

Abstract
A new and efficient method was developed for the synthesis of C3-substituted sialyl glycals that are useful for novel sialidase inhibitor discovery. This method was based on cross-coupling reactions of 3-iodo-sialyl glycal methyl ester with boronic acids, alkenes and alkynes to directly introduce various functional groups to the sialyl glycal C3-position. A series of C3-aryl, alkyl, alkenyl, and alkynyl derivatives of sialyl glycal were efficiently and conveniently synthesized for the first time by this method, which has demonstrated its wide application scopes.
Graphical Abstract

Sialyl glycal C3-aryl, alkyl, alkenyl, and alkynyl derivatives useful for sialylidase inhibitor discovery were efficiently synthesized by a new method based on cross-coupling of 3-iodo-sialyl glycal with boronic acids, alkenes, and alkynes.
Introduction
Flus caused by influenza viruses remain as one of the most common diseases and a great threat to the public health despite the extensive efforts to develop flu therapeutics and vaccines. Even with hard-driving and widespread vaccination programs, flu epidemics occur every year to result in about 40 million infections, half a million hospitalizations, and over 30 thousand deaths annually in the United States alone,1 and nearly half a million deaths worldwide.2 Therefore, new and effective flu therapeutics are highly demanded.
Inhibition of enzymes involved in virus replication and spread is one of the common strategies for antiviral drug development.3 Influenza viruses bind sialic acid (5-N-acetylneuraminic acid, Neu5NAc) on the host cell surface to assist their entrance into cells, where they replicate.4 Replicated virus is then secreted and released from the host cell. In this process, neuraminidase (also called sialidase) plays a vital role. Viral sialidase catalyses the hydrolysis of the glycosyl bond of sialic acid on host cells to facilitate the release and spread of replicated viruses.5 Thus, inhibition of the sialidase of influenza virus can block its release, spread, and replication cycle to prevent viral infection. Sialidase inhibitors, such as Zanamivir and Laninamivir (Figure 1), have been successfully utilized in clinic for the treatment of influenza flus.6 These inhibitors are sialyl glycal (Neu5Ac2en, Figure 1) derivatives, which mimic the key intermediate involved in sialidase-catalysed sialoside hydrolysis.6–7
Figure 1.

Structures of Zanamivir, Laninamivir and Neu5Ac2en
The success of Zanamivir, Laninamivir, and other related drugs has motivated the quest for more potent sialidase inhibitors and structure-activity relationship analysis of Neu5Ac2en analogs.8 Despite the extensive studies on manipulation of Neu5Ac2en at many positions, reports about C3-modified Neu5Ac2en analogs are few, probably due to the difficulty in modifying this position compared to other positions. On the other hand, structural studies on influenza sialidases disclosed that the binding site of these enzymes has a flexible hydrophobic loop (150-loop) that can be lock-open to interact with substituents on sialic acid C3.9 It was also revealed that C3-substituted Neu5Ac2en derivatives were potent inhibitors of human parainfluenza viruses.10 In addition, C3-allyl derivatives of Neu5Ac2en were not sensitive to enzyme mutations—the cause of drug resistance.9a These results suggest the promise of C3-modified Neu5Ac2en analogs as candidates of next generation anti-influenza drugs.
However, in the literature, there are only a few reports about the synthesis of C3-modified Neu5Ac2en derivatives. Starting with the fully protected Neu5Ac2en 1, the von Itzstein group prepared C3-allyl and azido derivatives 2 and 4, which could be modified further via cross metathesis9a, 11 and click reaction (Schemes 1A and 1B).12 Another method was based on the ring-opening of epoxide 6 (Scheme 1C), which was followed by 1,2-dehydration to give 3-O-substituted Neu5Ac2en derivatives.13 The Ye and Boutureira groups also explored Neu5Ac2en C3-perfluoroalkylation.14 The problems associated with reported synthetic methods are that they are not very efficient, give poor overall yields, afford restricted types of products, and involve dangerous or toxic reagents. Moreover, even for the relatively flexible methods as outlined in Schemes 1A and 1B, the product diversity is based on the allyl and azido groups in 2 and 4; this resulted in synthetic targets 3 and 5 carrying a common spacer between the Neu5Ac2en moiety and R group to not only limit the structural diversity but also influence their activities. To address this issue, we have developed herein a new, expedient, and versatile method for the synthesis of various C3-substituted Neu5Ac2en derivatives.
Scheme 1.

Reported syntheses of C3-substituted sialyl glycals
Results and Discussion
Our plan was to use 3-iodo-sialyl glycal 8 for the synthesis of C3-substituted Neu5Ac2en derivatives 9 (Scheme 2). Vinyl iodides are versatile substrates for many cross-coupling reactions, e.g., Suzuki,15 Heck,16 Stille,17 Sonogashira,18 Buchwald-Hartwig19 and Nozaki-Hiyama-Kishi reactions,20 thus we anticipated that 8 would allow for efficient and direct introduction of a range of functionalities onto the C3-position. Besides C-C bond forming reactions, direct amination and etherification of vinyl iodides are also possible.21 Furthermore, these coupling reactions are compatible with aqueous media,22 enabling the use of basically free glycal 8 in the reaction to directly afford the final synthetic targets. This would make the structural diversification to be the last synthetic step to circumvent global deprotection, rendering the method easily adoptable for high-throughput synthesis of C3-modified Neu5Ac2en derivatives
Scheme 2.

Proposed synthesis of C3-substituted sialyl glycals
Vinyl iodide 8 has not been reported previously. We started its synthesis from sialyl glycal 123 as outlined in Scheme 3. First, 1 was transformed into 1014b according to Vankar’s method24 with minor modifications, e.g., using 0.5 equiv of AgNO3. Although 10 was reported previously,14b it has not been studied for cross-coupling. We subjected 10 to Suzuki and Heck reactions under various conditions but, unfortunately, only a trace amount of the Suzuki coupling product was observed with MS. Next, we removed the O-acetyl groups in 10 with MeONa in MeOH to provide 8 in a nearly quantitative yield. Its coupling reaction with phenylboronic acid under conventional Suzuki-Miyaura conditions under the influence of K2CO3 and palladium tetrakis(triphenylphosphine) (5 mol%) in a 2/1 (v/v) mixture of dioxane and H2O went smoothly, with concomitant methyl ester hydrolysis, to afford 9a in an 82% isolated yield. TLC and MS results showed that the coupling reaction completed within 2 h while the methyl ester saponification was slow and took longer time, suggesting that the C1-ester did not have a major impact on the cross-coupling reaction. In this research, we eventually adopted the elongated time (overnight) for this and subsequent reactions to ensure complete saponification. It was surprising to discover that the acetyl groups in 10 had such a big impact on its reactivity. Since electron-withdrawing groups on the halides usually favour oxidative addition by metal catalysts, which is the initial step of these coupling reactions, we assume that the poor reactivity of 10 was due to steric effects caused by the acetyl groups in its structure.
Scheme 3.

Synthesis of 8 and C3-phenyl-Neu5Ac2en 9a by Suzuki reaction of 8 with phenylboronic acid
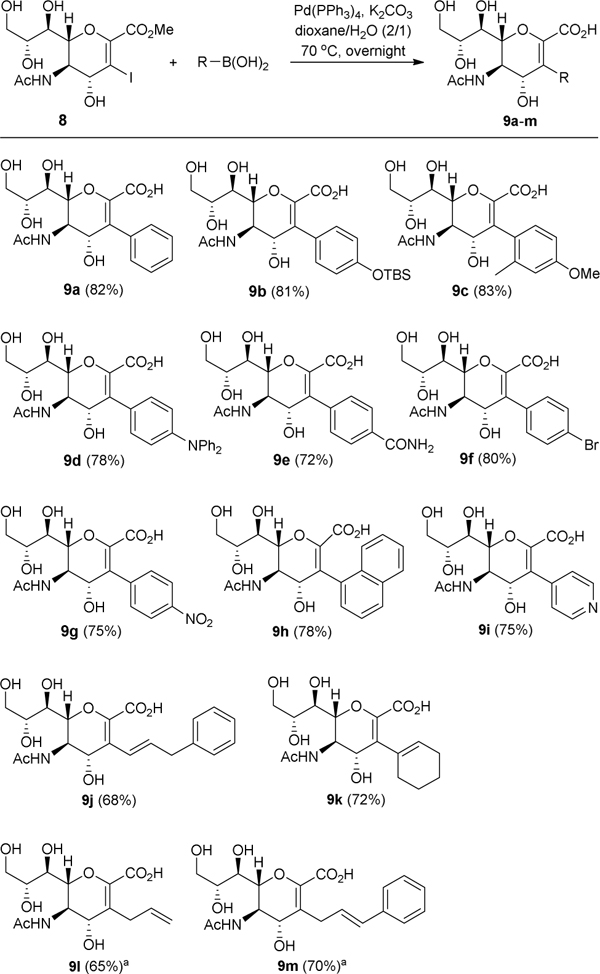
After the successful coupling of 8 and phenylboronic acid under above-established conditions, we explored its application scope employing other boronic acids. As shown in Table 1, differently substituted phenyl, vinyl, and alkyl groups were efficiently attached to the Neu5Ac2en C3-position by this new method to generate desired products 9b-m in very good yields (72–83%). Phenylboronic acid substrates, including those carrying mono-/di-substitutions and electron-donating/withdrawing groups, were examined. All reactions gave similar yields of the products 9b-g. This suggests that neither steric effect nor electronic effect had a significant impact on the reactivity of boronic acid substrates in the coupling reaction. Moreover, besides amido, carboxyl and hydroxyl groups, other functional groups such as silyl ether (9b) and amine (9d) were also compatible with the coupling reaction and its conditions. Boronic acids with fused and heterocyclic aromatic rings were also proved to be good substrates to afford 9h and 9i, respectively. It was noticed that the 1H and 13C NMR spectra of 9h showed a mixture of two rotamers (3:2), which was probably caused by the restricted rotation of naphthalenyl group due to steric hindrances from adjacent hydroxyl and carboxylic groups.
Table 1.
C3-Modified Neu5Ac2en derivatives 9a-m prepared by Suzuki-Miyaura coupling of 8 with boronic acids or borates
|
Boronic pinacolate was used as the alkylating reagent.
Vinylation and alkylation at the Neu5Ac2en C3-position by the same protocol afforded products 9j/k and 9l/m, respectively, in slightly reduced but still very good yields (65–72%). Boronic pinacolates were used for the synthesis of 9l and 9m due to commercial availability. It is worth noting that 9l and 9m were previously reported; from 1, they were prepared in five and six steps and 11% and 3.6% yields (Scheme 1A), respectively.9a, 11a Using the current method, 9l and 9m were derived from the same starting material 1 in only three steps and in 35% and 38% yields. Attempts to couple 8 with simple alkyl boronic acids (e.g., n-heptylboronic acid) under various conditions, including using elevated reaction temperature, more reactive trifluoroborates as substrates and different types of catalysts [palladium Xphos II, PdCl(C3H5)(dppb),25 Pd(dba)2/PPh3/Ag2O26 and PdCl2(dppf)27], were unsuccessful.
To further broaden the product scope derived from 8, we probed its coupling with alkenes (Heck reaction) and alkynes (Sonogashira reaction), which are more readily accessible substrates than the corresponding boronic acid derivatives. As listed in Table 2, three terminal alkenes (styrene, 1-octene, and methyl acrylate) were tested under Heck conditions. The reactions of 8 with styrene using palladium acetate together with triphenylphosphine as the catalyst gave 9n (E isomer only, JCH=CH = 16.3 ppm) in a good yield (85%). Compared to the conditions used for Suzuki coupling, a 4/1 (v/v) mixture of dioxane and H2O was used here to help dissolve nonpolar alkenes; moreover, after the coupling reactions were completed, aqueous NaOH solution was added to the reaction mixture at room temperature to accelerate ester saponification. 1-Octene did not give the desired product under the same condition, thus Ag2CO3 was used in place of K2CO3 as suggested in the literature28 to produce 9o with a long aliphatic chain E-specifically (JCH=CH = 15.7 ppm) in a good yield (67%). The coupling reaction between 8 and methyl acylate was carried out at a lower temperature (50 °C) to avoid potential side reactions and related complications. The subsequent saponification to unmask both carboxylic groups went smoothly to produce 9p (51%). The Sonogashira reaction of 8 and phenylacetylene under similar conditions but using Pd(OAc)2 as the catalyst29 gave 9q in a 78% yield. With three examples of alkene that represent different types of alkene and an alkyne example, we have shown that various alkenes and alkynes may be used for cross-coupling with 8 to further expand the pools of C3-modified Neu5Ac2en derivatives.
Table 2.
Synthesis of C3-modified Neu5Ac2en derivatives 9n-q by Heck and Sonogashira reactions

|
Ag2Co3 was used instead of K2Co3.
The reaction temperature was 50 °C instead of 70 °C.
No triphenylphosphine was added as catalyst ligand.
Conclusions
In summary, we have developed a new and efficient method to synthesize C3-substituted Neu5Ac2en derivatives. This method features Suzuki, Heck and Sonogashira couplings in aqueous media to achieve structural diversification and concomitant hydrolysis of carboxylate to access the synthetic targets from 3-iodo-sialyl glycal 8 in one step. In turn, the key intermediate 8 was obtained from sialyl glycal 1 in two steps on a large scale. This synthetic method proved to be compatible with various functionalities. Therefore, 17 novel sialyl glycal derivatives carrying diverse and representative aryl, alkyl, alkenyl, and alkynyl groups at the C3-position were synthesized by this method to exhibit not only its feasibility and flexibility but also its broad application scopes. Some synthetic targets, e.g., 9b and 9l, can be modified further to accomplish more structural diversities. Compared to the existing approaches to prepare C3-substituted sialyl glycals, the current method has several advantages. For example, it is by far the shortest and the most efficient method to synthesize C3-modified sialyl glycals from sialic acid. It leads to products with much more structural variations and diversities; in fact, most products described herein cannot be prepared by any existing approach. The essentially free, universal substrate 3-iodo-sialyl glycal 8 has enabled the concomitant coupling and carboxylate deprotection to afford final products directly without the tedious and often troublesome final-stage global deprotection. As a result, this synthetic method is especially suitable for the preparation of structurally diverse libraries of sialyl glycal derivatives useful for the discovery of new sialidase inhibitors, which is presently pursued in our laboratory.
Supplementary Material
Acknowledgements
This work is supported by NIH/NIGMS (Grant: 1R35 GM131686). ZG is also grateful to Steven and Rebecca Scott for the endowment. The MS instrument was funded by NIH (S10 OD021758).
Footnotes
Electronic Supplementary Information (ESI) available: Copies of the 1D and 2D NMR spectra of all new compounds. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.Centers for Disease Control and Prevention, Disease Burden of Influenza https://www.cdc.gov/flu/about/burden/index.html, accessed in september, 2021.
- 2.Paget J, Spreeuwenberg P, Charu V, Taylor RJ, Iuliano AD, Bresee J, Simonsen L, and Viboud C, J. Glob. Health 2019, 9, e020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraeusslich H-G and Bartenschlager R Editors, Handb. Exp. Pharmacol, Vol. 189. 2009; pp379. [PubMed] [Google Scholar]
- 4.(a) Gottschalk A, Biochim. Biophys. Acta 1957, 23, 645; [DOI] [PubMed] [Google Scholar]; (b) Klenk E, Faillard H, and Lempfrid H, Hoppe-Seyler’s Z. Physiol. Chem 1955, 301, 235. [PubMed] [Google Scholar]
- 5.(a) Compans RW, Dimmock NJ, and Meier-Ewert H, J. Virol 1969, 4, 528; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Palese P, Tobita K, Ueda M, and Compans RW, Virology 1974, 61, 397; [DOI] [PubMed] [Google Scholar]; (c) Griffin JA, Basak S, and Compans RW, Virology 1983, 125, 324. [DOI] [PubMed] [Google Scholar]
- 6.von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, and Oliver SW, Nature 1993, 363, 418. [DOI] [PubMed] [Google Scholar]
- 7.Moscona A, Engl N J. Med 2005, 353, 1363. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wen W-H, Wang S-Y, Tsai K-C, Cheng Y-SE, Yang A-S, Fang J-M, and Wong C-H, Bioorg. Med. Chem 2010, 18, 4074; [DOI] [PubMed] [Google Scholar]; (b) Das A, Adak AK, Ponnapalli K, Lin C-H, Hsu K-C, Yang J-M, Hsu T-A, and Lin C-C, Eur. J. Med. Chem 2016, 123, 397; [DOI] [PubMed] [Google Scholar]; (c) Cheng LP, Wang TC, Yu R, Li M, and Huang JW, Bioorg. Med. Chem. Lett 2018, 28, 3622; [DOI] [PubMed] [Google Scholar]; (d) Abdul Rahim AS and von Itzstein M, Annu. Rep. Med. Chem 2013, 48, 249; [Google Scholar]; (e) Adabala PJP, LeGresley EB, Bance N, Niikura M, and Pinto BM, J. Org. Chem 2013, 78, 10867; [DOI] [PubMed] [Google Scholar]; (f) Mohan S and Pinto BM, Can. J. Chem 2018, 96, 91; [Google Scholar]; (g) Han N and Mu Y, PLoS One 2013, 8, e73344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Rudrawar S, Dyason JC, Rameix-Welti M-A, Rose FJ, Kerry PS, Russell RJM, van der Werf S, Thomson RJ, Naffakh N, and von Itzstein M, Nat. Commun 2010, 1, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Greenway KT, LeGresley EB, and Mario Pinto B, PLoS One 2013, 8, e59873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guillon P, Dirr L, El-Deeb IM, Winger M, Bailly B, Haselhorst T, Dyason JC, and von Itzstein M, Nat. Commun 2014, 5, 5268. [DOI] [PubMed] [Google Scholar]
- 11.(a) Rudrawar S, Kerry PS, Rameix-Welti M-A, Maggioni A, Dyason JC, Rose FJ, van der Werf S, Thomson RJ, Naffakh N, Russell RJM, and von Itzstein M, Org. Biomol. Chem 2012, 10, 8628; [DOI] [PubMed] [Google Scholar]; (b) Rudrawar S, Pascolutti M, Bhatt B, Thomson RJ, and von Itzstein M, Tetrahedron Lett 2013, 54, 1198; [Google Scholar]; (c) Rudrawar S, Dyason JC, Maggioni A, Thomson RJ, and Itzstein M, Bioorg. Med. Chem 2013, 21, 4820. [DOI] [PubMed] [Google Scholar]
- 12.Pascolutti M, Dirr L, Guillon P, Van Den Bergh A, Ve T, Thomson RJ, and von Itzstein M, ACS Chem. Biol 2018, 13, 1544. [DOI] [PubMed] [Google Scholar]
- 13.Pascolutti M, Madge PD, Thomson RJ, and von Itzstein M, J. Org. Chem 2015, 80, 7746. [DOI] [PubMed] [Google Scholar]
- 14.(a) Wang B, Xiong D-C, and Ye X-S, Org. Lett 2015, 17, 5698; [DOI] [PubMed] [Google Scholar]; (b) Mestre J, Castillon S, and Boutureira O, J. Org. Chem 2019, 84, 15087. [DOI] [PubMed] [Google Scholar]
- 15.(a) Miyaura N, Yamada K, and Suzuki A, Tetrahedron Lett 1979, 3437; [Google Scholar]; (b) Miyaura N and Suzuki A, Chem. Rev 1995, 95, 2457. [Google Scholar]
- 16.Heck RF, Org. React 1982, 27, 345. [Google Scholar]
- 17.Stille JK, Angew. Chem 1986, 98, 504. [Google Scholar]
- 18.Sonogashira K, J. Organomet. Chem 2002, 653, 46. [Google Scholar]
- 19.(a) Paul F, Patt J, and Hartwig JF, J. Am. Chem. Soc 1994, 116, 5969; [Google Scholar]; (b) Guram AS and Buchwald SL, J. Am. Chem. Soc 1994, 116, 7901. [Google Scholar]
- 20.Jin H, Uenishi J, Christ WJ, and Kishi Y, J. Am. Chem. Soc 1986, 108, 5644. [Google Scholar]
- 21.Monnier F and Taillefer M, Angew. Chem., Int. Ed 2009, 48, 6954. [DOI] [PubMed] [Google Scholar]
- 22.(a) Herve G, Sartori G, Enderlin G, MacKenzie G, and Len C, RSC Adv 2014, 4, 18558; [Google Scholar]; (b) Chatterjee A and Ward TR, Catal. Lett 2016, 146, 820. [Google Scholar]
- 23.Ercegovic T and Magnusson G, J. Org. Chem 1995, 60, 3378. [Google Scholar]
- 24.Dharuman S and Vankar YD, Org. Lett 2014, 16, 1172. [DOI] [PubMed] [Google Scholar]
- 25.Fall Y, Doucet H, and Santelli M, Appl. Organomet. Chem 2008, 22, 503. [Google Scholar]
- 26.Chausset-Boissarie L, Ghozati K, LaBine E, Chen JLY, Aggarwal VK, and Crudden CM, Chem. Eur. J 2013, 19, 17698. [DOI] [PubMed] [Google Scholar]
- 27.Sato M, Miyaura N, and Suzuki A, Chem. Lett 1989, 1405. [Google Scholar]
- 28.Jeffery T, J. Chem. Soc., Chem. Commun 1991, 324. [Google Scholar]
- 29.Fan W, Chen Y, Lou Q, Zhuang L, and Yang Y, J. Org. Chem 2018, 83, 6171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.