical trial (NCT01897415) involving individuals with chemorefractory PDAC was undertaken.39 These autologous T cells were engineered using mRNA electroporation, inducing transient expression of a second-generation anti-MSLN CAR construct coupled to 4-1BB and CD3ζ.39 6 individuals with PDAC were administered CAR T cells intravenously 3 times a week for 3 weeks. MSLN expression varied between individuals, with the protein either confined to the cytoplasm or expressed on the cell surface. No individuals experienced severe adverse events (AEs) or dose-limiting toxicity. Clinical response to treatment was defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. The best response observed was stabilization of disease in 2 individuals for 3.8 and 5.4 months.
Another phase 1 trial (NCT02159716) investigated the safety and efficacy of lentivirally transduced CARs.40 Individuals with chemorefractory solid malignancies comprising pancreatic cancer, mesothelioma, and ovarian cancer were enrolled and administered anti-MSLN CAR T cells intravenously with or without lymphodepletion, comprising a dose of 1.5 g/m2 cyclophosphamide. The preconditioning chemotherapy was associated with an increase in CAR T cell expansion in peripheral blood compared with the “no lymphodepletion” group, but there was no significant difference in persistence after day 28 of treatment. Overall, CAR T cell expansion peaked on day 14 and became undetectable in individuals 6 months after administration. This is reflected in the responses observed where all individuals subsequently developed disease progression, although 11 of 15 individuals did achieve stable disease in the short term, according to RECIST v.1.1 and immune-related response criteria (irRCs). Most common AEs experienced were low-grade fatigue and nausea. Several enrolled individuals exhibited limited expression of MSLN in their primary tumor, with only 3 of 15 having greater than 75% MSLN expression. The authors noted that the murine-derived scFv in the CAR construct may potentially cause an immune response, eliminating the CAR. Phase 1 trials (NCT03054298, NCT03323944) are currently being conducted to evaluate a MSLN-directed CAR containing a human scFv along with criteria to screen for MSLN expression at baseline.
CD133
CD133, a transmembrane glycoprotein, is expressed by hemopoietic and epithelial cells.41,42 CD133 is highly expressed in PDAC cancer stem cells and has been found in other cancers, such as hepatocellular and gastric carcinomas.42,43 In a phase 1 clinical trial (NCT02541370), T cells were engineered using lentiviral vectors to target CD133.44 23 individuals with various solid tumors, including 7 with advanced pancreatic cancer, were enrolled in the clinical trial. All individuals’ tumors exhibited greater than 50% CD133 expression. Individuals were preconditioned using cyclophosphamide (30 mg/kg) and nab-paclitaxel (150 mg/m2) prior to CAR T cell infusion. Individuals received two to four cycles of CAR T cell therapy by intravenous infusion. Prior to treatment, 1 individual with stage IV PDAC had multiple metastases. After the first infusion cycle, the tumor was reduced by 40% for a period of 4 months. Repeated cell infusion cycles provided a greater period of disease control within the pancreatic cohort. Overall, 3 individuals achieved stable disease, 2 partial remission, and the remaining 2 disease progression (RECIST v.1.1). AEs within the pancreatic cancer cohort included grade 2 and 3 for leukopenia, thrombocytopenia, anemia, anorexia, nausea, and muscosal hyperemia, and 1 individual experienced grade 4 leukopenia. After treatment, CD133 expression was no longer observed in tissue biopsies, suggesting that CD133-positive cells had been eliminated.
EGFR
Epidermal growth factor receptor (EGFR) is a transmembrane protein that binds to the extracellular EGF family of proteins.45 EGFR has been detected in up to 90% of individuals with PDAC.46, 47, 48 A phase 1 study (NCT01869166) using EGFR-directed CAR T cells was undertaken in 16 individuals with PDAC with metastatic disease.45 All tumors had greater than 50% EGFR expression. The individuals received up to three cycles of EGFR-directed CAR T cell infusions within 6 months of undergoing preconditioning treatment: cyclophosphamide (35 mg/kg) and nab-paclitaxel (200 mg/m2). During treatment, some individuals received palliative radiotherapy for tumor-associated pain. Low-grade AEs were experienced by 58% of individuals, such as fever, fatigue, and nausea. Grade 3 and 4 lymphocytopenia was experienced by 38% of individuals. 2 individuals experienced grade 3 pleural effusions and pulmonary interstitial exudation toxicities. These AEs were also observed in other clinical trials utilizing the same CAR construct.49,50 EGFR is expressed on a variety of epithelial, mesenchymal, and neuronal tissues and is considered a tumor-associated antigen.51 The expression profile of EGFR in normal tissue may increase the risk of on-target/off-tumor toxicity, potentially accounting for the observed AEs. Of the 16 individuals, 8 achieved stable disease for 2–4 months, 4 were categorized as partial response for 2–4 months, and 2 exhibited disease progression (RECIST v.1.1). The remaining 2 individuals were lost during follow-up. Overall, the authors concluded that EGFR-directed CAR T cells were safe and showed modest efficacy in metastatic PDAC. However, the concept of safety when utilizing a target that is expressed in a wide range of tissues could pose potential adverse effects.
HER2
Human epidermal growth receptor 2 (HER2) is a cell surface transmembrane glycoprotein in the EGFR family of proteins that mediates cellular proliferation and differentiation.52 As an essential mediator of cellular activities, HER2 is expressed in epithelial, mesenchymal, and neuronal tissues.52,53 Up to 60% of individuals with PDAC exhibit HER2 overexpression.54 One of the first clinical trials (NCI-09-C-0041) using HER2-directed CAR T cells reported that, within 15 min of CAR T cell infusion, an individual suffered a severe on-target/off-tumor response resulting in death,55 the risk of on-target/off-tumor toxicity is high.
A phase 1 clinical trial (NCT01935843) using HER2-directed CAR T cells was undertaken to determine safety and feasibility as a target for immunotherapy.49 11 individuals were enrolled in the trial, 2 of which had PDAC. Participants were required to have greater than 50% HER2-positive tumor cells. Individuals were preconditioned using cyclophosphamide (35 mg/kg) and nab-paclitaxel (200 mg/m2) before receiving up to two cycles of anti-HER2 CAR T cell infusions. Low-grade AEs during preconditioning included nausea and fatigue as well as lymphopenia. All individuals experienced acute febrile syndrome related to CAR T cell infusion, in which an increase of 1.5- to 18-fold was observed in C-reactive protein (CRP) and IL-6 levels. No severe cytokine release syndrome (CRS) was reported, and most toxicities were reversible and treatable. At final assessment, both individuals with pancreatic cancer achieved stable disease for 5.3 and 8.3 months (RECIST v.1.1).
Current ‘Car barriers’ in pancreatic cancer
To improve the efficacy of CAR T cell therapy in solid tumors, a number of barriers have been identified (Figure 2). These include (1) the inability of T cells to efficiently traffic to tumor sites and infiltrate the tumor, (2) the limited array of targetable antigens and heterogeneous antigen expression, (3) the limited fitness and survival of CAR T cells prior to reaching the tumor site, and (4) the immunosuppressive tumor microenvironment (TME).56, 57, 58, 59 The first barrier is failure of CAR T cells to detect the tumor while traveling through the circulatory system. This results in inefficient infiltration, limiting activation and functional persistence and, therefore, decreasing clinical response.
Figure 2.

The four major barriers hindering cellular immunotherapies in PDAC and potential strategies to overcome them
(A) The extracellular matrix (ECM) and cancer-associated fibroblasts (CAFs) form a dense physical barrier, limiting the ability of CAR cells to infiltrate and target tumor cells. Intratumoral delivery, CAR design, and arming CARs with chemokine receptors can assist with trafficking and tumor infiltration. (B) The heterogeneity of tumor cells results in varying antigen expression, which limits CAR cell efficacy. Dual-targeting CARs or antibody-targeting CARs can target multiple antigens, potentially increasing efficacy by targeting more tumor cells. The TME consists of ECM; various structural and immune cells, such as myeloid-derived suppressor cells (MDSCs); tumor-associated macrophages (TAMs); monocytes; and regulatory T cells. (C) This results in an immunosuppressive environment that can inhibit CAR T cell fitness (exhaustion) and survival. Preconditioning regimens assist with altering the immune TME, and checkpoint inhibitors may help avoid CAR cell exhaustion. (D) Combination therapy, such as addition of oncolytic viruses with CAR cell therapy or targeting CAFs, can overcome the immunosuppressive TME to increase CAR cell therapy efficacy. References to key articles on these strategies are shown.149,150
The unique PDAC TME presents a multitude of challenges for CAR T cells. It accounts for up to 80% of the total pancreatic tumor mass and is comprised of the extracellular matrix and numerous cellular components, such as tumor-associated macrophages, cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), dendritic cells, B cell lymphocytes, T cell lymphocytes, and regulatory T cells.60, 61, 62 This results in a physical barrier preventing detection, trafficking, and infiltration of CAR T cells. This phenomenon has already been observed to limit chemotherapy delivery and activity.60,62 CAFs are responsible for producing the extracellular matrix and modulating tumor behavior.63 TME immune cells secrete and express molecules that suppress T cell activation, limiting CAR T cell antitumor response.64
Identifying an ideal target antigen in PDAC is difficult. Heterogeneity in the tumor as well as the TME may lead to reduced efficacy and emergence of resistance, limiting clinical success.63 A number of target antigens for cellular immunotherapy have been identified and tested in PDAC, preclinically and clinically. These include CEA, CD24, HER2, PSCA, MUC1, and MSLN, with most being expressed on the primary and secondary tumors. Thus, CAR T cell therapy may be effective in early- and late-stage disease settings.60,65, 66, 67 There are multiple ongoing clinical trials and studies testing different target antigens (Table 2).
Table 2.
Ongoing CAR cell therapy clinical trials in PDAC
| Target | Trial | Phase | Trial participants | Trial name | Progress | Institution (location) |
|---|---|---|---|---|---|---|
| CCT301-59 | NCT03960060 | I | 18 | A Study of CCT301-59 CAR T Therapy in Adult Subjects With Recurrent or Refractory Solid Tumors (CAR) | active, not yet recruiting | Shanghai Zhongshan Hospital (China) |
| CCT303-406 | NCT04511871 | I | 15 | A Phase I Trial of CCT303-406 in Patients With Relapsed or Refractory HER2 Positive Solid Tumors | recruiting | Shanghai Zhongshan Hospital (China) |
| CD22 | NCT04556669 | I | 30 | Anti-PD-L1 Armored Anti-CD22 CAR-T/CAR-TILs Targeting Patients With Solid Tumors | recruiting | Hebei Senlang Biotechnology Inc., Ltd. (China) |
| CD70 | NCT02830724 | I/II | 2 | Administering Peripheral Blood Lymphocytes Transduced With a CD70-Binding Chimeric Antigen Receptor to People With CD70 Expressing Cancers | suspended | National Cancer Institute (NCI) (USA) |
| CEA | NCT02349724 | I | 75 | A Clinical Research of CAR T Cells Targeting CEA Positive Cancer | unknown | Southwest Hospital (China) |
| CEA | NCT04348643 | I/II | 167 | Safety and Efficacy of CEA-Targeted CAR T Therapy for Relapsed/Refractory CEA+ Cancer | recruiting | Chongqing Precision Biotech Co., Ltd (China) |
| CEA | NCT04037241 | II/III | 167 | Study of Anti-CEA CAR T + Chemotherapy VS Chemotherapy Alone in Patients With CEA+ Pancreatic Cancer & Liver Metastases | not yet recruiting | Sorrento Therapeutics, Inc. (USA) |
| CEA | NCT03818165 | I | 6 | Phase 1b Study of CAR2Anti-CEA CAR T Cell Hepatic Infusions for Pancreatic Carcinoma Patients With CEA + Liver Metastases (AntiCEA_CART) | active, not yet recruiting | Sorrento Therapeutics, Inc. (USA) |
| CEA | NCT03682744 | I | 18 | CAR T Intraperitoneal Infusions for CEA-Expressing Adenocarcinoma Peritoneal Metastases or Malignant Ascites (IPC) | active, not yet recruiting | Sorrento Therapeutics, Inc. (USA) |
| CEA | NCT02850536 | I | 5 | CAR T Hepatic Artery Infusions or Pancreatic Venous Infusions for CEA-Expressing Liver Metastases or Pancreas Cancer (HITM-SURE) | active, not yet recruiting | University of Colorado, Denver (USA) |
| Claudin18.2 | NCT04404595 | I | 30 | Claudin18.2 CAR T (CT041) in Patients With Gastric or Pancreatic Cancer | recruiting | Carsgen Therapeutics, Ltd. |
| Claudin18.2 | NCT03874897 | I | 70 | Chimeric Antigen Receptor T Cells Targeting claudin18.2 in Solid Tumors. | recruiting | Peking University (China) |
| Claudin18.2 | NCT03159819 | NA | 24 | Clinical Study of CAR-CLD18 T Cells in Patients With Advanced Gastric Adenocarcinoma and Pancreatic Adenocarcinoma | recruiting | Changhai Hospital (China) |
| Claudin18.2 | NCT04404595 | I | 30 | Claudin18.2 CAR T (CT041) in Patients With Gastric or Pancreatic Cancer | recruiting | Carsgen Therapeutics, Ltd. |
| Claudin18.2 | NCT03890198 | I | 2 | A Phase 1 Study of LCAR-C182A Cells in the Treatment of Advanced Gastric Cancer and Pancreatic Ductal Adenocarcinoma | terminated | First Affiliated Hospital Xi'an Jiaotong University (China) |
| Claudin18.2/CD19/BCMA/GPC3 | NCT03302403 | NA | 18 | Clinical Study of Redirected Autologous T Cells With a Chimeric Antigen Receptor in Patients With Malignant Tumors | active, not yet recruiting | First Affiliated Hospital of Wenzhou Medical University (China) |
| EGFR | NCT03182816 | I/II | 40 | CTLA-4 and PD-1 Antibodies Expressing EGFR-CAR T Cells for EGFR Positive Advanced Solid Tumor | unknown | Shanghai Cell Therapy Research Institute (China) |
| EGFR | NCT01869166 | I/II | 60 | Treatment of Chemotherapy Refractory EGFR (Epidermal Growth Factor Receptor) Positive Advanced Solid Tumors (CART-EGFR) (CART-EGFR) | unknown | Chinese PLA General Hospital (China) |
| EpCAM | NCT03013712 | I/II | 60 | A Clinical Research of CAR T Cells Targeting EpCAM Positive Cancer (CARTEPC) | unknown | First Affiliated Hospital of Chengdu Medical College (China) |
| EpCam/TM4SF1 | NCT04151186 | NA | 72 | A Clinical Study on the Safety and Efficacy of CAR T Therapy for the TM4SF1- and EpCAM-positive Solid Tumors | not yet recruiting | Shanghai Biomed-union Biotechnology Co., Ltd. (China) |
| GD2 | NCT02992210 | I/II | 100 | Study on GD2 Positive Solid Tumors by 4SCAR-GD2 | unknown | Shenzhen Geno-Immune Medical Institute (China) |
| HER2 | NCT04650451 | I | 220 | Safety and Activity Study of HER2-Targeted Dual Switch CAR T Cells (BPX-603) in Subjects With HER2-Positive Solid Tumors | recruiting | Bellicum Pharmaceuticals (USA) |
| HER2 | NCT04660929 | I | 18 | CAR-macrophages for the Treatment of HER2 Overexpressing Solid Tumors | recruiting | Carisma Therapeutics Inc (USA) |
| HER2 | NCT03740256 | I | 45 | Binary Oncolytic Adenovirus in Combination With HER2-Specific Autologous CAR VST, Advanced HER2 Positive Solid Tumors (VISTA) | recruiting | Baylor College of Medicine (USA) |
| HER2 | NCT01935843 | I/II | 10 | Treatment of Chemotherapy Refractory Human Epidermal growth Factor Receptor-2(HER-2) Positive Advanced Solid Tumors (CART-HER-2) | unknown | Chinese PLA General Hospital (China) |
| HER2 | NCT04660929 | I | 18 | CAR-macrophages for the Treatment of HER2 Overexpressing Solid Tumors | recruiting | Carisma Therapeutics Inc (USA) |
| HER2 | NCT02713984 | I/II | NA | A Clinical Research of CAR T Cells Targeting HER2 Positive Cancer | withdrawn | Southwest Hospital (China) |
| MSLN | NCT02959151 | I/II | 20 | A Study of Chimeric Antigen Receptor T Cells Combined With Interventional Therapy in Advanced Liver Malignancy | unknown | Shanghai Cancer Hospital (China) |
| MSLN | NCT03497819 | I | 10 | Autologous CARTmeso/19 Against Pancreatic Cancer | unknown | First Affiliated Hospital of Wenzhou Medical University (China) |
| MSLN | NCT04203459 | NA | 80 | The Mechanism of Enhancing the Anti-tumor Effects of CAR T on PC by Gut Microbiota Regulation | recruiting | First Affiliated Hospital of Harbin Medical University (China) |
| MSLN | NCT03545815 | I | 10 | Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-knocked Out Mesothelin-directed CAR T Cells in Patients With Mesothelin Positive Multiple Solid Tumors. | recruiting | Chinese PLA General Hospital (China) |
| MSLN | NCT03323944 | I | 18 | CAR T cell Immunotherapy for Pancreatic Cancer | recruiting | University of Pennsylvania (USA) |
| MSLN | NCT03182803 | I/II | 40 | CTLA-4 and PD-1 Antibodies Expressing Mesothelin-CAR T Cells for Mesothelin Positive Advanced Solid Tumor | unknown | Shanghai Cell Therapy Research Institute (China) |
| MSLN | NCT02465983 | I | 4 | Pilot Study of Autologous T-cells in Patients With Metastatic Pancreatic Cancer | terminated | University of Pennsylvania (USA) |
| MSLN | NCT03638193 | NA | 10 | Study of Autologous T-cells in Patients With Metastatic Pancreatic Cancer | recruiting | The First Affiliated Hospital with Nanjing Medical University (China) |
| MSLN | NCT03747965 | I | 10 | Study of PD-1 Gene-knocked Out Mesothelin-directed CAR T Cells With the Conditioning of PC in Mesothelin Positive Multiple Solid Tumors | unknown | Chinese PLA General Hospital (China) |
| MSLN | NCT03497819 | I | 10 | Autologous CARTmeso/19 Against Pancreatic Cancer | unknown | First Affiliated Hospital of Wenzhou Medical University (China) |
| MSLN | NCT01583686 | I/II | 15 | CAR T cell Receptor Immunotherapy Targeting Mesothelin for Patients With Metastatic Cancer | terminated | National Cancer Institute (NCI) (USA) |
| MSLN | NCT03638206 | I/II | 73 | Autologous CAR-T/TCR-T Cell Immunotherapy for Malignancies | recruiting | The First Affiliated Hospital of Zhengzhou University (China) |
| MSLN | NCT02580747 | I | 20 | Treatment of Relapsed and/or Chemotherapy Refractory Advanced Malignancies by CART-meso | unknown | Chinese PLA General Hospital (China) |
| MSLN | NCT03030001 | I/II | 40 | PD-1 Antibody Expressing CAR T Cells for Mesothelin Positive Advanced Malignancies | unknown | Ningbo Cancer Hospital (China) |
| MSLN | NCT02706782 | I | 30 | A Study of Mesothelin Redirected Autologous T Cells for Advanced Pancreatic Carcinoma (meso-CART) | unknown | Shanghai GeneChem Co., Ltd. (China) |
| MSLN/CD19 | NCT03497819 | I | 10 | Autologous CARTmeso/19 Against Pancreatic Cancer | unknown | First Affiliated Hospital of Wenzhou Medical University (China) |
| MSLN/PSCA/CEA/HER2/MUC1/EGFRvIII | NCT03267173 | I | 10 | Evaluate the Safety and Efficacy of CAR T in the Treatment of Pancreatic Cancer. | unknown | First Affiliated Hospital of Harbin Medical University (China) |
| MUC1 | NCT02839954 | I/II | 10 | CAR-pNK Cell Immunotherapy in MUC1 Positive Relapsed or Refractory Solid Tumor | unknown | The First People's Hospital of Hefei (China) |
| MUC1 | NCT03179007 | I/II | 40 | CTLA-4 and PD-1 Antibodies Expressing MUC1-CAR T Cells for MUC1 Positive Advanced Solid Tumor | unknown | Shanghai Cell Therapy Research Institute (China) |
| MUC1 | NCT02587689 | I/II | 20 | Phase I/II Study of Anti-Mucin1 (MUC1) CAR T Cells for Patients With MUC1+ Advanced Refractory Solid Tumor | unknown | The First People's Hospital of Hefei (China) |
| MUC1 | NCT03633773 | I/II | 9 | Safety and Efficacy Evaluation of MUC-1 CART in the Treatment of Intrahepatic Cholangiocarcinoma | recruiting | Second Affiliated Hospital, School of Medicine, Zhejiang University (China) |
| PSCA | NCT02744287 | I/II | 151 | Safety and Activity Study of PSCA-Targeted CAR T Cells (BPX-601) in Subjects With Selected Advanced Solid Tumors | recruiting | Bellicum Pharmaceuticals (USA) |
CEA, carcinoembryonic antigen; BCMA, B cell maturation antigen; EGFR, epidermal growth factor receptor; EpCAM, epithelial cell adhesion molecule; GD2, disialoganglioside; GPC3, Glypican 3; HER2, human epidermal growth factor receptor 2; MSLN, mesothelin; PSCA, prostate stem cell antigen; MUC1, mucin 1.
CARs of the future: overcoming the barriers for effective cellular immunotherapy
Locoregional delivery
Overcoming the unique PDAC TME barrier could drastically improve outcomes. One potential method to navigate the TME is to employ physical means by locoregional or intratumoral delivery. This involves administration of CAR T cells directly to the tumor site, potentially bypassing the TME and avoiding trafficking issues. Promising results have been shown in an orthotopic mesothelioma mouse model, where intrapleural administration of MSLN-directed CAR T cells resulted in rapid antigen-induced T cell activation, leading to enhanced antitumor efficacy.57 Locoregional delivery resulted in a 30-fold increase in efficacy compared with intravenous delivery.57 This strategy is being tested in individuals with mesothelioma in a clinical trial (NCT04577326). Another study examined regional intraperitoneal delivery of anti-CEA CAR T cells for peritoneal carcinomas in mouse models.59 Regional delivery of the CEA-directed CAR T cells resulted in tumor regression of CEA-positive tumors and CAR T cell persistence.59 Although locoregional delivery has not been examined in PDAC, a number of clinical trials are testing this strategy in other solid tumors (NCT01373047, NCT02416466). One potential way this could be undertaken in PDAC is through ultrasound-guided endoscopic (EUS) administration, which has been trialed for delivery of chemotherapy,68 viral vectors,69 and immunotherapy70 in PDAC.
Armored CARs
Through a more comprehensive understanding of the TME and its components, CAR T cells can be designed to withstand and take advantage of the TME. Tumors secrete specific chemokines to recruit immune cells that suppress antitumor immunity.71 Armored CARs, such as fourth-generation CARs or TRUCKs, employ this technique. Recent studies have engineered CAR T cells to express receptors to these chemokines, aiding in recruitment to the site of the tumor, redirecting their trafficking. Preclinical studies with CAR T cells directed toward MSLN with the co-expressed chemokine receptors CCR2b and CCR4 used non-small cell lung cancer mouse models. Migration to and infiltration of the tumor were improved with these modified CAR T cells, which showed superior antitumor effects.72 IL-7 in CAR T cells plays an important role in maintaining the number of naive and memory T cells.73 IL-7 has been shown to promote persistence in solid tumors, with the absolute number of infiltrating cells being greater compared with conventional CAR T cells.73 Furthermore, addition of tumor-specific chemokine receptors can direct CAR T cells to the tumor site, improving CAR T cell infiltration. MSLN-directed CAR T cells expressing the chemokine receptor CXCR6 enhanced their infiltration and antitumor activity in an orthotopic human pancreatic cancer mouse model.74 CXCR6, along with trans-presentation of IL-15, has been shown to be critical for cytotoxic lymphocyte T cell survival and local expansion within the TME, illustrating that these chemokine receptors not only contribute to their infiltration but to other cellular functions to improve efficacy.75 CCL1 has also been shown to assist with homing of CCR8+ T cells toward tumor sites in a murine pancreatic cancer model.76 The study found that anti-MSLN CAR T cells, engineered to express CCR8, effectively recognized and eliminated target cells where CCR8+ T cells migrated toward the CCL1 gradient.
Multi-targeting CARs
Dual-targeting CAR T cells, in which T cells targeting two target antigens, have been suggested to overcome the heterogeneous nature of PDAC. One preclinical study used CAR T cells transduced with two constructs targeting CEA and MSLN.77 The dual-targeting CAR T cell exerted significant cytotoxicity when encountering both antigens on the tumor cell, with high-level activation of the CAR T cell resulting in reduction and elimination of tumor cells, compared with single antigen recognition. Another study tested infusing two separate CAR T cell products targeting CD19 and MSLN.78 However, the study experienced similar obstacles with lack of infiltration into the solid tumor, and persistence was transient. Overall, the study showed safety in individuals administered two CAR products simultaneously, but clinical outcomes were not improved.78
Another approach is to prime the tumor using tumor-targeting antibodies conjugated to a label, such as fluorescein isothiocyanate (FITC). This is followed by anti-FITC CAR T cells to target the antibody-bound tumor cells.79 This model has been validated in preclinical studies, with cytokine secretion of the CAR T cell validated against concentration of the antibody. By modulating the dose of the tumor-targeting antibody, AEs related to high-dose CAR T cells, such as CRS, may be mitigated. The advantage of this strategy is that multiple targeting antibodies labeled with FITC can be administered simultaneously with the FITC-targeted CAR T cells.80
Combination therapies
Because of the complexity of PDAC and its heterogeneity, a multifaceted treatment approach targeting different carcinogenic features in combination with cellular immunotherapies may be warranted. Effective combination therapies may elicit a synergistic effect, potentially increasing therapeutic antitumor activity, reducing primary resistance, and minimizing side effects.81,82 Chemotherapeutic agents, such as cyclophosphamide and nab-paclitaxel, have been utilized as preconditioning agents in PDAC prior to CAR T therapy. Recent studies have indicated that preconditioning regimens inhibit autoimmunity and remove suppressive cells, reduce tumor burden, sensitize tumor cells to immunotherapy, and improve CAR T cell persistence in vivo.82 In the aforementioned MSLN-directed CAR T clinical trial (NCT02159716) involving individuals with PDAC, an increase in CAR T cell expansion was initially observed with individuals who had preconditioning chemotherapy, but the difference did not persist beyond day 28.40
Tumors can upregulate immune checkpoint receptors to evade the immune system, leading to CAR T cell inhibition.83 A range of immune checkpoint inhibitors has been approved by the FDA for solid tumors, including monoclonal antibodies against programmed death protein 1 (PD-1; such as nivolumab, pembrolizumab, and pidilizumab) and programmed death ligand-1 (PD-L1; such as MDX-1105 and MPDL3280A).82,84,85 PD-1-mediated exhaustion in CAR T cells following treatment of solid cancers has prompted administration of PD-1 checkpoint blockade inhibitors in combination with CAR T cells.86 One study demonstrated that combination therapy of PD-1 blockade and anti-HER2 CAR T cell therapy was successful in treating HER2-positive tumors and correlated with an increase in CAR T cell function.87 Because systemic administration of immune checkpoint blockade is known to result in autoimmune-like toxicities, an oncolytic adenovirus expressing PD-L1 was combined with anti-HER2 CAR T cells in a subcutaneous prostate cancer mouse model.88 This resulted in local production of PD-L1 antibodies within the TME, limiting CAR T exhaustion, and was found to be more effective than systemic PD-L1 antibody administration alone. Alternatively, a PD-1 dominant-negative receptor (DNR) can be transduced into the CAR T cell, resulting in enhanced CAR T cell persistence in an orthotopic mesothelioma mouse model.86 CAR T cells can also be engineered to secrete checkpoint inhibitors to target PD-1. Secretion of anti-PD-1 enhanced antitumor activity of CAR T cells, and prolonged functional persistence in a humanized lung cancer mouse model has been observed.89 Third-generation anti-PD-1 and anti-PD-L1 CAR T cells were tested in PD-L1-overexpressing PDAC cells and in PDAC mouse models, resulting in both CAR T cells inducing tumor regression.90 This was correlated with reduced T cell exhaustion.
Administration of oncolytic virus therapy, which utilizes genetically engineered viruses, such as adenovirus or vaccinia virus, to replicate in tumor cells, may improve CAR T cell function by stimulating interferon genes, induce recruitment of T cells, and reverse local immunosuppression, which can enhance CAR T cell infiltration into the tumor.91, 92, 93, 94 One study engineered an oncolytic virus to express RANTES and IL-15 in combination with GD2-targeted CAR T cell therapy.95 In a preclinical neuroblastoma xenograft mouse model, the combined therapy demonstrated improved survival and higher CAR T cell infiltration rates compared with CAR T cell monotherapy. Furthermore, RANTES and IL-15 have been shown to be localized to the tumor, indicating that the oncolytic virus was specific to the tumor, and could be a potential strategy to circumvent cytokine toxicities associated with their systemic administration.95 Uninfected tumor cells are infected when neighboring infected tumor cells are killed by CAR T cell-mediated tumor lysis, causing viral particles to be released, promoting viral spread to the uninfected tumor cells and targeting by CAR T cells.96 Oncolytic viruses are known to upregulate checkpoint ligands by mediating release of type I interferons.97 Therefore, the combination of oncolytic viruses, CAR T cells, and checkpoint blockade may contribute to overcoming T cell exhaustion and achieving CAR T cell persistence.
Other cellular immunotherapies
Autologous CAR T cells present challenges for widespread implementation. These include the requirement for individual-specific manufacturing, their high cost and potential for inconsistent yield and function (depending on the immune system of the individual) with consequent risk of severe AEs.98,99 This has prompted research into exploring use of allogeneic CAR T cells as well as other cellular immunotherapies, such as CAR natural killer cells and tumor-infiltrating lymphocyte therapy.
Allogeneic CAR T cells
Allogeneic CAR T cells from healthy donors may offer advantages over use of autologous CAR T cells, including their immediate availability, standardization of product, and reduction in cost.100,101 T cells from healthy donors can be expanded exponentially and cryopreserved, allowing an off-the-shelf product without manufacturing or treatment delays.102 This standardized high-volume manufacturing provides an opportunity for affected individuals to receive more cycles with the same standard of product.103
However, allogeneic CAR T cell therapy introduces potential risks in the form of graft versus host disease (GvHD). Human leukocyte antigen (HLA) mismatch between donor and recipient can lead to an immune response that readily eliminates the allogeneic CAR T cells.101,104 High-resolution HLA typing with next-generation sequencing may mitigate the risks of potential HLA mismatch,104 but complete matching of donor-recipient HLA haplotype could diminish donor availability.105 Gene editing technologies have made it possible to eliminate T cell receptor (TCR) expression by editing the TRAC gene, making allogeneic CAR T cells less accessible to the host immune system in preclinical models, with clinical trials currently underway.100,106 GvHD is a life-threatening complication and has prompted research into alternatives such as other immune cells; for example, natural killer cells and tumor-infiltrating lymphocytes.
CAR natural killer cells
The natural killer (NK) cell has been identified as one of the immune cells that may be used as an alternative to allogeneic CAR T cells.98,107 NK cells are part of the innate immune system, having the ability to target foreign or damaged cells.108 However, unlike T cells, NK cells can recognize targets in a non-antigen-specific manner without the need for prior sensitization, making them a potential candidate for therapy against cancer.108, 109, 110
Initially, autologous NK cells were directed against tumors to prevent GvHD, but NK cells can recognize self and inhibit cytotoxic functions, diminishing their therapeutic utility.108 Allogeneic NK cells, derived from healthy individuals, exhibit greater cytotoxicity compared with autologous NK cells from individuals with cancer.108,111 One study used NK cells isolated from umbilical cord blood that were modified to express an anti-PSCA CAR construct with soluble IL-15.112 These PSCA-directed CAR NK cells were tested in a metastatic humanized pancreatic cancer mouse model. An increase in cytotoxic function, suppressed tumor growth, and prolonged survival were observed. On day 48, pancreatic biopsies revealed minimal tumor cells and a high number of NK cells, indicating persistence of the immune cells within the TME.112 Two clinical trials (NCT02839954 and NCT03941457) are currently examining allogeneic NK cell infusions in PDAC. A case study report from NCT03941457 found that allogeneic NK cell infusions targeting ROBO1 in PDAC were well tolerated and did not lead to serious toxicity.113 Although there is a lack of clinical results so far, allogeneic NK cell therapy could potentially lead to a feasible off-the-shelf’ product for PDAC.
Use of allogeneic NK cells has been demonstrated to be feasible, but there is a limited number of NK cells that can be collected from a given donor, prompting investigation of NK cell lines.110 A phase 1 clinical trial utilizing activated NK-92 cells was undertaken to address the practicality, safety, and activity against acute myeloid leukemia.114 The treatment was well tolerated with no grade 3 or 4 toxicity, demonstrating the potential of the cell line as an off-the-shelf therapy. CAR-engineered NK-92 cells targeting MSLN in ovarian cancer were evaluated for efficacy and therapeutic effects.115 MSLN-directed CAR NK-92 cells co-cultured with ovarian cancer cell lines killed MSLN-positive ovarian cancer cells in vitro and effectively eliminated all cancerous cells in subcutaneous and intraperitoneal tumor mouse models in vivo. A phase 1 clinical trial examined a second-generation CAR NK-92 cell directed against MUC1 and PD-1 in a range of cancers positive for both targets.116 Of the 13 individuals, 9 had stable disease, 1 showed progressive disease, and the remaining 3 were withdrawn from the study. No severe AEs were encountered during the trial. This indicates that allogeneic CAR NK cells should be the subject of further clinical trials.
An orthotopic pancreatic tumor model was used to study the synergistic efficacy of anti-ROBO1 CAR NK-92 cells in combination with brachytherapy, an internal radiation therapy where radioactive beads are placed in proximity to the tumor.117 ROBO1 is a member of the neural axon guidance receptor family and has been found to be overexpressed in PDAC.118 Tumor burden was reduced significantly in the brachytherapy-only arm, with further reductions observed in the brachytherapy and CAR NK combination arm. Second-generation anti-ROBO1 CAR NK-92 cells were administered as a case study in an individual with pancreatic cancer with liver metastases.113 The individual was treated with weekly systemic infusions and intratumoral injections to the liver metastasis. Stable disease was achieved for 5 months, and the only reported AE was fever after infusion. These promising results have led to initiation of three phase I/II clinical trials (NCT03941457, NCT03940820, NCT03931720) to assess the safety and efficacy of ROBO1 as a target for CAR NK-92 cell therapy in PDAC and other solid tumors. Use of CAR NK-92 cells is feasible and provides a foundation for further development of CAR NK cell therapy.
Checkpoint blockades and immunosuppression can limit CAR NK cell function and reduce persistence, resulting in the need for multiple infusions with consequent increased risk of rejection. To address this, CAR NK cells can be manufactured from induced pluripotent stem cells (iPSCs). CAR iPSC-NK cells are genetically edited to carry a CAR with immune suppression genes removed to prolong NK persistence and efficacy. Unlike allogeneic NK cells, CAR iPSC NK cells are derived from triple-homozygous HLA donors, reducing the risk of rejection over multiple infusions. TAG72 is an adenocarcinoma neoantigen. TAG72-targeted CAR iPSC-NK cells were generated and tested against multiple ovarian cancer cell lines.98 The study demonstrated on-target cytotoxic function in vitro. In an ovarian cancer xenograft mouse model, iPSC-NK cells reduced tumor burden and increased median survival compared with control mice.119,120 Although clinical studies are ongoing, CAR iPSC-NK cells could potentially enable on-demand production for each individual and provide consistent off-the-shelf capabilities to treat a variety of cancers using a single cell therapy product.98
Tumor-infiltrating lymphocytes
Tumor-infiltrating lymphocytes (TILs) are mononuclear immune cells that infiltrate tumor tissue during the initial immune response.121 Protocols have been established to isolate TILs using density centrifugation of mechanically dissociated tumor tissue.122 TIL therapy in PDAC is currently being tested in phase I and phase II clinical trials (NCT05098197, NCT03935893, and NCT03610490), with results yet to be published. TIL therapy has achieved positive clinical results in several phase I and phase II trials in other cancers.123, 124, 125, 126 TIL therapy is limited by IL-2 AEs because high-dose IL-2 is required after infusion.123,124 In a phase II clinical trial, 12 individuals with metastatic melanoma were administered low-dose subcutaneous IL-2 to examine whether a lower dose of the cytokine can achieve results similar to a high dose.123 The majority of AEs were attributed to IL-2 but were manageable (grades 1–2). Of the 12 individuals, 3 exhibited partial response, 6 had stable disease, and 3 had progressive disease. Interestingly, the study reported a T cell subpopulation in an individual’s peripheral blood, dominant in the infusion product, that was present in their peripheral blood 2 years after infusion, indicating TIL persistence.123 In another phase II clinical trial, 9 individuals with metastatic cervical cancer were enrolled for treatment with human papillomavirus (HPV)-targeted TIL therapy.126 TILs were separated from tumor fragments and expanded, selecting for reactivity against HPV-16 or HPV-18, generating HPV-targeting TILs. Individuals were administered TIL therapy with bolus injection of aldesleukin (recombinant IL-2). 2 individuals exhibited stable disease and 1 partial response, and the remaining individuals showed disease progression. The 3 individuals who demonstrated tumor responses had the highest frequency of HPV-reactive TILs in their infusion product. Common AEs were associated with lymphodepletion, and no acute toxicities were related to HPV-TIL infusion.126 The studies demonstrate that TIL infusion is feasible, safe, and clinically active.
PDAC models to evaluate cellular immunotherapy
Murine models
Preclinical models are used to examine CAR T therapy efficacy and safety before translation to human clinical trials. The majority of preclinical CAR T studies are performed in mice, with four main murine models used: syngeneic (immunocompetent allograft), xenograft, transgenic (immunocompetent), and humanized mice.
Syngeneic mouse models use mouse-derived CAR T cells, tumors, and target antigens. These mice have an intact immune system, allowing study of a functional immune response to CAR T cells, which can reveal potential on-target/off-tumor toxicities that may be conserved between species.127 A disadvantage of the syngeneic model is that it reflects mouse biology, failing to be a true representation of a human. Syngeneic models have been used in CAR T cell studies to target components of the TME, such as fibroblast activation protein (FAP) on CAFs.128 However, FAP is also strongly expressed in bone marrow stromal cells, resulting in considerable on-target/off-tumor bone-related toxicity and limited antitumor effects.
Xenograft mouse models involve use of immunocompromised mice, where an implanted human tumor is tested for effects following administration of human CAR T cells.129 The most commonly used mouse strain is the non-obese diabetic (NOD) severe combined immunodeficiency (SCID) gamma (NSG) mouse. Because the mice are immunocompromised, there are limited interactions with adoptive immune cells; therefore, on-target/off-tumor toxicity may be missed. Nevertheless, xenograft mice are a model for validation of proof-of-concept studies, such as testing CAR designs for efficacy, as well as investigation of human tumor biology.130 One study tested the efficacy of CAR constructs that also constitutively express human cytokines in systemic lymphoma.131 IL-7 and IL-21 were found to be superior in their effects to modulate antitumor activity. Another study utilized second- and third-generation anti-MSLN CAR T cells with either or both CD28 and 4-1BB co-stimulatory domains.132 Second-generation anti-MSLN 4-1BB CAR T cells have been shown to reduce tumor burden and eradicate tumors in some cases. The third-generation MSLN-CAR with 4-1BB and CD28 co-stimulatory domains enhanced T cell persistence. This study shows CAR biology within a system that mimics the nature of tumors and CAR T cell therapy.132
Immunocompetent transgenic mice involve expressing a human tumor antigen in immunocompetent mice and are often employed to predict treatment safety.129 Several studies have utilized immunocompetent transgenic mice to evaluate CAR T cells. Transgenic mice expressing human CEA in the intestines and lung tissue were used to test anti-CEA CAR T cells in an orthotopic PDAC mouse model.133 Long-term tumor eradication was achieved. Although CAR T cells were found in the intestines and lungs, they did not result in a local inflammatory response.
Humanized transgenic mice are immunocompromised mice with a human immune system, human tumors, and introduced CAR T cells. NSG mice transplanted with CD34+ human cells are a relatively simple model that is used routinely to recapitulate the human immunological environment.129 A more complicated model is the BLT SCID mouse model, where human fetal bone marrow, liver, and thymus tissues are transplanted for a more complete reconstruction of T cells in vivo, providing a wider variety of human immune cells and immune responses.129
Although, advancements are being made with preclinical mouse models, no model perfectly recapitulates the human immune system or reflects the unwanted side effects, such as CRS or on-target/off-tumor toxicity (Figure 3). Therefore, careful selection is required to evaluate CAR T cell efficacy and safety.
Figure 3.

Proposed workflow for implementing preclinical PDAC models to evaluate cellular immunotherapies
Pancreatic cancer tissue is sampled, and preclinical organoids and xenograft models are generated. CAR immune cells are screened for suitable CAR candidates in PDOs or used to identify individual-specific antigens and improve CAR design. Positive candidates can then be translated for personalized treatment (back to the same individual) or tested in clinical trials. PDX models can also be utilized for cytotoxicity validation (immunocompetent mouse models) and to examine potential on-target/off-tumor effects and CRS (humanized transgenic mouse models).
Organoids
Recent organoid technology has opened a new avenue in cancer models. Patient-derived organoids (PDOs) are three-dimensional structures maintaining the key cellular hierarchy and function of the host tumor. Importantly, organoids have been found to recapitulate the host tumor genetically and phenotypically and have the potential to predict therapeutic response.134 PDOs are rapidly replacing the standard patient-derived xenograft (PDX) models, in which human tissue is transplanted and grown in an immunocompromised mouse. It is thought that PDOs more faithfully recapitulate the pathogenic process and facilitate more timely and less costly establishment of cultures.135
PDAC PDOs were first established from normal and cancerous pancreatic tissue to interrogate the pathways of tumorigenesis.136 Orthotopically transplanted organoids were found to induce a TME and recapitulate tumor development beyond that achieved using cell lines. Transplanted PDAC PDOs allow modeling of tumor growth from early to metastatic stages.136, 137, 138 However, in vitro PDOs do not have the capacity to produce the TME. Co-cultured PDOs with pancreatic fibroblasts have been found to support PDO organoids, recapitulating parts of the TME in vitro.139 A triple co-culture system of PDAC PDOs established from biopsies, pancreatic fibroblasts, and T cells has been used to study immunotherapies.140 PDO platforms have been established for exploring and evaluating the efficacy of therapeutic targets, generate individual-specific data, and provide a disease model capable of predicting responses of affected individuals.141,142 Ultimately, PDOs have paved the way for development of tailored “precision” treatments.143, 144, 145
Testing cellular immunotherapies using PDOs is an emerging but promising strategy to enhance precision medicine. Several strategies and methodologies have been established and examined, but none have so far been tested in a clinical setting. A study showed that, in bladder cancer, surface antigens on primary tissue was also found on PDOs, so it could be used to identify an individual’s CAR-recognizable antigens and confirm this by testing the antigen specific CAR T cells in vitro.146 In addition, bioprinted (neuroblastoma) organoids could be used to preselect CAR T cell constructs.147 There is no standard cytotoxicity assay to test the efficacy of CARs with PDOs. One study established a luciferase-based endpoint assay and a live microscopy assay for continuous and cell-resolved analysis to monitor NK CAR-mediated cytotoxicity against colorectal cancer organoids.148 The study found that killing efficiency rates differed for different sized organoids, where smaller organoids were lysed more rapidly compared with larger organoids, potentially reflecting CAR responses to solid tumor masses. Although the study found that anti-EpCAM CAR NK-92 cells were able to migrate on the surface, they were incapable of deeply penetrating the Matrigel. This may be CAR NK cell specific because another study illustrated effective killing of HER2 CAR T cells in PDAC PDOs grown in Matrigel, measured using microscopy.67 There is a need to develop PDO assays to evaluate cellular immunotherapies for use in clinical settings. This may lead to PDO platforms to screen cellular immunotherapies or to confirm manufacturing of cellular immunotherapies prior to infusion into affected individuals. These should lead to improved selection of individuals for specific cellular immunotherapies and greater treatment options for those with PDAC (Figure 3).
Conclusion
The clinical efficacy of CAR T cell therapy in PDAC is currently limited, but it remains an active, viable, and promising field of research. The current challenges in translating successful CAR T cell therapies from hematological to solid tumors are slowly being overcome by several strategies designed to adapt and overcome the barriers within the TME. CAR therapies may be improved by increasing their efficacy against the chosen antigen through CAR design and overcoming tumor heterogeneity by selecting more than one targetable antigen. Selecting tumor-restricted antigens will minimize on-target/off-tumor toxicity. Development of potential off-the-shelf cellular immunotherapies, including allogeneic CAR T, CAR NK cells, and TILs, should standardize products and reduce manufacturing costs and the time to treatment administration. Although there are many strategies being tested in preclinical and clinical settings, an approach utilizing organoid models to identify the right treatment for the right individual may be required to improve outcomes in PDAC.
Acknowledgments
This work is supported by the Li Ka Shing Cell & Gene Therapy Initiative (to J.E.J.R.), Cancer Council NSW Project Grant RG20-07 (to J.E.J.R.), Cancer Council NSW Pathways Grant PW18-03 (to J.E.J.R.), National Health and Medical Research Council Investigator Grant 1177305 (to J.E.J.R.), and CSR Australia project funding (to D.Y. and J.E.J.R.). The funders had no role in the study design, decision to publish, or preparation of the manuscript. The graphical abstract and figures were created with BioRender.
Author contributions
D.Y. planned the manuscript. D.Y. and C.G. wrote the manuscript and designed the figures. D.Y., C.G., P.S., and J.E.J.R. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Declaration of interest
J.E.J.R. reports advisory roles in The Gene Technology Technical Advisory Committee, Office of the Gene Technology Regulator, Australian Government. J.E.J.R. also reports honoraria, speaker fees, or advisory roles for GSK, Takeda, Gilead, Cynata, Pfizer, Spark, Novartis, Celgene, Bluebird Bio, Shire, Avrobio, ATARA, and Bayer; stocks in Genea; and a consultant role for Rarecyte (stocks in lieu).
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Masiak-Segit W., Rawicz-Pruszynski K., Skorzewska M., Polkowski W.P. Surgical treatment of pancreatic cancer. Pol. Przegl. Chir. 2018;90:45–53. doi: 10.5604/01.3001.0011.7493. [DOI] [PubMed] [Google Scholar]
- 4.Loveday B., Lipton L., Thomson B. Pancreatic cancer: an update on diagnosis and management. Aust. J. Gen. Pract. 2019;48:826–831. doi: 10.31128/AJGP-06-19-4957. [DOI] [PubMed] [Google Scholar]
- 5.Landman A., Feetham L., Stuckey D. Working together to reduce the burden of pancreatic cancer. Lancet Oncol. 2020;21:334–335. doi: 10.1016/S1470-2045(20)30088-7. [DOI] [PubMed] [Google Scholar]
- 6.Balachandran V.P., Beatty G.L., Dougan S.K. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology. 2019;156:2056–2072. doi: 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H.S., Park S.W. Systemic chemotherapy in advanced pancreatic cancer. Gut Liver. 2016;10:340–347. doi: 10.5009/gnl15465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres C., Grippo P.J. Pancreatic cancer subtypes: a roadmap for precision medicine. Ann. Med. 2018;50:277–287. doi: 10.1080/07853890.2018.1453168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golan T., Hammel P., Reni M., Van Cutsem E., Macarulla T., Hall M.J., Park J.O., Hochhauser D., Arnold D., Oh D.Y., et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le D.T., Durham J.N., Smith K.N., Wang H., Bartlett B.R., Aulakh L.K., Lu S., Kemberling H., Wilt C., Luber B.S., et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pishvaian M.J., Blais E.M., Brody J.R., Lyons E., DeArbeloa P., Hendifar A., Mikhail S., Chung V., Sahai V., Sohal D.P.S., et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020;21:508–518. doi: 10.1016/S1470-2045(20)30074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.CAR T-cells: an exciting frontier in cancer therapy. Lancet. 2017;390:1006. doi: 10.1016/S0140-6736(17)32395-4. [DOI] [PubMed] [Google Scholar]
- 13.Abreu T.R., Fonseca N.A., Gonçalves N., Moreira J.N. Current challenges and emerging opportunities of CAR-T cell therapies. J. Control. Release. 2020;319:246–261. doi: 10.1016/j.jconrel.2019.12.047. [DOI] [PubMed] [Google Scholar]
- 14.Philipson B., Milone M.C. In: Second Generation Cell and Gene-Based Therapies. Vertès A.A., Smith D.M., Qureshi N., Dowden N.J., editors. Academic Press; 2020. Chapter 4 - T cell engineering and the rise of CAR-T cell therapies; pp. 69–90. [Google Scholar]
- 15.Sadelain M., Rivière I., Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadelain M., Brentjens R., Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.-A.N., Lanier B.J., et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eshhar Z., Waks T., Gross G., Schindler D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. U S A. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Stegen S.J.C., Hamieh M., Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allegra A., Innao V., Gerace D., Vaddinelli D., Musolino C. Adoptive immunotherapy for hematological malignancies: current status and new insights in chimeric antigen receptor T cells. Blood Cells Mol. Dis. 2016;62:49–63. doi: 10.1016/j.bcmd.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Chmielewski M., Abken H. TRUCKs: the fourth generation of CARs. Expert Opin. Biol. Ther. 2015;15:1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 22.Kueberuwa G., Kalaitsidou M., Cheadle E., Hawkins R.E., Gilham D.E. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol. Ther. Oncolytics. 2018;8:41–51. doi: 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma X., Shou P., Smith C., Chen Y., Du H., Sun C., Porterfield Kren N., Michaud D., Ahn S., Vincent B., et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020;38:448–459. doi: 10.1038/s41587-019-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Condomines M., Arnason J., Benjamin R., Gunset G., Plotkin J., Sadelain M. Tumor-targeted human T cells expressing CD28-based chimeric antigen receptors circumvent CTLA-4 inhibition. PLoS One. 2015;10:e0130518. doi: 10.1371/journal.pone.0130518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koneru M., Purdon T.J., Spriggs D., Koneru S., Brentjens R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagoya Y., Tanaka S., Guo T., Anczurowski M., Wang C.H., Saso K., Butler M.O., Minden M.D., Hirano N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018;24:352–359. doi: 10.1038/nm.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yip A., Webster R.M. The market for chimeric antigen receptor T cell therapies. Nat. Rev. Drug Discov. 2018;17:161–162. doi: 10.1038/nrd.2017.266. [DOI] [PubMed] [Google Scholar]
- 28.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W., et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abramson J.S., Palomba M.L., Gordon L.I., Lunning M.A., Wang M., Arnason J., Mehta A., Purev E., Maloney D.G., Andreadis C., et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 30.Munshi N.C., Anderson L.D., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 31.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang K., Pastan I., Willingham M.C. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int. J. Cancer. 1992;50:373–381. doi: 10.1002/ijc.2910500308. [DOI] [PubMed] [Google Scholar]
- 34.Castelletti L., Yeo D., van Zandwijk N., Rasko J.E.J. Anti-Mesothelin CAR T cell therapy for malignant mesothelioma. Biomarker Res. 2021;9:11. doi: 10.1186/s40364-021-00264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hassan R., Ho M. Mesothelin targeted cancer immunotherapy. Eur. J. Cancer. 2008;44:46–53. doi: 10.1016/j.ejca.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ordóñez N.G. Application of mesothelin immunostaining in tumor diagnosis. Am. J. Surg. Pathol. 2003;27:1418–1428. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Beatty G.L., Haas A.R., Maus M.V., Torigian D.A., Soulen M.C., Plesa G., Chew A., Zhao Y., Levine B.L., Albelda S.M., et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Y., Moon E., Carpenito C., Paulos C.M., Liu X., Brennan A.L., Chew A., Carroll R.G., Scholler J., Levine B.L., et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beatty G.L., O'Hara M.H., Lacey S.F., Torigian D.A., Nazimuddin F., Chen F., Kulikovskaya I.M., Soulen M.C., McGarvey M., Nelson A.M., et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155:29–32. doi: 10.1053/j.gastro.2018.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haas A.R., Tanyi J.L., O'Hara M.H., Gladney W.L., Lacey S.F., Torigian D.A., Soulen M.C., Tian L., McGarvey M., Nelson A.M., et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther. 2019;27:1919–1929. doi: 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith L.M., Nesterova A., Ryan M.C., Duniho S., Jonas M., Anderson M., Zabinski R.F., Sutherland M.K., Gerber H.P., Van Orden K.L., et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br. J. Cancer. 2008;99:100–109. doi: 10.1038/sj.bjc.6604437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmohl J.U., Vallera D.A. CD133, selectively targeting the root of cancer. Toxins. 2016;8:165. doi: 10.3390/toxins8060165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrandina G., Petrillo M., Bonanno G., Scambia G. Targeting CD133 antigen in cancer. Expert Opin. Ther. Targets. 2009;13:823–837. doi: 10.1517/14728220903005616. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y., Chen M., Wu Z., Tong C., Dai H., Guo Y., Liu Y., Huang J., Lv H., Luo C., et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7:e1440169. doi: 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y., Guo Y., Wu Z., Feng K., Tong C., Wang Y., Dai H., Shi F., Yang Q., Han W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: a phase I clinical trial. Cytotherapy. 2020;22:573–580. doi: 10.1016/j.jcyt.2020.04.088. [DOI] [PubMed] [Google Scholar]
- 46.Xiong H.Q., Rosenberg A., LoBuglio A., Schmidt W., Wolff R.A., Deutsch J., Needle M., Abbruzzese J.L. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: a multicenter phase II Trial. J. Clin. Oncol. 2004;22:2610–2616. doi: 10.1200/JCO.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 47.Fjällskog M.L., Lejonklou M.H., Oberg K.E., Eriksson B.K., Janson E.T. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin. Cancer Res. 2003;9:1469–1473. [PubMed] [Google Scholar]
- 48.Ueda S., Ogata S., Tsuda H., Kawarabayashi N., Kimura M., Sugiura Y., Tamai S., Matsubara O., Hatsuse K., Mochizuki H. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2004;29 doi: 10.1097/00006676-200407000-00061. e1–8. [DOI] [PubMed] [Google Scholar]
- 49.Feng K., Liu Y., Guo Y., Qiu J., Wu Z., Dai H., Yang Q., Wang Y., Han W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9:838–847. doi: 10.1007/s13238-017-0440-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo Y., Feng K., Liu Y., Wu Z., Dai H., Yang Q., Wang Y., Jia H., Han W. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin. Cancer Res. 2018;24:1277–1286. doi: 10.1158/1078-0432.CCR-17-0432. [DOI] [PubMed] [Google Scholar]
- 51.Yano S., Kondo K., Yamaguchi M., Richmond G., Hutchison M., Wakeling A., Averbuch S., Wadsworth P. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res. 2003;23:3639–3650. [PubMed] [Google Scholar]
- 52.Cho H.S., Mason K., Ramyar K.X., Stanley A.M., Gabelli S.B., Denney D.W., Jr., Leahy D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 53.Olayioye M.A., Neve R.M., Lane H.A., Hynes N.E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan M., Schwaederle M., Arguello D., Millis S.Z., Gatalica Z., Kurzrock R. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34:157–164. doi: 10.1007/s10555-015-9552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagner J., Wickman E., DeRenzo C., Gottschalk S. CAR T cell therapy for solid tumors: bright future or dark reality? Mol. Ther. 2020;28:2320–2339. doi: 10.1016/j.ymthe.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adusumilli P.S., Cherkassky L., Villena-Vargas J., Colovos C., Servais E., Plotkin J., Jones D.R., Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker A.J., Majzner R.G., Zhang L., Wanhainen K., Long A.H., Nguyen S.M., Lopomo P., Vigny M., Fry T.J., Orentas R.J., Mackall C.L. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther. 2017;25:2189–2201. doi: 10.1016/j.ymthe.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katz S.C., Point G.R., Cunetta M., Thorn M., Guha P., Espat N.J., Boutros C., Hanna N., Junghans R.P. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23:142–148. doi: 10.1038/cgt.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cutmore L.C., Brown N.F., Raj D., Chauduri S., Wang P., Maher J., Wang Y., Lemoine N.R., Marshall J.F. Pancreatic Cancer UK Grand Challenge: developments and challenges for effective CAR T cell therapy for pancreatic ductal adenocarcinoma. Pancreatology. 2020;20:394–408. doi: 10.1016/j.pan.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 61.Jang J.E., Hajdu C.H., Liot C., Miller G., Dustin M.L., Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017;20:558–571. doi: 10.1016/j.celrep.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li K.-Y., Yuan J.-L., Trafton D., Wang J.-X., Niu N., Yuan C.-H., Liu X.-B., Zheng L. Pancreatic ductal adenocarcinoma immune microenvironment and immunotherapy prospects. Chronic Dis. Transl. Med. 2020;6:6–17. doi: 10.1016/j.cdtm.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ligorio M., Sil S., Malagon-Lopez J., Nieman L.T., Misale S., Di Pilato M., Ebright R.Y., Karabacak M.N., Kulkarni A.S., Liu A., et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell. 2019;178:160–175.e27. doi: 10.1016/j.cell.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bailey P., Chang D.K., Forget M.A., San Lucas F.A., Alvarez H.A., Haymaker C., Chattopadhyay C., Kim S.H., Ekmekcioglu S., Grimm E.A., et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016;6:35848. doi: 10.1038/srep35848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akce M., Zaidi M.Y., Waller E.K., El-Rayes B.F., Lesinski G.B. The potential of CAR T cell therapy in pancreatic cancer. Front. Immunol. 2018;9:2166. doi: 10.3389/fimmu.2018.02166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raj D., Nikolaidi M., Garces I., Lorizio D., Castro N.M., Caiafa S.G., Moore K., Brown N.F., Kocher H.M., Duan X.B., et al. CEACAM7 is an effective target for CAR T-cell therapy of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2021;27:1538–1552. doi: 10.1158/1078-0432.CCR-19-2163. [DOI] [PubMed] [Google Scholar]
- 67.Raj D., Yang M.H., Rodgers D., Hampton E.N., Begum J., Mustafa A., Lorizio D., Garces I., Propper D., Kench J.G., et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut. 2019;68:1052–1064. doi: 10.1136/gutjnl-2018-316595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Levy M.J., Alberts S.R., Chari S.T., Farnell M.B., Haddock M.G., Kendrick M.L., Mukhopadhyay D., Petersen G.M., Takahashi N. 716 EUS guided intra-tumoral gemcitabine therapy for locally advanced and metastatic pancreatic cancer. Gastrointest. Endosc. 2011;73:AB144–AB145. [Google Scholar]
- 69.Hecht J.R., Bedford R., Abbruzzese J.L., Lahoti S., Reid T.R., Soetikno R.M., Kirn D.H., Freeman S.M. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin. Cancer Res. 2003;9:555–561. [PubMed] [Google Scholar]
- 70.Chang K.J., Nguyen P.T., Thompson J.A., Kurosaki T.T., Casey L.R., Leung E.C., Granger G.A. Phase I clinical trial of allogeneic mixed lymphocyte culture (cytoimplant) delivered by endoscopic ultrasound-guided fine-needle injection in patients with advanced pancreatic carcinoma. Cancer. 2000;88:1325–1335. doi: 10.1002/(sici)1097-0142(20000315)88:6<1325::aid-cncr8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 71.Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M., et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 72.Wang Y.A., Wang J., Yang X.Y., Yang J.L., Lu P.P., Zhao L., Li B.K., Pan H.Y., Jiang Z.T., Shen X.T., et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front. Immunol. 2021;12:628906. doi: 10.3389/fimmu.2021.628906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 74.Lesch S., Blumenberg V., Stoiber S., Gottschlich A., Ogonek J., Cadilha B.L., Dantes Z., Rataj F., Dorman K., Lutz J., et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021;21:1246–1260. doi: 10.1038/s41551-021-00737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Pilato M., Kfuri-Rubens R., Pruessmann J.N., Ozga A.J., Messemaker M., Cadilha B.L., Sivakumar R., Cianciaruso C., Warner R.D., Marangoni F., et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. 2021;184:4512–4530.e22. doi: 10.1016/j.cell.2021.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cadilha B.L., Benmebarek M.R., Dorman K., Oner A., Lorenzini T., Obeck H., Vanttinen M., Di Pilato M., Pruessmann J.N., Stoiber S., et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci. Adv. 2021;7:eabi5781. doi: 10.1126/sciadv.abi5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang E., Yang P., Gu J., Wu H., Chi X., Liu C., Wang Y., Xue J., Qi W., Sun Q., et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J. Hematol. Oncol. 2018;11:102. doi: 10.1186/s13045-018-0646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ko A.H., Jordan A.C., Tooker E., Lacey S.F., Chang R.B., Li Y., Venook A.P., Tempero M., Damon L., Fong L., et al. Dual targeting of mesothelin and CD19 with chimeric antigen receptor-modified T cells in patients with metastatic pancreatic cancer. Mol. Ther. 2020;28:2367–2378. doi: 10.1016/j.ymthe.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tamada K., Geng D., Sakoda Y., Bansal N., Srivastava R., Li Z., Davila E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012;18:6436–6445. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]
- 80.Lee Y.G., Marks I., Srinivasarao M., Kanduluru A.K., Mahalingam S.M., Liu X., Chu H., Low P.S. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. 2019;79:387–396. doi: 10.1158/0008-5472.CAN-18-1834. [DOI] [PubMed] [Google Scholar]
- 81.Moon E.K., Wang L.C., Dolfi D.V., Wilson C.B., Ranganathan R., Sun J., Kapoor V., Scholler J., Puré E., Milone M.C., et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu J., Wang Y., Shi J., Liu J., Li Q., Chen L. Combination therapy: a feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncol. Lett. 2018;16:2063–2070. doi: 10.3892/ol.2018.8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruella M., Kalos M. Adoptive immunotherapy for cancer. Immunol. Rev. 2014;257:14–38. doi: 10.1111/imr.12136. [DOI] [PubMed] [Google Scholar]
- 84.Henick B.S., Herbst R.S., Goldberg S.B. The PD-1 pathway as a therapeutic target to overcome immune escape mechanisms in cancer. Expert Opin. Ther. Targets. 2014;18:1407–1420. doi: 10.1517/14728222.2014.955794. [DOI] [PubMed] [Google Scholar]
- 85.Grosser R., Cherkassky L., Chintala N., Adusumilli P.S. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471–482. doi: 10.1016/j.ccell.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cherkassky L., Morello A., Villena-Vargas J., Feng Y., Dimitrov D.S., Jones D.R., Sadelain M., Adusumilli P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.John L.B., Devaud C., Duong C.P., Yong C.S., Beavis P.A., Haynes N.M., Chow M.T., Smyth M.J., Kershaw M.H., Darcy P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 88.Tanoue K., Shaw A.R., Watanabe N., Porter C., Rana B., Gottschalk S., Brenner M., Suzuki M. Armed oncolytic adenovirus–expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77:2040–2051. doi: 10.1158/0008-5472.CAN-16-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li S., Siriwon N., Zhang X., Yang S., Jin T., He F., Kim Y.J., Mac J., Lu Z., Wang S., et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res. 2017;23:6982–6992. doi: 10.1158/1078-0432.CCR-17-0867. [DOI] [PubMed] [Google Scholar]
- 90.Yang C.-Y., Fan M.H., Miao C.H., Liao Y.J., Yuan R.-H., Liu C.L. Engineering chimeric antigen receptor T cells against immune checkpoint inhibitors PD-1/PD-L1 for treating pancreatic cancer. Mol. Ther. Oncolytics. 2020;17:571–585. doi: 10.1016/j.omto.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ajina A., Maher J. Prospects for combined use of oncolytic viruses and CAR T-cells. J. Immunother. Cancer. 2017;5:90. doi: 10.1186/s40425-017-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim D.S., Dastidar H., Zhang C., Zemp F.J., Lau K., Ernst M., Rakic A., Sikdar S., Rajwani J., Naumenko V., et al. Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat. Commun. 2017;8:344. doi: 10.1038/s41467-017-00324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scott E.M., Duffy M.R., Freedman J.D., Fisher K.D., Seymour L.W. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol. Biosci. 2018;18:1700187. doi: 10.1002/mabi.201700187. [DOI] [PubMed] [Google Scholar]
- 94.Rezaei R., Esmaeili Gouvarchin Ghaleh H., Farzanehpour M., Dorostkar R., Ranjbar R., Bolandian M., et al. Combination therapy with CAR T cells and oncolytic viruses: a new era in cancer immunotherapy. Cancer Gene Ther. 2021 doi: 10.1038/s41417-021-00359-9. [DOI] [PubMed] [Google Scholar]
- 95.Nishio N., Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4:e988098. doi: 10.4161/21505594.2014.988098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park A.K., Fong Y., Kim S.-I., Yang J., Murad J.P., Lu J., Jeang B., Chang W.-C., Chen N.G., Thomas S.H. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020;12:eaaz1863. doi: 10.1126/scitranslmed.aaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dagher O., King T.R., Wellhausen N., Posey A.D. Combination therapy for solid tumors: taking a classic CAR on new adventures. Cancer Cell. 2020;38:621–623. doi: 10.1016/j.ccell.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 98.Boyd N.R., Tiedemann M., Cartledge K., Cao M., Evtimov V., Shu R., Nguyen T., Nguyen N., Nisbet I., Boyd R., Trounson A. Off-the-shelf ipsc derived car-nk immunotherapy for solid tumors. Cytotherapy. 2020;22:S199. [Google Scholar]
- 99.Hu Y., Tian Z.-g., Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol. Sin. 2018;39:167–176. doi: 10.1038/aps.2017.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 101.Aftab B.T., Sasu B., Krishnamurthy J., Gschweng E., Alcazer V., Depil S. Toward “off-the-shelf” allogeneic CAR T cells. Adv. Cell Gene Ther. 2020;3:e86. [Google Scholar]
- 102.Roddie C., O'Reilly M., Pinto J.D.A., Vispute K., Lowdell M. Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy. 2019;21:327–340. doi: 10.1016/j.jcyt.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 103.Caldwell K.J., Gottschalk S., Talleur A.C. Allogeneic CAR cell therapy-more than a pipe dream. Front. Immunol. 2021;11:618427. doi: 10.3389/fimmu.2020.618427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zeiser R., Blazar B.R. Acute graft-versus-host disease—biologic process, prevention, and therapy. N. Engl. J. Med. 2017;377:2167–2179. doi: 10.1056/NEJMra1609337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Howard C.A., Fernandez-Vina M.A., Appelbaum F.R., Confer D.L., Devine S.M., Horowitz M.M., Mendizabal A., Laport G.G., Pasquini M.C., Spellman S.R. Recommendations for donor human leukocyte antigen assessment and matching for allogeneic stem cell transplantation: consensus opinion of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) Biol. Blood Marrow Transpl. 2015;21:4–7. doi: 10.1016/j.bbmt.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu P., Liu M., Lyu C., Lu W., Cui R., Wang J., Li Q., Mou N., Deng Q., Yang D. Acute graft-versus-host disease after humanized anti-CD19-CAR T therapy in relapsed B-all patients after allogeneic hematopoietic stem cell transplant. Front. Oncol. 2020;10:573822. doi: 10.3389/fonc.2020.573822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Siegler E.L., Zhu Y., Wang P., Yang L. Off-the-Shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell. 2018;23:160–161. doi: 10.1016/j.stem.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 108.Geller M.A., Miller J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy. 2011;3:1445–1459. doi: 10.2217/imt.11.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Daher M., Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr. Opin. Immunol. 2018;51:146–153. doi: 10.1016/j.coi.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bergman H., Sissala N., Hagerstrand H., Lindqvist C. Human NK-92 cells function as target cells for human NK cells - implications for CAR NK-92 therapies. Anticancer Res. 2020;40:5355–5359. doi: 10.21873/anticanres.14543. [DOI] [PubMed] [Google Scholar]
- 111.Geller M.A., Cooley S., Judson P.L., Ghebre R., Carson L.F., Argenta P.A., Jonson A.L., Panoskaltsis-Mortari A., Curtsinger J., McKenna D., et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13:98–107. doi: 10.3109/14653249.2010.515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Froelich W. CAR NK cell therapy directed against pancreatic cancer. Oncol. Times. 2021;43:46. [Google Scholar]
- 113.Li C., Yang N., Li H., Wang Z. Robo1-specific chimeric antigen receptor natural killer cell therapy for pancreatic ductal adenocarcinoma with liver metastasis. J. Cancer Res. Ther. 2020;16:393–396. doi: 10.4103/jcrt.JCRT_190_20. [DOI] [PubMed] [Google Scholar]
- 114.Boyiadzis M., Agha M., Redner R.L., Sehgal A., Im A., Hou J.-Z., Farah R., Dorritie K.A., Raptis A., Lim S.H., et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19:1225–1232. doi: 10.1016/j.jcyt.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 115.Cao B.H., Liu M.T., Wang L., Liang B.X., Feng Y.F., Chen X.P., Shi Y.Y., Zhang J.L., Ye X.D., Tian Y., et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020;524:96–102. doi: 10.1016/j.bbrc.2020.01.053. [DOI] [PubMed] [Google Scholar]
- 116.Li Q., Wang Y., Lin M., Xia L., Bao Y., Sun X., Yang L. Abstract A014: phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunol. Res. 2019;7:A014. [Google Scholar]
- 117.Xia N., Haopeng P., Gong J.U., Lu J., Chen Z., Zheng Y., Wang Z., Sun Y.U., Yang Z., Hoffman R.M., Liu F. Robo1-specific CAR-NK immunotherapy enhances efficacy of (125)I seed brachytherapy in an orthotopic mouse model of human pancreatic carcinoma. Anticancer Res. 2019;39:5919–5925. doi: 10.21873/anticanres.13796. [DOI] [PubMed] [Google Scholar]
- 118.Wrona E., Borowiec M., Potemski P. CAR-NK cells in the treatment of solid tumors. Int. J. Mol. Sci. 2021;22:5899. doi: 10.3390/ijms22115899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hermanson D.L., Bendzick L., Pribyl L., McCullar V., Vogel R.I., Miller J.S., Geller M.A., Kaufman D.S. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells. 2016;34:93–101. doi: 10.1002/stem.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li Y., Hermanson D.L., Moriarity B.S., Kaufman D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–192.e5. doi: 10.1016/j.stem.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Underwood J. Lymphoreticular infiltration in human tumours: prognostic and biological implications: a review. Br. J. Cancer. 1974;30:538. doi: 10.1038/bjc.1974.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tan Y.S., Lei Y.L. Isolation of tumor-infiltrating lymphocytes by Ficoll-Paque density gradient centrifugation. Methods Mol. Biol. 2019;1960:93–99. doi: 10.1007/978-1-4939-9167-9_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nguyen L.T., Saibil S.D., Sotov V., Le M.X., Khoja L., Ghazarian D., Bonilla L., Majeed H., Hogg D., Joshua A.M., et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019;68:773–785. doi: 10.1007/s00262-019-02307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Andersen R., Donia M., Ellebaek E., Borch T.H., Kongsted P., Iversen T.Z., Hölmich L.R., Hendel H.W., Met Ö., Andersen M.H., et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin. Cancer Res. 2016;22:3734–3745. doi: 10.1158/1078-0432.CCR-15-1879. [DOI] [PubMed] [Google Scholar]
- 125.Yang C.J., McSherry F., Mayne N.R., Wang X., Berry M.F., Tong B., Harpole D.H., Jr., D'Amico T.A., Christensen J.D., Ready N.E., Klapper J.A. Surgical outcomes after neoadjuvant chemotherapy and ipilimumab for non-small cell lung cancer. Ann. Thorac. Surg. 2018;105:924–929. doi: 10.1016/j.athoracsur.2017.09.030. [DOI] [PubMed] [Google Scholar]
- 126.Stevanović S., Draper L.M., Langhan M.M., Campbell T.E., Kwong M.L., Wunderlich J.R., Dudley M.E., Yang J.C., Sherry R.M., Kammula U.S., et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015;33:1543–1550. doi: 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sanmamed M.F., Chester C., Melero I., Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016;27:1190–1198. doi: 10.1093/annonc/mdw041. [DOI] [PubMed] [Google Scholar]
- 128.Tran E., Chinnasamy D., Yu Z., Morgan R.A., Lee C.-C.R., Restifo N.P., Rosenberg S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013;210:1125–1135. doi: 10.1084/jem.20130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Siegler E.L., Wang P. Preclinical models in chimeric antigen receptor-engineered T-cell therapy. Hum. Gene Ther. 2018;29:534–546. doi: 10.1089/hum.2017.243. [DOI] [PubMed] [Google Scholar]
- 130.Walsh N.C., Kenney L.L., Jangalwe S., Aryee K.E., Greiner D.L., Brehm M.A., Shultz L.D. Humanized mouse models of clinical disease. Annu. Rev. Pathol. 2017;12:187–215. doi: 10.1146/annurev-pathol-052016-100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Markley J.C., Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508–3519. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Carpenito C., Milone M.C., Hassan R., Simonet J.C., Lakhal M., Suhoski M.M., Varela-Rohena A., Haines K.M., Heitjan D.F., Albelda S.M., et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. U S A. 2009;106:3360. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chmielewski M., Hahn O., Rappl G., Nowak M., Schmidt–Wolf I.H., Hombach A.A., Abken H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology. 2012;143:1095–1107.e2. doi: 10.1053/j.gastro.2012.06.037. [DOI] [PubMed] [Google Scholar]
- 134.Driehuis E., Kretzschmar K., Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020;15:3380–3409. doi: 10.1038/s41596-020-0379-4. [DOI] [PubMed] [Google Scholar]
- 135.Bhimani J., Ball K., Stebbing J. Patient-derived xenograft models-the future of personalised cancer treatment. Br. J. Cancer. 2020;122:601–602. doi: 10.1038/s41416-019-0678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boj S.F., Hwang C.I., Baker L.A., Chio I.I.C., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M.S., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tiriac H., Belleau P., Engle D.D., Plenker D., Deschenes A., Somerville T.D.D., Froeling F.E.M., Burkhart R.A., Denroche R.E., Jang G.H., et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018;8:1112–1129. doi: 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tiriac H., Plenker D., Baker L.A., Tuveson D.A. Organoid models for translational pancreatic cancer research. Curr. Opin. Genet. Dev. 2019;54:7–11. doi: 10.1016/j.gde.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsai S., McOlash L., Palen K., Johnson B., Duris C., Yang Q., Dwinell M.B., Hunt B., Evans D.B., Gershan J., James M.A. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. 2018;18:335. doi: 10.1186/s12885-018-4238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Boj S.F., Hwang C.I., Baker L.A., Engle D.D., Tuveson D.A., Clevers H. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol. Cell Oncol. 2016;3:e1014757. doi: 10.1080/23723556.2015.1014757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ponz-Sarvise M., Corbo V., Tiriac H., Engle D.D., Frese K.K., Oni T.E., Hwang C.I., Ohlund D., Chio I.I.C., Baker L.A., et al. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin. Cancer Res. 2019;25:6742–6755. doi: 10.1158/1078-0432.CCR-19-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Romero-Calvo I., Weber C.R., Ray M., Brown M., Kirby K., Nandi R.K., Long T.M., Sparrow S.M., Ugolkov A., Qiang W., et al. Human organoids share structural and genetic features with primary pancreatic adenocarcinoma tumors. Mol. Cancer Res. 2019;17:70–83. doi: 10.1158/1541-7786.MCR-18-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baker L.A., Tiriac H., Clevers H., Tuveson D.A. Modeling pancreatic cancer with organoids. Trends Cancer. 2016;2:176–190. doi: 10.1016/j.trecan.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aberle M.R., Burkhart R.A., Tiriac H., Damink S., Dejong C.H.C., Tuveson D.A., van Dam R.M. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br. J. Surg. 2018;105:E48–E60. doi: 10.1002/bjs.10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yu L., Li Z., Mei H., Li W., Chen D., Liu L., Zhang Z., Sun Y., Song F., Chen W., Huang W. Patient-derived organoids of bladder cancer recapitulate antigen expression profiles and serve as a personal evaluation model for CAR-T cells in vitro. Clin. Transl. Immunol. 2021;10:e1248. doi: 10.1002/cti2.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Grunewald L., Lam T., Andersch L., Klaus A., Schwiebert S., Winkler A., Gauert A., Heeren-Hagemann A.I., Astrahantseff K., Klironomos F., et al. A reproducible bioprinted 3D tumor model serves as a preselection tool for CAR T cell therapy optimization. Front. Immunol. 2021;12:689697. doi: 10.3389/fimmu.2021.689697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schnalzger T.E., de Groot M.H., Zhang C., Mosa M.H., Michels B.E., Roder J., Darvishi T., Wels W.S., Farin H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019;38:e100928. doi: 10.15252/embj.2018100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sridhar P., Petrocca F. Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers. 2017;9:92. doi: 10.3390/cancers9070092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cho J.H., Collins J.J., Wong W.W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426–1438.e11. doi: 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]